

Abstract
Pathologic skin scarring presents a vast economic and medical burden. Unfortunately, the molecular mechanisms underlying scar formation remain to be elucidated. We used a hypertrophic scarring (HTS) mouse model in which Jun is overexpressed globally or specifically in α-smooth muscle or collagen type I–expressing cells to cause excessive extracellular matrix deposition by skin fibroblasts in the skin after wounding. Jun overexpression triggered dermal fibrosis by modulating distinct fibroblast subpopulations within the wound, enhancing reticular fibroblast numbers, and decreasing lipofibroblasts. Analysis of human scars further revealed that JUN is highly expressed across the wide spectrum of scars, including HTS and keloids. CRISPR-Cas9–mediated JUN deletion in human HTS fibroblasts combined with epigenomic and transcriptomic analysis of both human and mouse HTS fibroblasts revealed that JUN initiates fibrosis by regulating CD36. Blocking CD36 with salvianolic acid B or CD36 knockout model counteracted JUN-mediated fibrosis efficacy in both human fibroblasts and mouse wounds. In summary, JUN is a critical regulator of pathological skin scarring, and targeting its downstream effector CD36 may represent a therapeutic strategy against scarring.
INTRODUCTION
The medical and economic burden of scars and their sequelae is extensive (1); in the United States alone, more than 100 million new scars are formed every year as a physiological response to cutaneous injury (2). The resulting fibrotic scar tissue is abnormal in both form and function and has the potential to cause devastating disfigurement and permanent functional loss (3). Fibrosis exists as a spectrum: from scarless healing in mammalian fetal skin (4) to normal scarring in healthy postnatal skin to excessive scarring in hypertrophic scars (HTSs) and keloids (5). Despite the prevalence of scars and an abundance of treatment options, no current molecular therapies effectively prevent or reverse scarring (6). Hence, there remains a critical need to elucidate the key mechanisms mediating scar formation.
Fibroblasts are chiefly responsible for dermal regeneration and repair and appear within wounded skin 2 to 5 days after injury (7). By 7 days, after a period of cellular replication and migration, fibroblasts become the dominant cell population within the wound and synthesize large volumes of extracellular matrix (ECM) (8). Recent literature has highlighted the existence of multiple fibroblast subpopulations with distinct roles in wound repair (9–11). We have previously described a profibrotic fibroblast lineage in the mouse dorsal dermis marked by embryonic expression of Engrailed-1, which produces ECM in both acute and chronic fibroses (10). However, precisely how specific fibroblast subpopulations change throughout the wound healing process remains unknown. Elucidating the contributions of fibroblast subpopulations to scarring would facilitate the therapeutic targeting of distinct cell subsets to potentially prevent or even reverse scarring.
JUN is a major component of the heterodimeric transcription factor AP-1 and a critical driver of tissue scarring (12). We recently demonstrated that Jun is predominantly expressed in fibroblasts, that fibroblasts are selectively responsive to Jun expression, and that JUN is a key driver of systemic scarring in multiple organs of mice and humans (12–15). Here, we explored the molecular mechanisms by which Jun promotes cutaneous scarring using several transgenic mouse models and human scar (hSc)–derived fibroblasts. We demonstrate that blocking the downstream target CD36 with either salvianolic acid B (SAB), short hairpin RNA (shRNA) knockdown, or a CD36 knockout (KO) mouse model counteracts JUN-mediated fibrosis in both human fibroblasts and mouse wounds. Identification of the transcriptional pathways through which Jun promotes fibrosis reveals potential therapeutic agents for the treatment of pathologic skin scarring.
RESULTS
Jun overexpression promotes scar formation
Following the observation that JUN is highly expressed in dermal wounds of mice (fig. S1A), we explored the profibrotic effects of JUN on wound healing by developing transgenic tetracycline-inducible JUN mice (c-JuntetO R26-rtTA) (12). These mice ubiquitously express the reverse tetracycline transactivator (rtTA) from the Rosa26 locus and the tetracycline operator minimal promoter (tetOP). Upon administration of tetracycline or tetracycline derivative such as doxycycline (“dox”), rtTA binds to the tetOP promoter and induces Jun expression (Fig. 1A and table S1). Control mice (Rosa26-rtTA) have the rtTA construct but lack c-JuntetO and thus exhibit physiologic JUN expression with or without dox induction.
Fig. 1. Jun overexpression induces scar formation.
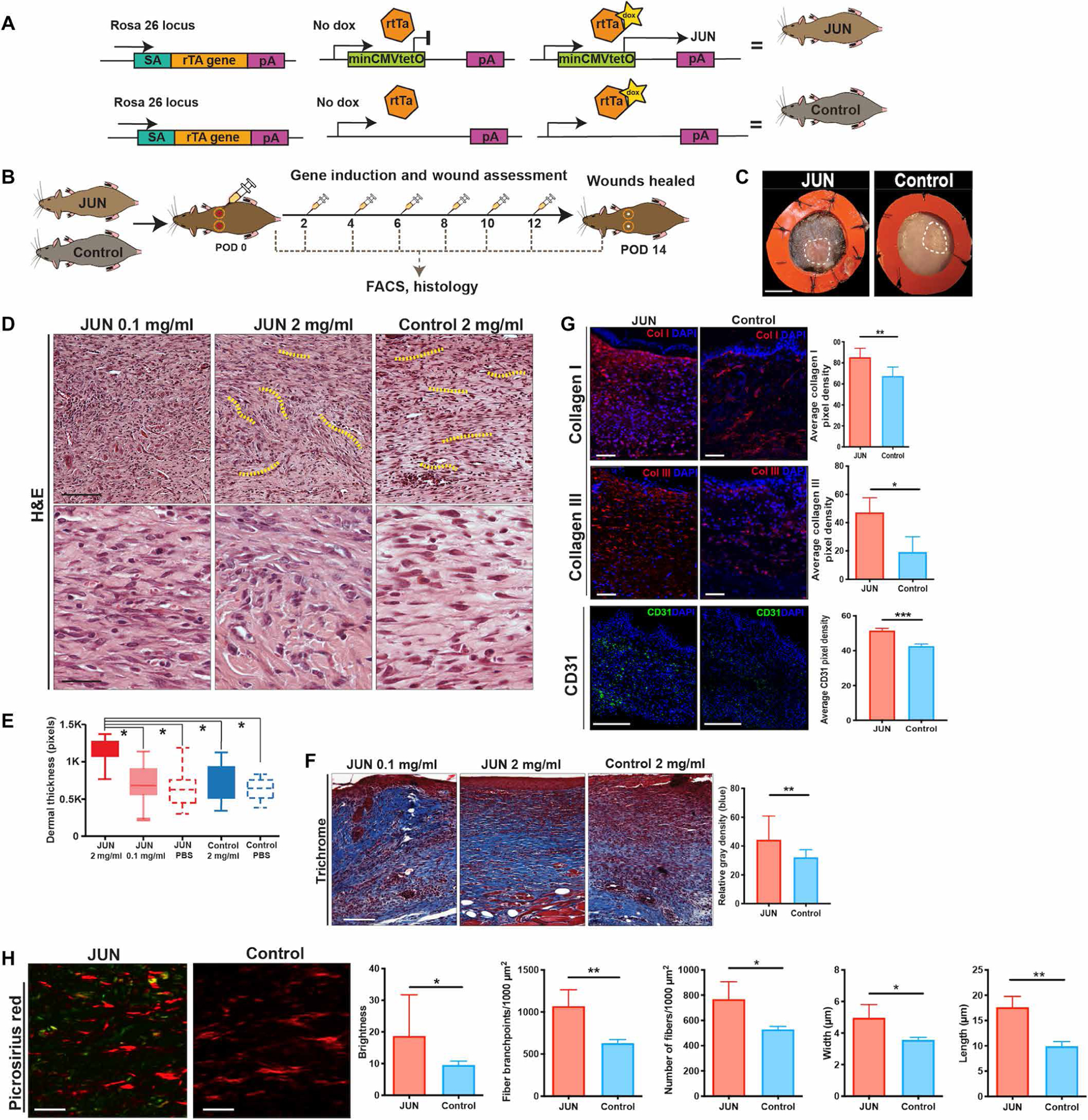
(A) Targeting construct of JUN doxycycline (dox)–inducible mice. In JUN mice, the construct at the Rosa 26 locus (rTtA) is coupled with the tetracycline operator minimal promoter (tetO) and leads to Jun overexpression in the presence of dox (top right) but no change to Jun expression without dox (top middle). In the Control mice, Jun is physiologically expressed due to the lack of the rtTA with (bottom right) and without dox (bottom middle). (B) Schematic showing experimental approach: Six-millimeter stented excisional dorsal wounds were created in JUN and Control mice. Dox (20 μl of 2 mg/ml) was administered on the day of surgery and on alternate postoperative days (POD) until complete wound closure (POD 14). Wounds were harvested for fluorescence-activated cell sorting (FACS) and histology (n = 18 mice per group per time point). (C) Representative gross photographs of healed (POD 14) wounds from JUN and Control mice receiving dox (2 mg/ml). White dotted line, healed scar. Scale bar, 0.25 cm. (D) Representative hematoxylin and eosin (H&E)–stained wounds of JUN and Control mice. Scale bars, 75 μm (top) and 25 μm (bottom). Yellow dotted lines show the whorl pattern of hypertrophic scars (HTSs). (E) Comparison of dermal thickness in wounds of JUN and Control mice at POD 14. (F) Representative Masson’s trichrome–stained wounds of JUN and Control mice at POD 14. Comparison of total collagen content (defined as relative mean gray density) from Masson trichrome staining. Scale bar, 100 μm. (G) Immunofluorescently labeled collagen type I (COL1) (red), COL3 (red), and CD31 (green) in JUN and Control mice on POD 14. Scale bars, 100 μm. DAPI, 4′,6-diamidino-2-phenylindole. (H) Picrosirius red–stained wounds of JUN and Control mice on POD 14. Scale bars, 25 μm. Computational quantification of collagen fiber networks evaluating length, branching, brightness, width, and number of fibers in JUN mice. All data are presented as means ± SEM. n = 3 independent experiments. *P < 0.05, **P < 0.01, and ***P < 0.001.
Full-thickness stented excisional wounds were created on the dorsum of JUN (c-JuntetO R26-rtTA) and Control (Rosa26-rtTA) mice. Wounds were injected with dox (20 μl, 2 mg/ml) on the day of wounding [postoperative day (POD) 0] and then on alternate days until POD 14 (Fig. 1B). Gross assessment of wound size revealed that JUN overexpression resulted in HTS formation at POD 14 but did not change the overall time to complete wound closure (Fig. 1C and fig. S1, B to D). Hematoxylin staining showed that JUN overexpression led to both dermal and epidermal thickening, with scars resembling the whorl pattern characteristic of human HTS (P < 0.05) (Fig. 1, D and E, and fig. S1E) (8). In addition, JUN wounds showed greater epithelization than Control wounds at POD 14 (fig. S1F). Dox dosing and titration regimens were optimized by evaluating the effect of dox at 0.1, 2, and 4 mg/ml on JUN wounds. Because of the toxicity observed at 4 mg/ml and the minimal effect of JUN induction with 0.1 mg/ml, a dosing of 2 mg/ml was implemented throughout the study (Fig. 1E).
