

Abstract
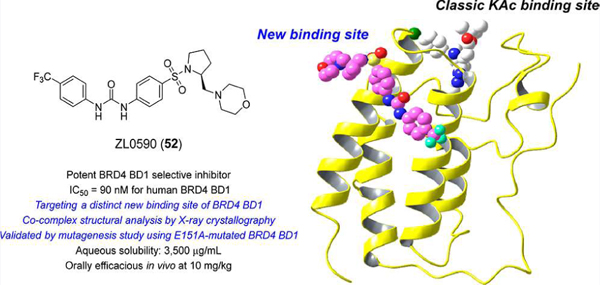
Bromodomain-containing protein 4 (BRD4) is an emerging epigenetic drug target for intractable inflammatory disorders. The lack of highly selective inhibitors among BRD4 family members has stalled the collective understanding of this critical system and the progress toward clinical development of effective therapeutics. Here we report the discovery of a potent BRD4 bromodomain 1 (BD1)-selective inhibitor ZL0590 (52) targeting a unique, previously unreported binding site, while exhibiting significant anti-inflammatory activities in vitro and in vivo. The X-ray crystal structural analysis of ZL0590 in complex with human BRD4 BD1 and the associated mutagenesis study illustrate a first-in-class nonacetylated lysine (KAc) binding site located at the helix αB and αC interface that contains important BRD4 residues (e.g., Glu151) not commonly shared among other family members and is spatially distinct from the classic KAc recognition pocket. This new finding facilitates further elucidation of the complex biology underpinning bromodomain specificity among BRD4 and its protein–protein interaction partners.
Graphical Abstract
INTRODUCTION
Chronic obstructive pulmonary disease (COPD), characterized by acute episodic decompensations and an inevitable decline in pulmonary function, is a highly prevalent (251 million cases globally in 2016) and progressive life-threatening lung disease.1 It is estimated that 3.17 million deaths, representing 5% of all deaths globally, were caused by COPD in 2015.2 Respiratory viral infections, responsible for over half of the acute decompensations in patients with COPD,3 trigger leukocytic inflammation, mucous production, and airway hyperreactivity.4 The limited treatments that are available can relieve symptoms, improve quality of life, and reduce the risk of death; however, COPD still lacks highly efficient therapeutic options, and novel target-based approaches are urgently needed to address various untenable aspects of this disease.
Recently, our team has explored the roles of bromodomain-containing protein 4 (BRD4) and NF-κB/RelA in mediating the relevant innate immune responses in both human small airway epithelial cells (hSAECs) and in multiple murine models of airway inflammation and pulmonary fibrosis.5–11 Specifically, activation of the NF-κB/RelA transcription factor activates atypical BRD4 histone acetyltransferase activity, RNA polymerase II phosphorylation, and secretion of neutrophilic chemokines during airway inflammation. BRD4 is one of the 46 bromodomain-containing proteins divided into eight families. BRD4 belongs to the bromodomain and extra-terminal domain (BET) family which also contains BRD2, BRD3, and bromodomain testis-specific protein BRDT. All BET family members share two bromodomains termed BD1 and BD2, which exhibit a high level of sequence conservation. As epigenetic readers for histone lysine acetylation (KAc), BET family proteins mediate signaling transductions to changes in gene regulatory networks.12–14 Disrupting the protein–protein interactions between BET proteins and histone acetylated lysines represents a valid and promising target for the treatment of human diseases including cancer,12,15–17 inflammation,18–21 viral infection,22–26 CNS disorders,27,28 and others.29,30 Currently, there are a dozen BET protein inhibitors in clinical trials and numerous others in the discovery stage (representative inhibitors 1–7 are shown in Figure 1) with a more focused pursuit of diverse cancer indications. RVX-208 (1),31 relatively more selective for BD2 of BET proteins, has finished phase III clinical trials on high-risk cardiovascular disease patients with type 2 diabetes mellitus and low levels of high-density lipoprotein but failed to meet the primary end point. BMS-986158 (2), developed by Bristol-Myers Squibb, is a potent and orally bioavailable BET inhibitor for the treatment of cancer.32 (+)-JQ1 (3) is the most widely used pharmacological BET inhibitor,33 and its analogue CPI-0610 ((S)-2-(6-(4-chlorophenyl)-1-methyl-4H-benzo[c]-isoxazolo[4,5-e]azepin-4-yl)acetamide) is undergoing phase I/II clinical trials for various indications including acute myelocytic leukemia, multiple myeloma, and lymphoma and is also under phase III studies for myelofibrosis (NCT04603495).34 ABBV-744 (4),35 with improved BET BD2 selectivity compared to ABBV-075,36–38 is in phase I clinical trials for acute myeloid leukemia as a monotherapy, and myelofibrosis via combination with Ruxolitinib or Navitoclax. Compounds GSK778 (5) and GSK046 (6) are recently reported BET BD1-selective and BET BD2-selective small molecule inhibitors with >130-fold and >300-fold selectivity over the other corresponding bromodomains, respectively, as determined by surface plasmon resonance (SPR) assays.39 Proteolysis targeting chimeras (PROTACs) represent an emerging therapeutic strategy that triggers BRD4 protein degradation, rather than inhibiting chromatin-binding activity and is currently being investigated for clinical applicability.40,41 Numerous chemical classifications of BET inhibitors have been reported, and the scientific community’s interest in targeting BRD4 for the treatment of inflammatory diseases has been comprehensively discussed in our review papers.29,42,43 Herein, we report the discovery of a new series of potent BRD4 BD1-selective inhibitors targeting a unique, previously unreported binding site, while exhibiting significant anti-inflammatory activities in vitro and in vivo. X-ray cocrystal structural analysis of such an inhibitor with human BRD4 BD1 and mutagenesis study on BRD4 BD1 illustrate a first-in-class non-KAc binding site located at the helix αB and αC surface, providing an additional pathway for modulating BRD4-associated protein–protein interactions.
Figure 1.
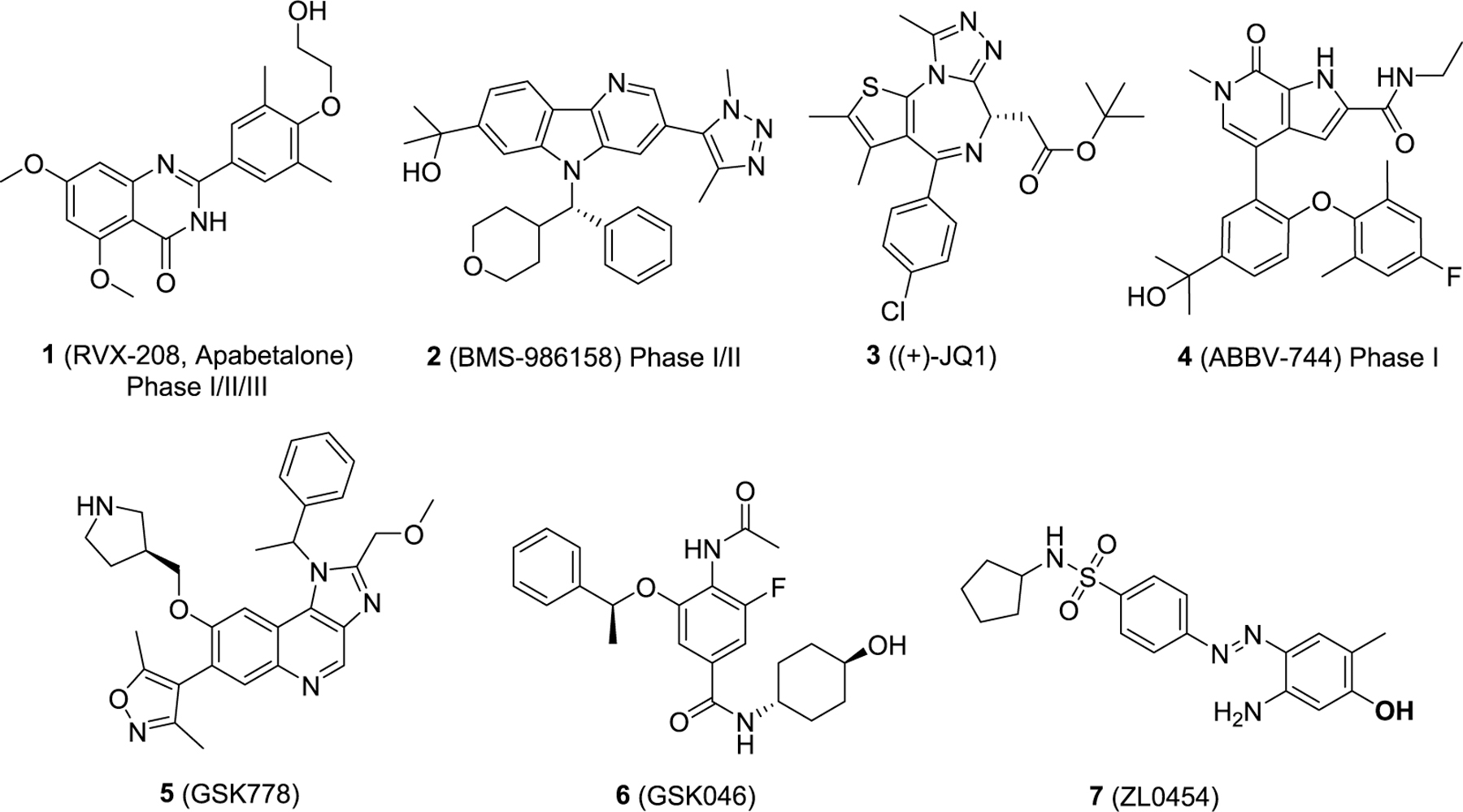
Chemical structures of representative BET inhibitors 1–7.
RESULTS AND DISCUSSION
Design.
Despite BET bromodomain selectivity has attracted significant attention,35,44 very few available BET inhibitors have achieved a satisfactory selectivity profile among BET family proteins due to their occupancy of the highly conserved KAc recognition pocket (Supporting Information, Figures S1 and S2).45 This lack of selectivity not only hinders elucidation of the biological function of BRD4 but may also lead to off-target effects for KAc-directed drug candidates.46 In our previous work investigating the role of BRD4 and NFκB/RelA in innate immune responses during airway inflammation and lung fibrosis, we identified lead compound 7 (ZL0454) as a potential therapeutic for inflammatory diseases that exhibited 16- to 57-fold BRD4 selectivity over the highly similar BRD2.8,9,41,47,48 While the in vivo efficacy of compound 7 in a murine model of acute airway inflammation (intraperitoneal injection (ip)) produced promising proof-of-concept results, further optimization of its pharmacokinetic (PK) profile is imperative, particularly oral bioavailability (e.g., compound 7 with fairly low Cmax of 36 ng/mL, AUC0–t of 104 ng·h/mL at the oral dose of 20 mg/kg, and oral bioavailability of 0.5%).47 To further enhance the drug metabolism and pharmacokinetics (DMPK) profile as well as improve selectivity among BRD4 bromodomains, two strategies were employed. One strategy was replacing the core structure with selected privileged scaffolds (e.g., quinazolin-4-one core and chromen-4-one), which led to the discovery of ZL0516 ((R)-2-(4-(2-hydroxy-3-(4-methylpiperazin-1-yl)propoxy)-3,5-dimethylphenyl)-5,7-di-methoxy-4H-chromen-4-one) that exhibits BRD4 BD1 selectivity through direct interaction with BRD4 BD1-unique residue Asn93, while providing improvements to the DMPK profile including good oral bioavailability (F = 35%).49 The second strategy, as presented in the current work, is to optimize compound 7 by replacing the metabolically labile pharmacophores using a property-based drug design approach. Indeed, by critically examining compound 7, we identified a number of key features that likely contribute to the overall poor PK profile including the exposed free amino phenol (P1 site) and the N=N azo linker (P2 site) that can be readily decomposed by colonic bacteria,50 as well as contribute poor aqueous solubility (12.8 μg/mL at pH = 7 for compound 7). To this end, we anticipate that modifying the amino phenol moiety that interacts directly with Asn140 in the KAc pocket to expand the chemical space and achieve key protein–ligand interactions. Meanwhile, replacing the N=N azo linker to improve the metabolic stability and in vivo PK profiles, as well as incorporating basic N-containing aliphatic heterocycles (e.g., piperidine and morpholine) into the P3 site may significantly enhance their aqueous solubility. As depicted in Figure 2, we designed three subseries of compounds to probe additional interactions for novel mode of action and to improve BRD4 selectivity, metabolic stability, as well as other important drug-like properties.
Figure 2.
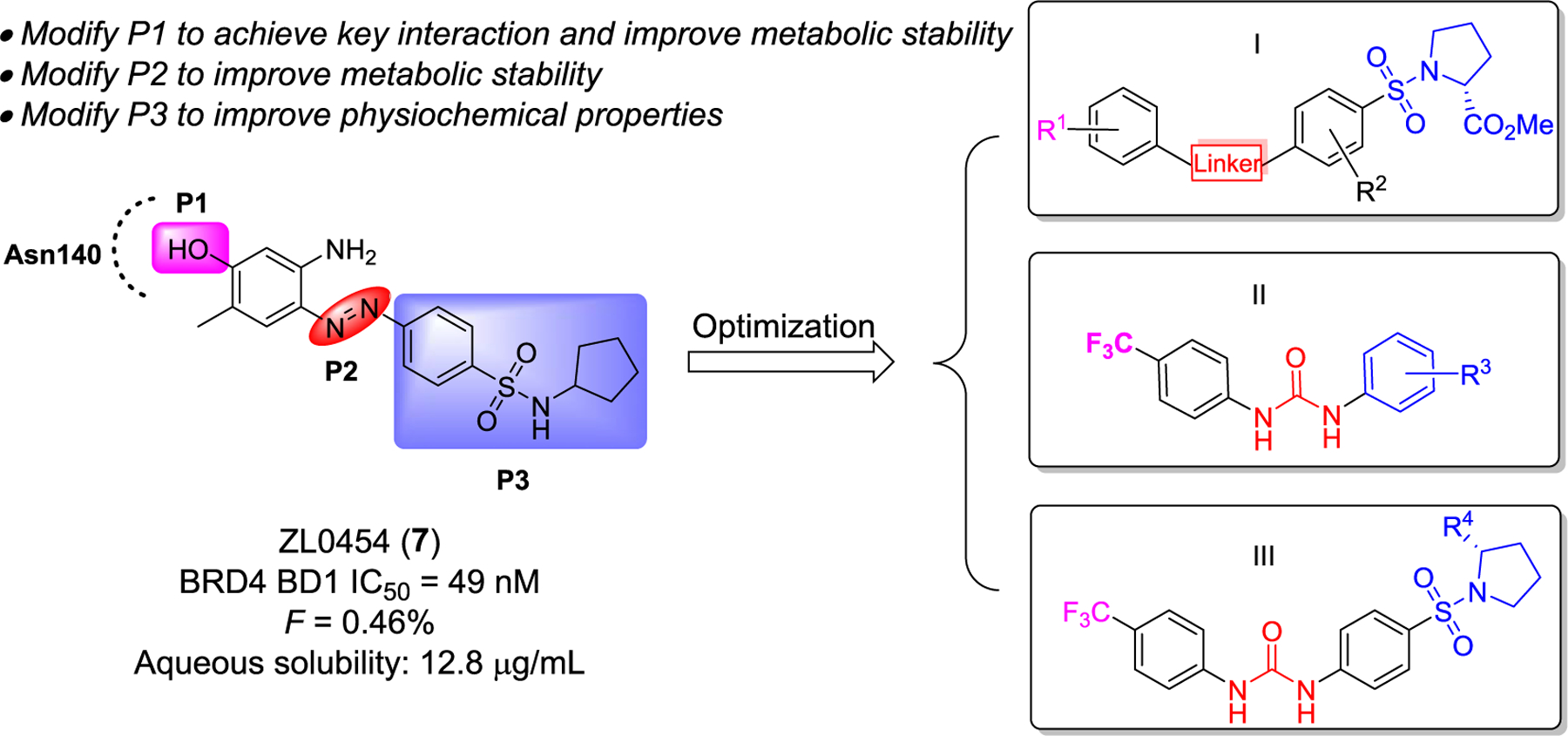
Design of three new subseries I, II, and III of compounds as potential novel BRD4 inhibitors based on our lead BRD4 inhibitor 7.
