

Abstract
Covalent labeling mass spectrometry allows for protein structure elucidation via covalent modification and identification of exposed residues. Diethylpyrocarbonate (DEPC) is a commonly used covalent labeling reagent that provides insight into structure through the labeling of lysine, histidine, serine, threonine, and tyrosine residues. We recently implemented a Rosetta algorithm that used binary DEPC labeling data to improve protein structure prediction efforts. In this work, we improved on our modeling efforts by accounting for the level of hydrophobicity of neighboring residues in the microenvironment of serine, threonine, and tyrosine residues to obtain a more accurate estimate of the hydrophobic neighbor count. This was incorporated into Rosetta functionality, along with considerations for solvent exposed histidine and lysine residues. Overall, our new Rosetta score term successfully identified best scoring models with less than 2 Å root-mean-squared deviations (RMSDs) for five of the seven benchmark proteins tested. We additionally developed a confidence metric to measure prediction success for situations in which a native structure is unavailable.
Graphical Abstract
INTRODUCTION
Knowledge of protein structure can enable understanding and manipulation of protein function. While techniques exist to explicitly determine protein structure, experimental limitations can impede structure elucidation. With low sample amount requirements, no size limitations, and no crystallization requirement, structural mass spectrometry (MS) is a valuable experimental technique that can provide insight into protein structure in the absence of higher resolution information.(1–3) Covalent labeling MS (CL-MS) employs reagents that covalently modify proteins in solution to glean structural and topological information, particularly solvent exposure for modified residues.(1) Some CL-MS experiments include hydroxyl radical protein footprinting, trifluoromethylation, carbenes, and diethylpyrocarbonate (DEPC) labeling.(1,2,4,5) DEPC is a commercially-available labeling reagent that produces a single product with +72.021 Da mass addition during the covalent modification of the protein N-terminus, Cys, Lys, His, Ser, Thr, and Tyr.(4,6–8) Knowledge of DEPC-labeled and unlabeled residues coupled with computational methods can be used for protein structure elucidation.(9)
Computational methods in combination with MS and other experimental data have been previously shown to support protein structure prediction efforts across different modeling platforms.(9–20) Hydrogen-deuterium exchange (HDX) data from MS and nuclear magnetic resonance and chemical cross-linking (XL) data have been used for successful protein modeling.(21–26) XL data provides insight into spatial proximity of residues, while HDX data can be used to infer solvent exposure of modified resides.(1) CL-MS data provides several advantages for structural modeling over XL and HDX data due to inherent advantages in the data. CL-MS employs irreversible labeling with reagents to target a variety of residue types that can be more precisely confirmed at the residue level during data analysis. Identifying cross-linked sites in XL is more challenging, and the surface distance restraints provided by XL are associated with high uncertainty. As commonly applied, HDX does not provide residue-level information and is prone to back exchange during MS analysis, often resulting in the loss of structural information.(4) Rosetta is a molecular modeling suite that can be used to examine structure prediction via generation of ab initio and homology models.(11,27–29) Functionality within Rosetta also exists to examine solvent exposure of protein models, which can be used to aid in structure prediction.(21,22) Additionally, Rosetta has been a successful venue for incorporation of MS data, including CL-MS data, into protein structure prediction efforts, allowing sparse experimental data to guide modeling when atomic-resolution structure elucidation methods fall short.(1,9,10,12,13,24–26,30–35)
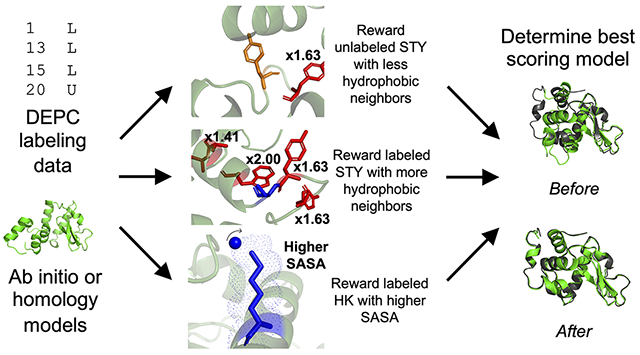
Previously, we incorporated the first DEPC-guided Rosetta score term based on the labeling sensitivity of Ser, Thr, and Tyr (STY) residues to a hydrophobic microenvironment and based on solvent exposure of His and Lys residues.(9) Labeled and unlabeled STY residues with low relative solvent accessible surface area (SASA) were rewarded based on the number of hydrophobic neighbors. Labeled residues with more hydrophobic neighbors and unlabeled residues with fewer hydrophobic residues were rewarded based on our previous work that indicated neighboring hydrophobic residues promoted an increased local concentration of DEPC and thus facilitated labeling.(8) Overall, we demonstrated that modeling guided by DEPC led to improved structure prediction with both ab initio and homology modeling, as the best scoring model root-mean-square deviation (RMSD) improved with score term usage. Here, we sought to build upon this previous work by accounting for the level of hydrophobicity of neighboring residues within the microenvironment. By using the normalized hydrophobicity of residues to dictate their contribution to the hydrophobic neighbor count (HNC), we have identified a more distinct difference in HNC distributions for labeled and unlabeled residues. This guided the development of a label status-based score term, depc_hydrophobicity, that combined the solvent exposure rewards for labeled His and Lys residues with the HNC rewards for STY residues based on the proximity of hydrophobic neighbors within the residue microenvironment. In a benchmark test of our algorithm, we used DEPC data for seven proteins of known structure to guide modeling. We identified accurate atomic detail in the best scoring models of five of the seven proteins using both ab initio and homology model sets.
