

Abstract
The heterogeneous response of acute myeloid leukemia (AML) to current anti‐leukemic therapies is only partially explained by mutational heterogeneity. We previously identified GPR56 as a surface marker associated with poor outcome across genetic groups, which characterizes two leukemia stem cell (LSC)‐enriched compartments with different self‐renewal capacities. How these compartments self‐renew remained unclear. Here, we show that GPR56+ LSC compartments are promoted in a complex network involving epithelial‐to‐mesenchymal transition (EMT) regulators besides Rho, Wnt, and Hedgehog (Hh) signaling. Unexpectedly, Wnt pathway inhibition increased the more immature, slowly cycling GPR56+CD34+ fraction and Hh/EMT gene expression, while Wnt activation caused opposite effects. Our data suggest that the crucial role of GPR56 lies in its ability to co‐activate these opposing signals, thus ensuring the constant supply of both LSC subsets. We show that CDK7 inhibitors suppress both LSC‐enriched subsets in vivo and synergize with the Bcl‐2 inhibitor venetoclax. Our data establish reciprocal transition between LSC compartments as a novel concept underlying the poor outcome in GPR56high AML and propose combined CDK7 and Bcl‐2 inhibition as LSC‐directed therapy in this disease.
Keywords: AML, CDK7 inhibition, GPR56, leukemia stem cell, self‐renewal
Subject Categories: Cancer, Signal Transduction
RNA‐ and ATAC‐seq profiling combined with functional in vitro and in vivo studies unravel the multi‐faceted roles of GPR56, a surface marker associated with high leukemia stem cell (LSC) burden and poor prognosis in acute myeloid leukemia (AML).
The paper explained.
Problem
Therapy response is highly heterogeneous in acute myeloid leukemia (AML), which is only partially explained by genetic risk groups. The adhesion G protein coupled receptor GPR56/ADGRG1 was previously identified as surface marker associated with poor outcome across genetic groups and characterizes two leukemia stem cell (LSC)‐enriched compartments with different self‐renewal capacities. Whether GPR56 played a functional role in this process and how this might be targeted remained unclear.
Results
GPR56 is functionally required for the maintenance of human LSC‐enriched compartments by regulating Wnt, Hedgehog (Hh), and EMT‐associated genes in addition to RhoA signaling. While reporter assays and functional tests show that these pathways are all enhanced by GPR56, Wnt and Hh/TGFb pathways reciprocally inhibit each other, and their target genes are differentially expressed in the CD34+GPR56+ versus CD34‐GPR56+ LSC‐enriched fractions. Cell sorting and in vivo experiments provide evidence for reciprocal transition between the two compartments. In particular, combination of a Wnt inhibitor with a Hh agonist and TGFb is sufficient to regenerate the CD34+GPR56+ from the CD34‐GPR56+ compartment in vitro. The CDK7/12/13 inhibitor THZ1 and more specific CDK7 inhibitors synergize with the Bcl‐2 antagonist venetoclax to suppress both GPR56+ compartments in primary human AML cells in vitro and in vivo.
Impact
The study unravels a complex network around GPR56 in AML, thus raising it from a simple marker that enriches for LSCs to a functional stemness regulator with a pivotal role in preventing exhaustion of the LSC pool. The study puts forward a model of reciprocal transition between functionally distinct LSC compartments, rapidly and slowly cycling, which is aberrant in AML compared with the healthy hematopoietic system. This model provides an explanation on how LSCs escape current chemotherapies and suggests that synergistically acting CDK7 and Bcl‐2 inhibitors might represent a novel therapeutic approach to target the GPR56‐associated network in AML.
Introduction
Acute myeloid leukemia (AML) is a hematologic malignancy affecting both young and elderly patients, for whom intensive therapies are often not an option (Döhner et al, 2015). Assessment of cytogenetic and molecular genetic aberrations has become the gold standard for risk stratification and for guiding therapeutic decisions for AML patients harboring targetable mutations (Döhner et al, 2017). Targeting mutated proteins by small molecules such as IDH1/2 or FLT3 inhibitors has considerably broadened the therapeutic repertoire in AML and improved survival (reviewed in refs. Kindler et al, 2010; Chaturvedi et al, 2013). However, when given as monotherapy, they usually delay leukemia progression rather than permanently eradicate the disease (Kindler et al, 2010). Moreover, there is large heterogeneity in patient outcome even within previously defined genetic groups (Cancer Genome Atlas Research Network et al, 2013) demonstrating the need to better understand how targetable and non‐targetable mutations together with the induced downstream pathways synergize to drive the disease. While RNA‐seq is powerful in detecting global gene expression changes in homogeneous populations, it may miss subtle changes in lowly abundant mRNAs such as transcription factors (TFs), especially in heterogeneous populations such as primary AML samples. Furthermore, expression levels of TFs are not necessarily indicative of their activity, which is often regulated posttranslationally (Filtz et al, 2014). We and others have shown that epigenetic analyses are often more powerful in detecting differences between heterogeneous primary samples, as they also reveal the epigenetic potential (cell fates) rather than only events that have already happened (cell states; Assi et al, 2019; Berest et al, 2019). Besides genetic subtyping, AML can be characterized by shared signaling pathways and aberrant immunophenotypes such as co‐expression of CD7, CD56 (Chang et al, 2004), or a CD34lowGPR56high profile, the latter of which we associated with co‐mutations in NPM1, DNMT3A, and FLT3‐ITD (Garg et al, 2019), and high LSC frequency (Pabst et al, 2016). In AML with an aberrant CD34lowGPR56high profile, both the CD34 positive and negative GPR56+ fractions contain LSCs. Besides establishing GPR56 as an LSC marker, we showed that high GPR56 expression is associated with poor prognosis in AML (Pabst et al, 2016). In line, GPR56 is part of a 17‐gene stemness signature associated with poor prognosis in AML (Ng et al, 2016). GPR56 belongs to the adhesion G‐protein coupled receptor (aGPCR) family characterized by a 7‐transmembrane (7TM) domain, flanked by an intracellular C‐terminus and a long extracellular N‐terminus, which contains the GPCR proteolytic site (GPS) within the GPCR autoproteolysis‐inducing (GAIN) domain (Chiang et al, 2011; Purcell & Hall, 2018). Splice variants have been described to possess overlapping but also non‐redundant functions, which might explain why different knock‐out strategies caused distinct phenotypes (Kim et al, 2010b; Rao et al, 2015; Li et al, 2020). The impact of GPR56 on Rho signaling and actin cytoskeleton regulation suggested a predominant role in adhesion as shown in the neuronal system (Iguchi et al, 2008) and certain solid tumors (Shashidhar et al, 2005). Despite the associations of GPR56 with poor prognosis in several different cancer entities (Shashidhar et al, 2005; Kausar et al, 2011) and endothelial‐to‐hematopoietic transition (Solaimani Kartalaei et al, 2015), it was unclear whether and how GPR56 was functionally involved in LSC activity in human AML and how its downstream pathways might be targeted.
Here, we explored chromatin accessibility and transcription factor activities through ATAC‐seq profiling of 35 primary human AML samples followed by RNA‐seq, functional assays, and preclinical models to dissect the role of GPR56 in the identified network. Our data suggest a model, in which GPR56 by co‐activating reciprocally inhibitory signals promotes oscillation of signaling pathways that drive either towards the CD34 positive or negative GPR56+ LSC compartment, which differ by LSC frequency and cycling characteristics (Pabst et al, 2016). Moreover, we identify CDK7/12/13 inhibitors as novel compound class that targets diverse routes in this network and synergizes with the Bcl‐2 inhibitor venetoclax to eradicate primary human AML cells.
Results
ATAC‐ and RNA‐seq profiling link GPR56 to increased TF activities associated with EMT, Wnt and Hh signaling
We applied ATAC‐seq to 35 primary human AML samples, which represented the full range from low to very high protein expression levels of the LSC marker GPR56, to identify leukemia‐driving pathways in GPR56high AML (Fig 1A, Dataset EV1). We identified 247,442 unique chromatin peaks, of which 24,026 were differentially accessible in GPR56high versus (vs) GPR56low AML (false discovery rate, FDR < 5%, see Fig 1A legend for grouping criteria). The shared peaks (32,406 peaks found in at least 30 samples) were enriched in promoters, while more sample‐specific peaks were enriched in intergenic/intronic regions (Fig 1B). As the latter often contain regulatory elements, we interrogated EnhancerAtlas 2.0 (Gao & Qian, 2020) and found enrichment for enhancers with known activity in CD34+, AML blasts, and CD8+ T‐cells among the GPR56high‐specific peaks, whereas enhancers associated with monocytes and macrophages were significantly enriched in the GPR56low group (Fig 1C). These results support previous observations that AML with high GPR56 expression represents a more immature, poorly differentiated type of AML (Pabst et al, 2016).
Figure 1. ATAC‐ and RNA‐seq profilings position GPR56 within a Rho/Wnt/Hh network.
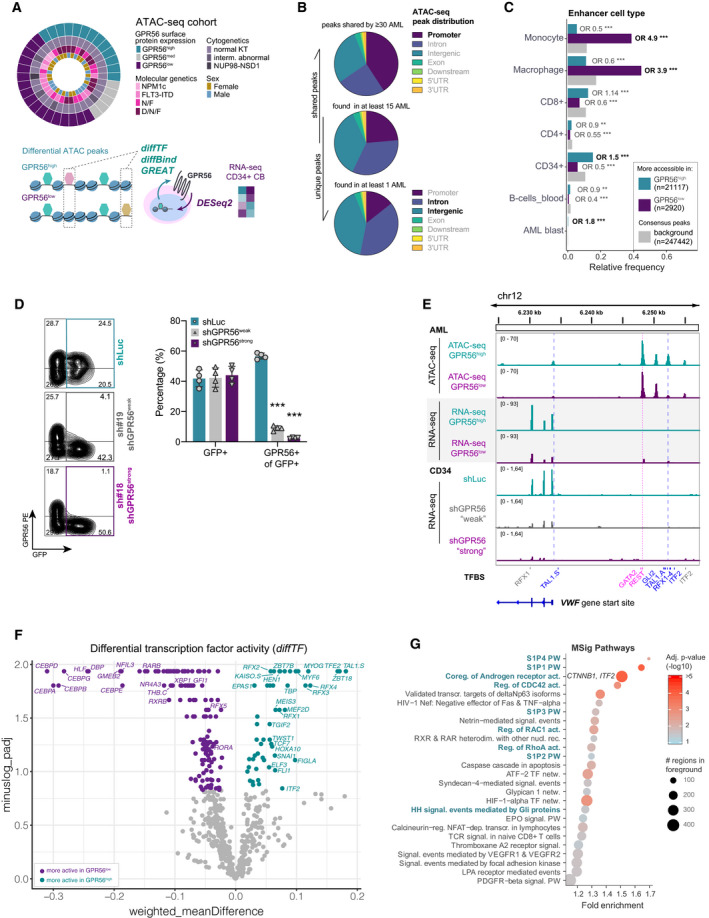
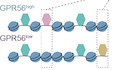
- Overview of the experimental and analytical setup. ATAC‐seq was performed on 35 AML samples comprising the mutational groups N: NPM1 mutated, F: FLT3‐ITD, N/F: NPM1/FLT3‐ITD, D/N/F: DNMT3A/NPM1/FLT3‐ITD. Indicated colors represent GPR56 protein expression grouped into high (> 70% GPR56+ cells per sample, n = 13), medium (30%–70% GPR56+, n = 5), and low (< 30% GPR56+, n = 17), cytogenetic and molecular genetic characteristics, and gender. ATAC‐seq data were subjected to the computational pipelines diffTF, diffBind, and GREAT. RNA‐seq was performed on cord blood (CB) CD34+ cells after GPR56 knockdown and analyzed by DESeq2. The combined information was used to identify differential TF and signaling pathway activities up‐ and downstream of GPR56.
- Pie charts showing the distribution of ATAC‐seq peaks shared by most samples (top), by at least 15 samples (middle), and those present in at least one sample. Colors indicate the different genomic regions. Chromatin regions that are rather differentially accessible (bottom) are enriched for introns and intergenic regions, which often contain regulatory elements, while shared peaks are more often located in promoter regions (top).
- ATAC‐seq peaks more accessible in GPR56low (violet) or GPR56high (turquoise) against the background (gray) are enriched for enhancers associated with specific hematopoietic cell types. P‐values and odds ratios are given for a pairwise, two‐sided Fisher’s Exact test comparing each category (GPR56low/high) against the background. Enhancer annotations are taken from EnhancerAtlas 2.0. ***P < 0.0005, **P < 0.005, *P < 0.05.
- Knockdown efficiency of two shRNAs against GPR56 (shGPR56weak and shGPR56strong) versus shLuc as negative control measured on protein level by flow cytometry in CD34+ CB cells. Shown are representative FACS plots (left) and the percentage of GPR56+ cells of transduced GFP+ cells on day 5 (right panel). Biological replicates N = 4, unpaired t‐test, bars and error bars represent mean and SD. ***P < 0.0005, **P < 0.005, *P < 0.05.
- Integrative Genome Viewer (IGV) snapshot showing ATAC‐seq peaks along and upstream of the VWF gene in GPR56high vs. lowsamples (top, average peak size of 10 GPR56high (turquoise) and 15 GPR56low samples (violet)), RNA‐seq reads of the same location in AML samples with high (n = 9) versus low (n = 11) GPR56 expression (two middle tracks), and RNA‐seq reads of shLuc versus GPR56 knockdown CD34+ cells (3 bottom tracks, one of two replicates shown for each condition). Track height was group‐scaled. Dashed vertical lines indicate binding sites for the annotated TFs. TFBS: transcription factor‐binding site derived from the HOCOMOCO v10 database; TSS: transcription start site. TFs in blue bind to differentially accessible chromatin regions.
- Volcano plot of differential TF motif accessibility (activity) in GPR56high (turquoise) vs. GPR56low (violet) samples and their corresponding adjusted P‐values determined with diffTF. Highlighted are TFs whose RNA expression was also positively or negatively affected by GPR56 KD in the RNA‐seq dataset.
- Pathway enrichment analysis for peaks that are more accessible in GPR56high AML. The GREAT algorithm was used to assign peaks to genes, the MSig database was used for pathway enrichment analysis (Pathway Interaction Database). Shown are all terms with adjusted P‐value < 0.05. PW: pathway, (Co‐)reg.: (Co‐) regulation, act.: activity, NR: nuclear receptor, transcr.: transcription(al), netw.: network, signal.: signaling.
To assess the role of GPR56 in normal hematopoietic stem and progenitor cells (HPSCs), we performed a knockdown (KD) of GPR56 in cord blood (CB) CD34+ cells using shRNAs against GRP56 or luciferase (shLuc) as negative control, followed by RNA‐seq (Fig 1D and E). The CB CD34+ cells are hereafter called shGPR56weak and shGPR56strong according to the shRNA’s different efficiency to suppress GPR56 surface expression on transduced GFP+ cells (Fig 1D, Appendix Fig S1A), which might be explained by the different shRNA target sequence localization (Appendix Fig S1B; Kim et al, 2010b). CB CD34+ cells were used as representative, non‐mutated CD34+GPR56+ model cell type (Pabst et al, 2016) to avoid AML sample‐specific results. GO term enrichment analysis of differentially expressed genes determined with Deseq2 revealed “GPCR signaling”, “adhesion” and “migration”, in line with the known role of GPR56 but also pointed toward platelet associated processes and phospholipase activity (Appendix Fig S1C and D, Dataset EV2).
