

Abstract
Analyses of genetic diversity can shed light on both the origins of biodiversity hotspots, as well as the conservation status of species that are impacted by human activities. With these objectives, we assembled a genomic dataset of 14,935 single nucleotide polymorphisms from 513 grey reef sharks (Carcharhinus amblyrhynchos) sampled across 17 locations in the tropical Indo-Pacific. We analysed geographic variation in genetic diversity, estimated ancient and contemporary effective population size (Ne) across sampling locations (using coalescent and linkage disequilibrium methods) and modelled the history of gene flow between the Coral Triangle and the Coral Sea. Genetic diversity decreased with distance away from the Coral Triangle and north-western Australia, implying that C. amblyrhynchos may have originated in this region. Increases in Ne were detected across almost all sampling locations 40,000–90,000 generations ago (approximately 0.6–1.5 mya, given an estimated generation time of 16.4 years), suggesting a range expansion around this time. More recent, secondary increases in Ne were inferred for the Misool and North Great Barrier Reef sampling locations, but joint modelling did not clarify whether these were due to population growth, migration, or both. Despite the greater genetic diversity and ancient Ne observed at sites around Australia and the Coral Triangle, remote reefs around north-western New Caledonia had the highest contemporary Ne, demonstrating the importance of using multiple population size assessment methods. This study provides insight into both the past and present demographics of C. amblyrhynchos and contributes to our understanding of evolution in marine biodiversity hotspots.
Subject terms: Population genetics, Population genetics
Introduction
The Coral Triangle (sensu Carpenter et al. 2011) and the Great Barrier Reef (GBR) are among the most species-rich marine regions in the world (McWilliam et al. 2018; Mouillot et al. 2014; Renema et al. 2008). The species assemblages of such biodiversity hotspots have been compared to less diverse regions to infer how processes like continental drift and climatic fluctuations have generated and maintained biodiversity (Keith et al. 2013; Leprieur et al. 2016; Pellissier et al. 2014; Renema et al. 2008). Yet, biodiversity begins at the population level: patterns of genetic diversity within and amongst populations are linked to their demographic histories (Fang et al. 2021, 2020), which are largely shaped by past geological and climatic events (Kotlík et al. 2018; Wei et al. 2020). The geographic distribution of genetic diversity—i.e., the level of biodiversity at which evolution takes place—can therefore shed light on the generation and maintenance of diverse populations in biodiversity hotspots.
Part of the Arafura Shelf connecting Australia and New Guinea (Fig. 1a) most recently re-submerged only ~7000 years ago to form the Torres Strait (Woodroffe et al. 2000). This reconnected the GBR (in the Pacific Ocean) with the western Coral Triangle and north-western Australia (in the Indian Ocean) for the first time in over 100,000 years. Before this reconnection (during previous Pleistocene climate fluctuations), western Australia and the Coral Triangle were refugia for coral reef-associated species (Cowman and Bellwood 2013; Greenstein and Pandolfi 2008; Pellissier et al. 2014). On the other side of the Torres Strait—following millennia of changing sea levels and intense sedimentation—the GBR only began growing into its current form and size around 10,000 years ago (Carter and Johnson 1986; Marshall and Davies 1984; Webster et al. 2018). Therefore, the most recent submergence of the Torres Strait could have aided the recolonization of the GBR by marine fauna from western Australian and Coral Triangle refugia and increased the genetic diversity of species with populations that persisted in both regions.
Fig. 1. Bathymetric map of our Indo-Pacific study region with Carcharhinus amblyrhynchos sampling location information.

Sampling locations are indicated by red circles, and the number of specimens collected at each location is indicated in parentheses. Grey shading denotes land area above mean sea level; light blue denotes area no deeper than 200 m; darker blue shading denotes area deeper than 200 m. Inset (a) is a bathymetric profile of southern Indonesia, New Guinea, and northern Australia during the last glacial maximum. Area that is currently below sea level but was exposed (i.e., land) during the last glacial maximum is coloured green. Inset (b) shows the wider Indian and Pacific Oceans and inset (c) shows the New Caledonia area in more detail.
The role played by the cyclical emergence and submergence of the Torres Strait in shaping genetic diversity of populations on either side of the strait remains unclear. Even when dispersal-related life history traits are accounted for across a variety of organisms, the level of genetic differentiation amongst populations of species present on opposite sides of the Torres Strait varies widely (Gaither and Rocha 2013; Keyse et al. 2018; Liggins et al. 2016; Mirams et al. 2011). With up to six-metre differences in the sea level on each side of the Torres Strait due to the different tidal regimes in adjacent basins (Wolanski et al. 1988), the corresponding eddies and currents can be both strong and highly variable. Furthermore, net water movement through the Torres Strait both switches directions and varies threefold in strength seasonally (Margvelashvili et al. 2008; Saint-Cast and Condie 2006). All available genetic and hydrological evidence therefore suggests that dispersal through the strait is highly stochastic.
Large marine predators such as sharks can undertake active migrations. Therefore, despite the irregular oceanographic conditions, sharks can likely move through the Torres Strait region more consistently than organisms with planktonic larval stages. Catch and acoustic tagging studies have demonstrated that several species of coastal and coral reef associated sharks have specific habitat requirements such that they tend to disperse mainly along continental shelves (Chin et al. 2012; Dudgeon et al. 2013; Espinoza et al. 2015a, 2015b, 2014). Accordingly, genetic connectivity amongst coastal and coral reef-associated shark species in this region is facilitated by shallow shelfal areas (like the Torres Strait), while hindered by deep oceanic expanses (Dudgeon et al. 2009; Momigliano et al. 2017; Ovenden et al. 2009; Vignaud et al. 2014). When the shallow Torres Strait became land, however, shark populations on both sides of Australia may have diverged, although this hypothesis has not yet been explicitly tested.
Many previous coastal and reef shark studies have relied mainly on mtDNA and microsatellites (Boissin et al. 2019; Dudgeon et al. 2009; Ovenden et al. 2009; Portnoy et al. 2016; Vignaud et al. 2014) which often may not provide the necessary statistical power to resolve detailed evolutionary history (although see Portnoy et al. (2016) for a counterexample). While these studies may provide valuable insights into the genetic diversity and connectedness of reef shark populations, recent studies have highlighted the improved resolution of genomic sequencing (e.g., Kraft et al. 2020). Recent genomic studies of the blacktip reef shark (Carcharhinus melanopterus) demonstrated how studies with fewer markers may miss the signature of an ancient range expansion (Maisano Delser et al. 2018, 2016). Further study of populations using genome-wide markers has become a priority in the management and conservation of reef sharks (Domingues et al. 2018; Dudgeon et al. 2012; Heupel et al. 2019), and the more intensive evolutionary analyses they allow could provide insight into the origin of species as well as their current distribution and patterns of genetic diversity.
In this study, we used genome-wide single nucleotide polymorphism (SNP) data from populations of the endangered grey reef shark (Carcharhinus amblyrhynchos) (Simpfendorfer et al. 2020) to shed light on both their historical demographics and contemporary effective population sizes (Ne). In particular, we aimed to reconstruct the demographic history of this species, as well as determine its centre of origin and the potential legacy that the Torres Strait land bridge has had on patterns of genetic diversity. If C. amblyrhynchos has undergone a range expansion similar to the one inferred for C. melanopterus (Maisano Delser et al. 2018, 2016), demographic history models could detect an initial expansion across multiple lineages of the species, bottlenecks (decreases in Ne) due to founder effects, and possibly a spatial gradient of genetic diversity. If other major demographic events have occurred since the inception of the species—such as populations on both sides of the Torres Strait intermittently exchanging migrants—then demographic history models may infer more recent changes in Ne and migration rates (and possibly erase genetic signatures of earlier events). Estimates of coalescent Ne from demographic history models were compared with estimates of contemporary Ne from a common conservation genetics method to assess both the ancient and recent past of this endangered species.
Materials and methods
Sampling and genotyping
The genetic data in this study come from 513 C. amblyrhynchos samples collected across 17 locations in the Indian and Pacific Oceans (Fig. 1b, c). The capture and tissue sampling procedures for all individuals have been described in previous studies (Boussarie 2019; Momigliano et al. 2017, 2015a). The sequencing and SNP discovery was performed by Diversity Arrays Technology Pty. Ltd. (Canberra, Australia) using the standard DArTseq protocol (Sansaloni et al. 2011). After calling and filtering, the dataset contained 7952 loci with a total of 14,935 SNPs across ~551,040 base pairs (bp). For more details about the bioinformatics and filtering, see the Supplementary Information.
