

SUMMARY
Regeneration is the holy grail of tissue repair, but skin injury typically yields fibrotic, non-functional scars. Developing pro-regenerative therapies requires rigorous understanding of the molecular progression from injury to fibrosis or regeneration. Here, we report the divergent molecular events driving skin wound cells toward scarring or regenerative fates. We profile scarring versus YAP-inhibition-induced wound regeneration at the transcriptional (single-cell RNA sequencing), protein (timsTOF proteomics), and tissue (extracellular matrix ultrastructural analysis) levels. Using cell-surface barcoding, we integrate these data to reveal fibrotic and regenerative “molecular trajectories” of healing. We show that disrupting YAP mechanotransduction yields regenerative repair by fibroblasts with activated Trps1 and Wnt signaling. Finally, via in vivo gene knockdown and overexpression in wounds, we identify Trps1 as a key regulatory gene that is necessary and partially sufficient for wound regeneration. Our findings serve as a multi-omic map of wound regeneration and could have therapeutic implications for pathologic fibroses.
Graphical abstract
In brief
Using integrated analysis of single-cell transcriptomic, proteomic, and tissue ultrastructural data, Mascharak, Talbott, Januszyk, et al. define divergent molecular trajectories of scarring versus wound regeneration in the context of YAP inhibition. They reveal a key role for Trps1 in driving wound regeneration and show that activated Wnt and Trps1 signaling are hallmarks of regenerative healing.
INTRODUCTION
Fibrosis, replacement of functional tissue with non-functional connective tissue, is the end result of damage to virtually every human organ. Collectively, fibroses are implicated in 45% of all deaths in the U.S. (Wynn, 2004). Scarring is fibrosis following dermal injury, a late evolutionary adaptation to prioritize speed in healing. Scars lack skin’s hair or glands and, thus, normal thermoregulatory or barrier function. Despite the enormous medical burden of fibrosis, current therapies remain lacking, due, in part, to limited understanding of the fundamental mechanisms differentiating regenerative from fibrotic healing (Foster et al., 2021; Griffin et al., 2020; Mascharak et al., 2020; Reish and Eriksson, 2008).
We previously reported that the En-1-positive fibroblast (EPF) lineage drives dorsal dermal fibrosis in mice (Rinkevich et al., 2015). More recently, we showed that EPFs can arise via En-1-negative fibroblasts (ENFs) activating En-1 in response to postnatal wound mechanics (Mascharak et al., 2021). Blocking mechanotransduction (verteporfin, Yes-associated protein [YAP] inhibitor) prevents this conversion and yields ENF-mediated regeneration, defined by the restoration of: (1) dermal appendages (hair follicles [HF], glands); (2) extracellular matrix (ECM) architecture identical to that of unwounded (UW) skin; and (3) tensile strength of UW skin.
Here, we use a multi-omic approach to compare regenerative and fibrotic skin repair over time at the transcriptomic, proteomic, and ECM ultrastructural levels. We apply cell barcoding to integrate data with single-mouse resolution and impute bulk (proteomic, ECM ultrastructure) onto single-cell (scRNA-seq) data. We reveal two repair trajectories defined by distinct fibroblast molecular motifs: one is fibrotic, dominated by mechanical signaling and the other is regenerative, characterized by developmental pathways. We identify genes that discriminate between these trajectories and, via in vivo gene knockdown (KD)/overexpression (OE), identify Trps1 as necessary and partially sufficient for wound regeneration. Our multi-omic roadmap identifies the biology behind a novel example of adult mammalian wound regeneration and can be a resource for future studies on regeneration and fibrosis.
RESULTS
Multimodal analysis of fibrotic and regenerative wounds
As injury repair comprises multiple phases, it is critical to examine the cellular and molecular dynamics of healing over time. We analyzed mouse wounds and mouse skin at seven time points: UW skin, postoperative day (POD) 2 (inflammation), POD 7 (fibroblast proliferation), POD 10, POD 14 (wounds re-epithelialized, fibroblasts producing ECM), POD 21, and POD 30 (fibroblasts remodeling ECM). We used a splinted excisional wound model, which prevents the rapid wound contraction typical of loose-skinned mice, thus inducing human-like healing kinetics (Foster et al., 2021; Mascharak et al., 2021). Immediately after wounding, the wound bed was injected with verteporfin or vehicle (phosphate-buffered saline [PBS]) control.
Control wounds formed distinct fibrotic scars, which grossly were bare areas and had dense, linearly aligned ECM fibers without secondary elements (HF, glands); in contrast, by POD 30, verteporfin-treated wounds regrew levels of HF and glands comparable with those in UW skin and had less-dense, more randomly oriented ECM fibers (Figures 1A and S1A; hairless regions surrounding wounds result from splinting and are unrelated to the experiment, Figures S1B and S1C top two panels). Confirming that regeneration did not reflect off-target effects of verteporfin, a comparable phenotype was observed with En-1 fibroblast lineage-targeted YAP knockout (En-1Cre-ERT;YAPfl/fl mice; Figure S1G–H top panels).
Figure 1. Multimodal interrogation of scarring and regenerative wound repair.
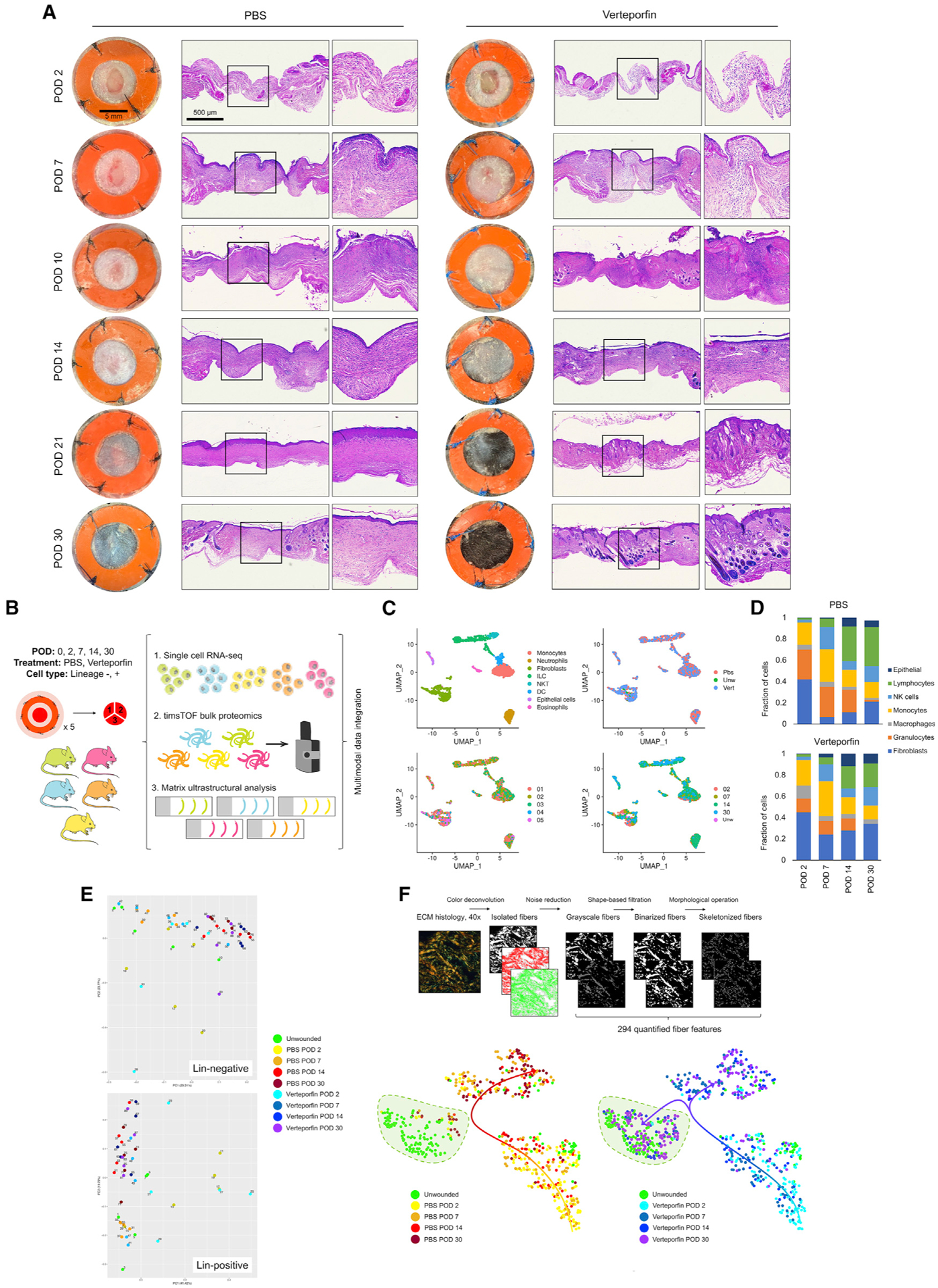
(A) Gross photos (left columns) and low-power hematoxylin and eosin (H&E) histology (right columns) of PBS and verteporfin-treated wounds at indicated PODs.
(B) Experimental strategy for mouse-paired multimodal analysis of wound healing over time.
(C) Manifold projection of scRNA-seq cells colored by cell type (top left), treatment group (top right), individual mouse (bottom left), and POD (bottom right).
(D) Proportion of cell types in PBS and verteporfin-treated wounds by time point.
(E) Principal components mapping of proteomic data for Lin− (fibroblast, top) and Lin+ (non-fibroblast, bottom) cells.
(F) Top, schematic outline of image processing and analysis pipeline to quantify 294 ECM parameters. Bottom, t-SNE plots of control (left) and verteporfin (right) wounds over time. Green shaded region, UW skin cluster; overlaid arrows, ECM evolution over time.
To study cellular and molecular dynamics of regenerative and fibrotic wounds, we wounded and analyzed five mice (two wounds pooled/mouse) from each of five key time points (UW, POD 2, 7, 14, and 30) and treatment conditions (control, verteporfin; Figure 1B). One-third of tissue per mouse was used for histology, and the remainder was cell-sorted per established fluorescence-activated cell sorting (FACS) strategy to isolate fibroblasts (Lin−) and other cells (primarily immune cells; Lin+). Half of the cells were subjected to proteomic analysis: peptides purified from the Lin− and Lin+ cells from each mouse underwent timsTOF mass spectrometry (high-resolution shotgun sequencing). The remaining cells were subjected to scRNA-seq; each sample was tagged with hashtag oligonucleotide (HTO; unique 12-base oligomers) cell-surface barcodes to enable the linkage of scRNA-seq cells with companion proteomic and histologic samples from the same mouse.
We analyzed a total of 2,986 cells by scRNA-seq. Cells formed distinct transcriptomic clusters in uniform manifold approximation and projection (UMAP)-space based on cell type; using data demultiplexing, we also analyzed the clusters based on mouse, treatment, and POD of origin (Figure 1C).
Fibroblasts were modestly increased in verteporfin-treated wounds; however, cell proportions were generally similar between the control and verteporfin wounds (Figure 1D), suggesting that verteporfin may induce regeneration by altering cell phenotypes rather than relative representation (or by altering the representation of subpopulations within cell types).
We next examined timsTOF profiles. On principal component analysis, Lin− and Lin+ proteomic specimens differed strongly by time point, whereas the differences between PBS and verteporfin specimens were more narrowly focused (Figures 1E and S2A–S2C), suggesting that proteomic analysis could capture coherent fibrotic changes among broader repair dynamics. Gene set enrichment analysis (GSEA) revealed that PBS wound fibroblasts were enriched for mechanical activation (e.g., cytoskeleton organization and actin filament terms), compared with verteporfin wounds that were associated with keratins and metabolic processes (Figures S2A and S2B). We also saw notable increases in multiple collagens (e.g., Col2a1 and Col6a2) by POD 30 and significant changes in other fibrotic markers, such as Col1a1 (Hua et al., 2020; Terashima et al., 2019), Diablo (Guicciardi et al., 2017), and Cdh11 (Pedroza et al., 2017; Schneider et al., 2012) that selectively changed in response to verteporfin (Figure S2C).
