

ABSTRACT
The bluegill sunfish Lepomis macrochirus and the closely related redear sunfish Lepomis microlophus have important ecological and recreational value and are widely used for research and aquaculture. While both species have been introduced outside of their native ranges, only the bluegill is considered invasive. Here, we report de novo transcriptome assemblies for these fish as a resource for sunfish biology. Comparative analyses of the transcriptomes revealed an unexpected, bluegill-specific expansion in the HSP70 and HSP90 molecular chaperone gene families. These expansions were not unique to the bluegill as expansions in HSP70s and HSP90s were identified in the genomes of other teleost fish using the NCBI RefSeq database. To determine whether gene family expansions are specific for thermal stress responses, GST and SOD gene families that are associated with oxidative stress responses were also analyzed. Species-specific expansions were also observed for these gene families in distinct fish species. Validating our approach, previously described expansions in the MHC gene family were also identified. Intriguingly, the number of HSP70 paralogs was positively correlated with thermotolerance range for each species, suggesting that these expansions can impact organismal physiology. Furthermore, fish that are considered invasive contained a higher average number of HSP70 paralogs than non-invasive fish. Invasive fish also had higher average numbers of HSP90, MHC and GST paralogs, but not SOD paralogs. Taken together, we propose that expansions in key cellular stress response gene families represent novel genetic signatures that correlate with invasive potential.
KEY WORDS: HSP70, HSP90, Heat shock proteins, Transcriptome, Invasive species, Bluegill sunfish
Summary: A comparative analysis of sunfish transcriptomes and 95 fish genomes reveals expansions in specific stress response gene families that correlate with invasive potential.
INTRODUCTION
The genus Lepomis in the order Perciformes and family Centrarchidae contains 13 extant freshwater ray-finned fish species native to North America commonly known as sunfish (Near et al., 2005). Sunfish have been widely used in research, including biomechanical models for suction feeding (Carroll et al., 2004; Wainwright, 1996) and feeding ecology (Brogowski et al., 2005; Osenberg et al., 1988), responses to environmental pollutants (van den Hurk et al., 2017), host species to study parasites (Boone et al., 2018), sexual dimorphisms (Hayward and Wang, 2006), sex-determination (Shen et al., 2016) and alternative reproductive strategies (Gross and Charnov, 1980; Partridge et al., 2016). Sunfish are abundant panfish that are valued for recreational fishing and have been used for aquaculture (Fuller et al., 1999; Takahara et al., 2013).
The bluegill sunfish Lepomis macrochirus Rafinesque 1819 and the redear sunfish Lepomis microlophus Günther 1859 are closely related fishes with overlapping native ranges in the southeastern USA, including subtropical regions such as Florida (Fig. 1, top panels). Ecological and morphological differences between these two species include feeding mechanisms and prey. The bluegill sunfish is a suction-feeding generalist with a small mouth and large buccal cavity, feeding mostly on water-column prey such as plankton and small crustaceans; whereas redear sunfish is a molluscivorous specialist with large pharyngeal jaws, feeding on benthic hard prey such as snails (Carroll et al., 2004; Fisher Huckins et al., 2000; Wainwright, 1996). While both of these fish species have been widely introduced outside of their native ranges, the current distribution of the bluegill sunfish is much broader than the redear sunfish in the USA and across the globe (Fig. 1, bottom panels). The bluegill sunfish can be found in North America, South America, Africa and Asia, while the redear sunfish is primarily found in North America. Similarly, the native range of bluegill extends through a much wider range of latitudes than redear, extending up to the northernmost regions of the eastern USA.
Fig. 1.

Distributions of the bluegill sunfish and redear sunfish reveal an expanded native range and distribution for bluegill. In the top panels, the native ranges are shown in tan and expanded ranges in the USA are shown in shades of red indicating various levels of evidence. The data is from the United States Geological Survey (USGS) Nonindigenous Aquatic Species Database (https://nas.er.usgs.gov/). In the bottom panels, the current global distributions are shown with data taken from the CABI (Centre for Agriculture and Bioscience International) Invasive Species Compendium (https://www.cabi.org/ISC/). The left panels correspond to the bluegill sunfish and the right panels correspond to the redear sunfish.
Importantly, only the bluegill sunfish has been considered a pest in some areas where it has been introduced, including South Africa, Germany, the UK, Japan and Korea (Kawamura et al., 2006; Toshikazu and Tetsuro, 2004). Therefore, the bluegill sunfish is classified as an invasive species by the Center for Invasive Species and Ecosystem Health (www.invasive.org) and in the CABI Invasive Species Compendium (https://www.cabi.org/ISC/). The prevalence of invasive species has become a global environmental problem with negative impacts on agriculture and the environment (Pimentel et al., 2001). Recent cost estimates from invasive species reach as high as US$162 billion annually (Diagne et al., 2021).
Myriad factors influence the ability of an organism to invade and overtake new territory (Pauchard et al., 2004; Ricciardi et al., 2017; Sakai et al., 2001; Thuiller et al., 2012; Turingan and Sloan, 2016). Factors known to influence invasive potential can be roughly split into two categories: external and inherent. External factors include predation, food abundance and anthropogenic influences. Inherent factors that promote invasiveness include rapid reproduction rates, a high mutation rate, facile adaptation to new environments, and robust stress responses that are able to withstand taxing introduction events. Expansions in particular gene families have been associated with some of these traits, revealing that inherent factors can be associated with specific genetic signatures. For example, an expansion in the MHC gene family, which contributes to immune responses, has been identified in some fish, including Atlantic cod and tilapia (Grimholt, 2016; Star et al., 2011). Furthermore, an expansion in HSP70, which affects thermotolerance, has been identified in Limnoperna fortune, an invasive mussel species (Uliano-Silva et al., 2014).
Thus far, the genetic basis of invasiveness potential for the bluegill sunfish has not been established. Unfortunately, there is limited genomic information available for any sunfish. Although the technology to obtain genomic information has advanced rapidly, the time and the expense associated with these approaches is still a significant barrier to research. In the past decade, transcriptomic analysis has emerged as an important genomic tool to study many aspects of fish biology, including developmental biology, immunology, aquatic toxicology, physiology and evolution (Qian et al., 2014). Recent improvements in sequencing technology that facilitate longer sequencing read lengths have improved the ability to collect transcriptomic information. In fact, current technology and bioinformatic tools allow for the analyses of transcriptomes even without the existence of completed genomic sequence information or annotation, termed de novo transcriptomics. In this manuscript, we use comparative genomic analysis based on transcriptomics in order to investigate genetic correlations with the expanded range and invasive potential of bluegill.