Genetic diversity
Species range expansions are expected to leave transient spatial patterns of genetic diversity. More recently colonized areas tend to have lower genetic diversity than the region of origin due to the sequential bottlenecks that characterise range expansion (Harpending and Rogers 2000; Mona 2017). This transient spatial pattern of decreasing genetic diversity is expected to persist until migration-drift equilibrium is reached. A simple test to identify a centre of origin was proposed by Ramachandran et al. (2005): genetic diversity is expected to be negatively correlated with distance from a putative centre of origin, with the most likely centre of origin being the location with the strongest negative correlation coefficient. Combining all samples from a given location, we computed the number of segregating sites (S), pairwise differences between sequences (π) and Tajima’s D directly from its site frequency spectrum (SFS) using the software package moments (Jouganous et al. 2017). We report values of π per site (i.e., divided by the total number of sequenced bases; Supplementary Information). A geographical lattice of 22,880 points (every 0.5 degrees between 70.25°E–179.75°E and 25.75°N–25.75°S) was constructed and then filtered down to include only points located in the ocean shallower than 200 metres since C. amblyrhynchos is very rarely observed away from continental shelves (with the exception of coral reefs in isolated seamounts) (Last and Stevens 2009). For each of the included 2028 points, the shortest distance by sea to each sampling location was calculated using the r packages ‘gDistance’ (van Etten 2018) and ‘marmap’ (Pante and Simon-Bouhet 2013). The distance from the point to each of these sites was then rank-correlated using Spearman’s rho (ρ) with the π at those sites using r version 3.6 (R Core Team 2019). A second, finer-scale run was performed in a narrower geographic area considering only 15 sampling locations (omitting the two oceanic archipelagos in the Indian Ocean—Cocos (Keeling) and Chagos) where an initial lattice of 125,584 points (every 0.125 degrees between 91.625°E–174.875°E and 20.875°N–25.875°S) was filtered the same way to produce 7521 points for the correlation analysis. Maps and figures were produced using r as described in the Supplementary Information.
Demographic history models
Studies on the demographic history of populations are often constrained by the necessity of using predefined models to test alternative demographic scenarios. Instead of using pre-defined models to detect up to one or two changes in the size of individual populations (e.g., Maisano Delser et al. 2016, 2018), we used stairway plot v2 (Liu and Fu 2015), a model-flexible method that calculates the composite likelihood of the observed SFS under several possible size change scenarios across the entire history of a population.
stairway plot infers the most likely timing and magnitude of changes in Ne at various epochs, with the transition between epochs corresponding to coalescent events in the population. stairway plot initially calculates the population-scaled mutation rate θ, from which Ne can be derived if μ (the germline mutation rate) is known (θ = 4•Ne•μ). Previous studies on the congener C. melanopterus used a uniform prior distribution of 8.05–8.54 • 10–9 for μ in an Approximate Bayesian Computation analyses, which they estimated using samples of Carcharhinus galapagensis from opposite sides of the Central American Isthmus (Maisano Delser et al. 2018, 2016). Here, we used the midpoint of this range (8.295 • 10–9). After producing a randomly-thinned, minor allele (folded) SFS for each population (Supplementary Information), stairway plot was run using the recommended (nseq − 2) ÷ 4 break points (where nseq is the number of haplotypes sampled) and 200 replicate runs for each population (except Cocos (Keeling) which had too few sequenced specimens to be considered in this analysis).
We further evaluated the joint demographic history of the Misool and North GBR populations using the method implemented in moments. This program is largely based on dadi (Gutenkunst et al. 2009), but uses direct computation of the frequency spectrum without solving the diffusion system. After creating the two-dimensional site frequency spectrum (2D-SFS) of the Misool and North GBR populations (Supplementary Information), we tested five types of gene flow models: the strict isolation model (SI), the isolation with migration model (IM), the two-epoch model (2EP), the secondary contact model (SC) and the ancient migration model (AM) (Sousa and Hey 2013). Combined with each type of gene flow scenario, we also modelled different plausible historical population growth scenarios, including ancestral expansions (AE) and bottlenecks (B). Visual representations of these models can be found in Fig. S1 and Table S1 contains a list of all model names and their parameters. The SI model assumes a split at time T1, with no migration. The IM model includes continuous, asymmetric migration following the population split at T1, while the 2EP model allows for migration rates to change at time T2. The SC model has a period of strict isolation starting at the time of split (T1) followed by gene flow starting at time T2, while the AM model assumes migration at the initial stages of divergence but strict contemporary isolation. If gene flow has not occurred between Misool and the North GBR since they were colonized (or stopped shortly after colonization), then the SI and AM models should fit the data well. On the other hand, if there is ongoing gene flow between these two regions, the IM, 2EP, or SC models should fit the data better. If this gene flow has been constant since both regions were colonized, the IM model should not fit the data any worse than the SC or 2EP models. If gene flow was interrupted or diminished by the presence of the Torres Strait land bridge, the SC or 2EP models should fit the data best. Since overlooked demographic events can in some cases bias demographic inference (Momigliano et al. 2021), and since both stairway plot and Tajima’s D values suggest the possibility of an ancestral population expansion, we also considered an additional version of each main model that included a population expansion at time TAE prior to the split (AE models). Since stairway plot also suggested a possible recent expansion in the North GBR (potentially corresponding with its regrowth following the last glacial maximum), we tested variations of each model that included exponential growth following a bottleneck starting at the time of split T1 (B models). We further tested variations of each model that included both ancestral expansion and exponential growth in the North GBR. For each basic scenario, we therefore tested four variants (e.g., for the IM model, we tested the following variants: IM, IMB, IMAE, IMAEB).
Model parameters were optimized in 10 independent optimization routines, following the pipeline developed by Portik et al. (2017) for dadi, adapted to moments by Momigliano et al. (2021), and further modified by Le Moan et al. (2021). Each independent routine was composed of four rounds of subsequent parameter optimization. In the first round a Broyden–Fletcher–Goldfarb–Shanno algorithm optimization (function “optimize.log”; max-iter = 10) was run for 30 sets of threefold randomly-perturbed parameters. The parameter values with the highest composite likelihoods were then chosen as starting parameters from a second round of optimization, this time run for 20 sets of two-fold randomly-perturbed parameters and increasing max-iter to 20. The same procedure applied to round two was used in round three and four, this time using two-fold randomly-perturbed parameters and one-fold randomly perturbed parameters. Convergence was checked using the best replicate of each of the 10 independent optimizations. Model choice was performed as described in Rougeux et al. (2017) and Momigliano et al. (2021). We calculated the Akaike information criterion (AIC) and ΔAIC for each model from the replicate with the highest likelihood. Then we calculated Akaike weight of evidence (wAIC) as outlined in Rougeux et al. (2017). Finally, we checked the fit of the best scoring models by plotting the residuals between the empirical 2D-SFS and the 2D-SFS produced by the models.
Contemporary effective population size
Contemporary Ne was estimated for each sampled site using the ldne linkage disequilibrium method (Waples and Do 2008) implemented in NeEstimator 2.1 (Do et al. 2014). This is a commonly used method to estimate recent Ne in population genetic studies for conservation purposes (e.g., Reid-Anderson et al. 2019). Simulation studies have shown that it can reliably detect changes in Ne from restriction-site associated sequencing data (Nunziata and Weisrock 2018). The mean linkage disequilibrium (r2) between all pairs of (putatively) unlinked loci in n sampled individuals from a given cohort is compared to the linkage disequilibrium that would be expected from sampling n individuals from an idealized (infinite) population (in which there is no actual linkage disequilibrium other than what is introduced by finite sampling bias). The difference between the observed and expected linkage disequilibrium indicates the level of genetic drift occurring in the sampled cohort, which is inversely proportional to its Ne. An estimate of infinite Ne is possible if the observed linkage disequilibrium between pairs of loci in the sampled cohort is indistinguishable from the expected linkage disequilibrium in an equivalent sample size from an idealized population. This method of estimating Ne is therefore most useful for small population sizes (tens to hundreds), and the upper 95% confidence interval limit on estimates can be infinite for estimates in the hundreds or low thousands (Lonsinger et al. 2018; Moran et al. 2019).
An adjustment of this method’s final estimate of Ne should be applied when a large number of loci are used. This depends on the size of the species’ genome, which has not yet been determined for C. amblyrhynchos. However, diploid chromosome numbers of 74–86 have been estimated for other species in the family Carcharhinidae (Gregory 2018), so an intermediate value of 40 haploid chromosomes was used for this correction. The recommended pcrit (minor allele frequency cut-off) of 0.05 was used to avoid upwardly biasing estimates of Ne (Waples et al. 2016). Since confounding factors such as overlapping generations (Waples et al. 2014) cannot be controlled for in this dataset (see Reid-Anderson et al. (2019) for additional considerations), the estimates provided here should not be taken as valid, standalone assessments of Ne, but rather as calculations used to compare the inferred Ne of each sampling location relative to each other and to the other methods of estimating Ne in this study.