METHODS
Benchmark Set of Proteins with DEPC Labeling Data
Our benchmark set consisted of seven proteins, including myoglobin (PDB 1DWR, 152 residues), human growth hormone (HGH, PDB 1HGU, 191 residues), β2-microglobulin (β2m, PDB 1JNJ, 100 residues), ubiquitin (PDB 1UBQ, 76 residues), carbonic anhydrase (PDB 1V9E, 259 residues), superfolder green fluorescent protein (sfGFP, PDB 2B3P, 244 residues), and lysozyme (PDB 2LYZ, 129 residues). Experimental data collection for myoglobin(8), β2m(8), ubiquitin(8), lysozyme(36), HGH(8), and carbonic anhydrase(37) has been described and published previously, with DEPC labeling status data for His, Lys, Ser, Thr, and Tyr residues for each protein.(9) Experimental data for sfGFP has been included in Table S1.
Benchmark protein ab initio and homology model generation
For all seven benchmark proteins, 3mer and 9mer fragments were generated using the Robetta fragment server based on the amino acid sequence of the proteins deposited in the Protein Data Bank.(38) Ab initio sets of 10,000 models per benchmark protein were generated with the Rosetta AbinitioRelax protocol using the fragment libraries and FASTA sequences. Models were scored with the Rosetta Energy Function 15 (Ref15) and ranked by score. Alpha carbon root-mean-square deviation (RMSD) values were calculated in Rosetta during scoring by providing the crystal structure. As such, residues that were not resolved in the crystal structure were not modeled and not included in RMSD calculations. The crystal structures were used exclusively for RMSD calculations. The best RMSD model generation was considered as a metric for homology modeling: if a model set’s best RMSD model generated had an RMSD of larger than 5 Å, homology modeling was pursued for model distribution generation.
Rosetta’s Comparative Modeling protocol was employed for homology model production for carbonic anhydrase, lysozyme, HGH, and sfGFP.(28) Five templates (Table S2) per benchmark protein with different sequence identities (23%-91%) and coverages (52%-100%) were used for generation of 3,500 models per template. Homology models were relaxed with the Rosetta Relax application prior to scoring with Ref15 and calculating RMSDs.(39)
Hydrophobicity Contribution in Hydrophobic Neighbor Counts
Hydrophobic neighbor count (HNC) was calculated for labeled and unlabeled STY residues from benchmark protein crystal structures by a custom Python script that utilized previously published normalized hydrophobicity values.(40) The HNC used a gradual contribution method, meaning that the contribution to the HNC increased as the distance between the STY residue of interest and the hydrophobic neighbor decreased. The distances (distij) between the beta carbons of hydrophobic residues (j) and the hydroxyl oxygens of STY residues (i) were determined and factored into the HNC, as shown in Eq 1.
| Eq. 1 |
The contribution of a particular hydrophobic neighbor was multiplied by a factor of 1.XX, in which XX represented the normalized hydrophobicity of the residue.(40) The multiplication factors were 2.00 for Phe, 1.99 for Ile, 1.97 for Trp and Leu, 1.63 for Tyr, and 1.41 for Ala. Based on DEPC molecule dimensions, an HNC midpoint value (8 Å) and steepness (2.0) were employed to maximize the HNC of hydrophobic residues within 6 Å.(8) The total contribution was calculated by summing all the hydrophobic neighbor contributions relevant to the STY residue. Additionally, we previously explored data accuracy and the tolerance towards false negatives in a covalent labeling data set. We found that such data sets can accommodate 35% false negative data points without losing their ability to meaningfully guide structural modeling, and our dataset for this work fell within that range.(9,12)
Determination and employment of SASA
Relative SASA calculations were performed with the Rosetta RelSASA application. Relative SASA was defined as the determined residue side chain SASA divided by the residue side chain SASA from a Gly-X-Gly tripeptide.
Different SASA ranges were used for both STY residues and labeled His and Lys (HK) residues. Labeled and unlabeled STY residues with 30-60% relative SASA and labeled HK residues with greater than 50% relative SASA were examined. Within these ranges, 23 labeled STY residues, 44 unlabeled STY residues, and 44 HK residues were included across the seven benchmark proteins.
Scoring model agreement with DEPC labeling data
The ab initio and homology models were scored with depc_hydrophobicity, our newly implemented score term. Our score term rewarded models based on agreement with the labeling data, resulting in models that demonstrated stronger agreement with the data receiving more favorable scores. The DEPC-guided score term was calculated based on contributions from labeled STY residues, unlabeled STY residues, and labeled HK residues, as shown in Eq. 2.
| Eq. 2 |
For the labeled and unlabeled STY portions of the equation, the HNC of the labeled or unlabeled STY residue, respectively, was calculated within the Rosetta score term according to Eq. 1. The labeled STY midpoint, 5.29, was the average labeled HNC value as calculated using the benchmark protein crystal structures. Labeled STY residues were rewarded for having a higher-than-average HNC, while unlabeled STY residues were rewarded for having a lower-than-average HNC. The unlabeled STY midpoint, 3.81, was the average unlabeled HNC as calculated using benchmark protein crystal structures. The labeled HK midpoint, 0.50, represented a relative SASA of 50%. Labeled HK residues were rewarded for having higher relative SASA values. The depc_hydrophobicity score was weighted and added to the initial Rosetta score, as demonstrated in Eq. 3:
where a weight of 11.0 was used. Improvements from the depc_hydrophobicity scoring protocol were evaluated by examining the best scoring model RMSD compared to scoring with just Rosetta Ref15. Best, or lowest, scoring models were examined because the best scoring model is thought to be the most native-like, so improvements in the best scoring model RMSD would indicate better model selection.