Finally, as ECM architecture/organization is a key determinant of physical tissue properties, we quantified the ECM ultrastructure from wounded and UW skin using a newly developed image-processing algorithm (Figure 1F top). We quantified 294 parameters from connective tissue histology sections throughout each sample and performed cluster analysis by t-distributed stochastic neighbor embedding (t-SNE). Control wound ECM diverged dramatically from UW ECM over time (Figure 1F bottom left), evidencing fibrotic scarring. In contrast, POD 14/30 verteporfin wounds overlapped with UW skin (Figure 1F, bottom right), suggesting that healing following verteporfin treatment yielded UW-like ECM. With either treatment, the greatest differences were seen between POD 7–14, consistent with ECM deposition largely occurring during this time. We also developed an ECM score ranging from −1 to +1 (−1, scar; +1, UW skin) by first using our algorithm to train a neural network on ECM histology from mouse scars (PBS) and UW skin and then using the trained neural network to score experimental histology. ECM scores revealed recovery of UW-like ECM in verteporfin wounds, most notable during the remodeling phase (POD 14–30; Figure S1C bottom).
Identifying molecular trajectories of repair
Having established our ability to profile wound dynamics with scRNA-seq, proteomic, and ECM ultrastructural analyses, we sought to apply these methods to identify divergent mechanisms of regeneration and fibrosis. As fibroblasts are highly mechanosensitive and the primary drivers of scarring (Aarabi et al., 2007; Barnes et al., 2018; Wong et al., 2011), we hypothesized that verteporfin promotes regeneration by altering fibroblast phenotypes and focused our scRNA-seq analysis on fibroblasts. We used Monocle3 to generate temporally informed, treatment-agnostic cell trajectories (Figure 2A). An optimal manifold was selected to maximally capture the changes among PODs in a treatment-agnostic fashion. The resulting trajectory had two bifurcating branches, with early (POD 2) wound fibroblasts near the branchpoint and late wound fibroblasts at the branch ends (Figure 2B first panel). Interestingly, UW skin fibroblasts were almost exclusively in the bottom branch (Figure 2B second panel), which was also relatively enriched with verteporfin wound fibroblasts (POD 30 wounds, 80.7% of verteporfin fibroblasts in bottom arm versus 40.0% of PBS fibroblasts; *p = 0.015, Fisher’s exact test). Thus, we postulated that the bottom arm represented a regenerative repair trajectory, with fibroblasts progressing toward an UW-like state, whereas the upper arm (enriched in PBS/lacking UW skin fibroblasts) was a comparatively fibrotic trajectory. Of note, some PBS wound fibroblasts were found in the regenerative arm (Figure 2B second panel), high-lighting the notion that regeneration and fibrosis may not be cell-ularly mutually exclusive; cells with pro-regenerative activity may exist in scarring wounds, but in the absence of intervention (e.g., YAP inhibition), pro-fibrotic molecular programing may override this and lead to the dominant scarring phenotype.
Figure 2. Transcriptomic trajectory analysis of fibroblasts in pseudotime.

(A) Schematic outlining manifold generation, optimization, and branchpoint analysis for fibroblasts in Monocle3.
(B) Fibroblast manifold colored by POD (left), treatment group (middle-left), pseudotime value (middle-right), and Louvain-based (Seurat) cluster (right).
(C–E) mRNA expression of selected genes (C) and functional enrichment analyses of genes (D) and proteins (E) positively correlated with pseudotime.
(F–H) As in (C–E), but negatively correlated with pseudotime.
(I and J) SCENIC analysis of regulons corresponding to each fibroblast cluster.
(K) RNA velocity analysis of fibroblasts from all conditions.
(L) Top, fibroblast manifold projection colored by CytoTRACE score; bottom, CytoTRACE scores for cells in each arm.
(M) IF histology for YAP (red) and Trps1 (green; left) or Dpp4 (CD26; far right) in UW skin and PBS- or verteporfin-treated wounds at indicated time points.
(N) Quantification of Trps1 nuclear localization by Biodock AI automated analysis.
We also analyzed two other wound-relevant cell populations: myeloid cells (granulocytes, monocytes, and macrophages) and lymphocytes (natural killer [NK], T, and B cells). Upon similar analysis of myeloid and lymphoid cells, no manifolds diverged by treatment condition (Figure S3). Myeloid cell pseudotime analysis (Figures S3A–S3C) showed enrichment for inflammatory genes (e.g., Ccl6, Ccl9, and Cxcl2) at an early stage (Figure S3D) and antigen presentation genes (e.g., H2 complex) at later PODs (Figure S3E). However, these genes were similarly expressed in PBS and verteporfin cells, indicating that the analysis did not differentiate between fibrotic and regenerative healing. Lymphoid cell pseudotime analysis (Figures S3F–S3H) also did not differentiate between fibrotic and regenerating wounds, instead reflecting early (Figure S3I) and late (Figure S3J) infiltration of NK and B/T cells, respectively. Collectively, these data were consistent with verteporfin promoting regeneration by modulating fibroblasts rather than immune cells.
We next sought to identify distinct signatures of the putative fibrotic and regenerative fibroblast trajectories. Louvain-based (Seurat) clustering identified six fibroblast clusters: clusters 0 and 4 made up the “regenerative” arm; 1, 2, and 5 made up the “fibrotic” arm; and 3 comprised mostly UW skin fibroblasts (Figures 2B last panel and 3A). We calculated pseudotemporal Pearson correlations for each gene along our primary trajectory of interest (Figure 2B third panel). The fibrotic arm (clusters 1, 2, and 5) was enriched for known pro-fibrotic markers like Dpp4 (marks scarring EPF lineage [Rinkevich et al., 2015]; notably, most Dpp4+ fibroblasts in the regenerative arm were from UW skin, where EPFs are the majority of dorsal fibroblasts), Fap, and Gpx3 (Figures 2C and 3B). Fibrotic trajectory cells also expressed more Fgf2, Il6, Ccl2, Myc, and Tead1, whose upregulation in fibroblasts has been implicated in focal adhesion kinase (FAK)-mediated organ fibrosis and tumor stroma deposition (Murphy et al., 2020; Wong et al., 2011). GSEA on the top 1% of genes positively correlated with pseudotime revealed ECM deposition (e.g., ECM structural constituent and glycosamino-glycan [GAG] binding), growth factor, mechanotransduction (cell adhesion molecule binding and integrin binding), and fibrosis-related Reactome terms like mitogen-activated protein kinase (MAPK)/Ras signaling and focal adhesion (Figures 2D and 3D), supporting a classical pro-fibrotic phenotype.
Figure 3. Pseudotime and CytoTRACE analysis of fibroblasts in scarring and regeneration.
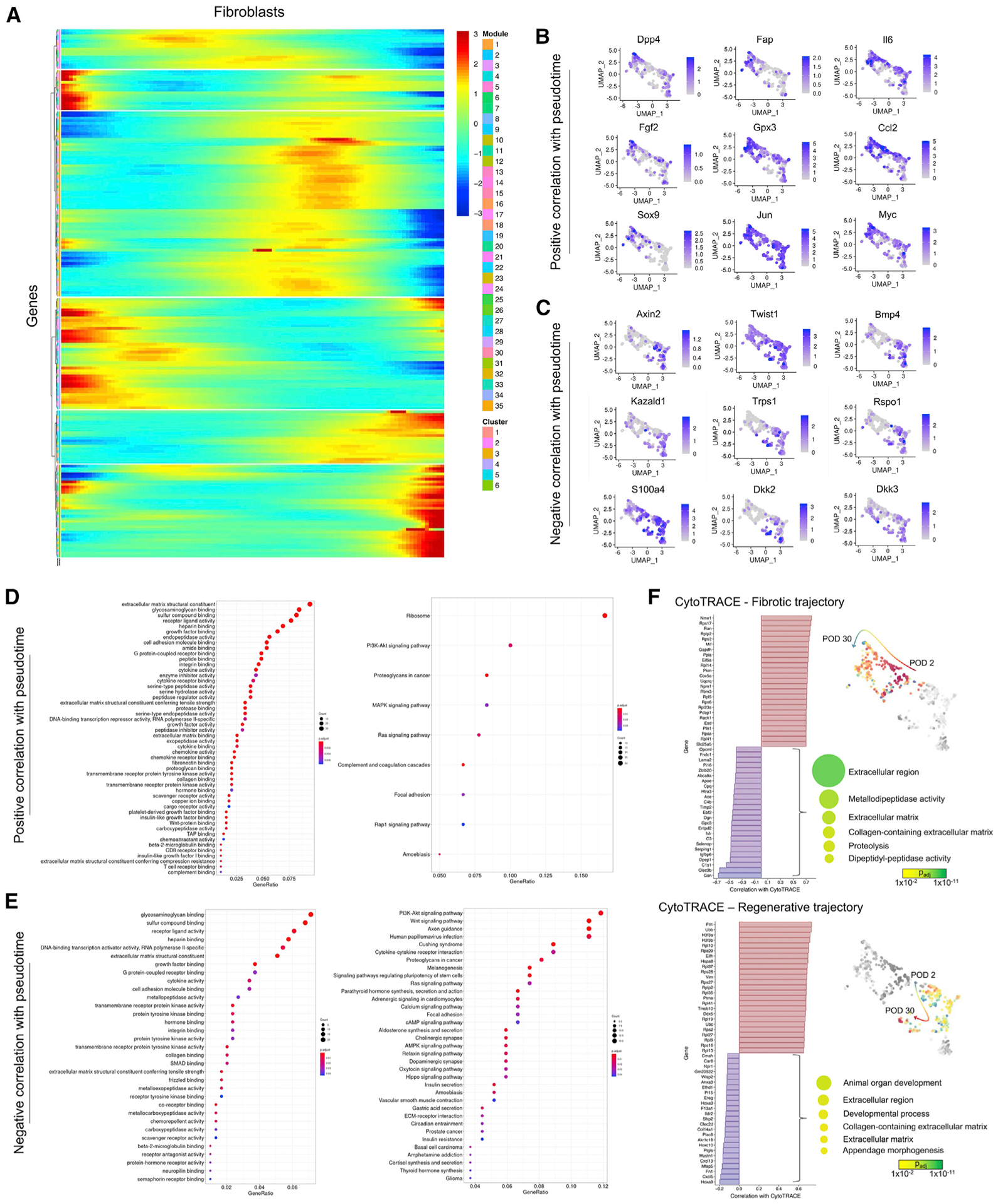
(A) Fibroblast clustering by pseudotemporal expression pattern.
(B and C) mRNA expression feature plots for selected genes positively (B) and negatively (C) correlated with pseudotime.
(D and E) EnrichR gene ontology analysis on top 1% of genes positively (D) or negatively (E) correlated with pseudotime.
(F) Waterfall plots showing genes positively (red) and negatively (blue) correlated with CytoTRACE score for cells in the fibrotic (top) and regenerative (bottom) trajectories. Right panels, functional enrichment results for genes correlated with decreasing CytoTRACE score (i.e., with fibroblast differentiation).