We further explored the ATAC‐seq data by applying our computational tool diffTF, which estimates differential TF activities on the basis that chromatin accessibility increases when TFs interact with chromatin at specific TF‐binding sites and thus contribute to the ATAC‐seq signal (Assi et al, 2019; Berest et al, 2019). In addition, we overlayed diffTF results with the RNA‐seq data to assess whether gene expression of differentially active TFs or their respective target genes was affected by GPR56 KD, which would suggest that they act downstream of GPR56. To illustrate how differential TF activities were identified in diffTF, we visualized one example region upstream of the VWF gene locus (Fig 1E) using the integrative genome viewer (IGV). Two differential peaks with stronger ATAC‐seq signal in GPR56high vs. GPR56low AML were identified upstream of and at the gene start site. These contained binding sites for TAL1, GLI2, RFX1‐4, and ITF2, while binding motifs for GATA2 and REST were similarly accessible in GPR56high and GPR56low AML (Fig 1E, upper row). Moreover, VWF mRNA expression was higher in GPR56high vs GPR56low AML (RNA‐seq from (Garg et al, 2019), Fig 1E, middle row). Finally, the shRNA‐seq dataset revealed a downregulation of VWF in CD34+ cells after GPR56 suppression with both shRNAs (Fig 1E, lower row). Together, these observations indicated that GPR56 functionally upregulates WVF expression, potentially upstream of RFX1‐4 and GLI2, which are known TFs in the HH pathway. When applying this approach on a genome‐wide level using diffTF, we identified 166 TFs with significantly different activities in the GPR56high vs low group at an FDR of 10% (Fig 1F and Dataset EV3). These comprised several TFs related to Wnt and Hh pathways such as TCF7 (Stemmer et al, 2008), TFE2/TCF3 (Zhao et al, 2019), and RFX1‐4 (Piasecki et al, 2010) in the GPR56high group, while the GPR56low group was characterized by higher CEBPA activity (ATAC‐seq), a TF essential for myeloid differentiation (Kandilci & Grosveld, 2009). The EMT‐associated TFs SNAIL1, TWIST1 (Nieto et al, 2016), and TGIF2 (Du et al, 2019) were also more active in GPR56high samples (Fig 1F) but were not affected by GPR56 suppression (Dataset EV2) suggesting that they act upstream of GPR56. In support of this hypothesis, suppression of SNAIL1 and ITF2/TCF4 reduced GPR56 mRNA in external RNA‐seq datasets (GSE70872, GSE38236, and Doostparast Torshizi et al, 2019). We subsequently confirmed suppression of GPR56 upon ITF2 knockdown in our own q‐RT‐PCR experiments (Appendix Fig S1E, Dataset EV4).
To gain more insight into the pathways more active in GPR56high AML, we applied the tool ‘GREAT’ (McLean et al, 2010) to the ATAC‐seq data, which identified enrichment for Rho signaling, a well‐established GPR56 downstream pathway (Iguchi et al, 2008), but also pointed toward the Wnt genes CTNNB1 (β‐catenin) and ITF2/TCF4 (Kolligs et al, 2002) contained in “Coregulation of Androgen receptor activity”, as well as “Hh signaling events mediated by Gli proteins” (Chen et al, 2009; Fig 1G, Dataset EV5). We therefore screened the RNA‐seq dataset for bona fide Wnt and Hh genes (Dataset EV6) and found significant downregulation upon GPR56 suppression for Wnt pathway genes such as DVL1, TNKS2, VEGFA, MYC, CCND1 (He et al, 1998; Shtutman et al, 1999; Corbit et al, 2005; Zhan et al, 2017), as well as SMO, the key Hh pathway effector (Corbit et al, 2005) (Appendix Fig S1F).
Together, these analyses pointed toward involvement of Wnt, Hh, and EMT regulators in GPR56high AML in addition to Rho.
GPR56 is required for in vitro and in vivo expansion of primary human AML cells
To further characterize the functional role of GPR56, we determined the effects of GPR56 KD on human CD34+ CB and AML cells. Both shRNAs significantly impaired CD34+ cell proliferation and colony formation capacity (Fig 2A, Dataset EV7). For the in vivo experiment, we used unsorted cells with comparable gene transfer in the three conditions prior to injection (40–50% GFP+, as shown in Fig 1D). Strong GPR56 suppression (shGPR56strong) significantly hampered short‐ (ST) and long‐term (LT) HSC engraftment in NSG mice (Fig 2B, Dataset EV7). Analysis of hematopoietic subpopulations at 20 weeks revealed that shGPR56strong had depleted the HSPC compartment in most mice and significantly affected the B lymphoid lineage stronger than the myeloid lineage (Fig 2C, Appendix Fig S2A, Dataset EV7), resulting in an altered CD19+/CD33+ ratio (Fig 2D). ShGPR56weak only showed a trend towards lower engraftment at four weeks and had no disadvantage in overall human CD45+GFP+ engraftment at later time points. However, it also significantly reduced the rare HSPC compartment (CD34+CD33−CD19−SSClow) (Fig 2C) and significantly reduced the CD19+/CD33+ ratio similarly to shGPR56strong (Fig 2D). These results suggested that high expression levels of GPR56 are functionally important for the maintenance of the more primitive human HS(P)C compartments, while low levels are sufficient to maintain committed progenitors and their progeny in vivo.
Figure 2. Functional role of GPR56 in healthy CD34+ and leukemic cells.
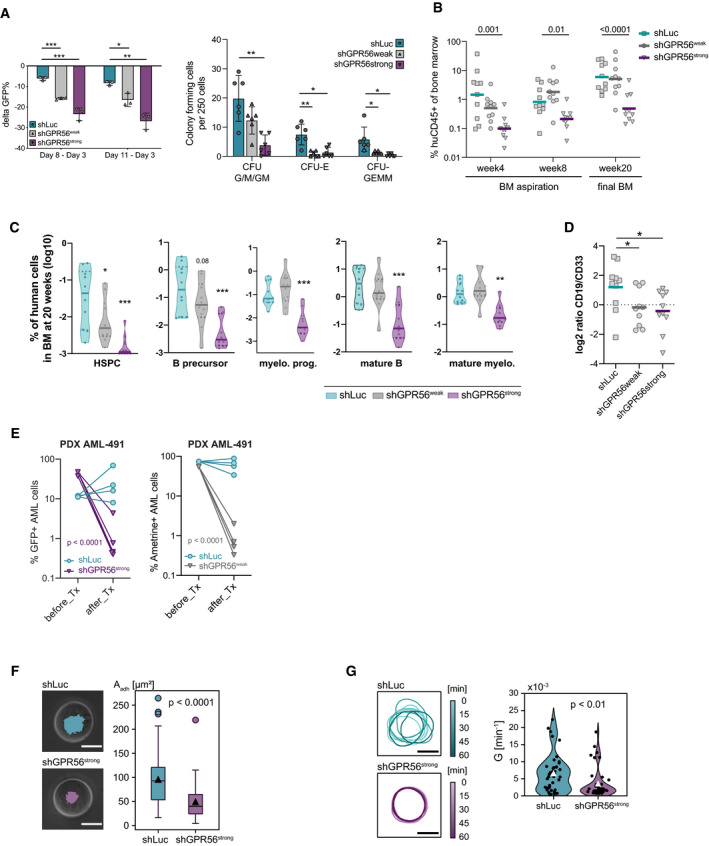
- Functional role of GPR56 in vitro. Left: CB CD34+ cells were transduced with shGPR56weak and shGPR56strong versus shLuc as negative control. The loss of the GFP+ fraction on day 8 and day 11 compared with day 3 post transduction (delta GFP+) correlates with the level of GPR56 KD. Three replicate wells were monitored per condition, unpaired t‐test, bars and error bars represent mean and SD, ***P < 0.0005, **P < 0.005, *P < 0.05. Right: Colony‐forming cell assay. Pooled CB CD34+ cells were transduced with shRNAs against GPR56 or shLuc control and GFP+ cells were directly sorted into methylcellulose 72 h post transduction. Two hundred fifty GFP+ cells were plated per well, and colonies were counted 10 days post plating. CFC: colony forming cell, CFU: colony forming unit, G: granulocyte, M: macrophage, GM: granulocyte/macrophage, E: erythrocyte, GEMM: granulocyte, erythrocyte, megakaryocyte, macrophage. Shown is one of two independent assays. Six wells were plated per condition, unpaired t‐test, bars and error bars represent mean and SD. *P < 0.05, **P < 0.005.
- Functional role of GPR56 in vivo. CB CD34+ cells were transduced with the two shRNAs against GPR56 versus shLuc as negative control. Gene transfer was approximately 50% at experiment start. Equal numbers of cells were injected in 10 recipient NSG mice per condition, and engraftment of human CD45+ (huCD45) cells in the bone marrow was assessed at 4 and 8 weeks by bone marrow aspiration and at 20 weeks by total bone marrow harvest. Strong GPR56 KD (shGPR56strong) significantly reduced overall human CD45+ engraftment, while incomplete KD (shGPR56weak) showed a trend toward lower engraftment at 4 weeks, but no difference at later time points indicating that low GPR56 surface levels might be sufficient for LT‐HSC engraftment. Unpaired t‐test of log10‐transformed values, symbols represent individual mice, horizontal bars represent geometric means, BM: bone marrow.
- Violin plots showing geometric mean (horizontal line) and individual values (circles) of the engraftment levels of different hematopoietic cell types in the bone marrow of NSG mice 20 weeks after transplantation of CB CD34+ cells following overnight‐infection with shGPR56strong (turqouise) and shGPR56weak (gray) versus shLuc control (violet). Shown are percentages of indicated populations, which are also co‐positive for human CD45 and GFP, relative to all harvested bone marrow cells. HSPC: CD34+SSClowCD33−CD19−, B precursor: CD34+CD19+CD33−, myeloid progenitor (myelo. prog.): CD34+CD33+CD19−, mature B cells: CD19+CD34−, mature myeloid cells: CD33+CD19−CD34−. Multiple unpaired t‐tests of log‐transformed values, P‐values were Benjamini and Hochberg corrected (P adj), *P adj < 0.05, **P adj < 0.005, ***P adj < 0.0005. See Appendix Fig S2A for gating strategy.
- Log2‐fold changes (log2FC) of the CD19+ (B lymphoid) versus CD33+ (myeloid) cell ratios within the human CD45+GFP+ fractions at final analysis 20 weeks after transplantation. Both shRNAs change the lymphoid/myeloid ratio in favor of myeloid cells. Unpaired t‐test of log2FC, symbols represent individual mice, horizontal bars represent average log2FC, *P < 0.05.
- GPR56 KD results in highly reduced engraftment of AML‐491 in mice. Left: Cells from two independent infections with shGPR56strong were injected in 4 NRGS mice. Shown are fractions of GFP+ cells before and 10 weeks after transplantation (Tx). Unpaired t‐tests of log(%GFP+) before versus after transplantation. Right: weak GPR56 KD results in highly reduced engraftment of AML‐491 in NSGW41 mice. Cells were injected in 4 recipient NSGW41 mice. Shown are fractions of positively transduced Ametrine (AM)+ cells before and 32 weeks after Tx. Unpaired t‐tests of log(%AM+) before versus after transplantation. No difference in overall leukemic engraftment (including non‐transduced cells) was observed excluding technical issues with injections (Appendix Fig S2C).
- Adhesion of control and GPR56 KD K562 cells on fibronectin‐functionalized substrates. Left: Overlay of phase contrast and microinterferometry images of control (above) and GPR56 KD cells (below). The area of tight adhesion extracted by microinterferometry is highlighted in turquoise and violet, respectively. Right: Comparison of adhesion areas extracted by microinterferometry, reduction of adhesion area by factor 1.9 in GPR56 KD cells (P = 1.0 × 10−10, two‐sided Mann‐Whitney test, box plots showing medians, quartiles, and outliers according to the Tukey method). Technical replicates N control = 71, Technical replicates N KD = 104, scale bar 10 µm.
- Active deformation of control and GPR56 KD cells. Left: Cell periphery of control (top) and GPR56 KD (bottom) K562 cells tracked over 60 min. Right: Comparison of deformation power of control and GPR KD cells, showing a reduction of deformation power by factor 1.8 (P = 0.005, two‐sided Mann‐Whitney test, white triangles of the violin plot represent mean). Technical replicates N control = 40, Technical replicates N KD = 40, scale bar 10 µm.
To assess the effect of GPR56 suppression on leukemic cells, we transduced eight AML cell lines and observed that four of the five lines most sensitive to GPR56 suppression harbored mutations in either NPM1 (OCI‐AML3), DNMT3A (OCI‐AML2, OCI‐AML3), FLT3‐ITD (MV4‐11), or had a MECOM/EVI1 overexpression (HNT34), which represent genetic groups that we had previously connected with high GPR56 expression (Pabst et al, 2016) (Appendix Fig S2B, Dataset EV8). To determine whether GPR56 suppression affected the leukemia initiating capacity, a hallmark of LSCs (Lapidot et al, 1994), we transplanted patient‐derived xenograft cells in immunocompromised mice after short overnight lentiviral transduction to avoid effects of GPR56 suppression before injection. We observed that both strong and weak GPR56 suppression significantly hampered leukemic engraftment in mice, while overall human CD45+ levels including non‐transduced cells were similarly high in all mice and thus excluded technical issues during transplantation (Fig 2E, Appendix Fig S2C, Dataset EV9).
Given the established connection between GPR56 and Rho signaling, we determined the effects of GPR56 suppression on adhesive and migratory properties of leukemic cells by using microinterferometry. These assays were established by us before and proven to reflect the adhesion properties of hematopoietic stem and leukemic cells (Burk et al, 2015). To obtain a sufficient number of transduced cells with comparable viability, we selected the K562 cell line for this assay, as these cells were less affected by GPR56 KD within the first days post transduction compared to other cell types (Appendix Fig S2B). This assay revealed a significant reduction of the tight adhesion area on fibronectin‐coated glass slides after GPR56 suppression (Fig 2F). Moreover, a high‐throughput assay utilizing pressure waves induced by pico‐second laser pulse (Yoshikawa et al, 2011; Burk et al, 2015) revealed a significant reduction of the critical pressure for detachment in GPR56 KD vs. control conditions (Appendix Fig S2D). In addition, we estimated the active deformation of cells by tracing the cell periphery over time (Burk et al, 2015; Lamas‐Murua et al, 2018), which was significantly reduced in GPR56 KD vs control cells (Fig 2G). In summary, these studies provided strong evidence for a functional role of GPR56 in healthy and leukemic HSPCs and confirmed a major impact of GPR56 on the adhesion and deformation capacity of human leukemic cells.
