

Abstract
Parathyroid cancer is a rare endocrine malignancy with only a few thousand cases reported worldwide. As a result, there exists considerable controversy regarding the various aspects of this disease, viz., etiology, diagnosis, and management. We hereby attempt to review the literature on parathyroid carcinoma (PC) and summarize the practices based on the current evidence available. The majority of the PC are sporadic although an association with hyperparathyroidism-jaw tumor syndrome, multiple endocrine neoplasia (MEN) 1 and 2, and isolated familial hyperparathyroidism has been shown. As preoperative diagnosis is challenging, PC should be suspected in patients presenting with a neck mass with signs and symptoms of invasion to surrounding structures. Skeletal and renal symptoms are often associated with PC as presenting complaints. The biochemical parameters are more pronounced in the case of PC compared with benign countpart. Due to its rarity, the American Joint Committee of cancer control (AJCC) acknowledges that as yet a clear distinct staging system to prognosticate the disease would be premature. Complete excision with negative margins at first surgery offers the best chance of cure. The role of radiotherapy (RT) is still unclear; however few series have suggested a better locoregional control with adjuvant RT. Recurrences are common and are most significantly associated with an incomplete clearance at initial surgery. Surgical salvage of recurrent/metastatic disease with medical management of hypercalcemia is the treatment of choice. Large prospective studies and trials need to be conducted to understand the pathology better and improve management protocols; however this is a challenge due to rarity of cases.
Keywords: Endocrine cancers, Hypercalcemia, Parathyroid neoplasms, Parathyroid neoplasms/ diagnosis, Parathyroid neoplasms/ surgery
Introduction
Parathyroid cancers (PC) are one of the rarest cancers, accounting for 0.005% of all cancers and less than 1% of all parathyroid disorders [1, 2]. Only a few thousand cases have been reported in the literature since the first case that was described by De Quervain in 1904 [3]. There is a lack of randomized trials, and large prospective studies make the understanding of these tumors limited resulting in a lack of standard guidelines. The evidence is mainly contributed by retrospective case series and databases, some larger ones from surveillance, epidemiology, and endresult (SEER) and national cancer database (NCDB). Although the majority of the cases are sporadic, its association with genetic syndromes like hyperparathyroidism-jaw tumor syndrome (HPT-JT), multiple endocrine neoplasia-1 and 2A, and familial isolated hyperparathyroidism has also been reported [4, 5]. The overlapping spectrum of clinical and biochemical parameters makes the differentiation between benign and malignant tumors difficult, and diagnosis often relies on postoperative histopathology. Due to its rarity and heterogeneity in the reported cases, the AJCC 8th edition mentions that identification of significant prognostic variables has been challenging and the proposal of a standardized TNM staging system would be premature [6].
Etiology and Pathogenesis
The majority of the PCs are sporadic, and rarely these can arise in hereditary/syndromic setting as well [4, 5]. The etiology of these tumors is largely obscure even after the fact that a century has elapsed from its first description. The literature reports coexisting adenoma and carcinoma, PC arising in cases of primary hyperparathyroidism, and rarely long-standing secondary and tertiary hyperparathyroidism, but evidence to support causal association is lacking [7]. Schantz and Castleman described the histopathological features of malignancy in these tumors and reported that in the study of 67 cases, none arose in the background of benign primary hyperparathyroidism [8]. Verdelli et al. studied the epigenetic alterations in parathyroid cancers and concluded that microRNA alterations and aberrant DNA methylation in PC are different from benign lesions suggesting it to be a distinct entity [9]. Few studies comprising of case reports have suggested that neck irradiation increases the risk of developing PC by inducing malignant transformation in the gland [10, 11].
Rarely when these tumors are caused by genetic factors, a mutation in hyperparathyroidism type 2 (HRPT2) or cell division cycle 73 (CDC73) gene is most common, which was first described in 2002 [12]. It has demonstrated a syndromic association and is also seen in sporadic cases. The HRPT2 gene is the tumor suppressor gene associated with hyperparathyroidism-jaw tumor syndrome HPT-JT [13, 14]. It is an autosomal dominant mutation that increases the lifetime risk of developing PC by 15% [5, 12, 15]. The CDC73 mutation has a much higher occurrence in sporadic PC (as high as 75%) compared with adenoma (< 2%) [16]. About a third of mutations in sporadic cases are germline pointing to the possibility of underlying HPT-JT syndrome. Apart from that, familial isolated hyperparathyroidism (FIHP), MEN 2A, and, rarely, MEN 1 have also been associated with PC [4, 5, 14], and the germline mutations are seen in the CDC73, MEN1 and rearranged during transfection (RET).
Tumor suppressor genes including retinoblastoma and breast carcinoma susceptibility (BRCA2) have also been studied, and there are no somatic mutations to support their direct role however decreased expression could be associated with the carcinogenesis secondarily [17]. Other mutations described are epigenetic modification, viz., DNA methylation and histone modification and microRNA misregulation [16]. The progression of genetic mutations leading to carcinogenesis has been suggested, and patterns of chromosomal imbalance reported are loss of 1p and 13 q which is seen in more than 40% of cases. The gain is seen involving 19p, Xc-q13, 9q33-qter, 1q31-q32, and 16p [18]. The study also showed that loss of 11q13 which was the commonest in adenoma was absent in carcinoma.