We next examined genes most strongly anticorrelated with pseudotime. Consistent with the regenerative phenotype over-represented in this branch, we found multiple development-/regeneration-related genes (Figures 2F and 3C), including genes previously implicated in regenerative injury responses like Bmp4 (mammalian digit tip regeneration [Han et al., 2003]) and Kazald1 (axolotl limb regeneration/blastema formation [Bryant et al., 2017]). We also identified several genes involved in Wnt signaling (important in HF growth and development [Ito et al., 2007]), including Wnt targets Axin2 and Twist1; Wnt agonist Rspo1; Dkk2 and Dkk3, involved in embryonic skin development (Song et al., 2018); and Wnt pathway regulator Trps1, implicated in HF morphogenesis (Fantauzzo and Christiano, 2012). By GSEA on the top 1% of genes anticorrelated with pseudotime, the regenerative arm was enriched for bone morphogenetic protein (BMP) and Wnt-related terms like Frizzled binding; enriched Reactome terms included multiple Wnt-related terms and “signaling pathways regulating pluripotency of stem cells” (Figures 2G and 3E). We next used the SCENIC platform to compare trajectories based on gene regulatory networks (Aibar et al., 2017) (Figure 2I). Trps1, Gli1, and Twist1 regulons were more activated in the regenerative arm (Figure 2J), further supporting Wnt activation. We also analyzed our scRNA-seq data using CellChat, which uses known ligand-receptor interactions to infer intercellular communication (Jin et al., 2021) (Figures 4A and 4B). CellChat highlighted increased pro-inflammatory signaling (e.g., CXCL and IL6) involving scar (PBS) fibroblasts and non-canonical Wnt signaling in the regenerative arm (Figures 4C–4E). These unique functional enrichment terms, combined with lower Dpp4 expression in this arm (Figure 2C, top left), implied a distinct molecular healing trajectory orchestrated by non-scarring ENFs.
Figure 4. CellChat analysis of fibroblast cell interactions.

(A) Strength of ligand-receptor interactions between cell population pairs. Edge width is proportional to the number of ligand-receptor pairs.
(B) As in (A), subset by cell population.
(C) Heatmaps showing relative importance of each cell group based on the computed network centrality measures of indicated signaling networks (ncWNT, non-canonical Wnt).
(D and E) Inferred outgoing (D) and incoming (E) communication patterns for secreting cells, showing correspondence between inferred latent patterns and cell groups (left) and signaling pathways (right).
We next applied RNA velocity analysis, which predicts cells’ future transcriptomic states based on relative prevalence of spliced/unspliced mRNA to infer the molecular dynamics among cells (Bergen et al., 2020). The fibrotic and regenerative arms had opposing RNA velocity directionalities, with vectors in the fibrotic arm directed toward increasing PODs and those in the regenerative arm directed toward earlier time point cells (Figure 2K). We also applied CytoTRACE, a computational pipeline that predicts individual cell differentiation states based on the distribution of unique mRNA transcripts (Gulati et al., 2020). UW skin fibroblasts were most differentiated (lower CytoTRACE scores), whereas wound fibroblasts were relatively less differentiated (higher scores; Figure 2L). Fibrotic arm fibroblasts were less differentiated overall and increased in differentiation (decreasing scores) with increasing POD, whereas regenerative arm cells decreased in differentiation along the trajectory, suggesting increased developmental potential as they progressed through healing. We next separately analyzed the clusters comprising each trajectory and examined the genes most correlated with CytoTRACE scores along each branch. Decreasing scores over time in the fibrotic arm were largely driven by ECM-related genes (Figure 3F top), consistent with differentiation toward a mature scar fibroblast fate. On the other hand, increasing scores over time in the regenerative arm were explained by de-differentiation away from a scarring fate (Figure 3F bottom), consistent with HF and ECM regeneration being most prominent later in healing (POD 14/30; Figures 1 and S1C). Collectively, CytoTRACE supported the opposing cell trajectories on RNA velocity analysis and independently supported fibrotic versus regenerative arm phenotypes.
Multimodal data integration to analyze wound repair trajectories
For cross-platform analysis, we first attempted to broadly correlate fibroblast (Lin−) proteomic data with associated scRNA-seq data. Consistent with previous literature (Brunner et al., 2021), individual genes were not directly predictive of protein copy number (average correlation < 0.5), but genes and proteins correlated at the pathway level; for example, fibrotic trajectory cells had similar pathway enrichment for RNA and timsTOF data (e.g., mechanical activation; Figure S2A). Next, by leveraging mouse-to-mouse variability and our HTO barcoding system (Figure 1B), we used imputation analysis to map protein expression onto cells in our scRNA-seq dataset based on shared mouse of origin. Consistent with scRNA-seq, the expression of mechanical signaling proteins like Ankrd1 (well-validated readout of YAP activity [Elster et al., 2018]) and Dag1 was higher in the fibrotic trajectory at POD 2/7 (Figure S2D). GSEA of proteins enriched in the fibrotic arm showed terms related to growth factor signaling, mechanical signaling, and contractility and strong enrichment of ECM deposition/organization and ECM components (collagens, fibulin [Fbln1], and fibronectin [Fn1]; Figure 2E). In contrast, the regenerative arm had a lack of mechanical signaling-, contractility-, or ECM-related terms, consistent with scRNA-seq (Figures 2H and S2E).
We next correlated scRNA-seq with variation in ECM architecture and HF/gland counts. For ECM, we used the score based on our ultrastructural quantification algorithm (−1, scar ECM; +1, UW skin). Overall, the regenerative arm correlated with UW-like and the fibrotic arm to scar-like ECM (Figure S1D bottom). HF and glands were associated with fibroblasts in both arms, but, notably, cluster 5 (fibrotic arm) was associated with absent HF/glands and scar-like ECM (Figure S1D top and middle). Cluster 5 cells also highly expressed Dpp4, Il6, and Ccl2, characteristic of scarring fibroblasts (Figure 3B).
We then performed unsupervised analysis of ECM parameters from PBS- and verteporfin-treated wounds at POD 0 (UW), 2, 7, 14, and 30. We identified seven distinct clusters of matrix features that differentiated samples (Figure S4A; each cluster represents a unique “module” of ECM features characteristic of images in that cluster) and analyzed the enrichment of modules across wound conditions (Figure S4B). Matrix clusters 5 and 6 were most enriched at POD 2, suggesting characteristic features of provisional wound matrix (Figure S4B, first row). Consistent with this identity, when we extracted images with high enrichment scores for these modules (i.e., exemplary of that set of ECM features), they had sparse, short, immature collagen fibers (Figure S4B, first row). Several parameter clusters appeared to differentiate fibrotic versus regenerative wounds at different phases. Matrix cluster 1 was enriched in POD 14/30 PBS wounds; representative images had a typical scar-like appearance, with dense, linearly aligned collagen fibers (Figure S4B, fourth row). Matrix clusters 3 and 4 were most characteristic of UW skin and also strongly enriched in POD 30 verteporfin wounds; representative cluster 3 images had short, broad ECM fibers with a basketweave pattern typical of regenerated superficial dermis (Figure S4B, bottom row left), whereas cluster 4 images had aligned, ribbon-shaped fibers consistent with deep dermis, which were notably absent from POD 30 scars (Figure S4B, bottom row right). Although matrix cluster 3 was enriched similarly in PBS versus verteporfin wounds, cluster 4 was much more strongly enriched in the latter, suggesting that deep dermal ECM ultrastructure may be most relevant for differentiating scarring and regeneration.
Although both proteomic and ECM data correlated with distinct fibroblast scRNA-seq clusters, such trends were not seen in other cell types. Imputation analysis of proteomic data for Lin+ cells (Figure S2F) onto myeloid/lymphoid cell scRNA-seq data revealed no apparent correlation of identified fibrotic and regenerative proteomic features with either myeloid (Figure S2G) or lymphoid cell trajectories (Figure S2H), consistent with our initial scRNA-seq analysis suggesting that fibroblasts are the primary drivers of healing differences between PBS and verteporfin treatment.
Interrogation of fibrotic and regenerative trajectory markers in wounds
Our previous analysis suggested that spatial localization—and not merely cell numbers—may dictate the differing phenotypes in PBS and verteporfin wounds, so we performed immunofluorescent (IF) staining for Trps1 (Wnt pathway regulator), YAP (mechanotransduction signaling), and Dpp4 (En-1 lineage scarring fibroblasts; Figure 2M). As previously reported (Mascharak et al., 2021), Dpp4+ cells (EPFs) were significantly increased in PBS versus verteporfin wounds, particularly in the deep dermis (Figures 2M right, S1E, and S1F). Consistent with verteporfin inhibiting mechanotransduction, YAP expression was greater in PBS wounds than in verteporfin wounds or UW skin (Figure 2M). Trps1 expression was minimal in control wounds but substantial in verteporfin wounds, and AI-based quantification of Trps1 nuclear localization in thousands of cells revealed higher nuclear-Trps1+ fibroblasts in verteporfin wounds (Figure 2N). Although UW skin Trps1 expression was limited to basal epithelium and dermal papilla, as previously described (Fantauzzo et al., 2008a) (Figure 2M, left), in verteporfin wounds clusters of high nuclear-Trps1+ cells were found around regenerating HF (Figure 2M, bottom). Verteporfin-treated wounds also had few Dlk1+ cells but increased Crabp1+ cells throughout the dermis (Figure S1K first two rows) and clusters of cells expressing neogenic HF marker Lef1 (Figure S1L, first two rows), a staining pattern previously suggested to be regenerative (Abbasi et al., 2020; Lim et al., 2018; Phan et al., 2021). Reconfirming that verteporfin acts via YAP inhibition, we saw similar IF staining in En-1Cre-ERT;YAPfl/fl wounds as in verteporfin-treated wounds, with increased nuclear Trps1+ fibroblasts (Figure S1I, top), decreased Dpp4 (Figure S1J, top), increased Crabp1, decreased Dlk1 expression (Figure S1K, third row), and Lef1+ cells around regenerating HF (Figure S1L, third row). Finally, we compared these results with IF staining patterns in wound-induced hair neogenesis (WIHN), a different mouse model wherein very large dorsal wounds heal by contraction and recovery of sparse central follicles (Figures S1G and S1H, bottom panels). Similar to verteporfin and En-1Cre-ERT;YAPfl/fl wounds, WIHN wounds contained Trps1+, Crabp1+, and Lef1+ fibroblasts; however, they also contained relatively more YAP+, Dpp4+, and Dlk1+ fibroblasts (Figures S1I–S1L, bottom rows).
A striking difference between PBS- and verteporfin-treated wounds was the emergence of regenerated HF in the latter within 30 days; control wounds formed bare areas. We wondered whether our fibroblast trajectory analysis could reveal a master regulator of HF/gland regeneration. Trps1, a zinc finger transcription factor strongly enriched in the regenerative arm (Figures 2F and 3C), emerged as a top candidate. Trps1 was previously reported as a novel “master regulator” of Wnt signaling, which is critical in HF development, growth, and proliferation (Fantauzzo and Christiano, 2012; Fantauzzo et al., 2012; Ito et al., 2007). Multiple studies have implicated Trps1 in HF morphogenesis, and aberrant Trps1 activity has been associated with human conditions of excess hair growth (hypertrichosis) (Fantauzzo and Christiano, 2012; Fantauzzo et al., 2012; Fantauzzo et al., 2008b), making it a promising candidate for driving wound HF regeneration. Supporting this hypothesis, Trps1+ cells were enriched in regenerating wounds and spatially clustered around neogenic HF (Figures 2M and 2N). Further, ECM imputation revealed that Trps1-expressing fibroblasts in Seurat cluster 4 correlated strongly with regenerative ECM (ECM score > 0; Figure S1D, bottom). Importantly, Trps1 is also reported to transcriptionally regulate YAP signaling by competing for shared YAP/TEA domain (TEAD) genomic binding sites (Elster et al., 2018). Given that the regenerative trajectory is characterized by decreased mechanotransduction and elevated Trps1, we postulated that Trps1 may be a key regulator of wound regeneration.