GPR56 enhances Wnt and Hedgehog pathways
As our ATAC‐ and RNA‐seq data pointed towards a role of GPR56 in Wnt and Hh pathways, we next sought to investigate these interactions mechanistically. To test whether and how GPR56 may enter the Wnt pathway (Fig 3A illustrating potential scenarios), we generated full length (FL) and constitutively active C terminal fragment (CTF) versions of GPR56 (Kishore et al, 2016) and used a standard Wnt luciferase reporter assay (“SuperTop”, modified Topflash) as readout (Fig 3B, Appendix Fig S3A). Adhesion GPCRs undergo autoproteolytic cleavage at the GPCR proteolytic site (GPS), after which the N‐terminus stays non‐covalently connected with the CTF. Conformational changes through ligand binding or mechanical stimuli can expose the most N‐terminal part of the CTF, also referred to as Stachel‐peptide, which may then act as a tethered agonist to induce signaling (Liebscher et al, 2014). CTF versions mimicking this process are therefore widely used to provide a ligand‐independent constitutive signal that can be assessed in reporter assays. Overexpression of GPR56‐CTF significantly enhanced the baseline and Wnt3a‐induced SuperTop signal (Fig 3C). We did not observe an increased Wnt reporter signal with MAL/MKL1, a strong Rho activating positive control (Miralles et al, 2003), ruling out that the GPR56‐induced enhancement of Wnt signaling occurs via its activating effect on RhoA (Appendix Fig S3B). Conversely, Wnt3a‐conditioned media did not enhance the signal in a Serum Response Factor Response Element (SRF) luciferase reporter (Miralles et al, 2003) used to detect RhoA signaling (Appendix Fig S3C). Furthermore, we observed significant reduction of the Wnt3a‐induced cytosolic accumulation of β‐catenin by GPR56 KD (Fig 3D) suggesting that GPR56 acted upstream of β‐catenin. In support of this, transfection of HEK293T cells with a β‐catenin expression plasmid led to a complete rescue of the shGPR56‐mediated reduction in the Wnt3a‐induced signal (Fig 3E). In line, there was no signal reduction by shRNAs against GPR56 upon usage of the GSK3 inhibitor CHIR99021 to induce the Wnt reporter signal (Appendix Fig S3D). Since our results suggested that GPR56 acted upstream of β‐catenin, we assessed the effect of GPR56 on the essential Wnt co‐receptor LRP6. We observed that the Wnt3a‐induced increase in LRP6 protein was significantly suppressed by GPR56 KD (Appendix Fig S3E). In addition, GPR56 CTF was unable to generate a Wnt signal in an LRP6−/− knockout (KO) HEK293 cell line (Appendix Fig S3F) indicating that the effect of GPR56 on Wnt signaling occurred on the receptor level. Finally, we introduced several missense mutations in the intracellular loops of GPR56 causing amino acid changes in potential phosphorylation sites and included also one mutation in the 7th transmembrane (TM) domain (L640R), which was reported to cause bilateral frontoparietal polymicrogyria (Piao et al, 2004) (Appendix Fig S3A). All mutants reduced the signal in both, SRF and Wnt reporter assays compared with non‐mutated CTF (Fig 3F, Appendix Fig S3G), suggesting that these positions have conserved roles in GPR56 receptor activation in Wnt and SRF (RhoA) signaling. Together, these results showed that GPR56 promoted Wnt signaling on the co‐receptor level.
Figure 3. GPR56 modulates Wnt and Hh signaling pathways.
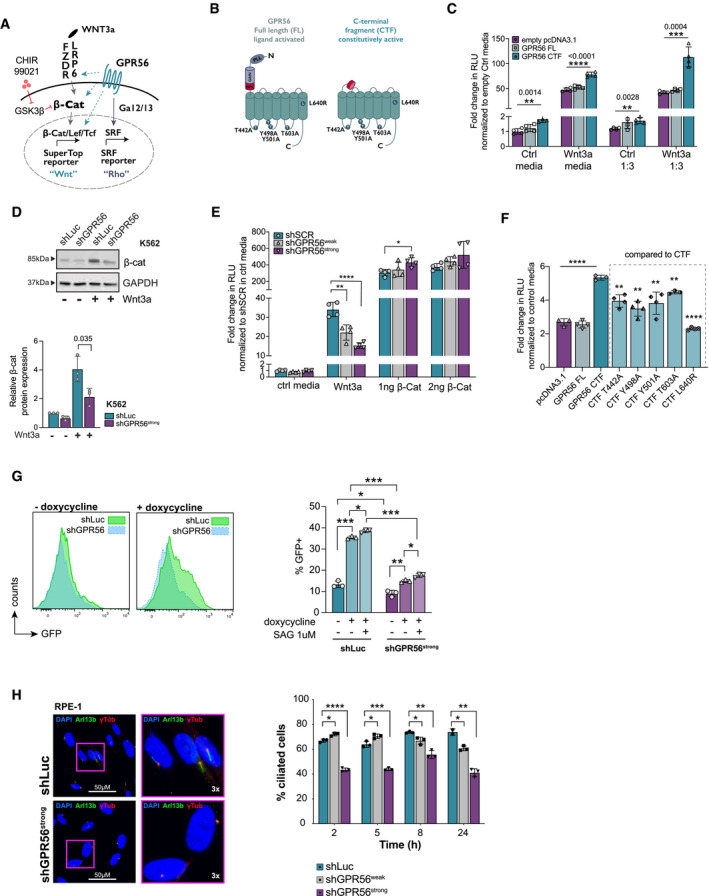
- Cartoon illustrating at which levels GPR56 may enhance canonical Wnt signaling (turquoise dashed arrows): at the level of ligand‐receptor/co‐receptor interaction, β‐catenin, downstream of β‐catenin, indirectly via activation of Rho/SRF through Gɑ12/13. CHIR99021 is a GSK3 inhibitor that prevents degradation of β‐catenin thus enhancing Wnt signaling.
- Cartoon illustrating GPR56 full length (FL, left) and the C‐terminal fragment (CTF, right). Amino acid changes introduced in the intracellular loops are indicated by circles and letters. GAIN: GPCR autoproteolysis‐inducing domain, GPS: GPCR proteolytic site, PLL: Pentraxin/Laminin/neurexin/sex‐hormone‐binding‐globulin‐Like (Salzman et al, 2016).
- SuperTop reporter assay showing fold change of relative luminescence units (RLU) in Wnt3a conditioned culture media normalized to empty pcDNA3.1+ in the respective control (Ctrl) media after transfection of HEK293T cells with empty vector (violet), GPR56 FL (light turquoise), and GPR56 CTF (turquoise). Shown are means, SD, and individual values of four technical replicates performed in either complete media or 1:3 diluted Wnt3a conditioned media. Unpaired t‐test, ****P < 0.0001, ***P < 0.0005, **P < 0.005.
- β‐catenin protein expression in K562 cells infected with shGPR56strong or shLuc negative control in presence and absence of Wnt3a (representative Western Blot (above) and quantification after normalization to GAPDH (below)). Shown is fold‐change compared with shLuc in control media, bars and error bars represent mean and SD of three biological replicates, unpaired t‐test.
- SuperTop reporter assay showing fold‐change of RLU in presence of Wnt3a normalized to a scrambled shRNA (shSCR) in control media after transfection with shSCR, shRNAs against GPR56 with or without additional transfection with 1 ng or 2 ng of β‐catenin plasmid. Overexpression of β‐catenin fully rescues the SuperTop signal reduction caused by shRNAs against GPR56. Unpaired t‐test, bars, and error bars represent mean and SD of four biological replicates, ****P < 0.0001, ***P < 0.0005, **P < 0.005, *P < 0.05.
- SuperTop reporter assay indicating fold change in RLU normalized to control media after transfection of HEK293T cells with empty vector, GPR56‐FL, or 5 GPR56‐CTF mutants. Unpaired t‐test, bars, and error bars represent mean and SD of four biological replicates, ****P < 0.0001, **P < 0.005.
- Left: SMO‐GFP reporter RPE cell line or RPE‐1 wild‐type cells were infected with shGPR56strong or shLuc control followed by administration of doxycycline or the SMO agonist SAG as indicated. GPR56 KD significantly reduces the baseline and doxycycline‐induced SMO‐GFP signal. Right: representative FACS histogram plots showing GFP intensity in shLuc versus shGPR56strong RPE cells with and without doxycycline. Unpaired t‐test, bars, and error bars represent mean and SD of three biological replicates, ***P < 0.0005, **P < 0.005, *P < 0.05.
- Left: Representative immunofluorescence (IF) images showing primary cilium formation in RPE cells upon starvation after infection with shLuc (above) or shGPR56strong (below). The non‐motile primary cilium is visualized using antibodies against γ‐tubulin, which stains the basal body, and the ciliary membrane marker ARL13B. DAPI indicates the nucleus. Images on the right show selected cells at 3× magnification. Brightness was increased in all images to enhance visibility of cilia. Right: Percentage of RPE‐1 cells with primary cilium in shLuc vs. shGPR56strong RPE‐1 cells at different time points after the start of serum starvation. Unpaired t‐test, bars, and error bars represent mean and SD of three biological replicate wells. ****P < 0.0001, ***P < 0.0005, **P < 0.005, *P < 0.05.
To characterize the role of GPR56 in the Hh pathway, we utilized the retinal pigment epithelial cell line RPE‐1, since the Hh pathway proteins have been localized to the primary cilium, which has best been studied and visualized in this cell line (Corbit et al, 2005; May‐Simera et al, 2018). In addition to the RPE‐1 wild type (wt) cells, we used a well‐established doxycycline inducible SMO‐GFP reporter RPE‐1 cell line (Joo et al, 2013); (May‐Simera et al, 2018), as visualization of endogenous SMO protein is hampered by the very low expression levels. First, we tested whether GPR56 suppression also downregulated SMO and β‐catenin in the RPE‐1 cell line and confirmed that the effects of GPR56 KD on Wnt and Hh pathways were conserved in this cell type (Appendix Fig S3H and I). Administration of doxycycline to the RPE‐1 SMO‐GFP cells significantly increased the 10% baseline SMO‐GFP expression, which was further enhanced by the SMO agonist SAG (Fig 3G). GPR56 suppression, in contrast, significantly decreased baseline and doxycycline‐stimulated SMO‐GFP signals. Furthermore, GPR56 suppression caused a significant reduction in the fraction of ciliated RPE‐1 cells (Fig 3H). Measurement of cilium length over time revealed significantly longer cilia early after serum‐starvation with both shRNAs (Appendix Fig S3J), which is a typical phenomenon in ciliopathies (Kim et al, 2010a). When combining basal body with ɑ‐tubulin staining, GPR56 suppression strongly altered the cell shape and the ɑ‐tubulin architecture from a horizontally aligned to a rather radially organized network surrounding the γ‐tubulin mark (Appendix Fig S3K). Interestingly, in a public RNA‐seq dataset (GSE147727, Dataset EV10), SMO suppression significantly reduced GPR56 mRNA levels, which might hint at positive feedback between GPR56 and Hh pathway. In summary, these mechanistic studies confirmed the ATAC‐/RNA‐seq‐driven hypotheses that GPR56 plays a functional role in Wnt and Hh signaling.
TGFb, HH, and Wnt pathway activity determine the balance between the GPR56+CD34+ and GPR56+CD34− LSC compartments
Wnt, Hh, and EMT regulators had been associated with self‐renewal in AML before (Dierks et al, 2008; Wang et al, 2010; Carmichael et al, 2020). To better understand why GPR56 enhances all three processes in parallel, we compared the GPR56 knockdown RNA‐seq results with our previously published gene expression study comparing ten sorted CD34+GPR56+ and CD34‐GPR56+ LSC compartments, which we showed before contain slowly versus more rapidly cycling LSCs, respectively (Pabst et al, 2016; Garg et al, 2019). This combined analysis revealed that key drivers of the three networks, which are all enhanced by GPR56, have divergent expression patterns in the two LSC compartments: While TGFB1, SRC, and the Hh targets GLI1/GLI2 are overexpressed in the CD34+GPR56+ fraction, the Wnt targets MYC and TNKS2 are more highly expressed in the CD34−GPR56+ compartment (Fig 4A, Appendix Fig S4C, summarized in Fig 4B). To gain insight into the mechanism that maintains the differential expression patterns in the two GPR56+ compartments, we used small molecule inhibitors and agonists to modulate the pathways in primary bulk and sorted AML cells. We found that Wnt/β‐catenin/CBP inhibition by the molecules PRI‐724 or iCRT3 increased the phenotypic CD34+GPR56+ LSC fraction in the PDX sample AML‐661 (Appendix Fig S4B).
Figure 4. TGFb, HH, and Wnt pathway activities determine the balance between the GPR56+CD34+ and GPR56+CD34− LSC compartments.
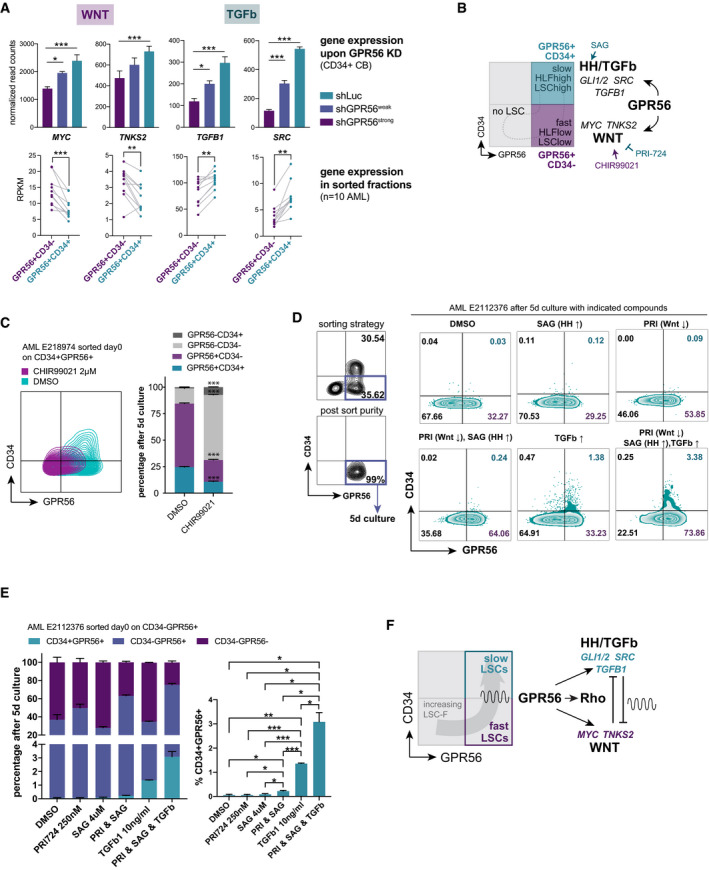
- RNA‐seq after GPR56 KD in CD34+ CB cells reveals that GPR56 enhances gene expression displayed as reads per kilobase per million mapped reads (RPKM) of MYC, TNKS2, TGFB1, and SRC (top). Previously published RNA‐seq of ten sorted CD34+GPR56+ versus CD34−GPR56+ fractions (Garg et al, 2019) shows a divergent expression pattern: downregulation of Wnt targets MYC and TNKS, but upregulation of TGFB1 and SRC in the CD34+GPR56+ fraction. Unpaired t‐test, bars, and error bars represent mean and SD of three biological replicates, ***P < 0.0005, **P < 0.005, *P < 0.05.
- Cartoon visualizing that GPR56 enhances genes and pathways differentially active in the CD34+GPR56+ fraction, which is characterized by slow cell cycle progression, high LSC frequency, and high expression of the stemness gene HLF versus the CD34−GPR56+ cells, which are more differentiated (lower LSC frequency), cycle faster, and have little HLF expression. Arrows and blocked arrows indicate activation or inhibition by the indicated small molecules, respectively, which were used in subsequent experiments.
- Contour FACS plots (left) and summary bar graph (right) showing CD34 and GPR56 expression after 5‐day culture of purified CD34+GPR56+ cells from AML sample E218974 with CHIR99021 or vehicle DMSO, Unpaired t‐test, bars, and error bars represent mean and SD of three technical replicates, ***P < 0.0005.