Confidence Metric
In order to assess the confidence in our modeling results, we implemented a size-normalized score confidence metric. Rosetta with DEPC data scores were isolated for the best scoring models. Scores were divided by the number of residues in the respective benchmark protein to obtain a size-normalized score. A lower size-normalized score corresponded to a higher confidence prediction.
RESULTS AND DISCUSSION
Incorporation of level of hydrophobicity contribution and parameterization of score term
The hydrophobic residue microenvironment has previously been shown to impact DEPC labeling of STY residues, as more hydrophobic residues were observed in the microenvironment of labeled STY residues than unlabeled STY residues.(8) In earlier efforts, we modeled protein structure based on DEPC labeling data, but we did not account for varying levels of hydrophobicity of neighboring residues. Here, we hypothesized that residues with higher levels of hydrophobicity would further facilitate DEPC labeling. We thus sought to determine whether accounting for the level of hydrophobicity of neighboring residues in the microenvironment could lead to improvements in protein structure prediction. To do this, we examined the residue microenvironment of seven proteins for which DEPC labeling data was available: β2m, carbonic anhydrase, HGH, lysozyme, myoglobin, sfGFP, and ubiquitin. We calculated the HNC of each labeled and unlabeled STY residue by implementing a gradual neighbor contribution method in which the contribution of hydrophobic neighbors to the HNC was scaled based on their distance to the STY residue. The HNC was weighted by the actual normalized hydrophobicity of the neighboring residue. When comparing the HNC distributions between labeled and unlabeled STY residues (Figure 1), labeled STY residues exhibited higher HNCs than unlabeled STY residues.
Figure 1.
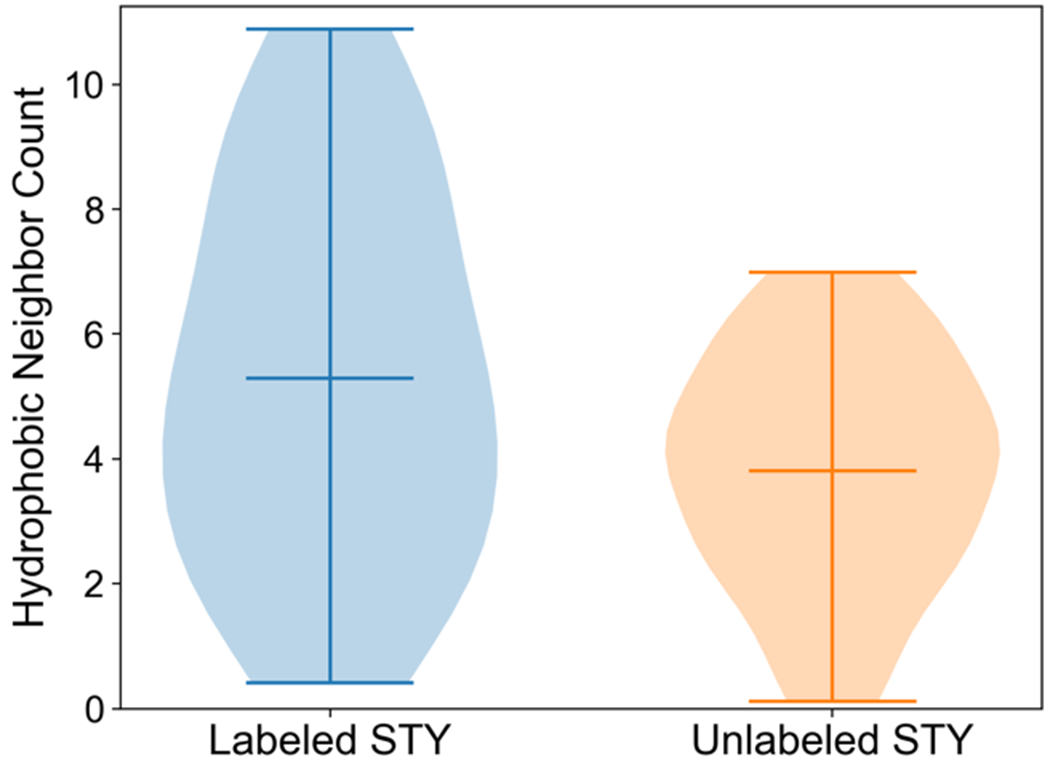
Comparison of relative frequencies of hydrophobic neighbor count values for labeled and unlabeled Ser, Thr, and Tyr residues with low to moderate exposure. Labeled residues (23) and unlabeled residues (44) from the seven benchmark proteins are included in the violin plot. Average, minimum, and maximum HNCs are shown.
The average HNC for labeled STY was 5.29 and for unlabeled STY it was 3.81, meaning that on average every labeled STY residue had 1.5 additional hydrophobic neighboring residues as compared to unlabeled STY residues. Of the unlabeled residues, 73% had HNC less than five. in contrast, 60% of labeled residues had HNC greater than four. Overall, this demonstrated that the hydrophobicity contribution incorporated into the HNC calculation led to notable HNC difference between labeled and unlabeled STY residues while capturing the effect of neighboring residue identity in the microenvironment.