Based on our integrated multimodal analysis, we sought to validate the relevance of key genes in distinct wound phenotypes. We selected four markers that distinguished, and represented pathways (e.g., mechanical activation and Wnt signaling) discriminating between, the fibrotic and regenerative arms: Ankrd1 (YAP target gene), Trps1 (Wnt regulator), Rspo1 (Wnt agonist), and Dpp4 (marker for EPF lineage; Figures 2C, 2F, 3B, and 3C). Collectively, these genes differentiated the mechanically activated/pro-fibrotic (Dpp4 and Ankrd1) and regenerative (Rspo1 and Trps1) pathways elucidated by our analyses. To probe wound spatial distribution of key genes, we performed multiplexed RNAscope in situ hybridization for Ankrd1, Dpp4, Trps1, and Rspo1. To focus on dermal fibroblasts, we performed dermis-restricted morphological reconstruction (excluding epidermis and HF; see STAR Methods). In POD 14 verteporfin wounds, fibroblasts containing Rspo1 and Trps1 RNA granules were found in the superficial dermis clustered around the regions of invaginating epithelium, consistent with neogenic HF (Figures 5A and 5C bottom). In contrast, Rspo1+ and Trps1+ cells in POD 14 PBS wounds were randomly dispersed through the dermis (Figures 5A and 5C top). At POD 30, the time of greatest phenotypic divergence between fibrotic/regenerating wounds, PBS wound fibroblasts had more Dpp4 and Ankrd1, whereas verteporfin wounds had more Rspo1 and Trps1 RNA granules, concentrated around regenerating HF (Figures 5B and 5D). Quantifying granules from thousands of cells confirmed these trends (Figures 5E and 5F), supporting the finding that ENF-mediated healing (low Dpp4) in YAP-inhibited (low Ankrd1) wounds yielded HF regeneration through Wnt activation (high Trps1 and Rspo1).
Figure 5. Spatial transcriptomic analysis of fibroblasts in scarring and regeneration.
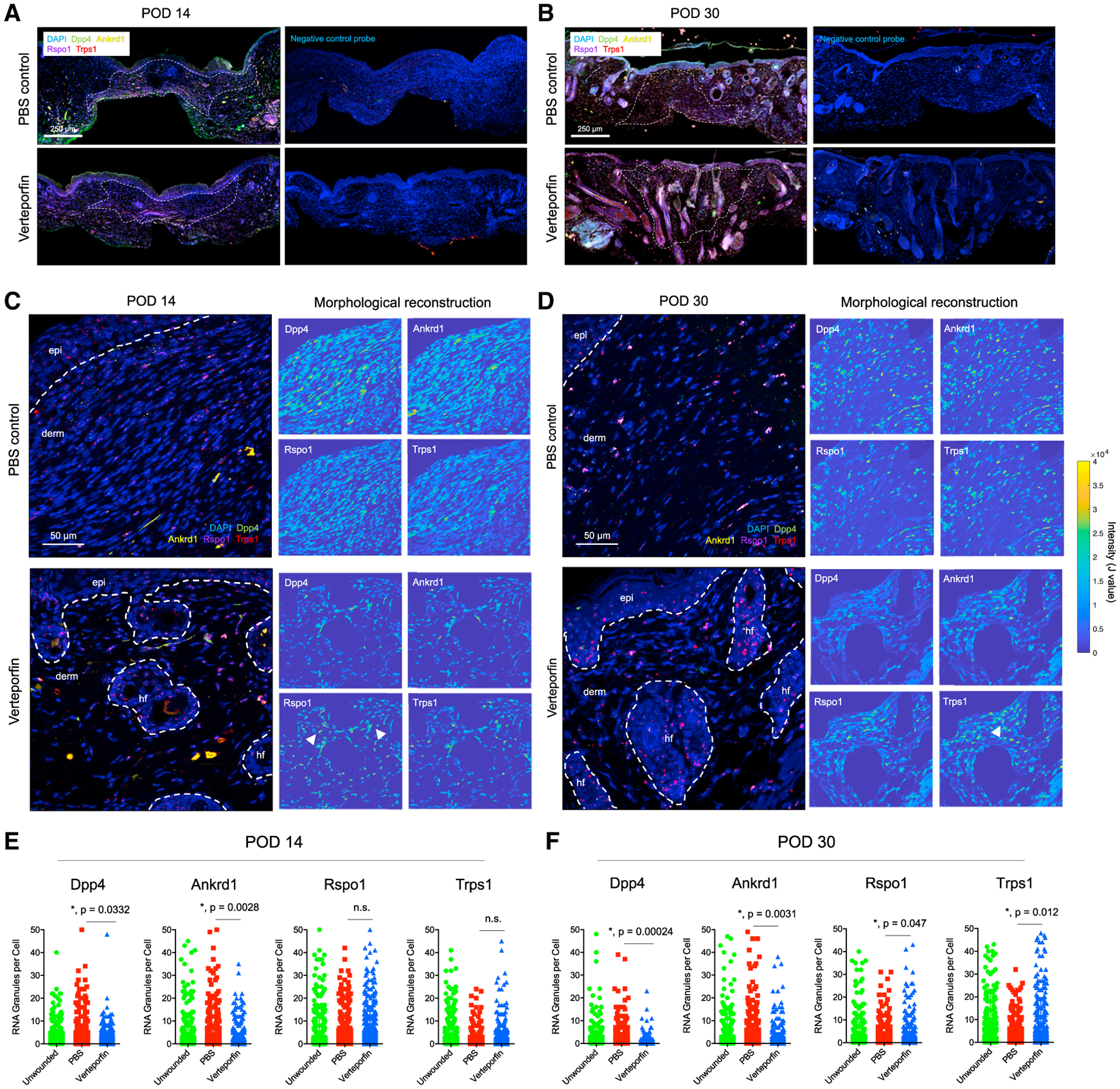
(A and B) Low-power tile scan of RNAscope multiplexed in situ hybridization for Dpp4 (green), Ankrd1 (yellow), Rspo1 (magenta), and Trps1 (red) in PBS- and verteporfin-treated wounds at POD 14 (A) and 30 (B). White dotted line, scar boundary. Right panels, neighboring slides hybridized with negative control scrambled probes.
(C and D) High-power images of RNAscope granules in PBS- and verteporfin-treated wounds at POD 14 (C) and 30 (D). White dotted line, quantified regions of interest in dermis. Right panels, morphological reconstructions (dermis only; epidermis and HF excluded); yellow, cells strongly expressing indicated gene.
(E and F) Quantification of Dpp4, Ankrd1, Rspo1, and Trps1 RNA puncta per cell in POD 14 (E) and 30 (F) specimens.
Determining the functional significance of Trps1 in regeneration
Given the importance of Trps1 in Wnt signaling, HF morphogenesis, and YAP regulation and the spatial relationship of Trps1+ fibroblasts and regenerating HF, we sought to determine the function of Trps1 in wound regeneration. We first performed fibroblast-specific Trps1 OE to assess if Trps1 was sufficient for wound regeneration under normally scarring conditions. We developed FLEXon Trps1:T2A:EGFP lentiviral vectors encoding a floxed mTrps1:EGFP transgene with proteolyzable T2A linker (successfully transduced fibroblasts are enhanced green fluorescent protein [EGFP]+), which, when injected intradermally into tamoxifen-induced Col1a1-CreERT;Ai9 mice (fibroblasts express tdTomato), enabled fibroblast-specific Trps1 OE. Confirming specificity, IF staining for non-fibroblast markers showed GFP signal only in fibroblasts and not in endothelial, adipocyte, epithelial, or immune cells (Figures S5A and S5B). To target distinct phases of repair, we overexpressed Trps1 both early (POD −3 and 7)—to investigate its role in early processes like inflammation, granulation, and early ECM synthesis—and late (POD 7 and 17)—for later processes like fibroblast migration, ECM deposition, and early remodeling, in scarring (PBS) wounds. Control wounds (Trps1-OEC) were injected with FLEXon EGFP lentivirus. All wounds were harvested at POD 30. Grossly, Trps1-OEC wounds formed scars, whereas Trps1-OEE and Trps1-OEL wounds had less prominent scars and regenerated HF (Figure 6A). Histology confirmed increased Trps1 expression (Figure 6C) and showed small numbers of regenerated adnexal structures and more UW-like ECM in Trps1-OEE and Trps1-OEL wounds (Figures 6B, 6D, right, and 6E); these findings were confirmed quantitatively, with significantly more HF/glands and significantly higher (i.e., more UW-like) ECM scores (Figure 6D). Regeneration was more prominent in Trps1-OEE versus Trps1-OEL wounds, suggesting that Trps1’s pro-regenerative effects may be greatest at early healing time points. Collectively, these findings suggest that Trps1 OE is partially sufficient for wound regeneration.
Figure 6. Fibroblast-targeted lentiviral Trps1 overexpression and knockdown.

(A) Gross photos, PBS-treated wounds at POD 30 (Col1a1-CreERT;Ai9 mice) injected with control lentivirus (Trps1-OEC, left), Trps1 OE lentivirus at POD −3/7 (Trps1-OEE, middle), or POD 7/17 (Trps1-OEL, right). Red dotted lines demarcate the region where the silicone splint was attached and later removed.
(B) H&E (top row) and IF (bottom row; GFP+Ai9+ cells, successfully transduced fibroblasts) histology of Trps1-OEC, Trps1-OEE, and Trps1-OEL wounds.
(C) IF staining for Trps1 in indicated wounds; white arrowheads, nuclear Trps1.
(D) Quantification of HF (left), glands (middle), and ECM score (right).
(E) Matrix ultrastructural analysis of UW skin and POD 30 Trps1-OEC, Trps1-OEE, and Trps1-OEL wounds.
(F–J) As in (A–E), but for verteporfin-treated wounds with control lentivirus (Trps1-KDC), Trps1 KD lentivirus at POD 3/7 (Trps1-KDE), or POD 7/17 (Trps1-KDL). In (D) and (I), error bars bars indicate mean ± standard error (SEM).
Next, to assess if Trps1 was necessary for verteporfin-induced wound regeneration, we developed FLEXon EGFP:miR30-mTrps1 lentiviral vectors encoding floxed miR30-based Trps1 shRNA, which, when injected intradermally in tamoxifen-induced Col1a1-CreERT;Ai9 mice, enabled fibroblast-specific Trps1 KD (fibroblasts express tdTomato; transduced cells express EGFP). We knocked down Trps1 early (POD −3/7; Trps1-KDE) and late (POD 7/17; Trps1-KDL) in regenerating (verteporfin) wounds to again target distinct repair phases. Control verteporfin-treated wounds (Trps1-KDC) received FLEXon EGFP:Scramble miR30 lentivirus. Wounds were harvested at POD 30. Grossly, Trps1-KDC wounds exhibited hair regeneration and absent scarring, whereas Trps1-KDE and Trps1-KDL wounds formed hairless scars (Figure 6F; decreased Trps1 expression confirmed by IF, Figure 6H). Histologically, Trps1-KDE and Trps1-KDL wounds had complete abrogation of ECM regeneration, developing scar ECM instead (Figures 6G, 6I right, and 6J). Interestingly, significant loss of adnexal structures was only observed in Trps1-KDL wounds (Figures 6G right and 6I), implying that Trps1 is specifically required later in healing for verteporfin-induced regeneration of adnexae (e.g., HF). Collectively, these data suggest that Trps1 is necessary for complete wound regeneration, with distinct roles in early and late healing.