- Left: FACS profile and sorting strategy for the CD34‐GPR56+ fraction (top), and post‐sort purity (bottom) of AML sample E2112376 on the day of thawing. Right: CD34 and GPR56 expression measured by flow cytometry 5 days post exposure of purified CD34‐GPR56+cells to the indicated molecules or their combinations. PRI: PRI‐724, arrows indicate whether the pathway is inhibited (↓) or activated (↑) by the compound.
- Left: Bar graph showing the distribution of CD34 and GPR56 fractions at the end of the 5‐day culture of purified CD34−GPR56+ cells from AML E2112376 with indicated compounds. Right: statistical analysis shown only for the CD34+GPR56+ fraction. See Dataset EV11 for full statistical analysis. PRI: PRI‐724, Unpaired t‐test, bars, and error bars represent mean and SD of three individual treatments, ***P < 0.0005, **P < 0.005, *P < 0.05.
- Cartoon visualizing the proposed mechanism by which both GPR56+ LSC‐enriched compartments are maintained: GPR56 enhances pathways, which reciprocally inhibit each other and are differentially active in the two fractions. This should result in a constant transition between the compartments and thus prevent exhaustion of the two populations.
Conversely, the Wnt agonist CHIR99021 or exposure to Wnt3a conditioned media caused a significant loss of this compartment (Appendix Fig S4B and C). In line, PRI‐724 increased SRC, TGFB1, and SNAI1 mRNA expression in AML‐661 (Appendix Fig S4D).
In further support of our hypothesis that Wnt antagonizes EMT associated genes in AML, we found that TGFB1, TWIST1, and SNAIL1 were exclusively suppressed by the Wnt/AhR agonist 6‐BIO, but not by the pure AhR agonist MeBIO, indicating that the suppression was predominantly caused by the Wnt agonist activity of 6‐BIO and not by AhR activation (Appendix Fig S4E).
To test whether modulation of the different pathways caused a real reciprocal shift between the compartments rather than enrichment of one over the other fraction, we performed several sorting experiments. When sorted CD34+GPR56+ cells from AML E218974 were stimulated with CHIR99021 for 5 days, the CD34+GPR56+ fraction was significantly reduced compared with the differentiation that naturally occurs in standard culture conditions (Fig 4C). Next, we sorted CD34+GPR56+ and CD34‐GPR56+ cells from two other primary AML samples and cultured the cells with Wnt antagonist PRI‐724, Hh agonist SAG, and TGFβ. The combination of all three molecules was most efficient in maintaining GPR56 expression during the 5‐day culture and even generated CD34+GPR56+ from the sorted CD34‐GPR56+ cells (Fig 4E, Appendix Fig 4F and G, Dataset EV11).
Together, these results suggest a scenario, in which the Wnt‐enhancing activity of GPR56 in the CD34+GPR56+ compartment supports the transition to the more differentiated and more rapidly cycling MYC high TGFB1 low GPR56+CD34‐ compartment. In turn, suppression of Wnt activity and activation of SMO/Hh‐ and EMT‐associated genes replenish the slowly proliferative MYC low TGFB1 high CD34+ fraction (scenario outlined in Fig 4F). This model provides an explanation why specific Wnt and Hh inhibitors are inefficient to permanently eradicate AML (Jiang et al, 2018; Cortes et al, 2019). Instead, it suggests that small molecules have to act more upstream, e.g., on the level of GPR56, to eradicate LSCs.
The CDK7/12/13 inhibitor THZ1 suppresses both GPR56+ LSC compartments in vitro and in vivo
The search for small molecules that might act upstream of the GPR56‐associated network led us to the CDK7/12/13 inhibitor THZ1, as it had been shown to overcome resistance to SMO antagonists in medulloblastoma by acting upstream and independent of SMO (Liu et al, 2019). We therefore tested its activity in primary AML cells and found that it transcriptionally repressed GPR56, as well as Hh, Wnt, and EMT‐associated genes (Appendix Fig S5A–C). The CDK7 inhibitor (CDK7i) LDC4297 caused similar, but weaker effects, and had higher half‐inhibitory concentrations (IC50) in different AML cell lines and primary AML cells (Appendix Fig S5D). We therefore used THZ1 for in vivo treatment of a GPR56high human PDX AML sample (04H112) and found that the molecule significantly suppressed leukemia development during the four‐week treatment period (Fig 5B and C). Immunophenotyping of the human cells engrafted in NSG mice revealed a significant reduction of both GPR56+ LSC‐enriched compartments (Fig 5D). These effects were still visible after two‐week drug withdrawal (Fig 5E). However, the CD34+GPR56+ compartment was replenished in some mice in which this compartment was completely suppressed before drug withdrawal, suggesting that the CD34+GPR56+ fraction was specifically vulnerable to THZ1. In support of this, there was an anti‐correlation between the CD34+GPR56+ percentage in eight normal karyotype AML specimens and corresponding half‐inhibitory concentrations (IC50) for THZ1, suggesting that THZ1 affected pathways active in this compartment more than pathways in other fractions (Fig 5F). To dissect whether the activity of THZ1 against the GPR56+ compartment was caused by CDK7 or CDK12/13 inhibition, we treated the GPR56+ AML cell line HEL (Fig 5G, Appendix Fig 5E) and healthy CB CD34+ cells (Appendix Fig 5F) with specific CDK7i (YKL‐5‐124) and CDK12/13i (THZ531) (Kwiatkowski et al, 2014; Zeng et al, 2018; Olson et al, 2019). These experiments revealed that only CDK7i, but not CDK12/13i, suppressed GPR56 in these cell types. Further characterization of the compounds revealed that only CDK7i (THZ1, YKL‐5‐124), but not CDK12/13i (THZ531), suppressed the GPR56 CTF‐induced SRF signal in the luciferase reporter assay (Fig 5H, Appendix Fig 5G), while only THZ1 and THZ531, but not YKL‐5‐124, suppressed the baseline and GPR56‐enhanced Wnt reporter signals (Appendix Fig 5H).
Figure 5. CDK7/12/13 inhibitors suppress the GPR56+ LSC compartment in vitro and in vivo .
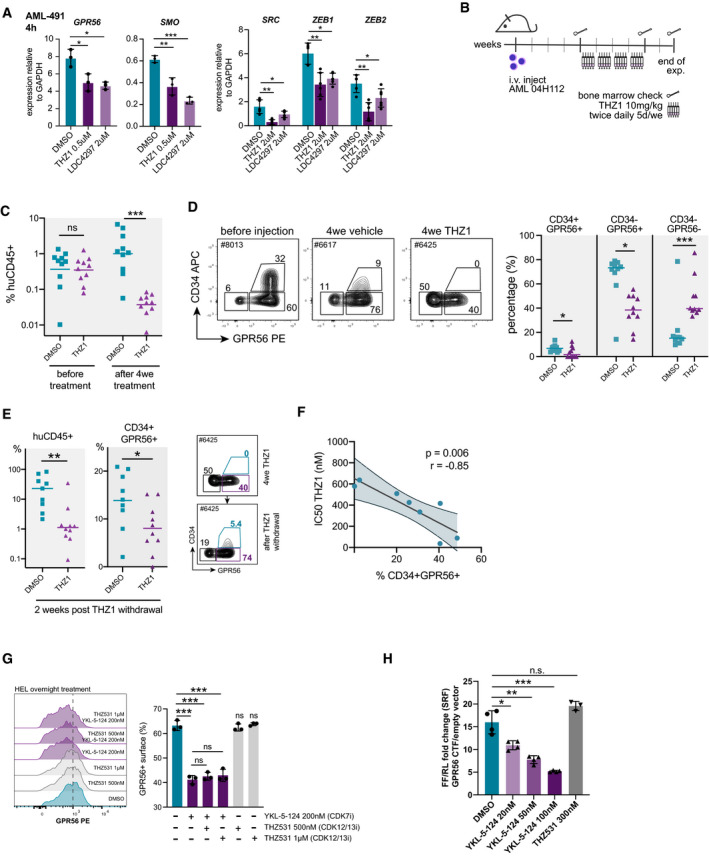
- Gene expression of GPR56, SMO (left), and TGFb targets SRC, ZEB1, and ZEB2 (right) normalized to GAPDH in AML‐491 cells determined by q‐RT‐PCR 4 h after treatment with THZ1 0.5 µM or LDC4297 2 µM. Unpaired t‐test. Mean, SD, and individual values from three individual treatments. ***P < 0.0005, **P < 0.005, *P < 0.05.
- Setup of in vivo drug treatment. NSG mice were injected with 105 AML 04H112 cells. Four weeks post injection, bone marrow (BM) was analyzed for human leukemic engraftment by BM aspiration. Treatment with either THZ1 or vehicle was started in the following week as indicated. BM was analyzed again after the end of the 4‐week treatment period.
- Overall percentage of human (huCD45+) leukemic cells in mice before and at the end of the 4‐week treatment period. Individual mice and mean engraftment are shown. Unpaired t‐test. ***P < 0.0005. N = 10 mice for each group.
- Left: Representative FACS plots showing the typical CD34lowGPR56high profile of sample 04H112 before injection and after the 4‐week treatment with THZ1. Right: both LSC compartments, the CD34‐GPR56+ and the CD34+GPR56+ fractions were significantly reduced in vivo in the THZ1 treatment group. Individual mice and mean engraftment are shown. Unpaired t‐test. ***P < 0.0005, **P < 0.005, *P < 0.05. N = 10 mice for each group.
- Left: Overall percentage of human (huCD45+) leukemic cells and the fraction of CD34+GPR56+ among human cells (mean, individual mice) two weeks after drug withdrawal. Right: FACS plot visualizing that the CD34+GPR56+ is re‐established upon drug removal. Unpaired t‐test, ***P < 0.0005, **P < 0.005, *P < 0.05.
- Correlation plot showing an anti‐correlation between the percentage of CD34+GPR56+ cells in AML samples and the corresponding IC50s for THZ1 determined in the respective samples. Pearson correlation. Each dot represents one sample. See Dataset EV12 for sample characteristics.
- FACS histogram plot (left) and summary bar graph from three replicate wells (right) showing reduction of GPR56 surface expression on the GPR56‐positive AML cell line HEL only in conditions that contain the more specific CDK7 inhibitor YKL‐5‐124, but not those that contain only the specific CDK12/13 inhibitor THZ531. Unpaired t‐test, bars, and error bars represent mean and SD of three individual treatments, ***P < 0.0005, **P < 0.005, *P < 0.05.
- SRF reporter assay showing dose‐dependent suppression of the GPR56‐CTF‐induced SRF signal by the CDK7 inhibitor YKL‐5‐124, but not by the CDK12/13 inhibitor THZ531. Four technical replicates. One of the four individual experiments. Unpaired t‐test, bars and error bars represent mean and SD, ***P < 0.0005, **P < 0.005, *P < 0.05.
These in vivo and in vitro results suggested that additional compounds are required to more efficiently eradicate both LSC compartments.
CDK7 inhibition synergizes with the Bcl‐2 inhibitor venetoclax in vitro and in vivo
As previous studies had already revealed a synergistic effect of THZ1 with venetoclax in other cancers (Cayrol et al, 2017), we tested the synergistic effect of THZ1 and the more specific CDK7i YKL‐5‐124 alone and in combination with 10 nM, 50 nM, and 500 nM of venetoclax in eleven primary AML samples (Fig 6A, Dataset EV12, Appendix Fig 6A). These included samples of different genetic groups, as well as patients refractory to salvage chemotherapies such as 5‐azacytidin with venetoclax, or quizartinib with high‐dose cytarabine and mitoxantrone. Samples not reaching a half‐maximum cell reduction at 500 nM venetoclax were considered venetoclax‐resistant. Normalized IC50s for THZ1 were significantly lower with increasing concentrations of venetoclax (Fig 6A, Dataset EV12). As synergism between drugs is not well reflected by individual IC50s, we used the R package ‘synergyfinder’ (He et al, 2018) to calculate different synergy scores including BLISS and ZIP scores (Bliss, 1939; Yadav et al, 2015; Fig 6A; Dataset EV12). There was in general strong synergism with venetoclax, however, BLISS scores for THZ1 were higher compared with the specific CDK7i YKL‐5‐124 in most samples. This might be because of the strong MCL‐1 suppressive activity of THZ1, which is also observed with CDK12/13i, but which is weaker or absent with CDK7i and not recapitulated by GPR56 KD (Appendix Fig 6B–D).
Figure 6. Synergism of CDK7 inhibitors with venetoclax.
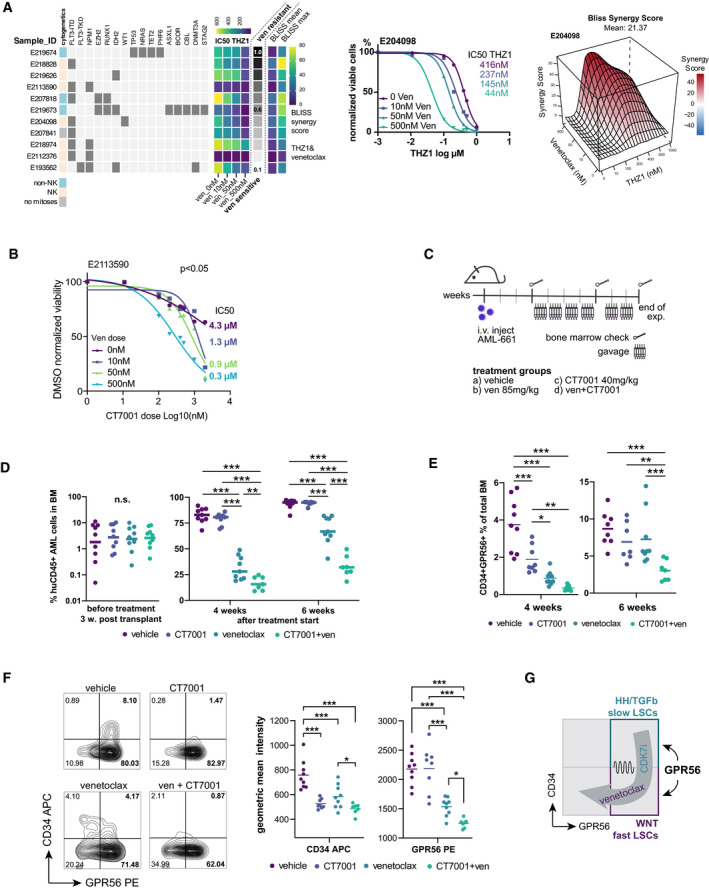
- Left: Eleven primary AML specimens with different cytogenetic and molecular genetic aberrations (see Dataset EV12 for details) were treated with THZ1 or YKL‐5‐124 alone or in combination with venetoclax. IC50 values in presence of increasing doses of venetoclax are visualized for THZ1. The gray‐scale bar indicates the fraction of viable cells in presence of a high dose of venetoclax (500 nM) compared with DMSO to provide an approximation whether a sample was rather sensitive or resistant against venetoclax alone. Note that the addition of venetoclax reduces the IC50 in all samples regardless of their baseline resistance against venetoclax alone. The last four columns provide the average and maximum BLISS scores reached by a combination of THZ1 and venetoclax (N = 11) Middle: representative dose‐response curves of THZ1 in presence of increasing doses of venetoclax for one primary AML specimen with intermediate venetoclax sensitivity. Right: representative 3D‐synergy plot showing the BLISS synergy scores at indicated concentrations of THZ1 and venetoclax for AML E204098.