MATERIALS AND METHODS
Specimen collection
Bluegill and redear sunfish were collected from freshwater ponds in Melbourne, Florida, which is within the native range for both species. After collection, the fish were acclimated in an aquarium overnight to minimize gene expression changes associated with handling stress. Then, fish were anesthetized by immersion in 100 mg tricaine methanesulfonate (MS 222) per liter of freshwater solution (Schreck and Moyle, 1990). Approximately 5 g muscle tissue was excised from the anterior head of the epaxialis muscle, flash frozen in liquid nitrogen and stored at −140°C. Fish handling procedures followed the Florida Institute of Technology animal care guidelines under IACUC permit #151001-01.
RNA purification and sequencing
Frozen muscle tissue was ground with a mortar and pestle. The resulting fine powder was subjected to two organic extractions using Trizol (Thermo Fisher Scientific) and precipitated with isopropanol. RNA sample quality was assessed via visualization on agarose gel and with an Agilent 2100 bioanalyzer and then quantified with a Qubit. RNA preparation (including rRNA subtraction), cDNA library construction and PE150 sequencing on an Illumina HiSeq was performed by Novogene. The quality of the sequencing was determined by measuring the sequencing error rates per nucleotide and by using Q20 and Q30 metrics, which correspond to the percentage of sequencing with scores better than 99% (Q20) or 99.9% (Q30) inferred base call accuracy of the clean reads.
De novo transcriptome assembly and annotation
As there are no reference genomes for bluegill or redear sunfish, sequence reads were assembled to generate a de novo transcriptome using Trinity RNA-Seq software (v. r20140413p1; https://github.com/trinityrnaseq/trinityrnaseq/wiki; Grabherr et al., 2011; Haas et al., 2013). Preliminary data analysis and de novo transcriptome assembly were performed by Novogene. The ‘SS_lib_type’ and ‘min_glue’ parameters used in the Trinity program were set to default while the ‘min_kmer_cov’ parameter was set to 2 to help remove low quality k-mer sequences. The longest assembled transcript from each Trinity component was selected as the unigene and used for downstream annotation and analysis. Each unigene was annotated by searching through seven databases: NR (NCBI non-redundant protein sequences), NT (NCBI nucleotide sequences), Pfam (Protein family), KOG (euKaryotic Orthologous Groups), Swiss-Prot, KEGG (Kyoto Encyclopedia of Genes and Genome), and GO (Gene Ontology) databases.
Gene identification in model organisms
Zebrafish orthologues for literature-annotated sets of human genes were identified using the Alliance of Genome Resources Database (https://alliancegenome.org), which contains an orthology identification tool that uses 12 different methodologies (Ensembl Compara, HGNC, Hieranoid, InParanoid, OMA, Orthofinder, OrthoInspector, PANTHER, PhylomeDB, Roundup, TreeFam and ZFIN) (The Alliance of Genome Resources Consortium et al., 2020). This database was also used to identify homologous genes across other model organisms (humans, mouse, rat, zebrafish, fruit fly, worm and yeast). The orthology searches were used with the ‘no filter’ setting so that an orthologue was included if it was identified using any of these tools.
Gene identification in fish
Coding sequences for zebrafish genes were obtained from Ensembl and used for nucleotide BLAST (BLASTn) searches against sunfish transcriptomes or fish RefSeq genomes as indicated. Discontiguous MegaBLAST with the default E-value threshold of 10−5 was used as recommended for interspecies comparisons. For the sunfish transcriptomes, the BLAST searches were completed in two stages. First, the zebrafish sequences were used as the query and searched against the TSA submission. Then, each of the sunfish sequences were used to BLAST against the reference zebrafish RNA sequences database (refseq_rna) to uniquely associate it with a zebrafish orthologue. When more than one sunfish unigene was associated with the same zebrafish gene, additional approaches were used to determine if they represented multiple paralogs. First, the sunfish unigenes were blasted against each other to ensure that they were not splice isoforms or multiple alleles of a single gene and excluded if the similarity was greater than 95%. Then, all sunfish unigene paralogs were mapped onto the zebrafish gene and excluded if they represented non-overlapping fragments.
RESULTS
Transcriptome assembly and annotation reveal largely similar genomes between bluegill and redear
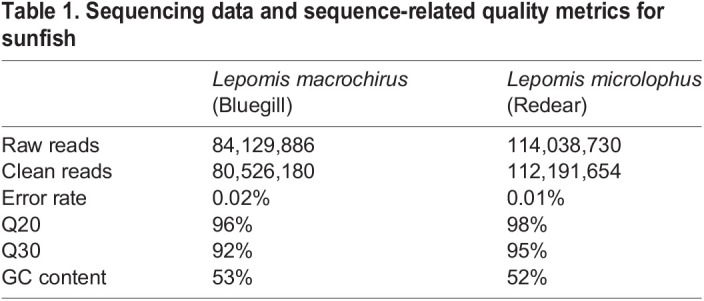
In order to rapidly and economically obtain genome-level information, we performed a comparative genetic analysis of the bluegill sunfish and the redear sunfish using RNA sequencing. Illumina PE150 RNA-seq generated more than 80 million clean reads (>12G clean bases) from bluegill and more than 112 million clean reads (>16G clean bases) from redear (Table 1). Good sequencing quality was indicated by low sequencing error rates (<0.02%), and the samples having >96% for Q20 and >92% for Q30 for the clean reads. The GC content of the transcriptome was similar for both species, at 52% for redear and 53% for bluegill. The sequences were deposited in the Short Read Archive (SRA) in the NCBI database for the bluegill sunfish (SRR5645631) and the redear sunfish (SRR5646640).
Table 1.
Sequencing data and sequence-related quality metrics for sunfish
As there are no existing genomes for these fish, de novo transcriptomes were assembled from the clean reads using Trinity (Moreton et al., 2016). This approach generated a set of unique transcripts, some of which could arise from multiple alleles or through alternative RNA processing. Therefore, overlapping transcripts were further collapsed into a set of unique unigenes containing the longest transcript. This approach generated 54,711 unigenes for bluegill with a median length of 343 nucleotides and a total of 32 million total nucleotides (Table 2). For redear, this approach generated 150,444 unigenes with a median length of 805 nucleotides and 207 million total nucleotides. Unexpectedly, the number of transcripts and unigenes were similar in the redear sunfish, indicating that this pipeline was suboptimal at collapsing alternative transcripts into a smaller set of unigenes for this species.
Table 2.