Systemic induction of JUN expression is known to induce severe multiorgan fibrosis (12). Here, we show that wounds in mice with inducible JUN expression healed with excess collagen deposition (P < 0.01) (Fig. 1F). We found a significantly greater density of collagen I and III staining in JUN wounds compared to Control wounds at POD 14 (P < 0.05) (Fig. 1G). Furthermore, treatment of dorsal dermal wounds in wild-type mice with a JUN inhibitor (T5224) supported the critical role of JUN in skin scarring; these wounds yielded significantly thinner dermal scars with less collagen deposition (P < 0.05) (fig. S1G). In addition, CD31 staining showed greater vessel formation after JUN induction (Fig. 1G). Vascular endothelial growth factor gene expression was significantly greater in fibroblasts derived from wounds of JUN mice compared to Control mice (P < 0.05) (fig. S1H), suggesting that Jun overexpression promoted the increased vessel formation in the wounds.
Computational analysis of wound ECM architecture from Picrosirius red imaging demonstrated differences in the collagen fiber organization and brightness in JUN mice. Picrosirius red staining showed brighter collagen fibers in JUN mice (Fig. 1H). Furthermore, collagen fibers were longer, showed greater branching, and were wider and greater in number in JUN mice, suggesting fibrotic ECM formation with JUN overexpression (P < 0.05) (Fig. 1H). In summary, these data support a role for JUN expression in promoting HTS in the mouse dermis.
Jun drives fibrosis through a specific reticular fibroblast subpopulation
Four functionally distinct fibroblast subpopulations with unique surface marker expression have previously been identified in the unwounded mouse dermis: papillary (superficial, PDGFRα+CD26+Blimp+Lrig+Sca1−), reticular (deep dermal, PDGFRα+Dlk1+Sca1−), zigzag (hair follicle associated, PDGFRα+CD26−Sox2−), and adipocyte precursors or “lipofibroblasts” (residing below the reticular layer, PDGFRα+CD26−Sca1+Dlk1+/) (16, 17). To explore the relative proportions of these putative fibroblast subgroups during the wound healing process, we evaluated the prevalence of cells with each surface marker profile using fluorescence-activated cell sorting (FACS) in JUN and Control mice on POD 0 (baseline), 1 (early inflammatory phase), 7 (proliferative phase with fibroblast migration into wound beds), and 14 (wound closure) (Fig. 2A and fig. S2, A and B) (9). There was a significant increase in reticular fibroblasts (and decrease in lipofibroblasts) in JUN compared to Control mice only at POD 7 (P < 0.001) (Fig. 2B); by POD 14, the contributions of lipofibroblasts and reticular fibroblasts were similar in both groups. The proportions of wound zigzag and papillary fibroblasts also did not differ between JUN and Control mice at POD 14 (fig. S2B). Myofibroblasts are marked by α–smooth muscle actin (α-SMA) and excessive ECM and have contractile properties (18). Immunofluorescence showed less staining for adiponectin and increased staining for α-SMA in JUN mice in POD 7 (Fig. 2C), suggesting that JUN may activate fibroblasts toward a profibrotic myofibroblast phenotype.
Fig. 2. Jun drives fibrosis through a specific reticular fibroblast subpopulation.
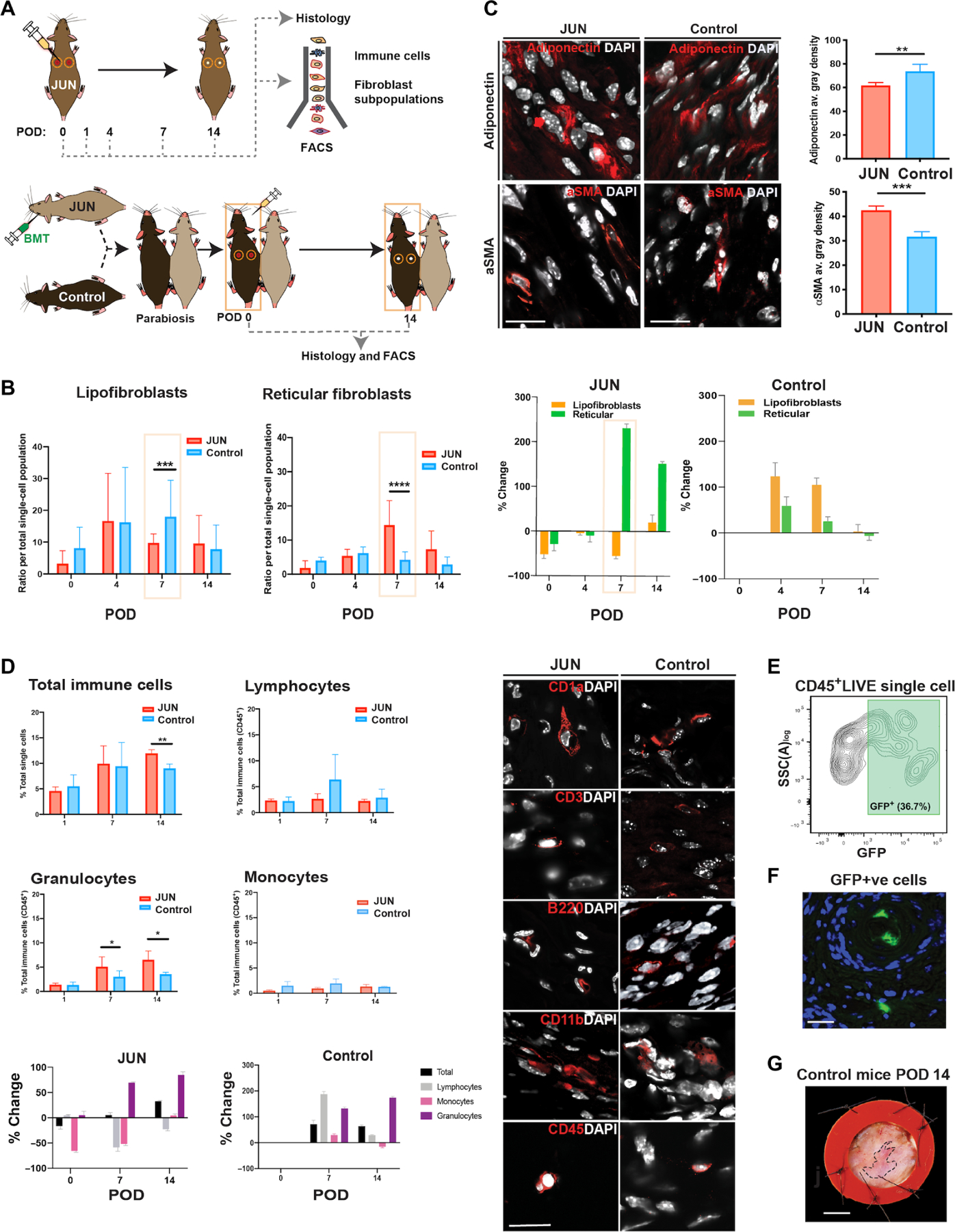
(A) Schematic showing experimental approach for immune and fibroblast cell contribution (top): Six-millimeter stented excisional dorsal wounds were created in the dorsum of JUN and Control mice. Dox (20 μl of 2 mg/ml) was administered on the day of surgery and on alternative days until complete wound closure on POD 14. Wounds were harvested for FACS to isolate fibroblasts and immune cell populations and to compare wounds histologically. Schematic showing experimental approach for parabiosis experiment and bone marrow transplantation (BMT) with 1 million whole bone marrow cells isolated from JUN mice and transduced to express GFP and luciferase. Six-millimeter stented excisional dorsal wounds were then created on the dorsum of Control parabionts. Dox (20 μl of 2 mg/ml) was administered on the day of surgery and on alternative days until POD 14. Wounds were harvested on POD 14 for FACS and histological assessment of healing wounds. (B) Bar graphs (left) showing the numbers of lipofibroblasts and reticular fibroblasts in JUN and Control mice throughout wound healing (POD 0, 4, 7, and 14). Bar graphs (right) showing % change in numbers of lipofibroblasts and reticular fibroblasts in JUN (compared to Control mice equivalent time point) and Control mice (compared to Control baseline). (C) Immunofluorescent staining for adiponectin (labeling lipofibroblasts and extracellular adiponectin) and α-smooth muscle actin (α-SMA; labeling myofibroblasts and vascular smooth muscle cells) in the wounds of JUN and Control mice on POD 14. Scale bars, 25 μm. (D) Bar graphs (left) showing the total percent of immune cells (CD45+), lymphocytes, granulocytes, and monocytes in JUN (compared to Control mice equivalent time point) and Control mice (compared to Control baseline) throughout wound healing (POD 0, 1, 7, and 14). Immunofluorescent images (right) illustrating the different immune cells in JUN and Control mice on POD 14: CD1a (Langerhans’ cells), CD3 (T cells), B220 (B cells), CD11b (or “MAC-1,” monocytes, and macrophages), and CD45 (all hematopoietic cells). Scale bar, 30 μm. Bar graph showing % change in numbers of immune cells in JUN and Control mice (bottom). (E) JUN/Control parabionts 3 weeks post-parabiosis surgery with representative FACS plot showing successful chimerism with 36.7% GFP+ CD45+ LIVE single cells in the peripheral circulation of Control parabiont 4 weeks post-parabiosis surgery. (F) Representative immunostaining showing GFP+ cells in wounds of the Control parabiont at POD 14 within blood vessels and the dermis. (G) A representative wound from a Control parabiont on POD 14, which resembles the wounds of Control mice in Fig. 1C. *P < 0.05, **P < 0.01, and ***P < 0.001. All data are presented as means ± SEM. n = 3 independent experiments. *P < 0.05, **P < 0.01, and ****P < 0.0001.
Resident and infiltrating immune effector cells are known to act in conjunction with dermal fibroblasts to orchestrate cutaneous tissue repair (19, 20). The effects of JUN overexpression on wound fibroblasts may be driven primarily within the fibroblasts themselves or mediated through their interactions with immune cells. Hence, wounds were analyzed at POD 0, 1, 7, and 14 by FACS to compare the immune profiles of wounds from JUN and Control mice (Fig. 2D and fig. S2C). There were significantly more immune cells in JUN mice on POD 14 (P < 0.01) (Fig. 2D). However, there were no changes in monocytes or lymphocytes (Fig. 2D). These results were confirmed by immunofluorescence staining (Fig. 2D). To determine whether the increase in total immune cells with JUN overexpression was responsible for the observed wound scarring, we conducted bone marrow transplantation assays on parabiotic mice (Fig. 2, A and E to G). Despite chimerism and the presence of JUN immune cells in the wounds of Control parabiont mice throughout wound healing (Fig. 2F), the wounds of Control parabionts did not heal with excessive fibrosis (Fig. 2G and fig. S2D) and appeared similar to the wounds of Control mice.