- Dose‐response curve for the CDK7i CT7001 in presence of increasing doses of venetoclax for primary AML E2113590. The indicated doses on the right represent the IC50 dose.
- Schematic visualizing the setup for the combinatory in vivo treatment. AML‐661 cells were injected in NSG mice. Bones indicate the timepoints of bone marrow aspirations to monitor engraftment of human leukemic cells in mice before treatment start and during treatment.
- Percentage of human CD45+ leukemic cells three weeks post injection, i.e. before treatment start (left), and 4 and 6 weeks after treatment start (right). Dots represent individual mice, horizontal lines represent means. Unpaired t‐test, ***P < 0.0005, **P < 0.005.
- Percentage of human CD34+GPR56+ cells in the mouse bone marrow at 4 and 6 weeks post treatment start with the indicated compounds. Dots represent individual mice, horizontal lines represent means. Unpaired t‐test, ***P < 0.0005, **P < 0.005, *P < 0.05.
- Left: Representative FACS plots showing CD34 and GPR56 expression on AML cells after 4‐week treatment with the indicated compounds. Right: Statistical analysis of the geometric mean intensity of CD34 APC (left) and GPR56 PE (right) in the four treatment groups. Horizontal lines represent means. Unpaired t‐test, ***P < 0.0005, **P < 0.005, *P < 0.05.
- Cartoon visualizing how CDK7i and venetoclax synergize to suppress both GPR56+ compartments in AML.
Because of the pleiotropic effects of THZ1 including very strong MCL1 suppression, more specific CDK7i have been developed for clinical trials. To assess synergism of CDK7i and venetoclax in vivo, we therefore selected the more specific CDK7i CT7001, an orally active drug, which has entered clinical trials for solid cancers (NCT03363893), after confirming synergism with venetoclax in vitro in two different primary AML samples (Fig 6B, Appendix Fig 6E). The combinatory treatment of CT7001 and venetoclax significantly reduced AML expansion more than venetoclax alone (Fig 6C and D). The low efficacy of CT7001 as monotherapy might be because of the low dosage applied by gavage to reduce side effects (40 mg/kg instead of 100 mg/kg as reported elsewhere (Clark et al, 2017)). While both drugs as monotherapy significantly reduced the CD34+GPR56+ compartment within total mouse bone marrow when assessed at the end of the 4‐week treatment, only the combination therapy was able to significantly suppress the CD34+GPR56+ compartment by the end of the experiment (Fig 6E). FACS profiles assessed at four weeks confirmed that CT7001 affected the CD34+GPR56+ more than the CD34‐GPR56+ fraction similar to what we had observed with THZ1 (Fig 5D), while venetoclax significantly suppressed overall GPR56+ expression (Fig 6F).
In summary, our data suggest that combined CDK7i/venetoclax treatment might represent an efficient strategy to suppress both GPR56+ LSC compartments in AML (Fig 6G).
Discussion
The rationale for applying ATAC‐seq profiling to primary human AML samples was the aim to detect subtle changes in lowly abundant TF activities that might not be detectable by RNA‐seq to identify the network underlying the very poor outcome of AML with high GPR56 expression (Pabst et al, 2016; Garg et al, 2019) including TFs upstream of GPR56. Our combined ATAC‐/RNA‐seq approach pointed towards EMT regulators, Wnt, Hh, and Rho signaling. Although these pathways have been linked to self‐renewal in AML before, targeting them with small molecule inhibitors has not been proven sufficient to permanently eradicate the disease neither in pre‐clinical models (Fukushima et al, 2016; Jiang et al, 2018), nor in the clinical setting (e.g. FDA approved Hh inhibitor glasdegib; Savona et al, 2018; Norsworthy et al, 2019). The inability of these inhibitors to induce durable complete remissions raises the question why targeting bona fide self‐renewal pathways seems to rather have transient effects on LSCs, but fails to eliminate them. Here, we propose a model that provides an explanation on how LSCs might escape pathway‐specific LSC‐directed therapies while attributing GPR56 a pivotal role in this process.
We first dissected in individual functional assays including luciferase reporter assays, IF imaging, and xenotransplantation that GPR56 is functionally important for engraftment of human AML cells in immunocompromised mice and activates not only RhoA signaling, as described before (Piao et al, 2004; Singer et al, 2013; Kishore et al, 2016), but also regulates Wnt and Hh signaling, and EMT‐associated gene expression. In particular, our mechanistic studies revealed that GPR56 enhances Wnt signaling in dependence of the co‐receptor LRP6. Another example of an aGPCR that modulates Wnt signaling at the co‐receptor level is GPR124, which facilitates binding of Wnt7 to Frizzled and thereby promotes Wnt ligand‐specific effects (Eubelen et al, 2018). Moreover, we identified ITF2/TCF4, a basic helix‐loop‐helix TF, which is induced by Wnt/β‐catenin to promote cancer growth (Kolligs et al, 2002), as upstream regulator of GPR56, suggesting positive feedback between GPR56 and Wnt. A similar loop was found for SMO/Hh in a way that GPR56 KD suppressed SMO mRNA and RFX2 target genes, while the GPR56 promoter in turn was shown before to be bound by several RFX TFs (Bae et al, 2014). Moreover, the EMT regulator SNAI1 was identified upstream of GPR56, while GPR56 itself positively regulated TGFB1, the key driver of EMT, and SRC, which is required for TGFB1 induced EMT (Galliher & Schiemann, 2006).
Despite these positive feedback loops, we found that the Wnt pathway seems to play a crucial antagonizing role in this network, raising the question why GPR56 concomitantly enhances pathways that inhibit each other. The principle of reciprocal inhibition is known from the neuronal system as the basis of continuous neuronal oscillation (Friesen, 1994), and antagonism of Wnt and Hh has also been observed in neurogenesis (Joksimovic et al, 2009). Integration of our novel functional data presented here with our previous gene expression studies (Garg et al, 2019) and sorting experiments (Pabst et al, 2016) finally suggests a scenario, in which GPR56 ensures the constant supply of LSC‐enriched compartments with different biological features: the CD34+GPR56+ fraction characterized by high TGFb/Hh activity, low WNT activity, and slow cell cycle progression (Hh/TGFbonWNToff), and the more rapidly expanding, CD34 ‐ GPR56+ compartment characterized by lower LSC frequency, low Hh/TGFb activity, and high WNT activity (Hh/TGFboffWNTon). Evidence for reciprocal transition between the two LSC compartments exists: we previously showed that CD34+GPR56+ cells expanded more slowly than CD34‐GPR56+ cells and were capable of generating all other fractions in vitro and in vivo (Pabst et al, 2016). Here, we provide evidence that the CD34+GPR56+ compartment can be regenerated by CD34‐ cells from sorted in vitro cells and also in mice, in which the CD34+GPR56+ compartment was completely depleted during THZ1 treatment, further corroborating that the two GPR56+ LSC compartments can replenish each other. In line with this scenario, GPR56‐regulated genes comprised CCNE1, which has been described to enable quiescent cells to re‐enter cell cycle (Campaner et al, 2013).
Reciprocal transition between slowly and rapidly cycling GPR56+ LSCs would also explain how GPR56high AML escapes standard chemotherapy treatment. Moreover, the interactions of multiple signaling pathways suggest that inhibiting one of these pathways individually, e.g. as tried with Hh inhibitors will be insufficient to fully block the GPR56‐associated network. Our data suggest that combined CDK7/12/13 inhibitors such as THZ1 might be required to overcome these issues. As observed in other cancers (Cayrol et al, 2017), THZ1 synergized with the Bcl‐2 inhibitor venetoclax most likely in part by suppressing MCL‐1. However, we also observed synergism of venetoclax with more specific CDK7i in vitro and in vivo, which had no or only a weak effect on MCL‐1 protein levels suggesting that the synergism between CDK7i and venetoclax was mediated also by other mechanisms than MCL‐1 suppression. The precise molecular processes underlying synergism of CDK7i and venetoclax will have to be addressed in future studies. Importantly, synergism of THZ1 and the more specific CDK7i YKL‐5‐124 occurred also in samples, which were highly resistant to venetoclax alone, and also in FLT3‐ITD samples from patients, who were refractory to FLT3‐inhibitors, and might therefore offer additional benefits compared to currently available venetoclax ‐based combination regimens. Together, these results propose combinatorial treatment of venetoclax with CDK7i as promising therapeutic approach to suppress the GPR56+ associated network in AML.
Materials and Methods
Patient and cord blood samples
Patient samples and cord blood units were collected after obtaining written informed consent in accordance with the Declaration of Helsinki and the Department of Health and Human Services Belmont Report. Cryopreserved AML patient samples were provided by the Medical Department V, Heidelberg University Hospital, Germany, the Department of Internal Medicine I, University Hospital of Dresden Carl Gustav Carus, Germany, the Department of Internal Medicine III, Ludwig‐Maximilians‐University, Munich, Germany, and the Leukemia Cell Bank of Quebec, Maisonneuve‐Rosemont Hospital, Montreal, Canada. Samples were provided according to ethically approved protocols by several biobanks. The project was approved by the Research Ethics Board of the Medical Faculty of Heidelberg University. Cord blood units were collected after obtaining written informed consent at the CHU Sainte‐Justine, Montreal, Canada, and the Department of Obstetrics at University Hospital Heidelberg, Germany. Patient‐derived xenograft AML‐491 and AML‐661 cells were generated as described before (Vick et al, 2015) and were kindly provided by I. Jeremias and B. Vick.
In vivo experiments
NOD. Cg‐PrkdcscidIl2rgtm1Wjl (NSG) and NOD. Rag1‐; γcnull‐SGM3 (NRGS) mice were purchased from Jackson Laboratories. NOD. Cg‐KitW‐41J Prkdcscid Il2rgtm1Wjl /WaskJ (NSGW41) mice were kindly provided by Dr. Claudia Waskow (Cosgun et al, 2014). Female or male mice aged 6–20 weeks were used in our study. They were bred and housed in the specific pathogen‐free animal facility at the German Cancer Research Center (DKFZ), Heidelberg. All animal experiments were approved and performed in accordance with the regulatory guidelines of the official committee (Regierungspräsidium Karlsruhe). For xenotransplantation assays, cells were injected in the mice via the tail vein after sublethal irradiation (1.75 Gy for NSG, 2 × 2.5 Gy for NRGS). Engraftment levels were analyzed at indicated time points by bone marrow aspiration, and total marrow was analyzed at final sacrifice. For CB CD34+ experiments, cells were injected after overnight transduction, and gene transfer was determined by flow cytometry in the remaining cultured cells 48 h later. For Fig 2E, 5 × 105 and 1 × 105 transduced AML‐491 cells were injected in NRGS and NSGW41 mice, respectively, after overnight transduction. Gene transfer was determined by flow cytometry 72 h post transduction. For the THZ1 in vivo treatment, 1 × 105 patient‐derived xenograft (PDX‐04H112) AML cells were suspended in 200 μl phosphate buffer solution and injected intravenously (i.v.) in NSG mice. Successful engraftment levels were confirmed at week 4 post injection. Intraperitoneal (i.p.) treatment was started subsequently with either vehicle (1% DMSO + 20% PEG300 + 30% corn oil in water) or THZ1 (10 mg/kg, twice daily, Monday to Friday) for 4 weeks. For the CT7001 and venetoclax in vivo treatment, 1 × 105 patient‐derived xenograft PDX AML‐661 cells were injected intravenously (i.v.) in sublethally irradiated NSG mice. At 3 weeks post injection, mice were randomly sorted into 4 treatment groups: vehicle (1% DMSO + 20% PEG300 + 30% corn oil in water), 85 mg/kg venetoclax, 40 mg/kg CT7001, or a combination of 85 mg/kg venetoclax and 40 mg/kg CT7001. Mice were dosed orally, once daily, 5 days a week, for 4 weeks. Bone marrow was analyzed by aspiration from one femur in the following week, and treatment was restarted for another two weeks, after which bone marrow was analyzed again.
RNA‐and ATAC‐seq analyses
ATAC‐seq library preparation details
We followed the Omni‐ATAC method outlined previously (Buenrostro et al, 2015) for sample preparation. The library was optimized for enrichment for 100–1,000 bp fragments using SPRI beads based size‐selection, and the quality of the purified DNA library was analyzed on a Bioanalyzer (2100 Expert software, Agilent Technologies) using High Sensitivity DNA Chips (Agilent Technologies Inc #5067‐4626). The appropriate concentration of sample was determined using the Qubit Fluorometer (Molecular Probes). Ten 40 nM samples were pooled and run on a NextSeq 500/550 High Output Kit (Illumina, Inc. San Diego, CA #20024907) and the NextSeq 500 Illumina Sequencer to obtain paired end reads of 75 bp.
ATAC‐seq data processing
The processing of ATAC‐seq data has been described in detail in a previous study by (Berest et al, 2019). Briefly, we used an in‐house Snakemake (Köster & Rahmann, 2012) pipeline that starts with raw fastq files and integrates multiple steps for quality control (FastQC), adaptor trimming (Trimmomatic (Bolger et al, 2014)), alignment (Bowtie2; Langmead & Salzberg, 2012), as well as general and ATAC‐Seq specific post‐alignment filtering and processing steps (Berest et al, 2019). Noteworthy, the filtering steps include: (i) removing mitochondrial reads and reads from non‐assembled contigs or alternative haplotypes, (ii) filtering reads with a mapping quality below 10, (iii) marking and removing duplicate reads with MarkDuplicates from the Picard toolset, (iv) adjusting read start sites as described previously (Buenrostro et al, 2013); and (v) removing reads with insertions or deletions using samtools. Finally, peak calling is done with MACS2 (parameters: ‐q 0.01 ‐g hg19 ‐‐nomodel ‐‐keep‐dup all) followed by filtering using bedtools subtract against the publicly available blacklist regions. Last, for quality control, we also obtained various summary statistics and additional files and plots (e.g., coverage files for visualization, transcription start site enrichment, sample‐specific fragment length distributions, library complexity measures, PCA, sample correlations).
Differential peak analysis and consensus peak generation
To identify differentially bound peaks between GPR56high and GPR56low samples, we used the DiffBind Bioconductor R package (Ross‐Innes et al, 2012) with the mutational status as a blocking factor. Consensus peaks were generated with the function dba.peakset using the parameter minOverlap to define the number of samples within which a peak should be present based on all 35 samples, therefore, also including the GPR56middle ones that were not used further subsequently except for consensus peak generation. We used different values for minOverlap and generated a consensus peakset out of the individual peak files as generated by the ATAC‐seq pipeline as outlined above for values of 1, 10, 15, and 30. The consensus peakset based on minOverlap = 10 consists of 109,803 (used as default for all subsequent analyses), for minOverlap = 1 247,442 (used as background for the GREAT analysis), for minOverlap = 15 87,410 and for minOverlap = 30 32,406 peaks.
ATAC‐seq PCA
We performed a principal‐component analysis (PCA) based on the top 500 variable peaks from the consensus peaks (based on the variance using rowVars) based on the variance‐stabilized data (see below for details). To summarize and quantify the contribution of each covariate to the overall variability, we then ran a linear regression for each covariate and each of the first 5 PCs and extracted the adjusted R‐squared value.
Enrichment analysis of differential ATAC‐seq peaks
For annotating the differentially bound peaks, we used multiple methods. First, we used three different consensus peaksets (based on minOverlap = 1, 10, and 30) as well as the differentially bound peaks only and overlapped them with known annotation categories (e.g., promoter, downstream, intragenic, UTR, etc) with the annotatePeak function of the ChIPseeker R/Bioconductor package using the default parameters. For overlaps with the enhancer annotation, we used the human data from the EnhancerAtlas 2.0 (http://www.enhanceratlas.org/; Gao & Qian, 2020).