In addition to STY residues, we aimed to account for labeling of His and Lys residues. Since residues with higher solvent exposure are more likely to be exposed to a labeling reagent, we examined the differences in relative SASAs between labeled and unlabeled HK residues. Figure S1 demonstrates the comparison of relative SASA values between labeled and unlabeled HK residues. Of 58 HK residues with 50-100% SASA in the benchmark proteins, 44 residues were labeled while only 14 were unlabeled. Labeled HK residues had an average SASA of 73%, which was higher than the unlabeled average SASA (66%). Consequently, we pursued rewarding labeled HK residues with higher SASA values.
Implementation of DEPC-guided term led to improvements in best scoring model RMSD
We developed a Rosetta score term in which label status, residue microenvironment hydrophobicity, solvent exposure, and residue type guided model scoring. HNC was calculated for labeled and unlabeled STY residues using the level of hydrophobicity of the immediate microenvironment. Labeled STY residues were rewarded for having a large number of hydrophobic neighbors while unlabeled STY residues were rewarded for having a small number of hydrophobic neighbors. Relative SASA was calculated for labeled HK residues, which was used to reward those HK residues with high solvent exposure. We generated 10,000 ab initio models of all seven benchmark proteins in order to evaluate our new term. However, for four of the benchmark proteins, the best RMSD model generated was larger than 5 Å, indicating that native-like models were not present within the model set. For those four proteins (carbonic anhydrase, HGH, lysozyme, and sfGFP), we pursued homology modeling using multiple templates with varying sequence identity and coverage.
Both ab initio and homology model sets were scored with Rosetta and the new depc_hydrophobicity term to determine a total score. Score versus RMSD plots and best scoring models aligned with the crystal structures for each benchmark protein are shown in Figure 2 for ab initio models and Figure 3 for homology models.
Figure 2.
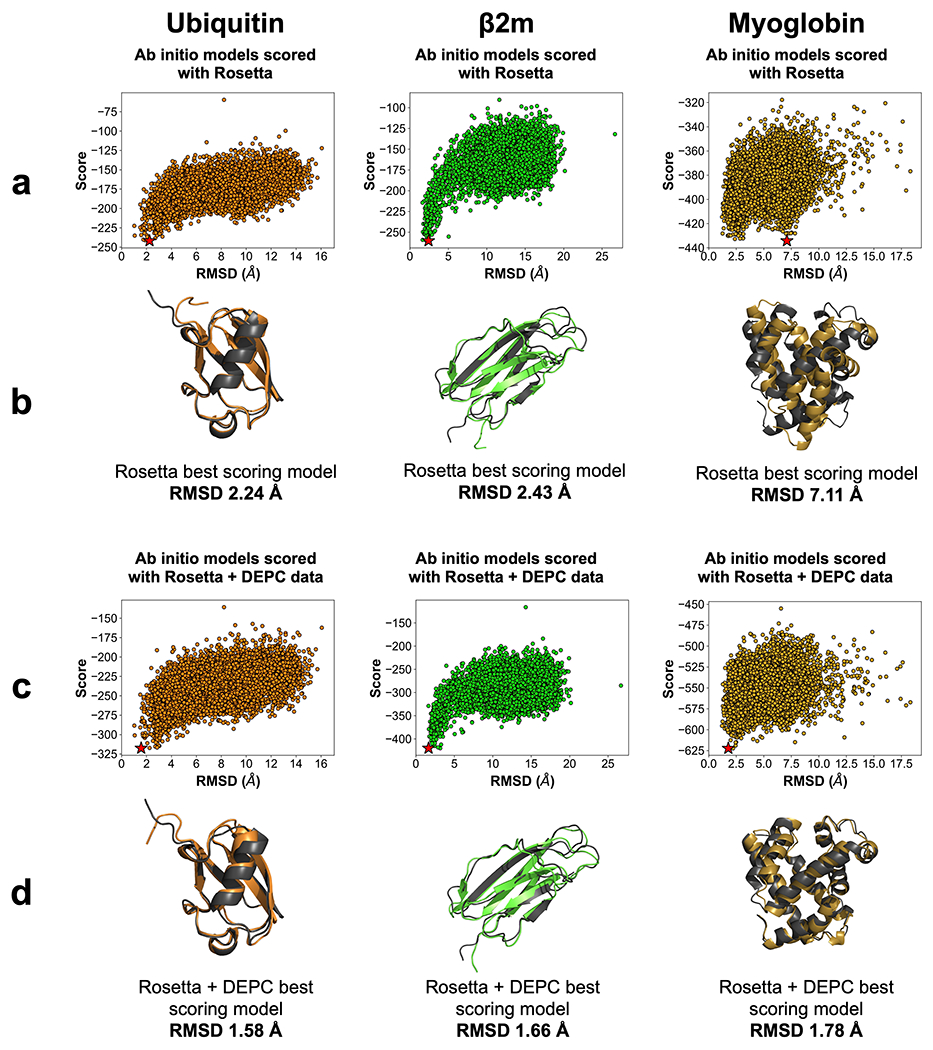
(a) Rosetta score versus RMSD plots for 10,000 ab initio models of ubiquitin (orange), β2m (lime), and myoglobin (gold). Best scoring models are denoted by a star (★). (b) Best scoring Rosetta model (color) aligned with respective crystal structure (dark grey). RMSD is listed below the alignments. (c) Rosetta and DEPC score versus RMSD plots for benchmark protein model sets. Best scoring models are denoted by a star (★). (d) Best scoring Rosetta and DEPC model (color) aligned with respective crystal structure (dark grey). RMSD is listed below the alignments.