Many cell types contribute to skin fibrosis after tissue injury, including fibroblasts, immune cells, endothelial cells, and keratinocytes (4). The parabiosis experiments above confirmed that JUN overexpression–induced dermal scarring was not attributable to changes in immune cells. In contrast, it is possible that JUN overexpression may have modulated keratinocytes within the healing wounds, because there was significantly greater wound epithelization in JUN mice compared to Control (P < 0.0001) (fig. S1F). Hence, to further confirm that the Jun overexpression–induced dermal fibrotic response was primarily due to wound fibroblasts, we generated a transgenic mouse that used fibroblast-specific JUN induction (fig. S3).
The fibroblast-specific marker α-SMA was chosen because of its role in inducing dermal scarring through excessive ECM deposition (21). Furthermore, JUN mice showed greater α-SMA protein expression than Control mice at POD 14 after wounding (fig. S3A). First, α-SMACreER transgenic mice were crossed with ROSA26mTmG reporter mice. Upon administration of tamoxifen, Cre recombinase removes the floxed Rosa26mT/mG cassette, resulting in expression of targeted enhanced green fluorescent protein (eGFP). The α-SMACreER.Rosa26mT/mG mice were then crossed with JUN mice to obtain triple-positive offspring (α-SMACreER.Rosa26mT/mG.JUN), which overexpressed JUN in α-SMA–positive cells after dox induction; these triple-positive mice are referred to as JUN-Fib (table S1). The JUN-Fib mice that received phosphate-buffered saline (PBS) are the corresponding control mice and are referred to as Control-Fib mice.
After 5 days of intraperitoneal tamoxifen injections, full-thickness excisional wounding on the dorsum of JUN-Fib mice was performed. Following the same protocol, dox (20 μl, 2 mg/ml) injections were administered on the day of wounding (POD 0) and then on alternate days until POD 14 to induce Jun overexpression (fig. S3B). The resulting fibroblast-specific JUN induction caused a similar HTS phenotype by gross photography as those observed in JUN mice (fig. S3C), which was not seen in Control-Fib mice. JUN-Fib mice demonstrated thicker scars with greater collagen deposition than Control-Fib wounds (*P < 0.05; fig. S3D and table S1). These data indicate that the JUN fibrotic response was primarily mediated by fibroblasts. There were similar increases in gene and protein expression of collagen I and III in JUN-Fib mice as those observed in JUN mice (fig. S3, E and F). Furthermore, the decrease in lipofibroblasts and increase in reticular fibroblasts observed at POD 7 in JUN mice were also observed in JUN-Fib at POD 7 and 14 (fig. S3G), without any change in the immune cells (fig. S3H).
Because α-SMA may also be expressed in vascular smooth cells, we performed the same analysis with an additional transgenic mouse that used collagen type I (COL1) as the specific fibroblast marker (fig. S4). First, Col1CreER.Rosa26mT/mG mice were crossed with JUN mice, with the triple-positive offspring (Col1CreER.Rosa26mT/mG.JUN) overexpressing JUN in COL1-positive cells, after dox and tamoxifen induction (fig. S4A). The COL1-specific JUN induction led to a similar hypertrophic wound healing response by gross photography and histology (fig. S4, B to G) as observed in JUN (Fig. 1) and JUN-Fib mice (fig. S3). Second, there was a similar decrease in lipofibroblasts and increase in reticular fibroblast number at POD 7 and 14 (fig. S4F) as those observed in JUN (Fig. 2B) and JUN-Fib mice (fig. S3G), without any change in immune cells (Fig. 2D and figs. S3H and S4H). In summary, these data demonstrate that the fibrotic effect of Jun overexpression in dermal wounds is due to fibroblasts.
Jun up-regulates fibrotic signaling pathways in reticular fibroblasts and PPARγ signaling in lipofibroblasts
To understand the molecular mechanisms underlying the cellular changes evident on POD 7, we profiled the gene expression and chromatin accessibility landscape of reticular and lipofibroblast subpopulations FACS-isolated from JUN and Control mouse wounds (Fig. 3A) and performed hierarchical clustering of biological replicates for both RNA sequencing (RNA-seq) and assay for transposase-accessible chromatin sequencing (ATAC-seq) data based on Jun induction (fig. S5A). Principal components analyses (PCAs) showed that Jun induction state was the main contributor driving the clustering of JUN and Control fibroblasts in both RNA- and ATAC-seq data (fig. S5B).
Fig. 3. Jun up-regulates fibrotic signaling pathways in reticular fibroblasts and PPARγ signaling in lipofibroblasts.
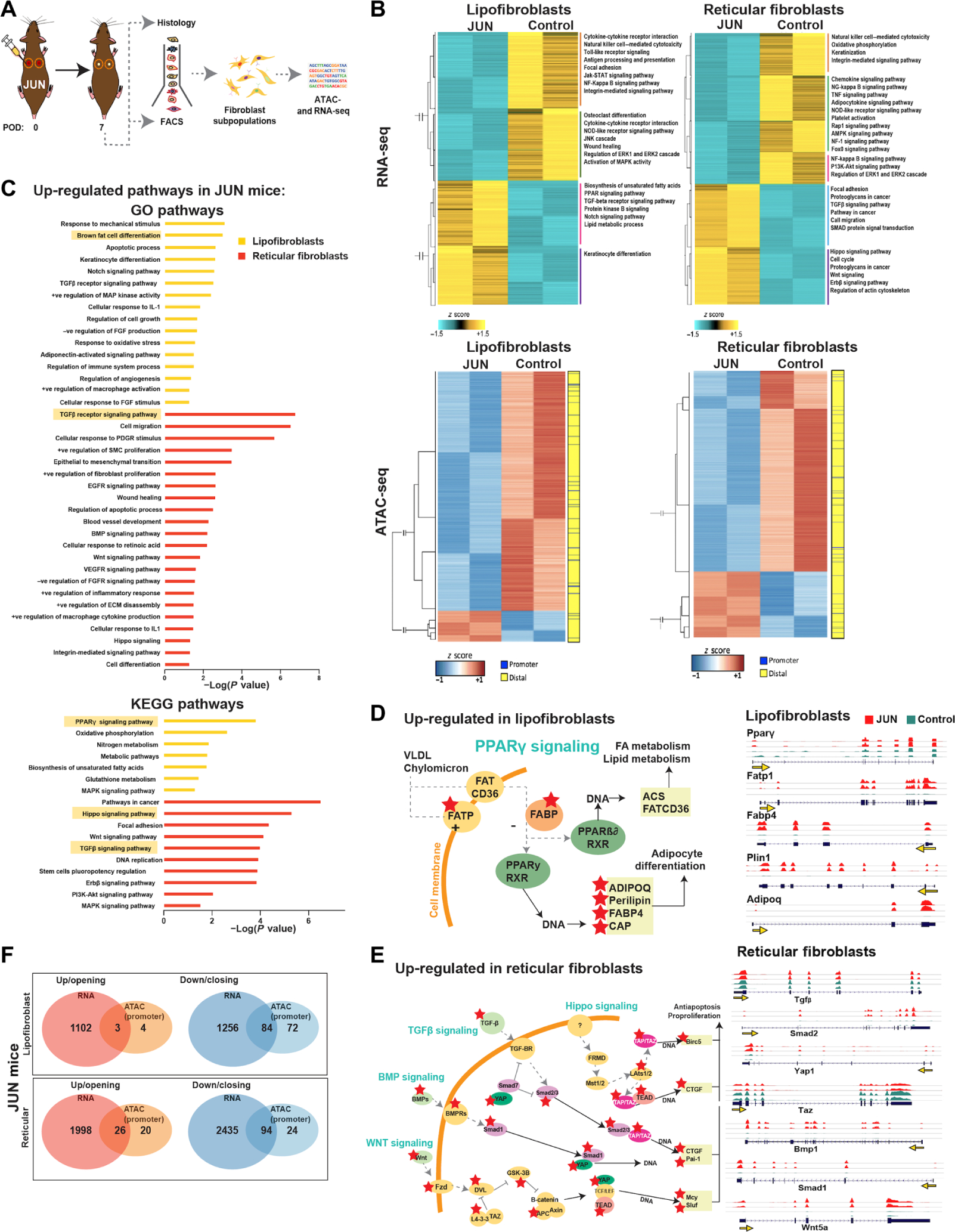
(A) Schematic showing experimental approach: Lipofibroblast and reticular fibroblasts were isolated by FACS on POD 7 for ATAC- and RNA-seq analysis. (B) Heatmap showing hierarchical clustering of differentially expressed genes (FDR < 0.01, fold change > 2) between JUN and Control in lipofibroblasts (top left) and reticular fibroblasts (top right). Heatmap showing chromatin regions with changes in accessibility (FDR < 0.01, fold change > 2) in JUN versus Control mice in lipofibroblasts (bottom left) and reticular fibroblasts (bottom right). (C) Gene set enrichment analysis showing the most up-regulated GO (top) and Kyoto Encyclopedia of Genes and Genomes (bottom) pathways (based on RNA-seq data) in JUN versus Control mice in lipofibroblasts (yellow) and reticular fibroblasts (red) on POD 7. (D) Schematic showing the up-regulated genes identified from the PPARγ signaling pathway in lipofibroblasts in JUN versus Control mice on POD 7; red stars show significantly up-regulated genes (P < 0.05). Gene expression track analysis of individual genes in the PPARγ signaling pathway up-regulated in lipofibroblasts in JUN versus Control mice on POD 7. Yellow arrow indicates the promoter. (E) Schematic showing significantly up-regulated genes identified from multiple converging fibrotic signaling pathways [Hippo, transforming growth factor–β (TGFβ), bone morphogenic protein (BMP), and wingless (WNT)] in reticular fibroblasts in JUN versus Control mice on POD 7; red stars show significantly up-regulated genes (P < 0.05). Gene expression track analysis of individual genes found in the fibrotic signaling pathways up-regulated in reticular fibroblasts in JUN versus Control mice on POD 7. Yellow arrow indicates the promoter. (F) Genes with significantly increased expression and opening promoter chromatin (distance from TSS <100 kb) (left, red) or decreased expression and closing promoter chromatin (right, blue) in lipofibroblasts (top) and reticular fibroblasts (bottom) in JUN versus Control mice on POD 7 (P < 0.05). Significant genes found to be both up-regulated and with opening chromatin in JUN versus Control mice in reticular (bottom, n = 26) and lipofibroblasts (top, n = 3) (*P < 0.05). All data are presented as means ± SEM.
RNA-seq analysis of lipofibroblasts indicated that there were similar numbers of up-regulated (1150) and down-regulated (1340) transcripts [false discovery rate (FDR) < 0.01, fold change > 2] (Fig. 3B). In addition, RNA-seq analysis of reticular fibroblasts (FDR < 0.01, fold change > 2) also demonstrated similar numbers of up-regulated (2024) and down-regulated (2529) transcripts in JUN as compared to Control (Fig. 3B).