Last, we also used GREAT (McLean et al, 2010) v3 via the rGREAT Bioconductor package with the consensus peakset based on minOverlap = 1 (see above) as background and as foreground all differentially bound peaks (FDR < 0.05) with either a positive or a negative fold‐change.
Differential TF activity analysis
We ran diffTF (Berest et al, 2019) in permutation mode with 1,000 permutations for 640 TFs based on in silico predicted TFBS (using PWMScan, see (Berest et al, 2019) for full details) using TF‐binding models from the HOCOMOCO v10 database (Kulakovskiy et al, 2013) with the consensus peakset for minOverlap = 10 as described above and the same design as for DiffBind (i.e., including the mutational status as covariate).
GPR56 KD RNA‐seq experiment
Frozen cord blood (CB) MNC from two female donors were thawed, CD34+ cells were isolated by magnetic bead separation as described above, pooled, and transduced with lentiviral particles at multiplicity of infection (MOI) 30 after 48 h pre‐stimulation in culture media at a density of 150,000 cells per well in a 12‐well plate, 1 ml volume, 2 individual infections per condition. Cells were washed on the next day and sorted for GFP positivity 72 h post infection. Cells were resuspended in Trizol for RNA isolation directly after sorting, and RNA was isolated according to the manufacturer's instructions followed by an additional purification step on RNeasy Mini columns. High RNA quality was confirmed by nanodrop and bioanalyzer (RIN between 9.8 and 10). Ovation human FFPE RNA‐Seq Library Systems (Nugen) was used for library preparation. Libraries were prepared using random and poly(T) priming, and unstranded, rRNA‐depleted cDNA libraries were sequenced on an Illumina NextSeq platform.
GPR56 KD RNA‐seq data processing
We first performed initial quality control before and after adaptor trimming (cutadapt, ‐m 30) (Martin, 2011) with FastQC. We then aligned the samples to hg38 using STAR (Dobin et al, 2013) with the parameters ‐‐alignSJoverhangMin 8 ‐‐alignSJDBoverhangMin 1 ‐‐alignMatesGapMax 1000000 ‐‐alignIntronMin 20 ‐‐alignIntronMax 1000000 ‐‐outFilterType BySJout ‐‐outFilterMultimapNmax 20 ‐‐outFilterMismatchNmax 999 ‐‐outFilterMismatchNoverReadLmax 0.04. For gene annotation, we used the Gencode (Harrow et al., 2012) v28 annotation. We then quantified gene counts using featureCounts from the Subread (Liao et al, 2014) package with the parameters ‐p ‐B ‐C ‐Q 10 ‐O ‐s 2 ‐t exon ‐g gene_id. For differential expression, we employed DESeq2 (Love et al, 2014) with the design formula “condition,” and size factor normalization to compare both the weak and the strong KD versus control. Finally, we computed and subsequently used the shrunken log2 fold‐changes via lfcShrink from DESeq2 (Love et al, 2014) with the apeglm (Zhu et al, 2019) method.
Visualization of ATAC‐seq and RNA‐seq data
For all heatmaps as well as the PCA, we variance‐stabilized the shown data using the vst function from DESeq2.
Cell culture
Frozen cryotubes were briefly thawed in a 37°C water bath and resuspended in warm thawing media containing Iscove´s modified Dulbecco´s medium (IMDM) (Thermo Scientific #21980065) supplemented with 20% Fetal bovine serum (FBS) (Sigma #F7524) and 100 µg/ml DNase I (Sigma #DN25). Primary AML cells were cultured in IMDM supplemented with 15% BIT (bovine serum albumin, insulin, transferrin, Stem Cell Technologies #09500), SCF 100 ng/ml (Shenandoah #100‐04), FLT3L 50 ng/ml (Shenandoah #100‐21), IL‐3 20 ng/ml (Shenandoah #100‐80), G‐CSF 20 ng/ml (Shenandoah #100‐72), β‐mercaptoethanol (10−4 M), Gentamicin (50 µg/ml), and Ciprofloxacin (10 µg/ml). Fresh cord blood samples were subjected to mononuclear cell (CB‐MNCs) isolation by Ficoll Hypaque (Thermo # GE17‐1440‐02) density gradient. CD34+ cells were isolated from CB using MACS microbeads (Miltenyi #130‐100‐453) for 1–2 rounds and checked for CD34+ purity by FACS. CD34+ cells were cultured in IMDM supplemented with 20% BIT (Stem Cell Technologies #09500), SCF 100 ng/ml (Miltenyi #130‐096‐695), FLT3L 100 ng/ml (Miltenyi #130‐096‐479), TPO 50 ng/ml (Miltenyi #130‐095‐752), β‐mercaptoethanol 10−4 M (Gibco #21985023), Gentamicin 50 µg/ml (Thermo #15750060), and UM171 35 nM (Stem cell technologies #72912). For large‐scale cultures, cells were cultured in T25 flasks (TPP #90026), 6‐well plates (Sarstedt #833.920.500), 12‐well plates (Sarstedt #833.921.300) at a density of 3–5 × 105 per ml. Wnt3a and control conditioned media were generated as described (Willert et al, 2003).
Cell lines
MV4‐11 cells (#ACC 102), OCI‐AML2 (#ACC 99), OCI‐AML3 (#ACC 582), HL‐60 (#ACC 3), Kasumi1 (#ACC 220), K562 (#ACC 10), KG1a (#ACC 421), UCSD‐AML1 (#ACC 691), HEL (#ACC 11), and HNT34 (#ACC 600) were purchased from Leibniz Institute DSMZ‐German Collection of Microorganisms and Cell Cultures, Braunschweig, Germany and cultured according to the company´s recommendations. hTERT‐immortalized retinal pigment epithelial cells (RPE‐1, ATCC CRL‐4000) were maintained in DMEM, 10% FBS prior to serum starvation. RPE‐1 cells carrying murine SMO‐eGFP were generated as described (Viol et al, 2020).
Reporter assays and reporter cell lines
Wnt and SRF reporter assays
WT or LRP6 KO (LRP6−/−) (Berger et al, 2017) HEK293T cells were cultured in DMEM supplemented with 10% FBS. For the SuperTop Wnt reporter assay, cells were plated in 96‐well plates in 3–4 replicates and transfected with 50 ng SuperTop reporter plasmid and 2.5 ng pTK‐Renilla control (Promega) as previously described (Cruciat et al, 2010) together with 50 ng of plasmid DNA carrying either shRNA against GPR56 or cDNA of GPR56 FL, GPR56 CTF, or GPR56 CTF mutants (pcDNA3.1) as indicated using TurboFect transfection reagent (Thermo #R0532) following the supplier’s protocol. Cells were stimulated with Wnt3a‐conditioned media overnight one day post transfection when indicated in figures. For detection of Rho pathway activation, the previously described 3DA. Luc SRF reporter system was used together with MAL/MKL1 (Megakaryocytic Acute Leukemia) expression plasmid (Miralles et al, 2003) as positive control and pTK‐Renilla (Promega) for normalization. For bioluminescence detection, which was performed 48‐h post transfection, cells were lysed in a 96‐well plate format using 1× passive lysis buffer provided in Dual‐Luciferase Reporter Assay System (Promega #E1910), and 25 μl of lysates were transferred into white 96‐well plates (Thermo #9502887). Firefly and Renilla luminescence were measured in a Tecan Microplate reader (SPARK). To normalize for transfection efficiency, Firefly luciferase activity was divided by Renilla luciferase activity to obtain Relative Luminescence Units (RLU).
SMO‐GFP RPE‐1 cell line
Doxycycline‐inducible Tet3G‐Smo‐EGFP RPE cells were generated as described (Viol et al, 2020). For GFP detection, 1 × 104 SMO‐GFP RPE cells were plated in a 48‐well plate format followed by lentiviral transduction of GPR56 shRNAs and shLuc on the second day. Seventy‐two hours post‐infection, cells were treated overnight with 5 ng/ml doxycycline with or without 1 μM Smoothened Agonist (SAG) HCl (Selleckchem #S7779). On the next day, cells were trypsinized and analyzed by flow cytometry for GFP expression or by q‐RT‐PCR for gene expression changes.
Lentivirus production and transduction
The production of high‐titer lentiviral particles was carried out following previously described protocols (Garg et al, 2019). In brief, TurboFect transfection reagent (Thermo #R0532) was used to transiently package 7.5 μg lentiviral vector with 15 μg psPAX2 packaging plasmid and 4.5 µg VSV‐G plasmid. psPAX2 was a gift from Didier Trono (Addgene plasmid #12260; http://n2t.net/addgene:12260; RRID:Addgene_12260). HEK293T cells were transfected, cultured in DMEM supplemented with 3% heat‐inactivated FBS, and lentiviral supernatant was harvested 48 and 72 h post transfection. Ultracentrifugation was performed through a 20% sucrose cushion to concentrate viral supernatants using a Sorvall WX Ultra 100 ultracentrifuge for 2 h at 4°C and 29,000 rpm. The high‐titer lentiviral particles were reconstituted in Opti‐MEM and stored at −80°C until use. For transduction of shRNAs, cells were first incubated with protamine sulfate for 30 min, followed by adding lentiviral particles directly into culture media for 12–16 h. Forty‐eight to 72 h post infection, gene transfer was checked using flow cytometry.
Colony assays
After lentiviral transduction, CB CD34+ cells were sorted into methylcellulose (Methocult, Stem Cell Technologies #4035) supplemented with EPO 3 IU/ml (Cedarlane #102‐04‐2000 IU) and gentamicin 50 µg/ml (Thermo #15750060) 72 h post infection and plated at 2 ml per well in 12‐well plates (Sarstedt #833.921.300). Colonies were analyzed 10–14 days after plating using an inverted microscope and 10× and 20× magnification.
Flow cytometry and cell sorting
Cells were stained using the following antibodies: CD45‐Pacific blue (Biolegend #304029), GPR56‐PE (Biolegend #358204), CD11b‐PECy5 (Biolegend #301308), CD34‐APC (BD biosciences #555824), CD14‐APC‐Cy7 (Biolegend #325620), CD45RA‐PE (BD bioscience # 555489), CD11b‐PECy5 (Biolegend #301308), CD38‐PECy7 (BD biosciences #560677), in vivo engraftment levels were analyzed with anti‐human CD45‐APC (BD biosciences #555485), CD33‐PECy5 (BD biosciences #551377) and CD19‐PECy7 (BD biosciences #557835). Cell sorting was performed on BD FACS Aria II. Data were acquired on a BD LSRII or BD Celesta flow cytometer equipped with a High throughput sampler (HTS) device and analyzed using BD FACS Diva 4.0 and Flowjo X (Treestar Inc.) software.
Quantitative real‐time polymerase chain reaction (q‐RT‐PCR)
Total RNA was extracted using Trizol reagent (Invitrogen/Life Technologies) according to the manufacturer's instructions. cDNA was obtained from total RNA by reverse transcription using M‐MLV RT (Thermo #28025103) in 25 μl reactions. Subsequently, q‐RT‐PCR was performed on a Biorad q‐RT‐PCR machine (CFX96 Touch Real Time PCR detection system). GAPDH was used as endogenous control. The q‐RT‐PCR primers used in the study are listed in Appendix Table S1.
Immunofluorescence and microscopy
The antibodies used were mouse anti‐γ‐tubulin (Sigma #T6557), rabbit polyclonal anti‐Arl13b (Proteintech #17711‐1AP), rabbit polyclonal anti α‐tubulin (MBL #PM054). For cilia analysis, RPE cells were grown on coverslips coated with fibronectin, followed by lentiviral transduction of shRNAs 24 h post plating. Forty‐eight hours after infection, cells were cultured in serum‐free DMEM culture media for 24 h to stimulate cilia growth. Cells were then washed and fixed in 4% paraformaldehyde at 4°C followed by iced methanol for 5 min. After blocking in 0.1% Triton X100/PBS supplied with 1% BSA for 1 h, fixed cells were incubated with indicated primary antibodies (1:200) at 4°C overnight. After washing, cells were incubated with either mouse or rabbit Alexa fluor 488, Alexa fluor 594, and Alexa fluor 647 secondary antibodies (1:500, Thermo). DNA was stained with DAPI. Ciliated cell samples were observed using Axio Observer Z1 fluorescence motorized microscope (Zeiss) equipped with 40× NA 1.4 Plan‐Apochromat oil immersion objective. Raw images were processed in ImageJ.
Compound testing and calculation of synergism
The CDK12/13 inhibitor THZ531 (Selleckchem #S6595), the CDK7/12/13 inhibitor THZ1 (Selleckchem #S7549), and the CDK7 inhibitors YKL‐5‐124 (Selleckchem #S8863) and LDC4297 (Selleckchem #S7992) were dissolved in DMSO at 50 mM stock concentration and diluted in media so that final DMSO concentration was 0.1% in all conditions. SAG (Selleckchem #S7779) was dissolved in water at 20 mM stock concentration. PRI‐724 (#S8968), CHIR‐99021 (#S2924), iCRT3 (#S8647), and venetoclax (#8048) were purchased from Selleckchem and dissolved in DMSO. For in vivo THZ1 treatment, THZ1(#HY‐80013) was purchased from MedChemExpress and dissolved in DMSO at 300 mg/ml stock concentration. For the in vivo combinatorial treatment, CT7001 (Chemietek #CT‐CT7001) and venetoclax (Hycultec #HY‐15531) were dissolved in vehicle solution (1% DMSO + 20% PEG300 + 30% corn oil in water).
For IC50 calculation and synergism calculation, cells were plated in 384‐well plates and incubated with the combinations of different drugs for 3–5 days as indicated in figures. Each treatment condition was performed in at least 3 replicates. Cell viability was measured based on propidium iodide (PI) staining using a BD LSRII or BD Celesta flow cytometer equipped with a High throughput sampler (HTS) device. The resulting dose‐response matrix data and Bliss scores were analyzed with the synergyfinder R package (Yadav et al, 2015; He et al, 2018). IC50s were calculated with GraphPad Prism v09.
Western blot
For isolation of total lysates, cells were pelleted after 1× PBS wash and lysed in RIPA lysis buffer (Thermo #89900) supplied with protease inhibitor cocktail (Sigma #11836170001). Lysates were collected in new pre‐chilled tubes and protein concentration was measured using Bradford reagent (Bio‐Rad #500‐025) and BSA standards (Bio‐Rad #500‐027). Equal amount of protein was mixed with 4× gel loading dye (Thermo #NP007) and analyzed by pre‐casted gel (Thermo #NP0036) and Western blotting. Final western blots were detected on a GE Healthcare Life Sciences, Amersham Imager 600, followed by analyzing band intensities with ImageJ (U.S. National Institutes of Health, Bethesda, MD, USA).
Antibodies used for western blotting were as follows: anti‐LRP6 (#S2560) primary antibody was purchased from Cell Signaling Technology, antibody against anti‐GAPDH (#GTX627408) was from GeneTex, anti‐Vinculin (#sc‐736914) primary antibody was from Santa Cruz, antibody against β‐catenin (#610153) was from BD Biosciences, anti‐MCL‐1 (#ab32087), anti‐BCL‐2 (#ab692) primary antibodies were from Abcam.