We also performed Trps1 OE and KD early (POD −3/7) and harvested wounds at POD 17, during the early scar maturation phase. At POD 17, Trps1 OE wounds had early regenerating HF similar to verteporfin-treated controls, whereas Trps1 KD in verteporfin wounds abrogated HF regeneration and instead resulted in scar formation (Figures S6A and S6B). Trps1-OEE wounds had increased Trps1 and elevated Lef1 expression (like verteporfin-treated controls), consistent with regenerating HF and possible early neogenic dermal papillae, whereas YAP expression was similar among all wound conditions, suggesting that modulating Trps1 did not directly affect YAP expression (Figures S6C and S6D).
Collectively, our integrated multimodal analysis differentiated two trajectories of repair. The first is EPF-mediated (Dpp4+) and characterized by mechanical activation (high nuclear YAP, Ankrd1+, Trps1−) and results in scar ECM. The second is ENF-mediated (Dpp4−), lacks mechanical activation (low nuclear YAP, Ankrd1−, Trps1+), and results in the regeneration of normal ECM and HF via Wnt activation.
DISCUSSION
In this study, we sought to delineate divergent mechanisms of fibrotic versus regenerative healing by leveraging our recent finding that verteporfin (a mechanotransduction inhibitor) prevents postnatal activation of mechanosensitive fibroblasts, yielding wound regeneration by pro-regenerative fibroblasts (ENFs) (Mascharak et al., 2021). Using multi-omic analysis over the course of healing, we demonstrated that fibrotic wound repair is characterized by the activation of canonical mechanotransduction (YAP) signaling, leading to EPF-mediated scarring; in contrast, regenerative repair (verteporfin) exhibits disrupted mechanical signaling and enrichment of HF development-related pathways in ENFs. Transcriptional divergence between these two trajectories may occur as early as POD 2, with proteomic and histologic divergence following on POD 2/7 and 14, respectively. Integration of three distinct datasets yielded interesting biological insights. We saw only minimal correlation between individual gene mRNA and protein levels, consistent with emerging literature (Brunner et al., 2021); variable translation/degradation and differences in splice variants may complicate such analysis. However, we observed correlations at the pathway level, indicating that higher-order analyses may be useful in integrating these distinct data types.
Although we found differences in the relative representation of cell populations by phenotype (e.g., verteporfin wound fibroblasts were more enriched in the late “regenerative” trajectory), these differences were modest. Spatial interrogation of markers for the divergent trajectories suggests that differential cell localization/activity may instead drive divergent healing. We were interested to find that verteporfin treatment may also induce epidermal changes such as increased Trps1 in keratinocytes (Figure 2M), given previous reports implicating epithelial Wnt signaling in regeneration (Ito et al., 2007; Myung et al., 2013). However, these effects on the epidermis were recapitulated with fibroblast-restricted Trps1 modulation (Figure 6C), suggesting that verteporfin’s epidermal effects are fundamentally from fibroblast-mediated mechanisms via epithelial-fibroblast crosstalk (Fantauzzo et al., 2008a).
A recent study (Abbasi et al., 2020) also concluded that regeneration and fibrosis may not be entirely mutually exclusive but pro-fibrotic factors in wounds may normally dominate an underlying regenerative competency. This is consistent with our finding that Trps1 OE was only partially sufficient for regeneration; without the suppression of mechanical signaling, OE of pro-regenerative genes may be insufficient to fully override fibrotic programing. Incomplete lentiviral KD/OE efficiency (50%–75% transduction in our experiments) could also contribute to findings of only partial necessity/sufficiency. Abbasi et al. identified mechanisms of regeneration both convergent with and divergent from those reported here. Both studies show Wnt-driven regeneration and neogenic HF with Trps1+/Lef1+ mesenchymal cells by POD 30. Several studies suggest that the WIHN model used by Abbasi et al. induces Wnt signaling in a fibroblast subset that gives rise to inductive dermal condensate cells, which may be marked by Trps1 (Abbasi et al., 2020; Lim et al., 2018; Phan et al., 2021). The Trps1+ fibroblasts identified in our study could reflect a similar expansion of a subset of fibroblasts that acquired neogenic HF fates in a distinct regeneration-permissive environment. Abbasi et al.’s finding that increasing fibroblast proliferation positively influenced regeneration is consistent with modestly increased fibroblasts in verteporfin-treated wounds (Figure 1D). However, verteporfin wounds had minimal Dpp4+ (EPF), Dlk1+, and YAP+ cells, but WIHN wounds had more Dlk1+ and numerous Dpp4+ and YAP+ cells (Figures S1I–S1K, bottom). Furthermore, UW-like ECM was restored in verteporfin-treated wounds by POD 30, whereas WIHN wounds regenerated sparse HF against a background of fibrotic, scar-like ECM, indicating that normal skin ultrastructure was not reestablished (Figure S1H), possibly due to large numbers of EPFs. A key source of divergent results may be the different models used. Our study compared smaller (6-mm diameter), splinted wounds, whereas Abbasi et al. (and most previous studies reporting wound regeneration) employed the WIHN model, which involves very large (>2.25 cm2), un-splinted wounds (Figures S1G and S1H bottom panels). Given these significant experimental differences, we did not expect a complete overlap of findings.
Our studies provide a framework for leveraging cell-surface barcoding with multi-omic interrogation to dramatically enhance the analysis of tissue dynamics and suggest a potential mechanism driving regeneration in ENF-mediated repair. Trps1, which competes with YAP for shared target sites, was associated with a less-fibrotic transcriptional trajectory and spatially linked to regenerating HF. Interestingly, our findings suggest that Trps1 and downstream Wnt signaling also occur in scarring wound fibroblasts but, in the absence of YAP inhibition, are phenotypically dominated by YAP signaling. This may indicate that regeneration is a “default” repair pathway superseded by a tendency toward mechanically activated fibrosis.
Limitations of study
Gene/protein correlation is imprecise because of the limited capacity for simultaneous measurements, so conclusions from these comparisons should be tempered. That the two trajectories in this study are based on high-dimensional-omic profiles precludes prospective isolation of cells, so further analysis (e.g., ChIP-seq) is not currently feasible. Finally, there may be some contraction of splinted wound sites despite silicone ring stenting, introducing trace intact HF; however, we expect these contributions to be small relative to the phenotypes observed.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Michael T. Longaker (longaker@stanford.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
The scRNA-seq data generated during this study have been deposited in a GEO repository (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE186527). These data are publicly available as of the date of publication. The accession number is listed in the key resources table.
Original scripts for the ECM ultrastructure and RNAscope analysis algorithms used in this study have been deposited in a Github repository (https://github.com/shamikmascharak/Mascharak-et-al-ENF) and are publicly available as of the date of publication. DOIs are listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
All studies were conducted in accordance with Stanford University Animal Care and Use Committee guidelines. Mice were housed at the Stanford University Comparative Medicine Pavilion under the care of the Department of Comparative Medicine in the Veterinary Service Center (VSC), in accordance with Stanford APLAC guidelines (APLAC-11048). Animal were housed in a controlled environment with optimal temperature and fed rodent chow ad libitum.
Ten C57BL/6J mice were acquired from Jackson Laboratory (8 weeks of age) and wounded (see section below) for each experimental time point (POD 0, 2, 7, 14, 30; PBS- or verteporfin-treated).
Mice strains
C57/BL/6J, En1Cre-ERT, YAPfl/fl, Col1a1Cre-ERT, and Ai9 mice were obtained from The Jackson Laboratory (Bar Harbor, ME). En-1Cre-ERT;YAPfl/fl mice were generated by crossing En-1Cre-ERT with YAPfl/fl mice to obtain En-1Cre-ERT;YAPfl/+ mice; these mice were then again bred to YAPfl/fl mice to yield En-1Cre-ERT;YAPfl/fl mice. Col1a1Cre-ERT;Ai9 mice were generated by crossing Col1a1Cre-ERT with homozygous Ai9 mice in order to drive Cre expression and fluorescent reporter expression in fibroblasts (Col1a1-expressing). Equal numbers of female and male mice ranging from the age of 8–12 weeks were allocated to experimental groups. Sample size for any given experiment is detailed in the figure legend.
METHOD DETAILS
Mouse Wounding
Experimental mice were wounded in accordance with established protocols for dorsal splinted excisional wounding ((Galiano et al., 2004); Mascharak et al., 2021) and large unsplinted excisional wounding (WIHN model) (Abbasi et al., 2020; Ito et al., 2007). For both wounding models, mice were anesthetized (2% isoflurane), their dorsal hair was removed with depilatory cream, and the dorsal skin was prepped with alternating betadine and alcohol wipes. Following wounding (see below), postoperative analgesia was accomplished with buprenorphine SR 0.05 mg/kg every four hours for three doses, and then as indicated.
For dorsal splinted wounding, two 6 mm full-thickness circular wounds were placed (extending through the panniculus carnosus) on the dorsum of each animal, roughly 6 mm below the ears and 4 mm lateral to the midline on each side. The wounds were stented open with silicone rings, which were secured around the wound perimeter with adhesive and eight simple interrupted 6–0 nylon sutures (Ethicon) per ring. For mice receiving mechanotransduction inhibitor (verteporfin), 30 μL of verteporfin (1 mg/mL) per wound was injected locally into the wound base; PBS was injected into wounds for vehicle controls. Dressings were changed every other day under anesthesia. At the indicated post-operative day (POD; 2, 7, 14, or 30), the wounds were harvested by excising a 2 mm ring of tissue around each wound using a 10 mm biopsy punch. One-third of each wound was processed for histology as described below; the remaining two-thirds of each wound were processed for FACS as described below. For unwounded (POD 0) samples, uninjured skin was harvested from the dorsum from a comparable region to wound samples and split between histology and FACS as for wounds.
For WIHN wounding, large square full-thickness wounds were created centrally on the dorsum, measuring ≥1.5 cm per side (≥2.25 cm2 area). Wounds were dressed with sterile dressings (Tegaderm) and dressings were changed every other day under anesthesia until wounds were re-epithelialized. At indicated postoperative days, wounds were harvested by excising a ≥5mm margin around the wound border, then processed for histology as described below.
Fluorescence Activated Cell Sorting
The dorsal skin was harvested using dissecting scissors by separation along fascial planes. Next, the subcutaneous fat was trimmed with a scalpel, and the skin was rinsed in betadine, followed by 5 rinses in cold PBS. To achieve a cell suspension, the harvested skin was finely minced using sharp scissors, enzymatically digested (Liberase DL, 0.5 mg/mL, 1 hour), and filtered through a 40 μm nylon mesh. Cells were isolated from experimental mice via a previously reported FACS strategy. Briefly, a lineage gate (Lin) for hematopoietic (CD45, Ter-119), endothelial (CD31, Tie2), and epithelial (CD326, CD324) cell markers was used as a negative gate to isolate fibroblasts (Lin−) and non-fibroblasts (Lin+).
Single-cell Transcriptomic Analysis
Cell suspensions were labeled with TotalSeq Series B hashtag oligonucleotide-labeled antibodies (BioLegend). Single-cell RNA-seq (scRNA-seq) was then performed at the Stanford Functional Genomics Facility (SFGF) for droplet-based microfluidics using the 10x Chromium Single Cell platform (Single Cell 3’ v3, 10x Genomics, USA). Droplets of the cellular suspensions, reverse transcription master mix, and partitioning oil were mixed, loaded onto a single cell chip, and processed on the Chromium Controller. Reverse transcription was performed, and cDNA was amplified using a BioRad C1000 Touch thermocycler, with cDNA size selected using SpriSelect beads (Beckman Coulter, USA). An Agilent Bioanalyzer High Sensitivity DNA chip was used to analyze cDNA for qualitative control purposes; cDNA was then fragmented using the proprietary fragmentation enzyme blend for 5min at 32°C, followed by end repair and A-tailing at 65°C for 30 min. DNA was double-sided size selected using SpriSelect beads. Sequencing adaptors were ligated to the cDNA at 20°C for 15min. cDNA was amplified using a sample-specific index oligo as primer, followed by another round of double-sided size selection using SpriSelect beads. Final libraries were analyzed on an Agilent Bioanalyzer High Sensitivity DNA chip for qualitative control purposes. Libraries were sequenced on a HiSeq 4000 Illumina platform targeting 50,000 reads per cell.