Transcriptome data and transcriptome-related quality metrics for sunfish

The quality of the transcriptomes was assessed using several distinct metrics. The first was an analysis of the N50 and N90 values, which indicate the minimum length of contigs that contain 50% or 90% of the total sequence content of the unigenes. The N50 and N90 values are 838 and 250 for the bluegill and 2185 and 583 for redear respectively (Table 2). The higher N values for redear are consistent with the greater depth of sequencing information collected. Additionally, transcriptome quality was assessed by searching for genes that are typically found only in a single copy at different levels of phylogeny using BUSCO version 5 (Benchmarking Universal Single-Copy Orthologs) (Manni et al., 2021; Simão et al., 2015). This analysis suggested that compared with the typical Eukaryota, the bluegill transcriptome is 63% complete, 23% fragmented and has approximately 15% missing genes, and the redear transcriptome is 90% complete, 7% fragmented and 3% missing (Table 3). The relatively low completeness of the bluegill transcriptome indicates that additional transcriptomic or genomic sequencing of bluegill could be beneficial. In contrast, the redear is more complete, but it has a much higher level of potential duplication artifacts, estimated at approximately 45%. This likely contributes to an artificially high unigene number for redear. Nevertheless, these metrics indicate that both transcriptomes are of good quality. Therefore, the transcriptomes were deposited as transcript shotgun assembly (TSA) projects at DDBJ/ENA/GenBank under the accession numbers GFOV00000000 (bluegill) and GFPH00000000 (redear). The versions described in this paper are the first versions, GFOV00000000.1 and GFPH00000000.1.
Table 3.
BUSCO analysis of universal, single copy orthologues at various levels of taxonomy for sunfish transcriptomes

In order to make functional predictions for the unigenes, each unigene was annotated by BLAST searches through seven widely used databases: NR, NT, Pfam, KOG, Swiss-Prot, KEGG and GO. Most of the unigenes (72% for bluegill and 63% for redear) were annotated in at least one database. The best match for the majority of bluegill sequences were nucleotide sequences from other fish species, including Larimichthys crocea (Richardson, 1846) (41.1%), Stegastes partitus (Poey 1868) (18.7%), Notothenia coriiceps Richardson 1844 (7.1%), Oreochromis niloticus (Linnaeus 1758) (4.7%), and Haplochromis burtoni (Günther 1894) (2.8%) (Fig. S1). Consistent with current phylogenies, the best matches to bluegill are from the Percomorpha, which includes the Centrarchiformes (sunfishes) (Near et al., 2012). As expected, analysis of the distribution of matches from redear revealed a similar closest-match species distribution to that observed for bluegill.
Gene analysis uncovers expansion of HSP70 and HSP90 gene families in bluegill
Typically, transcriptomic analyses are used to detect differential gene expression patterns in one species that have been exposed to different conditions. However, de novo transcriptomics contains a wealth of information that can be used for gene-level analysis rather than transcript-level analysis. In order to analyze species-specific differences in gene composition between the bluegill sunfish and the redear sunfish, we first looked at gene categories by comparing the gene classifications from the KEGG database (Fig. S2). This analysis revealed nearly identical gene family distributions between the two sunfish.
Having observed no striking differences in gene classifications, we next examined specific gene families that could be associated with the expanded range of the bluegill sunfish. Temperature has been shown to have dramatic influences on fish physiology (Brett, 1971; Qin et al., 2018). Therefore, we first analyzed the six different families of heat shock proteins (HSPs): HSP110s, HSP90s, HSP70s, HSP40s chaperonins and small HSPs (Kampinga et al., 2009). Many HSPs function as molecular chaperones that maintain protein folding homeostasis, or proteostasis, and constitute one of the primary cellular defenses against proteotoxic stress (Balchin et al., 2016; Guisbert and Morimoto, 2012). HSPs are important for thermal stress responses and required for development in many organisms including fish (Basu et al., 2002; Deane and Woo, 2011; Iwama et al., 1998).
HSP homologs were identified in the bluegill and redear sequences using BLAST starting from the well-annotated zebrafish genome. This analysis revealed that the distribution of HSPs in the sunfish transcriptome varies among the different HSP families (Fig. 2A, Table S1). For the small HSP gene family, we found substantially fewer HSP unigenes in both bluegill (7) and redear (6) than in zebrafish (14). This is likely due to a limitation of our transcriptome since many small HSPs have restricted, tissue-specific expression patterns and we analyzed only muscle tissue. In addition, our approach cannot identify genes that are not expressed and many small HSPs are expressed at very low levels in the absence of stress. For the chaperonin, HSP40 and HSP110 gene families, we identified similar or only slightly fewer unigenes in bluegill and redear compared with zebrafish. In contrast, for the HSP70 gene family, there were 37.5% more unigenes in bluegill (22) and 12.5% more in redear (18) than found in zebrafish (16). For the HSP90 gene family, there were 60% more in bluegill (8) and 40% more in redear (7) than found in zebrafish (5).
Fig. 2.

Analysis of stress response genes in sunfish indicate a bluegill-specific expansion in HSP70s and HSP90s. (A) Number of stress response genes found in sunfish transcriptomes using BLAST searches starting with zebrafish coding regions for selected gene families. Sunfish Unigene numbers are compared with the zebrafish genome counts. (B) Number of HSP70 and HSP90 genes found in model organism genome databases compared with sunfish transcriptomes.
To determine the specificity of these expansions, several additional gene families were analyzed (Fig. 2A, Table S1). The major histocompatibility (MHC) gene family was included as expansions in this family have been previously described in some fish including Atlantic cod (Grimholt, 2016). MHC genes encode cell surface proteins that present antigens to the cells in the immune system among other functions. We identified 26 MHC class I and 25 MHC class II genes in the zebrafish genome, yet only a small number in bluegill (3 MHC class I and 3 MHC class II) and redear (11 MHC class I and 4 MHC class II). We also analyzed two stress response gene families associated with resistance to oxidative damage and xenobiotic stresses: the superoxide dismutase (SOD) and glutathione S-transferase (GST) families. SOD genes encode for proteins that detoxify oxygen radicals and GST genes encode for proteins that can reverse oxidative damage and maintain redox homeostasis. There are 4 SOD and 26 GST paralogues in zebrafish, but we identified only 3 SOD and 14 GST orthologues in both bluegill and redear. Together, the analysis of the MHC, SOD, and GST gene families further indicates that the expansions found in bluegill HSP70s and HSP90s are specific to these gene families.
In order to investigate whether expansions in HSP70 and HSP90 gene families occur in other taxa, we analyzed these gene families in model organisms including mouse (Mus musculus), rat (Rattus norvegicus), zebrafish (Danio rerio), worms (Caenorhabditis elegans), fruit flies (Drosophila melanogaster) and yeast (Saccharomyces cerevisiae) using the Alliance of Genome Resources Database (https://alliancegenome.org). This analysis revealed that the number of HSP70 genes in each species was relatively constant, ranging from 11 members in mice to 16 members in zebrafish (Fig. 2B). The number of HSP90 genes was also relatively constant, ranging from 2 in yeast to 5 in zebrafish and rats. While in both cases, zebrafish has more gene family members than other model organisms, this analysis further indicated that the 22 HSP70 paralogs and 8 HSP90 paralogs in bluegill were unusual.