Results
Genetic diversity
Overall, there were only slight differences in nucleotide diversity between sampling locations. While Entrecasteaux and several other New Caledonian sampling locations had the highest number of segregating sites (Table S2), Scott Reef and the three locations closest to it—Misool, Rowley Shoals, and Ningaloo Reef—all had the highest nucleotide diversity with π > 0.00147 (Table S3). The Indian Ocean archipelago locations (Chagos and Cocos (Keeling)) had both the lowest number of segregating sites and lowest nucleotide diversity, while Matthew had the next lowest nucleotide diversity. Tajima’s D was negative at all locations.
In the diversity-distance correlation analysis, the most likely centres of origin were located slightly east of Misool in the Cenderawasih Bay (Fig. 2). The ρ of −0.87 for these locations is indicative of a strong negative correlation in genetic diversity with distance away from the Coral Triangle, particularly towards the east. The second run without Indian Ocean archipelago sampling locations confirmed this: a steep gradient in ρ was observed through the Torres Strait (Fig. S2), and the correlation between nucleotide diversity and degrees East (ρ = −0.839) was only slightly weaker than the strongest correlation found at any point (ρ = −0.857).
Fig. 2. Spatial distribution of the Spearman’s ρ (rho) coefficient between genetic diversity and distance by sea from all 17 Carcharhinus amblyrhynchos sampling locations (black crosses) for 2028 points throughout the Indo-Pacific region shallower than 200 m.

The lowest values of rho (indicating the most likely centre of origin) were −0.87 and located east of Misool in the Cenderawasih Bay of Western New Guinea (white X’s).
Demographic history models
A strong signal of expansion 40,000–90,000 generations ago was inferred by stairway plot for all locations except Chagos (Fig. 3). Immediately after this ancestral expansion, the Ne of the three locations in western Australia were inferred to be at least 20% larger than all others. More recent expansions were inferred at the Misool and North GBR populations. The steady decrease after the initial expansion inferred for Matthew is consistent with what has previously been interpreted as a founder effect in studies on a closely-related reef shark C. melanopterus (Maisano Delser et al. 2018). Less extreme, more recently-initiated declines were inferred for Walpole, Grand Astrolabe, and Ningaloo Reef.
Fig. 3. Carcharhinus amblyrhynchos demographic histories inferred by stairway plot.

Sampled locations are grouped into three different regions, and those with trends distinct from others in their region are labelled. Cocos (Keeling) has been removed due to insufficient sample size. The dashed vertical lines at 40,000 and 90,000 generations ago on the x-axis span the period during which an expansion was inferred across nearly all sampled locations.
Two-population models that included contemporary migration (IM, SC and 2EP models) always had lower AIC scores (better fit) than models with no contemporary gene flow (Fig. 4). The SI models performed worst, and they are not represented in Fig. 4 since their very high AIC would obscure the differences between the rest of the models (Table S1). In general, models that included growth in the North GBR performed better than models that did not, and models that included an ancestral expansion performed better than models that assumed an ancestral population at drift-equilibrium at the time of split between the North GBR and Misool (Fig. 4). The model with the lowest AIC was SCB (secondary contact coinciding with exponential growth in the GBR), however Akaike weights for the SCB, IMAEB, SCAE, and SCAEB models were fairly similar.
Fig. 4. Modelling results from moments for the Misool and North GBR populations of Carcharhinus amblyrhynchos.

SI = strict isolation, IM = isolation with migration, twoep = two periods with different levels of migration, SC = secondary contact, AM = ancestral migration (see Fig. S2 for visual representations of relevant models). a AIC from the best 3 optimizations for all tested models, with the exclusion of SI gene flow (GF) scenarios (as SI models had such high AIC that their inclusion in the graph would have obscured differences among better fitting models). The x-axis groups models by whether or not they include an ancestral expansion (AE), exponential growth in the North GBR following a bottleneck (B), both (AEB), or neither (basic); (b) Weight of evidence (wAIC) for each model, in decreasing order on the x-axis. Four models had wAIC above 0.1: SCB, IMAEB, SCAE and SCAEB. See Table S1 for more detailed results and all parameter estimates for each model.
The four best models (i.e., those with the lowest AIC values: SCB, IMAEB, SCAE, and SCAEB) all show a good fit to the data, with small and normally-distributed residuals (Fig. 5). However, they each represent a fairly different demographic history. The second, third, and fourth best models (respectively IMAEB, SCAE and SCAEB) inferred an ancestral population expansion between 79,000 and 87,000 generations ago, in line with the results from stairway plot. These three models (IMAEB, SCAE and SCAEB) indicate the Misool and North GBR groups diverged between 12,200 and 13,500 generations ago, and the SC models indicate that secondary contact was established again 1400–2400 generations ago. The SCB model suggests a much older divergence time of ~145,000 generations ago—even older than the oldest coalescence event inferred within any single population by stairway plot (~120,000 generations ago).
Fig. 5. Graphical representation of the empirical 2D-SFS for Carcharhinus amblyrhynchos in Misool and the North GBR, as well as the inferred parameters and residuals from the four best models in moments.

Time parameters are given as thousands of generations ago (kga).
Contemporary effective population size
Effective population sizes were estimated to be largest in the most northern and western locations sampled in New Caledonia (Fig. 6). Chesterfield had the largest Ne at 5529, followed by adjacent sampling sites Northern Lagoon and Entrecasteaux at 4974 and 4401, respectively (Table S3). The Ne at each of these three sites was more than six times larger than all others east of the Torres Strait, and even the lower limit of their jackknife 95% confidence intervals overlapped only with the Ne estimated for Ningaloo Reef (3885), which was twice as large as any other location west of the Torres Strait (Fig. 6, Table S3). The only other locations with Ne > 1000 were Scott Reef and Misool which, along with Ningaloo Reef, were the three locations with the highest nucleotide diversity (Fig. 6, Table S3). Matthew had the second lowest Ne at 161 and was the only location for which a finite upper 95% confidence interval limit was estimated (547), while the South GBR had the lowest Ne at only 70 (Fig. 6, Table S3).
Fig. 6. Effective population size estimates at each Carcharhinus amblyrhynchos sampling location using the linkage-disequilibrium (ldne) method implemented in NeEstimator.
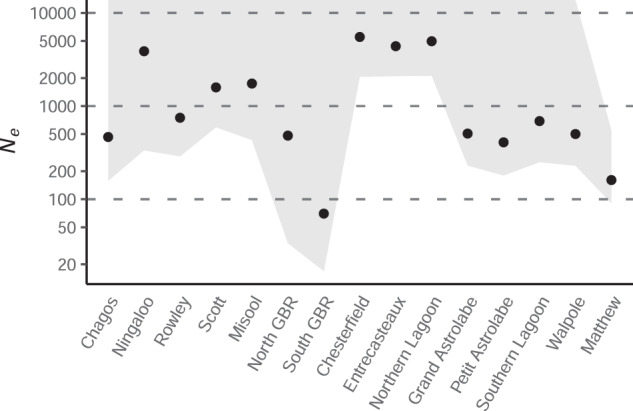
Locations are listed in increasing order of longitude on x-axis and Ne is represented on the y-axis (on a log2 scale). Shaded area indicates jackknife 95% confidence intervals (all upper limits were infinite except for Matthew).
Discussion
This genomic analysis of 513 C. amblyrhynchos individuals from 17 locations indicates that a range expansion and glacial cycles may have affected the distribution of diversity within the species. However, these ancient events do not appear to have had much of an effect on modern populations, as their estimated contemporary effective population sizes do not correspond with levels of genetic diversity or coalescent history.