Base calls were converted to reads using the Cell Ranger (10X Genomics; version 3.1) implementation mkfastq and then aligned against the Cell Ranger mm10 reference genome, available at: http://cf.10xgenomics.com/supp/cell-exp/, using Cell Ranger’s count function with SC3Pv3 chemistry and 5,000 expected cells per sample. Cell barcodes representative of quality cells were differentiated from apoptotic cell barcodes or background RNA based on a threshold of having at least 200 unique transcripts profiled, less than 100,000 total transcripts, and less than 10% of their transcriptome of mitochondrial origin. Unique molecular identifiers (UMIs) from each cell barcode were retained for all downstream analysis, normalized with a scale factor of 10,000 UMIs per cell, and subsequently natural log transformed with a pseudocount of 1 using the R package Seurat (version 3.1.1) ((Chen et al., 2013)). Hashtag oligos (HTOs) for samples were demultiplexed using Seurat’s implementation HTODemux. Briefly, k-medoid clustering is performed on the normalized HTO values, after which a ‘negative’ HTO distribution is calculated. For each HTO, the cluster with the lowest average value is treated as the negative group and a negative binomial distribution is fit to this cluster. Using the 0.99 quantile of this distribution as a threshold, each cell is classified as positive or negative for each HTO. Cells that arepositive formore thanone HTOsare annotatedasdoublets and removed. Cells thatare not positive for any HTO are also removed. The first 15 principal components of the aggregated data were then used for uniform manifold approximation and projection (UMAP) analysis (Gulati et al., 2020). Cell annotations were ascribed using SingleR (version 3.11) against the Mouse-RNAseq reference dataset, available at https://rdrr.io/github/dviraran/SingleR/man/mouse.rnaseq.html.
Cell-type marker lists were generated with two separate approaches: first, we employed Seurat’s native FindMarkers function with a log fold change threshold of 0.25 using the ROC test to assign predictive power to each gene; second, we employed a characteristic direction analysis to better account for the mutual information contained within highly correlated predictive genes (Chen et al., 2013).
Pseudotime Analysis
Pseudotime analysis was performed using the Monocle 3 package in R (version 3 0.2.0) ((Trapnell et al., 2014)). Counts for individual cells were preprocessed using principal component analysis (PCA) with 15 dimensions following log-normalization. Dimensional reduction was performed using a UMAP reduction with min_dist = 0.5, n_neighbors = 30, and repulsion.strength = 2.0. Cells were then clustered using Monocle 3’s Louvain implementation with a resolution of 1e-5. A principal graph was then learned from the reduced dimension space using reversed graph embedding with default parameters, and cell order selection was made from the two elements at either end of the trajectory. Pseudotime trajectory heatmaps were created using the Monocle 2 package in R.
For fibroblast, myeloid, and lymphoid cell subsets, we identified a root starting point to calculate pseudotime values for each cell within the dataset. Then, we then used the R function cor to correlate pseudotime values with gene expression levels, protein expression z scores, and matrix histology levels. These correlation values were used to determine the genes and proteins that either increased (positive correlation) or decreased (negative correlation) with pseudotime. Correlation values in the top 1% of positively or negatively correlated genes were chosen for downstream analyses. Integration of timsTOF proteomic data onto pseudotime scRNA-seq manifolds was performed pairwise between modalities on a per-mouse basis without replacement for missing values.
CytoTRACE Analysis
We utilized the recently developed bioinformatics tool CytoTRACE to compare differentiation states among cells in our dataset (https://cytotrace.stanford.edu/) (Gulati et al., 2020). This tool analyzes the number of uniquely expressed genes per cell, as well as other factors like distribution of mRNA content and number of RNA copies per gene, to calculate a score assessing the differentiation and developmental potential of each cell (lowest differentiation and highest developmental potential at 1; highest differentiation and lowest developmental potential at 0). Cells are then ordered by their predicted differentiation status. CytoTRACE analysis was performed using default parameters for each fibroblast in our dataset.
SCENIC Analysis
To examine the role of transcriptional regulators in scarring and regenerative healing, we used SCENIC ((Van de Sande et al., 2020)) in conjunction with the mm9 RcisTarget database. The unique molecular identifier (UMI) counts for all transcriptionally defined fibroblasts were provided as input. Based on these matrices, we constructed co-expression modules between transcription factors and potential target genes filtered by importance. Genes with significant motif enrichment were considered direct targets for a given transcription factor using default parameters and defined as a regulon. Each regulon was then scored according to active value using ranked genes. Cells were then re-clustered using a matrix of regulon values, and regulons were judged to be “active” in cells according to default threshold parameters.
CellChat Receptor-Ligand Analysis
To evaluate the potential for cell-cell interactions between our fibroblast populations and other cell types, including immune cells, we applied the recently developed CellChat platform (Jin et al., 2021). This was implemented using our scRNA-seq Seurat object in R, in conjunction with the standalone CellChat Shiny App for its Cell-Cell Communication Atlas Explorer. Cells were binned according to the SingleR-defined cell type classifications, with fibroblast cells subsetted based on their location within either the scarring or regenerative pseudotime arms. Default parameterizations were used throughout, and Secreted Signaling, ECM-Receptor, and Cell-Cell Contact relationships were considered.
RNA Velocity Analysis
RNA velocity analysis was performed using the scVelo package (Bergen et al., 2020). scVelo uses a likelihood-based dynamical model to solve the full transcriptional dynamics of spliced and unspliced mRNA kinetics of each gene. RNA velocity analysis allowed us to identify transient cellular states in our dataset and to predict the directional progression of transcriptomic signatures along the identified trajectories. These predictions are based on gene-specific rates of transcription, splicing, and degradation of mRNA to estimate each cell’s position in their own underlying differentiation process. The RNA velocity across all genes is then projected as a stream of arrows on the UMAP embedding.
timsTOF Shotgun Proteomic Analysis
Cells for proteomic analysis were pelleted using a swinging-bucket rotor centrifuge (500 × g, 5 minutes, 4 °C). Supernatant was removed, and pellets were snap frozen on dry ice, then stored at −80 °C until protein purification. Peptides were purified from pellets using the PreOmics iST kit (Martinsried, Germany) following the kit protocol. Samples were homogenized by passing through QIAshredder spin columns (QIAGEN) following lysis. Peptide concentration was quantified with a plate reader using the Pierce Quantitative Colorimetric Peptide Assay (Thermo Scientific). Finally, peptide samples were diluted to normalize concentration and 50 μg of each was loaded into the timsTOF analyzer. The timsTOF Pro (Bruker) was operated in PASEF mode using Compass Hystar 5.0.36.0. All raw files were analyzed by MaxQuant v1.6.6 software using the Andromeda search engine. Functional enrichment and network analyses were performed on positively and negatively correlated pseudotime genes using EnrichR.
Histology and Immunofluorescence
Samples were placed into histology cassettes and fixed by incubation in 5% buffered formalin phosphate for 16 hours at 4 °C. The samples were washed with PBS, dehydrated through serial ethanol washes, cleared with xylene, infiltrated with paraffin through serial incubations, and embedded in paraffin. Sections were cut at a thickness of 8 μm and incubated at 37 °C overnight to affix sections to slides prior to staining. Picrosirius Red staining was performed per standard protocols from the manufacturer (Abcam).
For immunofluorescent staining, samples were cleared with xylene, re-hydrated, and treated with antigen retrieval buffer per established protocols (Abcam ab970). Samples were then permeabilized in 0.25% Triton-X (15 minutes), blocked for 2 hrs (Power-block), and stained overnight with primary antibodies in 0.1x Powerblock (YAP Santa Cruz Biotechnology sc-101199, Trps1 Protein-tech 20003–1-AP). Samples were then washed 3 times with 0.1x Powerblock, stained for 1 hr with secondary antibodies (Invitrogen), washed another 3 times, and finally mounted with Fluoromount-G containing DAPI. Slides were imaged using a Leica TCS SP5 confocal microscope. Nuclear localization analysis of Trps1 was performed using the BioDock AI Nuclear Segmentation module (www.biodock.ai) ((Stauffer et al., 2018)).
Quantitative Analysis of Connective Tissue Ultrastructure
Picrosirius Red-stained histologic sections derived from unwounded skin and wounds (5 PBS- and 5 verteporfin-treated, 2 wounds per mouse, 3 sections per slide) were imaged in tiles at 40x magnification. Next, images were normalized using the RGB histogram method (Stain Normalization Toolbox, University of Warwick) and color deconvoluted using the algorithm previously described by Ruifrok et al. ((Ruifrok and Johnston, 2001); (Ruifrok et al., 2003)), wherein each pure stain is characterized by absorbances within three RGB channels. Ortho-normal transformation of the histology images produced individual images corresponding to each color’s individual contribution to the image. Applied to birefringent Picrosirius Red images (green to red color under polarized light depending on packing of fiber bundles), this technique produced deconvoluted red and green images corresponding to mature and immature connective tissue fibers, which were then analyzed independently. Noise reduction of deconvoluted fibers was achieved using an adaptive Wiener filter in Matlab 2019a (wiener2 function), which tailors itself to the local image variance within a pre-specified neighborhood (3-by-3 pixels in our application). The filter preferentially smooths regions with low variance, thereby preserving sharp edges of fibers. Smooth images were then binarized using the im2bw command and processed through erosion filters with diamond-shaped structuring elements to select for fiber-shaped objects. Finally, the fiber network was “skeletonized” using the bwmorph command and various parameters of the digitized map (fiber length, width, persistence, alignment, etc.; see Table S1 for full list and descriptions of parameters) were measured using the regionprops command. Dimensionality reduction of quantified fiber network properties by t-distributed stochastic neighbor embedding (t-SNE) was achieved using the default tsne (distance metric specified as Euclidian distance) command in Matlab. Hierarchical clustering was performed by first filtering low variance parameters (bottom 25%) and then calculating Euclidean distances. Matlab scripts containing our fiber quantification pipeline are available at the following Github repository: https://github.com/shamikmascharak/Mascharak-et-al-ENF.
To facilitate correlation of mouse-to-mouse variation at the scRNA-seq level to variation in matrix features, we envisioned a unified “ECM score” based on quantified ultrastructural values (−1 resembles scar, +1 resembles unwounded skin). To calculate this score, we utilized the Machine Learning toolbox in Matlab to train a neural network on Picrosirius red matrix histology from mouse scars and unwounded skin (10-fold cross validated, AUC > 0.95). The trained neural network was then used to score an external test set of histology from PBS- and verteporfin-treated wounds across all PODs.
RNAScope Spatial Transcriptomic Analysis
RNAscope was used to evaluate spatial distributions of mechanically-activated fibrotic (Ankrd1+, CD26+) and non-activated regenerative (Trps1+, Rspo1+) fibroblasts. Paraffin-embedded slides were processed in house by the manufacturer (ACD, Newark CA) for multiplexed in-situ hybridization. Slides were then imaged on a Leica TCS SP8 confocal microscope at 40x magnification. Spatial transcriptomic patterns were analyzed using a custom Matlab pipeline. Briefly, confocal images were split into separate fluorescent channels and images of nuclei (DAPI) and RNA puncta (Dpp4 Opal 520, Ankrd1 Opal 570, Rspo1 Opal 620, Trps1 Opal 690) were binarized using shaped-based filtration. Next, regions of interest were traced around nuclei with an additional 10-pixel berth to capture somatic transcripts. Finally, RNA puncta per cell were quantified across all cells and images for each mouse. Morphological reconstruction was achieved using the imreconstruct function on non-binarized RNA puncta and nuclei. Analysis was restricted to the dermis by manually defining regions of interest with the createMask function prior to morphological reconstruction, thereby excluding epidermal elements. Matlab scripts containing our RNAscope analysis pipeline are available at the following Github repository: https://github.com/shamikmascharak/Mascharak-et-al-ENF.