Gene family expansions in fish correlate with invasiveness
To analyze whether HSP70 and HSP90 gene family expansions occur in other fish species, we conducted a comprehensive analysis of their distribution in all of the 95 teleost fish genomes contained in the RefSeq Reference Sequence Database (https://www.ncbi.nlm.nih.gov/refseq/about/). RefSeq is a comprehensive, non-redundent database of well-annotated, high-quality genomes maintained by NCBI. Similar to the analysis of the transcriptomes, zebrafish HSP genes were used to identify HSP homologs with BLAST. Our analysis found that zebrafish provided a good baseline for this approach since it was rare for any fish species to have fewer orthologues than zebrafish (Fig. S3, Table S2). In contrast, we found multiple fish species that contained gene family expansions in each of the HSP70, HSP90, MHC, GST and SOD gene families. For example, the HSP70 family was expanded more than 2.6 times in green swordtail, Xiphophorus helleri Heckel 1848, the HSP90 gene family was expanded 4.2 times in Paramormyrops kingsleyae (Günther 1896), the GST gene family was expanded 2 times in the clown anemonefish Amphiprion ocellaris Cuvier 1830, the MHC class I gene family was expanded 4.8 times in Atlantic cod, Gadus morhua Linnaeus 1758, and the SOD gene family was expanded 2.3 times in ploughfish, Gymnodraco acuticeps Boulenger 1902 (Fig. 3). This approach correctly identified the previously described MHC expansion in Atlantic cod (Star et al., 2011). Taken together, this analysis indicates the presence of specific stress response gene expansions in various teleost fishes.
Fig. 3.

Identification of stress response gene expansions in well-annotated fish genomes. BLAST searches were conducted starting with zebrafish coding regions for selected stress response gene families across all fish in the RefSeq database. Shown are selected species of fish that contain expansions in HSP70, HSP90, GST, MHC and SOD gene families. Data are plotted as a percentage relative to zebrafish levels.
The presence of gene family expansions does not indicate whether the gene duplications are functionally relevant. The HSP70 gene family has been strongly associated with thermotolerance. Therefore, to explore whether expansions in the HSP70 gene family expansions could affect organismal physiology, we compared the number of HSP70 genes with the thermotolerance range for each species. Thermotolerance ranges have been measured or estimated for a wide variety of fish (Dahlke et al., 2020a,b). We found a positive correlation between the number of HSP70 genes and the thermotolerance range (Fig. 4, Table S3). A regression trendline (least squares) for this data revealed a slope of 0.17 and an R2 value of 0.16. A strong correlation was not observed for expansions in the other gene families that were analyzed (Fig. S4). Together, this analysis suggests a correlation exists between the number of HSP70 gene family members and organismal thermotolerance.
Fig. 4.
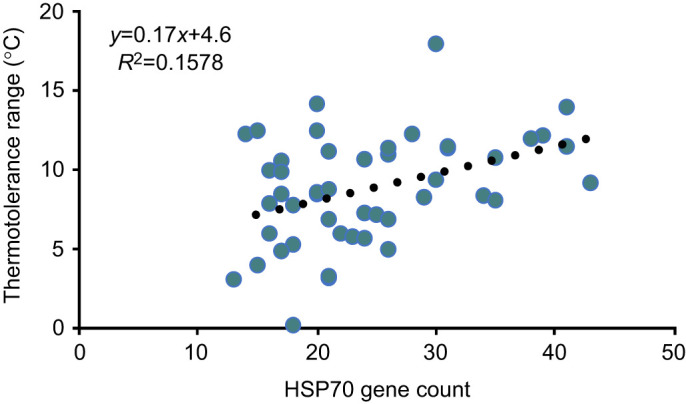
Positive correlation between the HSP70 gene count and thermotolerance range. The number of HSP70 paralogs identified in the RefSeq genome in each species was plotted relative to the thermotolerance range for each fish species included in both databases. The dotted line represents a least squares linear regression of the data. The equation describing the line and the R2 correlation are included.
Thermotolerance is a trait that has been associated with invasive potential. Therefore, we tested whether the number of HSP70 paralogs would also correlate with invasiveness. We used a database from the Center for Invasive Species and Ecosystem Health to determine which of the fish species with RefSeq genomes have been identified as invasive in North America. Then, we compared the number of HSP70 genes in the fish listed as invasive (21 species) versus species that are not listed as invasive. We found a striking, nearly 40% increase in the average number of HSP70 genes in invasive fish (30) compared with noninvasive fish (22) (Fig. 5, Table S3). As expected, we also found a significant increase in the number of MHC genes in invasive fish (72 compared with 43). The role of HSP90 in phenotypic plasticity suggested that it could also contribute to invasive potential. In support of this hypothesis, we also found an increase in HSP90 genes in invasive compared with noninvasive fish (10 compared with 7). Finally, we tested whether the GST and SOD genes correlate with invasive potential as they have roles in the defense against oxidative damage. Here, we only found an increase in GST genes (48 compared with 36) in invasive fish. Together, these data reveal that gene family expansions for a variety of stress response genes including HSP70, HSP90, GST, and MHC correlate with fish invasiveness and therefore could contribute to invasive potential.
Fig. 5.
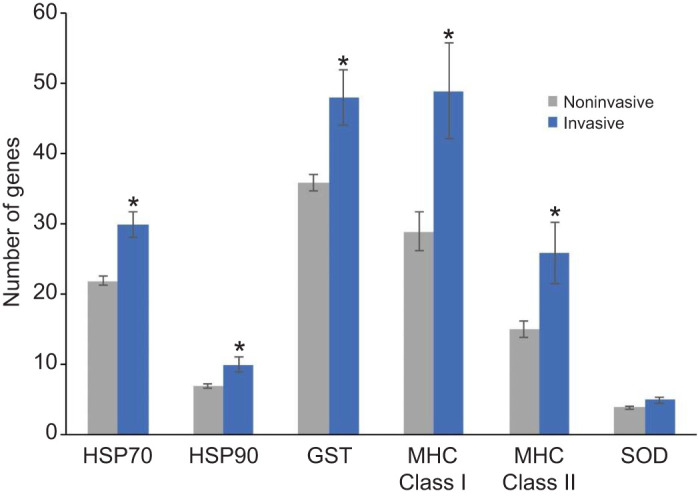
Positive correlation between stress response gene counts and invasive potential. All fish with RefSeq genomes were divided into two groups, those listed as invasive in the CABI database (blue) and those that were not (gray). Shown are the average number of stress response genes for each group. Error bars represent s.e.m. Asterisk indicates P<0.05 with the Student's t-test.