Clinical Presentation
The clinical spectrum overlaps with the benign counterpart and it is not uncommon that the diagnosis of the PC comes after surgery on histopathology. Parathyroid cancers usually present in the fourth–fifth decade without the sex predilection which is at variance with parathyroid adenoma which is seen a decade later and with female preponderance [19, 20]. Approximately 90% of the PCs are functionally active and present with signs and symptoms of hypercalcemia [19, 21]. These include neurological symptoms like fatigue, depression, and anxiety; renal manifestations like nephrolithiasis and impaired renal function; skeletal manifestations like bony pains, pathological fractures, and browns tumor; and GI dysfunction like bloating, constipation, or pancreatitis. The severity of hypercalcemia and the associated manifestations are usually more pronounced in PC compared with benign parathyroid disorders [21]. Also, the renal and skeleton involvement is more common in PC, and features of both may be present at the time of clinical presentation which is contrary to adenoma. Non-functional PCs are much less common amounting to just 10% [22]. Due to a lack of the above symptoms, these tumors usually present late and at an advanced stage. They present with locally invasive symptoms like a large palpable mass, hoarseness, and fixation to the adjacent structures. The involvement of surrounding structures in the order of decreasing frequency reported is thyroid, straps, recurrent laryngeal nerve (RLN), esophagus, and trachea [11]. Lymph node involvement may occur in 6–19% and metastasis eventually develop in about 30% of cases [19, 23, 24]. The presence of regional and distant metastasis is pathognomonic of malignancy. The common site of distant metastasis is lung, bone, brain, skin, liver, mediastinum, adrenal gland, and pancreas [6].
Besides, when these tumors arise in the syndromic association, there are other features as well. The HPT-JT has been associated with fibro-osseous lesions of the mandible and maxilla. MEN 2 cases present with medullary thyroid carcinoma and pheochromocytoma in addition to other manifestations like mucosal neuroganglioma, marfanoid habitus, Hirschsprung disease, and cutaneous lichen amyloidosis, whereas MEN 1 may have additional features of pituitary and pancreatic tumors.
Evaluation
Patients presenting with signs and symptoms of hypercalcemia with or without a palpable neck mass should be evaluated for a parathyroid pathology. There are no stringent cut-offs between benign disorders and malignancies of the parathyroid, but PCs usually have more elevated laboratory values [7, 21]. Serum calcium of more than 14 mg/dl and parathormone (PTH) values more than 5 times normal should raise the suspicion of malignancy rather than an adenoma [4]. Alkaline phosphatase values are usually > 300 IU/L with PC [25]. The imaging helps in localizing the disease as well as corroborates the diagnosis of malignancy. It also helps to detect recurrence and metastasis. A combination of ultrasound (USG) neck and Technitium 99 sestamibi or a 4-dimensional computed tomography (CT) scan are the imaging modalities of choice to characterize the lesion as well to localize the tumor (Fig. 1). Studies have shown that the PC tends to be larger, inhomogeneous, hypoechoic, and lobulated masses compared with adenoma on ultrasonography [26]. Another recently published study has shown that PC significantly has a higher incidence of being larger in size, higher depth/width ratio, heterogenous echotexture with cystic changes, irregular in shape with non-circumscribed margins and indistinct border, intraparenchymal microcalcification, suspicious nodes, and local invasion [27, 28]. In cases where local invasion to surrounding structures is suspected based on preliminary examination and investigations, contrast-enhanced computed tomography (CECT) should be done to assess invasiveness [29] (Fig. 2). Imaging for distant metastasis in the form of FDG-18 positron emission tomography (PET) scan is not usually recommended but may be done in cases where there is a strong suspicion of PC with local invasion and high lymph nodal burden. The utility of a PET scan in PC has been reported in a few case series but mostly in the post-operative period to detect recurrences [30–32]. Fine needle aspiration cytology/Biopsy (FNAC/FNAB) is not recommended because it will not alter the management as it cannot differentiate benign from a malignant disorder and carries the additional risk of tumor seeding [33]. However, FNAC of the metastatic node diagnoses the malignancy.
Fig. 1.
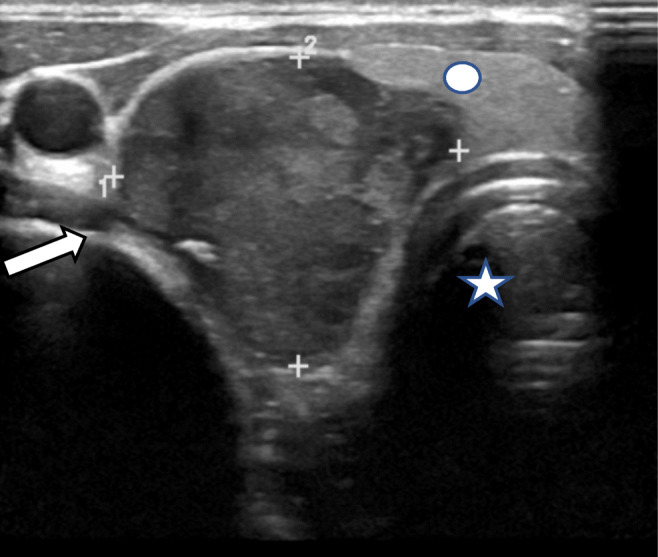
High frequency ultrasound of the neck shows a well-defined hypoechogenic mass (white arrow) arising at the inferior pole of thyroid gland compressing adjacent thyroid parenchyma (Circle).The mass extends into the tracheo-esophageal groove.(Trachea marked with star)
Fig. 2.

Right inferior parathyroid Carcinoma in a 33-year-old male patient. a Axial Contrast Enhanced CT (CECT)image of the neck shows a relatively well defined heterogeneously enhancing lesion(white arrow) abutting the right lobe of thyroid extending into the right tracheoesophageal groove(Star). b Sagittal Reformatted CECT image shows parathyroid lesion (white arrow) with lytic lesion involving vertebral body of 10th Dorsal vertebra (yellow arrow) with collapse and associated soft tissue component, suggestive of Brown’s Tumor
Pathology
Parathyroid carcinoma is a clinical as well as a histopathological quandary. Large size (as compared to the normal weight of Parathyroid glands), and peroperative adherence to adjacent structures are some of the important characteristics of PC. T-size greater than 3 cm in a hypercalcemic patient with hyperparathyroidism is highly suggestive of PC, whereas a size greater than 4 cm is associated with an increased risk of death (Hazard Ratio 1.91, 95% Confidence Interval 1.35–2.69) [34]. In 1973, Schantz and Castleman proposed the criteria for defining parathyroid carcinoma [8] that included fibrous bands arranged in a trabecular design, mitotic activity, capsular invasion, and vascular invasion. The author reported that all the features may not be present in every case; however, several of these features together help in diagnosis, and the presence of mitosis within the parenchyma was the most specific criteria in this study (Fig. 3). The salient histological findings of this series and other large volume series of parathyroid carcinoma published till date are elucidated in Table 1.