Although ATAC-seq analysis showed many differential accessible regions between JUN and Control lipofibroblasts, there were far more closing regions (2590 peaks) in JUN than opening regions (336 peaks) (FDR < 0.01, fold change > 2) (Fig. 3B). ATAC-seq analysis also showed a larger number of closing regions (3151 peaks) in JUN reticular fibroblasts than opening regions (1040) (FDR < 0.01, fold change > 2) (Fig. 3B). Jun is an AP-1 family member that dimerizes with other AP-1 family transcription factors (12). Overexpression of Jun in reticular and lipofibroblasts may perturb the natural pairing of such transcription factors and inhibit them, resulting in the observed greater number of closing than opening regions (22). Most of the differential accessible regions between JUN and Control in both lipofibroblasts and reticular fibroblasts were distal (>1-kb distance) from transcription start sites (TSSs) (fig. S5C).
Gene set enrichment analysis of differentially expressed genes highlighted a number of pathways up-regulated in JUN lipofibroblasts and reticular fibroblasts (Fig. 3, C to E). For lipofibroblasts, these included “brown fat differentiation” and “peroxisome proliferator–activated receptor (PPARγ) signaling” (Fig. 3D). Within the PPARγ signaling pathway up-regulated in JUN lipofibroblasts, we found up-regulation of surface proteins (fatty acid transport protein), as well as intracellular [fatty acid–binding protein (FABP)] and downstream mediators [adiponectin (ADIPOQ), perilipin (PLIN1), and catabolite activator protein] (Fig. 3D). JUN overexpression resulted in a profibrotic phenotype at the transcriptional level in reticular fibroblasts with up-regulation of the “transforming growth factor–β (TGFβ) receptor,” “Hippo,” “phosphoinositide 3-kinase–Akt/protein kinase B,” “wingless-related integration sites (WNT),” “bone morphogenic protein (BMP),” and “focal adhesion kinase (FAK)” signaling pathways (Fig. 3E and fig. S5D). Pathways down-regulated in JUN mice related to immune cell function, such as “graft-versus-host diseases” and “tumor necrosis factor (TNF) signaling” in both lipofibroblasts and reticular fibroblasts (fig. S5, E and F).
To further identify molecular mechanisms contributing to the observed JUN-induced fibrotic effect, we identified genes that exhibited both differential expression and changes in chromatin accessibility in JUN versus Control fibroblasts. JUN lipofibroblasts had 3 genes with both increased expression and opening promoter chromatin regions and 84 with decreased expression and closed promoter regions of chromatin (Fig. 3F and fig. S5G). Similarly, JUN reticular fibroblasts had 26 genes with both increased expression and opening promoter chromatin regions, and 94 genes had decreased expression and closed promoter regions of chromatin (Fig. 3F and fig. S5G). Analysis of genes associated to the distal differential accessible peaks (>1-kb distance from TSS) demonstrated an increase in the number of common genes exhibiting both differential chromatin accessibility and differential expression. Specifically, in the lipofibroblasts, 38 common genes were increasing in accessibility and expression, and 213 genes were decreasing in accessibility and expression (fig. S5H). In reticular fibroblasts, the overlapping genes increase by 233 and 555, respectively (fig. S5H).
JUN is highly expressed in human HTS fibroblasts
To derive clinically useful therapies, it is essential to determine whether JUN also drives scarring within human HTS fibroblasts. Hence, we compared JUN protein expression in human normal skin (hNS), scar (hSc), HTS (hHTS), and keloids (hKel) (Fig. 4A and table S2). In patient tissue protein arrays, JUN was highly expressed in the skin, especially in hHTS, and localized to the fibroblast nucleus (Fig. 4B). There was also increased COL1, JUN, α-SMA, and JUN/α-SMA coexpression in hHTS compared to hSc and hNS (fig. S6A). The degree of JUN overexpression in JUN mice (fig. S6B) was compared to that of human tissue scars (Fig. 4B) and fibroblasts isolated from non-keloid scars (hSc) (fig. S6A). JUN protein expression in hSc showed a similar increase compared to hNS (~20-fold) (fig. S6B).
Fig. 4. CD36 antagonism minimizes JUN-dependent fibroproliferative activity.
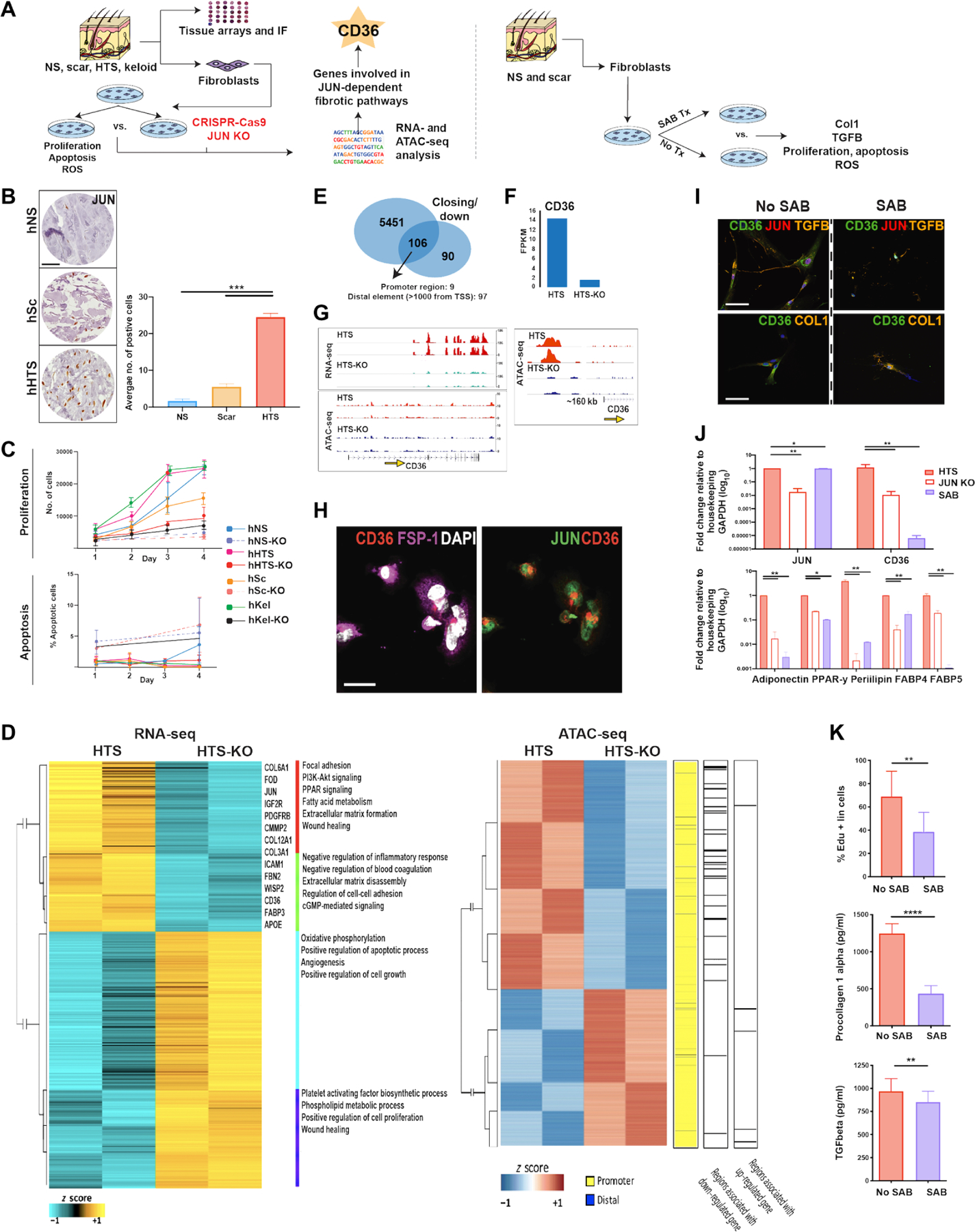
(A) Schematic of experimental approach. Left: Human normal skin (NS), scar, HTS, and keloid specimens were assessed for expression of JUN and fibrogenic fibroblast markers using tissue protein arrays and immunofluorescence (IF). Primary cultures of fibroblasts were derived from skin specimens, and CRISPR-Cas9 was used to delete JUN expression. Human fibroblasts derived from HTSs (hHTSs) with (KO) and without (non-KO) JUN deletion were compared for proliferation, apoptosis, and by RNA- and ATAC-seq. Right: hHTS-derived human fibroblasts were treated with salvianolic acid (SAB; 100 μM) for 48 hours before analysis of COL1 and TGFβ protein secretion, proliferation, apoptosis, and production of reactive oxygen species (ROS). (B) Tissue array showing JUN expression in hNS, non-keloid scar (hSc), and hHTS. Scale bar, 10 μm. (C) Apoptosis and proliferation analysis of HTS fibroblasts with and without KO of JUN over 4 days, assessed using annexin V and 5-Ethynyl-2′-deoxyuridine (EdU) labeling, respectively. (D) Heatmaps from RNA- and ATAC-seq comparative analysis showing differently expressed genes (FDR < 0.01, fold change > 2) and chromatin accessibility regions in HTS and HTS-KO fibroblasts. (E) Bar chart demonstrating the genes that were significantly down-regulated (90), exhibited a decrease in chromatin accessibility (5451), and exhibited both a decrease in chromatin accessibility and were down-regulated (106) in KO versus non-KO HTS fibroblasts (P < 0.05). (F) Bar graph showing the FPKM (fragments per kilobase of exon model per million reads mapped) of CD36 in HTS versus HTS-KO fibroblasts. (G) RNA-seq expression and ATAC-seq tracks for CD36 in HTS fibroblasts. Yellow arrow indicates the promoter. (H) Plated hHTS fibroblasts immunostained with (left) fibroblast-specific protein 1 (FSP-1, purple)/CD36 (red)/DAPI (white) and (right) JUN (green)/CD36 (red). Scale bar, 10 μm. (I) Plated hHTS fibroblasts immunostained with CD36 (green)/JUN (red)/TGF-β (orange, top row) and CD36 (green)/COL1 (orange, bottom row) without (left) and with (right) SAB treatment. Scale bars, 10 μm. (J) RT-qPCR analysis (top); JUN and CD36 expression in hHTS fibroblasts at baseline, after SAB treatment, and in hHTS-KO fibroblasts. Bottom: Adipogenic-associated genes [Adipoq (adiponectin), peroxisome proliferator–activated receptor-γ (Ppar-γ), Perilipin, Fatty acid–binding protein 4 (Fabp4), and Fabp5] in hHTS fibroblasts at baseline, after SAB treatment, and in hHTS-KO fibroblasts (*P < 0.05 and **P < 0.01). (K) hHTS fibroblasts with and without 48 hours of SAB treatment compared for proliferation by EdU staining (top), COL1 secretion (middle), and TGFβ (bottom) secretion. All data are presented as means ± SEM. n = 3 independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
We next explored the role of JUN within hSc-derived fibroblasts using in vitro assays. After primary culture of hNS, hSc, hHTS, and hKel fibroblasts, JUN expression was deleted using CRISPR-Cas9. Genotyping showed successful mutation induced by Cas9 (fig. S6C), and Western blotting revealed successful elimination of JUN protein expression (fig. S6C). Cellular proliferation, apoptosis, and cytokine expression were compared between cells with (“KO”) and without (non-KO) JUN deletion (Fig. 4C). JUN deletion decreased proliferation and increased apoptosis in hHTS and hNS fibroblasts (Fig. 4C).