DISCUSSION
Species can be ‘hard-wired’ to be broadly successful in a wide variety of ecological environments by their genomes. We report here that invasive fish contain specific expansions in several stress response gene families, including HSP70s, HSP90s and GSTs. This association was first identified upon generation of de novo transcriptomes for the invasive bluegill sunfish and the closely related but noninvasive redear sunfish. Then, the association was validated and expanded using all 95 well-annotated fish genomes available in the NCBI RefSeq database. Taken together, this analysis suggests that duplications in stress response gene families represent novel genetic mechanisms that contribute to invasive potential (Fig. 6).
Fig. 6.
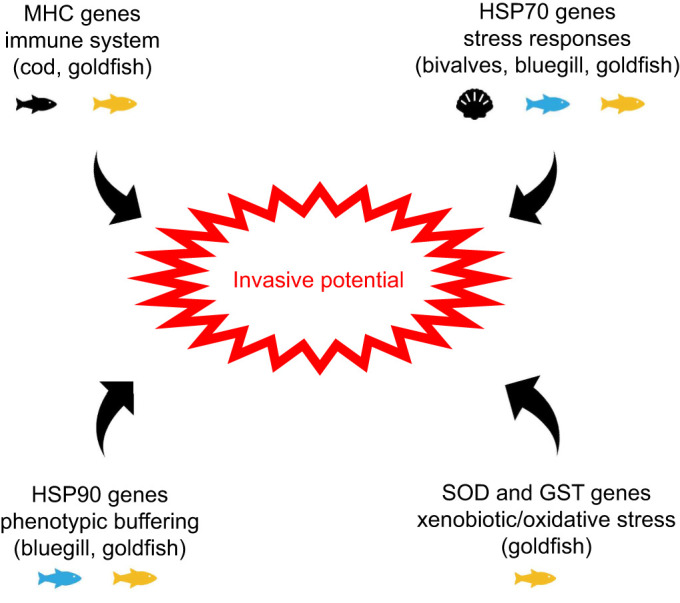
Model figure proposing a link between expansions in stress response and immunity genes and the invasive potential of an organism.
To our knowledge, this work represents the first description of an HSP70 gene family expansion in a vertebrate. HSP70 proteins are a class of molecular chaperones that maintain protein folding homeostasis or proteostasis. They are the cellular workhorses responsible for promoting the proper folding of a large fraction of the proteome as well as clearing the bolus of denatured proteins that are induced upon stress conditions. An expansion in HSP70 chaperones could promote organismal survival in a broad range of conditions or during stressful events such as introduction into new habitats. Remarkably, the number of HSP70 chaperones is stable in model organisms from yeast to humans. Notably, an expansion of the HSP70 family has been documented in mollusks (Cheng et al., 2016; Uliano-Silva et al., 2014; Zhang et al., 2012). Mollusks occupy the intertidal region and are subject to dramatic daily fluctuations in their environment. Similarly, the HSP70 expansion in bluegill and other fish could enable them to enjoy a wide habitable range. In support of this, we found a correlation between the number of HSP70s and the thermal range for fish. In fishes, a broad habitable range has been associated with invasive potential (Kolar, 2002). Therefore, we hypothesize that the expansion in HSP70 chaperones is a genetic mechanism that accounts for the broad habitable range of bluegill and also contributes to its invasive potential.
The HSP90 family has been postulated to function as an evolutionary capacitor that increases the tolerable load of cryptic variation available for natural selection (Lindquist, 2009; Rutherford and Lindquist, 1998; Sangster et al., 2004). Accordingly, HSP90 is proposed to act as a buffer for morphological evolution and phenotypic variation. This role of HSP90 has been borne out in organisms as diverse as E. coli, yeast, Arabidopsis, Tetrahymena, cavefish and more (Frankel et al., 2001; Queitsch et al., 2002; Rohner et al., 2013; Tyedmers et al., 2008). Similar to HSP70s, the number of HSP90s is remarkably stable in model organisms from yeast to humans. Since increased genetic diversity or faster rates of mutation are a feature of invasive species, we hypothesize that the expansion of HSP90s in bluegill is a second genetic mechanism that could contribute to its invasive potential.
The discovery of HSP70 and HSP90 transcriptome expansion was possible through the combination of two approaches: targeted analysis of specific gene families and de novo transcriptomics. Interestingly, the HSP70 and HSP90 expansions were not apparent from traditional annotation efforts using the standard databases. Standard functional classifications of gene annotations including KEGG, KOG and GO showed that the two species are largely similar. This highlights an important limitation of first-level analyses for comparative genomics and reinforces the value of manual curation of genomic data. Furthermore, our research also highlights the value of the massive amounts of sequencing information in the database, and how these databases provide an important tool for re-evaluation of molecular pathways to gain new insights into biology. As we have shown, the identification of these expansions would not have been possible with approaches limited to model organisms.
Our research supports the generation of de novo transcriptomes for gene analysis as a rapid, cost-effective and facile approach to rapidly identify genetic signatures in non-model organisms. Transcriptome level analyses are also widely accessible because of publicly available, user-friendly computation programs and resources. Furthermore, by employing transcriptomic rather than genomic sequencing, the focus is immediately on the ∼1–5% of the genome that encodes proteins, which greatly simplifies complexity and cost.
Approaches using de novo transcriptomes are not without their limitations. For example, the valuable information carried in non-coding regions of the genome is sacrificed, which limits its use for population genetics and regulatory analyses that depend on non-coding regions. Additionally, transcriptomic analyses can only evaluate genes that are expressed in the tissue examined. Furthermore, this approach generates unigene gene models that cannot carry the same weight as genes annotated from a whole-genome sequencing effort, illustrated by a significant percentage of fragmented genes and likely duplications identified by the BUSCO analysis. Despite these drawbacks, we posit that our approach provides a valuable alternative to whole genome sequencing and can be used to motivate future deep sequencing efforts.