Chemistry.
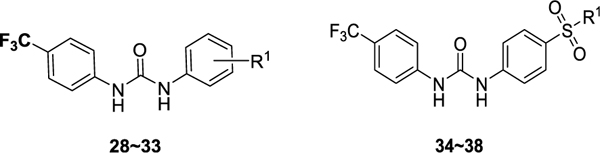
The first series of compounds was designed to explore more metabolically stable substituents and linkers on the P1 and P2 sites (Scheme 1). Compounds 11–16, 20, and 21 were obtained through coupling of various isocyanatobenzenes with intermediate 10 in good yields. An alternative method to form the isocyanates directly from amines using triphosgene was applied to afford compounds 17–19. 4-(Trifluoromethyl)benzoyl chloride was substituted by methyl ((4-aminophenyl)sulfonyl)-l-prolinate to yield compound 22 with an amide linker, while the same intermediate was isopropylated with acetone and then coupled with 1-isocyanato-4-(trifluoromethyl)benzene to give compound 23 with a bulky urea linker for structure–activity relationship (SAR) comparison. Compound 24 with a longer urea linker was synthesized following a similar procedure for synthesizing compounds 17–19. Intermediate 10 was treated with sodium nitrite and 10% hydrochloric acid to generate the corresponding aryldiazonium chlorides which were directly coupled with 3-oxo-3-(4-(trifluoromethoxy)phenyl)propanenitrile, affording compound 25 in a yield of 82% for two steps.51 The second series of compounds (Scheme 2) retaining −CF3 and the urea linker was employed to investigate diverse substitutions on the P3 site. Anilines 27 substituted with different hydrophilic aliphatic rings were reacted with 1-isocyanato-4-(trifluoromethyl)benzene 26 to generate target compounds 28–38 in yields of 41–97%.
Scheme 1. Synthesis of Compounds Series I with Modifications on the P1 and P2 Sitesa.
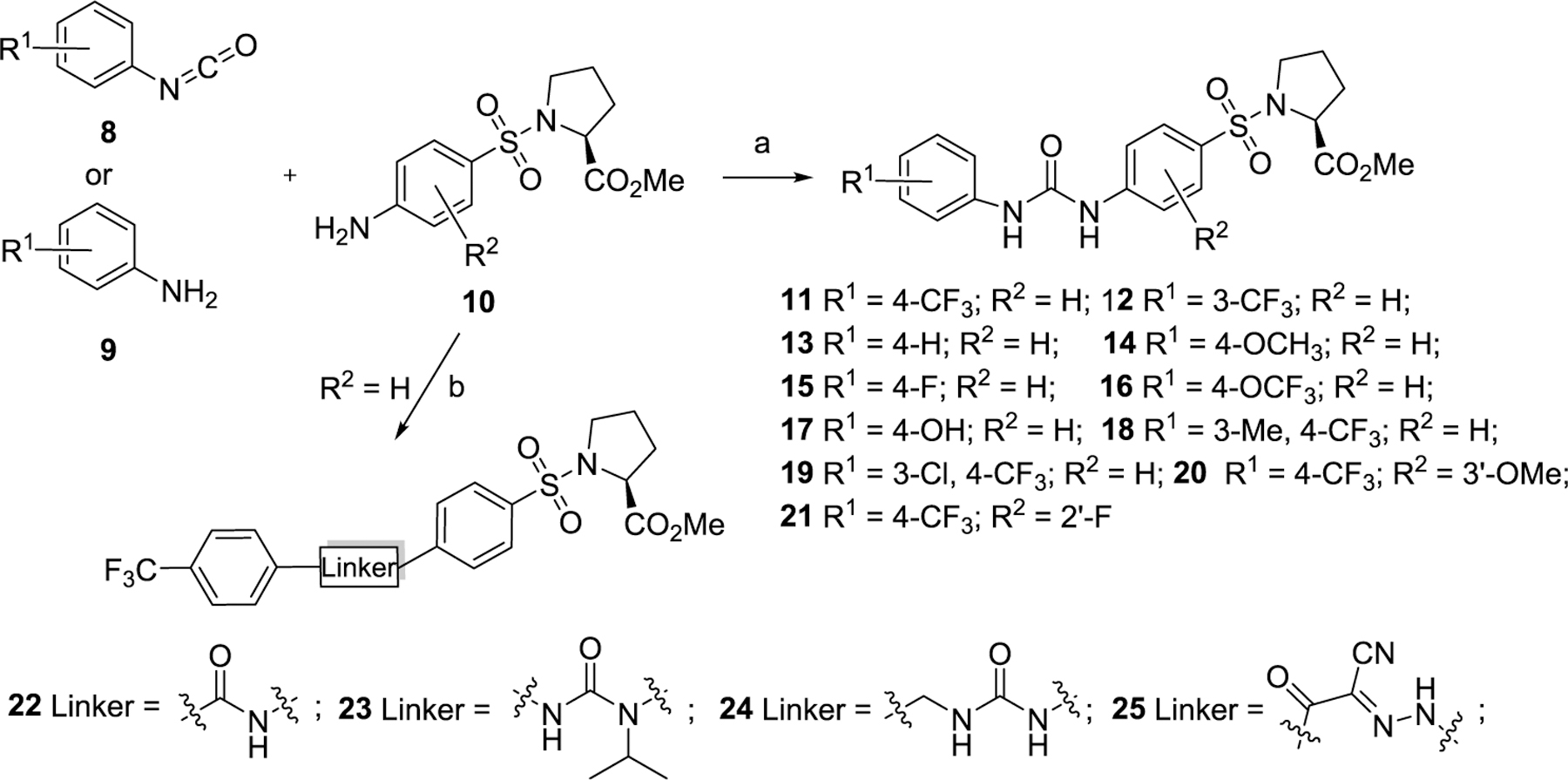
aReagents and conditions: (a) For 11–16, 20, and 21, CH2Cl2, reflux, overnight, 66%–quant. For 17–19, triphosgene, toluene, 120 °C, overnight, 30%–44%. (b) For 22, 4-(trifluoromethyl) benzoyl chloride, Et3N, CH2Cl2, 0–25 °C, quant. For 23, (i) acetone, H2, Pd/C, rt, 76%, (ii) 1-isocyanato-4-(trifluoromethyl)benzene, CH2Cl2, rt, 49%. For 24, (4-(trifluoromethyl)phenyl) methanamine, triphosgene, toluene, 120 °C, overnight, 64%. For 25, (i) 10% HCl, NaNO2, H2O, rt, (ii) 3-oxo-3-(4-(trifluoromethoxy)phenyl)propanenitrile, NaOAc, EtOH, rt, 5 min, 82%.
Scheme 2. Synthesis of Compounds Series II with Modifications on the P3 Sitea.
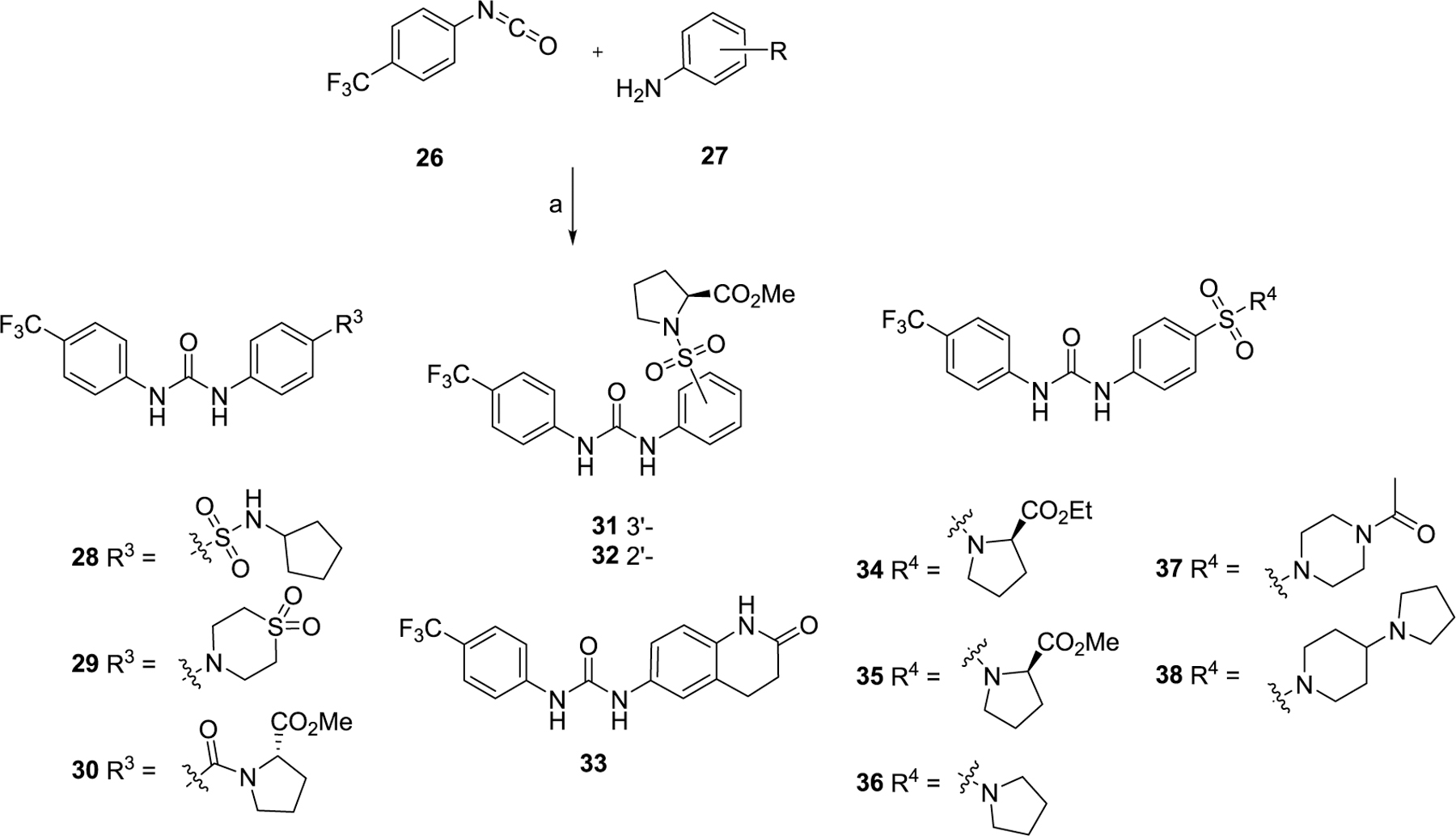
aReagents and conditions: (a) CH2Cl2, reflux, overnight, 41–97%.
The synthetic route of series III with further optimizations to the P3 site is outlined in Scheme 3. Compound 42 was prepared following the synthetic procedures of series I in a yield of 89%. The key intermediate methyl ((4-nitrophenyl)-sulfonyl)-l-prolinate 41 was hydrolyzed in the presence of LiOH to produce 43 in a quantitative yield. Intermediate 43 was then coupled with various amines in the presence of HBTU and DIPEA to afford 44, followed by the reduction and coupling with 1-isocyanato-4-(trifluoromethyl)benzene 26, leading to target compounds 45–48 in 39% to quantitative yield. Hydrolysis and reduction of compound 11 resulted in 49 and 50, respectively. Compound 50 was sulfonylated by TsCl to yield compound 51, which was then substituted by aliphatic amines in the presence of K2CO3 producing new compounds 52 and 53 in a yield of 57% and 44%, respectively.
Scheme 3. Synthesis of Compounds Series III with Further Modifications on the P3 Sitea.
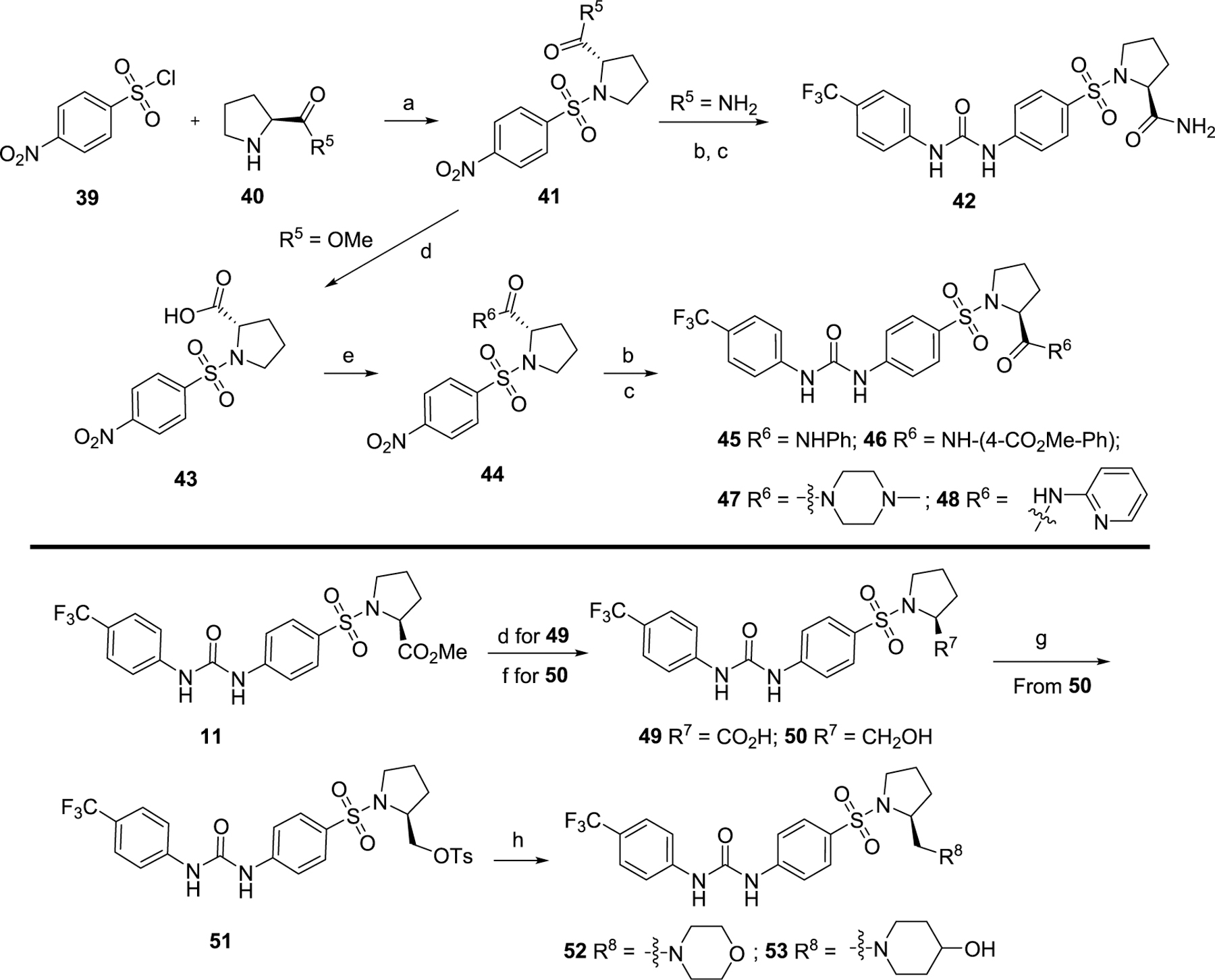
aReagents and conditions: (a) DIPEA, CH2Cl2, 0–25 °C, 89%; (b) Zn, NH4Cl, EtOH/H2O, 80 °C, quant.; (c) 26, CH2Cl2, 39%–quant.; (d) LiOH, CH3OH/H2O, rt, 4 h, quant.; (e) HBTU, DIPEA, CH2Cl2, rt, 66%–84% (f) LiAlH4, THF, 0 °C, 30 min, quant.; (g) TsCl, CH2Cl2, rt, 82%; (h) K2CO3, CH3CN, 50 °C, overnight, 44–57%.