Grey reef shark genetic diversity decreased with distance away from the Coral Triangle and north-western Australia. A similar genetic diversity gradient was observed in C. melanopterus (Maisano Delser et al. 2018). While these gradients are typically inferred to indicate a species’ centre of origin, in this case, they could also indicate a centre of overlap or accumulation of diversity from the adjacent Indian and Pacific Oceans. Maisano Delser et al. (2018) found further support for the centre of origin hypothesis in C. melanopterus by calculating directionality indices (Peter and Slatkin 2013), demonstrating that significantly higher proportions of shared derived alleles were found outside of north-western Australia. This region could be both a centre of origin and a centre of accumulation for C. amblyrhynchos as these hypotheses are not mutually exclusive (Bowen et al. 2013; Briggs and Bowen 2013; Cowman and Bellwood 2013; Gaither and Rocha 2013). Further evidence that C. amblyrhynchos originated from the Coral Triangle comes from the detection of an initial expansion across almost all sampled locations 40,000–90,000 generations ago by stairway plot, followed by larger post-expansion Ne west of the Torres Strait (in the Coral Triangle and north-western Australia). Tajima’s D was negative across all locations, which also supports the hypothesis of an expansion. However, this last result should be interpreted with caution as our sample sizes are not homogeneous and Tajima’s D can be affected by sample size (Subramanian 2016). The early and steady decline inferred by stairway plot for Matthew matches the founder effects observed in many Pacific island populations of C. melanopterus using a similar ABC-skyline approach (Maisano Delser et al. 2018). Indeed, our modelling results are consistent with previous studies of a C. melanopterus range expansion: the C. melanopterus expansion inferred to have occurred around 59,000 generations ago (Maisano Delser et al. 2016) is in the middle of the expansion range inferred in this study, and the highest Nm (the product of Ne and migration rate) values through time were also found in populations from northern and western Australia (Maisano Delser et al. 2018). The two closest relatives of C. melanopterus (Carcharhinus cautus and Carcharhinus fitzroyensis) are geographically restricted to Western Australia, Queensland, and New Guinea (Lyle 1987). Accordingly, Maisano Delser et al. (2018) suggest that the cautus-melanopterus-fitzroyensis clade may have originated in this region. The results of our study indicate that a third, more distantly related species of the same genus shares a similar demographic history. This is consistent with species assemblage studies showing that north-western Australia and the Coral Triangle were refugia for species-level diversity during the Pleistocene (Greenstein and Pandolfi 2008; Ludt and Rocha 2015; Pellissier et al. 2014), and that the Coral Triangle is a centre of speciation (Bowen et al. 2013; Briggs 1999; Cowman and Bellwood 2013).
While the geographic distribution of genetic diversity in C. amblyrhynchos is similar to that of C. melanopterus (strongly correlated with distance away from the Coral Triangle), the differences between locations were less extreme in C. amblyrhynchos. Maisano Delser et al. (2018) found genetic diversity differences of greater than 310% between continental and island C. melanopterus populations, and as much as a 38% difference between continental Australian populations. Furthermore, putative founder effects (localized bottlenecks) were detected across almost all C. melanopterus populations outside of Australia (Maisano Delser et al. 2018). Only a few bottlenecks were observed in this study, while the greatest disparity in nucleotide diversity between locations was 208% overall and only 8.7% between continental Australian locations. Different sequencing and associated bioinformatic approaches (for restriction site-associated sequencing data here versus target gene capture in Maisano Delser et al. (2016, 2018)) may make our results less comparable. However, the more uniform genetic diversity in C. amblyrhynchos relative to C. melanopterus could also be explained by the two species’ differing dispersal potential. Migration increases local genetic diversity and decreases structuring across the range of a species, leading towards migration-drift equilibrium during a range expansion (Mona et al. 2014). Although both C. amblyrhynchos and C. melanopterus generally exhibit site fidelity (Bonnin et al. 2021; Chin et al. 2013; Espinoza et al. 2015a, 2015b; Mourier et al. 2012; Papastamatiou et al. 2009; Speed et al. 2011), C. amblyrhynchos has a larger average dispersal distance (Dwyer et al. 2020), activity space (Speed et al. 2016), and utilizes offshore resources more frequently (McCauley et al. 2012; Papastamatiou et al. 2018). Furthermore, only C. amblyrhynchos has been observed to undertake repeated migrations of several hundred kilometres (Bonnin et al. 2019)—sometimes more than 900 kilometres (White et al. 2017)—across oceanic expanses.
In single population models, inferred changes in Ne may also be due to changes in migration, which, as discussed above, brings additional diversity into the population via gene flow. While Maisano Delser et al. (2016, 2018) accounted for this by using metapopulation structure in their single population models, we followed up single population modelling with joint population modelling to explicitly consider different historical gene flow scenarios between locations on each side of the Torres Strait. Three of the best four models were of isolation followed by secondary contact between Misool and the North GBR, implying not only that migrant exchange between these two locations is ongoing, but also that they have previously been isolated from each other. However, the inferred ages of demographic changes were quite different between models. The SCB model had the lowest AIC, yet yielded a very old divergence time of ~143,000 generations ago. This seems unrealistic given that it is ten times older than the divergence times inferred in the next three best models and is even older than all coalescent events inferred by stairway plot. Furthermore, it was the only model of the best four to not include an ancestral population expansion (as indicated by stairway plot and negative Tajima’s D values at all locations). Momigliano et al. (2021) showed that when true demographic scenarios include both an ancestral expansion and subsequent growth in a newly diverged population, models including only new growth (like the SCB model) can erroneously produce a very good fit and misestimate parameters. Thus, given the multiple lines of evidence in this study that suggest an ancestral expansion has occurred, we believe it is likely that the SCB model is providing an oversimplified scenario.
To compare the timing of geological events (in years) to the timing of demographic events inferred by coalescent models (in generations, when a mutation rate is given), an estimated generation time for the sampled organism is required. Maisano Delser et al. (2016, 2018) used seven years as the generation time for C. melanopterus. If the same value was used as the generation time in our best two-population models (excluding SCB), the divergence of the Misool and North GBR populations would be inferred to have occurred during the last glacial period, and the SC models would suggest that secondary contact occurred around when the Torres Strait resubmerged 7000 years ago (Woodroffe et al. 2000). However, this figure of 7 years used to calibrate previous models was actually female maturation time, not generation time. Generation time also factors in the timing of subsequent pups throughout the female’s lifespan and can effectively be considered the average difference in age of a parent and all of its offspring. Therefore, the generation time of iteroparous organisms (that reproduce more than once during their lifespan) is somewhere between their age of sexual maturity and death (typically closer to the latter age as older, larger individuals are generally more fecund). In C. amblyrhynchos, female maturity was observed after a minimum of 11 years, and as fecundity increased with age, generation time was estimated to be 16.4 years (Robbins 2006). Using this generation time to calibrate our models, all events are inferred to have occurred before the last glacial maximum (more than 21,000 years ago). Note that because of uncertainties in estimating generation times and mutation rates, these dates should be regarded as approximations.
Regarding the grey reef shark populations on either side of the Torres Strait, we can confidently draw two conclusions. The first is that there is likely ongoing gene flow between them (as indicated by the poor performance of the ancient migration and strict isolation models). The second is that, as shown by stairway plot, the sizes of these two populations, the level of gene flow between them, or both, have increased since the initial expansion of the species. Four of the top five joint history models and stairway plot indicate that the North GBR population expanded since its divergence from the Misool population. This may correspond with the expansion of the GBR habitat following the last glacial maximum (Carter and Johnson 1986; Marshall and Davies 1984; Webster et al. 2018). However, the Ne increase in Misool inferred by stairway plot instead seems more likely to be due to resumed gene flow through the Torres Strait. If it were instead due to an expansion in habitat availability similar to that of the GBR (or increased gene flow from a different sampled region), we would have expected to observe similar increases in Ne in western Australian populations around the same time (given their similar bathymetry and history as refugia). The possibility of migrant exchange between the Coral Triangle and the Coral Sea via northern New Guinea and the Bismarck Sea could confound these modelling results. However, the most consistent genetic break identified amongst a variety of organisms with larval stages in the Coral Triangle was in northern New Guinea: southeast of Halmahera Eddy near the Cenderawasih Bay (Carpenter et al. 2011). This implies that connectivity through this region is low for organisms with larval dispersal, yet a similar analysis of organisms without larval dispersal such as sharks has not been completed. To fill this knowledge gap, further sampling in northern New Guinea, the Bismarck Sea, and the Solomon Islands could reveal unknown lineages with different demographic histories, as well as how gene flow differs between larval and non-larval organisms near barriers such as the Halmahara Eddy and the Torres Strait.
The large estimates and infinite 95% confidence interval upper bounds for most linkage-disequilibrium estimates of Ne imply that most populations are large, and that their sizes cannot be confidently estimated or statistically distinguished from one another. Despite this, the largest estimated population sizes were upwards of 60 times larger than the smallest, arguably warranting further consideration. Despite the higher genetic diversity and ancient Ne inferred via coalescent modelling for locations west of the Torres Strait, linkage-disequilibrium estimates of Ne show that the largest populations are currently found around remote reefs and atolls in the north-western part of the New Caledonian archipelago. These locations, due to their isolation from centres of human population, are also likely among the least anthropogenically-impacted worldwide (Cinner et al. 2018; Januchowski‐Hartley et al. 2020; Juhel et al. 2019, 2018; MacNeil et al. 2020) and are fully protected by strict no-entry rules within the Coral Sea Nature Park. This supports previous findings that reef shark abundance and diversity is higher in isolated coral reef ecosystems (Juhel et al. 2019, 2018; MacNeil et al. 2020; Sandin et al. 2008). On the other end of the spectrum, the Ne estimated for the South GBR is considerably smaller than all other estimates in this study. This is likely the result of several factors, possibly linked to the habitat structure from which these individuals were sampled (Momigliano et al. 2017, 2015a). Reef habitat is less contiguous towards the southern end of the GBR with greater gaps between reefs, unlike further north where most reefs are highly connected and likely perceived as contiguous habitat by sharks across a much wider range (Espinoza et al. 2015a; Momigliano et al. 2015b). This site is also at the extreme southern end of the C. amblyrhynchos distribution on the GBR, further limiting potential population size. Habitat area is likely a major limiting factor in the oceanic islands of Chagos, Cocos (Keeling), Walpole, and Matthew as well—which in addition to having low diversity and inferred Ne in all analyses performed here—are also furthest from the inferred point of origin for C. amblyrhynchos.