Fig. 3.

Histomorphological and immunohistochemical features of parathyroid carcinoma. a Low power view highlights the nodular architecture of parathyroid carcinoma with broad bands of fibrosis separating the nodules of tumor cells. H and E, ×40. b Infiltration of skeletal muscle by parathyroid carcinoma. H and E, ×100. This is one of the diagnostic features of Parathyroid carcinoma; other being Lymphovascular space invasion [(D), Hand E, ×40). c Macronucleoli and tumor cell necrosis (shown by an arrow). H and E, ×200. Immunohistochemistry findings shows that the tumor is diffusely and strongly positive for Chromogranin [(E), DAB, ×100)] and is negative for Thyroid transcription factor 1[(F), DAB, ×100)]
Table 1.
Salient histological features of Parathyroid carcinomas in large volume series published in the literature. NR: Not reported
| Histological criteria | Schantz et al. [8] (1973) | Smith et al. [35] (1984) | Bondeson et al. [36] (1993) | Busaidy et al. [37] (2004) | Quinn et al. [38] (2015) | Asawari et al. [34] (2020) |
|---|---|---|---|---|---|---|
| Fibrous bands/Nodular architecture | 60/67 (90%) | 19/20 (95%) | 45/56(80%) | 12/27 (44%) | 94.4% | 14/20(70%) |
| Capsular invasion | 45/67(67%) | NR | NR | 7/27 (26%) | 100% | 20/20(100%) |
| Invasion of surrounding structures | NR | 10/20 (50%) | NR | 18/27 (66.7%) | 77.8% | 10/20(50%) |
| Angioinvasion | 8/67(12%) | NR | NR | 10/27 (37%) | 66.7% | 19/20(95%) |
| Mitoses | 54/67(81%) | 19/20 (95%) | 38/50 (74%) | 11/27 (40%) | NR | 17/20(85%) |
| Necrosis | NR | NR | 20/56 (36%) | NR | 22.2% | 4/20(20%) |
| Metastasis (locoregional and distant) | 18/67(26.9%) | 4/20(20%) | 21/56 (37%) | 6/27 (22.2%) | 11.8% | 2/16(12%) |
| Others | – | Trabecular (19/20; 95%) | Invasion of thyroid, muscle, large blood vessels (41/56), Trabecular growth(11/56, 20%) | Trabeculae (3/27;11%), Lymphatic invasion (3/27;11) | Macronucleoli (22.2%) | Prominent nucleoli (4/20; 20%) |
Capsular Invasion, Invasion of Surrounding Structures, or Both
The diagnosis of PC, according to the current WHO guidelines, requires infiltration of surrounding structures,or vascular or perineural invasion, and/or documented metastases [40]. Capsular invasion has not been alluded to; however in the WHO 2004 edition, “capsular penetration with growth into adjacent tissue” was required to diagnose PC. The extent of capsular invasion and capsular invasion without infiltration into adjacent structures had not been clarified [41]. The abstruse importance of capsular invasion was addressed by the recent study from our institute which has shown that capsular invasion was present in all cases of PC; infact it was the only feature that was common amongst all 20 cases of PC, thus reiterating the need to document capsular invasion, and perform additional sampling/sectioning to rule out a PC in cases with capsular invasion without invasion of the surrounding tissues [39].
Angioinvasion
This is the single most important criterion to diagnose malignancy in a parathyroid lesion. It has been observed in 70–90% cases of PC [40]. The use of immunohistochemistry markers (CD31, CD34, D2–40) for vascular/lymphatic markers to identify angioinvasion is usually not required, but may be employed in certain cases.
Nodular Growth Pattern
Nodules of varying sizes with intratumoural dense broad fibrotic bands are the characteristic pattern of PCs. Approximately 60–80% cases of PC reported in literature show this pattern making it the most common pattern of growth in PCs [8].
Necrosis and Atypical Mitosis
Necrosis is reported to a varying extent from 5 to 20% cases. Brisk mitosis, though uncommon, does not differentiate parathyroid adenoma from PC. However, the presence of atypical mitosis is highly suggestive of PC [8, 35, 37].
Other important features include macronucleoli and tumor cell spindling. Bondeson et al. suggested that the histologic triad of macronucleoli, > 5 mitoses per 50 high-power fields, and necrosis is associated with aggressive behavior due to recurrent disease, though this system is not widely used [36].
Utility of Frozen Section
As the “malignant nature” of the parathyroid neoplasm is usually unsuspected pre-operatively, an intra-operative frozen section may be sought for confirming/ruling out a diagnosis of malignancy. However, the utility of frozen section has largely met with disrepute due to a reported low accuracy of a diagnosis of PC [42, 43]. This notion has recently been challenged by the results of the study from our institute where 8 out of 9 cases were accurately suspected on frozen section to be PC [39]. The authors conclude that a dedicated oncology center with expertise to treat and diagnose PC coupled with experienced head and neck pathologists significantly impact the accuracy of intra-operative diagnosis.