Figure 3.
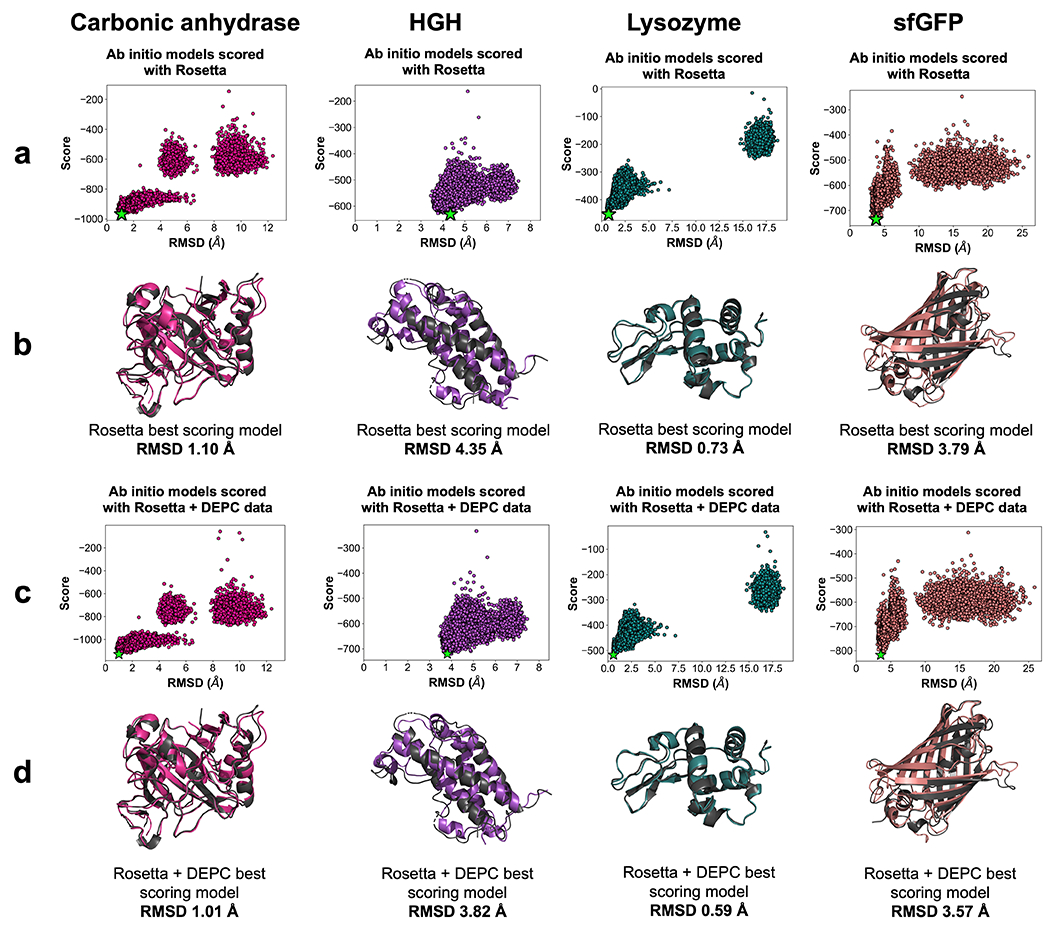
(a) Rosetta score versus RMSD plots for homology models of carbonic anhydrase (deep pink), HGH (light purple), lysozyme (teal), and sfGFP (light coral). Best scoring models are denoted by a star (★). (b) Best scoring Rosetta model (color) aligned with respective crystal structure (dark grey). RMSD is listed below the alignments. (c) Rosetta and DEPC score versus RMSD plots for benchmark protein model sets. Best scoring models are denoted by a star (★). (d) Best scoring Rosetta and DEPC model (color) aligned with respective crystal structure (dark grey). RMSD is listed below the alignments. The disconnected point clouds observed for carbonic anhydrase, lysozyme, and sfGFP are a result of using multiple templates for homology modeling. These point clouds represent similarity with particular templates employed in the homology modeling process.
Overall, scoring with the depc_hydrophobicity term consistently improved the best scoring model RMSD from scoring with Rosetta for ab initio model sets. The best scoring model RMSD of ubiquitin improved from 2.24 Å with Rosetta to 1.58 Å with Rosetta and DEPC-guided scoring. Because of the possible flexibility of the C-terminal loop of ubiquitin, it was impossible to confirm that the best scoring model from Rosetta and DEPC-guided scoring indeed had better C-terminal agreement. Improvements were also noted for β2m (from 2.43 Å to 1.66 Å). Myoglobin demonstrated notable changes, improving from 7.11 Å with Rosetta scoring to 1.78 Å with Rosetta and DEPC scoring.
Of the homology modeling sets, consistent best scoring RMSD improvements were also observed. HGH exhibited improvements, with the best scoring model RMSD changing from 4.35 Å to 3.82 Å. The carbonic anhydrase best scoring model RMSD decreased from 1.10 Å to 1.01 Å. sfGFP had improvement from 3.79 Å with Rosetta to 3.57 Å with Rosetta including DEPC data. Finally, lysozyme improved from 0.73 Å to 0.59 Å. Since the crystallographic resolution for lysozyme (PDB 2LYZ) was 2 Å, both of these models were considered perfect predictions.