Lentiviral Trps1 Overexpression and Knockdown
Trps1 overexpression and knockdown studies were conducted in Col1a1Cre-ERT;Ai9 mice (fibroblasts express tdTomato). For Trps1 overexpression, we utilized FLEXon Trps1:T2A:EGFP lentiviral vectors encoding a floxed mTrps1:EGFP transgene with a proteolyzable T2A linker (control lentivirus contained EGFP alone; successfully transduced fibroblasts expressing transgene are EGFP+). For Trps1 knockdown, we utilized FLEXon EGFP:miR30-mTrps1 lentiviral vectors encoding a floxed miR30-based Trps1 shRNA (control lentivirus contained scrambled miR30 sequence; successfully transduced fibroblasts expressing transgene are EGFP+). Vector designs may be accessed at the following links:
pLV-CMV>LL:rev(mTrps1(ns):T2A:EGFP):rev(LL): https://en.vectorbuilder.com/vector/VB210107-1237pqp.html
pLV-Puro-CMV>LL:rev(EGFP:miR30-mTrps1):rev(LL): https://en.vectorbuilder.com/vector/VB210112-1008hzm.html
Ultra-purified overexpression or knockdown Trps1 lentivirus was delivered via four 15 ul intradermal injections at the wound site (>109 TU/ml). For the early transduction experiments, two applications were delivered at three days prior to wounding (POD −3) and seven days post wounding (POD 7). For late transduction experiments, two applications were delivered at seven and seventeen days post wounding (POD 7 and 17).
QUANTIFICATION AND STATISTICAL ANALYSIS
All statistical analyses were performed using GraphPad Prism 8.0.2 (GraphPad Software, La Jolla, California). Unless otherwise specified, all values are expressed as mean ± standard deviation (SD). Statistical significance between groups was determined using an unpaired Student’s t test assuming two-tailed distribution and unequal variances if not stated in the figure legends. The value of n refers to the number of mice used in the mice study part. A p value < 0.05 was considered statistically significant. For all figures, asterisks denote statistical significance and the associated p values are shown adjacently.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| TotalSeq Series B anti-mouse hashtag antibodies 1–10 | BioLegend | Cat# 155831, 155833, 155835, 155837, 155839, 155841, 155843, 155845, 155847, 155849; RRID: AB_2814067, AB_2814068, AB_2814069, AB_2814070, AB_2814071, AB_2814072, AB_2814073, AB_2814074, AB_2814075, AB_2814076 |
| Anti-Mouse Monoclonal eFluor 450, CD45 | Thermo Fisher Scientific | Cat# 48-0451-82; RRID: AB_1518806 |
| Anti-Mouse Monoclonal eFluor 450, Ter-119 | Thermo Fisher Scientific | Cat# 48-5921-82; RRID: AB_1518808 |
| Anti-Mouse Monoclonal eFluor 450, CD31 | Biolegend | Cat# 303114; RRID: AB_2114316 |
| Anti-Mouse Monoclonal eFluor 450, Tie-2 | Thermo Fisher Scientific | Cat# 13-5987-82; RRID: AB_466848 |
| Anti-Mouse Monoclonal eFluor 450, CD326 | Thermo Fisher Scientific | Cat# 48-5791-82; RRID: AB_10717090 |
| Anti-Mouse Monoclonal eFluor 450, CD324 | Thermo Fisher Scientific | Cat# 13-3249-82; RRID: AB_1659688 |
| Goat Anti-Rabbit Alexa Fluor 488 | Thermo Fisher Scientific | Cat# A32731; RRID: AB_2633280 |
| Goat Anti-Rabbit Alexa Fluor 568 | Thermo Fisher Scientific | Cat# A-11011; RRID: AB_143157 |
| Goat Anti-Rabbit Alexa Fluor 647 | Thermo Fisher Scientific | Cat# A-21245; RRID: AB_2535813 |
| Rabbit Anti-Trps1 | Proteintech | Cat# 20003-1-AP; RRID: AB_10666999 |
| Mouse Anti-YAP | Santa Cruz Biotechnology | Cat# sc-101199; RRID: AB_1131430 |
| Rabbit Anti-DPP4 | Abcam | ab187048 |
| Rabbit Anti-Crabp1 | Cell Signaling Technology | Cat# #13163; RRID: AB_2750569 |
| Mouse Anti-Dlk1 | Thermo Fisher Scientific | Cat# MA5-15915; RRID: AB_11155588 |
| Rabbit Anti-Lef1 | Abcam | ab137872; RRID: AB_2892647 |
| Rabbit Anti-Perilipin | Abcam | ab3526; RRID: AB_2167274 |
| Rabbit Anti-CD45 | Abcam | ab10558; RRID: AB_442810 |
| Rabbit Anti-CD31 | Abcam | ab281583 |
| Rabbit Anti-EpCAM | Abcam | ab221552 |
| Bacterial and virus strains | ||
| pLV-CMV>LL:rev(mTrps1(ns): T2A:EGFP):rev(LL) (VectorBuilder, LVMP(VB210107-1237pqp)) | VectorBuilder | Custom |
| pLV-CMV>LL:rev(EGFP):rev(LL) (VectorBuilder, LVLP(VB210218-1358cza)) | VectorBuilder | Custom |
| pLV-Puro-CMV>LL:rev(EGFP:miR30-mTrps1):rev(LL) (VectorBuilder, LVMP(VB210112-1008hzm)) | VectorBuilder | Custom |
| pLV-Puro-CMV>LL:rev(EGFP:Scramble):rev(LL) (VectorBuilder, LVLP(VB210218-1356jhw)) | VectorBuilder | Custom |
| Chemicals, peptides, and recombinant proteins | ||
| Verteporfin | Millipore Sigma | Cat# SML0534-5MG |
| Fluoromount-G with DAPI | Thermo Fisher Scientific | Cat# 00-4959-52 |
| Permount | Fisher Chemicals | Cat# SP15 |
| Liberase DL Research Grade | Millipore Sigma | Cat# 5466202001 |
| Biogenex Laboratories Power Block | Fisher Scientific | Cat# NC9495720 |
| Trypsin antigen retrieval solution | Abcam | Cat# ab970 |
| Tissue-Tek OCT Compound | VWR | Cat# 25608-930 |
| Ethanol | GoldShield | Cat# 64175 |
| Xylene | Sigma | Cat# 534056 |
| Triton-X 100 | Sigma | Cat# X100 |
| Phosphate Buffered Saline | Sigma | Cat# P5368 |
| DAPI | BioLegend | Cat# 422801 |
| Hematoxylin | Sigma | Cat# H3136 |
| Eosin | Sigma | Cat# HT1101128 |
| Picro Sirius Red stain kit | Abcam | Cat# ab150681 |
| Critical commercial assays | ||
| PreOmics iST peptide purification kit | PreOmics (Martinsried, Germany) | Cat# P.O.00027 |
| Pierce Quantitative Colorimetric Peptide Assay | Thermo Fisher Scientific | Cat# 23275 |
| 10X Genomics Chromium Genetic Analyzer | 10X Genomics | RRID:SCR_019326 |
| Agilent Bioanalyzer | Agilent | RRID:SCR_018043 |
| Deposited data | ||
| Hashed single cell RNA sequencing data | GEO: GSE186527 | https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE186527 |
| Experimental models: Organisms/strains | ||
| C57/BL/6J | The Jackson Laboratory | Stock No. 000664 |
| En1 Cre-ERT | The Jackson Laboratory | Stock No. 007917 |
| YAP fl/fl | The Jackson Laboratory | Stock No. 027929 |
| Col1a1 Cre-ERT | The Jackson Laboratory | Stock No. 016241 |
| Ai9 | The Jackson Laboratory | Stock No. 007909 |
| Oligonucleotides | ||
| RNAscope Probe - Mm-Ankrd1-Opal 520 | ACD | Cat# 833651 |
| RNAscope Probe - Mm-Trps1-Opal 620 | ACD | Cat# 879008 |
| RNAscope Probe - Mm-Rspo1-Opal 690 | ACD | Cat# 479598 |
| RNAscope Probe - Mm-Dpp4-Opal 570 | ACD | Cat# 412578 |
| Polr2a-Opal 520/Ppib-Opal 570/Ubc-Opal 620/Hrpt1-Opal 690 (4-plex positive control) | ACD | Custom |
| dapB-Opal 520/dapB-Opal 570/dapB-Opal 620/dapB-Opal 690 (4-plex negative control) | ACD | Custom |
| Software and algorithms | ||
| ImageJ | National Institutes of Health | RRID:SCR_003070 |
| MATLAB | MathWorks | RRID:SCR_001622 |
| Rstudio | RStudio | RRID:SCR_000432 |
| Matrix ultrastructural analysis | This paper | https://github.com/shamikmascharak/Mascharak-et-al-ENF |
| RNAscope analysis pipeline | This paper | https://github.com/shamikmascharak/Mascharak-et-al-ENF |
| Other | ||
| 6-0 nylon sutures | Ethicon | Cat# 1698G |
| Tegaderm dressings | 3M | Cat# 1624W |
| QIAshredder spin columns | QIAGEN | Cat# 79656 |
Highlights.
Verteporfin (YAP inhibitor) treatment yields wound regeneration in mice
Divergent molecular pathways of scarring versus regeneration are defined by differential mechanical versus Wnt signaling
Trps1 drives wound regeneration in the context of blocked mechanical signaling
ACKNOWLEDGMENTS
We thank the Stanford Functional Genomics Facility, Stanford Cell Sciences Imaging Facility, and Stanford Shared FACS Facility Cores. Funding: this work was supported by the Hagey Laboratory for Pediatric Regenerative Medicine (to M.T.L., D.C.W., and G.C.G.); the Gunn/Olivier Research Fund, Stinehart/Reed Award, Scleroderma Research Foundation, Wu Tsai Human Performance Alliance, and Pitch and Catherine Johnson Fund (to M.T.L.); and NIH grants U24-DE029463 (to M.T.L., G.C.G., and D.C.W.), R01-GM136659 and R01-GM116892 (to M.T.L.), and R01-DE027346 (to D.C.W. and M.T.L.).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.stem.2021.12.011.
DECLARATION OF INTERESTS
M.T.L., S.M., H.E.T., and M.F.D. are inventors on patent 62/879,369 held by Stanford University that covers the use of YAP inhibition for wound healing. S.M., H.E.T., and M.T.L. are inventors on patent application PCT/US2020/043717 that covers a machine-learning algorithm for analysis of connective tissue networks in scarring and chronic fibroses. The authors declare no other competing interests.