Immunohistochemistry (IHC)
The use of immunohistochemistry is restricted to certain unusual, ectopic, and difficult parathyroid lesions. Differentiating an unsuspected parathyroid lesion from a thyroid neoplasm is a common conundrum, which can be resolved by the use of neuroendocrine markers like Synaptophysin, Chromogranin, and Parathormone, which will be positive in all parathyroid lesions (benign to malignant), and thyroid transcription factor1(TTF1) and PAX8, which will be negative in parathyroid and positive in thyroid lesions [44] (Fig. 3). A relatively new marker of neuroendocrine differentiation, insulinoma-associated protein 1 (INSM1), is positive in neuroendocrine/neuroectodermal tumors of head and neck, and lung, however, is negative in parathyroid lesions. This may help in diagnosing a parathyroid lesion and differentiating it from other neuroendocrine tumors at an ectopic, unsuspected location, e.g., pleura and mediastinum. The more desired use of immunohistochemistry in parathyroid lesions is to distinguish between parathyroid hyperplasia, parathyroid adenoma, and parathyroid carcinoma. Though no one marker has been found to achieve this distinction, a panel of markers has been employed in various studies which include a combination of bcl-1, Ki-67, and p27 [45]. In addition, parafibromin, a CDC73 protein, the inactivation of which, whether resulting from sporadic or germline mutation, has been identified to drive tumorigenesis in parathyroid. The expression of parafibromin is decreased in PC, and its loss (weak or negative) is associated with 94.4% specificity in diagnosing PC [46]. Despite these promising results in certain studies, the difficulty in standardization, interobserver variability, and the cost of these markers has prevented clinically validated use of these markers.
Molecular Basis
Besides CDC73, CCND1, PRUNE2, PIK3CA, HMT2D, ADCK1, MTOR, THRAP3, and CDKN2C have been identified by next-generation sequencing studies to be putative drivers of parathyroid carcinogenesis [47, 48].
Staging
AJCC 8th edition has incorporated a staging for PC; however, the panel feels that it is premature as the published literature has mainly small retrospective studies and is heterogeneous making the identification of prognostic factors difficult. The rationale behind the proposed staging system is for the uniform capturing of data which will help in refining the staging in the future. The T stage for PC is based on the extension of disease beyond the parathyroid capsule and invasion of the surrounding structures irrespective of the size. Similarly, the N stage is based on the level of involvement with no size criteria. Metastasis is based on the absence or presence as in all other head and neck subsites. There is no prognostic stage grouping proposed due to the lack of robust data [6].
Management
Surgery
It is prudent to involve a multidisciplinary team in the management of these rare tumors for the optimal outcomes [49]. Surgery is the mainstay of treatment for parathyroid neoplasm; however the extent of resection is more radical in PC compared with adenoma. It is at times difficult to differentiate between carcinoma and adenoma preoperatively. The diagnosis comes as a histopathological surprise postoperatively in such cases. Therefore, the surgeon should be vigilant to identify the sinister features of malignancy intraoperatively to perform adequate surgery at the time of initial management. Carcinoma is usually bigger, irregular whitish mass, with a fibrous capsule that may adhere or infiltrate the surrounding fibrofatty tissue or thyroid lobe [34, 50]. The presence of metastatic nodes is pathognomonic of malignancy.
There is a lack of consensus regarding the extent of surgery for PC. Sandelin et al. in a study reported the outcomes of 95 cases of PC in which 42 patients underwent tumor resection and 40 tumor resections plus partial or total thyroidectomy. The results of the study showed that patients with extensive surgery had longer survival and longer relapse-free interval after adjusting for other factors [51]. According to the NCDB database, survival outcomes were better in patients who underwent complete excision as compared with those with a incomplete or piecemeal resection [19]. Contrary to this, Young et al. in their study analyzed the impact of the extent of surgery on survival in 136 patients. Sixty patients underwent parathyroidectomy alone, 58 en bloc resection, and 18 parathyroidectomy followed by delayed thyroid resection. The authors reported that the overall survival rates did not differ with the extent of surgery [52]. Similarly, Harari et al. analyzed the survival outcomes in 37 cases of PC and reported that the extent of surgical resection was not associated with mortality. Inadequate surgery may lead to disease recurrence, and re-surgery is associated with high rates of complications [23]. The important point borne out by the study was that initial surgery performed at a dedicated center prolongs survival and decreases the complication rates. Therefore, the initial resection should be en bloc resection targeting complete removal of disease with gross and microscopic negative margins. Caution should be exercised not to rupture the tumor capsule to avoid tumor spillage and seeding of the surgical field. The surrounding structures like strap muscles if involved by the disease should be sacrificed. The incidence of central compartment metastasis is reported to be between 3 and 19% of cases [24]. Prophylactic neck dissection is not warranted due to the low incidence of regional metastasis. Regional lymph node dissection should be performed if metastasis is suspected clinicoradiologically or intraoperatively.
Parathyroid carcinoma may come as a histopathological surprise and further management in this scenario becomes complex. In such instances, a decision regarding observation with close surveillance versus a re-operation is made based on pathological characteristics, serum calcium, and PTH levels. A patient who is normocalcemic with PTH levels within the normal range, in whom the diagnosis was made under the microscopic demonstration of invasion and with no other high-risk features, can be kept under surveillance and close follow-up with serial ultrasound and serum PTH levels [7, 53]. On the contrary, symptomatic patients with hypercalcemia, elevated serum PTH, and histopathology showing extensive vascular/capsular invasion should prompt the surgeon to evaluate thoroughly with appropriate imaging to localize the disease and remove locoregional disease completely along with involved structures to obtain cure [7].
Unlike parathyroid adenoma or multiglandular hyperplasia, where intraoperative rapid PTH (IOPTH) assay monitoring helps ascertain resection of the diseased glands, it has limited utility in PC. If available, it should be used as a significant fall in the value within the normal range, which is a reassuring sign of complete disease clearance. An inadequate fall in the value could be due to many reasons including incomplete resection, coexistent adenomas in other glands, or metastatic disease. Decision-making in such cases then becomes difficult as unguided 4 gland exploration might not improve outcome but will increase the morbidity [54].
Post-operative refractory and prolonged hypocalcemia have often been observed in patients of PC. Post-surgical correction of hyperparathyroidism causes rapid mineralization of bone, resulting in the redistribution of calcium from the blood to bone thus causing hypocalcemia, a phenomenon known as Hungry bone syndrome. The more severe the bone disease before surgery, the more severe the hypocalcemia post-surgery. Therefore, the serum calcium should be closely monitored in these patients and appropriately supplemented. The requirement goes down as the remaining parathyroids take over the function and the bones are remineralized.