Confidence in modeling efforts was established via size-normalized score metric
Scoring with DEPC data improved predictions for all benchmark proteins and led to the identification of near-native-like (< 2 Å RMSD) models for five of the seven benchmark proteins pursued. We developed a confidence metric as a method to establish confidence in results when RMSD cannot be calculated. In the absence of a native model, generally the lowest scoring model is identified as the predicted structure, as a lower Rosetta score indicates a more energetically favorable conformation that is thought to be the most native-like model. In the absence of experimental data in model generation based on Monte Carlo sampling, these best scoring models may constitute outliers such as the myoglobin best scoring model with Rosetta Ref15 scoring. In order to independently evaluate models predicted by our DEPC-guided scoring approach, we implemented a confidence metric, the size-normalized score, to test prediction accuracy in the absence of a solved structure. Depending on the system, confidence can be inferred from the steepness of the point funnel from score versus RMSD plots, where deeper funnels correspond to higher confidence. However, a metric such as the size-normalized score contributed an additional layer of confidence for when funnels could not be attained due to lack of a native structure for RMSD calculations. The size-normalized score was determined using the top scoring model when scored with Rosetta and DEPC data. Each model’s score was divided by the number of residues in the respective protein. A lower size-normalized score was considered more native-like, thus establishing more confidence in the prediction. Figure 4 illustrates the top scoring model RMSD versus its size-normalized score when scoring with Rosetta including DEPC data.
Figure 4.
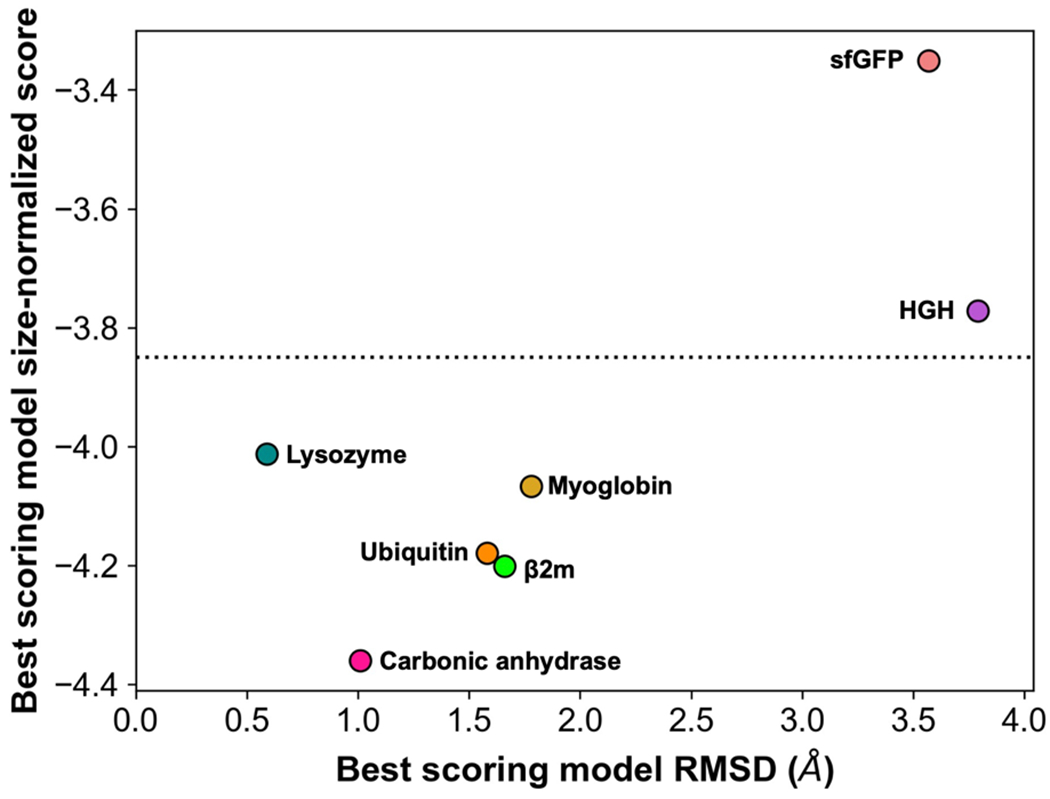
RMSD versus size-normalized score for best scoring benchmark protein models when scored with Rosetta + DEPC data. Points are labeled by protein. Models of high confidence fall below the dotted line, and models of lower confidence appear above the dotted line.
A size-normalized score value of −3.85 was established as the cutoff between high and low confidence modeling, reinforced by the 5 perfectly predicted benchmark proteins all having size-normalized score values less than −3.85. Two proteins, HGH and sfGFP, were of lower confidence than the remaining proteins in the benchmark set. Their best models generated in the set had RMSDs of 3.46 Å and 2.92 Å, respectively, and thus were correctly labeled as less accurate. The absence of near-native, high-confidence models for HGH and sfGFP was a direct consequence of a lack of high-quality templates available for homology modeling. Overall, the confidence metric effectively confirmed the prediction accuracy with DEPC-MS data and can be applied to systems in which RMSD calculations are unattainable.
CONCLUSION
In an effort to further account for the effect of the residue microenvironment in DEPC labeling, we incorporated an attribute into our hydrophobic neighbor count calculation that accounts for the normalized hydrophobicity of the neighboring residue. HNCs were calculated for both labeled and unlabeled STY residues and were subsequently used to guide scoring. Labeled STY residues with higher HNC and unlabeled STY residues with lower HNC were rewarded. Additionally, exposed HK residues were rewarded. Our score term was tested on a benchmark set of seven proteins, including β2m, carbonic anhydrase, HGH, lysozyme, myoglobin, sfGFP, and ubiquitin. Implementation of our DEPC data-guided score term improved best scoring model RMSD for all benchmark protein model sets, demonstrating the score term’s success with both ab initio and homology model sets. Additionally, five of the seven benchmark proteins had best scoring model RMSDs less than 2 Å. Our work further demonstrates the power of DEPC labeling data in combination with computational modeling efforts, as accounting for the level of hydrophobicity led to improvements with structural modeling. As such, we conclude that more hydrophobic microenvironments facilitate DEPC labeling of STY residues.