Most alarmingly, studies using multiple assessment methods have detected reef shark population declines on the GBR (Hisano et al. 2011; Robbins et al. 2006), with catch per unit effort of reef sharks in this region having decreased by 82% between 1962 and 2016 (Roff et al. 2018). This figure is similar to the much lower abundance and diversity of reef sharks in southern New Caledonia (near the capital city Nouméa) relative to remote north-western New Caledonia (Juhel et al. 2019, 2018). These apparent localized declines (detected using the linkage disequilibrium method) have not resulted in a loss of genetic diversity though as π in the south GBR and Southern Lagoon is greater than the π observed in Northern Lagoon (which is estimated to have a contemporary Ne of nearly 5000 in this study). This is consistent with recent findings that human-induced extinction can exceed the pace at which losses of genetic diversity are detectable—even with genome-wide sequencing (Roycroft et al. 2021). Furthermore, our coalescent analyses (stairway plot) also did not detect any recent declines, inferring the history of the south GBR and Southern Lagoon populations to be similar to those in north-western New Caledonia (with the largest contemporary Ne observed in this study). Simulation studies have shown that the linkage disequilibrium method of estimating Ne detects declines (in populations of 250–1000) two times sooner than coalescent methods (i.e., following only half as many generations since the initiation of a decline (Nunziata and Weisrock 2018)). Thus, it seems likely that the reef shark population declines previously documented in this area (Hisano et al. 2011; Robbins et al. 2006; Roff et al. 2018) are not yet detectable using coalescent methods, but that future sampling efforts may detect these declines. Overall, these contrasting results show that the various methods employed in this study, which are routinely used to infer population declines and range expansions, are not interchangeable. Researchers and managers looking to assess the current status as well as the histories of populations should therefore consider the appropriate methods to achieve their goals.
Conclusions
While reef shark populations are declining worldwide, the scientific community has called for increased use of genomic tools to assess their status (Domingues et al. 2018; Dudgeon et al. 2012; Heupel et al. 2019). By analysing the diversity and Ne of C. amblyrhynchos across 17 locations using genome-wide SNP data, we identified the Coral Triangle and north-western Australia as the most likely region from which this species originated. Although joint demographic history modelling of populations on both sides of the Torres Strait yielded inconclusive results, single population history models indicate that the intermittent land barrier here may have obstructed gene flow. We also reinforce the conclusions of previous studies, this time using genetic methods, that the largest current reef shark populations are found in isolated, protected coral reef ecosystems (Juhel et al. 2019, 2018; MacNeil et al. 2020; Robbins et al. 2006; Sandin et al. 2008). This study demonstrates which methods can be used to disentangle long term Ne from contemporary population sizes and illustrates how genetic diversity in marine biodiversity hotspots is shaped by ancient demographic processes.
Supplementary information
Acknowledgements
Thanks to Emilie Boulanger, Kosmas Hench, Raphaël Leblois, Miguel Navascués, as well as the students, faculty, and administration of the Erasmus Mundus Master Programme in Evolutionary Biology (MEME) for the support provided to C.A.J.W. during this project. Thanks to Brian Bowen, Andrea Schmidt, Joanne Collier and the anonymous peer reviewers for their comments and suggestions that helped improve this manuscript. We thank the crew of Research Vessel Amborella, the many volunteers that contributed to field trips in New Caledonia, and the support of Laurent Millet of the Plateforme du Vivant lab at IRD Nouméa for help in the processing of genetic samples. Sampling and genetic analyses in New-Caledonia were funded by research grants of Laurent Vigliola (APEX programme, Government of New Caledonia, Pew Charitable Trust, Total Foundation, IRD core funding), and David Mouillot (APEX programme, Total Foundation). Sampling in other countries was supported by the Great Barrier Reef Marine Park Authority, Cooperative Research Centre (CRC) Reef, Australian Coral Reef Society, and PADI Aware. Cameron A.J. Walsh was funded by an Erasmus Mundus Joint Master Degree scholarship from the Erasmus Mundus Master Programme in Evolutionary Biology and an internship stipend from David Mouillot and Stéphanie Manel. Paolo Momigliano was funded by the Academy of Finland (grant 316294). Germain Boussarie and Lucas Bonnin were funded by the École Normale Supérieure de Lyon. The writing of this paper occurred while Stéphanie Manel was funded by the project SEAMOUNTS. Genetic samples in New Caledonia were collected under permits to Laurent Vigliola by the Government of New Caledonia (permit #2015-1351/GNC), the Northern Province of New Caledonia (permit #60912-1508-2015/JJC) and the Southern Province of New Caledonia (permits #479-2016/ARR/DENV and #2093-2016/ARR/DENV). Great Barrier Reef Marine Park permits include G01/356, G02/021 and G03/8048.1.
Author contributions
C.A.J.W., P.M., G.B., D.M., L.V., and S.M. conceived the project; P.M., G.B., W.D.R., L.B., J.J.K., and L.V. designed and conducted the sampling; P.M., G.B. and C.F. performed lab work and organized sequencing data; C.A.J.W. and P.M. performed the analyses; C.A.J.W., P.M., and S.M. wrote the first draft of the manuscript and all authors contributed with revisions.
Data availability
Raw sequence data from sites in New Caledonia is on Zenodo (10.5281/zenodo.5935133) and data from other sites is available on GenBank Sequence Read Archive (Bioproject PRJNA795958). The code needed to reproduce the analyses performed here can be found in GitHub repositories (https://github.com/cajwalsh/grey_reef_gen_demog and https://github.com/Nopaoli/Shark-Demographic-Models).
Competing interests
The authors declare no competing interests.
Footnotes
Associate editor Paul Sunnucks.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Cameron A. J. Walsh, Paolo Momigliano.
These authors jointly supervised this work: Laurent Vigliola, Stéphanie Manel.
Supplementary information
The online version contains supplementary material available at 10.1038/s41437-022-00514-4.