Inhibitory Effects of Newly Synthesized Compounds on Poly(I:C)-Induced Expression of Inflammatory Genes CIG5 and IL-6 in hSAECs.
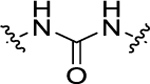
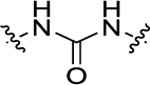
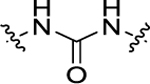
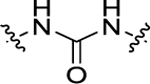
For all the newly synthesized compounds, we first evaluated their in vitro cellular effects on inhibiting poly(I:C)-induced inflammatory genes at hSAECs (residual activity rates (%) of tested compounds on expression of poly(I:C)-induced inflammatory genes CIG5 and IL-6 provided in Tables 1–3 and fold changes of poly(I:C)-induced CIG5 and IL-6 gene expression listed in Supporting Information, Table S1). hSAECs, which have many characteristics of representative primary lower-airway epithelial cells,7 were first pretreated with BRD4 inhibitors at the final concentration of 10 μM for 24 h and followed by adding poly(I:C) into culture medium (10 μg/mL, 4 h) before harvesting the cells. Expression of inflammatory genes CIG5 and IL-6 was determined by quantitative real-time PCR (qRT-PCR) analysis. Expression activity rates (%) were calculated based on the poly(I:C)-induced CIG5 and IL-6 expression without inhibitors, respectively. This reliable cellular assay allows us to efficiently evaluate the in vitro efficacy of compounds at an early stage to exclude those compounds likely with poor cellular permeability and inconclusive in vitro cellular inhibitory effects to conserve resources for thoroughly characterizing our most promising candidates. Previously reported BRD4 inhibitors, compounds 3 and 7, were included as positive controls. We evaluated the first series of compounds with more stable substitutions on the P1 site and diverse linkers on the P2 site (Table 1). The sulfonyl-l-prolinate group on the P3 site of 7 was retained given that it was tolerable in previous studies and facilitated further robust optimization. 4-CF3 in place of 4-OH on the P1 site and urea as the linker to replace the azo (compound 11) displayed equivalent inhibitory activity compared to positive controls 3 and 7. The shift of 4-CF3 to 3-CF3 (12) led to an obvious loss of the inhibition of IL-6. The absence of −CF3 moiety (13), replacement by −OCH3 (14), −F (15), or −OH (17) all resulted in a complete loss of cellular activity. Inserting an O atom (16) displayed slightly decreased inhibition against both CIG5 and IL-6 compared to 11. Our iterative evaluation and chemical modification process identified the 4-CF3 group as a highly favorable substituent on the phenyl ring, which was subsequently kept intact while we examined whether adding a hydrophobic substituent like a methyl moiety (18) or chlorine (19) would further improve bioactivity. However, in both cases, improvements were not realized. Additionally, we explored small-size substitutions on the second aromatic ring, and the results indicated that neither electron-donating OMe (20) nor electron-withdrawing F (21) substituents were beneficial. Shorter (22), bulkier (23), and longer linkers (24) were investigated, but none of them displayed better activity than 11 against both CIG5 and IL-6 expression. Interestingly, elongation of the P1 and linker portion (25) was found to retain inhibitory activity, although not as robustly as 11, indicating that a nonlinear linker lacking sufficient H-bond formation may produce an unfavorable alignment of the tail portion.
Table 1.
Residual Activity Rates of Series I Compounds on Poly(I:C)-Induced Expression of Inflammatory Genes CIG5 and IL-6 in hSAECsa
| |||||
|---|---|---|---|---|---|
| Compd | R1 | Linker | R2 | CIG5 (%) | IL-6 (%) |
| 3 | - | - | - | 2.60 ± 0.22 | 8.10 ± 0.68 |
| 7 | - | - | - | 15.6 ± 1.3 | 0.91 ± 0.08 |
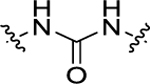
| 11 | 4-CF3 |
|
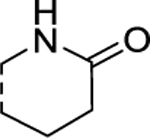
H | 2.19 ± 0.16 | 3.42 ± 0.30 |
| 12 | 3-CF3 |
|
H | 4.0 ± 0.39 | 66.7 ± 5.2 |
| 13 | H |
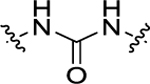
|
H | 75.3 ± 6.4 | 237 ± 22 |
| 14 | 4-OCH3 |
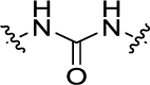
|
H | 73.3 ± 5.2 | 418 + 30.3 |
| 15 | 4-F |
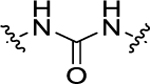
|
H | 28.8 ± 2.5 | 149 ± 11.3 |
| 16 | 4-OCF3 |
|
H | 20.5 ± 1.5 | 14.3 ± 1.1 |
| 17 | 4-OH |
|
H | 326 ± 24 | 45.2 ± 3.6 |
| 18 | 3-Me, 4-CF3 |
|
H | 64.7 + 5.7 | 42.5 ± 3.6 |
| 19 | 3-C1, 4-CF3 |
|
H | 74.7 ± 6.1 | 37.9 ± 2.8 |
| 20 | 4-CF3 |
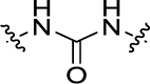
|
3’-OMe | 59.5 ± 5.2 | 22.5 ± 1.6 |
| 21 | 4-CF3 |
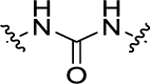
|
2’-F | 137 ± 11.4 | 51.9 ± 3.5 |
| 22 | 4-CF3 |
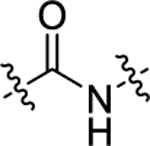
|
H | 105 ± 7.6 | 71 ± 6.7 |
| 23 | 4-CF3 |
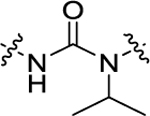
|
H | 461 ± 32.1 | 62.8 + 4.5 |
| 24 | 4-CF3 |
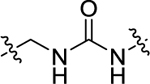
|
H | 150 ± 11.9 | 40.4 ± 3.4 |
| 25 | 4-OCF3 |
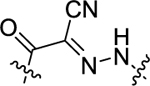
|
H | 1.02 ± 0.08 | 17.5 ± 2.0 |
hSAECs were first pretreated with compounds (at the final concentration of 10 μM) and followed by adding poly(I:C). The treated cells were harvested and expression of inflammatory gene CIG5 and IL-6 was determined by quantitative real-time PCR (qRT-PCR) analysis. Residual activity rates (%) of tested compounds, compared to the positive control group (poly(I:C) alone, at 100%), are presented as the average from three independent measurements (mean ± SD, n = 3).
Table 3.
Residual Activity Rates of Series III Compounds on Poly(I:C)-Induced Expression of Inflammatory Genes CIG5 and IL-6 in hSAECsa
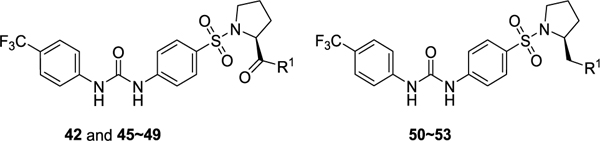
| |||
|---|---|---|---|
| Compd | R1 | CIG5 (%) | IL-6 (%) |
| 11 | OMe | 2.19 ± 0.16 | 3.42 ± 0.30 |
| 42 | NH2 | 22.6 ± 1.55 | 12.7 ± 1.01 |
| 45 |
|
0.62 ± 0.005 | 13.7 ± 1.06 |
| 46 |
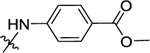
|
4.58 ± 0.33 | 23.7 ± 2.15 |
| 47 |
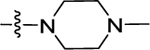
|
5.47 ± 0.41 | 6.63 ± 0.48 |
| 48 |
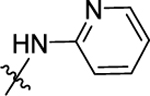
|
1.12 ± 0.09 | 32.8 ± 2.84 |
| 49 | -OH | 214 ± 17.1 | 25.3 ± 2.13 |
| 50 | -OH | 0.39 ± 0.03 | 1.71 ± 0.13 |
| 51 | -OTs | 0.63 ± 0.05 | 6.98 ± 0.63 |
| 52 |
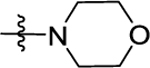
|
0.34 ± 0.025 | 0.35 ± 0.03 |
| 53 |
|
0.21 ± 0.02 | 0.40 ± 0.03 |
Residual activity rates (%) of tested compounds (at the final concentration of 10 μM) comparing to the positive control group (poly(I:C) alone, at 100%) are presented as the average from three independent measurements (mean ± SD, n = 3).
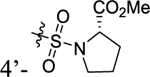
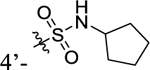
According to the results of first-round optimization, we retained the favorable 4-CF3 and urea linker on the P1 and P2 sites and investigated diverse moieties on the P3 site (Table 2). This site was found to be broadly tolerated, including the N-cyclopentylsulfonic amide modification to compound 7 (28), which displayed residual activity rates of 28% and 190% against CIG5 and IL-6, respectively. Compounds with directly attached hydrophobic dioxidothiomorpholine (29) and fused ring (33) from (E)-6-((2-amino-4-hydroxy-5-methylphenyl)-diazenyl)-3,4-dihydroquinolin-2(1H)-one (ZL0420)47 displayed moderate inhibitory effects against IL-6, while molecules with amide (30) and sulfonamide (11, 34–35, and 37–38) were found to be more potent with residual activity rates ranging from 2.2% to 16.5%. After a chemically and pharmacologically favorable group in the P3 site was identified (methyl (4-phenyl)sulfonyl-l-prolinate of 11), we then explored the positions of the sulfonyl-l-prolinate moiety on the phenyl ring, resulting in an activity order of para- (11) > ortho- (32) > meta- (31). The introduction of an ethyl to replace methyl ester (34) or unnatural d-proline (35) had little impact on the potency. Removal of the branched ester (36) was found detrimental, likely due to the requirement of a larger size for the binding site. Similarly, the inhibitory activity of compounds with rings of larger size 4-acetylpiperazine (37) and 4-(pyrrolindin-1-yl)piperidine (38) were dramatically enhanced compared to that of 36.
Table 2.
Residual Activity Rates of Series II Compounds on Poly(I:C)-Induced Expression of Inflammatory Genes CIG5 and IL-6 in hSAECsa
| |||
|---|---|---|---|
| Compd | R1 | CIG5 (%) | IL-6(%) |
| 3 | - | 2.6±0.22 | 8.1±0.68 |
| 7 | - | 15.6±1.3 | 0.91±0.08 |
| 11 |
|
2.19±0.16 | 3.42±0.30 |
| 28 |
|
28.0±2.2 | 190±13.9 |
| 29 |
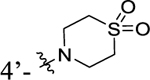
|
6.14±0.51 | 73.6±6.2 |
| 30 |
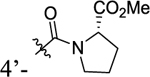
|
5.79±0.44 | 16.5±1.4 |
| 31 |
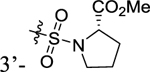
|
163±11.9 | 19.9±1.7 |
| 32 |
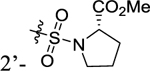
|
12.6±1.1 | 2.63±0.22 |
| 33 |
|
3.34±0.26 | 106±8.7 |
| 34 |
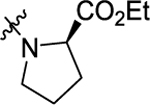
|
4.74±0.37 | 0.57±0.05 |
| 35 |
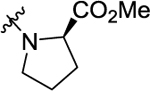
|
2.21±0.18 | 0.47±0.04 |
| 36 |
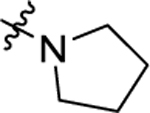
|
24.3±2.1 | 102±7.3 |
| 37 |
|
20.0±1.7 | 15.2±1.3 |
| 38 |
|
10±0.79 | 0.31±0.03 |
Residual activity rates (%) of tested compounds (at the final concentration of 10 μM) compared to the positive control group (poly(I:C) alone, at 100%) are presented as the average from three independent measurements (mean ± SD, n = 3).
Finally, we further optimized the substituents on the P3 site by taking advantage of the sulfonyl-l-prolinate moiety (Table 3). The methyl ester may be readily metabolized in vivo, and it was thus hydrolyzed and reduced to introduce more stable and soluble fragments to improve its overall physiochemical properties given that this portion is available for optimization. Interestingly, the final target molecules with alkyl amine moieties (compounds 50–53) were found more active in general than those compounds with the amide fragments (compounds 42–49). These results suggested that compounds with the alkyl amine moieties possessing a less constrained conformation may increasingly accommodate diverse binding site topography compared to compounds with the amide moieties. Moreover, by keeping the basicity of the nitrogen, the target molecules with alkyl amines are predicted to display better aqueous solubility. Such compounds 50–53 were thus selected for further determination of corresponding IC50 values to their in vitro efficacy.
Considering the residual activity rates of all new compounds at a single concentration, we selected compounds 11, 45, and 50–53 for further determination of IC50 values of their in vitro inhibitory effects, which would reflect their inhibitory activities against poly(I:C)-induced expression of inflammatory genes CIG5 and IL-6 more accurately (Table 4 and Figure 3). IC50 values were determined from their inhibitory curves at eight concentrations of each inhibitor using the Four Parameters Regression method (https://www.aatbio.com/tools/ic50-calculator/). Most compounds have nanomolar to submicromolar inhibition against poly(I:C)-induced gene expression of CIG5 and IL-6 in this cellular assay. Compound 52 has the most promising IC50 value of 200 nM against CIG5, which is about 8.7- and 4.9-fold more potent than positive controls 1 and 3, respectively. Meanwhile, compound 53 showed the most potent inhibition against poly(I:C)-induced gene expression of IL-6 with an IC50 value of 220 nM, which is 15.9- and 4.8-fold more potent than compounds 1 and 3, respectively.
Table 4.
IC50 Values of Selected Compounds against Poly(I:C)-Induced Expression of Inflammatory Genes CIG5 and IL-6 in hSAECsa
| compd | CIG5 (μM) | IL-6 (μM) |
|---|---|---|
| 1 | 1.73 ± 0.19 | 3.49 ± 0.38 |
| 3 | 0.97 ± 0.11 | 1.06 ± 0.09 |
| 11 | 1.35 ± 0.17 | 1.83 ± 0.19 |
| 45 | 2.06 ± 0.24 | 1.88 ± 0.21 |
| 50 | 0.86 ± 0.12 | 2.29 ± 0.27 |
| 51 | 10.8 ± 1.24 | 4.31 ± 0.53 |
| 52 | 0.20 ± 0.03 | 0.37 ± 0.07 |
| 53 | 0.39 ± 0.05 | 0.22 ± 0.04 |
IC50 values are reported as the mean ± SD derived from three independent measurements. Each one is determined from 8 different concentrations, and IC50 values are calculated using Four Parameters Regression method (https://www.aatbio.com/tools/ic50-calculator/).
Figure 3.