Role of Adjuvant Radiotherapy
Recurrence can occur in more than 50% of cases of PC after surgery [4]. While the locoregional recurrence can be salvaged by re-surgery, at times reported as high as 9 procedures in a study, it is associated with higher morbidity [51]. Therefore, investigators tried to assess the benefit of adjuvant treatment in these patients to prevent locoregional recurrences. The role of adjuvant radiotherapy however remains controversial in the management of these tumors [55–57]. Munson et al. analyzed the risk factors for locoregional disease recurrence and the role of adjuvant RT in preventing it. The authors reported that the factors determining the recurrence are the margin status and the institution of the first surgery. The study concluded that adjuvant radiotherapy may lower the risk of locoregional recurrence and improve cause-specific survival [55]. A recently published NCDB study of 885 patients failed to show survival improvement with adjuvant radiotherapy. The authors also pointed out that about 10.5% of the patients with complete clearance of disease at surgery received adjuvant RT with no OS benefit [56]. However, what can be stated in the lack of consensus is that the adjuvant radiotherapy has some role in preventing locoregional recurrence and in recurrent cases. It should be considered in patients at high risk of such recurrences as positive margins, regional metastasis, and a gross invasion of surrounding structures.
Recurrent and Metastatic Disease
Most of the recurrences are locoregional and occurring within 2–3 years of initial surgery, although relapse as late as 23 years later has also been reported [4, 7]. Patients usually present with gradually increasing levels of PTH and serum calcium. The treatment of choice for recurrent disease is control of hypercalcemia followed by surgical re-excision after appropriate localization of the disease. Surgery entails the removal of all functioning tumor tissue with wide margins [29]. Reoperation in this area can be challenging because of scar tissue and loss of anatomical planes. Thus, the morbidity reported in redo surgery is much higher with an approximately 38% incidence of RLN palsy through the course of all surgeries [23, 58]. Intraoperative localization using methylene blue, a dye that is selectively taken up by the parathyroid tissue, or technetium 99 and a gamma probe has been described, but their advantage to an experienced parathyroid surgeon in PC is still doubtful [29, 59].
Approximately half of the patients with recurrence also have distant metastasis [60]. Even though metastatic, all resectable diseases should be removed surgically as this enables better control and medical management of hypercalcemia which is the most important cause of death in these patients [29]. The medical management of hypercalcemia in the form of intravenous hydration, loop diuretics, bisphosphonates, and calcium-sensing receptor agonists, i.e., cinacalcet, is an important aspect of treatment. In the case of unresectable disease, image-guided percutaneous ethanol ablation is an effective form of palliation as it improves hypercalcemia [61].
Although there is no role of chemotherapy in a curative setting, however, the limited role has been explored to provide palliation. There are small case reports that show anecdotal benefits of chemotherapy in recurrent and metastatic cases. There are no standard regimes, and various drugs including dacarbazine have been tried either alone or in combination showing some benefit [62, 63].
Prognosis and Outcomes
PC is a slow but progressive disease with a variable prognosis. There is no standardized validated prognostication system to date as the outcomes do not correlate with tumor size and nodal metastasis consistently. The median overall survival is 14.3 years but ranges from around 85% at 5 years to 35–79% at 10 years [19, 23, 64]. The most important factor determining outcomes is the completeness of resection at initial surgery. En bloc resection results in 8% local recurrence and 89% long-term survival compared with around 51% and 53%, respectively, with initial incomplete surgery [11]. Other negative prognostic factors include lymph node and distant metastasis at diagnosis, and non-functioning tumors [11]. In a large population-level analysis of the NCDB database of 1022 PC patients by Sadler et al., the 5-year OS was 81.1% and in multivariate analysis, positive lymph nodes and older age were significantly associated with poor OS [65].
Conclusion
Parathyroid carcinoma is a rare endocrine malignancy. Preoperative diagnosis is challenging but should be suspected in a neck mass with signs and symptoms suggestive of local invasion along with hypercalcemia. Complete surgical excision at the initial surgery offers the best possible chance of cure. The role of adjuvant treatment in the form of radiation and chemotherapy is not well established. Recurrence is common and surgical salvage of recurrent/metastatic disease along with medical management of hypercalcemia is the treatment of choice. Although challenging due to the given rarity of disease, prospective studies and multicentric trials need to be designed to establish management protocols and improve the outcomes.
