

Conspectus
Photocatalytic CO2 reduction is a critical objective in the field of artificial photosynthesis because it can potentially make a total solution for global warming and shortage of energy and carbon resources. We have successfully developed various highly efficient, stable, and selective photocatalytic systems for CO2 reduction using transition metal complexes as both photosensitizers and catalysts. The molecular architectures for constructing selective and efficient photocatalytic systems for CO2 reduction are discussed herein. As a typical example, a mixed system of a ring-shaped Re(I) trinuclear complex as a photosensitizer and fac-[Re(bpy)(CO)3{OC2H4N(C2H4OH)2}] as a catalyst selectively photocatalyzed CO2 reduction to CO with the highest quantum yield of 82% and a turnover number (TON) of over 600. Not only rare and noble metals but also earth abundant ones, such as Mn(I), Cu(I), and Fe(II) can be used as central metal cations. In the case using a Cu(I) dinuclear complex as a photosensitizer and fac-Mn(bpy)(CO)3Br as a catalyst, the total formation quantum yield of CO and HCOOH from CO2 was 57% and TONCO+HCOOH exceeded 1300.
Efficient supramolecular photocatalysts for CO2 reduction, in which photosensitizer and catalyst units are connected through a bridging ligand, were developed for removing a diffusion control on collisions between a photosensitizer and a catalyst. Supramolecular photocatalysts, in which [Ru(N∧N)3]2+-type photosensitizer and Re(I) or Ru(II) catalyst units are connected to each other with an alkyl chain, efficiently and selectively photocatalyzed CO2 reduction in solutions. Mechanistic studies using time-resolved IR and electrochemical measurements provided molecular architecture for constructing efficient supramolecular photocatalysts. A Ru(II)–Re(I) supramolecular photocatalyst constructed according to this molecular architecture efficiently photocatalyzed CO2 reduction even when it was fixed on solid materials. Harnessing this property of the supramolecular photocatalysts, two types of hybrid photocatalytic systems were developed, namely, photocatalysts with light-harvesting capabilities and photoelectrochemical systems for CO2 reduction.
Introduction of light-harvesting capabilities into molecular photocatalytic systems should be important because the intensity of solar light shone on the earth’s surface is relatively low. Periodic mesoporous organosilica, in which methyl acridone groups are embedded in the silica framework as light harvesters, was combined with a Ru(II)–Re(I) supramolecular photocatalyst with phosphonic acid anchoring groups. In this hybrid, the photons absorbed by approximately 40 methyl acridone groups were transferred to one Ru(II) photosensitizer unit, and then, the photocatalytic CO2 reduction commenced.
To use water as an abundant electron donor, we developed hybrid photocatalytic systems combining metal-complex photocatalysts with semiconductor photocatalysts that display high photooxidation powers, in which two photons are sequentially absorbed by the metal-complex photosensitizer and the semiconductor, resulting in both high oxidation and reduction power. Various types of dye-sensitized molecular photocathodes comprising the p-type semiconductor electrodes and the supramolecular photocatalysts were developed. Full photoelectrochemical cells combining these dye-sensitized molecular photocathodes and n-type semiconductor photoanodes achieved CO2 reduction using only visible light as the energy source and water as the reductant. Drastic improvement of dye-sensitized molecular photocathodes is reported.
The results presented in this Account clearly indicate that we can construct very efficient, selective, and durable photocatalytic systems constructed with the metal-complex photosensitizers and catalysts. The supramolecular-photocatalyst architecture in which the photosensitizer and the catalyst are connected to each other is useful especially on the surface of solid owing to rapid electron transfer from the photosensitizer to the catalyst. On basis of these findings, we successfully constructed hybrid systems of the supramolecular photocatalysts with photoactive solid materials. These hybridizations can add new functions to the metal-complex photocatalytic systems, such as water oxidation and light harvesting.
Key References
Takeda H.; Kamiyama H.; Okamoto K.; Irimajiri M.; Mizutani T.; Koike K.; Sekine A.; Ishitani O.. Highly Efficient and Robust Photocatalytic Systems for CO2 Reduction Consisting of a Cu(I) Photosensitizer and Mn(I) Catalysts. J. Am. Chem. Soc. 2018, 140, 17241–17254.1A new binuclear Cu(I) complex with an excellent photosensitizing capability was developed. A mixed system with this Cu(I) photosensitizer and a Mn(I)-complex catalyst photocatalyzed CO2 reduction with the total quantum yield of CO2 reduction products was 57%, the turnover number was over 1300, and the selectivity of CO2 reduction was 95%.
Saito D.; Yamazaki Y.; Tamaki Y.; Ishitani O.. Photocatalysis of a Dinuclear Ru(II)–Re(I) Complex for CO2 Reduction on a Solid Surface. J. Am. Chem. Soc. 2020, 142, 19249–19258.2On the Al2O3particles, superiority in photocatalysis of a supramolecular photocatalyst consisting Ru(II) photosensitizer and Re(I) catalyst units was much reinforced compared to a mixed system of the corresponding mononuclear Ru(II) and Re(I) complexes.
Ueda Y.; Takeda H.; Yui T.; Koike K.; Goto Y.; Inagaki S.; Ishitani O.. A Visible-Light Harvesting System for CO2 Reduction Using a RuII–ReI Photocatalyst Adsorbed in Mesoporous Organosilica. ChemSusChem 2015, 8, 439–442.3In a hybrid system, that is, a Ru(II)–Re(I) supramolecular photocatalyst fixed on periodic mesoporous organosilica (PMO), acridone groups embedded in PMO worked as light harvester and transferred excitation energy to the Ru(II) photosensitizer unit, and CO2 reduction efficiently proceeded on the supramolecular photocatalyst.
Kuttassery F.; Kumagai H.; Kamata R.; Ebato Y.; Higashi M.; Suzuki H.; Abe R.; Ishitani O.. Supramolecular photocatalysts fixed on the inside of the polypyrrole layer in dye sensitized molecular photocathodes: application to photocatalytic CO2 reduction coupled with water oxidation. Chem. Sci. 2021, 12, 13216–13232.4A dye-sensitized molecular photocathode with polypyrrole layer that involves Ru(II) photosensitizers and catalysts on NiO efficient photocatalyzed CO2reduction for 24 h. This photocathode was combined with semiconductor photoanodes to work as photocatalytic systems for CO2reduction with water oxidation.
Introduction
Photoelectron transfer has been extensively studied in various research fields. For example, the Marcus theory provides concrete kinetic and thermodynamic information,5 and in redox photosensitized reactions (also known as photoredox catalytic reactions), photoelectron transfer is initiated by an excited photosensitizer.6,7 In these photochemical reactions, in principle, one-time excitation of a molecule causes a single one-electron transfer.
In CO2 reduction, however, multielectron reduction, typically two-electron, is required because one-electron CO2 reduction is a highly endothermic reaction (eq 1) and two-electron reduction coupled with chemical reactions with protons (eqs 2 and 3 at pH 7) or another CO2 molecule (eq 4) can drastically reduce the endothermicity of CO2 reduction. For this reason, two-component systems are generally used, comprising a redox photosensitizer that initiates photochemical one-electron transfer from an electron donor to a catalyst, and the catalyst that obtains multiple electrons, reacts with CO2, and releases reduction products.7,8
 |
1 |
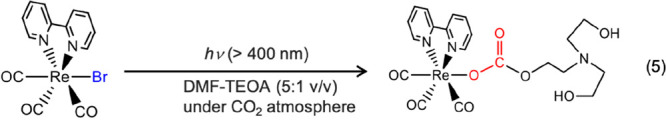
Transition metal complexes have various advantages, both as redox photosensitizers and catalysts for CO2 reduction. For example, visible-colored and emissive metal complexes, such as [Ru(N∧N)3]2+, [Os(N∧N)3]2+, [Re(N∧N)(CO)2(PR3)2]+, and [Ir(ppy)2(N∧N)]+ (N∧N = diimine ligand; ppy = 2-phenylpyridinato-C2,N ligand) have been reported as photosensitizers, and their colors can be systematically changed by changing the ligands.7,9 Their excited states have sufficiently long lifetimes for photoelectron transfer with the electron donor owing to rapid intersystem crossing to the lowest triplet excited state. In terms of the catalyst, ligands, such as N∧N, can function as a stable electron pool in the reduced metal complex, and the metal ion can fix CO2 onto itself, which results in low overpotential and high selectivity for CO2 reduction against hydrogen evolution. In typical photocatalytic CO2 reduction systems, Tanaka and Ishida et al. employed [Ru(bpy)3]2+ (bpy = 2,2′-bipyridine), which is the frequently used photosensitizer, and [Ru(bpy)2(CO)2]2+10,11 or Ru(bpy)(CO)2Cl212 as the catalyst. Visible light irradiation of a mixed system of [Ru(bpy)3]2+ and one of these catalysts in the presence of an electron donor resulted in CO2 reduction to HCOOH or CO.12 Lehn and Ziessel et al. reported that fac-Re(bpy)(CO)3X (X = Cl, Br) functions as a “single-handed” photocatalyst for CO2 reduction selectively to CO when triethanolamine (TEOA) was used as an electron donor.13 Recently, however, we established that the fac-Re(bpy)(CO)3Br complex is largely transformed to a carbonic-acid-ester complex in the initial stage of the photocatalytic reaction (eq 5).14 Therefore, a mixed system comprising the carbonic-acid–ester complex functioning as a catalyst and the remaining fac-Re(bpy)(CO)3Br operating as a photosensitizer is largely responsible for CO formation. In fact, a mixed system of [Ru(bpy)3]2+ and fac-Re(bpy)(CO)3Cl exhibited superior photocatalytic activity than fac-Re(bpy)(CO)3Cl alone.
 |
5 |
Although photocatalytic systems based solely on metal complexes can attain high selectivities and low overpotentials for CO2 reduction as described above, they lack several functionalities important for practical use, such as water oxidation capabilities for supplying electrons, and light-harvesting capacities. In this Account, we discuss our results on four interconnected research topics for photocatalytic CO2 reduction: (1) efficient photocatalysis using mixed systems comprising photosensitizer and catalyst complexes; (2) constructing efficient supramolecular photocatalysts consisting of both the photosensitizer and catalyst complexes in one molecule; (3) imparting the photocatalyst with light-harvesting properties; and (4) photocatalytic reduction of CO2 using water as a reductant and visible light as energy. Transition metal complexes play the central role in all of the above topics.