Future work will emphasize dynamics involved in DEPC labeling. Additionally, we aim to incorporate DEPC labeling data into structure prediction for protein complexes.
Supplementary Material
ACKNOWLEDGMENT
The authors thank members of the Lindert and Vachet labs for useful discussions and the Ohio Supercomputer Center for resources.(41) The authors additionally thank the Rosetta Commons community and the Rosetta Commons Research Experience for Undergraduates, supported by NSF (DBI 1950697). Research reported in this publication was supported by NSF (CHE 1750666) to S.L. and by the NIH (R01 GM075092) to R.W.V.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
DEPC labeling data for sfGFP, table of templates used for homology modeling generation, and violin plot comparing relative SASA between labeled and unlabeled HK residues (PDF)
REFERENCES
- 1.Biehn SE, and Lindert S (2022) Protein Structure Prediction with Mass Spectrometry Data. Annu. Rev. Phys. Chem 73, 1.1–1.19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Liu XR, Zhang MM, and Gross ML (2020) Mass Spectrometry-Based Protein Footprinting for Higher-Order Structure Analysis: Fundamentals and Applications. Chemical Reviews 120, 4355–4454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Seffernick JT, and Lindert S (2020) Hybrid methods for combined experimental and computational determination of protein structure. The Journal of Chemical Physics 153, 240901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Limpikirati P, Liu T, and Vachet RW (2018) Covalent labeling-mass spectrometry with non-specific reagents for studying protein structure and interactions. Methods 144, 79–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McKenzie-Coe A, Montes NS, and Jones LM (2021) Hydroxyl Radical Protein Footprinting: A Mass Spectrometry-Based Structural Method for Studying the Higher Order Structure of Proteins. Chemical Reviews [DOI] [PubMed] [Google Scholar]
- 6.Mendoza VL, and Vachet RW (2009) Probing protein structure by amino acid-specific covalent labeling and mass spectrometry. Mass Spectrometry Reviews 28, 785–815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mendoza VL, and Vachet RW (2008) Protein surface mapping using diethylpyrocarbonate with mass spectrometric detection. Analytical Chemistry 80, 2895–2904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Limpikirati P, Pan X, and Vachet RW (2019) Covalent Labeling with Diethylpyrocarbonate: Sensitive to the Residue Microenvironment, Providing Improved Analysis of Protein Higher Order Structure by Mass Spectrometry. Analytical Chemistry 91, 8516–8523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Biehn SE, Limpikirati P, Vachet RW, and Lindert S (2021) Utilization of hydrophobic microenvironment sensitivity in diethylpyrocarbonate labeling for protein structure prediction. Analytical Chemistry 93, 8188–8195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Biehn SE, and Lindert S (2021) Accurate protein structure prediction with hydroxyl radical protein footprinting data. Nature Communications 12, 341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Leman JK, Weitzner BD, Lewis SM, Adolf-Bryfogle J, Alam N, Alford RF, Aprahamian M, Baker D, Barlow KA, and Barth P (2020) Macromolecular modeling and design in Rosetta: recent methods and frameworks. Nature Methods, 1–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Aprahamian ML, and Lindert S (2019) Utility of Covalent Labeling Mass Spectrometry Data in Protein Structure Prediction with Rosetta. Journal of Chemical Theory and Computation 15, 3410–3424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Aprahamian ML, Chea EE, Jones LM, and Lindert S (2018) Rosetta protein structure prediction from hydroxyl radical protein footprinting mass spectrometry data. Analytical Chemistry 90, 7721–7729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Benesch JL, and Ruotolo BT (2011) Mass spectrometry: come of age for structural and dynamical biology. Current Opinion in Structural Biology 21, 641–649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jia R, Martens C, Shekhar M, Pant S, Pellowe GA, Lau AM, Findlay HE, Harris NJ, Tajkhorshid E, and Booth PJ (2020) Hydrogen-deuterium exchange mass spectrometry captures distinct dynamics upon substrate and inhibitor binding to a transporter. bioRxiv [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Murcia Rios A, Vahidi S, Dunn SD, and Konermann L (2018) Evidence for a Partially Stalled γ Rotor in F1-ATPase from Hydrogen–Deuterium Exchange Experiments and Molecular Dynamics Simulations. Journal of the American Chemical Society 140, 14860–14869 [DOI] [PubMed] [Google Scholar]
- 17.Hall Z, Politis A, and Robinson CV (2012) Structural modeling of heteromeric protein complexes from disassembly pathways and ion mobility-mass spectrometry. Structure 20, 1596–1609 [DOI] [PubMed] [Google Scholar]
- 18.Eschweiler JD, Farrugia MA, Dixit SM, Hausinger RP, and Ruotolo BT (2018) A structural model of the urease activation complex derived from ion mobility-mass spectrometry and integrative modeling. Structure 26, 599–606. e593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mistarz UH, Chandler SA, Brown JM, Benesch JL, and Rand KD (2019) Probing the dissociation of protein complexes by means of gas-phase h/d exchange mass spectrometry. Journal of The American Society for Mass Spectrometry 30, 45–57 [DOI] [PubMed] [Google Scholar]