Declarations
Conflict of Interest
No conflict of interest.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Lee PK, Jarosek SL, Virnig BA, Evasovich M, Tuttle TM. Trends in the incidence and treatment of parathyroid cancer in the United States. Cancer. 2007;109:1736–1741. doi: 10.1002/cncr.22599. [DOI] [PubMed] [Google Scholar]
- 2.Hakaim AG, Esselstyn CB., Jr Parathyroid carcinoma: 50-year experience at the Cleveland Clinic Foundation. Cleve Clin J Med. 1993;60:331–335. doi: 10.3949/ccjm.60.4.331. [DOI] [PubMed] [Google Scholar]
- 3.De Quervain F. Parastruma maligna aberrata [in German] Deutsche Zeitschrift fur Chirurgie. 1909;100:334–353. [Google Scholar]
- 4.Kebebew E. Parathyroid carcinoma. Curr Treat Options in Oncol. 2001;2(4):347–354. doi: 10.1007/s11864-001-0028-2. [DOI] [PubMed] [Google Scholar]
- 5.Sharretts JM, Simonds WF. Clinical and molecular genetics of parathyroid neoplasms. Best Pract Res Clin Endocrinol Metab. 2010;24(3):491–502. doi: 10.1016/j.beem.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Amin MB, Greene FL, Edge SB, Compton CC, Gershenwald JE, Brookland RK, Meyer L, Gress DM, Byrd DR, Winchester DP. The eighth edition AJCC Cancer staging manual: continuing to build a bridge from a population-based to a more “personalized” approach to cancer staging. CA Cancer J Clin. 2017;67(2):93–99. doi: 10.3322/caac.21388. [DOI] [PubMed] [Google Scholar]
- 7.Shane E. Clinical review 122: parathyroid carcinoma. J Clin Endocrinol Metab. 2001;86:485–493. doi: 10.1210/jcem.86.2.7207. [DOI] [PubMed] [Google Scholar]
- 8.Schantz A, Castleman B. Parathyroid carcinoma. A study of 70 cases. Cancer. 1973;31(3):600–605. doi: 10.1002/1097-0142(197303)31:3<600::aid-cncr2820310316>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 9.Verdelli C, Corbetta S. Epigenetic alterations in parathyroid cancers. Int J Mol Sci. 2017;18(2):310. doi: 10.3390/ijms18020310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Obara T, Fujimoto Y. Diagnosis and treatment of patients with parathyroid carcinoma: an update and review. World J Surg. 1991;15(6):738–744. doi: 10.1007/BF01665308. [DOI] [PubMed] [Google Scholar]
- 11.Koea JB, Shaw JH. Parathyroid cancer: biology and management. Surg Oncol. 1999;8(3):155–165. doi: 10.1016/s0960-7404(99)00037-7. [DOI] [PubMed] [Google Scholar]
- 12.Newey PJ, Bowl MR, Cranston T, Thakker RV. Cell division cycle protein 73 homolog (CDC73) mutations in the hyperparathyroidism-jaw tumor syndrome (HPT-JT) and parathyroid tumors. Hum Mutat. 2010;31(3):295–307. doi: 10.1002/humu.21188. [DOI] [PubMed] [Google Scholar]
- 13.Haven CJ, van Puijenbroek M, Tan MH, Teh BT, Fleuren GJ, van Wezel T, Morreau H. Identification of MEN1 and HRPT2 somatic mutations in paraffin-embedded (sporadic) parathyroid carcinomas. Clin Endocrinol. 2007;67:370–376. doi: 10.1111/j.1365-2265.2007.02894.x. [DOI] [PubMed] [Google Scholar]
- 14.Shattuck TM, Valimaki S, Obara T, et al. Somatic and germ-line mutations of the HRPT2 gene in sporadic parathyroid carcinoma. N Engl J Med. 2003;349:1722–1729. doi: 10.1056/NEJMoa031237. [DOI] [PubMed] [Google Scholar]
- 15.Gill AJ. Understanding the genetic basis of parathyroid carcinoma. Endocr Pathol. 2014;25(1):30–34. doi: 10.1007/s12022-013-9294-3. [DOI] [PubMed] [Google Scholar]
- 16.Cardoso L, Stevenson M, Thakker RV. Molecular genetics of syndromic and non-syndromic forms of parathyroid carcinoma. Hum Mutat. 2017;38(12):1621–1648. doi: 10.1002/humu.23337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shattuck TM, Kim TS, Costa J, Yandell DW, Imanishi Y, Palanisamy N, Gaz RD, Shoback D, Clark OH, Monchik JM, Wierman ME, Hollenberg A, Tojo K, Chaganti RSK, Arnold A. Mutational analyses ofandas candidate tumour suppressor genes in parathyroid carcinoma. Clin Endocrinol. 2003;59:180–189. doi: 10.1046/j.1365-2265.2003.01814.x. [DOI] [PubMed] [Google Scholar]
- 18.Kytola S, Farnebo F, Obara T, et al. Patterns of chromosomal imbalances in parathyroid carcinomas. Am J Pathol. 2000;157:579–586. doi: 10.1016/S0002-9440(10)64568-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hundahl SA, Fleming ID, Fremgen AM, Menck HR. Two hundred eighty-six cases of parathyroid carcinoma treated in the U.S between 1985-1995: a National Cancer Data Base Report. The American College of Surgeons Commission on Cancer and the American Cancer Society. Cancer. 1999;86(3):538–544. doi: 10.1002/(sici)1097-0142(19990801)86:3<538::aid-cncr25>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- 20.Rahbari R, Kebebew E. Parathyroid tumors. In: DeVita VT Jr, Lawrence TS, Rosenberg SA, editors. Cancer: principles and practice of oncology. Philadelphia: Wolters Kluwer Health/Lippincott Williams & Wilkins; 2011. [Google Scholar]
- 21.Levin KE, Galante M, Clark OH. Parathyroid carcinoma versus parathyroid adenoma in patients with profound hypercalcemia. Surgery. 1987;101:649–660. [PubMed] [Google Scholar]