Photosensitizer/Catalyst Mixed Systems
To improve CO2-reduction photocatalysis by metal-complex systems in homogeneous solutions, three important objectives should be fulfilled: (1) the development of a photosensitizer that absorbs a wide range of visible light well, is highly oxidative in the excited state, highly reductive in the reduced state, and stable in both the excited and reduced states; (2) the development of a catalyst capable of rapid CO2 reduction with a low overpotential, weak absorption of visible light, and is stable during the photocatalytic reaction; and (3) rapid electron transfer from the reduced photosensitizer to the catalyst. We have successfully developed highly efficient mixed photocatalytic systems by improving the photosensitizer and catalyst properties as described below.
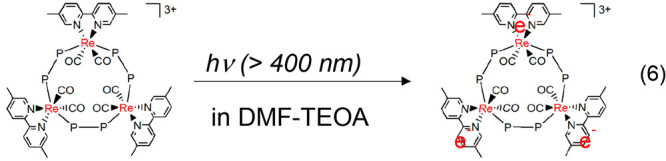
Some ring-shaped Re(I) multinuclear complexes (Re-rings) display fascinating photosensitizing properties.15−17 For example, Re-ring-1 can absorb visible light to give a triplet metal-to-ligand charge-transfer (3MLCT) excited state having a long lifetime (τ = 5.4 μs) and high oxidation capacity (Figure 1).15Re-ring-1 can accept three electrons in one molecule, and the reduced state is highly stable in solution, even at room temperature (eq 6). The one-electron reduced state (OERS) of Re-ring-1 is highly reductive (E1/2 = −1.87 V vs Ag/AgNO3).18
 |
6 |
Figure 1.

UV–vis absorption, emission, and structure of Re-ring-1.
We found that fac-[Re(bpy)(CO)3{OC2H4N(C2H4OH)2}] can capture CO2 to be converted into a carbonic-acid-ester complex (Re-CO2-TEOA in eq 7).14,19 Although this CO2 insertion into the Re–O bond is an equilibrium reaction, the equilibrium constant is very large (K = 1.7 × 103 M–1); in other words, this reaction supplies CO2 to the metal-complex catalyst before the photocatalytic reaction commences. This CO2 capturing reaction enables direct reduction of low concentrations of CO2, such as 1%, in electrocatalytic19 and photocatalytic reactions.20
 |
7 |
A mixed photocatalytic system of Re-ring-1 and Re-CO2-TEOA selectively reduced CO2 to CO with the highest quantum yield (ΦCO = 82%) among the reported systems (eq 8).15 Herein, we used the formal definition of the quantum yield, that is, the product amount divided by the number of absorbed photons (eq 9). The turnover number (the product amount divided by the number of the photosensitizer or catalyst, TON) of produced CO was 526.
 |
8 |
| 9 |
Photocatalytic systems using only earth-abundant elements have recently been increasingly investigated for photocatalytic CO2 reduction.21 Because the lack of efficient rare- or noble-metal-free photosensitizers was a significant concern, we developed new binuclear Cu(I) complexes with excellent photosensitizing capabilities, in which two quadridentate ligands are complexed with two Cu(I) ions (Cu2Xph in Chart 1).22−24 Although mononuclear Cu(I) complexes having both diimine and bisphosphine ligands have been reported as photosensitizers,25 they are unstable in polar solvents even at room temperature owing to ligand detachment. However, Cu2Xph is highly stable under the same conditions, and the excited state and OERS of Cu2Xph are also stable with long lifetimes (several microseconds in acetonitrile at 25 °C). A mixed system comprising Cu2Hph with fac-Mn(MeO-bpy)(CO)3Br1 or Fe(phen)2(SCN)222 as catalysts efficiently photocatalyzed CO2 reduction, as shown in Figure 2. The analogues bearing CF3 or Ph groups as X in Cu2Xph (Chart 1) were more efficient photosensitizers than Cu2Hph because of stronger oxidation power in the excited state or stronger absorption in visible region.24
Chart 1. Cu2Xph.
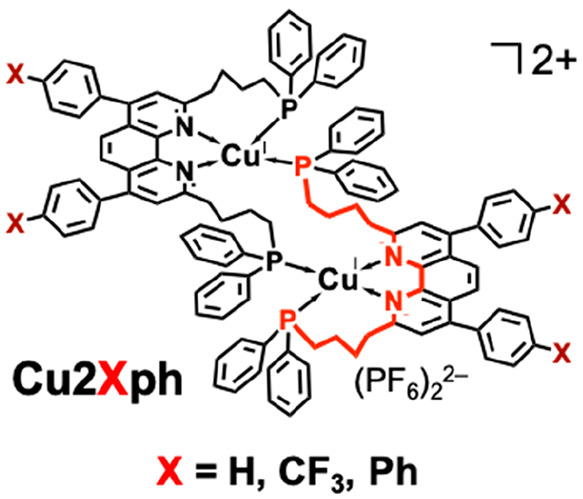
Figure 2.
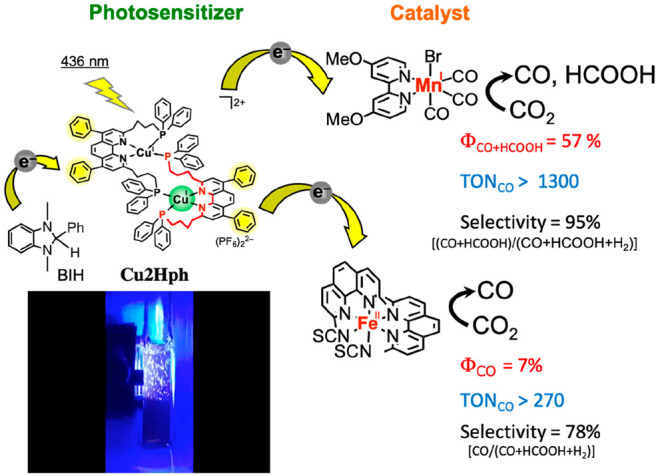
Photocatalysis by Cu2Hph with fac-Mn(MeO-bpy)(CO)3Br or Fe(phen)2(SCN)2 as catalysts. The photo shows active CO bubbles being produced during the photocatalytic reaction using fac-Mn(MeO-bpy)(CO)3Br.
The heavy metal effect of Os increases the probability of direct excitation to the triplet excited state (S-T absorption), which is ordinarily a forbidden transition. We applied this phenomenon to create a panchromatic photosensitizer, Os(L)(L′) capable of utilizing the entire visible light range (Figure 3).26 Irradiation of a N,N-dimethylacetamide (DMA) solution containing Os(L)(L′), Ru(bpy)(CO)2Cl2 as a catalyst, and 1,3-dimethyl-2-(o-hydroxyphenyl)-2,3-dihydro-1H-benzo[d]imidazole (BI(OH)H) as a sacrificial electron donor, induced photocatalytic CO2 reduction to HCOOH, even at λex > 770 nm.
Figure 3.
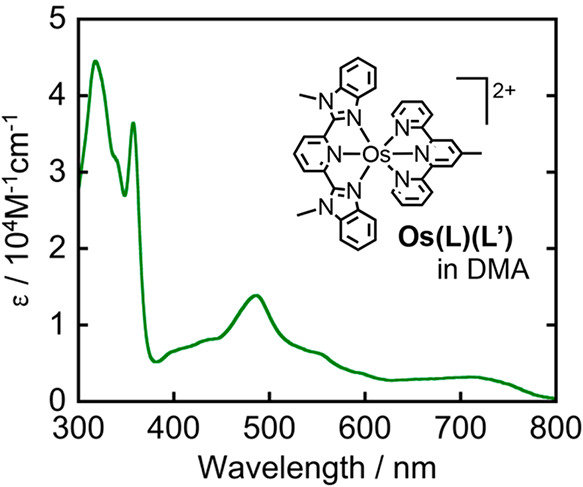
UV–vis absorption spectrum of Os(L)(L′) in DMA.
Supramolecular Photocatalysts
In the mixed systems of a photosensitizer and catalyst, the electron transfer from the OERS of the photosensitizer to the catalyst is limited by diffusion collisions between them. To accelerate electron transfer and improve photocatalytic activity, multinuclear metal complexes consisting of photosensitizer and catalyst have been developed;27 these are referred to as “supramolecular photocatalysts”.28 In 2005, we reported the successful supramolecular photocatalysts for CO2 reduction for the first time.29 The supramolecular photocatalysts comprising [Ru(N∧N)3]2+-type photosensitizer and fac-Re(N∧N)(CO)3Cl-type catalyst units achieved both TONs ≫ 1 and better photocatalytic activities than a mixed system of the corresponding mononuclear complexes.