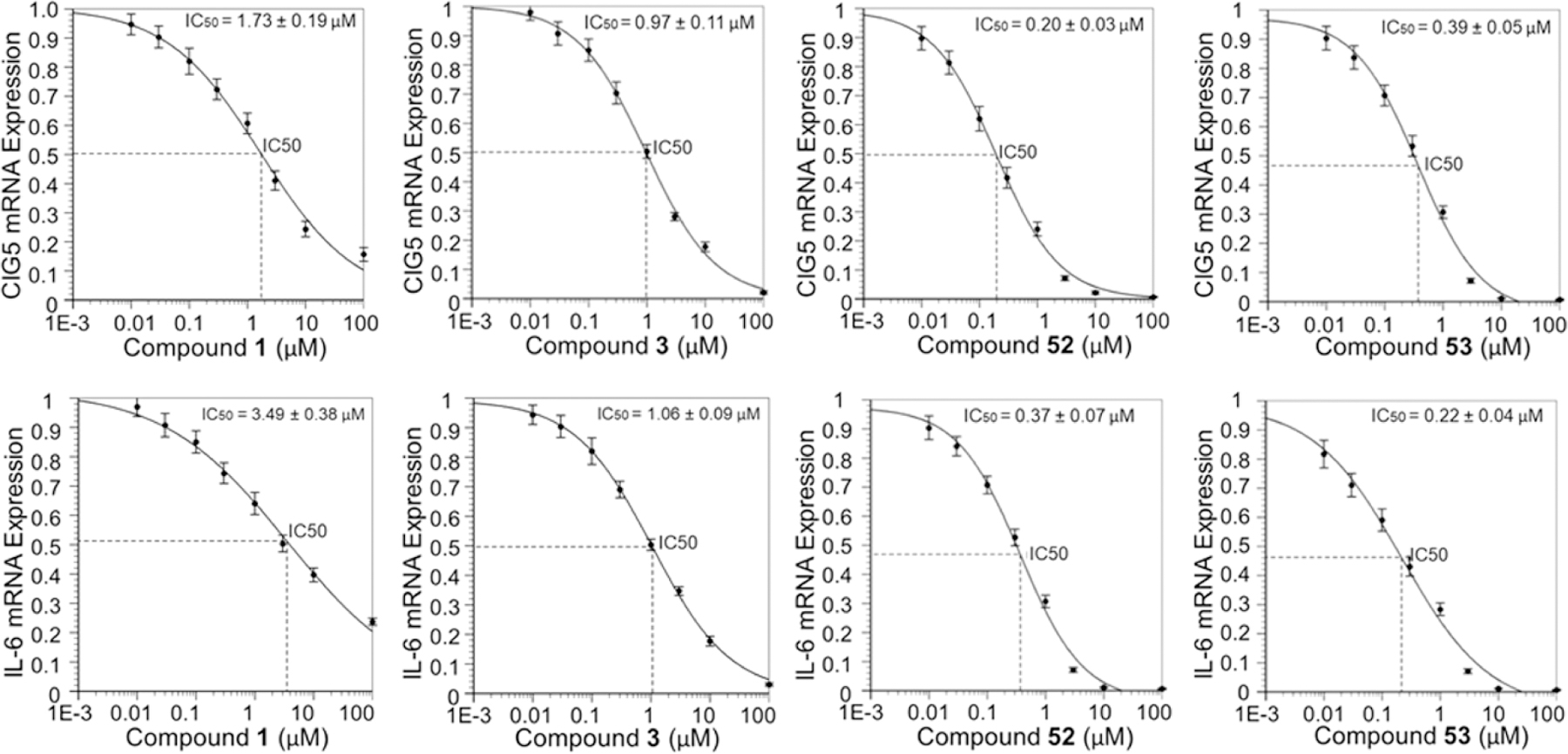
Dose–response curves and IC50 values of selected compounds 52 and 53 against poly(I:C)-induced expression of inflammatory genes CIG5 and IL-6 (with positive control compounds 1 and 3) in hSAECs. The IC50 values of each inhibitor are respectively determined on poly(I:C)-induced GIG5 (upper panel) or IL-6 (lower panel) expression. The results are derived from three independent experiments (n = 3), and IC50 values are calculated using the Four Parameters Regression method (https://www.aatbio.com/tools/ic50-calculator/).
Binding Affinities of Selected Active Compounds for BET Bromodomains.
Active compounds 50, 52, and 53 were then selected for binding affinity evaluations by time-resolved fluorescence resonance energy transfer (TR-FRET) assay following the detailed experimental procedures in the Methods section based on previously reported protocols for characterizing the binding affinities of BET inhibitors.33,52 We tested BRD4 BD1 and BRD4 BD2 as well as BRD2 BD1 and BRD2 BD2 to get a general sense of selectivity. Similar to the previously reported results, compound 3 had no apparent preference among them, and compound 1 displayed a relative selectivity for BD2 over BD1. While our compounds exhibited similar nanomolar level binding affinities for BRD4 BD1, they displayed a better selectivity over BRD4 BD2. Compounds 52 and 53 have promising BRD4 BD1 affinities with IC50 values of 90 and 93 nM, respectively, which are about 10-fold more selective over BRD4 BD2, BRD2 BD1, and BRD2 BD2 (Table 5, Figure 4, and Supporting Information, Table S7). Additional binding affinities for other bromodomain-containing proteins were investigated for compounds 52 and 53 to evaluate their selectivity over other BET members (e.g., BRD3 and BRDT) and non-BET protein (e.g., CBP). For compound 52, we also conducted non-BET protein panel screening through DiscoverX (Supporting Information, Table S2) and an additional comprehensive screening against more than 40 drug targets from the NIMH psychoactive drug screening program as described in our previous publications53,54 to evaluate the potential off-target effects (Supporting Information, Table S3). Excitingly, this extensive screening endeavor indicated that 52 maintains an overall excellent drug target specificity profile with no or weak binding affinities toward these screened drug targets.
Table 5.
Binding Affinities of Selected Active Compounds for the BET Bromodomains and Non-BET Protein CBP (IC50, nM)a
| bromodomains | 1 | 3 | 50 | 52 | 53 |
|---|---|---|---|---|---|
| BRD4 BD1 | 1,151 ± 132 | 92 ± 11 | 138 ± 14 | 90 ± 9 | 93 ± 12 |
| BRD4 BD2 | 132 ± 14 | 62 ± 9 | 746 ± 89 | 1,093 ± 136 | 1,168 ± 152 |
| BRD2 BD1 | 5,697 ± 675 | 77 ± 8 | 1,254 ± 143 | 835 ± 117 | 742 ± 89 |
| BRD2 BD2 | 246 ± 35 | 54 ± 7 | 1,207 ± 129 | 1,308 ± 149 | 1,376 ± 157 |
| BRD3 BD1 | 3,878 ± 437 | 79 ± 11 | NDb | 2,183 ± 275 | 2,104 ± 272 |
| BRD3 BD2 | 217 ± 26 | 72 ± 8 | NDb | 1,996 ± 257 | 2,018 ± 214 |
| BRDT BD1 | 5,027 ± 639 | 178 ± 19 | NDb | 2,416 ± 284 | 2,217 ± 253 |
| BRDT BD2 | 696 ± 81 | 224 ± 26 | NDb | 1,902 ± 225 | 1,987 ± 226 |
| CBP | >10000 | 9,546 ± 673 | NDb | >10000 | >10000 |
Binding affinity was measured using a TR-FRET assay with the isolated bromodomain. Reported as mean of three separate assay runs and IC50 values are calculated using Four Parameters Regression method (https://www.aatbio.com/tools/ic50-calculator/).
ND, not determined.
Figure 4.
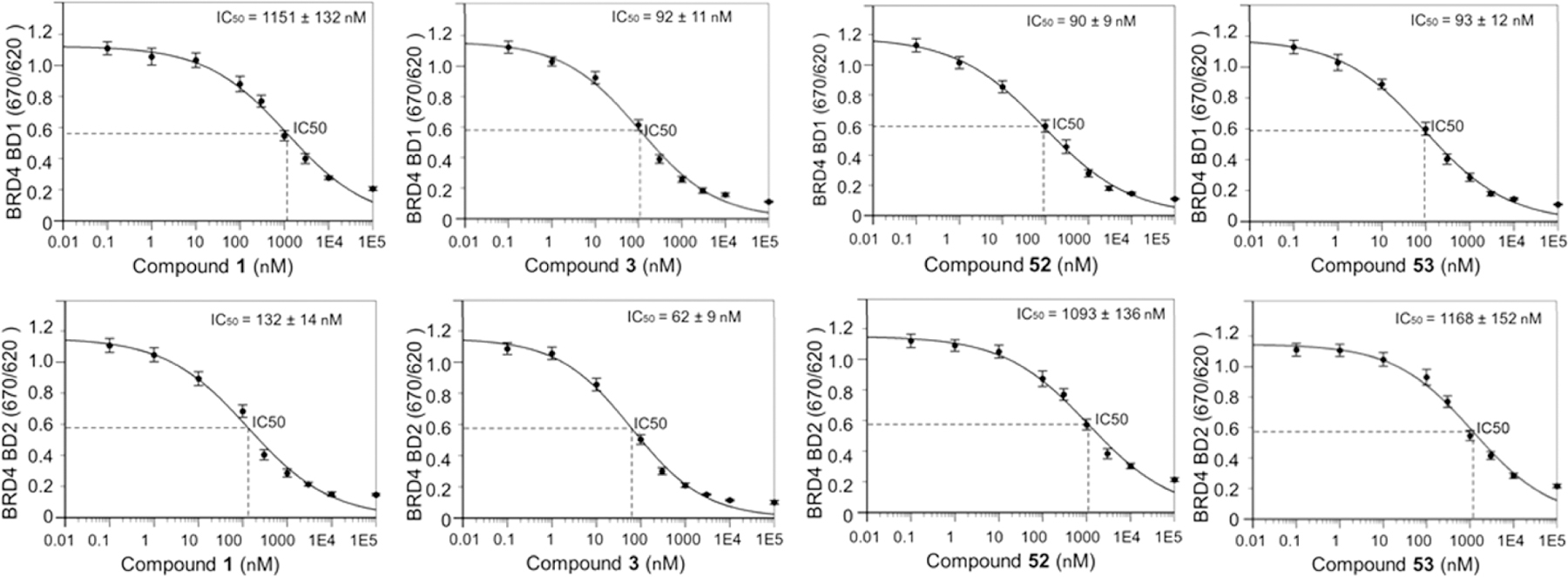
Dose–response curves and binding affinities of selected compounds 52 and 53 for the BRD4 bromodomains (with the positive controls compounds 1 and 3). TR-FRET assays were performed using recombinant BRD4 BD1 and BD2, and IC50 values are calculated using the Four Parameters Regression method (https://www.aatbio.com/tools/ic50-calculator/).
Cocrystal Structural Analysis of Compound 52 in Complex with Human BRD4 BD1.
It is well-known that almost all currently available BET inhibitors mimic an acetylated lysine to competitively bind to BET proteins.55,56 This well-defined classic KAc binding pocket (taking BET BD1 as an example, Supporting Information, Figure S1) located at the end of the four-helix bundle is extensively validated by X-ray crystallography and characterized by residues Asn140, Tyr97, and a WPF shelf (Trp81, Pro82, and Phe83 located at the ZA loop).57 With over 90% conserved identity for critical residues around the KAc site (Supporting Information, Figure S2), it is quite challenging to identify BET family protein-selective small molecule inhibitors, let alone to achieve BET member bromodomain (BD1 and BD2) selectivity (e.g., BRD4 BD1).45 With respect to the selectivity profile of compound 52, we envisioned that it either occupies a spatially distinct binding site or extends to new regions of the pocket to form distinct, distal interactions similar to the recently identified orthogonal site.58
To test our hypothesis and unambiguously understand the exact binding modes of our BRD4 inhibitors, the crystal structure of human BRD4 BD1 in cocomplex with 52 was solved with 1.4 Å resolution (Supporting Information, Table S4). Excitingly, with a single solvent DMSO molecule occupying the classic KAc site, compound 52 binds to a spatially distinct, new shallow pocket comprised of helices αB, αC, and the BC loop (Figure 5A). Overlay of known crystal structure of BRD4 BD1 with BRD4 inhibitor MS436 (PDB 4nud) and our structure of compound 52 (PDB 6U0D) clearly illustrates different locations of these two nearby binding sites with the classic KAc binding site in gray and the new recognition site in light blue (Figure 5B and Supporting Information, Figure S3). The distinct new binding site of compound 52 (magenta) in ribbon representation is shown in Figure 5C, while a solvent DMSO molecule occupies in the traditional KAc binding pocket. The unbiased omit map for the KAc pocket, calculated without the coordinates for the DMSO molecule, is depicted in Figure 5D, clearly indicating that other than DMSO, there is no molecule of compound 52 occupying the classic KAc binding pocket. To accommodate compound 52, several key residues of BRD4 BD1 as indicated are available to form stable molecular interactions (Figure 5E). The basic amine moiety of the morpholine group is likely protonated and forms a salt bridge with the carboxylate moiety of Glu151. The sulfonamide directly forms a hydrogen bond with the backbone amide of Gly143. The other oxygen in the sulfonamide forms a hydrogen bond with a water molecule that mediates an interaction with the backbone carbonyl of Asp144. The urea linker interacts via two water molecules with the backbone carbonyl of Tyr137 and the side chain carboxylate group of Glu154. In comparison with the traditional KAc pocket that BET proteins commonly share, the critical residues of this newly identified binding site are less conserved (Supporting Information, Table S5). The most similar sequence around the novel binding site is BRD2 BD1 (residues 147–169: TMFTNCYIYNKPTDDIVLMAQTL) which has three divergent residues compared with BRD4 BD1 (residues 131–153: TMFTNCYIYNKPGDDIVLMAEAL). Among the aforementioned five critical amino acid residues, only Tyr137 is conserved among BRD4 BD1, BRD4 BD2, and BRD2 BD1. Such amino acid residue difference within this new and unique binding site between BRD4 BD1 and BRD4 BD2 results in an 11-fold tighter binding of compound 52 to BRD4 BD1 over BRD4 BD2 (Figure 5F), providing a starting point to begin increasing the selectivity beyond this promising, yet suboptimal difference in affinity. Different from Glu151 in BRD4 BD1, Arg444 in BRD4 BD2 at the same position is not able to form a salt bridge with compound 52. Moreover, the only other human bromodomain with a glutamate equivalent to BRD4 BD1 Glu151 is the FALZ bromodomain (Supporting Information, Figure S4). It is therefore likely that the compound scaffold of compound 52 maintains a reasonable level of specificity for BRD4 BD1 over the other bromodomain-containing proteins as depicted in Table 5. While allosteric binding is not rare in the epigenetic machinery,59 the exact action mode could be complicated.60–62 We should be cautious as to whether this distinct new binding site is an allosteric site. For example, the crystal structure of human CREBBP bromodomain in complex with a small size 3,5-dimethylisoxazol ligand (PDB 3SVH) was demonstrated to have more than one protein molecule in the unit cell, and such an observation was taken as crystallographic artifacts. Compound 52 is much larger and interacts with BRD4 BD1 with multiple strong interactions including salt bridges and hydrogen bonds at a distinct site similar to that of traditional protein–protein interaction (PPI) inhibitors.
Figure 5.