Cloning procedures
Small hairpin (sh)‐oligos against GPR56, Luciferase, or scrambled were cloned into the plko.1 system (Sigma), in which the puromycin resistance cassette had been replaced by eGFP (pLKO‐U6‐shOligo‐hPGK‐eGFP) or Ametrine (pLKO‐U6‐shOligo‐hPGK‐Ametrine), the shRNA oligos used are listed in Appendix Table S2.
GPR56 full‐length cDNA was obtained from OriGene Technologies (NM_005682.4) and cloned into pcDNA3.1 and the previously described pCCL‐c‐MNDU3‐eGFP backbone (Garg et al, 2019). The primers used for generating GPR56 CTF and mutant constructs are listed in Appendix Table S3.
Statistical analyses
For the RNA‐ and ATAC‐seq analyses, statistical tests are described in each of the relevant analyses and p‐values are given in text or Figures. Individual assays were analyzed with Graph Pad Prism 8 by unpaired t‐tests to compare two conditions if not otherwise stated in Figure legends. When multiple groups were compared, P‐values were Benjamini Hochberg corrected. Asterisks indicate the following P‐value levels if not otherwise specified in the text: *P < 0.05, **P < 0.005, ***P < 0.0005, ****P < 0.0001.
Author contributions
Caroline Pabst: Conceptualization; Formal analysis; Supervision; Funding acquisition; Visualization; Writing—original draft; Writing—review and editing. Lixiazi He: Data curation; Formal analysis; Validation; Visualization; Writing—original draft; Writing – review and editing. Christian Arnold: Data curation; Formal analysis; Methodology. Judith Thoma: Formal analysis; Writing – review and editing. Christian Rohde: Software; Formal analysis; Methodology. Maksim Kholmatov: Formal analysis. Swati Garg: Data curation; Formal analysis; Validation. Cheng‐Chih Hsiao: Data curation; Formal analysis. Linda Viol: Data curation; Formal analysis; Visualization; Writing – original draft. Kaiqing Zhang: Data curation; Formal analysis. Rui Sun: Formal analysis. Christina Schmidt: Formal analysis; Visualization. Maike Janssen: Software; Formal analysis; Writing – original draft. Tara MacRae: Data curation; Formal analysis. Karin Huber: Data curation; Writing – original draft. Christian Thiede: Resources. Josée Hébert: Resources. Guy Sauvageau: Resources; Writing – original draft. Julia Spratte: Resources. Herbert Fluhr: Resources; Supervision. Gabriela Aust: Resources; Writing – original draft. Carsten Müller‐Tidow: Resources; Writing – original draft. Christof Niehrs: Resources; Writing – original draft. Gislene Pereira: Resources; Supervision. Jörg Hamann: —Resources; Supervision. Motomu Tanaka: Supervision; Writing – original draft; Writing – review and editing. Judith B Zaugg: Supervision; Writing – original draft.
In addition to the CRediT author contributions listed above, the contributions in detail are:
LH performed experiments, analyzed data, generated figures, and wrote the manuscript. CA performed computational analyses, generated figures, and wrote the manuscript. LH and CA contributed equally to this work. JT performed dynamic phenotyping experiments and analyses, MK analyzed data and generated figures, SG contributed to in vivo experiments, C‐CH generated plasmids, LV performed IF imaging and provided reagents, KZ and RS performed Wnt reporter assays, CS and MJ performed synergism experiments and analyses, TM performed and analyzed in vivo experiments, CR performed computational analyses, KH helped with in vivo experiments and edited the manuscript, CT, JH, and GS provided primary AML samples and edited the manuscript, JS and HF generated CB units and edited the manuscript, GA supported SRF reporter assays, CM‐T edited the manuscript, CN provided reporter assays and supported Wnt experiments, GP provided expertise and reagents for primary cilium detection and analyses, JH provided plasmids and expertise on structure‐function analyses, MT supervised and analyzed dynamic phenotyping experiments and edited the manuscript, JBZ supervised all computational analyses, wrote the manuscript, and co‐supervised the project, CP directed the project, performed analyses, and wrote the manuscript.
Disclosure and competing interests statement
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Dataset EV1
Dataset EV2
Dataset EV3
Dataset EV4
Dataset EV5
Dataset EV6
Dataset EV7
Dataset EV8
Dataset EV9
Dataset EV10
Dataset EV11
Dataset EV12
Acknowledgments
We thank V. Eckstein for support with cell sorting, R. Schneider for helping with cell culture experiments, and M. Frechette for assistance with in vivo experiments. We thank the teams of the Banque de Cellules Leucémiques du Québec (BCLQ), the Biomaterial banks of Medical Department V, Heidelberg University Hospital, the Department of Internal Medicine I, University Hospital of Dresden Carl Gustav Carus, Germany, the Department of Internal Medicine III, Ludwig‐Maximilians‐University, Munich, Germany, the Leukemia Cell Bank of Quebec, Maisonneuve‐Rosemont Hospital, Montreal, Canada, the CHU Sainte‐Justine, Montreal, Canada, the Department of Obstetrics at University Hospital Heidelberg, Germany, and all donors for providing primary human cells. We thank G. Posern for providing the SRF reporter system and positive control plasmids. We thank I. Jeremias and B. Vick, Helmholtz Zentrum München, for providing PDX AML‐491 cells. The work of G.P. and L.V. was supported by the collaborative SFB873 (project A14) and Heisenberg (PE1883‐3/4) programs of the DFG. This work was supported by Deutsche Forschungsgemeinschaft (DFG) FOR2149 grants PA 2815/2‐1 and HA‐4663/1‐1 to C.P. and J.H., respectively and by SFB1324 (project 331351713). C.P. is supported by a Max‐Eder‐Grant of German Cancer Aid (70111531) and by the Olympia‐Morata‐Program of the Medical Faculty of Heidelberg. The authors gratefully acknowledge the data storage service SDS@hd supported by the Ministry of Science, Research and the Arts Baden‐Württemberg (MWK) and the German Research Foundation (DFG) through grant INST 35/1314‐1 FUGG and INST 35/1503‐1 FUGG. Open Access funding enabled and organized by Projekt DEAL.
EMBO Mol Med (2022) 14: e14990.
Data availability
Sequencing data generated for this study are accessible through GEO database (https://www.ncbi.nlm.nih.gov/geo/browse/) with the accession numbers: GSE150175 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE150175) for the GPR56 KD RNA‐seq data, GSE150868 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE150868) for the ATAC‐seq data.
GPR56 expression following KD of TCF4 via shRNA in neuronal progenitors derived from iPSC was derived from Dataset EV4 of PMID31535015 (Doostparast Torshizi et al, 2019). For the integration with our human data, we created a mapping between the Ensembl IDs from human and mouse using biomaRt (Durinck et al, 2009). Public datasets used in this study were downloaded from GEO and comprise GSE147727 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE147727), GSE38236 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE38236), GSE70872 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE70872), and GSE48843 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE48843). We used the raw data whenever available (GSE111669; http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE111669) and processed them through DESeq2, in analogy to what has been described above. If no raw data were available or the data were also analyzed with DESeq2 (all other), we used the processed data (i.e., log2 fold‐changes and adjusted P‐values).
References
- Assi SA, Imperato MR, Coleman DJL, Pickin A, Potluri S, Ptasinska A, Chin PS, Blair H, Cauchy P, James SR et al (2019) Subtype‐specific regulatory network rewiring in acute myeloid leukemia. Nat Genet 51: 151–162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bae B‐I, Tietjen I, Atabay KD, Evrony GD, Johnson MB, Asare E, Wang PP, Murayama AY, Im K, Lisgo SN et al (2014) Evolutionarily dynamic alternative splicing of GPR56 regulates regional cerebral cortical patterning. Science 343: 764–768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berest I, Arnold C, Reyes‐Palomares A, Palla G, Rasmussen KD, Giles H, Bruch P‐M, Huber W, Dietrich S, Helin K et al (2019) Quantification of differential transcription factor activity and multiomics‐based classification into activators and repressors: diffTF. Cell Rep 29: 3147–3159 [DOI] [PubMed] [Google Scholar]
- Berger BS, Acebron SP, Herbst J, Koch S, Niehrs C (2017) Parkinson’s disease‐associated receptor GPR37 is an ER chaperone for LRP6. EMBO Rep 18: 712–725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bliss CI (1939) The toxicity of poisons applied jointly. Ann Appl Biol 26: 585–615 [Google Scholar]
- Bolger AM, Lohse M, Usadel B (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30: 2114–2120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ (2013) Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA‐binding proteins and nucleosome position. Nat Methods 10: 1213–1218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro JD, Wu B, Chang HY, Greenleaf WJ (2015) ATAC‐seq: a method for assaying chromatin accessibility genome‐wide. Curr Protoc Mol Biol 109: 21.29.1–21.29.9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burk AS, Monzel C, Yoshikawa HY, Wuchter P, Saffrich R, Eckstein V, Tanaka M, Ho AD (2015) Quantifying adhesion mechanisms and dynamics of human hematopoietic stem and progenitor cells. Sci Rep 5: 9370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campaner S, Viale A, De Fazio S, Doni M, De Franco F, D’Artista L, Sardella D, Pelicci PG, Amati B (2013) A non‐redundant function of cyclin E1 in hematopoietic stem cells. Cell Cycle 12: 3663–3672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network , Ley TJ, Miller C, Ding L, Raphael BJ, Mungall AJ, Robertson AG, Hoadley K, Triche TJ, Laird PW et al (2013) Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med 368: 2059–2074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmichael CL, Wang J, Nguyen T, Kolawole O, Benyoucef A, De Mazière C, Milne AR, Samuel S, Gillinder K, Hediyeh‐zadeh S et al (2020) The EMT modulator SNAI1 contributes to AML pathogenesis via its interaction with LSD1. Blood 136: 957–973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cayrol F, Praditsuktavorn P, Fernando TM, Kwiatkowski N, Marullo R, Calvo‐Vidal MN, Phillip J, Pera B, Yang SN, Takpradit K et al (2017) THZ1 targeting CDK7 suppresses STAT transcriptional activity and sensitizes T‐cell lymphomas to BCL2 inhibitors. Nat Commun 8: 14290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang H, Salma F, Yi Q, Patterson B, Brien B, Minden MD (2004) Prognostic relevance of immunophenotyping in 379 patients with acute myeloid leukemia. Leuk Res 28: 43–48 [DOI] [PubMed] [Google Scholar]
- Chaturvedi A, Araujo Cruz MM, Jyotsana N, Sharma A, Yun H, Görlich K, Wichmann M, Schwarzer A, Preller M, Thol F et al (2013) Mutant IDH1 promotes leukemogenesis in vivo and can be specifically targeted in human AML. Blood 122: 2877–2887 [DOI] [PubMed] [Google Scholar]
- Chen M‐H, Wilson CW, Li Y‐J, Law KKL, Lu C‐S, Gacayan R, Zhang X, Hui C, Chuang P‐T (2009) Cilium‐independent regulation of Gli protein function by Sufu in Hedgehog signaling is evolutionarily conserved. Genes Dev 23: 1910–1928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang N‐Y, Hsiao C‐C, Huang Y‐S, Chen H‐Y, Hsieh I‐J, Chang G‐W, Lin H‐H (2011) Disease‐associated GPR56 mutations cause bilateral frontoparietal polymicrogyria via multiple mechanisms. J Biol Chem 286: 14215–14225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark K, Ainscow E, Peall A, Thomson S, Leishman A, Elaine S, Ali S, Coombes R, Barrett A, Bahl A (2017) CT7001, a novel orally Bio‐available CDK7 inhibitor, is highly active in in vitro and in vivo models of AML. Blood 130(Supplement 1): 2645 [Google Scholar]
- Corbit KC, Aanstad P, Singla V, Norman AR, Stainier DYR, Reiter JF (2005) Vertebrate Smoothened functions at the primary cilium. Nature 437: 1018–1021 [DOI] [PubMed] [Google Scholar]
- Cortes JE, Heidel FH, Hellmann A, Fiedler W, Smith BD, Robak T, Montesinos P, Pollyea DA, DesJardins P, Ottmann O et al (2019) Randomized comparison of low dose cytarabine with or without glasdegib in patients with newly diagnosed acute myeloid leukemia or high‐risk myelodysplastic syndrome. Leukemia 33: 379–389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosgun KN, Rahmig S, Mende N, Reinke S, Hauber I, Schäfer C, Petzold A, Weisbach H, Heidkamp G, Purbojo A et al (2014) Kit regulates HSC engraftment across the human‐mouse species barrier. Cell Stem Cell 15: 227–238 [DOI] [PubMed] [Google Scholar]
- Cruciat C‐M, Ohkawara B, Acebron SP, Karaulanov E, Reinhard C, Ingelfinger D, Boutros M, Niehrs C (2010) Requirement of prorenin receptor and vacuolar H+‐ATPase‐mediated acidification for Wnt signaling. Science 327: 459–463 [DOI] [PubMed] [Google Scholar]
- Dierks C, Beigi R, Guo G‐R, Zirlik K, Stegert MR, Manley P, Trussell C, Schmitt‐Graeff A, Landwerlin K, Veelken H et al (2008) Expansion of Bcr‐Abl‐positive leukemic stem cells is dependent on Hedgehog pathway activation. Cancer Cell 14: 238–249 [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR (2013) STAR: ultrafast universal RNA‐seq aligner. Bioinformatics 29: 15–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Döhner H, Estey E, Grimwade D, Amadori S, Appelbaum FR, Büchner T, Dombret H, Ebert BL, Fenaux P, Larson RA et al (2017) Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood 129: 424–447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Döhner H, Weisdorf DJ, Bloomfield CD (2015) Acute myeloid leukemia. N Engl J Med 373: 1136–1152 [DOI] [PubMed] [Google Scholar]
- Doostparast Torshizi A, Armoskus C, Zhang H, Forrest MP, Zhang S, Souaiaia T, Evgrafov OV, Knowles JA, Duan J, Wang K (2019) Deconvolution of transcriptional networks identifies TCF4 as a master regulator in schizophrenia. Sci Adv 5: eaau4139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du R, Shen W, Liu Y, Gao W, Zhou W, Li J, Zhao S, Chen C, Chen Y, Liu Y et al (2019) TGIF2 promotes the progression of lung adenocarcinoma by bridging EGFR/RAS/ERK signaling to cancer cell stemness. Signal Transduct Target Ther 4: 60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durinck S, Spellman PT, Birney E, Huber W (2009) Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat Protoc 4: 1184–1191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eubelen M, Bostaille N, Cabochette P, Gauquier A, Tebabi P, Dumitru AC, Koehler M, Gut P, Alsteens D, Stainier DYR et al (2018) A molecular mechanism for Wnt ligand‐specific signaling. Science 361: eaat1178 [DOI] [PubMed] [Google Scholar]