- 22.Wilkins BJ, Lewis JS., Jr Non-functional parathyroid carcinoma: a review of the literature and report of a case requiring extensive surgery. Head Neck Pathol. 2009;3:140–149. doi: 10.1007/s12105-009-0115-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Harari A, Waring A, Fernandez-Ranvier G, Hwang J, Suh I, Mitmaker E, Shen W, Gosnell J, Duh QY, Clark O. Parathyroid carcinoma: a 43-year outcome and survival analysis. J Clin Endocrinol Metab. 2011;96:3679–3686. doi: 10.1210/jc.2011-1571. [DOI] [PubMed] [Google Scholar]
- 24.McClenaghan F, Qureshi YA. Parathyroid cancer. Gland Surg. 2015;4(4):329–338. doi: 10.3978/j.issn.2227-684X.2015.05.09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bae JH, Choi HJ, Lee Y, Moon MK, Park YJ, Shin CS, Park DJ, Jang HC, Kim SY, Kim SW. Preoperative predictive factors for parathyroid carcinoma in patients with primary hyperparathyroidism. J Korean Med Sci. 2012;27(8):890–895. doi: 10.3346/jkms.2012.27.8.890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hara H, Igarashi A, Yano Y, Yashiro T, Ueno E, Aiyoshi Y, Ito K, Obara T. Ultrasonographic features of parathyroid carcinoma. Endocr J. 2001;48(2):213–217. doi: 10.1507/endocrj.48.213. [DOI] [PubMed] [Google Scholar]
- 27.Liu J, Zhan WW, Zhou JQ, Zhou W (2020) Role of ultrasound in the differentiation of parathyroid carcinoma and benign parathyroid lesions. Clin Radiol 75(3):179-184. 10.1016/j.crad.2019.10.004 [DOI] [PubMed]
- 28.Nam M, Jeong HS, Shin JH. Differentiation of parathyroid carcinoma and adenoma by preoperative ultrasonography. Acta Radiol. 2017;58(6):670–675. doi: 10.1177/0284185116666418. [DOI] [PubMed] [Google Scholar]
- 29.Wei CH, Harari A. Parathyroid carcinoma: update and guidelines for management. Curr Treat Options in Oncol. 2012;13(1):11–23. doi: 10.1007/s11864-011-0171-3. [DOI] [PubMed] [Google Scholar]
- 30.Evangelista L, Sorgato N, Torresan F, Boschin IM, Pennelli G, Saladini G, Piotto A, Rubello D, Pelizzo MR. FDG-PET/CT and parathyroid carcinoma: review of literature and illustrative case series. World J Clin Oncol. 2011;2(10):348–354. doi: 10.5306/wjco.v2.i10.348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Neumann DR, MacIntyre WJ, Esselstyn CB, Kohse LM, Siciliano D, Licata AA. Preoperative imaging of parathyroid carcinoma by positron emission tomography. Ann Otol Rhinol Laryngol. 1994;103(9):741–745. doi: 10.1177/000348949410300916. [DOI] [PubMed] [Google Scholar]
- 32.Arslan N, Rydzewski B. Detection of a recurrent parathyroid carcinoma with FDG positron emission tomography. Clin Nucl Med. 2002;27(3):221–222. doi: 10.1097/00003072-200203000-00022. [DOI] [PubMed] [Google Scholar]
- 33.Agarwal G, Dhingra S, Mishra SK, Krishnani N. Implantation of parathyroid carcinoma along fine needle aspiration track. Langenbeck's Arch Surg. 2006;391(6):623–626. doi: 10.1007/s00423-006-0095-8. [DOI] [PubMed] [Google Scholar]
- 34.Sali AP, Motghare P, Bal M, Mittal N, Rane S, Kane S, Patil A. Parathyroid Carcinoma: A Single-Institution Experience with an Emphasis on Histopathological Features. Head and Neck Pathology. 2021;15:544–554. doi: 10.1007/s12105-020-01244-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Smith JF, Coombs RR. Histological diagnosis of carcinoma of the parathyroid gland. J Clin Pathol. 1984;37:1370–1378. doi: 10.1136/jcp.37.12.1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bondeson L, Sandelin K, Grimelius L. Histopathological variables and DNA cytometry in parathyroid carcinoma. Am J Surg Pathol. 1993;17:820–829. doi: 10.1097/00000478-199308000-00007. [DOI] [PubMed] [Google Scholar]
- 37.Busaidy NL, Jimenez C, Habra MA, Schultz PN, El-Naggar AK, Clayman GL, et al. Parathyroid carcinoma: a 22-year experience. Head Neck. 2004;26:716–726. doi: 10.1002/hed.20049. [DOI] [PubMed] [Google Scholar]
- 38.Quinn CE, Healy J, Lebastchi AH, Brown TC, Stein JE, Prasad ML, Callender GG, Carling T, Udelsman R. Modern experience with aggressive parathyroid tumors in a high-volume New England referral center. J Am Coll Surg. 2015;220:1054–1062. doi: 10.1016/j.jamcollsurg.2014.10.007. [DOI] [PubMed] [Google Scholar]
- 39.Sali AP, Motghare P, Bal M, Mittal N, Rane S, Kane S, Patil A (2020) Parathyroid carcinoma: a single-institution experience with an emphasis on histopathological features. Head Neck Pathol [DOI] [PMC free article] [PubMed]
- 40.DeLellis R, Arnold A, Bilezikian J, Eng C, Larsson C, Llyod RV, et al. Parathyroid carcinoma. In: Llyod RV, Osamura RY, Kloppel G, Rosai J, et al., editors. WHO classification of tumours of endocrine organs. 4. Lyon: IARC; 2017. pp. 147–152. [Google Scholar]
- 41.Bondeson L, Grimelius L, DeLellis RA, Lloyd R, Akerstrom G, Larsson C, et al. Parathyroid carcinoma. In: DeLellis RA, Llyod RV, Heitz PU, Eng C, et al., editors. Pathology and genetics of tumours of endocrine organs. Lyon: IARC; 2004. pp. 124–127. [Google Scholar]
- 42.Fingeret AL (2021) Contemporary Evaluation and Management of Parathyroid Carcinoma. JCO Oncol Pract 17(1):17-21.10.1200/JOP.19.00540 [DOI] [PubMed]
- 43.Wang P, Xue S, Wang S, Lv Z, Meng X, Wang G, Meng W, Liu J, Chen G. Clinical characteristics and treatment outcomes of parathyroid carcinoma: a retrospective review of 234 cases. Oncol Lett. 2017;14:7276–7282. doi: 10.3892/ol.2017.7076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Erickson LA, Mete O. Immunohistochemistry in Diagnostic Parathyroid Pathology. Endocr Pathol. 2018;29:113–129. doi: 10.1007/s12022-018-9527-6. [DOI] [PubMed] [Google Scholar]