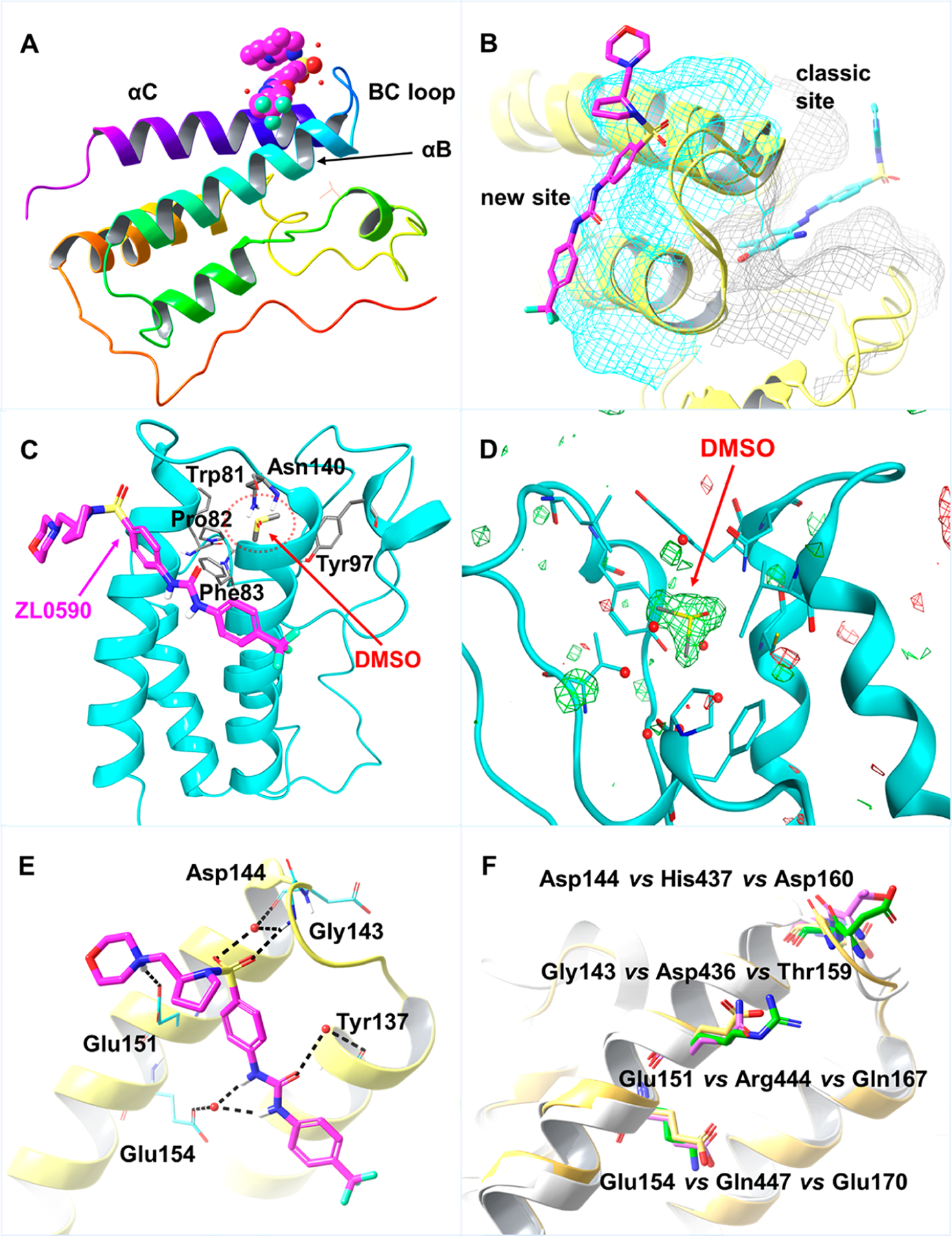
(A) Ribbon representation of crystal structure (PDB 6U0D) of BRD4 BD1 (sequence with residues Ser42 to Glu168) in complex with compound 52 (CPK representation in magenta). (B) Overlay of crystal structures BRD4 BD1 (in yellow) in complex with compound 52 (in magenta) and MS436 (in light-blue, PBD 4nud). The surface of traditional KAc pocket is colored in gray, and the new binding site is highlighted in light-blue. (C) New binding site of compound 52 (magenta) in ribbon representation. A solvent DMSO molecule occupies in the traditional KAc-binding pocket (red dashed circle area) with key residues of the classic KAc site shown in sticks. (D) Unbiased omit map for the KAc pocket, calculated without the coordinates for the DMSO molecule. The Fo-FcWT difference map electron density is shown as a green (positive density) or red (negative density) mesh, respectively, contoured at 3σ. (E) Detailed interactions of compound 52 with BRD4 BD1. Salt bridge with Glu151, direct hydrogen bond with Gly143, and indirect hydrogen bonds with Glu154, Tyr137, and Asp144 are highlighted in dashed line (black). (F) Overlay of BRD4 BD1 (PBD 4nud), BRD4 BD2 (PBD 6c7q), and BRD2 BD1 (PBD 3aqa). Key residues from BRD4 BD1 are in yellow, residues from BRD4 BD2 are in green, and residues from BRD2 BD2 are in magenta.
To further determine the binding affinity and selectivity of compound 52 for BRD4 BD1 vs BRD4 BD2, we employed in vitro thermal shift assay (TSA)63 to analyze the increased thermal stability of BRD4 BD1/BD2 upon binding to 52. Using wild-type (WT) BRD4 BD1 (44–168) and BD2 (349–461) expressed in and purified from bacteria (Figure 6A), we found that addition of 52 enhanced thermal stability of WT BRD4 BD1, leading to a dose-dependent increase in melting temperature (ΔTm) similar to that observed with pan-BET inhibitor JQ1. In contrast, 52 had no significant effect on WT BRD4 BD2 even at very high concentrations (Figure 6B). This result indicates that 52 has a higher selectivity for binding to BRD4 BD1 over BRD4 BD2. As shown in TR-FRET assay (Table 5), 52 selectively inhibits BD1 binding to acetyl histone peptide (IC50 = 90 nM) at least 10-fold better than BD2 (IC50 = 1,093 nM). In TSA that measures compound binding-enhanced protein stability resistant to temperature-induced protein unfolding (Figure 6B), 52 also consistently shows at least 10-time more temperature shift with BD1 (ΔTm ∼15 °C) versus BD2 (ΔTm ∼1 °C), whereas the orthosteric inhibitor JQ1 induces comparable but slightly better thermostability to BD1 (ΔTm ∼12 °C) than BD2 (ΔTm ∼7 °C). While TSA is only a semiquantitative binding assay, it indeed shows 52 binding selectivity to BD1. The weak binding of 52 to BD2, if any, does not significantly induce BD2 conformational change to expose its hydrophobic region for SYPRO Orange dye binding. To further demonstrate the newly identified binding site that is distinct from the classical KAc-binding pocket of BRD4, we also performed mutagenesis on the key residue Glu151 located at 52 binding site of BRD4 BD1. The selection was based on the cocrystal structure of 52 in complex with human BRD4 BD1, which suggests that Glu151 with a polar side chain plays a critical role in forming a key salt bridge with the morpholine moiety of 52. We purified a BRD4 BD1 mutant containing Ala-substituted Glu151 (E151A) to distinguish the binding modes by TSA (Figure 6A,C). As shown in Figure 6D, while JQ1 maintained a similar binding activity to E151A as to WT BRD4 BD1, 52 showed a complete loss of its binding affinity to E151A mutant. This result clearly indicates that 52 targets the newly identified binding site via interaction with Glu151, which differs significantly from that of JQ1. This distinct new binding site may play an important role in biological modulation of BRD4-associated protein–protein interactions. Given the complicated action modes of allosteric modulators,54,60,64–67 further studies are needed to elucidate the possible allosteric binding mechanism of 52.
Figure 6.
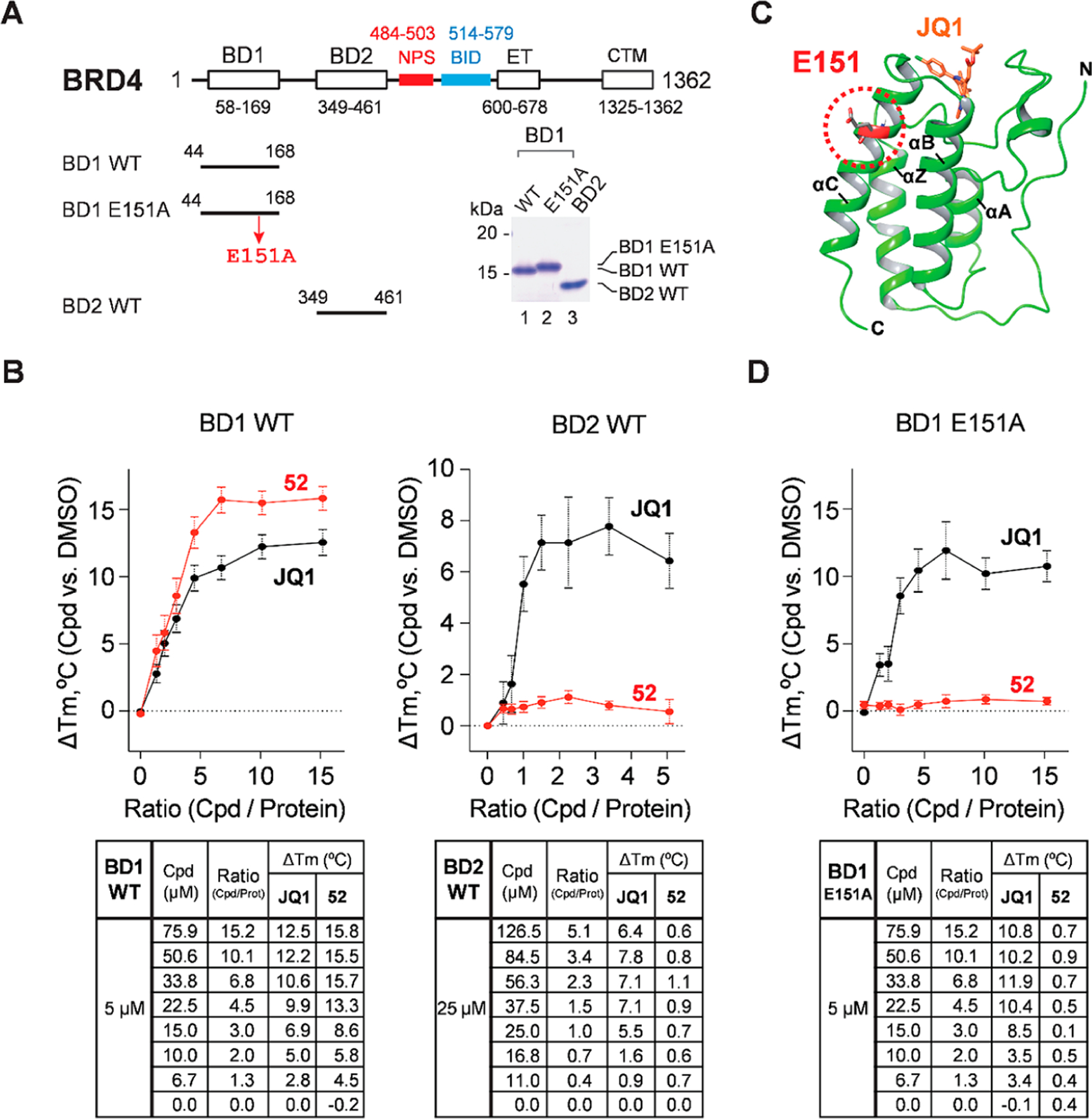
52 binds selectively to WT BRD4-BD1 but not WT BRD4-BD2 nor E151-mutated BRD4-BD1. (A) Purified human BRD4 BD1 and BD2 and the domain features of human BRD4 protein. E151A, glutamate 151 to alanine mutation. Numbers indicate the positions of amino acid residues in the full-length protein. The lower right panel is a Coomassie Brilliant Blue-stained SDS-PAGE gel image showing the purity of purified BD1 and BD2 domains used for TSA analysis. (B) TSA of 52 and JQ1 binding to WT BRD4 BD1 (left panel) and BRD4 BD2 (right panel). The graphs (top) show ΔTm upon compound binding (versus DMSO) at different compound/protein ratios (mean ± SEM) and the tables (bottom) show ΔTm values deduced from JQ1 or 52 binding at different compound/protein ratios with experimental replicates n = 9 for BD1 and n = 7 for BD2. Each experimental replicate was also analyzed in triplicate. (C) Superimposed BD1 structures showing the locations of E151 and JQ1-binding pocket (see PDB 3MXF and 6U0D). (D) TSA showing JQ1 but not 52 binding to E151A mutant of BRD4 BD1 similarly conducted as described in (B). Experimental replicates n = 9, each in triplicate.
DMPK Profiles of Compounds 52 and 53.
With promising in vitro activities in hand, we then assessed aqueous solubility for compounds 52 and 53 and subsequently evaluated their in vivo PK profiles in rats. As shown in Table 6, while compound 53 showed little improvement on oral bioavailability (0.4%) in comparison with lead compound 7, it exhibited an excellent half-life with a t1/2 value of 12 h. Compound 52 displayed good t1/2 value (3.7 h), a decent AUC value (602 ng·h/mL) when taken orally, and a better Cmax value of 86.9 ng/mL. The ultimate oral bioavailability of compound 52 (14%) has approximately 30-fold improvement compared to that of compound 7. Their aqueous solubilities were determined using the reported HPLC analysis methods.68 One-point calibration was performed against standards with known concentrations of the sample compounds to determine concentrations of the indicated compounds in samples. As expected, compound 52 exhibited a much better aqueous solubility than that of compound 7 (12.8 μg/mL) with a value of 3,500 μg/mL. Additionally, in vitro metabolic stability was evaluated in mouse and human microsomes as well as inhibitory effects against CYP450 enzymes (Supporting Information, Table S6).
Table 6.
In Vivo PK Profiles of Compounds 52 and 53 in Ratsa
| compd | dose | t1/2 (h)b | Cmax (ng/mL)c | AUC0–t (ng·h/mL)d | VSS (L/kg)e | CL (L/h/kg)f | F (%)g |
|---|---|---|---|---|---|---|---|
| 52 | po (20 mg/kg) | 3.7 ± 0.5 | 86.9 ± 37.5 | 602 ± 225 | 170 ± 57 | NA | 13.9 ± 5.2 |
| iv (10 mg/kg) | 2.5 ± 0.5 | 1,921 ± 429 | 2,172 ± 445 | 16.0 ± 2.7 | 4.5 ± 0.7 | ||
| 53 | po (20 mg/kg) | NAh | 2.0 ± 0.5 | 11.8 ± 2.2 | NA | NA | 0.4 ± 0.1 |
| iv (10 mg/kg) | 12 ± 2.2 | 1,592 ± 408 | 1,358 ± 72 | 105 ± 18 | 6.2 ± 0.3 |
Compounds were formulated in 10%DMSO/60%PEG400/30%Saline.
t1/2, half-life.
Cmax, maximum plasma concentration achieved.
AUC0–t, total exposure following single dose.
VSS, volume of distribution at steady state.
CL, total clearance.
F, oral bioavailability.
NA, not applicable.
In Vivo Efficacy of Compound 52.
To validate the in vivo efficacy of this novel inhibitor targeting a distinct new binding site of BRD4, compound 52 was evaluated in our established murine model of poly(I:C)-induced BRD4-dependent acute airway inflammation via oral administration (po).5 Vehicle alone and poly(I:C)-treated groups were established as a healthy control and pathological control, respectively. Also, compound 3 was included as a positive control. Histologically, we observed that poly(I:C)-induced neutrophilic inflammation around the small- and medium-sized airways was completely inhibited by compound 52 via oral administration (Figure 7A). Secretion of total cells, neutrophils (Figure 7B), and inflammatory cytokines (IL-6, KC, MCP-1, and RANTES, Figure 7C) in bronchoalveolar lavage fluid (BALF) as well as inflammatory gene expression (IL-6, KC, ISG54, and CIG5) in lung tissues of mice (Figure 7D) were also significantly attenuated by compound 52. Meanwhile, the positive control compound 3, a well-known BET inhibitor, also significantly blocked the poly(I:C)-induced BRD4-dependent acute airway inflammation, while our BRD4 inhibitor 52 is more efficacious (Figure 7).
Figure 7.