HSP70 and HSP90 expansions were first identified in the bluegill de novo transcriptome and later validated in well-annotated genomes of multiple other fish species using the RefSeq database. Because all fish appear to have slightly more HSP70s than standard model organisms, this suggests that one source of the additional paralogs is a teleost-specific whole genome duplication (WGD) that occurred in the common ancestor of extent teleost lineages (Glasauer and Neuhauss, 2014). However, not every WGD results in more HSP70s since the two rounds of WGD in the common ancestor of extant vertebrates did not result in more HSP70s in humans and mice compared with yeast, worms and flies (Fig. 2B). In fact, the number of HSP70s is remarkably well-conserved across phyla from yeast to humans. Interestingly, analysis of the RefSeq genomes enabled identification of expansions in other stress response gene families, including GSTs and SODs involved in oxidative stress. However, each gene family presented with a largely unique pattern of expansions across fish species. In some cases, fish species such as goldfish (Carassius auratus) contain multiple gene family expansions, likely reflecting a recent whole genome duplication within the cyprinid lineage.
Identification of gene family expansions alone does not reveal whether these additional genes are functionally relevant. Importantly, HSP70 expansions were found to correlate with a larger thermal range. Furthermore, HSP70, HSP90, MHC and GST expansions each individually correlated with invasive potential. Intriguingly, combinations of stress response gene family expansions were observed in some species. For example, the expansion of both the HSP70 and HSP90 chaperone families in bluegill. This leads to the hypothesis that multiple expansions could play synergistic roles. Robust responses to stressful environments from the HSP70 expansion coupled to increase in phenotypic plasticity and rapid adaptation to new environments from the HSP90 expansions could function together to increase invasive potential of the bluegill. Remarkably, all the stress response gene families were expanded in goldfish. These expansions may contribute to the hardiness that makes goldfish well-suited for use as pets, but also enhances the dangers of releasing these pets into new habitats where they can become invasive. The discovery of these expansions and correlations will now set the stage for in depth analysis of the functional roles of these expansions and experimental tests of these hypotheses.
Given the large number of invasive species currently damaging ecosystems around the world, a rapid, cost-effective method to interrogate the genomic basis driving invasive ecology should prove valuable. The ability to define these genetic signatures that facilitate invasive potential is now possible with the advent of next-generation, massively parallel sequencing technology. Unfortunately, the cost, technical expertise and computational resources required for whole-genome sequencing remains a significant barrier for genomic research. In this manuscript, we identify and validate the association of stress response gene expansions with invasive potential, which represents a new molecular mechanism driving invasiveness and could provide a novel predictor of invasive potential. Conversely, these signatures could also inform ecological predictions of how species will fare with changing environmental conditions and facilitate the identification of robust fish suitable for development of aquaculture.
Supplementary Material
Acknowledgements
The authors would like to thank the members of their laboratories and the Florida Fish and Wildlife Commission, Freshwater Fisheries Laboratory for their assistance.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
Conceptualization: K.S.K., E.G., R.G.T.; Methodology: K.S.K., D.J.C., E.G.; Investigation: T.R.S., K.S.K., S.M.P., M.O., I.K., N.R.H., A.L., R.G.T., E.G.; Data curation: M.O., I.K., N.R.H., A.L.; Writing - original draft: K.S.K., E.G.; Writing - review & editing: T.R.S., E.G., K.S.K.; Visualization: T.R.S., E.G.; Supervision: K.S.K., M.M.S., D.J.C., R.G.T., E.G.; Project administration: E.G.; Funding acquisition: M.M.S., D.J.C., E.G.
Funding
Funding was provided by the National Science Foundation (REU Biomath Program Grant 1359341) and the National Institutes of Health (R15 CA227573). Deposited in PMC for release after 12 months.
Data availability
The datasets supporting the conclusions of this article are included within the article and its additional files or are available in NCBI databases. The raw reads were uploaded to the NCBI Short Read Archive (SRA) for both bluegill (SRR5645631) and the redear sunfish (SRR5646640). The assembled transcripts were uploaded to the Transcriptome Shotgun Assembly (TSA) database: GFOV00000000 (bluegill) and GFPH00000000 (redear).
References
- Balchin, D., Hayer-Hartl, M. and Hartl, F. U. (2016). In vivo aspects of protein folding and quality control. Science 353, aac4354. 10.1126/science.aac4354 [DOI] [PubMed] [Google Scholar]
- Basu, N., Todgham, A. E., Ackerman, P. A., Bibeau, M. R., Nakano, K., Schulte, P. M. and Iwama, G. K. (2002). Heat shock protein genes and their functional significance in fish. Gene 295, 173-183. 10.1016/S0378-1119(02)00687-X [DOI] [PubMed] [Google Scholar]
- Boone, E. C., Laursen, J. R., Colombo, R. E., Meiners, S. J., Romani, M. F. and Keeney, D. B. (2018). Infection patterns and molecular data reveal host and tissue specificity of Posthodiplostomum species in centrarchid hosts. Parasitology 145, 1458-1468. 10.1017/S0031182018000306 [DOI] [PubMed] [Google Scholar]
- Brett, J. R. (1971). Energetic responses of salmon to temperature. A study of some thermal relations in the physiology and freshwater ecology of sockeye salmon (Oncorhynchus nerkd). Am. Zool 11, 99-113. 10.1093/icb/11.1.99 [DOI] [Google Scholar]
- Brogowski, Z., Siewert, H. and Keplinger, D. (2005). Feeding and growth responses of bluegill fish (Lepomis macrochirus) at various pH levels. Pol. J. Environ. Stud. 14, 517-519. [Google Scholar]
- Carroll, A. M., Wainwright, P. C., Huskey, S. H., Collar, D. C. and Turingan, R. G. (2004). Morphology predicts suction feeding performance in centrarchid fishes. J. Exp. Biol 207, 3873-3881. 10.1242/jeb.01227 [DOI] [PubMed] [Google Scholar]