Chart 2 shows the structures and abbreviations of the Ru(II)–Re(I) multinuclear complexes, and Table 1 summarizes their photocatalytic activities. Ru–Re1, in which the [Ru(N∧N)3]2+ photosensitizer unit and the fac-Re(N∧N)(CO)3Cl catalyst unit are bridged by an alkyl chain between two N∧N moieties, photocatalytically reduced CO2 selectively to CO in the presence of a sacrificial electron donor, 1-benzyl-1,4-dihydronicotinamide (BNAH), under visible-light irradiation (λex > 500 nm).29 CO produced with a high quantum yield (Φ) and the system was highly durable (entry 1 in Table 1: ΦCO = 0.12, TONCO = 170), being superior to the mixed system of [Ru(dmb)3]2+ and fac-Re(dmb)(CO)3Cl (entry 2: ΦCO = 0.062, TONCO = 101). The reaction mechanism of photocatalytic CO2 reduction using Ru–Re1 was partially clarified as follows. [Process 1] Selective photon absorption by the Ru(II) photosensitizer unit forms the 1MLCT excited state, which then relaxes to the 3MLCT excited state via rapid intersystem crossing. [Process 2] The 3MLCT excited Ru unit is reduced by BNAH, producing the OERS. [Process 3] The unpaired electron on the Ru unit transfers intramolecularly to the Re(I) unit. [Process 4] CO2 is reduced to CO on the Re unit via a second one-electron reduction (Figure 4).
Chart 2. Ru(II)–Re(I) Complexes.

Table 1. Photocatalytic Performances of Ru(II)–Re(I) Complexesa.
| entry | photocatalyst | ΦCO | TONCO |
|---|---|---|---|
| 1 | Ru–Re1 | 0.12 | 170 |
| 2 | [Ru(dmb)3]2+ + fac-Re(dmb)(CO)3Cl | 0.062 | 101 |
| 3 | Ru–Re2 | 50 | |
| 4 | Ru–Re3 | 3 | |
| 5 | Ru–Re4 | 28 |
CO2-saturated DMF–TEOA (5:1 v/v) solutions containing the complex and BNAH (0.1 M) were irradiated at λex > 500 nm. dmb = 4,4′-dimethyl-2,2′-bipyridine.
Figure 4.

Reaction mechanism of photocatalytic reduction of CO2 using Ru−Re1.
The peripheral ligands of the Ru(II) unit and the bridging ligand strongly affected the photocatalytic activity. Ru–Re2 (entry 3: TONCO = 50) and Ru–Re3 (entry 4: TONCO = 3) with 2,2′-bipyridine or 4,4′-bis(trifluoromethyl)-2,2′-bipyridine as peripheral ligands exhibited significantly lower photocatalytic activities than Ru–Re1. This was because of slow intramolecular electron transfer [process 3] in the cases of the OERS of Ru–Re2 and Ru–Re3, whereby the unpaired electron localizes predominantly on the peripheral ligands of the Ru unit rather than the bridging ligands, and the intramolecular electron transfer becomes endergonic: in the case of Ru–Re3, for example, E1/2red(RuI/II) = −1.23 V while E1/2red(Re0/I) = −1.76 V vs Ag/AgNO3. On the other hand, both the Ru and Re units of Ru–Re1 are reduced by one electron at an almost equal potential (E1/2red = −1.77 V), and intramolecular electron transfer proceeds readily. This intramolecular electron transfer process will be discussed in detail later.
Ru–Re4 exhibited a low photocatalytic activity (entry 5: TONCO = 28), even though the intramolecular electron transfer from the one-electron reduced Ru unit to the Re unit should proceed rapidly as it is highly exothermic. The reason is that the conjugation of the bridging ligand lowers the reducing power of the OERS of the Re catalyst unit (E1/2red = −1.10 V), which inhibits CO2 activation on the Re unit. We noted a strong relationship between the photocatalytic performances of mononuclear Re(I) complexes, namely, fac-[Re(4,4′-X2-bpy)(CO)3(PR3)]+, and their first reduction potentials (Table 2):30 overall, the Re(I) complexes should have a more negative reduction potential than E1/2red = −1.4 V to function as effective catalysts for the reduction of CO2.
Table 2. Photocatalytic Performances of fac-[Re(4,4′-X2-bpy)(CO)3(PR3)]+ Complexes and Their First Reduction Potentials.
|
fac-[Re(4,4′-X2-bpy)(CO)3(PR3)]+ |
||||
|---|---|---|---|---|
| X | R | ΦCOa | TONCOa | E1/2redb/V |
| Me | OEt | 0.18 | 4.1 | –1.55 |
| H | OiPr | 0.20 | 6.2 | –1.44 |
| H | OEt | 0.16 | 5.9 | –1.43 |
| H | OMe | 0.17 | 5.5 | –1.41 |
| H | Et | 0.024 | 0.83 | –1.39 |
| H | nBu | 0.013 | 0.65 | –1.39 |
| CF3 | OEt | 0.005 | 0.10 | –1.03 |
A DMF-TEOA (5:1 v/v, 4 mL) solution of the complex (2.6 mM) was irradiated at 365 nm under CO2 atmosphere.
Redox potentials measured in MeCN containing 0.1 M nBu4NClO4 using Ag/AgNO3 (10 mM) as a reference electrode.
To investigate the influence of the alkyl chain on the speed of intramolecular electron transfer from the OERS of the photosensitizer unit to the catalyst unit, we measured the rate constants of intramolecular electron transfers in the OERS of Ru(II)–Re(I) supramolecular photocatalysts bearing varying alkyl chain lengths conducting time-resolved IR (TR-IR) measurements of the stretching bands of the CO ligands (νCO), as shown in Figure 5.31,32 Although there should be a ground state (Ru–Re in Figure 5) and two intermediates (3Ru*–Re and Ru•––Re) in solution after irradiation in the presence of an electron donor before the intramolecular electron transfer proceeds, they should have similar νCO values owing to the alkyl chain separating the excited or reduced Ru unit from the Re unit. Only the product of intramolecular electron transfer (Ru–Re•–) shows lower-energy shifts of its νCO bands owing to reinforcement by π-back-donation from the Re center. Figure 5 shows the FT-IR spectrum of Ru–C2–Re, which has an ethylene chain connecting the Ru and Re units, and the TR-IR spectra obtained after selective excitation of the Ru photosensitizer unit in the presence of BIH as a sacrificial electron donor. We can calculate the rate constant of the intramolecular electron transfer at ke = 7.1 × 108 s–1 using increments in the νCO bands at 1872 and 1934 cm–1. It is noteworthy that this electron transfer is sufficiently rapid not to be the rate-limiting process in the photocatalytic reaction.
Figure 5.
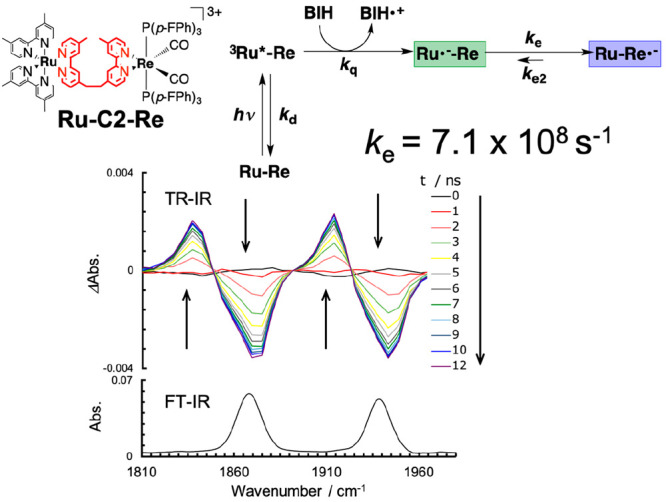
FT-IR and TR-IR spectra of Ru–C2–Re in DMF-TEOA (5:1 v/v) solution containing BIH (0.3 M): the excitation wavelength for TR-IR measurements was λex = 532 nm.
When a longer alkyl chain was used in the bridging ligand (Ru–Cn−Re, n = 2, 4, 6 in Figure 6), the electron transfer rate ke was lower, as shown in Figure 6, and a linear relationship was observed between the logarithm of ke and the distance between Ru and Re with an apparent decay coefficient factor (β) of 0.74 Å–1. In addition, Ru–RC2–Re, in which two ethylene chains connect the Ru and Re units, induced significantly faster intramolecular electron transfer than Ru–C2–Re bearing one ethylene chain. These results indicate that the intramolecular electron transfer from the OERS of the Ru unit to the Re unit proceeds via the through-bond mechanism.
Figure 6.

Relationship between the intramolecular electron transfer rate constant (ke) and the distance between Ru and Re.
From the results described above, the molecular architecture necessary to construct effective supramolecular photocatalysts can be obtained: [Rule 1] The photosensitizer unit must have equal to or more negative reduction potential than the catalyst unit to promote intramolecular electron transfer from the reduced photosensitizer unit to the catalyst unit. [Rule 2] The bridging ligand should not be conjugated to maintain the reducing power of the catalyst unit. On the basis of these rules, that is, employing dmb as peripheral ligands on the Ru photosensitizer unit and a bridging ligand with a −C2H4– chain, the supramolecular photocatalysts involving the Ru(II) carbonyl complexes as a catalyst unit instead of the Re(I) catalyst were developed (Chart 3).33,34
Chart 3. Ru(II)–Ru(II) and Os(II)–Re(I) Complexes.