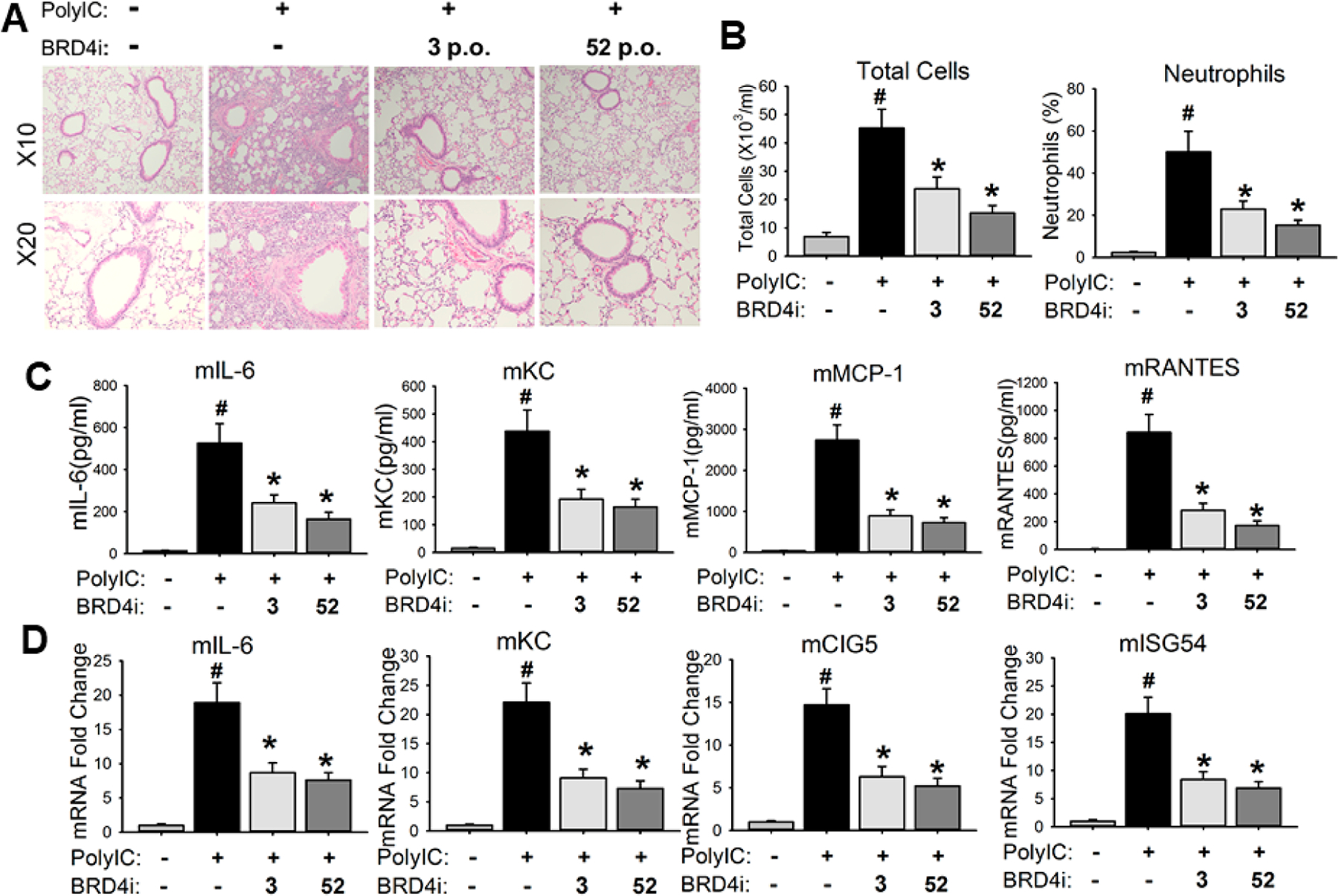
Oral administration of compound 52 blocked poly(I:C)-induced acute airway inflammation in mice. C57BL/6J mice were pretreated (10 mg/kg, po) with compound 52 before intranasal administration (i.n.) of poly(I:C). Compound 3 was included as a positive control. (A) Lung histology images of hematoxylin and eosin (H&E) staining were performed on paraffin-embedded lung sections and were shown at magnitude of ×10 and ×20, respectively. (B) Compound 52 blocked the secretion of poly(I:C)-induced inflammatory cells in bronchoalveolar lavage fluid (BALF). (C) Compound 52 blocked poly(I:C)-induced inflammatory cytokines in BALF. (D) Compound 52 blocked poly(I:C)-induced expression of inflammatory genes in lung tissues of mice. #p < 0.01, compared to control, *p < 0.01, compared to poly(I:C) only, n = 5.
To further confirm that the in vivo efficacy was induced specifically by BRD4 inhibition, we examined the level of H3K122Ac that reflects the HAT activity of BRD4, which has been demonstrated as an important regulator of immune responses.8,9 As shown in Figure 8, immunofluorescence staining of H3K122Ac demonstrated that poly(I:C) induced significant increase of BRD4 activity in both hSAECs and lung tissue of mice, and this poly(I:C)-induced H3K122Ac accumulation was significantly blocked by both BRD4 inhibitors 52 and 53. Meanwhile, compound 3 also blocked poly(I:C)-induced H3K122Ac accumulation in both hSAECs and lung tissue of mice, despite appearing marginally less effective than our BRD4 inhibitors 52 and 53 (Figure 8).
Figure 8.
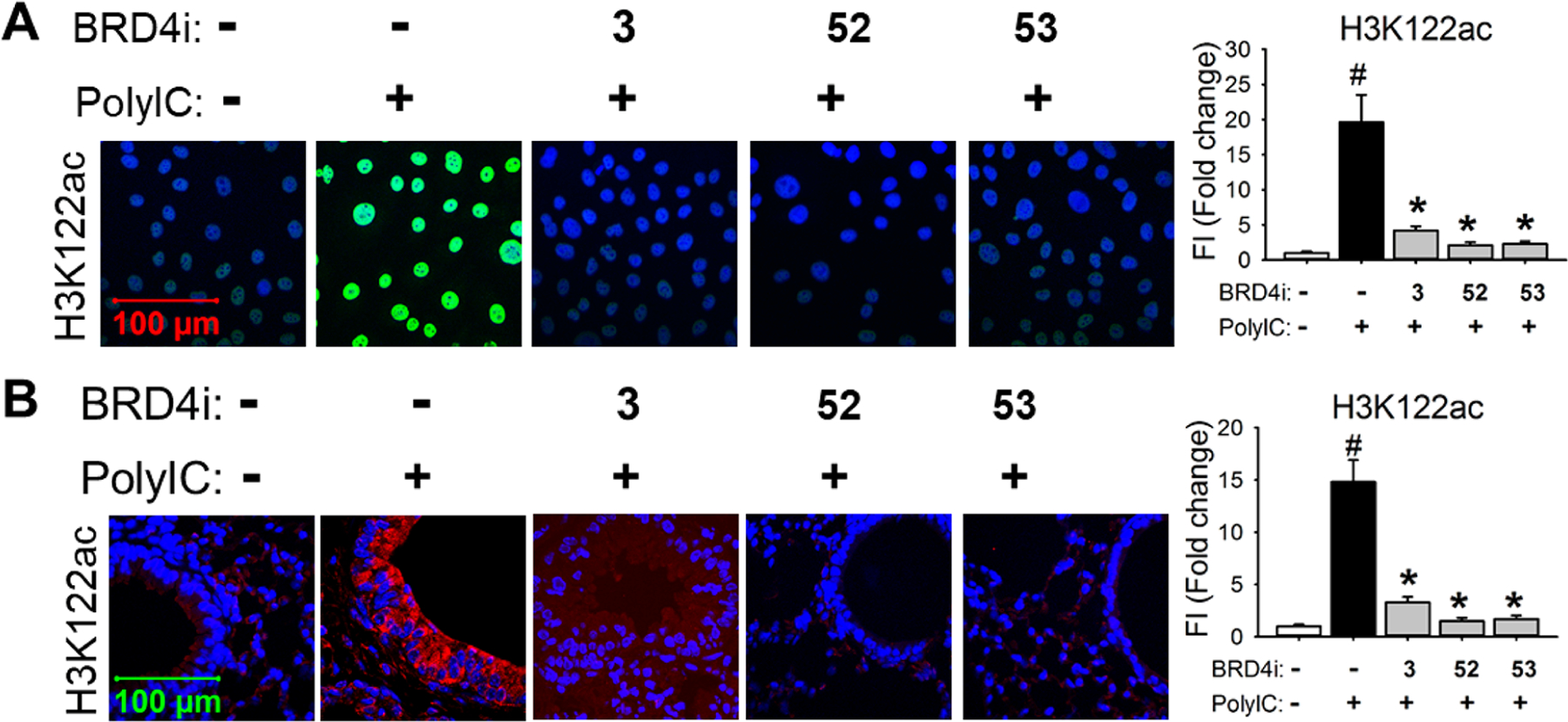
BRD4 inhibitors 52 and 53 significantly blocked poly(I:C)-induced H3K122Ac levels in hSAECs (A) and in lung tissue of mice (B). (A) Immunofluorescence staining of H3K122Ac (green color) was performed in hSAECs. (B) Immunofluorescence staining of H3K122Ac (red) was performed on paraffin-embedded lung sections of mice. Right panel, quantification of total fluorescence intensity shown as fold changes on immunofluorescence. #p < 0.01, compared with control; *p < 0.01 compared with poly(I:C) only, n = 5. Compound 3 was included as a positive control.
CONCLUSION
In summary, we have discovered a nanomolar range BRD4 BD1-selective inhibitor compound 52 (ZL0590) with a distinct new binding site on BRD4 and a favorable PK profile. The crystal structure of human BRD4 BD1 in complex with compound 52 and the relevant mutagenesis study on BRD4 BD1 reveal a first-in-class non-KAc binding site, which forms a unique structural basis for bromodomain targeting and ligand selectivity. This new binding site encompasses helices αB, αC, and BC loop, while the classic KAc recognition pocket is located at the end of the four-helix bundle. Multiple critical interactions including a salt bridge and hydrogen bonds were observed between compound 52 and critical amino acid residues Tyr137, Gly143, Asp144, Glu151, and Glu154. All of these key residues are not commonly shared among BET family proteins, and this offers a novel binding site whereby compound 52 displays improved bromodomain selectivity. By occupying this binding site of BRD4 BD1, compound 52 can modulate critical protein–protein interactions between BRD4 and associated partners, suppress poly(I:C)-induced expression of inflammatory genes in hSAECs, and block inflammatory responses in a murine model of acute airway inflammation. Notably, targeting this new binding site of BRD4 BD1 may provide a paradigm shift in understanding the unique and different biological functions mediated by the two BRD4 bromodomains. For example, we recently discovered that compound 45 (ZL0580), a structurally close analogue of compound 52, can epigenetically suppress HIV,40,69 while compound 3 was reported to activate latent HIV.22,70 Our further efforts will be focused on extensive SAR study based on compound 52 as an advanced chemical lead for the preclinical development of relevant epigenetic therapeutics and the investigation of potential new biological functions targeting this distinct recognition site of protein–protein interactions as well as the elucidation of the possible underlying allosteric modulation mechanisms with these novel chemical probes.
EXPERIMENTAL SECTION
General Chemistry Information.
All commercially available starting chemical reagents and solvents were ACS reagent grade and used without further purification unless otherwise specified. Reactions were performed under a nitrogen or argon atmosphere in dry glassware with magnetic stirring. Preparative column chromatography was performed using silica gel 60, particle size 0.063–0.200 mm (70–230 mesh, flash). Analytical TLC was carried out employing silica gel 60 F254 plates (Merck, Darmstadt). Visualization of the developed chromatograms was performed with detection by UV (254 nm). NMR spectra were recorded on a Bruker-600 or Bruker-300 (1H, 600 and 300 MHz; 13C, 150 and 75 MHz) spectrometer. 1H and 13C NMR spectra were recorded with TMS as an internal reference. Chemical shifts were expressed in ppm, and J values were given in Hz. High-resolution mass spectra (HRMS) were obtained from Thermo Fisher LTQ Orbitrap Elite mass spectrometer. Parameters include the following: nano ESI spray voltage was 1.8 kV, capillary temperature was 275 °C and the resolution was 60 000, and ionization was achieved by positive mode. Purities of final compounds were established by analytical HPLC, which was carried out on a Shimadzu HPLC system (model: CBM-20A LC-20AD SPD-20A UV/vis). HPLC analysis conditions: Waters μBondapak C18 (300 × 3.9 mm2), flow rate 0.5 mL/min, UV detection at 270 and 254 nm, linear gradient from 10% acetonitrile in water to 100% acetonitrile in water in 20 min followed by 30 min of the last-named solvent (0.1% TFA was added into both acetonitrile and water). All biologically evaluated compounds are >95% pure.
Methyl ((4-(3-(4-(Trifluoromethyl)phenyl)ureido)phenyl)-sulfonyl)-l-prolinate (11).
A solution of methyl ((4-aminophenyl)-sulfonyl)-l-prolinate (50 mg, 0.176 mmol) and 1-isocyanato-4-(trifluoromethyl)benzene (40 mg, 0.212 mmol) in 5 mL of CH2Cl2 was stirred at room temperature overnight. Then the mixture was concentrated directly, and the residue was purified by PTLC (CH2Cl2/MeOH = 100:1) to give the desired product 11 (40 mg, 96%) as a white solid. HPLC purity 99.9% (tR = 19.33 min). 1H NMR (300 MHz, CDCl3) Δ 8.02 (s, 1H), 7.96 (s, 1H), 7.77 (d, J = 8.8 Hz, 2H), 7.68–7.51 (m, 6H), 4.27 (dd, J = 7.7 Hz, 5.1 Hz, 1H), 3.72 (s, 3H), 3.52 (m, 1H), 3.32–3.19 (m, 1H), 2.13–1.90 (m, 3H), 1.77 (dd, J = 10.8, 5.4 Hz, 1H). 13C NMR (75 MHz, CDCl3) Δ 173.0, 152.2, 144.0, 141.7, 129.9, 128.6, 126.2 (q, J = 3.75 Hz), 125.1 (q, J = 33 Hz), 119.1, 119.0, 60.7, 52.7, 48.8, 30.9, 24.7. HRMS (ESI+): m/z calcd for C20H21F3N3O5S [M + H]+, 472.1154; found, 472.1166.
Methyl ((4-(3-(3-(Trifluoromethyl)phenyl)ureido)phenyl)-sulfonyl)-l-prolinate (12).
Compound 12 was obtained in a quantitative yield following the procedure of 11 as a yellow oil. HPLC purity 99.5% (tR = 12.86 min). 1H NMR (300 MHz, CDCl3) Δ 8.11 (s, 1H), 8.01 (s, 1H), 7.75 (d, J = 8.9 Hz, 3H), 7.63 (d, J = 8.9 Hz, 3H), 7.38 (t, J = 7.9 Hz, 1H), 7.27 (d, J = 6.5 Hz, 1H), 4.25 (dd, J = 7.4, 5.4 Hz, 1H), 3.71 (s, 3H), 3.50 (dt, J = 9.6, 6.1 Hz, 1H), 3.29–3.17 (m, 1H), 2.10–1.89 (m, 3H), 1.80–1.66 (m, 1H). 13C NMR (75 MHz, CDCl3) Δ 173.1, 152.6, 144.0, 139.0, 131.2 (q, J = 32 Hz), 129.8, 129.5, 128.6, 123.9 (q, J = 123.92), 122.8, 119.9 (q, J = 3.75 Hz), 119.0, 116.3 (q, J = 3.75 Hz), 60.7, 52.7, 48.7, 30.9, 24.6. HRMS (ESI+): m/z calcd for C20H21F3N3O5S [M + H]+, 472.1154; found, 472.1143.
Methyl ((4-(3-Phenylureido)phenyl)sulfonyl)-l-prolinate (13).