REFERENCES
- Aarabi S, Bhatt KA, Shi Y, Paterno J, Chang EI, Loh SA, Holmes JW, Longaker MT, Yee H, and Gurtner GC (2007). Mechanical load initiates hypertrophic scar formation through decreased cellular apoptosis. FASEB J 21, 3250–3261. 10.1096/fj.07-8218com. [DOI] [PubMed] [Google Scholar]
- Abbasi S, Sinha S, Labit E, Rosin NL, Yoon G, Rahmani W, Jaffer A, Sharma N, Hagner A, Shah P, et al. (2020). Distinct regulatory programs control the latent regenerative potential of dermal fibroblasts during wound healing. Cell Stem Cell 27, 396. e6–412.e6.. 10.1016/j.stem.2020.07.008 [DOI] [PubMed] [Google Scholar]
- Aibar S, González-Blas CB, Moerman T, Huynh-Thu VA, Imrichova H, Hulselmans G, Rambow F, Marine JC, Geurts P, Aerts J, et al. (2017). SCENIC: single-cell regulatory network inference and clustering. Nat. Methods 14, 1083–1086. 10.1038/nmeth.4463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes LA, Marshall CD, Leavitt T, Hu MS, Moore AL, Gonzalez JG, Longaker MT, and Gurtner GC (2018). Mechanical forces in cutaneous wound healing: emerging therapies to minimize scar formation. Adv. Wound Care (New Rochelle) 7, 47–56. 10.1089/wound.2016.0709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergen V, Lange M, Peidli S, Wolf FA, and Theis FJ (2020). Generalizing RNA velocity to transient cell states through dynamical modeling. Nat. Biotechnol 38, 1408–1414. 10.1038/s41587-020-0591-3. [DOI] [PubMed] [Google Scholar]
- Brunner A-D, Thielert M, Vasilopoulou CG, Ammar C, Coscia F, Mund A, Hoerning OB, Bache N, Apalategui A, Lubeck M, et al. (2021). Ultra-high sensitivity mass spectrometry quantifies single-cell proteome changes upon perturbation. bioRxiv. 10.1101/2020.12.22.423933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant DM, Johnson K, DiTommaso T, Tickle T, Couger MB, Payzin-Dogru D, Lee TJ, Leigh ND, Kuo TH, Davis FG, et al. (2017). A tissue-mapped axolotl de novo transcriptome enables identification of limb regeneration factors. Cell Rep 18, 762–776. 10.1016/j.celrep.2016.12.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV, Clark NR, and Ma’ayan A (2013). Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics 14, 128. 10.1186/1471-2105-14-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elster D, Tollot M, Schlegelmilch K, Ori A, Rosenwald A, Sahai E, and von Eyss B (2018). TRPS1 shapes YAP/TEAD-dependent transcription in breast cancer cells. Nat. Commun 9, 3115. 10.1038/s41467-018-05370-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fantauzzo KA, Bazzi H, Jahoda CA, and Christiano AM (2008a). Dynamic expression of the zinc-finger transcription factor Trps1 during hair follicle morphogenesis and cycling. Gene Expr. Patterns 8, 51–57. 10.1016/j.modgep.2007.10.006. [DOI] [PubMed] [Google Scholar]
- Fantauzzo KA, and Christiano AM (2012). Trps1 activates a network of secreted Wnt inhibitors and transcription factors crucial to vibrissa follicle morphogenesis. Development 139, 203–214. 10.1242/dev.069971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fantauzzo KA, Kurban M, Levy B, and Christiano AM (2012). Trps1 and its target gene Sox9 regulate epithelial proliferation in the developing hair follicle and are associated with hypertrichosis. PLoS Genet 8, e1003002. 10.1371/journal.pgen.1003002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fantauzzo KA, Tadin-Strapps M, You Y, Mentzer SE, Baumeister FA, Cianfarani S, Van Maldergem L, Warburton D, Sundberg JP, and Christiano AM (2008b). A position effect on TRPS1 is associated with Ambras syndrome in humans and the Koala phenotype in mice. Hum. Mol. Genet 17, 3539–3551. 10.1093/hmg/ddn247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster DS, Januszyk M, Yost KE, Chinta MS, Gulati GS, Nguyen AT, Burcham AR, Salhotra A, Ransom RC, Henn D, et al. (2021). Integrated spatial multiomics reveals fibroblast fate during tissue repair. Proc. Natl. Acad. Sci. USA 118, e2110025118. 10.1073/pnas.2110025118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galiano RD, Michaels JT, Dobryansky M, Levine JP, and Gurtner GC (2004). Quantitative and reproducible murine model of excisional wound healing. Wound Repair Regen 12, 485–492. 10.1111/j.1067-1927.2004.12404.x. [DOI] [PubMed] [Google Scholar]
- Griffin MF, desJardins-Park HE, Mascharak S, Borrelli MR, and Longaker MT (2020). Understanding the impact of fibroblast heterogeneity on skin fibrosis. Dis. Model. Mech 13, dmm044164. 10.1242/dmm.044164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guicciardi ME, Krishnan A, Bronk SF, Hirsova P, Griffith TS, and Gores GJ (2017). Biliary tract instillation of a SMAC mimetic induces TRAIL-dependent acute sclerosing cholangitis-like injury in mice. Cell Death Dis 8, e2535. 10.1038/cddis.2016.459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulati GS, Sikandar SS, Wesche DJ, Manjunath A, Bharadwaj A, Berger MJ, Ilagan F, Kuo AH, Hsieh RW, Cai S, et al. (2020). Single-cell transcriptional diversity is a hallmark of developmental potential. Science 367, 405–411. 10.1126/science.aax0249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han M, Yang X, Farrington JE, and Muneoka K (2003). Digit regeneration is regulated by Msx1 and BMP4 in fetal mice. Development 130, 5123–5132. 10.1242/dev.00710. [DOI] [PubMed] [Google Scholar]
- Hua X, Wang YY, Jia P, Xiong Q, Hu Y, Chang Y, Lai S, Xu Y, Zhao Z, and Song J (2020). Multi-level transcriptome sequencing identifies COL1A1 as a candidate marker in human heart failure progression. BMC Med 18, 2. 10.1186/s12916-019-1469-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito M, Yang Z, Andl T, Cui C, Kim N, Millar SE, and Cotsarelis G (2007). Wnt-dependent de novo hair follicle regeneration in adult mouse skin after wounding. Nature 447, 316–320. 10.1038/nature05766. [DOI] [PubMed] [Google Scholar]
- Jin S, Guerrero-Juarez CF, Zhang L, Chang I, Ramos R, Kuan CH, Myung P, Plikus MV, and Nie Q (2021). Inference and analysis of cell-cell communication using CellChat. Nat. Commun 12, 1088. 10.1038/s41467-021-21246-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim CH, Sun Q, Ratti K, Lee SH, Zheng Y, Takeo M, Lee W, Rabbani P, Plikus MV, Cain JE, et al. (2018). Hedgehog stimulates hair follicle neogenesis by creating inductive dermis during murine skin wound healing. Nat. Commun 9, 4903. 10.1038/s41467-018-07142-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mascharak S, desJardins-Park HE, Davitt MF, Griffin M, Borrelli MR, Moore AL, Chen K, Duoto B, Chinta M, Foster DS, et al. (2021). Preventing Engrailed-1 activation in fibroblasts yields wound regeneration without scarring. Science 372, eaba2374. 10.1126/science.aba2374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mascharak S, desJardins-Park HE, and Longaker MT (2020). Fibroblast heterogeneity in wound healing: hurdles to clinical translation. Trends Mol. Med 26, 1101–1106. 10.1016/j.molmed.2020.07.008. [DOI] [PubMed] [Google Scholar]
- Murphy JM, Rodriguez YAR, Jeong K, Ahn EE, and Lim SS (2020). Targeting focal adhesion kinase in cancer cells and the tumor microenvironment. Exp. Mol. Med 52, 877–886. 10.1038/s12276-020-0447-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myung PS, Takeo M, Ito M, and Atit RP (2013). Epithelial Wnt ligand secretion is required for adult hair follicle growth and regeneration. J. Invest. Dermatol 133, 31–41. 10.1038/jid.2012.230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedroza M, Welschhans RL, and Agarwal SK (2017). Targeting of cadherin-11 decreases skin fibrosis in the tight skin-1 mouse model. PLoS One 12, e0187109. 10.1371/journal.pone.0187109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phan QM, Sinha S, Biernaskie J, and Driskell RR (2021). Single-cell transcriptomic analysis of small and large wounds reveals the distinct spatial organization of regenerative fibroblasts. Exp. Dermatol 30, 92–101. 10.1111/exd.14244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reish RG, and Eriksson E (2008). Scar treatments: preclinical and clinical studies. J. Am. Coll. Surg 206, 719–730. 10.1016/j.jamcoll-surg.2007.11.022. [DOI] [PubMed] [Google Scholar]
- Rinkevich Y, Walmsley GG, Hu MS, Maan ZN, Newman AM, Drukker M, Januszyk M, Krampitz GW, Gurtner GC, Lorenz HP, et al. (2015). Skin fibrosis. Identification and isolation of a dermal lineage with intrinsic fibro-genic potential. Science 348, aaa2151. 10.1126/science.aaa2151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruifrok AC, and Johnston DA (2001). Quantification of histochemical staining by color deconvolution. Anal Quant Cytol. Histol 23, 291–299. [PubMed] [Google Scholar]
- Ruifrok AC, Katz RL, and Johnston DA (2003). Comparison of quantification of histochemical staining by hue-saturation-intensity (HSI) transformation and color-deconvolution. Appl. Immunohistochem. Mol. Morphol 11, 85–91. [DOI] [PubMed] [Google Scholar]
- Schneider DJ, Wu M, Le TT, Cho S-H, Brenner MB, Blackburn MR, and Agarwal SK (2012). Cadherin-11 contributes to pulmonary fibrosis: potential role in TGF-β production and epithelial to mesenchymal transition. FASEB J 26, 503–512. 10.1096/fj.11-186098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Y, Boncompagni AC, Kim SS, Gochnauer HR, Zhang Y, Loots GG, Wu D, Li Y, Xu M, and Millar SE (2018). Regional control of hairless versus hair-bearing skin by Dkk2. Cell Rep 25, 2981.e3. 2991.e3. 10.1016/j.celrep.2018.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stauffer W, Sheng H, and Lim HN (2018). EzColocalization: An ImageJ plugin for visualizing and measuring colocalization in cells and organisms. Sci. Rep 8, 15764. 10.1038/s41598-018-33592-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terashima H, Aonuma M, Tsuchida H, Sugimoto K, Yokoyama M, and Kato M (2019). Attenuation of pulmonary fibrosis in type I collagen-targeted reporter mice with ALK-5 inhibitors. Pulm. Pharmacol. Ther 54, 31–38. 10.1016/j.pupt.2018.11.005. [DOI] [PubMed] [Google Scholar]
- Trapnell C, Cacchiarelli D, Grimsby J, Pokharel P, Li S, Morse M, Lennon NJ, Livak KJ, Mikkelsen TS, and Rinn JL (2014). The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nature biotechnology 32, 381–386. 10.1038/nbt.2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van de Sande B, Flerin C, Davie K, De Waegeneer M, Hulselmans G, Aibar S, Seurinck R, Saelens W, Cannoodt R, Rouchon Q, et al. (2020). A scalable SCENIC workflow for single-cell gene regulatory network analysis. Nat. Protoc 15, 2247–2276. 10.1038/s41596-020-0336-2. [DOI] [PubMed] [Google Scholar]
- Wong VW, Rustad KC, Akaishi S, Sorkin M, Glotzbach JP, Januszyk M, Nelson ER, Levi K, Paterno J, Vial IN, et al. (2011). Focal adhesion kinase links mechanical force to skin fibrosis via inflammatory signaling. Nat. Med 18, 148–152. 10.1038/nm.2574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wynn TA (2004). Fibrotic disease and the T(H)1/T(H)2 paradigm. Nat. Rev. Immunol 4, 583–594. 10.1038/nri1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The scRNA-seq data generated during this study have been deposited in a GEO repository (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE186527). These data are publicly available as of the date of publication. The accession number is listed in the key resources table.
Original scripts for the ECM ultrastructure and RNAscope analysis algorithms used in this study have been deposited in a Github repository (https://github.com/shamikmascharak/Mascharak-et-al-ENF) and are publicly available as of the date of publication. DOIs are listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.