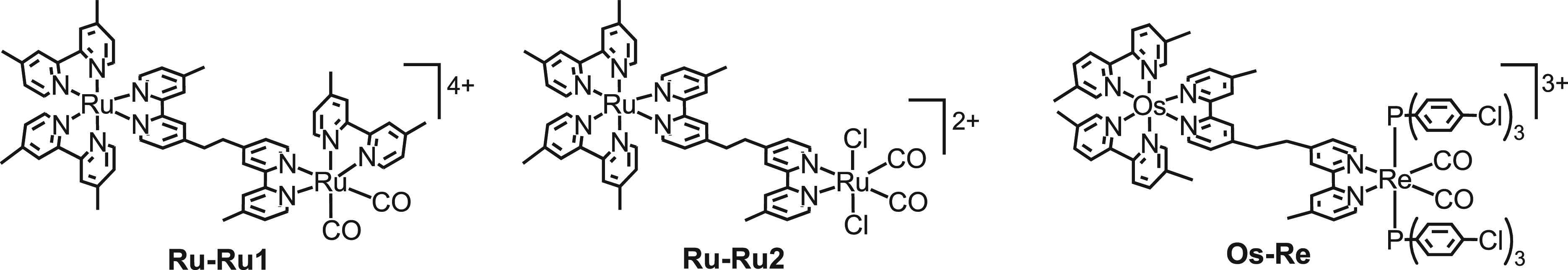
cis-[Ru(N∧N)2(CO)2]2+35,36 and cis,trans-Ru(N∧N)(CO)2Cl236 are well-known electrochemical catalysts for the reduction of CO2 to HCOOH with high selectivity under basic conditions. Ru–Ru1 with a [Ru(N∧N)(dmb)(CO)2]2+ unit photocatalyzed CO2 reduction to HCOOH with high selectivity and durability in the presence of BNAH under visible-light irradiation (ΦHCOOH = 0.038, TONHCOOH = 315, selectivity = 90%).33,37Ru–Ru2 with a cis,trans-Ru(N∧N)(CO)2Cl2 unit also functioned as a similarly active photocatalyst to reduce CO2 to HCOOH.34
The photosensitizer unit was changed from the Ru complex to the corresponding [Os(N∧N)2(BL)]2+-type complex. This complex (Os–Re, Chart 3) photocatalyzed CO2 reduction under irradiation even at λex > 620 nm, whereas the supramolecular photocatalysts containing the Ru photosensitizer unit do not absorb light at this wavelength and therefore did not induce CO2 reduction under the same conditions.38
To impart additional functions to photocatalytic systems consisting of metal complexes, hybrid systems comprising photocatalysts and photofunctional solid materials such as semiconductor photocatalysts are good candidates. To fix the supramolecular photocatalysts on the solid, phosphonic acid anchoring groups were introduced at the peripheral ligands of the Ru(II) photosensitizing unit via −CH2– to maintain the reduction power of the Ru(II) unit (Chart 4).
Chart 4. Supramolecular Photocatalysts Bearing Phosphonic Acid Anchoring Groups.

The advantage of supramolecular photocatalysts over the mixed systems consisting of mononuclear complexes is more pronounced when the metal complexes are fixed on a solid surface. When the mononuclear complexes of a photosensitizer and catalyst are fixed on a solid surface, the electron transfer from the photosensitizer to the catalyst should become extremely slow or not occur at all depending on the distance between these complexes (Figure 7b). In the case of a supramolecular photocatalyst, on the other hand, this electron transfer occurs intramolecularly and should therefore be faster (Figure 7a).
Figure 7.
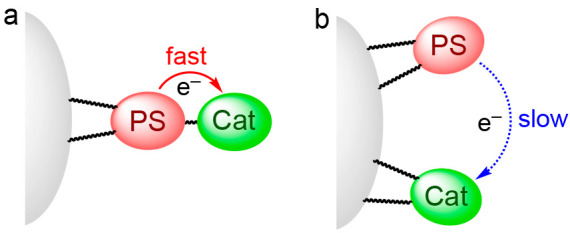
Conceptual images of a (a) supramolecular photocatalyst and (b) mixed system of mononuclear complexes on the surface of heterogeneous materials. PS and Cat represent photosensitizer and catalyst, respectively.
In fact, PRu-Re and the corresponding mononuclear complexes bearing anchoring groups were fixed on insulator Al2O3 particles, and their photocatalytic performances were compared using BNAH as a sacrificial electron donor.25 The Al2O3/PRu-Re composite photocatalyzed CO2 reduction to CO effectively even when the average distance between the supramolecular photocatalysts was considerably longer (6.3 nm) than the size of PRu-Re (maximum length = 2.4 nm). In contrast, in the case of Ru(II) and Re(I) mononuclear complexes, significantly less CO was produced at a comparable absorption density on Al2O3. Therefore, we decided to use a supramolecular photocatalyst for fabricating hybrid photocatalytic systems, as described below.
Hybrid Photocatalysts with Light-Harvesting Functionality
Because the intensity of solar light is low and the absorption cross sections of molecular photosensitizers are small, introduction of light-harvesting capabilities is important for molecular photocatalytic systems.
We successfully combined a supramolecular photocatalyst with periodic mesoporous organosilica (PMO), in which abundant organic molecules (R) are embedded in the silica framework as light harvesters (Figure 8a).3 Methyl acridone groups (ACR), which are capable of absorbing visible light, were used as the organic molecules in the PMO framework. The silica moiety interacts strongly with the phosphonic acid groups of PRu-Re (Chart 4, left) and confines PRu-Re mainly inside the mesopores, generating a hybrid (Figure 8b). The photons absorbed by approximately 40 ACR groups were transferred to a Ru(II) photosensitizer unit of one PRu-Re molecule in this hybrid, and then the photocatalytic reduction of CO2 commenced. This light-harvesting function of the PMO hybrid enhanced the photocatalytic formation rate of CO by a factor of 10 compared to that of PRu-Re-adsorbed mesoporous silica without a light harvester. In the photocatalytic reaction using BIH as the electron donor, the TONCO was >600.
Figure 8.
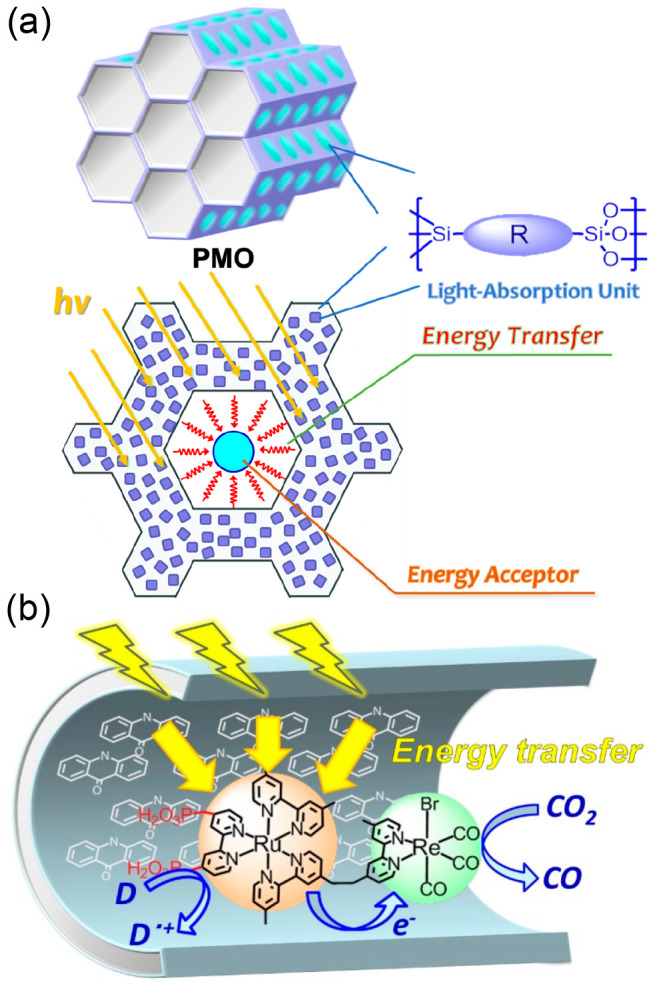
(a) Conceptual structure of PMO. (b) PRu-Re confined inside the mesopores of PMO with ACR, and photocatalytic CO2 reduction by this hybrid capable of light harvesting.
Hybrid Photocatalytic Systems Using Water As a Reductant
In terms of practical photocatalytic CO2 reduction, water is an ideal electron donor because of its abundance, low cost, and harmlessness. However, the low photooxidation power of metal-complex photosensitizers and the requirement of four-electron oxidation of two molecules of water lead difficulty in use of water in photocatalytic systems consisting of only metal complexes. To address these problems, we developed hybrid photocatalytic systems combining metal-complex photocatalysts with semiconductor photocatalysts that display high photooxidation powers, in which two photons are sequentially absorbed by the metal-complex photosensitizer and the semiconductor to obtain both high reduction and oxidation power.39 The photocatalytic units for the CO2 reduction and water oxidation were designed as photocathode and photoanode, respectively (Figure 9). The desired direction of photoelectron transfer from the semiconductor to the metal complex can be achieved by the appropriate coupling of these two photoelectrodes. In addition, the cell allows for separate product generation while avoiding reverse reactions.
Figure 9.
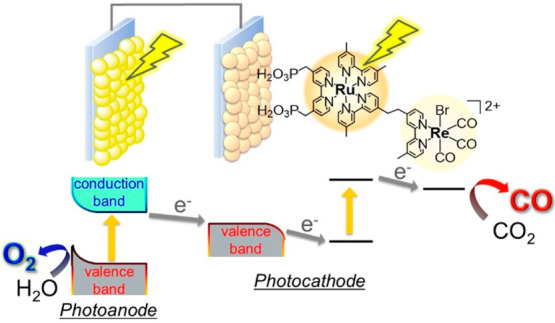
Hybrid photoelectrochemical cell constructed by combining a metal complex photocatalyst and a semiconductor photocatalyst for CO2 reduction/water oxidation.