Compound 13 was obtained in a yield of 99% as a colorless oil following the procedure of 11. HPLC purity 97.7% (tR = 17.58 min). 1H NMR (300 MHz, CDCl3) Δ 8.01 (s, 1H), 7.78–7.68 (m, 3H), 7.55 (d, J = 8.8 Hz, 2H), 7.40 (d, J = 7.8 Hz, 2H), 7.28 (d, J = 2.8 Hz, 2H), 7.05 (t, J = 7.3 Hz, 1H), 4.24 (t, J = 6.3 Hz, 1H), 3.71 (s, 3H), 3.55–3.43 (m, 1H), 3.22 (m, 1H), 2.02–1.92 (m, 3H), 1.78–1.67 (m, 1H). 13C NMR (75 MHz, CDCl3) Δ 172.9, 153.0, 144.0, 138.0, 130.0, 129.1, 128.6, 123.9, 120.3, 118.8, 60.6, 52.6, 48.7, 30.9, 24.6. HRMS (ESI+): m/z calcd for C19H22N3O5S [M + H]+, 404.1280; found, 404.1269.
Methyl ((4-(3-(4-Methoxyphenyl)ureido)phenyl)sulfonyl)-l-prolinate (14).
Compound 14 was obtained in a quantitative yield as a yellow oil following the procedure of 11. HPLC purity 98.9% (tR = 17.41 min). 1H NMR (300 MHz, CDCl3) Δ 8.00 (s, 1H), 7.69 (d, J = 8.8 Hz, 2H), 7.62 (s, 1H), 7.50 (d, J = 8.8 Hz, 2H), 7.24 (d, J = 8.9 Hz, 2H), 6.79 (d, J = 9.0 Hz, 2H), 4.23 (dd, J = 7.2, 5.3 Hz, 1H), 3.71 (d, J = 6.1 Hz, 6H), 3.53–3.41 (m, 1H), 3.27–3.15 (m, 1H), 1.98 (m, 3H), 1.73 (dd, J = 11.8, 6.7 Hz, 1H). 13C NMR (75 MHz, CDCl3) Δ 172.9, 156.6, 153.6, 144.0, 130.5, 130.1, 128.6, 123.1, 118.7, 114.3, 60.6, 55.5, 52.6, 48.7, 30.9, 24.6. HRMS (ESI+): m/z calcd for C20H24N3O6S [M + H]+, 434.1386; found, 434.1369.
Methyl ((4-(3-(4-Fluorophenyl)ureido)phenyl)sulfonyl)-l-prolinate (15).
Compound 15 was obtained in a quantitative yield as a yellow oil following the procedure of 11. HPLC purity 99.2% (tR = 17.83 min). 1H NMR (300 MHz, CDCl3) Δ 8.04 (s, 1H), 7.78 (s, 1H), 7.72 (d, J = 8.8 Hz, 2H), 7.55 (d, J = 8.8 Hz, 2H), 7.38–7.30 (m, 2H), 6.95 (t, J = 8.6 Hz, 2H), 4.24 (dd, J = 7.4, 5.2 Hz, 1H), 3.70 (s, 3H), 3.48 (m, 1H), 3.28–3.15 (m, 1H), 1.99 (m, 3H), 1.79–1.66 (m, 1H). 13C NMR (75 MHz, CDCl3) Δ 173.0, 166.6, 153.1, 144.1, 134.0, 129.9, 128.6, 122.2 (d, J = 3.25 Hz), 118.8, 115.6 (d, J = 22.5 Hz), 60.7, 52.7, 48.7, 30.9, 24.6. HRMS (ESI+): m/z calcd for C19H21FN3O5S [M + H]+, 422.1186; found, 422.1167.
Methyl ((4-(3-(4-(Trifluoromethoxy)phenyl)ureido)phenyl)-sulfonyl)-l-prolinate (16).
Compound 16 was obtained in a yield of 94% following the procedure of 11 as a white solid. HPLC purity 99.2% (tR = 19.26 min). 1H NMR (300 MHz, CDCl3) Δ 8.00 (s, 1H), 7.84 (s, 1H), 7.74 (d, J = 8.5 Hz, 2H), 7.59 (d, J = 8.5 Hz, 2H), 7.47 (d, J = 8.6 Hz, 2H), 7.14 (d, J = 8.5 Hz, 2H), 4.25 (dd, J = 7.6, 4.9 Hz, 1H), 3.71 (s, 3H), 3.51 (m, 1H), 3.23 (q, J = 7.8 Hz, 1H), 2.02 (m, 3H), 1.75 (m, 1H). 13C NMR (75 MHz, CDCl3) Δ 173.0, 152.6, 144.8, 144.8, 144.0, 137.0, 129.9, 128.6, 122.2, 121.7, 121.0, 118.9, 60.7, 52.7, 48.7, 30.9, 24.6. HRMS (ESI+): m/z calcd for C20H21F3N3O6S [M + H]+, 488.1103; found, 488.1094.
Methyl ((4-(3-(4-Hydroxyphenyl)ureido)phenyl)sulfonyl)-l-prolinate (17).
To a solution of triphosgene (25 mg, 0.08 mmol) in 5 mL of toluene under N2, methyl ((4-aminophenyl)sulfonyl)-l-prolinate (71 mg, 0.25 mmol) was added. After stirring at 120 °C for 2 h, 4-hydroxyl aniline (27 mg, 0.25 mmol) was added. Then the solution was allowed to stir at 120 °C for overnight before concentration. The residue was purified by PTLC to get 17 (46 mg, 44%) as a brown solid. HPLC purity 99.7% (tR = 19.50 min). 1H NMR (300 MHz, MeOD) Δ 7.77 (d, J = 8.8 Hz, 2H), 7.66 (d, J = 8.9 Hz, 2H), 7.24 (d, J = 8.8 Hz, 2H), 6.77 (d, J = 8.8 Hz, 2H), 4.24 (dd, J = 8.0, 4.1 Hz, 1H), 3.74 (s, 3H), 3.53–3.43 (m, 1H), 3.32–3.22 (m, 1H), 1.98 (m, 3H), 1.72 (m, 1H). 13C NMR (75 MHz, MeOD) Δ 173.1, 153.8, 153.6, 144.4, 130.1, 130.1, 128.4, 122.1, 117.9, 115.0, 60.5, 51.5, 48.4, 30.5, 24.2. HRMS (ESI+): m/z calcd for C19H22N3O6S [M + H]+, 420.1229; found, 420.1229.
Methyl ((4-(3-(3-Methyl-4-(trifluoromethyl)phenyl)ureido)-phenyl)sulfonyl)-l-prolinate (18).
Compound 18 was obtained in a yield of 30% following the procedure of 17. HPLC purity 95.7% (tR = 19.47 min). 1H NMR (300 MHz, CDCl3) Δ 7.98 (s, 1H), 7.80 (s, 1H), 7.74 (d, J = 8.4 Hz, 2H), 7.65 (s, 1H), 7.56 (dd, J = 16.6, 8.4 Hz, 3H), 7.18 (d, J = 8.3 Hz, 1H), 4.26 (t, J = 6.4 Hz, 1H), 3.72 (s, 3H), 3.50 (dt, J = 11.7, 5.6 Hz, 1H), 3.24 (q, J = 7.8 Hz, 1H), 2.39 (s, 3H), 2.00 (m, 3H), 1.75 (m, 1H). 13C NMR (75 MHz, CDCl3) Δ 172.9, 152.6, 143.9, 136.1, 132.5, 131.5, 130.1, 129.4, 129.0, 128.6, 126.0, 123.0, 122.4, 118.8, 117.5 (q, J = 6 Hz), 60.7, 52.6, 48.7, 30.9, 24.6, 18.6. HRMS (ESI+): m/z calcd for C21H23F3N3O5S [M + H]+, 486.1311; found, 486.1316.
Methyl ((4-(3-(3-Chloro-4-(trifluoromethyl)phenyl)ureido)-phenyl)sulfonyl)-l-prolinate (19).
Compound 19 was obtained in a yield of 41% as a white solid following the procedure of 17. HPLC purity 95.0% (tR = 19.88 min). 1H NMR (300 MHz, MeOD) Δ 7.91 (d, J = 2.1 Hz, 1H), 7.85–7.79 (m, 2H), 7.74–7.66 (m, 3H), 7.49 (dd, J = 8.6, 2.0 Hz, 1H), 4.26 (dd, J = 8.0, 4.0 Hz, 1H), 3.75 (s, 3H), 3.54–3.43 (m, 1H), 3.29 (m, 1H), 2.07–1.91 (m, 3H), 1.73 (m, 1H). 13C NMR (75 MHz, MeOD) Δ 173.1, 152.4, 143.7, 143.7, 132.2, 130.9, 128.4, 128.0, 127.93, 127.86, 128.0, 127.7, 125.0, 121.5, 121.4, 121.1, 120.2, 118.3, 116.1, 60.5, 51.5, 48.4, 30.5, 24.2. HRMS (ESI+): m/z calcd for C20H20ClF3N3O5S [M + H]+, 506.0764; found, 506.0752.
Methyl ((2-Methoxy-4-(3-(4-(trifluoromethyl)phenyl)ureido)-phenyl)sulfonyl)-l-prolinate (20).
Compound 20 was obtained in a quantitative yield following the procedure of 11 as a yellow solid. HPLC purity 99.8% (tR = 18.80 min). 1H NMR (300 MHz, CDCl3) Δ 8.12 (s, 1H), 8.01 (s, 1H), 7.77 (t, J = 5.5 Hz, 2H), 7.64 (d, J = 8.6 Hz, 2H), 7.55 (d, J = 8.8 Hz, 2H), 6.79 (dd, J = 8.7, 1.8 Hz, 1H), 4.65 (dd, J = 8.1, 4.1 Hz, 1H), 3.90 (s, 3H), 3.67 (s, 3H), 3.61–3.51 (m, 1H), 3.26–3.16 (m, 1H), 2.21–1.99 (m, 3H), 1.91–1.79 (m, 1H). 13C NMR (75 MHz, CDCl3) Δ 173.3, 157.8, 152.2, 146.2, 141.9, 132.4, 126.1 (q, J = 3.75 Hz), 124.9 (q, J = 32.25 Hz), 122.5, 119.0, 118.1, 110.0, 102.3, 61.3, 56.1, 52.6, 48.1, 30.8, 24.9. HRMS (ESI+): m/z calcd for C21H23F3N3O6SNa [M + Na]+, 524.1079; found, 524.1070.
Methyl ((3-Fluoro-4-(3-(4-(trifluoromethyl)phenyl)ureido)-phenyl)sulfonyl)-l-prolinate (21).
Compound 21 was obtained in a yield of 66% following the procedure of 11 as an off-white solid. HPLC purity 98.0% (tR = 19.56 min). 1H NMR (300 MHz, CDCl3) Δ 8.52 (dd, J = 8.7, 7.9 Hz, 1H), 8.13 (s, 1H), 7.94 (d, J = 3.5 Hz, 1H), 7.64 (m, 4H), 7.55 (d, J = 8.8 Hz, 2H), 4.31 (dd, J = 7.8, 4.6 Hz, 1H), 3.73 (s, 3H), 3.54 (dt, J = 9.2, 6.0 Hz, 1H), 3.29 (dt, J = 9.1, 7.1 Hz, 1H), 2.13–1.94 (m, 3H), 1.82 (m, 1H). 13C NMR (75 MHz, CDCl3) Δ 172.8, 151.7, 151.2 (d, J = 245 Hz) 141.4, 132.5 (d, J = 9.75 Hz), 130.5 (d, J = 6 Hz), 130.5, 126.2 (q, J = 3.75 Hz), 125.3 (q, J = 32.25 Hz), 124.4 (d, J = 2.25 Hz), 122.4, 120.6, 119.0, 114.3 (d, J = 22.5 Hz), 60.7, 52.7, 48.7, 30.9, 24.6. HRMS (ESI+): m/z calcd for C20H19F4N3O5SNa [M + Na]+, 512.0879; found, 512.0858.
Methyl ((4-(4-(Trifluoromethyl)benzamido)phenyl)sulfonyl)-l-prolinate (22).
Compound 22 was resynthesized in house following our previously reported procedure.47
Methyl ((4-(1-Isopropyl-3-(4-(trifluoromethyl)phenyl)ureido)-phenyl)sulfonyl)-l-prolinate (23).
Compound 23 was obtained in a yield of 49% as a white solid following the procedure of 11. HPLC purity 98.9% (tR = 19.84 min). 1H NMR (300 MHz, CDCl3) Δ 8.07–8.00 (m, 2H), 7.52–7.39 (m, 6H), 5.99 (s, 1H), 4.95 (m, 1H), 4.50 (dd, J = 8.5, 3.1 Hz, 1H), 3.64 (s, 3H), 3.63–3.47 (m, 2H), 2.31–2.18 (m, 1H), 2.11–1.95 (m, 3H), 1.17 (dd, J = 6.8, 1.8 Hz, 6H). 13C NMR (75 MHz, CDCl3) Δ 172.3, 153.0, 141.9, 141.5, 139.8, 131.5, 129.1, 126.0 (q, J = 3.75 Hz), 118.7, 60.2, 52.3, 48.3, 47.7, 31.0, 24.6, 21.44, 21.41. HRMS (ESI+): m/z calcd for C23H27F3N3O5S [M + H]+, 514.1624; found, 514.1610.
Methyl ((4-(3-(4-(Trifluoromethyl)benzyl)ureido)phenyl)-sulfonyl)-l-prolinate (24).
Compound 24 was obtained in a yield of 64% as a white solid following the procedure of 11. HPLC purity 97.3% (tR = 18.33 min). 1H NMR (300 MHz, CDCl3) Δ 8.00 (s, 1H), 7.66 (d, J = 8.6 Hz, 2H), 7.58–7.49 (m, 4H), 7.40 (d, J = 8.0 Hz, 2H), 6.25 (t, J = 6.0 Hz, 1H), 4.46 (d, J = 5.8 Hz, 2H), 4.18 (dd, J = 7.6, 5.1 Hz, 1H), 3.68 (s, 3H), 3.48–3.37 (m, 1H), 3.18 (dt, J = 9.4, 7.0 Hz, 1H), 1.96 (m, 3H), 1.69 (m, 1H). 13C NMR (75 MHz, CDCl3) Δ 173.0, 166.6, 155.2, 144.5, 143.2, 129.4 (q, J = 31.5 Hz), 129.4, 128.6, 127.5, 125.4 (q, J = 3.75 Hz), 122.3, 118.3, 60.6, 52.5, 48.7, 43.4, 30.8, 24.6. HRMS (ESI+): m/z calcd for C21H23F3N3O5S [M + H]+, 486.1311; found, 486.1316.
Methyl (E)-((4-(2-(1-Cyano-2-oxo-2-(4-(trifluoromethoxy)-phenyl)ethylidene)hydrazinyl) phenyl)sulfonyl)-l-prolinate (25).
To a solution of methyl ((4-aminophenyl)sulfonyl)-l-prolinate (62 mg, 0.22 mmol) in 1 mL of H2O and 1 mL of CH3CN, 10% HCl (0.88 mmol) was added at 0 °C. After stirring at room temperature for 30 min, NaNO2 (18 mg, 0.26 mmol) in 1 mL of H2O were added. After stirring at 0 °C for 5 min, NaOAc (108 mg, 1.32 mmol) and 3-oxo-3-(4-(trifluoromethoxy)phenyl)propanenitrile (50 mg, 0.22 mmol) in 2 mL of EtOH was added. The mixture was extracted with CH2Cl2 and purified by silica gel column (PE/EA = 20/1–5/1) to give the desired product 25 (94 mg, 82% for two steps) as a yellow solid. HPLC purity 99.6% (tR = 15.01 min). 1H NMR (300 MHz, DMSO-d6) Δ 12.65 (s, 1H), 8.05 (d, J = 8.6 Hz, 2H), 7.85 (d, J = 8.6 Hz, 2H), 7.56 (d, J = 8.5 Hz, 4H), 4.20 (dd, J = 8.1, 4.1 Hz, 1H), 3.65 (s, 3H), 3.16 (dd, J = 16.4, 7.0 Hz, 1H), 2.00–1.72 (m, 3H), 1.66–1.51 (m, 1H). 13C NMR (75 MHz, DMSO-d6) Δ 186.3, 172.6, 151.5, 146.2, 135.3, 133.1, 132.8, 129.5, 120.7, 117.1, 115.8, 111.5, 60.7, 52.6, 48.9, 30.9, 24.7. HRMS (ESI+): m/z calcd for C22H20F3N4O6S [M + H]+, 525.1056; found, 525.1050.