- Cheng, J., Xun, X., Kong, Y., Wang, S., Yang, Z., Li, Y., Kong, D., Wang, S., Zhang, L., Hu, X.et al. (2016). Hsp70 gene expansions in the scallop Patinopecten yessoensis and their expression regulation after exposure to the toxic dinoflagellate Alexandrium catenella. Fish Shellfish Immunol. 58, 266-273. 10.1016/j.fsi.2016.09.009 [DOI] [PubMed] [Google Scholar]
- Dahlke, F., Wohlrab, S., Butzin, M.Pörtner, H.-O. (2020a). Experimental data compilation, thermal tolerance and thermal responsiveness of fish species and life stages. PANGAEA. 10.1594/PANGAEA.917796 [Google Scholar]
- Dahlke, F. T., Wohlrab, S., Butzin, M. and Pörtner, H.-O. (2020b). Thermal bottlenecks in the life cycle define climate vulnerability of fish. Science 369, 65-70. 10.1126/science.aaz3658 [DOI] [PubMed] [Google Scholar]
- Deane, E. E. and Woo, N. Y. S. (2011). Advances and perspectives on the regulation and expression of piscine heat shock proteins. Rev. Fish Biol. Fish 21, 153-185. 10.1007/s11160-010-9164-8 [DOI] [Google Scholar]
- Diagne, C., Leroy, B., Vaissière, A.-C., Gozlan, R. E., Roiz, D., Jarić, I., Salles, J.-M., Bradshaw, C. J. A. and Courchamp, F. (2021). High and rising economic costs of biological invasions worldwide. Nature 592, 571-576. 10.1038/s41586-021-03405-6 [DOI] [PubMed] [Google Scholar]
- Fisher Huckins, C. J., Osenberg, C. W. and Mittelbach, G. G. (2000). Species introductions and their ecological consequences: an example with congeneric sunfish. Ecol. Appl. 10, 612-625. 10.1890/1051-0761(2000)010[0612:SIATEC]2.0.CO;2 [DOI] [Google Scholar]
- Frankel, J., Williams, N. E., Nelsen, E. M. and Keeling, P. J. (2001). An evaluation of Hsp90 as a mediator of cortical patterning in tetrahymena. J. Eukaryot. Microbiol 48, 147-160. 10.1111/j.1550-7408.2001.tb00297.x [DOI] [PubMed] [Google Scholar]
- Fuller, P. L., Nico, L. G. and Williams, J. D. (1999). Nonindigenous fishes introduced into inland waters of the United States. Am. Fish. Soc., pp. 353-376. [Google Scholar]
- Glasauer, S. M. K. and Neuhauss, S. C. F. (2014). Whole-genome duplication in teleost fishes and its evolutionary consequences. Mol. Genet. Genomics MGG 289, 1045-1060. 10.1007/s00438-014-0889-2 [DOI] [PubMed] [Google Scholar]
- Grabherr, M. G., Haas, B. J., Yassour, M., Levin, J. Z., Thompson, D. A., Amit, I., Adiconis, X., Fan, L., Raychowdhury, R., Zeng, Q.et al. (2011). Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol 29, 644-652. 10.1038/nbt.1883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimholt, U. (2016). MHC and evolution in teleosts. Biology 5, 6. 10.3390/biology5010006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross, M. R. and Charnov, E. L. (1980). Alternative male life histories in bluegill sunfish. Proc. Natl. Acad. Sci. USA 77, 6937-6940. 10.1073/pnas.77.11.6937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guisbert, E. and Morimoto, R., (2012). The regulation and function of the heat shock response In: Protein Quality Control in Neurodegenerative Diseases. Springer Berlin Heidelberg, pp. 1-18. [Google Scholar]
- Haas, B. J., Papanicolaou, A., Yassour, M., Grabherr, M., Blood, P. D., Bowden, J., Couger, M. B., Eccles, D., Li, B., Lieber, M.et al. (2013). De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc 8, 1494-1512. 10.1038/nprot.2013.084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayward, R. S. and Wang, H.-P. (2006). Rearing male bluegills indoors may be advantageous for producing food-size sunfish. J. World Aquac. Soc 37, 496-508. 10.1111/j.1749-7345.2006.00063.x [DOI] [Google Scholar]
- Iwama, G. K., Thomas, P. T., Forsyth, R. B. and Vijayan, M. M. (1998). Heat shock protein expression in fish. Rev. Fish Biol. Fish 8, 35-56. 10.1023/A:1008812500650 [DOI] [Google Scholar]
- Kampinga, H. H., Hageman, J., Vos, M. J., Kubota, H., Tanguay, R. M., Bruford, E. A., Cheetham, M. E., Chen, B. and Hightower, L. E. (2009). Guidelines for the nomenclature of the human heat shock proteins. Cell Stress Chaperones 14, 105-111. 10.1007/s12192-008-0068-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawamura, K., Yonekura, R., Katano, O., Taniguchi, Y. and Saitoh, K. (2006). Origin and dispersal of bluegill sunfish, Lepomis macrochirus, in Japan and Korea: Origin of bluegill sunfish in JAPAN and KOREA. Mol. Ecol 15, 613-621. 10.1111/j.1365-294X.2006.02823.x [DOI] [PubMed] [Google Scholar]
- Kolar, C. S. (2002). Ecological predictions and risk assessment for alien fishes in North America. Science 298, 1233-1236. 10.1126/science.1075753 [DOI] [PubMed] [Google Scholar]
- Lindquist, S. (2009). Protein folding sculpting evolutionary change. Cold Spring Harb. Symp. Quant. Biol 74, 103-108. 10.1101/sqb.2009.74.043 [DOI] [PubMed] [Google Scholar]
- Manni, M., Berkeley, M. R., Seppey, M., Simão, F. A. and Zdobnov, E. M. (2021). BUSCO update: novel and streamlined workflows along with broader and deeper phylogenetic coverage for scoring of eukaryotic, prokaryotic, and viral genomes. Mol. Biol. Evol 38, 4647-4654. 10.1093/molbev/msab199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreton, J., Izquierdo, A. and Emes, R. D. (2016). Assembly, assessment, and availability of De novo generated eukaryotic transcriptomes. Front. Genet 6, 361. 10.3389/fgene.2015.00361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Near, T. J., Bolnick, D. I. and Wainwright, P. C. (2005). Fossil calibrations and molecular divergence time estimates in centrarchid fishes (Teleostei: Centrarchidae). Evol. Int. J. Org. Evol 59, 1768-1782. 10.1111/j.0014-3820.2005.tb01825.x [DOI] [PubMed] [Google Scholar]
- Near, T. J., Eytan, R. I., Dornburg, A., Kuhn, K. L., Moore, J. A., Davis, M. P., Wainwright, P. C., Friedman, M. and Smith, W. L. (2012). Resolution of ray-finned fish phylogeny and timing of diversification. Proc. Natl. Acad. Sci. USA 109, 13698-13703. 10.1073/pnas.1206625109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osenberg, C. W., Werner, E. E., Mittelbach, G. G. and Hall, D. J. (1988). Growth Patterns in Bluegill (Lepomis macrochirus) and Pumpkinseed (L . gibbosus) Sunfish: Environmental Variation and the Importance of Ontogenetic Niche Shifts. Can. J. Fish. Aquat. Sci 45, 17-26. 10.1139/f88-003 [DOI] [Google Scholar]