References
- Boissin E, Thorrold SR, Braun CD, Zhou Y, Clua EE, Planes S. Contrasting global, regional and local patterns of genetic structure in gray reef shark populations from the Indo-Pacific region. Sci Rep. 2019;9:15816. doi: 10.1038/s41598-019-52221-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnin L, Mouillot D, Boussarie G, Robbins WD, Kiszka JJ, Dagorn L, Vigliola L. Recent expansion of marine protected areas matches with home range of grey reef sharks. Sci Rep. 2021;11:14221. doi: 10.1038/s41598-021-93426-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnin L, Robbins WD, Boussarie G, Kiszka JJ, Dagorn L, Mouillot D, Vigliola L (2019) Repeated long-range migrations of adult males in a common Indo-Pacific reef shark. Coral Reefs. 10.1007/s00338-019-01858-w
- Boussarie G (2019) Apports de l’analyse de l’ADN environnemental et de la génomique du paysage pour la conservation des requins de récif (PhD Thesis). Université Montpellier
- Bowen BW, Rocha LA, Toonen RJ, Karl SA. The origins of tropical marine biodiversity. Trends Ecol Evol. 2013;28:359–366. doi: 10.1016/j.tree.2013.01.018. [DOI] [PubMed] [Google Scholar]
- Briggs JC. Coincident biogeographic patterns: Indo-West Pacific Ocean. Evolution. 1999;53:326–335. doi: 10.1111/j.1558-5646.1999.tb03769.x. [DOI] [PubMed] [Google Scholar]
- Briggs JC, Bowen BW. Marine shelf habitat: biogeography and evolution. J Biogeogr. 2013;40:1023–1035. doi: 10.1111/jbi.12082. [DOI] [Google Scholar]
- Carpenter KE, Barber PH, Crandall ED, Ablan-Lagman MCA, Ambariyanto, Mahardika GN, Manjaji-Matsumoto BM, Juinio-Meñez MA, Santos MD, Starger CJ, Toha AHA. Comparative Phylogeography of the Coral Triangle and Implications for Marine Management. J Mar Biol. 2011;2011:e396982. doi: 10.1155/2011/396982. [DOI] [Google Scholar]
- Carter RM, Johnson DP. Sea-level controls on the post-glacial development of the Great Barrier Reef, Queensland. Mar Geol. 1986;71:137–164. doi: 10.1016/0025-3227(86)90036-8. [DOI] [Google Scholar]
- Chin A, Tobin A, Simpfendorfer C, Heupel M. Reef sharks and inshore habitats: patterns of occurrence and implications for vulnerability. Mar Ecol Prog Ser. 2012;460:115–125. doi: 10.3354/meps09722. [DOI] [Google Scholar]
- Chin A, Tobin AJ, Heupel MR, Simpfendorfer CA (2013) Population structure and residency patterns of the blacktip reef shark Carcharhinus melanopterus in turbid coastal environments. J Fish Biol 82:1192–1210. 10.1111/jfb.12057 [DOI] [PubMed]
- Cinner JE, Maire E, Huchery C, MacNeil MA, Graham NAJ, Mora C, et al. Gravity of human impacts mediates coral reef conservation gains. PNAS. 2018;115:E6116–E6125. doi: 10.1073/pnas.1708001115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowman PF, Bellwood DR. The historical biogeography of coral reef fishes: global patterns of origination and dispersal. J Biogeogr. 2013;40:209–224. doi: 10.1111/jbi.12003. [DOI] [Google Scholar]
- Do C, Waples RS, Peel D, Macbeth GM, Tillett BJ, Ovenden JR. NeEstimator v2: re-implementation of software for the estimation of contemporary effective population size (Ne) from genetic data. Mol Ecol Resour. 2014;14:209–214. doi: 10.1111/1755-0998.12157. [DOI] [PubMed] [Google Scholar]
- Domingues RR, Hilsdorf AWS, Gadig OBF. The importance of considering genetic diversity in shark and ray conservation policies. Conserv Genet. 2018;19:501–525. doi: 10.1007/s10592-017-1038-3. [DOI] [Google Scholar]
- Dudgeon CL, Blower DC, Broderick D, Giles JL, Holmes BJ, Kashiwagi T, Krück NC, Morgan JAT, Tillett BJ, Ovenden JR. A review of the application of molecular genetics for fisheries management and conservation of sharks and rays. J Fish Biol. 2012;80:1789–1843. doi: 10.1111/j.1095-8649.2012.03265.x. [DOI] [PubMed] [Google Scholar]
- Dudgeon CL, Broderick D, Ovenden JR (2009) IUCN classification zones concord with, but underestimate, the population genetic structure of the zebra shark Stegostoma fasciatum in the Indo-West Pacific. Mol Ecol 18:248–261. 10.1111/j.1365-294X.2008.04025.x [DOI] [PubMed]
- Dudgeon CL, Lanyon JM, Semmens JM (2013) Seasonality and site fidelity of the zebra shark, Stegostoma fasciatum, in southeast Queensland, Australia. Anim Behav 85:471–481. 10.1016/j.anbehav.2012.12.013
- Dwyer RG, Krueck NC, Udyawer V, Heupel MR, Chapman D, Pratt HL, Garla R, Simpfendorfer CA. Individual and Population Benefits of Marine Reserves for Reef Sharks. Curr Biol. 2020;30:480–489.e5. doi: 10.1016/j.cub.2019.12.005. [DOI] [PubMed] [Google Scholar]
- Espinoza M, Cappo M, Heupel MR, Tobin AJ, Simpfendorfer CA. Quantifying Shark Distribution Patterns and Species-Habitat Associations: Implications of Marine Park Zoning. PLOS ONE. 2014;9:e106885. doi: 10.1371/journal.pone.0106885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espinoza M, Heupel MR, Tobin AJ, Simpfendorfer CA. Residency patterns and movements of grey reef sharks (Carcharhinus amblyrhynchos) in semi-isolated coral reef habitats. Mar Biol. 2015;162:343–358. doi: 10.1007/s00227-014-2572-x. [DOI] [Google Scholar]
- Espinoza M, Lédée EJI, Simpfendorfer CA, Tobin AJ, Heupel MR (2015b) Contrasting movements and connectivity of reef-associated sharks using acoustic telemetry: implications for management. Ecological Applications 2101–2118. 10.1890/14-2293.1@10.1002/(ISSN)1939-5582.AcousticTelemetryinFisheriesManagement [DOI] [PubMed]
- Fang B, Kemppainen P, Momigliano P, Feng X, Merilä J. On the causes of geographically heterogeneous parallel evolution in sticklebacks. Nat Ecol Evolution. 2020;4:1105–1115. doi: 10.1038/s41559-020-1222-6. [DOI] [PubMed] [Google Scholar]
- Fang B, Kemppainen P, Momigliano P, Merilä J (2021) Population structure limits parallel evolution in sticklebacks. Molecular Biology and Evolution [DOI] [PMC free article] [PubMed]
- Gaither MR, Rocha LA. Origins of species richness in the Indo-Malay-Philippine biodiversity hotspot: evidence for the centre of overlap hypothesis. J Biogeogr. 2013;40:1638–1648. doi: 10.1111/jbi.12126. [DOI] [Google Scholar]
- Greenstein BJ, Pandolfi JM. Escaping the heat: range shifts of reef coral taxa in coastal Western Australia. Glob Change Biol. 2008;14:513–528. doi: 10.1111/j.1365-2486.2007.01506.x. [DOI] [Google Scholar]
- Gregory TR (2018) Animal Genome Size Database [WWW Document]. URL http://www.genomesize.com/
- Gutenkunst RN, Hernandez RD, Williamson SH, Bustamante CD. Inferring the joint demographic history of multiple populations from multidimensional SNP frequency data. PLOS Genet. 2009;5:e1000695. doi: 10.1371/journal.pgen.1000695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harpending H, Rogers A. Genetic perspectives on human origins and differentiation. Annu Rev Genomics Hum Genet. 2000;1:361–385. doi: 10.1146/annurev.genom.1.1.361. [DOI] [PubMed] [Google Scholar]
- Heupel MR, Papastamatiou YP, Espinoza M, Green ME, Simpfendorfer CA (2019) Reef shark science – key questions and future directions. Front Mar Sci 6. 10.3389/fmars.2019.00012
- Hisano M, Connolly SR, Robbins WD. Population growth rates of reef sharks with and without fishing on the great barrier reef: robust estimation with multiple models. PLOS ONE. 2011;6:e25028. doi: 10.1371/journal.pone.0025028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Januchowski‐Hartley FA, Vigliola L, Maire E, Kulbicki M, Mouillot D. Low fuel cost and rising fish price threaten coral reef wilderness. Conserv Lett. 2020;13:e12706. doi: 10.1111/conl.12706. [DOI] [Google Scholar]
- Jouganous J, Long W, Ragsdale AP, Gravel S. Inferring the joint demographic history of multiple populations: beyond the diffusion approximation. Genetics. 2017;206:1549–1567. doi: 10.1534/genetics.117.200493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juhel J-B, Vigliola L, Mouillot D, Kulbicki M, Letessier TB, Meeuwig JJ, Wantiez L. Reef accessibility impairs the protection of sharks. J Appl Ecol. 2018;55:673–683. doi: 10.1111/1365-2664.13007. [DOI] [Google Scholar]