The cross-talk between fibroblasts and immune cells is integral to the inflammatory phase of wound repair (4). Hence, we explored the effect of JUN KO on cytokine production. JUN KO decreased the expression of numerous cytokines including plasminogen activator inhibitor-1 (PAI-1), platelet-derived growth factor (PDGF-AA), interleukin-6 (IL-6), monocyte chemoattractant protein (MCP-1), and thymic stromal lymphopoietin (TSLP) (fig. S6D). In bleomycin-induced lung fibrosis, IL-6 is both proinflammatory and profibrotic (13, 23). Collectively, these findings suggest that in human cells, JUN expression modifies the communication between fibroblasts and immune cells.
JUN regulates profibrotic pathways in human HTS fibroblasts at the chromatin level
To complement our mouse data, we compared gene expression and chromatin accessibility landscapes between JUN KO and non-KO hHTS (Fig. 4D). Data quality assessment revealed high correlation between replicates of each condition (figs. S6E and S7, A to D) and clear distinction between JUN KO and non-KO fibroblasts in RNA-seq data (fig. S6F). ATAC-seq data quality was assessed by TSS enrichment (fig. S7A), insert size distribution (fig. S7B), and correlation between replicates. Replicates also had high correlation (fig. S7C), and PCA indicated that the main driver of differences was fibroblast type, followed by JUN KO state (fig. S7D). The hNS, hSc, hHTS, and hKel KO and non-KO fibroblasts exhibited differently expressed genes (Fig. 4D and fig. S6, G to I) and chromatin accessibility (Fig. 4D and fig. S7E).
The hSc, hHTS, and hKel fibroblasts exhibited overlapping differentially expressed genes in JUN-KO versus non-KO, but many differentially expressed genes were unique to each fibroblast type (fig. S6J). Gene ontology (GO) analysis of up-regulated (fig. S6K) and down-regulated genes (fig. S6L) in hHTS versus hNS fibroblasts highlighted pathways associated with JUN expression and fibrosis. In addition, hNS, hSc, hHTS, and hKel scars also showed differential expression of collagen subtypes (fig. S6M). Given that JUN expression was highly expressed in hHTS fibroblasts (fig. S6, G, N, and O) and consistent with JUN protein expression (Fig. 4B), we focused on HTS fibroblasts (Fig. 4D).
RNA-seq analysis revealed that a total of 196 genes were down-regulated and 294 genes were up-regulated in JUN-KO (FDR < 0.01, fold change > 2) (Fig. 4D). GO analysis indicated that down-regulated genes were involved in scarring-related pathways (fig. S6P). In addition, chromatin accessibility analysis indicated that JUN KO decreased accessibility of a large number of chromatin regions (5557 peaks) in hHTS fibroblasts (Fig. 4D and fig. S7E). GREAT and HOMER analysis indicated that most differential accessible regions were located distal from TSS (fig. S7, F and G). Motif analysis showed enrichment of 2 transcription factor motifs in opening peaks and 10 transcription factor motifs in closing regions (fig. S7H). One of the less accessible motifs in JUN KO hHTS fibroblasts was NFIX (nuclear factor I-C), which links PDGF and TGFβ1 signaling to wound healing in the skin, thus linking TGFβ signaling and JUN (24). In addition, GO analysis of chromatin regions exhibiting less accessibility in hHTS JUN-KO fibroblasts showed enrichment of terms associated with wound healing, scarring, and the PDGF signaling pathway (fig. S7I). Further analysis of the RNA- and ATAC-seq data revealed that 106 genes were both down-regulated and exhibited a decrease in chromatin accessibility (Fig. 4E), and 64 genes were up-regulated and exhibited increased chromatin accessibility in JUN-KO compared to non-KO hHTS fibroblasts (fig. S8, A and B). For example, collagen 12 type alpha (COL12A) had less accessibility after JUN KO (fig. S7J). These data suggest that, as in mouse fibroblasts, JUN is also important in mediating human dermal scarring including keloid and HTS.
To further evaluate the potential overlap between our human and mouse findings, we used the CIBERSORTx platform to deconvolve human bulk RNA-seq data into phenotypes based on our mouse RNA data groups (fig. S8C). The human fibroblasts derived from non-keloid scars and JUN reticular fibroblasts showed significant overlap in their transcriptional profiles (P < 0.05), suggesting that the Jun mouse model has translational relevance for the study of human skin fibrosis.
CD36 is a potential cell surface receptor target to block JUN overexpression
To identify a cell surface marker that could antagonize the effect of JUN-mediated dermal fibrosis, we examined the 106 genes that were both transcriptionally down-regulated and showed closing peaks after JUN KO in human fibroblasts (Fig. 4, F and G, and fig. S6Q). Of the seven identified cell surface markers (CD36, IL-1 receptor-1, receptor tyrosine kinase–like orphan receptor 2, oncostatin M receptor, epidermal growth factor receptor, coagulation factor II receptor, and insulin-like growth factor receptor-2), CD36 was the only marker that was found to be highly expressed in human scarring fibroblasts in vitro (fig. S8D).
To further investigate CD36 as a cell surface target to overcome JUN fibrosis, we used chromatin immunoprecipitation sequencing (ChIP-seq) (fig. S8E). We used ChIP analysis of human-derived fibroblasts to confirm that the genes showing overlap in both the ATAC- and RNA-seq mouse data are direct targets of JUN, including Cxcl1 (25), Wisp1 (26), Glis1 (27), and Ddah1 (fig. S8E) (28). Furthermore, CD36 was also shown to be a target of JUN in human fibroblasts (fig. S8E).
CD36 is a transmembrane glycoprotein and class B scavenger receptor expressed in a variety of cells including monocytes (29), macrophages (30), platelets (30), and endothelial cells (31). CD36 is the only fatty acid transporter well known to bind components in the ECM (32), including the ligands thrombospondin-1 (TSP-1) (30) and collagen (33). CD36 is thought to mediate TGFβ1 activation through TSP-1 (34). The pivotal profibrotic role of the TGFβ1 isoform has been highlighted by many reviews. HTS fibroblasts have higher concentrations of TGFβ mRNA than fibroblasts from normal skin (35). Activated TGFβ binds its surface receptor triggering intracellular SMAD-dependent and SMAD-independent signaling pathways (36, 37). In the SMAD-dependent pathway, phosphorylated SMAD proteins translocate to the nucleus to regulate the transcription of a number of ECM-related genes, including collagen genes and PAI-1 (38). We observed a loss of PAI-1 in the supernatant of cells after JUN KO (fig. S6D). The SMAD-independent pathways include the TGFβ/mitogen-activated protein kinase pathway, one of the most common scarring pathways, which also involves the JUN N-terminal kinase (JNK) pathway(39). This evidence highlighted that CD36 is a potentially important cell surface marker target to reverse skin scarring.
CD36 antagonism minimizes JUN-dependent fibroproliferative activity in human HTS fibroblasts
We aimed to investigate the relationship between JUN and CD36 to determine whether CD36 had potential as an antifibrotic therapeutic target. Immunofluorescence confirmed the association between JUN and CD36 in unwounded skin fibroblasts (Fig. 4H). A recent high-throughput screening for CD36 antagonists identified SAB, a small water-soluble molecule isolated from the root of red-rooted salvia, as an effective agent (40). SAB has also been linked to scarring, antioxidant, anti-inflammatory, and antiapoptotic activities (41–43). SAB inhibits expression of many matrix metalloproteinases (44) and inflammatory factors and is known to inhibit the TGFβ/SMAD pathway (45). Furthermore, SAB also inhibits TNF-α–induced PAI-1 secretion in human endothelial cells (43), consistent with our finding of reduced PAI-1 production after JUN KO in hHTS fibroblasts (fig. S6D). However, the relationship between SAB and JUN in the context of scarring has not been elucidated.
As a first step, we investigated whether SAB treatment (100 μg/ml) could antagonize the HTS fibroblast response and expression of JUN similarly to that after JUN deletion. Results indicated that SAB treatment in vitro was as effective as JUN deletion in reducing CD36 gene and protein expression (Fig. 4, I and J). Furthermore, SAB reduced protein expression of COL1 and reduced cell proliferation (Fig. 4K). TGFβ1 activity in fibroblasts is associated with scar formation (46). SAB treatment decreased TGFβ1 gene expression and protein secretion in fibroblasts derived from hSc (P < 0.05) (Fig. 4K and fig. S8F). Given the known role of SAB with reactive oxygen species (ROS) (47) and the impact of ROS on scarring, it was interesting to find reduced ROS after SAB treatment in a manner equivalent with JUN KO (fig. S8G).
To confirm the specificity of SAB for CD36, we evaluated the effect of CD36 shRNA knockdown on human-derived fibroblasts. After CD36 shRNA knockdown, COL1 and TGFβ1 gene and protein expression were reduced in hSc (P < 0.05) (fig. S9, A to C). The reduction in COL1 and TGFβ1 gene expression was comparable to that observed after SAB treatment, supporting the specificity of SAB for CD36 (fig. S9B). In addition, TGFβ1 gene and COL1 protein secretion were reduced to a similar extent as with SAB treatment (fig. S9, A and C). By contrast, SAB had no effect on COL1 and TGFβ1 expression in CD36-deficient cells (fig. S9, A and C).
To further clarify the relationship between JUN and CD36, we analyzed gene and protein expression in hSc in vitro over a 3-day period, after inhibition of JUN (with T5224 AP1) and CD36 (with SAB) (fig. S9, D and E). After T5224 AP1 inhibition, JUN gene and protein expression was down-regulated at all time points, and CD36 was down-regulated from day 2 onward (fig. S9, D and E). The inhibition of CD36 with SAB similarly showed a significant decrease in gene and protein expression at all time points, with no changes in JUN expression (P < 0.05) (fig. S9, D and E). These data confirm that JUN regulates CD36.
Our previous RNA-seq analysis revealed enhanced PPARγ signaling within lipofibroblasts after JUN overexpression (Fig. 3D). CD36 is known to act both as a surface receptor for very-low-density lipoprotein (VLDL) and chylomicrons and as a more downstream mediator in the PPARγ pathway (48). This is consistent with our immunofluorescence staining for CD36 in fibroblasts, which shows that CD36 is expressed both on the cell surface and in a perinuclear manner (Fig. 4H). We observed an increase in expression of PPARγ pathway genes in lipofibroblasts from JUN mice (Fig. 3D). To further confirm the relationship between CD36 and JUN, we compared the expression of genes in the PPARγ pathway between SAB- and KO-treated hHTS. Quantitative reverse transcription polymerase chain reaction (RT-qPCR) analysis demonstrated significantly decreased expression of a number of adipogenic genes involved in the PPARγ pathway in hHTS after both SAB treatment and JUN KO (P < 0.05) (Fig. 4J). Furthermore, SAB significantly reduced the protein expression of PPARγ, PLIN1, and ADIPOQ in JUN and Control mouse wounds at POD 14 (P < 0.01) (fig. S9F). These data parallel the increased expression of PPARγ pathway genes (Plin1, Fabp4, and Fabp5) seen in JUN lipofibroblasts and further support CD36 as a mediator of JUN-dependent scarring in HTS fibroblasts.