We developed a dye-sensitized molecular photocathode comprising PRu-Re (Chart 4, left) coupled with the p-type semiconductor NiO electrode.40,41 The 3MLCT excited state of the Ru photosensitizer unit of PRu-Re adsorbed on the electrode can obtain an electron from the valence band of NiO, instead of the ordinal sacrificial electron donor, to drive CO2 reduction. The electron flow through this NiO/PRu-Re photocathode corresponds to the cathodic photocurrent, and PRu-Re produces CO as the photocatalytic reduction product of CO2 under irradiation at λ > 460 nm and with appropriate external bias both in an organic solvent (DMF–TEOA (5:1, v/v)),40 as well as in aqueous solution41 with high selectivity (∼100% and 91%, respectively). Here, the downward band bending at the solid–liquid interface of the p-type semiconductor is beneficial for the reductive quenching of the excited photosensitizer unit and subsequent charge separation. These results revealed that the PRu-Re photocatalyst can function as the CO2 reduction side of the hybrid photoelectrochemical system.
We next combined the NiO/PRu-Re photocathode and an n-type semiconductor TaON photoanode for water oxidation to construct a full photoelectrochemical cell. TaON absorbs visible light (λ < 500 nm) and CoOx cocatalyst was loaded onto it to improve the activity and stability for water oxidation.42 Under irradiation at λ > 400 nm for both electrodes in an aqueous solution, the photoelectrochemical cell produced CO and O2 from the photocathode and photoanode, respectively. The TONCO based on the PRu-Re adsorbed on the electrode was 17. In this system, electrical and chemical biases (totally 0.4 V) were required to progress the photocatalytic reaction. The light-to-energy conversion efficiency of the reaction was 1.6 × 10–3%. The applicability of the hybrid cell was demonstrated by replacing the photoanode material with a Ta/N-codoped TiO2.43 This cell also required an external bias of 0.5 V between the two photoelectrodes and provided a conversion efficiency of 1.1 × 10–3% with considering the bias applied.
These hybrid photoelectrochemical cells suffer several drawbacks. First, an external applied bias is necessary to drive the photocatalytic reaction, owing to the energy loss during electron transfer, even though potential of photoexcited electrons in the semiconductor photoanode is sufficiently negative to transfer an electron to the excited Ru photosensitizer unit in PRu-Re. Therefore, we replaced NiO with another p-type semiconductor, CuGaO2 of which flat band potential is 0.16 V more positive than that of NiO.44 The onset potential of the cathodic photocurrent of the photocathode comprising CuGaO2 (CuGaO2/PRu-Re) shifted 0.4 V more positive from that of NiO/PRu-Re (Figure 10). This large positive shift arises from reduced energy loss in the electron transfer process from the valence band of the p-type semiconductor electrode to the excited Ru photosensitizer unit. Owing to the improved photocathode, the hybrid photoelectrochemical cell consisting of the CuGaO2/PRu-Re photocathode and the CoOx/TaON photoanode achieved photocatalytic reduction of CO2 and water oxidation without the application of an external bias (Figure 10c). The products were CO (232 nmol, TONCO = 22) and H2 (311 nmol) at the photocathode site and O2 (232 nmol) at the photoanode site. This is the first self-driven photoelectrochemical cell constructed using a metal complex photocatalyst that achieves CO2 reduction using only visible light as the energy source and water as the reductant.
Figure 10.

Current–potential curves of (a) CuGaO2/PRu-Re and (b) NiO/PRu-Re. (c) The photoelectrochemical cell consisting of a CuGaO2/PRu-Re photocathode and CoOx/TaON photoanode. From ref (44) with permission. Copyright 2017 Royal Society of Chemistry.
Another problem with the dye-sensitized molecular photocathodes with metal complex photocatalyst described above is their low efficiency and stability. The external quantum efficiency (termed the incident-photon-to-current efficiency, IPCE) was very low. In addition, the photocurrent decayed rapidly within several hours of irradiation. One of the main reasons for this is inadequate adsorption of the metal complex photocatalyst onto the electrode surface; the PRu-Re molecules are adsorbed only via hydrogen bonding involving the phosphonic acid groups to form a unimolecular layer on the surface of the p-type semiconductors. Therefore, the amount of adsorbed PRu-Re was only ∼4 nmol cm–2, which limits light absorption and in turn the activity of the photocathodes. In addition, the phosphonate-acid anchor groups were weakly absorbed; thus, PRu-Re gradually detached and the photocathode activity decayed. We adopted electrochemical polymerization to reinforce the attachment of a larger amount of the supramolecular photocatalyst onto the electrode surface.45 We used two types of metal complexes bearing vinyl groups on the bipyridine ligands of the Ru(II) photosensitizer unit, that is, a mononuclear Ru(II) diimine-type photosensitizer (PRuV) and a Ru(II)–Re(I) supramolecular photocatalyst (RuReV, middle in Scheme 1a). PRuV has another bipyridine ligand bearing methyl phosphonic acid groups for its first attachment to the NiO electrode to form NiO/PRuV (the left side of Scheme 1a). NiO/PRuV was soaked in electrolyte containing RuReV, and then a negative potential was applied to the electrode to give the polyethylene-modified photocathode (NiO/PRu-polyEt-Ru–Re), which contained a larger amount of Ru photosensitizer units (∼10 nmol cm–2) than PRu-Re adsorbed only via the methyl phosphonic acid anchor groups, and was more stable against detachment in the aqueous electrolyte. This molecular photocathode NiO/PRu-polyEt-Ru–Re produced approximately 2.5-fold more CO, and its total Faradaic efficiency of the reduction products also enhanced from 57% (NiO/PRu-Re) to 85%. The maximum IPCE was also improved to 0.93% at −0.5 V vs Ag/AgCl under 480 nm irradiation. However, the stability of the NiO/PRu-polyEt-Ru–Re was still unsatisfactory: the Re–C bonds were formed via the side reaction of the vinyl group with the Re center during reductive polymerization and cleaved during the photocatalytic reaction.
Scheme 1. Preparation of (a) NiO/PRu-polyEt-Ru–Re and (b) NiO/PRu-polyEt-Ru-RuCAT.

To address this issue, a three-step method for constructing dye-sensitized molecular photocathodes was developed (Scheme 1b).46 In the first step, the NiO/PRuV electrode was synthesized as described above. In the second step, electrochemical polymerization was performed between the adsorbed PRuV and another Ru(II) mononuclear complex bearing diimine ligands with a vinyl group and a noncoordinated diimine moiety (VRu-N∧N) to form NiO/PRu-polyEt-Ru–N∧N. In the last step, a catalyst complex unit Ru(N∧N)(CO)2Cl2 was formed via the reaction of the noncoordinated diimine moiety of NiO/PRu-polyEt-Ru–N∧N with [Ru(CO)2Cl2]n. The produced photocathode (NiO/PRu-polyEt-Ru-RuCAT) was highly stable: photoelectrochemical CO2 reduction proceeded for over 100 h under visible light, affording CO and HCOOH without noticeable degradation (Figure 11, the Faraday efficiency of the reduction product decreased due to slow air leakage). H2 formation was very low even in aqueous media (selectivity toward CO2 reduction was over 90%), and the TON(CO+HCOOH) reached 1200, which is the highest among the reported molecular photocathodes to date. The maximum incident photon to current conversion efficiency (IPCE) was 1.2% at −0.7 V vs Ag/AgCl under 480 nm irradiation in an aqueous solution (pH 6.6). The full cell consisting of the NiO/PRu-polyEt-Ru-RuCAT photocathode and a CoOx-modified BiVO4 semiconductor photoanode enabled both CO2 reduction and water oxidation without any external bias under visible light irradiation with a light energy conversion efficiency of 0.017%.
Figure 11.
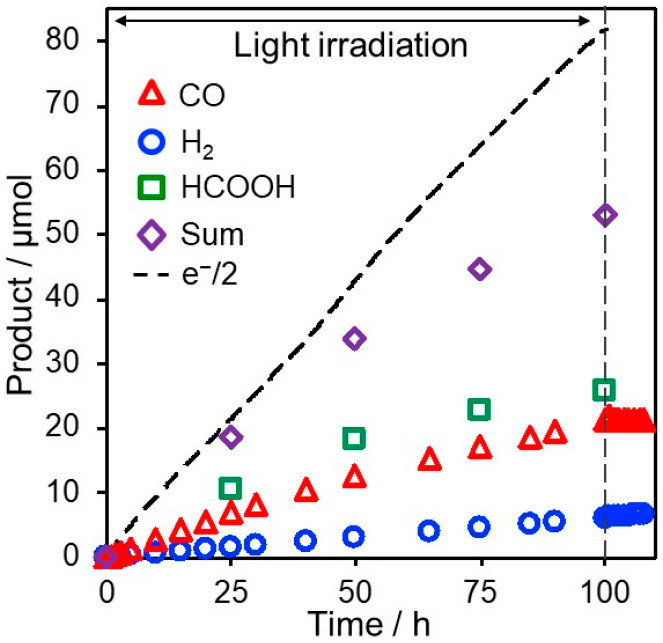
Time courses of the products from the NiO/PRu-polyEt-Ru-RuCAT photocathode during 100 h irradiation (460 nm < λ < 650 nm) at E = −0.7 V vs Ag/AgCl in a CO2-purged NaHCO3 aqueous solution. The dotted line shows a half number of the flowed electrons. Reproduced from ref (46) with permission. Copyright 2021 Royal Society of Chemistry.