- Juhel J-B, Vigliola L, Wantiez L, Letessier TB, Meeuwig JJ, Mouillot D. Isolation and no-entry marine reserves mitigate anthropogenic impacts on grey reef shark behavior. Sci Rep. 2019;9:2897. doi: 10.1038/s41598-018-37145-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keith SA, Baird AH, Hughes TP, Madin JS, Connolly SR. Faunal breaks and species composition of Indo-Pacific corals: the role of plate tectonics, environment and habitat distribution. Proc R Soc Lond B: Biol Sci. 2013;280:20130818. doi: 10.1098/rspb.2013.0818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keyse J, Treml EA, Huelsken T, Barber PH, DeBoer T, Kochzius M, Nuryanto A, Gardner JPA, Liu L-L, Penny S, Riginos C (2018) Historical divergences associated with intermittent land bridges overshadow isolation by larval dispersal in co-distributed species of Tridacna giant clams. J Biogeogr 45:848–858. 10.1111/jbi.13163
- Kotlík P, Marková S, Konczal M, Babik W, Searle JB. Genomics of end-Pleistocene population replacement in a small mammal. Proc R Soc B: Biol Sci. 2018;285:20172624. doi: 10.1098/rspb.2017.2624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraft DW, Conklin EE, Barba EW, Hutchinson M, Toonen RJ, Forsman ZH, Bowen BW (2020) Genomics versus mtDNA for resolving stock structure in the silky shark (Carcharhinus falciformis). PeerJ 8:e10186. 10.7717/peerj.10186 [DOI] [PMC free article] [PubMed]
- Last PR, Stevens JD. Sharks and rays of Australia. 2nd edn. Collingwood. Victoria: CSIRO Publishing; 2009. [Google Scholar]
- Leprieur F, Descombes P, Gaboriau T, Cowman PF, Parravicini V, Kulbicki M, Melián CJ, Santana CN, de, Heine C, Mouillot D, Bellwood DR, Pellissier L. Plate tectonics drive tropical reef biodiversity dynamics. Nat Commun. 2016;7:ncomms11461. doi: 10.1038/ncomms11461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Moan A, Bekkevold D, Hemmer-Hansen J. Evolution at two time frames: ancient structural variants involved in post-glacial divergence of the European plaice (Pleuronectes platessa) Heredity. 2021;126:668–683. doi: 10.1038/s41437-020-00389-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liggins L, Treml EA, Possingham HP, Riginos C. Seascape features, rather than dispersal traits, predict spatial genetic patterns in co-distributed reef fishes. J Biogeogr. 2016;43:256–267. doi: 10.1111/jbi.12647. [DOI] [Google Scholar]
- Liu X, Fu Y-X. Exploring population size changes using SNP frequency spectra. Nat Genet. 2015;47:555–559. doi: 10.1038/ng.3254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lonsinger RC, Adams JR, Waits LP (2018) Evaluating effective population size and genetic diversity of a declining kit fox population using contemporary and historical specimens. Ecology and Evolution. 10.1002/ece3.4660 [DOI] [PMC free article] [PubMed]
- Ludt WB, Rocha LA. Shifting seas: the impacts of Pleistocene sea-level fluctuations on the evolution of tropical marine taxa. J Biogeogr. 2015;42:25–38. doi: 10.1111/jbi.12416. [DOI] [Google Scholar]
- Lyle JM (1987) Observations on the biology of Carcharhinus cautus (Whitley), C. melanopterus (Quoy & Gaimard) and C. fitzroyensis (Whitley) from Northern Australia. Mar Freshw Res 38:701–710. 10.1071/mf9870701
- MacNeil MA, Chapman DD, Heupel M, Simpfendorfer CA, Heithaus M, Meekan M, et al. Global status and conservation potential of reef sharks. Nature. 2020;583:801–806. doi: 10.1038/s41586-020-2519-y. [DOI] [PubMed] [Google Scholar]
- Maisano Delser P, Corrigan S, Duckett D, Suwalski A, Veuille M, Planes S, Naylor GJP, Mona S (2018) Demographic inferences after a range expansion can be biased: the test case of the blacktip reef shark (Carcharhinus melanopterus). Heredity. 10.1038/s41437-018-0164-0 [DOI] [PMC free article] [PubMed]
- Maisano Delser P, Corrigan S, Hale M, Li C, Veuille M, Planes S, Naylor G, Mona S (2016) Population genomics of C. melanopterus using target gene capture data: demographic inferences and conservation perspectives. Sci Rep. 6:33753. 10.1038/srep33753 [DOI] [PMC free article] [PubMed]
- Margvelashvili N, Saint-Cast F, Condie S. Numerical modelling of the suspended sediment transport in Torres Strait. Continental shelf Res, Mar Resour, biophysical Process, Environ Manag a tropical shelf Seaw; Torres Strait, Aust. 2008;28:2241–2256. doi: 10.1016/j.csr.2008.03.037. [DOI] [Google Scholar]
- Marshall JF, Davies PJ. Last interglacial reef growth beneath modern reefs in the southern Great Barrier Reef. Nature. 1984;307:44–46. doi: 10.1038/307044a0. [DOI] [Google Scholar]
- McCauley DJ, Young HS, Dunbar RB, Estes JA, Semmens BX, Micheli F. Assessing the effects of large mobile predators on ecosystem connectivity. Ecol Appl. 2012;22:1711–1717. doi: 10.1890/11-1653.1. [DOI] [PubMed] [Google Scholar]
- McWilliam M, Hoogenboom MO, Baird AH, Kuo C-Y, Madin JS, Hughes TP (2018) Biogeographical disparity in the functional diversity and redundancy of corals. PNAS 201716643. 10.1073/pnas.1716643115 [DOI] [PMC free article] [PubMed]
- Mirams AGK, Treml EA, Shields JL, Liggins L, Riginos C. Vicariance and dispersal across an intermittent barrier: population genetic structure of marine animals across the Torres Strait land bridge. Coral Reefs. 2011;30:937–949. doi: 10.1007/s00338-011-0767-x. [DOI] [Google Scholar]
- Momigliano P, Florin A-B, Merilä J (2021) Biases in demographic modelling affect our understanding of recent divergence. Molecular Biology and Evolution. 10.1093/molbev/msab047 [DOI] [PMC free article] [PubMed]
- Momigliano P, Harcourt R, Robbins WD, Jaiteh V, Mahardika GN, Sembiring A, Stow A (2017) Genetic structure and signatures of selection in grey reef sharks (Carcharhinus amblyrhynchos). Heredity 119:142–153. 10.1038/hdy.2017.21 [DOI] [PMC free article] [PubMed]
- Momigliano P, Harcourt R, Robbins WD, Stow A (2015a) Connectivity in grey reef sharks (Carcharhinus amblyrhynchos) determined using empirical and simulated genetic data. Sci Rep. 5:13229. 10.1038/srep13229 [DOI] [PMC free article] [PubMed]
- Momigliano P, Harcourt R, Stow A (2015b) Conserving coral reef organisms that lack larval dispersal: are networks of Marine Protected Areas good enough? Front. Mar. Sci. 2. 10.3389/fmars.2015.00016
- Mona S. On the role played by the carrying capacity and the ancestral population size during a range expansion. Heredity. 2017;118:143–153. doi: 10.1038/hdy.2016.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mona S, Ray N, Arenas M, Excoffier L. Genetic consequences of habitat fragmentation during a range expansion. Heredity. 2014;112:291–299. doi: 10.1038/hdy.2013.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran BM, Hench K, Waples RS, Höppner MP, Baldwin CC, McMillan WO, Puebla O (2019) The evolution of microendemism in a reef fish (Hypoplectrus maya). Molecular Ecology. 10.1111/mec.15110 [DOI] [PubMed]
- Mouillot D, Villéger S, Parravicini V, Kulbicki M, Arias-González JE, Bender M, Chabanet P, Floeter SR, Friedlander A, Vigliola L, Bellwood DR. Functional over-redundancy and high functional vulnerability in global fish faunas on tropical reefs. PNAS. 2014;111:13757–13762. doi: 10.1073/pnas.1317625111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mourier J, Vercelloni J, Planes S. Evidence of social communities in a spatially structured network of a free-ranging shark species. Anim Behav. 2012;83:389–401. doi: 10.1016/j.anbehav.2011.11.008. [DOI] [Google Scholar]
- Nunziata SO, Weisrock DW. Estimation of contemporary effective population size and population declines using RAD sequence data. Heredity. 2018;120:196–207. doi: 10.1038/s41437-017-0037-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ovenden JR, Kashiwagi T, Broderick D, Giles J, Salini J. The extent of population genetic subdivision differs among four co-distributed shark species in the Indo-Australian archipelago. BMC Evolut Biol. 2009;9:40. doi: 10.1186/1471-2148-9-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pante E, Simon-Bouhet B. marmap: A Package for Importing, Plotting and Analyzing Bathymetric and Topographic Data in R. PLOS ONE. 2013;8:e73051. doi: 10.1371/journal.pone.0073051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papastamatiou YP, Lowe CG, Caselle JE, Friedlander AM. Scale-dependent effects of habitat on movements and path structure of reef sharks at a predator-dominated atoll. Ecology. 2009;90:996–1008. doi: 10.1890/08-0491.1. [DOI] [PubMed] [Google Scholar]