CD36 antagonism reverses hypertrophic dermal scarring
Last, to explore CD36 antagonism in vivo, wounded JUN and Control mice were administered either SAB (100 μg/ml in PBS) or PBS via intraperitoneal injection daily until complete wound closure (POD 14) (Fig. 5A and fig. S9G). At POD 14, SAB-treated wounds were less raised and erythematous than PBS-treated wounds in both JUN and Control mice (Fig. 5, B and H), although the wound closure end points were similar (Fig. 5, B and H). Histologically, SAB-treated wounds were thinner and composed of fewer immune cells (Fig. 5, C and I) and had less Picrosirius red (Fig. 5, D and J) and collagen I and III staining (Fig. 5, F and L). Furthermore, there was reduced immunostaining of CD36 after SAB treatment at POD 14 in JUN mice (fig. S9H). Moreover, SAB treatment significantly increased the number of lipofibroblasts and reduced the number of reticular fibroblasts in both JUN and Control mice (P < 0.05) (Fig. 5, E and K). To ensure that SAB caused an antifibrotic effect without anti-inflammatory effects, we analyzed the number of immune cells in Control and JUN mice after SAB and PBS treatment. Using FACS, we showed that there were no significant changes in total immune cells at POD 14 in Control or JUN mice (P > 0.05) (Fig. 5, G and M).
Fig. 5. CD36 antagonism reverses hypertrophic dermal scarring.
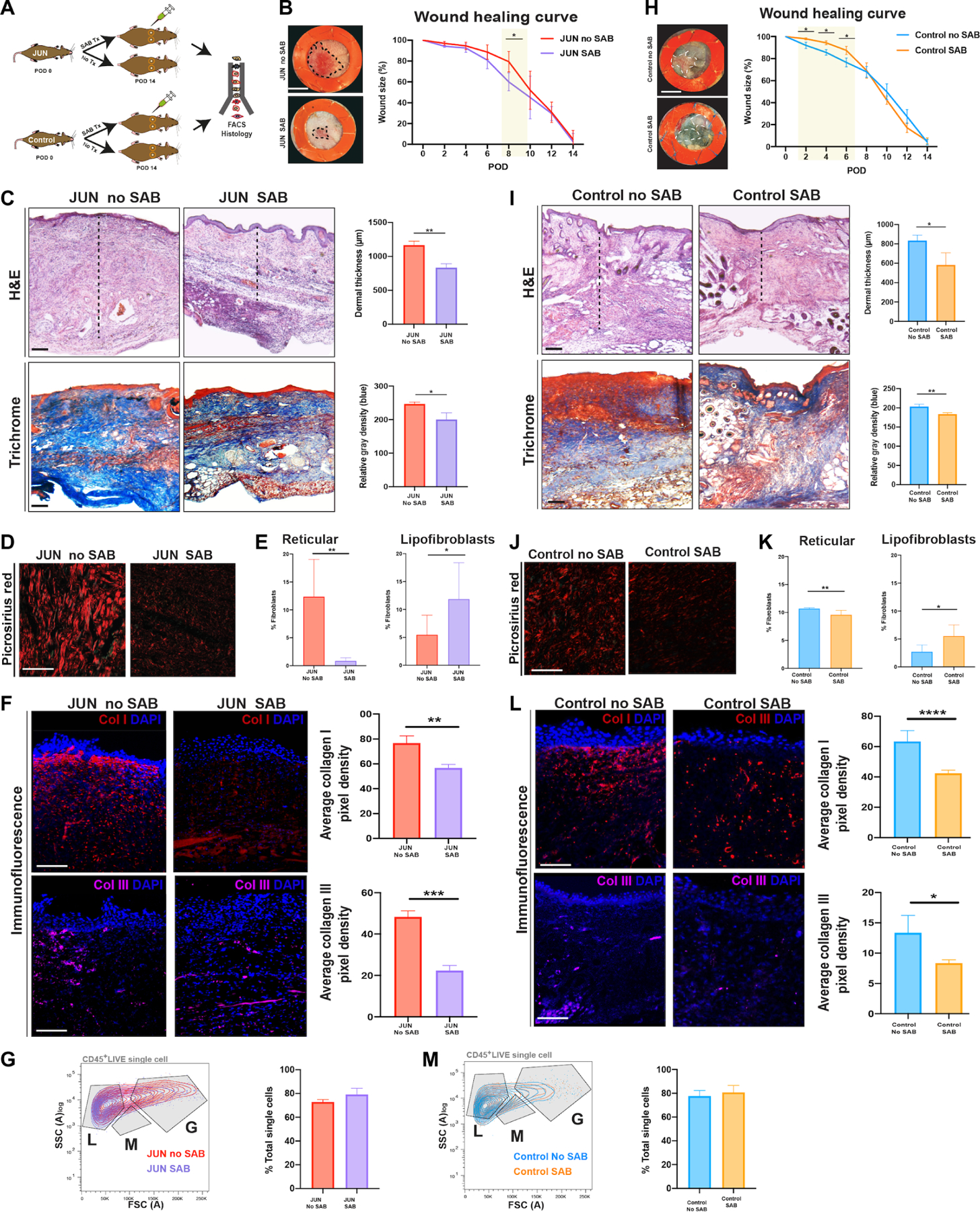
(A) Schematic of the experimental approach: Six-millimeter stented excisional dorsal wounds were created in JUN and Control mice. Dox (20 μl of 2 mg/ml) and SAB (100 μM) were administered on the day of surgery and on alternate PODs until complete wound closure (POD 14). Wounds were harvested for FACS and histology (n = 18 mice). Representative gross photographs of healed (POD 14) wounds of JUN (B) and Control (H) mice. Scale bars, 0.25 cm. Representative H&E- and Masson trichrome–stained wounds of JUN (C) and Control (I) mice. Scale bars, 150 μm. Picrosirius red–stained wounds of JUN (D) and Control (J) mice on POD 14. Scale bars, 25 μm. Sections are not adjacent slides but are from the same experimental group. Immunofluorescently labeled Col1 (red) and Col3 (purple) in JUN (F) and Control (L) mice on POD 14. Scale bars, 100 μm. Bar graph showing the numbers of lipofibroblasts and reticular fibroblasts in JUN (E) and Control (K) mice at POD 14. Bar graph showing the total percent of immune cells in JUN (G) and Control (M) mice at POD 14. All data are presented as means ± SEM. n = 3 independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
Because the small-molecule SAB inhibitor may have off-target effects, we characterized the effect of CD36 antagonism in vivo using CD36 KO mice (fig. S10A). Specifically, we asked whether CD36 KO wounds would heal with reduced scarring, congruent with wounds treated with SAB. At POD 14, CD36 KO wounds were minimally raised and erythematous (fig. S10B). Furthermore, CD36 KO wounds showed similar dermal thickness (fig. S10C), collagen deposition (fig. S10C), and Picrosirius red staining (fig. S10C) compared to unwounded skin. Last, there were minimal changes in COL1, COL3, and α-SMA staining compared to unwounded skin, further supporting reduced scar formation (fig. S10D). In summary, these data demonstrate that CD36 antagonism using SAB treatment can reduce skin scarring during wound repair in mice (fig. S10E).
DISCUSSION
Elucidation of the key mechanisms mediating pathological wound healing has been greatly hindered by a lack of animal models capable of recapitulating human scarring. To date, skin fibrosis has been linked to increased mechanical strain (3, 49), up-regulation of specific growth factors including TGF-B1 (38), and deposition of excess ECM by specific fibroblast lineage(s) (10). In contrast, knowledge of the key molecular pathways responsible for skin fibrosis is limited. Up-regulation of JUN overexpression has shown to contribute to skin scarring in mice, although the mechanism by which this occurs has not been elucidated (12, 50). Several human fibrotic diseases have also demonstrated the up-regulation of JUN in fibroblasts including lung fibrosis, bone marrow fibrosis, and scleroderma (12). Identifying the mechanism by which JUN induces skin fibrosis could allow for the identification of therapeutic targets to overcome skin scarring. To understand the effect of JUN overexpression on skin fibrosis, we have used a unique mouse model of hypertrophic skin scarring driven by induction of JUN overexpression upon wounding. Using our model, we show that JUN overexpression drives scarring by acting within distinct fibroblast subpopulations. An increase in the proportion of profibrotic reticular fibroblasts, with up-regulation of key fibrotic signaling pathways, leads to excessive deposition of ECM, resulting in scar formation. We translate these findings by ascertaining their relevance in humans via loss-of-function experiments with CRISPR-Cas9, showing that JUN is required for scarring in human skin.
To gain an understanding of the key molecular pathways acting within dermal fibroblasts after wounding, we performed RNA- and ATAC-seq. Transcriptional analysis demonstrated that JUN deletion in hSc-derived fibroblasts resulted in a loss of surface receptor expression. Antagonism of CD36 in human scarring fibroblasts in vitro demonstrated that SAB can be used to counteract scarring mediated by JUN. Furthermore, SAB treatment or a CD36 KO mouse model reduced HTS in mice. Fatty acid translocase CD36 is a multifunctional membrane protein that facilitates the uptake of long-chain fatty acid (51) several studies have shown that CD36 expression may contribute to organ fibrosis (52). Yang et al. (52) showed that knockdown of CD36 decreased renal tubule fibrosis by suppressing TGFB1. CD36-deficient mice have also demonstrated reduced proinflammatory and oxidative pathways in obstructed kidneys resulting in reduced collagen deposition (53). Down-regulation of CD36 through lysophosphatidic acid has also reduced liver fibrosis in mice (54) in the skin, CD36 expression has not been widely investigated. Hakvoort et al. (55) reported the up-regulation of CD36 in keratinocytes in human hypertrophic skin scars. Our study shows that JUN drives HTS, which can be alleviated through CD36 antagonism. Furthermore, we have provided evidence that JUN promotes fibrosis throughout the spectrum of human cutaneous scarring including keloid.
A key finding from this study was the effect of JUN overexpression on the relative proportions of lipofibroblasts and reticular fibroblasts during skin scarring. We report a decrease in lipofibroblasts and an increase in reticular fibroblast numbers at POD 7 after wounding in JUN mice. JUN overexpression up-regulated CD36 expression and consequentially changed the fibroblast subpopulation proportions within wounds of JUN mice. After up-regulation of CD36 expression, there was a decrease in lipofibroblasts and increase in scarring reticular fibroblasts. Furthermore, targeting CD36 using SAB or a CD36 KO mouse model reverses this alteration to reduce dermal scarring. The targeted modulation of CD36 antagonism may be an effective mechanism to limit fibrosis. Nevertheless, our study has limitations. First, we have performed most of the CD36 knockdown experiments in mice. It is necessary to further study the consequences of targeting CD36 in in vivo human scarring conditions and models to define the function and safety of CD36 antagonism. In addition, as a proof of concept, we used SAB to deliver CD36 antagonism in animal models. However, the safety of SAB needs to be fully explored before clinical translation. Next steps would be to validate our mouse data in a large animal porcine model and then perform safety and efficacy trials of various CD36 inhibitors and doses to optimize the potential antifibrotic effect and translation to the clinical bedside.