Although the reductive polymerization approach using vinyl groups is highly effective for improving both the efficiency and stability of the dye-sensitized molecular photocathode, the saturated hydrocarbon chain formed by the vinyl polymerization possibly causes low conductivity in the polymer layer, which limits the improvement of its photocatalysis. Therefore, we developed another dye-sensitized molecular photocathode produced by oxidative polymerization of pyrrole groups to afford a polypyrrole phase containing the metal complexes.4 The synthesis method of this photoelectrode (NiO/PRu-polyPyr-Ru-RuCAT) is similar to that of NiO/PRu-polyEt-Ru-RuCAT, except for the use of Ru(II) complexes containing pyrrole groups (PRuPyr and PyrRu-N∧N in Scheme 2) instead of PRuV and VRu-N∧N, and the application of the potential scan in the positive instead of the negative direction. The produced NiO/PRu-polyPyr-Ru-RuCAT contained both the photosensitizer and catalyst units in a ratio of ∼3:1, and the photocurrent reached a maximum at Eapp = −0.1 V vs Ag/AgCl (Figure 12a), whereas in the case of NiO/PRu-polyEt-Ru-RuCAT, an Eapp of −0.5 V was required. This suggests an improvement in the conductivity of the polymer phase in the NiO/PRu-polyPyr-Ru-RuCAT photoelectrode. The maximum IPCE was 4.7% (Figure 12b), which is the highest value reported for dye-sensitized molecular photocathodes. Visible light irradiation of NiO/PRu-polyPyr-Ru-RuCAT at Eapp = −0.7 V induced continuous photocatalytic CO2 reduction for 24 h to produce CO and HCOOH with a small amount of H2 (selectivity for CO2 reduction was 92%) in CO2-purged 50 mM aqueous NaHCO3 solution. The TONs for CO and HCOOH were 566 and 185, respectively, after 24 h of irradiation. A full photoelectrochemical cell comprising this photocathode and the CoOx/BiVO4 photoanode induced simultaneous photocatalytic CO2 reduction and water oxidation in the respective photoelectrodes (Figure 13a). For the same setup described in Figure 10c, the light energy conversion efficiency was 0.083% with Faraday efficiencies of 99% (photocathode) and 87% (photoanode). Another full cell in tandem configuration, wherein the photocathode was illuminated first and the photoanode absorbed the permeated light in the same compartment cell (Figure 13b), exhibited a solar-to-chemical conversion efficiency of 0.048% under simulated sunlight (AM1.5G). This state-of-the-art achievement suggests the possibility of solar energy conversion to useful chemicals with the use of a supramolecular photocatalyst as the photocatalytic center for CO2 reduction. There are still many opportunities for improving the photocatalytic efficiency of full cells comprising dye-sensitized molecular photocathodes, such as strengthening the absorption of the photosensitizer units, especially at longer wavelengths, and enhancing the conductivities in p-type semiconductors and polymers, as well as between them.
Scheme 2. Preparation of NiO/PRu-polyPyr-Ru-RuCAT.

Figure 12.

(a) Current–potential curves of NiO/PRu-PolyPyr-Ru-RuCAT under irradiation at λex = 460–650 nm (28.2 mW cm–2) in CO2-purged 50 mM aqueous NaHCO3. (b) Dependence of the IPCE of NiO/PRu-PolyPyr-Ru-RuCAT by the irradiated wavelength at −0.3 V vs Ag/AgCl with absorption spectrum of NiO/PRu-PolyPyr-Ru-RuCAT subtracted with the absorbance of the NiO electrode (red line).
Figure 13.

(a) Photoelectrochemical cell comprising the NiO/PRu-polyPyr-Ru-RuCAT photocathode and the CoOx/BiVO4 photoanode. (b) Tandem-type configuration in a single compartment cell irradiated by simulated sunlight (AM1.5G, 100 mW cm–2).
Conclusions and Perspectives
The results presented in this account clearly indicate that we can construct very efficient, selective, and durable photocatalytic systems consisting of metal-complex photosensitizers and catalysts according to the “rules”. The supramolecular-photocatalyst architecture in which the photosensitizer and the catalyst are connected to each other is useful especially on the solid surface owing to rapid electron transfer between the photosensitizer and the catalyst. Based on these findings, we successfully constructed hybrid systems of the supramolecular photocatalysts with photoactive solid materials such as the mesoporous organosilica and the semiconductors. These hybridizations can add new functions, that is, light harvesting and water oxidation to the metal-complex photocatalytic systems.
For developing practical systems of photocatalytic CO2 reduction, we have to add many functions to the systems as summarized in Figure 14. Although our approaches achieved to combine some of these functions,20 we have to add all of them in one system. Many additional efforts and new ideas should be directed to building “artificial photosynthesis” for CO2 utilization.
Figure 14.
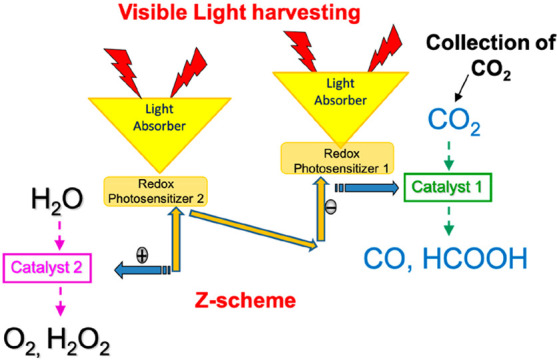
Required functions of artificial photosynthesis for CO2 reduction.
Acknowledgments
The authors acknowledge support by JST CREST (Grant Number JPMJCR13L1) and by JSPS KAKENHI (Grant Numbers JP20H00396, JP20K20367, JP17H06440).
Biographies
Hiromu Kumagai is an assistant professor in the Institute of Multidisciplinary Research for Advanced Materials at Tohoku University. His current research interests include photoelectrochemistry, photocatalysis, and inorganic materials for energy conversion.
Yusuke Tamaki is an assistant professor at Tokyo Institute of Technology. His research interests involve photochemistry of transition metal complexes, photofunctional metal complexes and organic molecules, and photocatalytic CO2 reduction.
Osamu Ishitani is a professor of Department of Chemistry at Tokyo Institute of Technology. His research interests are photochemistry and photofunctions of transition metal complexes, and artificial photosynthesis.
The authors declare no competing financial interest.
Special Issue
Published as part of the Accounts of Chemical Research special issue “CO2 Reductions via Photo and Electrochemical Processes”.
References
- Takeda H.; Kamiyama H.; Okamoto K.; Irimajiri M.; Mizutani T.; Koike K.; Sekine A.; Ishitani O. Highly Efficient and Robust Photocatalytic Systems for CO2 Reduction Consisting of a Cu(I) Photosensitizer and Mn(I) Catalysts. J. Am. Chem. Soc. 2018, 140, 17241–17254. 10.1021/jacs.8b10619. [DOI] [PubMed] [Google Scholar]
- Saito D.; Yamazaki Y.; Tamaki Y.; Ishitani O. Photocatalysis of a Dinuclear Ru(II)–Re(I) Complex for CO2 Reduction on a Solid Surface. J. Am. Chem. Soc. 2020, 142, 19249–19258. 10.1021/jacs.0c09170. [DOI] [PubMed] [Google Scholar]
- Ueda Y.; Takeda H.; Yui T.; Koike K.; Goto Y.; Inagaki S.; Ishitani O. A Visible-Light Harvesting System for CO2 Reduction Using a RuII-ReI Photocatalyst Adsorbed in Mesoporous Organosilica. ChemSusChem 2015, 8, 439–442. 10.1002/cssc.201403194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuttassery F.; Kumagai H.; Kamata R.; Ebato Y.; Higashi M.; Suzuki H.; Abe R.; Ishitani O. Supramolecular photocatalysts fixed on the inside of the polypyrrole layer in dye sensitized molecular photocathodes: application to photocatalytic CO2 reduction coupled with water oxidation. Chem. Sci. 2021, 12, 13216–13232. 10.1039/D1SC03756K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcus R. A. On the Theory of Oxidation-Reduction Reactions Involving Electron Transfer. I. J. Chem. Phys. 1956, 24, 966–978. 10.1063/1.1742723. [DOI] [Google Scholar]
- Prier C. K.; Rankic D. A.; MacMillan D. W. C. Visible Light Photoredox Catalysis with Transition Metal Complexes: Applications in Organic Synthesis. Chem. Rev. 2013, 113, 5322–5363. 10.1021/cr300503r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki Y.; Takeda H.; Ishitani O. Photocatalytic reduction of CO2 using metal complexes. J. Photochem. Photobiol. C 2015, 25, 106–137. 10.1016/j.jphotochemrev.2015.09.001. [DOI] [Google Scholar]