- 45.Stojadinovic A, Hoos A, Nissan A, Dudas ME, Cordon-Cardo C, Shaha AR, Brennan MF, Singh B, Ghossein RA. Parathyroid neoplasms: clinical, histopathological, and tissue microarray-based molecular analysis. Hum Pathol. 2003;34(1):54–64. doi: 10.1053/hupa.2003.55. [DOI] [PubMed] [Google Scholar]
- 46.Kim HK, Oh YL, Kim SH, Lee DY, Kang HC, Lee JI, Jang HW, Hur KY, Kim JH, Min YK, Chung JH, Kim SW. Parafibromin immunohistochemical staining to differentiate parathyroid carcinoma from parathyroid adenoma. Head Neck. 2012;34(2):201–206. doi: 10.1002/hed.21716. [DOI] [PubMed] [Google Scholar]
- 47.Pandya C, Uzilov AV, Bellizzi J, et al. Genomic profiling reveals mutational landscape in parathyroid carcinomas. JCI Insight. 2017;2(6):61–65. doi: 10.1172/jci.insight.92061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kasaian K, Wiseman SM, Thiessen N, Mungall KL, Corbett RD, Qian JQ, Nip KM, He A, Tse K, Chuah E, Varhol RJ, Pandoh P, McDonald H, Zeng T, Tam A, Schein J, Birol I, Mungall AJ, Moore RA, Zhao Y, Hirst M, Marra MA, Walker BA, Jones SJM. Complete genomic landscape of a recurring sporadic parathyroid carcinoma. J Pathol. 2013;230(3):249–260. doi: 10.1002/path.4203. [DOI] [PubMed] [Google Scholar]
- 49.Clayman GL, Gonzalez HE, el-Naggar A, Vassilopoulou-Sellin R. Parathyroid carcinoma: evaluation and interdisciplinary management. Cancer. 2004;100(5):900–905. doi: 10.1002/cncr.20089. [DOI] [PubMed] [Google Scholar]
- 50.Clark O. Parathyroid carcinoma. In: Doherty GM, Way L, editors. Current surgical diagnosis and treatment. Michigan: McGraw-Hill Medical; 2006. [Google Scholar]
- 51.Sandelin K, Auer G, Bondeson L, Grimelius L, Farnebo LO. Prognostic factors in parathyroid cancer: a review of 95 cases. World J Surg. 1992;16(4):724–731. doi: 10.1007/BF02067369. [DOI] [PubMed] [Google Scholar]
- 52.Young S, Wu JX, Li N et al More extensive surgery may not improve survival over parathyroidectomy alone in parathyroid carcinoma. Ann Surg Oncol 23:2898–2904 [DOI] [PubMed]
- 53.Fujimoto Y, Obara T, Ito Y, Kodama T, Nobori M, Ebihara S. Localisation and surgical resection of metastatic parathyroid carcinoma. World J Surg. 1986;10:539–547. doi: 10.1007/BF01655520. [DOI] [PubMed] [Google Scholar]
- 54.Givi B, Shah JP. Parathyroid carcinoma. Clin Oncol (R Coll Radiol) 2010;22(6):498–507. doi: 10.1016/j.clon.2010.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Munson ND, Foote RL, Northcutt RC, Tiegs RD, Fitzpatrick LA, Grant CS, van Heerden JA, Thompson GB, Lloyd RV. Parathyroid carcinoma: is there a role for adjuvant radiation therapy? Cancer. 2003;98(11):2378–2384. doi: 10.1002/cncr.11819. [DOI] [PubMed] [Google Scholar]
- 56.Limberg J, Stefanova D, Ullmann TM, Thiesmeyer JW, Bains S, Beninato T, Zarnegar R, Fahey TJ, 3rd, Finnerty BM. The use and benefit of adjuvant radiotherapy in parathyroid carcinoma: a National Cancer Database Analysis. Ann Surg Oncol. 2020;13:1–10. doi: 10.1245/s10434-020-08825-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Asare EA, Sturgeon C, Winchester DJ, Liu L, Palis B, Perrier ND, Evans DB, Winchester DP, Wang TS. Parathyroid Carcinoma: An Update on Treatment Outcomes and Prognostic Factors from the National Cancer Data Base (NCDB) Ann Surg Oncol. 2015;22:3990–3995. doi: 10.1245/s10434-015-4672-3. [DOI] [PubMed] [Google Scholar]
- 58.Kebebew E, Arici C, Duh QY, Clark OH. Localization and reoperation results for persistent and recurrent parathyroid carcinoma. Arch Surg. 2001;136(8):878–885. doi: 10.1001/archsurg.136.8.878. [DOI] [PubMed] [Google Scholar]
- 59.Placzkowski K, Christian R, Chen H. Radioguided parathyroidectomy for recurrent parathyroid cancer. Clin Nucl Med. 2007;32(5):358–360. doi: 10.1097/01.rlu.0000259623.79805.2d. [DOI] [PubMed] [Google Scholar]
- 60.Sandelin K, Tullgren O, Farnebo LO. Clinical course of metastatic parathyroid cancer. World J Surg. 1994;18(4):594–598. doi: 10.1007/BF00353773. [DOI] [PubMed] [Google Scholar]
- 61.Montenegro FL, Chammas MC, Juliano AG, et al. Ethanol injection under ultrasound guidance to palliate unresectable parathyroid carcinoma. Arq Bras Endocrinol Metabol. 2008;52:707–711. doi: 10.1590/s0004-27302008000400019. [DOI] [PubMed] [Google Scholar]
- 62.Chahinian AP. Chemotherapy for meta- static parathyroid carcinoma. Arch Intern Med. 1984;144(9):1889. [PubMed] [Google Scholar]
- 63.Bukowski RM, Sheeler L, Cunningham J, Esselstyn C. Successful combination chemotherapy for metastatic parathyroid carcinoma. Arch Intern Med. 1984;144(2):399–400. [PubMed] [Google Scholar]
- 64.Mohebati A, Shaha A, Shah J. Parathyroid Carcinoma. Hematology/Oncology Clinics of North America. 2012;26:1221–1238. doi: 10.1016/j.hoc.2012.08.009. [DOI] [PubMed] [Google Scholar]
- 65.Sadler C, Gow KW, Beierle EA, Doski JJ, Langer M, Nuchtern JG, Vasudevan SA, Goldfarb M. Parathyroid carcinoma in more than 1,000 patients: a population-level analysis. Surgery. 2014;156(6):1622–1629. doi: 10.1016/j.surg.2014.08.069. [DOI] [PubMed] [Google Scholar]