In summary, our results provide a comprehensive transcriptomic and epigenomic characterization of scarring with an in-depth analysis of HTS, highlighting how JUN mediates scarring. JUN induces scarring by modifying the contribution of specific fibroblast subpopulations. Future work should explore targeted modulation of JUN using CD36 antagonists, such as SAB, to modify specific fibroblast subpopulations in other pathological fibrotic conditions.
MATERIALS AND METHODS
Study design
The goals of this study were (i) to identify HTS-promoting genes, (ii) to understand the physiological role of JUN in HTS scarring, and (iii) to identify the potential therapeutic targets to treat HTS scarring diseases. Human skin tissue samples were obtained from patients undergoing plastic surgery procedures (abdominoplasty and scar revision procedures). All tissue handling was in accordance with guidelines set by the Institutional Review Board (IRB) under protocols for human tissue sampling approved by the Stanford University Ethics Board (IRB #45219). A range of in vitro assays were performed to evaluate proliferation, apoptosis, collagen production, gene expression, and chromatin accessibility of hSc-derived fibroblasts. Murine in vivo models were used to identify HTS scar–promoting genes. Age-matched animals were randomly assigned to control and treatment groups. Samples sizes were calculated on the basis of previous experience. No blinding was performed during the experimental administration of treatments to mice. All mice were maintained and humanely euthanized at predefined study end points in the Stanford University Comparative Medicine Pavilion in accordance with guidelines set by the Stanford University Animal Care and Use Committee and National Institutes of Health guidelines (APLAC#30911 and APLAC#30912).
Statistical analysis
All quantitative data were expressed as the means ± SEM. Comparisons were performed with Student’s t test or one-way analysis of variance (ANOVA) with post hoc analysis using the Bonferroni correction. All statistical tests were conducted using Prism GraphPad (www.graphpad.com/scientific-software/prism/) with significance set to P < 0.05. Graphs were generated using Prism and Microsoft Excel (v.16.33).
Supplementary Material
Acknowledgments:
We acknowledge P. Lovelace and C. Crumpton at the Shared FACS Facility in the Lokey Stem Cell Institute and V. Shokoohi at the Stanford Functional Genomics Facility for technical help with RNA- and ATAC-seq library preparation.
Funding:
This work was supported by the Plastic Surgery Research Foundation (to M.R.B.), the U.K. Fulbright Commission (to M.F.G.), the Hagey Laboratory for Pediatric Regenerative Medicine (to M.T.L.), the Sarnoff Cardiovascular Research Foundation (to A.H.S.), the Gunn/Olivier Research Fund (to M.T.L.), Stinehart-Reed Fund (to M.T.L.), the Wu Tsai Human Performance Alliance, Stanford University (to M.T.L), NIH grant R01 GM116892 (to H.P.L. and M.T.L.), NIH grant RO1GM136659 (to M.T.L.), NIH grant P50-HG007735 (to H.Y.C.), Howard Hughes Medical Institute (to H.Y.C.), NIH grant ROI DE 027346 (to D.C.W. and M.T.L.), Scleroderma Research Foundation Fund (to M.T.L., H.Y.C., and G.W.), F.P Johnson Jr Fund (to G.C.G. and M.T.L.), Scleroderma Research Foundation grant (to G.W.), NIH NHLBI grant SPO 1K08HL143143-01 (to G.W.), Ludwig Institute Fund (to G.W.), Stinehart-Reed Fund (to G.W.), DTRF Foundation Fund (to G.W.), and Esther Ehrman Lazard Faculty Scholarship (to G.W.).
Footnotes
Competing interests: H.Y.C. is a cofounder of Accent Therapeutics and Boundless Bio and advisor to 10x Genomics, Arsenal Bioscience, and Spring Discovery. All other authors declare that they have no competing interests.
SUPPLEMENTARY MATERIALS
Data and materials availability:
All data associated with this study are present in the paper or the Supplementary Materials. The RNA- and ATAC-seq data generated during this study have been deposited in the NCBI’s Gene Expression Omnibus (GEO) and is accessible through GEO series accession number GSE178562, and ChIP-seq data have been deposited in the GEO database under accession code GSE114844.
REFERENCES AND NOTES
- 1.Sen CK, Gordillo GM, Roy S, Kirsner R, Lambert L, Hunt TK, Gottrup F, Gurtner GC, Longaker MT, Human skin wounds: A major and snowballing threat to public health and the economy. Wound Repair Regen. 17, 763–771 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bayat A, McGrouther DA, Ferguson MW, Skin scarring. BMJ 326, 88–92 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gurtner GC, Werner S, Barrandon Y, Longaker MT, Wound repair and regeneration. Nature 453, 314–321 (2008). [DOI] [PubMed] [Google Scholar]
- 4.Larson BJ, Longaker MT, Lorenz HP, Scarless fetal wound healing: A basic science review. Plast. Reconstr. Surg 126, 1172–1180 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Alster TS, Tanzi EL, Hypertrophic scars and keloids. Am. J. Clin. Dermatol 4, 235–243 (2003). [DOI] [PubMed] [Google Scholar]
- 6.Reish RG, Eriksson E, Scar treatments: Preclinical and clinical studies. J. Am. Coll. Surg 206, 719–730 (2008). [DOI] [PubMed] [Google Scholar]
- 7.Singer AJ, Clark RA, Cutaneous wound healing. N. Engl. J. Med 341, 738–746 (1999). [DOI] [PubMed] [Google Scholar]
- 8.Stadelmann WK, Digenis AG, Tobin GR, Physiology and healing dynamics of chronic cutaneous wounds. Am. J. Surg 176, 26S–38S (1998). [DOI] [PubMed] [Google Scholar]
- 9.Driskell RR, Lichtenberger BM, Hoste E, Kretzschmar K, Simons BD, Charalambous M, Ferron SR, Herault Y, Pavlovic G, Ferguson-Smith AC, Watt FM, Distinct fibroblast lineages determine dermal architecture in skin development and repair. Nature 504, 277–281 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rinkevich Y, Walmsley GG, Hu MS, Maan ZN, Newman AM, Drukker M, Januszyk M, Krampitz GW, Gurtner GC, Lorenz HP, Weissman IL, Longaker MT, Identification and isolation of a dermal lineage with intrinsic fibrogenic potential. Science 348, aaa2151 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Januszyk M, Chen K, Henn D, Foster DS, Borrelli MR, Bonham CA, Sivaraj D, Wagh D, Longaker MT, Wan DC, Gurtner GC, Characterization of diabetic and non-diabetic foot ulcers using single-cell RNA-sequencing. Micromachines 11, 815 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wernig G, Chen S-Y, Cui L, Van Neste C, Tsai JM, Kambham N, Vogel H, Natkunam Y, Gilliland DG, Nolan G, Weissman IL, Unifying mechanism for different fibrotic diseases. Proc. Natl. Acad. Sci 114, 4757–4762 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cui L, Chen SY, Lerbs T, Lee JW, Domizi P, Gordon S, Kim YH, Nolan G, Betancur P, Wernig G, Activation of JUN in fibroblasts promotes pro-fibrotic programme and modulates protective immunity. Nat. Commun 11, 2795 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lerbs T, Cui L, King ME, Chai T, Muscat C, Chung L, Brown R, Rieger K, Shibata T, Wernig G, CD47 prevents the elimination of diseased fibroblasts in scleroderma. JCI Insight 5, e140458 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Foster DS, Marshall CD, Gulati GS, Chinta MS, Nguyen A, Salhotra A, Jones RE, Burcham A, Lerbs T, Cui L, King ME, Titan AL, Ransom RC, Manjunath A, Hu MS, Blackshear CP, Mascharak S, Moore AL, Norton JA, Kin CJ, Shelton AA, Januszyk M, Gurtner GC, Wernig G, Longaker MT, Elucidating the fundamental fibrotic processes driving abdominal adhesion formation. Nat. Commun 11, 4061 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sorrell JM, Caplan AI, Fibroblast heterogeneity: More than skin deep. J. Cell Sci 117, 667–675 (2004). [DOI] [PubMed] [Google Scholar]
- 17.Driskell RR, Giangreco A, Jensen KB, Mulder KW, Watt FM, Sox2-positive dermal papilla cells specify hair follicle type in mammalian epidermis. Development 136, 2815–2823 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schulz J-N, Plomann M, Sengle G, Gullberg D, Krieg T, Eckes B, New developments on skin fibrosis-Essential signals emanating from the extracellular matrix for the control of myofibroblasts. Matrix Biol. 68, 522–532 (2018). [DOI] [PubMed] [Google Scholar]
- 19.Naik S, Larsen SB, Cowley CJ, Fuchs E, Two to tango: Dialog between immunity and stem cells in health and disease. Cell 175, 908–920 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Larouche J, Sheoran S, Maruyama K, Martino MM, Immune regulation of skin wound healing: Mechanisms and novel therapeutic targets. Adv. Wound Care 7, 209–231 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu J, Zhao B, Zhu H, Pan Q, Cai M, Bai X, Li X, Hu X, Zhang M, Shi J, Zheng Z, Yang A, Hu D, Wnt4 negatively regulates the TGF-β1-induced human dermal fibroblast-to-myofibroblast transition via targeting Smad3 and ERK. Cell Tissue Res. 379, 537–548 (2020). [DOI] [PubMed] [Google Scholar]
- 22.Lynn RC, Weber EW, Sotillo E, Gennert D, Xu P, Good Z, Anbunathan H, Lattin J, Jones R, Tieu V, Nagaraja S, Granja J, de Bourcy CFA, Majzner R, Satpathy AT, Quake SR, Monje M, Chang HY, Mackall CL, c-Jun overexpression in CAR T cells induces exhaustion resistance. Nature 576, 293–300 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vaughan DE, PAI-1 and atherothrombosis. J. Thromb. Haemost 3, 1879–1883 (2005). [DOI] [PubMed] [Google Scholar]
- 24.Plasari G, Calabrese A, Dusserre Y, Gronostajski RM, McNair A, Michalik L, Mermod N, Nuclear factor I-C links platelet-derived growth factor and transforming growth factor β1 signaling to skin wound healing progression. Mol. Cell. Biol 29, 6006–6017 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Qiao Y, He H, Jonsson P, Sinha I, Zhao C, Dahlman-Wright K, AP-1 is a key regulator of proinflammatory cytokine TNFα-mediated triple-negative breast cancer progression. J. Biol. Chem 291, 5068–5079 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Maiese K, WISP1: Clinical insights for a proliferative and restorative member of the CCN family. Curr. Neurovasc. Res 11, 378–389 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shimamoto K, Tanimoto K, Fukazawa T, Nakamura H, Kanai A, Bono H, Ono H, Eguchi H, Hirohashi N, GLIS1, a novel hypoxia-inducible transcription factor, promotes breast cancer cell motility via activation of WNT5A. Carcinogenesis 41, 1184–1194 (2020). [DOI] [PubMed] [Google Scholar]