- a Kojima T. Photocatalytic CO2 Reduction Using Nickel Complexes as Catalysts. ChemPhotoChem. 2021, 5, 512–520. 10.1002/cptc.202000263. [DOI] [Google Scholar]; b Boutin E.; Merakeb L.; Ma B.; Boudy B.; Wang M.; Bonin J.; Anxolabéhère-Mallart E.; Robert M. Molecular catalysis of CO2 reduction: recent advances and perspectives in electrochemical and light-driven processes with selected Fe, Ni and Co aza macrocyclic and polypyridine complexes. Chem. Soc. Rev. 2020, 49, 5772–5809. 10.1039/D0CS00218F. [DOI] [PubMed] [Google Scholar]
- Balzani V.; Juris A.; Venturi M.; Campagna S.; Serroni S. Luminescent and redox-active polynuclear transition metal complexes. Chem. Rev. 1996, 96, 759–834. 10.1021/cr941154y. [DOI] [PubMed] [Google Scholar]
- Ishida H.; Tanaka K.; Tanaka T. Photoreduction of CO2 in the [Ru(bpy)2(CO)2]2+/[Ru(bpy)3]2+ or [Ru(phen)3]2+/Triethanolamine/N,N-Dimethylformamide System. Chem. Lett. 1987, 16, 1035–1038. 10.1246/cl.1987.1035. [DOI] [Google Scholar]
- Ishida H.; Terada T.; Tanaka K.; Tanaka T. Photochemical CO2 Reduction Catalyzed by [Ru(bpy)2(CO)2]2+ Using Triethanolamine and 1-Benzyl-1,4-dihydronicotinamide as an Electron Donor. Inorg. Chem. 1990, 29, 905–911. 10.1021/ic00330a004. [DOI] [Google Scholar]
- Kuramochi Y.; Fukaya K.; Yoshida M.; Ishida H. trans-(Cl)-[Ru(5,5-diamide-2,2-bipyridine)(CO)2Cl2]: Synthesis, Structure, and Photocatalytic CO2 Reduction Activity. Chem.—Eur. J. 2015, 21, 10049–10060. 10.1002/chem.201500782. [DOI] [PubMed] [Google Scholar]
- Hawecker J.; Lehn J. M.; Ziessel R. Photochemical and Electrochemical Reduction of Carbon-Dioxide to Carbon-Monoxide Mediated by (2,2′-Bipyridine)Tricarbonylchlororhenium(I) and Related Complexes as Homogeneous Catalysts. Helv. Chim. Acta 1986, 69, 1990–2012. 10.1002/hlca.19860690824. [DOI] [Google Scholar]
- Morimoto T.; Nakajima T.; Sawa S.; Nakanishi R.; Imori D.; Ishitani O. CO2 Capture by a Rhenium(I) Complex with the Aid of Triethanolamine. J. Am. Chem. Soc. 2013, 135, 16825–16828. 10.1021/ja409271s. [DOI] [PubMed] [Google Scholar]
- Morimoto T.; Nishiura C.; Tanaka M.; Rohacova J.; Nakagawa Y.; Funada Y.; Koike K.; Yamamoto Y.; Shishido S.; Kojima T.; Saeki T.; Ozeki T.; Ishitani O. Ring-Shaped Re(I) Multinuclear Complexes with Unique Photofunctional Properties. J. Am. Chem. Soc. 2013, 135, 13266–13269. 10.1021/ja406144h. [DOI] [PubMed] [Google Scholar]
- Asatani T.; Nakagawa Y.; Funada Y.; Sawa S.; Takeda H.; Morimoto T.; Koike K.; Ishitani O. Ring-Shaped Rhenium(I) Multinuclear Complexes: Improved Synthesis and Photoinduced Multielectron Accumulation. Inorg. Chem. 2014, 53, 7170–7180. 10.1021/ic501196q. [DOI] [PubMed] [Google Scholar]
- Rohacova J.; Sekine A.; Kawano T.; Tamari S.; Ishitani O. Trinuclear and Tetranuclear Re(I) Rings Connected with Phenylene, Vinylene, and Ethynylene Chains: Synthesis, Photophysics, and Redox Properties. Inorg. Chem. 2015, 54, 8769–8777. 10.1021/acs.inorgchem.5b01397. [DOI] [PubMed] [Google Scholar]
- It was reported that Ag/AgNO3 (10 mM) in MeCN = +0.55 V versus NHE.; Pavlishchuk V. V.; Addison A. W. Conversion constants for redox potentials measured versus different reference electrodes in acetonitrile solutions at 25 °C. Inorg. Chim. Acta 2000, 298, 97–102. 10.1016/S0020-1693(99)00407-7. [DOI] [Google Scholar]; The readers should be careful that this relationship is changeable depending on the solvent used.
- Kumagai H.; Nishikawa T.; Koizumi H.; Yatsu T.; Sahara G.; Yamazaki Y.; Tamaki Y.; Ishitani O. Electrocatalytic reduction of low concentration CO2. Chem. Sci. 2019, 10, 1597–1606. 10.1039/C8SC04124E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajima T.; Tamaki Y.; Ueno K.; Kato E.; Nishikawa T.; Ohkubo K.; Yamazaki Y.; Morimoto T.; Ishitani O. Photocatalytic Reduction of Low Concentration of CO2. J. Am. Chem. Soc. 2016, 138, 13818–13821. 10.1021/jacs.6b08824. [DOI] [PubMed] [Google Scholar]
- a Takeda H.; Cometto C.; Ishitani O.; Robert M. Electrons, Photons, Protons and Earth-Abundant Metal Complexes for Molecular Catalysis of CO2 Reduction. ACS Catal. 2017, 7, 70–88. 10.1021/acscatal.6b02181. [DOI] [Google Scholar]; b Steinlechner C.; Roesel A. F.; Oberem E.; Päpcke A.; Rockstroh N.; Gloaguen F.; Lochbrunner S.; Ludwig R.; Spannenberg A.; Junge H.; Francke R.; Beller M. Selective Earth-Abundant System for CO2 Reduction: Comparing Photo- and Electrocatalytic Processes. ACS Catal. 2019, 9, 2091–2100. 10.1021/acscatal.8b03548. [DOI] [Google Scholar]; c Zhang X.; Cibian M.; Call A.; Yamauchi K.; Sakai K. Photochemical CO2 Reduction Driven by Water-Soluble Copper(I) Photosensitizer with the Catalysis Accelerated by Multi-Electron Chargeable Cobalt Porphyrin. ACS Catal. 2019, 9, 11263–11273. 10.1021/acscatal.9b04023. [DOI] [Google Scholar]; d Wang Y.; Gao X.-W.; Li J.; Chao D. Merging an organic TADF photosensitizer and a simple terpyridine–Fe(III) complex for photocatalytic CO2 reduction. Chem. Commun. 2020, 56, 12170–12173. 10.1039/D0CC05047D. [DOI] [PubMed] [Google Scholar]
- Takeda H.; Ohashi K.; Sekine A.; Ishitani O. Photocatalytic CO2 Reduction Using Cu(I) Photosensitizers with a Fe(II) Catalyst. J. Am. Chem. Soc. 2016, 138, 4354–4357. 10.1021/jacs.6b01970. [DOI] [PubMed] [Google Scholar]
- Takeda H.; Monma Y.; Sugiyama H.; Uekusa H.; Ishitani O. Development of Visible-Light Driven Cu(I) Complex Photosensitizers for Photocatalytic CO2 Reduction. Front. Chem. 2019, 7, 418. 10.3389/fchem.2019.00418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda H.; Monma Y.; Ishitani O. Highly Functional Dinuclear CuI-Complex Photosensitizers for Photocatalytic CO2 Reduction. ACS Catal. 2021, 11, 11973–11984. 10.1021/acscatal.1c03336. [DOI] [Google Scholar]
- Rosas-Hernández A.; Steinlechner C.; Junge H.; Beller M. Earth-abundant photocatalytic systems for the visible-light-driven reduction of CO2 to CO. Green Chem. 2017, 19, 2356–2360. 10.1039/C6GC03527B. [DOI] [Google Scholar]
- Irikura M.; Tamaki Y.; Ishitani O. Development of a panchromatic photosensitizer and its application to photocatalytic CO2 reduction. Chem. Sci. 2021, 12, 13888–13896. 10.1039/D1SC04045F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Kimura E.; Bu X.; Shionoya M.; Wada S.; Maruyama S. A New Nickel(II) Cyclam (Cyclam = 1,4,8,11-Tetraazacyclotetradecane) Complex Covalently Attached to Ru(phen)32+ (phen = 1,10-Phenanthroline). A New Candidate for the Catalytic Photoreduction of Carbon Dioxide. Inorg. Chem. 1992, 31, 4542–4546. 10.1021/ic00048a020. [DOI] [Google Scholar]; b Komatsuzaki N.; Himeda Y.; Hirose T.; Sugihara H.; Kasuga K. Synthesis and Photochemical Properties of Ruthenium-Cobalt and Ruthenium-Nickel Dinuclear Complexes. Bull. Chem. Soc. Jpn. 1999, 72, 725–731. 10.1246/bcsj.72.725. [DOI] [Google Scholar]; c Gabrielsson A.; Lindsay Smith J. R.; Perutz R. N. Remote site photosubstitution in metalloporphyrin-rhenium tricarbonylbipyridine assemblies: photo-reactions of molecules with very short lived excited states. Dalton Trans. 2008, 4259–4269. 10.1039/b806267f. [DOI] [PubMed] [Google Scholar]; d Aukauloo A.; Gotico P.; Tran T.-T.; Baron A.; Vauzeilles B.; Lefumeux C.; Ha-Thi M.-H.; Pino T.; Halime Z.; Quaranta A.; Leibl W. Tracking Charge Accumulation in a Functional Triazole-Linked Ru-Re Dyad Towards Photocatalytic CO2 Reduction. ChemPhotoChem. 2021, 5, 654–664. 10.1002/cptc.202100010. [DOI] [Google Scholar]; e Brown C. M.; Auvray T.; DeLuca E. E.; Ezhova M. B.; Hanan G. S.; Wolf M. O. Controlling photocatalytic reduction of CO2 in Ru(II)/Re(I) dyads via linker oxidation state. Chem. Commun. 2020, 56, 10750–10753. 10.1039/D0CC04597G. [DOI] [PubMed] [Google Scholar]; f Kiyosawa K.; Shiraishi N.; Shimada T.; Masui D.; Tachibana H.; Takagi S.; Ishitani O.; Tryk D. A.; Inoue H. Electron Transfer from the Porphyrin S-2 State in a Zinc Porphyrin-Rhenium Bipyridyl Dyad having Carbon Dioxide Reduction Activity. J. Phys. Chem. C 2009, 113, 11667–11673. 10.1021/jp901548y. [DOI] [Google Scholar]; g Tamaki Y.; Ishitani O. Supramolecular Photocatalysts for the Reduction of CO2. ACS Catal. 2017, 7, 3394–3409. 10.1021/acscatal.7b00440. [DOI] [Google Scholar]; h Pirzada B. M.; Dar A. H.; Shaikh M. N.; Qurashi A. Reticular-Chemistry-Inspired Supramolecule Design as a Tool to Achieve Efficient Photocatalysts for CO2 Reduction. ACS Omega 2021, 6, 29291–29324. 10.1021/acsomega.1c04018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balzani V.; Ceroni P.; Juris A.: Photochemistry and Photophysics: Concepts, Research, Applications; Wiley-VCH: Weinheim, 2014. [Google Scholar]