- 20.Busch F, VanAernum ZL, Ju Y, Yan J, Gilbert JD, Quintyn RS, Bern M, and Wysocki VH (2018) Localization of protein complex bound ligands by surface-induced dissociation high-resolution mass spectrometry. Analytical Chemistry 90, 12796–12801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Marzolf DR, Seffernick JT, and Lindert S (2021) Protein Structure Prediction from NMR Hydrogen–Deuterium Exchange Data. Journal of Chemical Theory and Computation 17, 2619–2629 [DOI] [PubMed] [Google Scholar]
- 22.Nguyen TT, Marzolf DR, Seffernick JT, Heinze S, and Lindert S (2021) Protein structure prediction using residue-resolved protection factors from hydrogen-deuterium exchange NMR. Structure [DOI] [PubMed] [Google Scholar]
- 23.Zhang Y, Majumder EL-W, Yue H, Blankenship RE, and Gross ML (2014) Structural analysis of diheme cytochrome c by hydrogen–deuterium exchange mass spectrometry and homology modeling. Biochemistry 53, 5619–5630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kahraman A, Herzog F, Leitner A, Rosenberger G, Aebersold R, and Malmström L (2013) Cross-link guided molecular modeling with ROSETTA. PloS One 8, e73411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Piotrowski C, Moretti R, Ihling CH, Haedicke A, Liepold T, Lipstein N, Meiler J, Jahn O, and Sinz A (2020) Delineating the Molecular Basis of the Calmodulin–bMunc13-2 Interaction by Cross-Linking/Mass Spectrometry—Evidence for a Novel CaM Binding Motif in bMunc13-2. Cells 9, 136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hofmann T, Fischer AW, Meiler J, and Kalkhof S (2015) Protein structure prediction guided by crosslinking restraints–A systematic evaluation of the impact of the crosslinking spacer length. Methods 89, 79–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Alford RF, Leaver-Fay A, Jeliazkov JR, O’Meara MJ, DiMaio FP, Park H, Shapovalov MV, Renfrew PD, Mulligan VK, and Kappel K (2017) The Rosetta all-atom energy function for macromolecular modeling and design. Journal of Chemical Theory and Computation 13, 3031–3048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Song Y, DiMaio F, Wang RY-R, Kim D, Miles C, Brunette T, Thompson J, and Baker D (2013) High-resolution comparative modeling with RosettaCM. Structure 21, 1735–1742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Leelananda SP, and Lindert S (2016) Computational methods in drug discovery. Beilstein Journal of Organic Chemistry 12, 2694–2718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Seffernick JT, Harvey SR, Wysocki VH, and Lindert S (2019) Predicting protein complex structure from surface-induced dissociation mass spectrometry data. ACS Central Science 5, 1330–1341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Seffernick JT, Canfield SM, Harvey SR, Wysocki VH, and Lindert S (2021) Prediction of protein complex structure using surface-induced dissociation and cryo-EM. Analytical Chemistry 93, 7596–7605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Turzo SBA, Seffernick JT, Rolland AD, Donor MT, Heinze S, Prell JS, Wysocki V, and Lindert S (2021) Protein shape sampled by ion mobility mass spectrometry consistently improves protein structure prediction. bioRxiv, 2021.2005.2027.445812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Harvey SR, Seffernick JT, Quintyn RS, Song Y, Ju Y, Yan J, Sahasrabuddhe AN, Norris A, Zhou M, Behrman EJ, Lindert S, and Wysocki VH (2019) Relative interfacial cleavage energetics of protein complexes revealed by surface collisions. Proceedings of the National Academy of Sciences 116, 8143–8148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lössl P, Kölbel K, Tänzler D, Nannemann D, Ihling CH, Keller MV, Schneider M, Zaucke F, Meiler J, and Sinz A (2014) Analysis of nidogen-1/laminin γ1 interaction by cross-linking, mass spectrometry, and computational modeling reveals multiple binding modes. PLoS One 9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kalkhof S, Haehn S, Paulsson M, Smyth N, Meiler J, and Sinz A (2010) Computational modeling of laminin N-terminal domains using sparse distance constraints from disulfide bonds and chemical cross-linking. Proteins: Structure, Function, and Bioinformatics 78, 3409–3427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu T, Marcinko TM, and Vachet RW (2020) Protein-ligand affinity determinations using covalent labeling-mass spectrometry. Journal of the American Society for Mass Spectrometry [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pan X, Limpikirati P, Chen H, Liu T, and Vachet RW (2020) Higher-Order Structure Influences the Kinetics of Diethylpyrocarbonate Covalent Labeling of Proteins. Journal of the American Society for Mass Spectrometry 31, 658–665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kim DE, Chivian D, and Baker D (2004) Protein structure prediction and analysis using the Robetta server. Nucleic Acids Research 32, W526–W531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Conway P, Tyka MD, DiMaio F, Konerding DE, and Baker D (2014) Relaxation of backbone bond geometry improves protein energy landscape modeling. Protein Science 23, 47–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Monera OD, Sereda TJ, Zhou NE, Kay CM, and Hodges RS (1995) Relationship of Sidechain Hydrophobicity and α -Helical Propensity on the Stability of the Single-stranded Amphipathic α- Helix. J. Pept. Sci 1, 319–329 [DOI] [PubMed] [Google Scholar]
- 41.(1987) Ohio Supercomputer Center.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.