- 28.Li G, Guo GL, Farnesoid X receptor, the bile acid sensing nuclear receptor, in liver regeneration. Acta Pharm. Sin. B 5, 93–98 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Talle M, Rao P, Westberg E, Allegar N, Makowski M, Mittler R, Goldstein G, Patterns of antigenic expression on human monocytes as defined by monoclonal antibodies. Cell. Immunol 78, 83–99 (1983). [DOI] [PubMed] [Google Scholar]
- 30.Asch AS, Barnwell J, Silverstein RL, Nachman RL, Isolation of the thrombospondin membrane receptor. J. Clin. Invest 79, 1054–1061 (1987). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Swerlick RA, Lee KH, Wick TM, Lawley TJ, Human dermal microvascular endothelial but not human umbilical vein endothelial cells express CD36 in vivo and in vitro. J. Immunol 148, 78–83 (1992). [PubMed] [Google Scholar]
- 32.Febbraio M, Hajjar DP, Silverstein RL, CD36: A class B scavenger receptor involved in angiogenesis, atherosclerosis, inflammation, and lipid metabolism. J. Clin. Invest 108, 785–791 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tandon NN, Kralisz U, Jamieson G, Identification of glycoprotein IV (CD36) as a primary receptor for platelet-collagen adhesion. J. Biol. Chem 264, 7576–7583 (1989). [PubMed] [Google Scholar]
- 34.Kim YM, Huh JS, Lim Y, Cho M, Soy isoflavone glycitin (4′-hydroxy-6-methoxyisoflavone-7-D-glucoside) promotes human dermal fibroblast cell proliferation and migration via TGF-β signaling. Phytother. Res 29, 757–769 (2015). [DOI] [PubMed] [Google Scholar]
- 35.Tredget EE, Nedelec B, Scott PG, Ghahary A, Hypertrophic scars, keloids, and contractures. Surg. North Am 77, 701–730 (1997). [DOI] [PubMed] [Google Scholar]
- 36.Li Y, Liu H, Liang Y, Peng P, Ma X, Zhang X, DKK3 regulates cell proliferation, apoptosis and collagen synthesis in keloid fibroblasts via TGF-β1/Smad signaling pathway. Biomed. Pharmacother 91, 174–180 (2017). [DOI] [PubMed] [Google Scholar]
- 37.Wang Y, Huang G, Wang Z, Qin H, Mo B, Wang C, Elongation factor-2 kinase acts downstream of p38 MAPK to regulate proliferation, apoptosis and autophagy in human lung fibroblasts. Exp. Cell Res 363, 291–298 (2018). [DOI] [PubMed] [Google Scholar]
- 38.Lu J, Liu Q, Wang L, Tu W, Chu H, Ding W, Jiang S, Ma Y, Shi X, Pu W, Zhou X, Jin L, Wang J, Wu W, Increased expression of latent TGF-β-binding protein 4 affects the fibrotic process in scleroderma by TGF-β/SMAD signaling. Lab. Investig 97, 591–601 (2017). [DOI] [PubMed] [Google Scholar]
- 39.Xu X, Chen Z, Zhu X, Wang D, Liang J, Zhao C, Feng X, Wang J, Zou H, Sun L, S100A9 aggravates bleomycin-induced dermal fibrosis in mice via activation of ERK1/2 MAPK and NF-κB pathways. Iran. J. Basic Med. Sci 21, 194–201 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bao Y, Wang L, Xu Y, Yang Y, Wang L, Si S, Cho S, Hong B, Salvianolic acid B inhibits macrophage uptake of modified low density lipoprotein (mLDL) in a scavenger receptor CD36-dependent manner. Atherosclerosis 223, 152–159 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ma L, Tang L, Yi Q, Salvianolic acids: Potential source of natural drugs for the treatment of fibrosis disease and cancer. Front. Pharmacol 10, 97 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yu F, Lu Z, Chen B, Wu X, Dong P, Zheng J, Salvianolic acid B-induced microRNA-152 inhibits liver fibrosis by attenuatingDNMT1-mediated Patched1 methylation. J. Cell. Mol. Med 19, 2617–2632 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu Q, Chu H, Ma Y, Wu T, Qian F, Ren X, Tu W, Zhou X, Jin L, Wu W, Wang J, Salvianolic acid B attenuates experimental pulmonary fibrosis through inhibition of the TGF-β signaling pathway. Sci. Rep 6, 27610 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lin SJ, Lee IT, Chen YH, Lin FY, Sheu LM, Ku HH, Shiao MS, Chen JW, Chen YL, Salvianolic acid B attenuates MMP-2 and MMP-9 expression in vivo in apolipoprotein-E-deficient mouse aorta and in vitro in LPS-treated human aortic smooth muscle cells. J. Cell. Biochem 100, 372–384 (2007). [DOI] [PubMed] [Google Scholar]
- 45.Wang Q-L, Tao Y-Y, Yuan J-L, Shen L, Liu C-H, Salvianolic acid B prevents epithelial-to-mesenchymal transition through the TGF-β1 signal transduction pathway in vivo and in vitro. BMC Cell Biol. 11, 31 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Akhmetshina A, Palumbo K, Dees C, Bergmann C, Venalis P, Zerr P, Horn A, Kireva T, Beyer C, Zwerina J, Schneider H, Sadowski A, Riener M-O, MacDougald OA, Distler O, Schett G, Distler JH, Activation of canonical Wnt signalling is required for TGF-β-mediated fibrosis. Nat. Commun 3, 735 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schneiderhan W, Schmid-Kotsas A, Zhao J, Grünert A, Nüssler A, Weidenbach H, Menke A, Schmid RM, Adler G, Bachem MG, Oxidized low-density lipoproteins bind to the scavenger receptor, CD36, of hepatic stellate cells and stimulate extracellular matrix synthesis. Hepatology 34, 729–737 (2001). [DOI] [PubMed] [Google Scholar]
- 48.Wang P, Xu S, Li W, Wang F, Yang Z, Jiang L, Wang Q, Huang M, Zhou P, Salvianolic acid B inhibited PPARγ expression and attenuated weight gain in mice with high-fat diet-induced obesity. Cell. Physiol. Biochem 34, 288–298 (2014). [DOI] [PubMed] [Google Scholar]
- 49.Mascharak S, desJardins-Park HE, Davitt MF, Griffin M, Borrelli MR, Moore AL, Chen K, Duoto B, Chinta M, Foster DS, Shen AH, Januszyk M, Kwon SH, Wernig G, Wan DC, Lorenz HP, Gurtner GC, Longaker MT, Preventing Engrailed-1 activation in fibroblasts yields wound regeneration without scarring. Science 372, eaba2374 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Avouac J, Palumbo K, Tomcik M, Zerr P, Dees C, Horn A, Maurer B, Akhmetshina A, Beyer C, Sadowski A, Schneider H, Shiozawa S, Distler O, Schett G, Allanore Y, Distler JH, Inhibition of activator protein 1 signaling abrogates transforming growth factor β-mediated activation of fibroblasts and prevents experimental fibrosis. Arthritis Rheum. 64, 1642–1652 (2012). [DOI] [PubMed] [Google Scholar]
- 51.Zhong S, Zhao L, Wang Y, Zhang C, Liu J, Wang P, Zhou W, Yang P, Varghese Z, Moorhead JF, Chen Y, Ruan XZ, Cluster of differentiation 36 deficiency aggravates macrophage infiltration and hepatic inflammation by upregulating monocyte chemotactic protein-1 expression of hepatocytes through histone deacetylase 2-dependent pathway. Antioxid. Redox Signal 27, 201–214 (2017). [DOI] [PubMed] [Google Scholar]
- 52.Yang YL, Lin SH, Chuang LY, Guh JY, Liao TN, Lee TC, Chang WT, Chang FR, Hung MY, Chiang TA, Hung CY, CD36 is a novel and potential anti-fibrogenic target in albumin-induced renal proximal tubule fibrosis. J. Cell. Biochem 101, 735–744 (2007). [DOI] [PubMed] [Google Scholar]
- 53.Okamura DM, Pennathur S, Pasichnyk K, Lopez-Guisa JM, Collins S, Febbraio M, Heinecke J, Eddy AA, CD36 regulates oxidative stress and inflammation in hypercholesterolemic CKD. J. Am. Soc. Nephrol 20, 495–505 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lua I, Balog S, Yanagi A, Tateno C, Asahina K, Loss of lysophosphatidic acid receptor 1 in hepatocytes reduces steatosis via down-regulation of CD36. Prostaglandins Other Lipid Mediat. 156, 106577 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hakvoort TE, Altun V, Ramrattan RS, van der Kwast TH, Benner R, van Zuijlen PP, Vloemans AF, Prens EP, Epidermal participation in post-burn hypertrophic scar development. Virchows Arch. 434, 221–226 (1999). [DOI] [PubMed] [Google Scholar]
- 56.Suga H, Rennert RC, Rodrigues M, Sorkin M, Glotzbach JP, Januszyk M, Fujiwara T, Longaker MT, Gurtner GC, Tracking the elusive fibrocyte: Identification and characterization of collagen-producing hematopoietic lineage cells during murine wound healing. Stem Cells 32, 1347–1360 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Galiano RD, Michaels J, Dobryansky M, Levine JP, Gurtner GC, Quantitative and reproducible murine model of excisional wound healing. Wound Repair Regen. 12, 485–492 (2004). [DOI] [PubMed] [Google Scholar]
- 58.Zeng Z, Zhang H, Wang X, Liu K, Li T, Sun S, Li H, Salvianolic acid B suppresses cell proliferation and induces apoptosis in osteosarcoma through p38-mediated reactive oxygen species generation. Oncol. Lett 15, 2679–2685 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Morrison EJ, Champagne DP, Dzieciatkowska M, Nemkov T, Zimring JC, Hansen KC, Guan F, Huffman DM, Santambrogio L, D’Alessandro A, Parabiosis incompletely reverses aging-induced metabolic changes and oxidant stress in mouse red blood cells. Nutrients 11, 1337 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zarnegar BJ, Flynn RA, Shen Y, Do BT, Chang HY, Khavari PA, irCLIP platform for efficient characterization of protein–RNA interactions. Nat. Methods 13, 489–492 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ, Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 10, 1213–1218 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Buenrostro JD, Wu B, Chang HY, Greenleaf WJ, ATAC-seq: A method for assaying chromatin accessibility genome-wide. Curr. Protoc. Mol. Biol 109, 21.29.1–21.29.9 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Love MI, Huber W, Anders S, Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Huang DW, Sherman BT, Lempicki RA, Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 37, 1–13 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Huang DW, Sherman BT, Stephens R, Baseler MW, Lane HC, Lempicki RA, David gene ID conversion tool. Bioinformation 7, 428–430 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK, Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, Wenger AM, Bejerano G, GREAT improves functional interpretation of cis-regulatory regions. Nat. Biotechnol 28, 495–501 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data associated with this study are present in the paper or the Supplementary Materials. The RNA- and ATAC-seq data generated during this study have been deposited in the NCBI’s Gene Expression Omnibus (GEO) and is accessible through GEO series accession number GSE178562, and ChIP-seq data have been deposited in the GEO database under accession code GSE114844.