- Papastamatiou YP, Watanabe YY, Demšar U, Leos-Barajas V, Bradley D, Langrock R, Weng K, Lowe CG, Friedlander AM, Caselle JE. Activity seascapes highlight central place foraging strategies in marine predators that never stop swimming. Mov Ecol. 2018;6:9. doi: 10.1186/s40462-018-0127-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellissier L, Leprieur F, Parravicini V, Cowman PF, Kulbicki M, Litsios G, Olsen SM, Wisz MS, Bellwood DR, Mouillot D. Quaternary coral reef refugia preserved fish diversity. Science. 2014;344:1016–1019. doi: 10.1126/science.1249853. [DOI] [PubMed] [Google Scholar]
- Peter BM, Slatkin M. Detecting Range Expansions from Genetic Data. Evolution. 2013;67:3274–3289. doi: 10.1111/evo.12202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portik DM, Leaché AD, Rivera D, Barej MF, Burger M, Hirschfeld M, Rödel M-O, Blackburn DC, Fujita MK. Evaluating mechanisms of diversification in a Guineo-Congolian tropical forest frog using demographic model selection. Mol Ecol. 2017;26:5245–5263. doi: 10.1111/mec.14266. [DOI] [PubMed] [Google Scholar]
- Portnoy DS, Hollenbeck CM, Bethea DM, Frazier BS, Gelsleichter J, Gold JR (2016) Population structure, gene flow, and historical demography of a small coastal shark (Carcharhinus isodon) in US waters of the Western Atlantic Ocean. ICES J Mar Sci 73:2322–2332. 10.1093/icesjms/fsw098
- R Core Team (2019) R: A language and environment for statistical computing
- Ramachandran S, Deshpande O, Roseman CC, Rosenberg NA, Feldman MW, Cavalli-Sforza LL. Support from the relationship of genetic and geographic distance in human populations for a serial founder effect originating in Africa. PNAS. 2005;102:15942–15947. doi: 10.1073/pnas.0507611102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid-Anderson S, Bilgmann K, Stow A (2019) Effective population size of the critically endangered east Australian grey nurse shark Carcharias taurus. Mar Ecol Prog Ser 610:137–148. 10.3354/meps12850
- Renema W, Bellwood DR, Braga JC, Bromfield K, Hall R, Johnson KG, Lunt P, Meyer CP, McMonagle LB, Morley RJ, O’Dea A, Todd JA, Wesselingh FP, Wilson MEJ, Pandolfi JM. Hopping Hotspots: Global Shifts in Marine Biodiversity. Science. 2008;321:654–657. doi: 10.1126/science.1155674. [DOI] [PubMed] [Google Scholar]
- Robbins WD (2006) Abundance, demography and population structure of the grey reef shark (Carcharhinus amblyrhynchos) and the white tip reef shark (Triaenodon obesus) (Fam. Charcharhinidae) (PhD Thesis). James Cook University
- Robbins WD, Hisano M, Connolly SR, Choat JH. Ongoing Collapse of Coral-Reef Shark Populations. Curr Biol. 2006;16:2314–2319. doi: 10.1016/j.cub.2006.09.044. [DOI] [PubMed] [Google Scholar]
- Roff G, Brown CJ, Priest MA, Mumby PJ. Decline of coastal apex shark populations over the past half century. Commun Biol. 2018;1:223. doi: 10.1038/s42003-018-0233-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rougeux C, Bernatchez L, Gagnaire P-A (2017) Modeling the Multiple Facets of Speciation-with-Gene-Flow toward Inferring the Divergence History of Lake Whitefish Species Pairs (Coregonus clupeaformis). Genome Biol Evol 9:2057–2074. 10.1093/gbe/evx150 [DOI] [PMC free article] [PubMed]
- Roycroft E, MacDonald AJ, Moritz C, Moussalli A, Miguez RP, Rowe KC (2021) Museum genomics reveals the rapid decline and extinction of Australian rodents since European settlement. PNAS 118. 10.1073/pnas.2021390118 [DOI] [PMC free article] [PubMed]
- Saint-Cast F, Condie SA. Circulation modelling in Torres Strait. Canberra: Geoscience Australia; 2006. [Google Scholar]
- Sandin SA, Smith JE, DeMartini EE, Dinsdale EA, Donner SD, Friedlander AM, Konotchick T, Malay M, Maragos JE, Obura D, Pantos O, Paulay G, Richie M, Rohwer F, Schroeder RE, Walsh S, Jackson JBC, Knowlton N, Sala E. Baselines and Degradation of Coral Reefs in the Northern Line Islands. PLOS ONE. 2008;3:e1548. doi: 10.1371/journal.pone.0001548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sansaloni C, Petroli C, Jaccoud D, Carling J, Detering F, Grattapaglia D, Kilian A (2011) Diversity Arrays Technology (DArT) and next-generation sequencing combined: genome-wide, high throughput, highly informative genotyping for molecular breeding of Eucalyptus. BMC Proc 5:P54. 10.1186/1753-6561-5-S7-P54
- Simpfendorfer C, Fahmi Bin Ali A, Utzurrum JAT, Sehya L, Maung A, Bineesh KK, Yuneni RR, Sianipar A, Haque AB, Tanay D, Gautama DA, Vo VQ, (2020) IUCN Red List of Threatened Species: Carcharhinus amblyrhynchos. IUCN Red List of Threatened Species
- Sousa V, Hey J. Understanding the origin of species with genome-scale data: modelling gene flow. Nat Rev Genet. 2013;14:404–414. doi: 10.1038/nrg3446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speed CW, Meekan MG, Field IC, McMahon CR, Harcourt RG, Stevens JD, Babcock RC, Pillans RD, Bradshaw CJA. Reef shark movements relative to a coastal marine protected area. Regional Stud Mar Sci. 2016;3:58–66. doi: 10.1016/j.rsma.2015.05.002. [DOI] [Google Scholar]
- Speed CW, Meekan MG, Field IC, McMahon CR, Stevens JD, McGregor F, Huveneers C, Berger Y, Bradshaw CJA. Spatial and temporal movement patterns of a multi-species coastal reef shark aggregation. Mar Ecol Prog Ser. 2011;429:261–275. doi: 10.3354/meps09080. [DOI] [Google Scholar]
- Subramanian S. The effects of sample size on population genomic analyses – implications for the tests of neutrality. BMC Genomics. 2016;17:123. doi: 10.1186/s12864-016-2441-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Etten J, (2018) gdistance: Distances and Rutes on Geographical Grids
- Vignaud TM, Mourier J, Maynard JA, Leblois R, Spaet JLY, Clua E, Neglia V, Planes S (2014) Blacktip reef sharks, Carcharhinus melanopterus, have high genetic structure and varying demographic histories in their Indo-Pacific range. Mol Ecol 23:5193–5207. 10.1111/mec.12936 [DOI] [PubMed]
- Waples RK, Larson WA, Waples RS. Estimating contemporary effective population size in non-model species using linkage disequilibrium across thousands of loci. Heredity. 2016;117:233–240. doi: 10.1038/hdy.2016.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waples RS, Antao T, Luikart G. Effects of Overlapping Generations on Linkage Disequilibrium Estimates of Effective Population Size. Genetics. 2014;197:769–780. doi: 10.1534/genetics.114.164822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waples RS, Do C. ldne: a program for estimating effective population size from data on linkage disequilibrium. Mol Ecol Resour. 2008;8:753–756. doi: 10.1111/j.1755-0998.2007.02061.x. [DOI] [PubMed] [Google Scholar]
- Webster JM, Braga JC, Humblet M, Potts DC, Iryu Y, Yokoyama Y, Fujita K, Bourillot R, Esat TM, Fallon S, Thompson WG, Thomas AL, Kan H, McGregor HV, Hinestrosa G, Obrochta SP, Lougheed BC. Response of the Great Barrier Reef to sea-level and environmental changes over the past 30,000 years. Nat Geosci. 2018;11:426. doi: 10.1038/s41561-018-0127-3. [DOI] [Google Scholar]
- Wei S, Li Z, Momigliano P, Fu C, Wu H, Merilä J. The roles of climate, geography and natural selection as drivers of genetic and phenotypic differentiation in a widespread amphibian Hyla annectans (Anura: Hylidae) Mol Ecol. 2020;29:3667–3683. doi: 10.1111/mec.15584. [DOI] [PubMed] [Google Scholar]
- White TD, Carlisle AB, Kroodsma DA, Block BA, Casagrandi R, De Leo GA, Gatto M, Micheli F, McCauley DJ. Assessing the effectiveness of a large marine protected area for reef shark conservation. Biol Conserv. 2017;207:64–71. doi: 10.1016/j.biocon.2017.01.009. [DOI] [Google Scholar]
- Wolanski E, Ridd P, Inoue M. Currents through Torres Strait. J Phys Oceanogr. 1988;18:1535–1545. doi: 10.1175/1520-0485(1988)018<1535:CTTS>2.0.CO;2. [DOI] [Google Scholar]
- Woodroffe CD, Kennedy DM, Hopley D, Rasmussen CE, Smithers SG. Holocene reef growth in Torres Strait. Mar Geol. 2000;170:331–346. doi: 10.1016/S0025-3227(00)00094-3. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw sequence data from sites in New Caledonia is on Zenodo (10.5281/zenodo.5935133) and data from other sites is available on GenBank Sequence Read Archive (Bioproject PRJNA795958). The code needed to reproduce the analyses performed here can be found in GitHub repositories (https://github.com/cajwalsh/grey_reef_gen_demog and https://github.com/Nopaoli/Shark-Demographic-Models).