- Gholamkhass B.; Mametsuka H.; Koike K.; Tanabe T.; Furue M.; Ishitani O. Architecture of Supramolecular Metal Complexes for Photocatalytic CO2 Reduction: Ruthenium-Rhenium Bi- and Tetranuclear Complexes. Inorg. Chem. 2005, 44, 2326–2336. 10.1021/ic048779r. [DOI] [PubMed] [Google Scholar]
- Koike K.; Hori H.; Ishizuka M.; Westwell J. R.; Takeuchi K.; Ibusuki T.; Enjouji K.; Konno H.; Sakamoto K.; Ishitani O. Key Process of the Photocatalytic Reduction of CO2 Using [Re(4,4′-X2-bipyridine)(CO)3PR3]+ (X = CH3, H, CF3; PR3 = Phosphorus Ligands): Dark Reaction of the One-Electron-Reduced Complexes with CO2. Organometallics 1997, 16, 5724–5729. 10.1021/om970608p. [DOI] [Google Scholar]
- Koike K.; Grills D. C.; Tamaki Y.; Fujita E.; Okubo K.; Yamazaki Y.; Saigo M.; Mukuta T.; Onda K.; Ishitani O. Investigation of excited state, reductive quenching, and intramolecular electron transfer of Ru(II)-Re(I) supramolecular photocatalysts for CO2 reduction using time-resolved IR measurements. Chem. Sci. 2018, 9, 2961–2974. 10.1039/C7SC05338J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki Y.; Ohkubo K.; Saito D.; Yatsu T.; Tamaki Y.; Tanaka S.; Koike K.; Onda K.; Ishitani O. Kinetics and Mechanism of Intramolecular Electron Transfer in Ru(II)-Re(I) Supramolecular CO2-Reduction Photocatalysts: Effects of Bridging Ligands. Inorg. Chem. 2019, 58, 11480–11492. 10.1021/acs.inorgchem.9b01256. [DOI] [PubMed] [Google Scholar]
- Tamaki Y.; Morimoto T.; Koike K.; Ishitani O. Photocatalytic CO2 reduction with high turnover frequency and selectivity of formic acid formation using Ru(II) multinuclear complexes. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 15673–15678. 10.1073/pnas.1118336109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamaki Y.; Ishitani O. Supramolecular Photocatalysts for the Reduction of CO2. ACS Catal. 2017, 7, 3394–3409. 10.1021/acscatal.7b00440. [DOI] [Google Scholar]
- Ishida H.; Tanaka K.; Tanaka T. Electrochemical CO2 Reduction Catalyzed by [Ru(bpy)2(CO)2]2+ and [Ru(bpy)2(CO)CI]+. The Effect of pH on the Formation of CO and HCOO–. Organometallics 1987, 6, 181–186. 10.1021/om00144a033. [DOI] [Google Scholar]
- Ishida H.; Fujiki K.; Ohba T.; Ohkubo K.; Tanaka K.; Terada T.; Tanaka T. Ligand Effects of Ruthenium 2,2′-Bipyridine and 1,10-Phenanthroline Complexes on the Electrochemical Reduction of CO2. J. Chem. Soc., Dalton Trans. 1990, 2155–2160. 10.1039/DT9900002155. [DOI] [Google Scholar]
- Tamaki Y.; Koike K.; Ishitani O. Highly efficient, selective, and durable photocatalytic system for CO2 reduction to formic acid. Chem. Sci. 2015, 6, 7213–7221. 10.1039/C5SC02018B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamaki Y.; Koike K.; Morimoto T.; Yamazaki Y.; Ishitani O. Red-Light-Driven Photocatalytic Reduction of CO2 using Os(II)–Re(I) Supramolecular Complexes. Inorg. Chem. 2013, 52, 11902–11909. 10.1021/ic4015543. [DOI] [PubMed] [Google Scholar]
- The other hybrid photocatalytic systems combining metal complex with semiconductor that display high photooxidation powers, have been also reported. As typical examples:; a Suzuki T. M.; Yoshino S.; Takayama T.; Iwase A.; Kudo A.; Morikawa T. Z-Schematic and visible-light-driven CO2 reduction using H2O as an electron donor by a particulate mixture of a Ru-complex/(CuGa)1–xZn2xS2 hybrid catalyst, BiVO4 and an electron mediator. Chem. Commun. 2018, 54, 10199–10202. 10.1039/C8CC05505J. [DOI] [PubMed] [Google Scholar]; b Sekizawa K.; Sato S.; Arai T.; Morikawa T. Solar-Driven Photocatalytic CO2 Reduction in Water Utilizing a Ruthenium Complex Catalyst on p-Type Fe2O3 with a Multiheterojunction. ACS Catal. 2018, 8, 1405–1416. 10.1021/acscatal.7b03244. [DOI] [Google Scholar]; c Woo S.-J.; Choi S.; Kim S.-Y.; Kim P. S.; Jo J. H.; Kim C. H.; Son H.-J.; Pac C.; Kang S. O. Highly Selective and Durable Photochemical CO2 Reduction by Molecular Mn(I) Catalyst Fixed on a Particular Dye-Sensitized TiO2 Platform. ACS Catal. 2019, 9, 2580–2593. 10.1021/acscatal.8b03816. [DOI] [Google Scholar]; d Sekizawa K.; Maeda K.; Domen K.; Koike K.; Ishitani O. Artificial Z-Scheme Constructed with a Supramolecular Metal Complex and Semiconductor for the Photocatalytic Reduction of CO2. J. Am. Chem. Soc. 2013, 135, 4596–4599. 10.1021/ja311541a. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Sato S.; Morikawa T.; Saeki S.; Kajino T.; Motohiro T. Visible-Light-Induced Selective CO2 Reduction Utilizing a Ruthenium Complex Electrocatalyst Linked to a p-Type Nitrogen-Doped Ta2O5 Semiconductor. Angew. Chem., Int. Ed. 2010, 49, 5101–5105. 10.1002/anie.201000613. [DOI] [PubMed] [Google Scholar]
- Sahara G.; Abe R.; Higashi M.; Morikawa T.; Maeda K.; Ueda Y.; Ishitani O. Photoelectrochemical CO2 reduction using a Ru(II)–Re(I) multinuclear metal complex on a p-type semiconducting NiO electrode. Chem. Commun. 2015, 51, 10722–10725. 10.1039/C5CC02403J. [DOI] [PubMed] [Google Scholar]
- Sahara G.; Kumagai H.; Maeda K.; Kaeffer N.; Artero V.; Higashi M.; Abe R.; Ishitani O. Photoelectrochemical Reduction of CO2 Coupled to Water Oxidation Using a Photocathode With a Ru(II)-Re(I) Complex Photocatalyst and a CoOx/TaON Photoanode. J. Am. Chem. Soc. 2016, 138, 14152–14158. 10.1021/jacs.6b09212. [DOI] [PubMed] [Google Scholar]
- Higashi M.; Domen K.; Abe R. Highly stable water splitting on oxynitride TaON photoanode system under visible light irradiation. J. Am. Chem. Soc. 2012, 134, 6968–6971. 10.1021/ja302059g. [DOI] [PubMed] [Google Scholar]
- Nakada A.; Uchiyama T.; Kawakami N.; Sahara G.; Nishioka S.; Kamata R.; Kumagai H.; Ishitani O.; Uchimoto Y.; Maeda K. Solar Water Oxidation by a Visible-Light-Responsive Tantalum/Nitrogen-Codoped Rutile Titania Anode for Photoelectrochemical Water Splitting and Carbon Dioxide Fixation. ChemPhotoChem. 2019, 3, 37–45. 10.1002/cptc.201800157. [DOI] [Google Scholar]
- Kumagai H.; Sahara G.; Maeda K.; Higashi M.; Abe R.; Ishitani O. Hybrid photocathode consisting of a CuGaO2 p-type semiconductor and a Ru(II)-Re(I) supramolecular photocatalyst: non-biased visible-light-driven CO2 reduction with water oxidation. Chem. Sci. 2017, 8, 4242–4249. 10.1039/C7SC00940B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamata R.; Kumagai H.; Yamazaki Y.; Sahara G.; Ishitani O. Photoelectrochemical CO2 Reduction Using a Ru(II)-Re(I) Supramolecular Photocatalyst Connected to a Vinyl Polymer on a NiO Electrode. ACS Appl. Mater. Interfaces 2019, 11, 5632–5641. 10.1021/acsami.8b05495. [DOI] [PubMed] [Google Scholar]
- Kamata R.; Kumagai H.; Yamazaki Y.; Higashi M.; Abe R.; Ishitani O. Durable photoelectrochemical CO2 reduction with water oxidation using a visible-light driven molecular photocathode. J. Mater. Chem. A 2021, 9, 1517–1529. 10.1039/D0TA07351B. [DOI] [Google Scholar]