- Partridge, C. G., MacManes, M. D., Knapp, R. and Neff, B. D. (2016). Brain transcriptional profiles of male alternative reproductive tactics and females in bluegill sunfish. PLOS ONE 11, e0167509. 10.1371/journal.pone.0167509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauchard, A., Cavieres, L. A. and Bustamante, R. O. (2004). Comparing alien plant invasions among regions with similar climates: where to from here?: alien plant invasions and climate. Divers. Distrib 10, 371-375. 10.1111/j.1366-9516.2004.00116.x [DOI] [Google Scholar]
- Pimentel, D., McNair, S., Janecka, J., Wightman, J., Simmonds, C., O'Connell, C., Wong, E., Russel, L., Zern, J., Aquino, T.et al. (2001). Economic and environmental threats of alien plant, animal, and microbe invasions. Agric. Ecosyst. Environ 84, 1-20. 10.1016/S0167-8809(00)00178-X [DOI] [Google Scholar]
- Qian, X., Ba, Y., Zhuang, Q. and Zhong, G. (2014). RNA-Seq technology and its application in fish transcriptomics. OMICS J. Integr. Biol 18, 98-110. 10.1089/omi.2013.0110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin, G., Johnson, C., Zhang, Y., Zhang, H., Yin, J., Miller, G., Turingan, R. G., Guisbert, E. and Lin, Q. (2018). Temperature-induced physiological stress and reproductive characteristics of the migratory seahorse Hippocampus erectus during a thermal stress simulation. Biol. Open 7, bio032888. 10.1242/bio.032888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Queitsch, C., Sangster, T. A. and Lindquist, S. (2002). Hsp90 as a capacitor of phenotypic variation. Nature 417, 618-624. 10.1038/nature749 [DOI] [PubMed] [Google Scholar]
- Ricciardi, A., Blackburn, T. M., Carlton, J. T., Dick, J. T. A., Hulme, P. E., Iacarella, J. C., Jeschke, J. M., Liebhold, A. M., Lockwood, J. L., MacIsaac, H. J.et al. (2017). Invasion science: a horizon scan of emerging challenges and opportunities. Trends Ecol. Evol. 32, 464-474. 10.1016/j.tree.2017.03.007 [DOI] [PubMed] [Google Scholar]
- Rohner, N., Jarosz, D. F., Kowalko, J. E., Yoshizawa, M., Jeffery, W. R., Borowsky, R. L., Lindquist, S. and Tabin, C. J. (2013). Cryptic Variation in morphological evolution: HSP90 as a capacitor for loss of eyes in cavefish. Science 342, 1372-1375. 10.1126/science.1240276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutherford, S. L. and Lindquist, S. (1998). Hsp90 as a capacitor for morphological evolution. Nature 396, 336-342. 10.1038/24550 [DOI] [PubMed] [Google Scholar]
- Sakai, A. K., Allendorf, F. W., Holt, J. S., Lodge, D. M., Molofsky, J., With, K. A., Baughman, S., Cabin, R. J., Cohen, J. E., Ellstrand, N. C.et al. (2001). The Population Biology of Invasive Species. Annu. Rev. Ecol. Syst. 32, 305-332. 10.1146/annurev.ecolsys.32.081501.114037 [DOI] [Google Scholar]
- Sangster, T. A., Lindquist, S. and Queitsch, C. (2004). Under cover: causes, effects and implications of Hsp90-mediated genetic capacitance. BioEssays 26, 348-362. 10.1002/bies.20020 [DOI] [PubMed] [Google Scholar]
- Schreck, C. B. and Moyle, P. B. (eds), (1990). Methods for fish biology. Am. Fish. Soc., pp. 214-272. [Google Scholar]
- Shen, Z.-G., Wang, H.-P., Yao, H., O'Bryant, P., Rapp, D. and Zhu, K.-Q. (2016). Sex determination in bluegill sunfish lepomis macrochirus: effect of temperature on sex ratio of four geographic strains. Biol. Bull 230, 197-208. 10.1086/BBLv230n3p197 [DOI] [PubMed] [Google Scholar]
- Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. and Zdobnov, E. M. (2015). BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210-3212. 10.1093/bioinformatics/btv351 [DOI] [PubMed] [Google Scholar]
- Star, B., Nederbragt, A. J., Jentoft, S., Grimholt, U., Malmstrøm, M., Gregers, T. F., Rounge, T. B., Paulsen, J., Solbakken, M. H., Sharma, A.et al. (2011). The genome sequence of Atlantic cod reveals a unique immune system. Nature 477, 207-210. 10.1038/nature10342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahara, T., Minamoto, T. and Doi, H. (2013). Using environmental DNA to estimate the distribution of an invasive fish species in ponds. PLoS ONE 8, e56584. 10.1371/journal.pone.0056584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Alliance of Genome Resources Consortium, Agapite, J., Albou, L.-P., Aleksander, S., Argasinska, J., Arnaboldi, V., Attrill, H., Bello, S. M., Blake, J. A., Blodgett, O.et al. (2020). Alliance of genome resources portal: unified model organism research platform. Nucleic Acids Res. 48, D650-D658. 10.1093/nar/gkz813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thuiller, W., Gassó, N., Pino, J. and Vilà, M. (2012). Ecological niche and species traits: key drivers of regional plant invader assemblages. Biol. Invasions 14, 1963-1980. 10.1007/s10530-012-0206-0 [DOI] [Google Scholar]
- Toshikazu, M. and Tetsuro, U. (2004). Invasive alien species in japan: the status quo and the new regulation for prevention of their adverse effects. Glob. Environ. Res. 8, 171-191. [Google Scholar]
- Turingan, R. and Sloan, T. (2016). Thermal resilience of feeding kinematics may contribute to the spread of invasive fishes in light of climate change. Biology 5, 46. 10.3390/biology5040046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyedmers, J., Madariaga, M. L. and Lindquist, S. (2008). Prion switching in response to environmental stress. PLoS Biol. 6, e294. 10.1371/journal.pbio.0060294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uliano-Silva, M., Americo, J. A., Brindeiro, R., Dondero, F., Prosdocimi, F. and de Freitas Rebelo, M. (2014). Gene discovery through transcriptome sequencing for the invasive mussel Limnoperna fortunei. PLoS ONE 9, e102973. 10.1371/journal.pone.0102973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Hurk, P., Gerzel, L. E., Calomiris, P. and Haney, D. C. (2017). Phylogenetic signals in detoxification pathways in Cyprinid and Centrarchid species in relation to sensitivity to environmental pollutants. Aquat. Toxicol. Amst. Neth 188, 20-25. 10.1016/j.aquatox.2017.04.004 [DOI] [PubMed] [Google Scholar]
- Wainwright, P. C. (1996). Ecological explanation through functional morphology: the feeding biology of sunfishes. Ecology 77, 1336-1343. 10.2307/2265531 [DOI] [Google Scholar]
- Zhang, G., Fang, X., Guo, X., Li, L., Luo, R., Xu, F., Yang, P., Zhang, L., Wang, X., Qi, H.et al. (2012). The oyster genome reveals stress adaptation and complexity of shell formation. Nature 490, 49-54. 10.1038/nature11413 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.