- Filtz TM, Vogel WK, Leid M (2014) Regulation of transcription factor activity by interconnected post‐translational modifications. Trends Pharmacol Sci 35: 76–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friesen WO (1994) Reciprocal inhibition: a mechanism underlying oscillatory animal movements. Neurosci Biobehav Rev 18: 547–553 [DOI] [PubMed] [Google Scholar]
- Fukushima N, Minami Y, Kakiuchi S, Kuwatsuka Y, Hayakawa F, Jamieson C, Kiyoi H, Naoe T (2016) Small‐molecule Hedgehog inhibitor attenuates the leukemia‐initiation potential of acute myeloid leukemia cells. Cancer Sci 107: 1422–1429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galliher AJ, Schiemann WP (2006) Beta3 integrin and Src facilitate transforming growth factor‐beta mediated induction of epithelial‐mesenchymal transition in mammary epithelial cells. Breast Cancer Res 8: R42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao T, Qian J (2020) EnhancerAtlas 2.0: an updated resource with enhancer annotation in 586 tissue/cell types across nine species. Nucleic Acids Res 48: D58–D64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garg S, Reyes‐Palomares A, He L, Bergeron A, Lavallée V‐P, Lemieux S, Gendron P, Rohde C, Xia J, Jagdhane P et al (2019) Hepatic leukemia factor is a novel leukemic stem cell regulator in DNMT3A, NPM1, and FLT3‐ITD triple‐mutated AML. Blood 134: 263–276 [DOI] [PubMed] [Google Scholar]
- He L, Kulesskiy E, Saarela J, Turunen L, Wennerberg K, Aittokallio T, Tang J (2018) Methods for high‐throughput drug combination screening and synergy scoring. Methods Mol Biol 1711: 351–398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He TC, Sparks AB, Rago C, Hermeking H, Zawel L, da Costa LT, Morin PJ, Vogelstein B, Kinzler KW (1998) Identification of c‐MYC as a target of the APC pathway. Science 281: 1509–1512 [DOI] [PubMed] [Google Scholar]
- Iguchi T, Sakata K, Yoshizaki K, Tago K, Mizuno N, Itoh H (2008) Orphan G protein‐coupled receptor GPR56 regulates neural progenitor cell migration via a G alpha 12/13 and Rho pathway. J Biol Chem 283: 14469–14478 [DOI] [PubMed] [Google Scholar]
- Jiang X, Mak PY, Mu H, Tao W, Mak DH, Kornblau S, Zhang Q, Ruvolo P, Burks JK, Zhang W et al (2018) Disruption of Wnt/β‐catenin exerts antileukemia activity and synergizes with FLT3 inhibition in FLT3‐mutant acute myeloid leukemia. Clin Cancer Res 24: 2417–2429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joksimovic M, Yun BA, Kittappa R, Anderegg AM, Chang WW, Taketo MM, McKay RDG, Awatramani RB (2009) Wnt antagonism of Shh facilitates midbrain floor plate neurogenesis. Nat Neurosci 12: 125–131 [DOI] [PubMed] [Google Scholar]
- Joo K, Kim CG, Lee M‐S, Moon H‐Y, Lee S‐H, Kim MJ, Kweon H‐S, Park W‐Y, Kim C‐H, Gleeson JG et al (2013) CCDC41 is required for ciliary vesicle docking to the mother centriole. Proc Natl Acad Sci USA 110: 5987–5992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandilci A, Grosveld GC (2009) Reintroduction of CEBPA in MN1‐overexpressing hematopoietic cells prevents their hyperproliferation and restores myeloid differentiation. Blood 114: 1596–1606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kausar T, Sharma R, Hasan MR, Tripathi SC, Saraya A, Chattopadhyay TK, Gupta SD, Ralhan R (2011) Clinical significance of GPR56, transglutaminase 2, and NF‐κB in esophageal squamous cell carcinoma. Cancer Invest 29: 42–48 [DOI] [PubMed] [Google Scholar]
- Kim J‐E, Han JM, Park CR, Shin K‐J, Ahn C, Seong JY, Hwang J‐I (2010b) Splicing variants of the orphan G‐protein‐coupled receptor GPR56 regulate the activity of transcription factors associated with tumorigenesis. J Cancer Res Clin Oncol 136: 47–53 [DOI] [PubMed] [Google Scholar]
- Kim J, Lee JE, Heynen‐Genel S, Suyama E, Ono K, Lee K, Ideker T, Aza‐Blanc P, Gleeson JG (2010a) Functional genomic screen for modulators of ciliogenesis and cilium length. Nature 464: 1048–1051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kindler T, Lipka DB, Fischer T (2010) FLT3 as a therapeutic target in AML: still challenging after all these years. Blood 116: 5089–5102 [DOI] [PubMed] [Google Scholar]
- Kishore A, Purcell RH, Nassiri‐Toosi Z, Hall RA (2016) Stalk‐dependent and stalk‐independent signaling by the adhesion G protein‐coupled receptors GPR56 (ADGRG1) and BAI1 (ADGRB1). J Biol Chem 291: 3385–3394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolligs FT, Nieman MT, Winer I, Hu G, Van Mater D, Feng Y, Smith IM, Wu R, Zhai Y, Cho KR et al (2002) ITF‐2, a downstream target of the Wnt/TCF pathway, is activated in human cancers with β‐catenin defects and promotes neoplastic transformation. Cancer Cell 1: 145–155 [DOI] [PubMed] [Google Scholar]
- Köster J, Rahmann S (2012) Snakemake–a scalable bioinformatics workflow engine. Bioinformatics 28: 2520–2522 [DOI] [PubMed] [Google Scholar]
- Kulakovskiy IV, Medvedeva YA, Schaefer U, Kasianov AS, Vorontsov IE, Bajic VB, Makeev VJ (2013) HOCOMOCO: a comprehensive collection of human transcription factor binding sites models. Nucleic Acids Res 41: D195–D202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwiatkowski N, Zhang T, Rahl PB, Abraham BJ, Reddy J, Ficarro SB, Dastur A, Amzallag A, Ramaswamy S, Tesar B et al (2014) Targeting transcription regulation in cancer with a covalent CDK7 inhibitor. Nature 511: 616–620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamas‐Murua M, Stolp B, Kaw S, Thoma J, Tsopoulidis N, Trautz B, Ambiel I, Reif T, Arora S, Imle A et al (2018) HIV‐1 Nef disrupts CD4+ T lymphocyte polarity, extravasation, and homing to lymph nodes via Its Nef‐Associated Kinase Complex Interface. J Immunol 201: 2731–2743 [DOI] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL (2012) Fast gapped‐read alignment with Bowtie 2. Nat Methods 9: 357–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapidot T, Sirard C, Vormoor J, Murdoch B, Hoang T, Caceres‐Cortes J, Minden M, Paterson B, Caligiuri MA, Dick JE (1994) A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature 367: 645–648 [DOI] [PubMed] [Google Scholar]
- Li T, Chiou B, Gilman CK, Luo R, Koshi T, Yu D, Oak HC, Giera S, Johnson‐Venkatesh E, Muthukumar AK et al (2020) A splicing isoform of GPR56 mediates microglial synaptic refinement via phosphatidylserine binding. EMBO J 39: e104136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, Shi W (2014) featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30: 923–930 [DOI] [PubMed] [Google Scholar]
- Liebscher I, Schön J, Petersen SC, Fischer L, Auerbach N, Demberg LM, Mogha A, Cöster M, Simon K‐U, Rothemund S et al (2014) A tethered agonist within the ectodomain activates the adhesion G protein‐coupled receptors GPR126 and GPR133. Cell Rep 9: 2018–2026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F, Jiang W, Sui Y, Meng W, Hou L, Li T, Li M, Zhang L, Mo J, Wang J et al (2019) CDK7 inhibition suppresses aberrant hedgehog pathway and overcomes resistance to smoothened antagonists. Proc Natl Acad Sci USA 116: 12986–12995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S (2014) Moderated estimation of fold change and dispersion for RNA‐seq data with DESeq2. Genome Biol 15: 550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M (2011) Cutadapt removes adapter sequences from high‐throughput sequencing reads. Embnet J 17: 10 [Google Scholar]
- May‐Simera HL, Wan Q, Jha BS, Hartford J, Khristov V, Dejene R, Chang J, Patnaik S, Lu Q, Banerjee P et al (2018) Primary cilium‐mediated retinal pigment epithelium maturation is disrupted in ciliopathy patient cells. Cell Rep 22: 189–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, Wenger AM, Bejerano G (2010) GREAT improves functional interpretation of cis‐regulatory regions. Nat Biotechnol 28: 495–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miralles F, Posern G, Zaromytidou A‐I, Treisman R (2003) Actin dynamics control SRF activity by regulation of its coactivator MAL. Cell 113: 329–342 [DOI] [PubMed] [Google Scholar]
- Ng SWK, Mitchell A, Kennedy JA, Chen WC, McLeod J, Ibrahimova N, Arruda A, Popescu A, Gupta V, Schimmer AD et al (2016) A 17‐gene stemness score for rapid determination of risk in acute leukaemia. Nature 540: 433–437 [DOI] [PubMed] [Google Scholar]
- Nieto MA, Huang RY‐J, Jackson RA, Thiery JP (2016) EMT: 2016. Cell 166: 21–45 [DOI] [PubMed] [Google Scholar]
- Norsworthy KJ, By K, Subramaniam S, Zhuang L, Del Valle PL, Przepiorka D, Shen Y‐L, Sheth CM, Liu C, Leong R et al (2019) FDA approval summary: glasdegib for newly diagnosed acute myeloid leukemia. Clin Cancer Res 25: 6021–6025 [DOI] [PubMed] [Google Scholar]
- Olson CM, Liang Y, Leggett A, Park WD, Li L, Mills CE, Elsarrag SZ, Ficarro SB, Zhang T, Düster R et al (2019) Development of a selective CDK7 covalent inhibitor reveals predominant cell‐cycle phenotype. Cell Chem Biol 26: 792–803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pabst C, Bergeron A, Lavallée V‐P, Yeh J, Gendron P, Norddahl GL, Krosl J, Boivin I, Deneault E, Simard J et al (2016) GPR56 identifies primary human acute myeloid leukemia cells with high repopulating potential in vivo . Blood 127: 2018–2027 [DOI] [PubMed] [Google Scholar]
- Piao X, Hill RS, Bodell A, Chang BS, Basel‐Vanagaite L, Straussberg R, Dobyns WB, Qasrawi B, Winter RM, Innes AM et al (2004) G protein‐coupled receptor‐dependent development of human frontal cortex. Science 303: 2033–2036 [DOI] [PubMed] [Google Scholar]
- Piasecki BP, Burghoorn J, Swoboda P (2010) Regulatory Factor X (RFX)‐mediated transcriptional rewiring of ciliary genes in animals. Proc Natl Acad Sci USA 107: 12969–12974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purcell RH, Hall RA (2018) Adhesion G protein‐coupled receptors as drug targets. Annu Rev Pharmacol Toxicol 58: 429–449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao TN, Marks‐Bluth J, Sullivan J, Gupta MK, Chandrakanthan V, Fitch SR, Ottersbach K, Jang YC, Piao X, Kulkarni RN et al (2015) High‐level Gpr56 expression is dispensable for the maintenance and function of hematopoietic stem and progenitor cells in mice. Stem Cell Res 14: 307–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross‐Innes CS, Stark R, Teschendorff AE, Holmes KA, Ali HR, Dunning MJ, Brown GD, Gojis O, Ellis IO, Green AR et al (2012) Differential oestrogen receptor binding is associated with clinical outcome in breast cancer. Nature 481: 389–393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salzman GS, Ackerman SD, Ding C, Koide A, Leon K, Luo R, Stoveken HM, Fernandez CG, Tall GG, Piao X et al (2016) Structural basis for regulation of GPR56/ADGRG1 by its alternatively spliced extracellular domains. Neuron 91: 1292–1304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savona MR, Pollyea DA, Stock W, Oehler VG, Schroeder MA, Lancet J, McCloskey J, Kantarjian HM, Ma WW, Shaik MN et al (2018) Phase Ib study of glasdegib, a hedgehog pathway inhibitor, in combination with standard chemotherapy in patients with AML or high‐risk MDS. Clin Cancer Res 24: 2294–2303 [DOI] [PubMed] [Google Scholar]
- Shashidhar S, Lorente G, Nagavarapu U, Nelson A, Kuo J, Cummins J, Nikolich K, Urfer R, Foehr ED (2005) GPR56 is a GPCR that is overexpressed in gliomas and functions in tumor cell adhesion. Oncogene 24: 1673–1682 [DOI] [PubMed] [Google Scholar]
- Shtutman M, Zhurinsky J, Simcha I, Albanese C, D’Amico M, Pestell R, Ben‐Ze’ev A (1999) The cyclin D1 gene is a target of the beta‐catenin/LEF‐1 pathway. Proc Natl Acad Sci USA 96: 5522–5527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer K, Luo R, Jeong S‐J, Piao X (2013) GPR56 and the developing cerebral cortex: cells, matrix, and neuronal migration. Mol Neurobiol 47: 186–196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solaimani Kartalaei P, Yamada‐Inagawa T, Vink CS, de Pater E, van der Linden R, Marks‐Bluth J, van der Sloot A, van den Hout M, Yokomizo T, van Schaick‐Solernó ML et al (2015) Whole‐transcriptome analysis of endothelial to hematopoietic stem cell transition reveals a requirement for Gpr56 in HSC generation. J Exp Med 212: 93–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stemmer V, de Craene B, Berx G, Behrens J (2008) Snail promotes Wnt target gene expression and interacts with beta‐catenin. Oncogene 27: 5075–5080 [DOI] [PubMed] [Google Scholar]
- Vick B, Rothenberg M, Sandhöfer N, Carlet M, Finkenzeller C, Krupka C, Grunert M, Trumpp A, Corbacioglu S, Ebinger M et al (2015) An advanced preclinical mouse model for acute myeloid leukemia using patients’ cells of various genetic subgroups and in vivo bioluminescence imaging. PLoS One 10: e0120925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viol L, Hata S, Pastor‐Peidro A, Neuner A, Murke F, Wuchter P, Ho AD, Giebel B, Pereira G (2020) Nek2 kinase displaces distal appendages from the mother centriole prior to mitosis. J Cell Biol 219: e201907136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Krivtsov AV, Sinha AU, North TE, Goessling W, Feng Z, Zon LI, Armstrong SA (2010) The Wnt/beta‐catenin pathway is required for the development of leukemia stem cells in AML. Science 327: 1650–1653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willert K, Brown JD, Danenberg E, Duncan AW, Weissman IL, Reya T, Yates JR, Nusse R (2003) Wnt proteins are lipid‐modified and can act as stem cell growth factors. Nature 423: 448–452 [DOI] [PubMed] [Google Scholar]
- Yadav B, Wennerberg K, Aittokallio T, Tang J (2015) Searching for drug synergy in complex dose‐response landscapes using an interaction potency model. Comput Struct Biotechnol J 13: 504–513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshikawa HY, Rossetti FF, Kaufmann S, Kaindl T, Madsen J, Engel U, Lewis AL, Armes SP, Tanaka M (2011) Quantitative evaluation of mechanosensing of cells on dynamically tunable hydrogels. J Am Chem Soc 133: 1367–1374 [DOI] [PubMed] [Google Scholar]
- Zeng M, Kwiatkowski NP, Zhang T, Nabet B, Xu M, Liang Y, Quan C, Wang J, Hao M, Palakurthi S et al (2018) Targeting MYC dependency in ovarian cancer through inhibition of CDK7 and CDK12/13. Elife 7: e39030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan T, Rindtorff N, Boutros M (2017) Wnt signaling in cancer. Oncogene 36: 1461–1473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H, Zhao C, Li H, Zhang D, Liu G (2019) E2A attenuates tumor‐initiating capacity of colorectal cancer cells via the Wnt/beta‐catenin pathway. J Exp Clin Cancer Res 38: 276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu A, Ibrahim JG, Love MI (2019) Heavy‐tailed prior distributions for sequence count data: removing the noise and preserving large differences. Bioinformatics 35: 2084–2092 [DOI] [PMC free article] [PubMed] [Google Scholar]