N-Cyclopentyl-4-(3-(4-(trifluoromethyl)phenyl)ureido)-benzenesulfonamide (28).
1-Isocyanato-4-(trifluoromethyl)benzene (47 mg, 0.25 mmol) and 4-amino-N-cyclopentylbenzenesulfonamide (50 mg, 0.21 mmol) were dissolved in CH2Cl2. The mixture was stirred at room temperature for overnight. Then the solution was concentrated and purified by silica gel column (CH2Cl2:CH3OH = 100:1 to 50:1) to give 28 (69 mg, 77%) as a white solid. HPLC purity 97.7% (tR = 19.68 min). 1H NMR (300 MHz, DMSO-d6) Δ 9.24 (d, J = 2.4 Hz, 2H), 7.72 (t, J = 5.5 Hz, 2H), 7.66 (dd, J = 6.8, 5.4 Hz, 6H), 7.48 (d, J = 7.1 Hz, 1H), 3.42–3.36 (m, 1H), 1.56 (ddd, J = 12.7, 11.2, 6.4 Hz, 4H), 1.43–1.23 (m, 4H). 13C NMR (75 MHz, DMSO-d6) Δ 152.5, 143.6, 143.3, 134.8, 128.2, 126.6 (q, J = 3.75 Hz), 122.6 (q, J = 31.5 Hz), 118.6, 118.4, 54.9, 32.9, 23.3. HRMS (ESI+): m/z calcd for C19H20F3N3NaO3S [M + Na]+, 450.1075; found, 450.1060.
1-(4-(1,1-Dioxidothiomorpholino)phenyl)-3-(4-(trifluoromethyl)-phenyl)urea (29).
Compound 29 was obtained in a yield of 69% as a light-purple solid following the procedure of 11. HPLC purity 99.6% (tR = 17.90 min). 1H NMR (300 MHz, DMSO-d6) Δ 9.01 (s, 1H), 8.59 (s, 1H), 7.68–7.57 (m, 4H), 7.35 (d, J = 9.0 Hz, 2H), 6.99 (d, J = 9.0 Hz, 2H), 3.73–3.64 (m, 4H), 3.17–3.08 (m, 4H). 13C NMR (75 MHz, DMSO-d6) Δ 152.9, 144.1, 143.9, 132.3, 126.9, 126.5 (q, J = 3.75 Hz), 122.0 (q, J = 3.75 Hz), 120.6, 118.2, 117.3, 50.4, 48.1. HRMS (ESI+): m/z calcd for C18H19F3N3O3S [M + H]+, 414.1093; found, 414.1099.
Methyl (4-(3-(4-(Trifluoromethyl)phenyl)ureido)benzoyl)-L-prolinate (30).
Compound 30 was obtained in a yield of 86% as a white solid following the procedure of 11. HPLC purity 99.98% (tR = 18.15 min). 1H NMR (300 MHz, CDCl3) Δ 8.46 (s, 1H), 8.33 (s, 1H), 7.54 (q, J = 8.8 Hz, 4H), 7.40 (d, J = 8.4 Hz, 2H), 7.15 (d, J = 8.5 Hz, 2H), 4.67 (dd, J = 8.3, 5.3 Hz, 1H), 3.68 (s, 4H), 3.59 (m, 1H), 2.43–2.31 (m, 1H), 2.07 (dq, J = 11.2, 5.9 Hz, 2H), 1.97–1.87 (m, 1H). 13C NMR (75 MHz, CDCl3) Δ 172.4, 170.7, 152.7, 142.3, 141.7, 128.7, 128.1, 126.1, 126.0 (q, J = 3.75 Hz), 124.3 (q, J = 33 Hz), 122.5, 118.6, 118.5, 59.8, 52.4, 50.4, 29.4, 25.4. HRMS (ESI+): m/z calcd for C21H21F3N3O4 [M + H]+, 436.1484; found, 436.1485.
Methyl ((3-(3-(4-(Trifluoromethyl)phenyl)ureido)phenyl)-sulfonyl)-l-prolinate (31).
Compound 31 was obtained in a yield of 78% as a white solid following the procedure of 11. HPLC purity 99.6% (tR = 19.05 min). 1H NMR (300 MHz, CDCl3) Δ 8.02 (s, 1H), 7.94 (dt, J = 7.7, 1.8 Hz, 1H), 7.90–7.83 (m, 2H), 7.59 (d, J = 8.6 Hz, 2H), 7.54–7.39 (m, 4H), 4.33 (dd, J = 8.0, 4.6 Hz, 1H), 3.69 (s, 3H), 3.60–3.50 (m, 1H), 3.28 (dt, J = 9.4, 7.0 Hz, 1H), 2.11–1.90 (m, 3H), 1.80–1.67 (m, 1H). 13C NMR (75 MHz, CDCl3) Δ 172.9, 152.5, 141.8, 140.1, 137.3, 130.2, 126.1 (q, J = 3.75 Hz), 124.8 (q, J = 33 Hz), 124.0, 122.4, 121.1, 118.9, 117.6, 60.9, 52.7, 48.8, 30.9, 24.6. HRMS (ESI+): m/z calcd for C20H21F3N3O5S [M + H]+, 472.1154; found, 472.1145.
Methyl ((2-(3-(4-(Trifluoromethyl)phenyl)ureido)phenyl)-sulfonyl)-l-prolinate (32).
Compound 32 was obtained in a yield of 79% as a white solid following the procedure of 11. HPLC purity 99.6% (tR = 20.55 min). 1H NMR (300 MHz, DMSO-d6) Δ 10.14 (s, 1H), 8.67 (s, 1H), 8.04 (dd, J = 8.4, 1.2 Hz, 1H), 7.82 (dd, J = 8.0, 1.6 Hz, 1H), 7.75–7.62 (m, 5H), 7.28 (ddd, J = 8.2, 7.4, 1.2 Hz, 1H), 4.34 (dd, J = 8.7, 3.8 Hz, 1H), 3.57 (s, 3H), 3.41 (ddd, J = 9.3, 7.1, 5.0 Hz, 1H), 3.23 (dt, J = 9.4, 7.0 Hz, 1H), 2.11–1.98 (m, 1H), 1.95–1.68 (m, 3H). 13C NMR (75 MHz, DMSO-d6) Δ 172.4, 152.32, 143.8, 137.1, 134.3, 129.8, 127.0, 126.8, 126.6 (q, J = 3.75 Hz), 125.0, 123.8, 122.7 (q, J = 32.25 Hz), 118.7, 60.5, 52.7, 48.7, 30.8, 24.7. HRMS (ESI+): m/z calcd for C20H21F3N3O5S [M + H]+, 472.1154; found, 472.1156.
1-(2-Oxo-1,2,3,4-tetrahydroquinolin-6-yl)-3-(4-(trifluoromethyl)-phenyl)urea (33).
Compound 33 was resynthesized in house following our previously reported procedure.47
Ethyl ((4-(3-(4-(Trifluoromethyl)phenyl)ureido)phenyl)sulfonyl)-D-prolinate (34).
Compound 34 was obtained in a yield of 97% following the procedure of 11 as a white foam. HPLC purity 99.9% (tR = 19.40 min). 1H NMR (300 MHz, CDCl3) Δ 8.05 (s, 1H), 8.00 (s, 1H), 7.77 (d, J = 8.8 Hz, 2H), 7.65–7.57 (m, 4H), 7.53 (d, J = 8.8 Hz, 2H), 4.25 (m, 1H), 4.15 (dd, J = 14.9, 7.7 Hz, 2H), 3.51 (dt, J = 9.3, 6.1 Hz, 1H), 3.29–3.18 (m, 1H), 2.08–1.92 (m, 3H), 1.74 (dt, J = 12.4, 5.7 Hz, 1H), 1.25 (t, J = 7.1 Hz, 3H). 13C NMR (75 MHz, CDCl3) Δ 172.5, 152.1, 143.9, 141.7, 130.1, 128.6, 126.1 (q, J = 32.25 Hz), 125.1 (q, J = 3.75 Hz), 122.4, 119.0, 119.0, 61.8, 60.9, 48.7, 30.9, 24.6, 14.0. HRMS (ESI+): m/z calcd for C21H23F3N3O5SNa [M + Na]+, 508.1130; found, 508.1103.
Methyl ((4-(3-(4-(Trifluoromethyl)phenyl)ureido)phenyl)-sulfonyl)-d-prolinate (35).
Compound 35 was obtained in a yield of 85% as a white solid following the procedure of 11. HPLC purity 99.6% (tR = 18.99 min). 1H NMR (300 MHz, CDCl3) Δ 7.99 (s, 1H), 7.93 (s, 1H), 7.80–7.72 (m, 2H), 7.65–7.57 (m, 4H), 7.54 (d, J = 8.9 Hz, 2H), 4.28 (dd, J = 7.7, 5.0 Hz, 1H), 3.72 (s, 3H), 3.52 (qd, J = 6.9, 5.0 Hz, 1H), 3.25 (dt, J = 9.4, 7.1 Hz, 1H), 2.12–1.91 (m, 3H), 1.78 (tt, J = 6.7, 3.7 Hz, 1H). 13C NMR (75 MHz, CDCl3) Δ 173.0, 152.2, 143.9, 141.6, 130.0, 128.6, 126.2 (q, J = 3.75 Hz), 126.0, 125.1 (q, J = 32.25 Hz), 122.4, 119.0, 119.0, 60.7, 52.7, 48.7, 30.9, 24.7. HRMS (ESI+): m/z calcd for C20H21F3N3O5S [M + H]+, 472.1154; found, 472.1156.
1-(4-(Pyrrolidin-1-ylsulfonyl)phenyl)-3-(4-(trifluoromethyl)-phenyl)urea (36).
Compound 36 was obtained in a yield of 34% as a white solid following the procedure of 11. HPLC purity 99.5% (tR = 19.51 min). 1H NMR (300 MHz, MeOD) Δ 7.69 (ddd, J = 29.7, 21.9, 8.9 Hz, 8H), 3.28–3.20 (m, 4H), 1.81–1.72 (m, 4H). 13C NMR (75 MHz, MeOD) Δ 152.8, 143.7, 142.5, 129.7, 128.5, 125.7 (q, J = 3.75 Hz), 124.1 (q, J = 32.25 Hz), 118.4, 118.2, 47.7, 24.8. HRMS (ESI+): m/z calcd for C18H18F3N3NaO3S [M + H]+, 436.0919; found, 436.0909.
1-(4-((4-Acetylpiperazin-1-yl)sulfonyl)phenyl)-3-(4-(trifluoromethyl)phenyl)urea (37).
Compound 37 was obtained in a yield of 60% as a white solid following the procedure of 11. HPLC purity 99.5% (tR = 18.46 min). 1H NMR (300 MHz, DMSO-d6) Δ 9.33 (s, 1H), 9.24 (s, 1H), 7.75–7.62 (m, 8H), 3.51 (d, J = 4.4 Hz, 4H), 2.95–2.79 (m, 4H), 1.94 (s, 3H). 13C NMR (75 MHz, DMSO-d6) Δ 173.6, 157.2, 149.2, 148.2, 134.2, 132.5, 131.3 (q, J = 3.75 Hz), 131.3, 127.9, 127.5 (q, J = 31.5 Hz), 123.4, 123.3, 51.2, 51.0, 50.1, 26.2. HRMS (ESI+): m/z calcd for C20H22F3N4O4S [M + H]+, 471.1314; found, 471.1314.
1-(4-((4-(Pyrrolidin-1-yl)piperidin-1-yl)sulfonyl)phenyl)-3-(4-(trifluoromethyl)phenyl)urea (38).
Compound 38 was obtained in a yield of 45% as white solid following the procedure of 11. HPLC purity 98.3% (tR = 17.62 min). 1H NMR (300 MHz, DMSO-d6) Δ 9.31 (d, J = 19.9 Hz, 2H), 7.66 (s, 8H), 3.50 (s, 2H), 2.39 (s, 6H), 1.97–1.76 (m, 3H), 1.60 (s, 4H), 1.41 (d, J = 9.2 Hz, 2H). 13C NMR (75 MHz, DMSO-d6) Δ 152.5, 144.1, 143.5, 129.2, 128.6, 126.6 (q, J = 3.75 Hz), 126.5, 123.2, 122.7 (q, J = 31.5 Hz), 118.7, 118.4, 60.0, 51.2, 44.8, 30.5, 23.3. HRMS (ESI+): m/z calcd for C23H28F3N4O3S [M + H]+, 497.1834; found, 497.1827.
(S)-1-((4-(3-(4-(Trifluoromethyl)phenyl)ureido)phenyl)sulfonyl)-pyrrolidine-2-carboxamide (42).
Compound 42 was obtained in a yield of 89% as a white solid following the procedure of 11. HPLC purity 98.5% (tR = 17.59 min). 1H NMR (300 MHz, MeOD) Δ 7.84 (d, J = 8.9 Hz, 2H), 7.78–7.71 (m, 2H), 7.68 (d, J = 8.6 Hz, 2H), 7.61 (d, J = 8.8 Hz, 2H), 4.08 (dd, J = 8.1, 3.9 Hz, 1H), 3.63–3.51 (m, 1H), 3.30–3.20 (m, 1H), 2.03–1.78 (m, 3H), 1.71–1.57 (m, 1H). 13C NMR (75 MHz, MeOD) Δ 176.3, 152.7, 144.1, 142.5, 129.4, 129.4, 128.8, 126.2, 125.7 (q, J = 3.75 Hz), 124.1 (q, J = 32.25 Hz), 122.6, 118.3, 118.2, 62.0, 49.3, 30.7, 24.1. HRMS (ESI+): m/z calcd for C19H20F3N4O4S [M + H]+, 457.1157; found, 457.1162.
(S)-N-Phenyl-1-((4-(3-(4-(trifluoromethyl)phenyl)ureido)phenyl)-sulfonyl)pyrrolidine-2-carboxamide (45).
Compound 45 was resynthesized in house following our previously reported procedure.69
(S)-4-(1-((4-(3-(4-(Trifluoromethyl)phenyl)ureido)phenyl)-sulfonyl)pyrrolidine-2-carboxamido)phenyl acetate (46).
Compound 46 was obtained in a quantitative yield as a white powder following the procedure of 11. HPLC purity 95.7% (tR = 19.22 min). 1H NMR (300 MHz, DMSO-d6) Δ 10.00 (s, 1H), 9.31 (d, J = 20.4 Hz, 2H), 7.81 (d, J = 7.9 Hz, 2H), 7.74–7.61 (m, 8H), 7.13–7.04 (m, 2H), 4.18 (t, J = 7.3 Hz, 1H), 3.48 (s, 1H), 3.23 (s, 1H), 2.68 (d, J = 2.0 Hz, 1H), 2.25 (d, J = 3.3 Hz, 3H), 1.88 (m, 3H), 1.55 (s, 1H). 13C NMR (75 MHz, DMSO-d6) Δ 170.5, 169.7, 152.5, 146.6, 144.3, 143.5, 136.7, 130.0, 129.1, 126.7, 126.6, 126.5, 122.4, 121.0, 118.6, 118.4, 62.3, 49.6, 31.5, 24.8, 21.3. HRMS (ESI+): m/z calcd for C27H26F3N4O6S [M + H]+, 591.1525; found, 591.1531.
(S)-1-(4-((2-(4-Methylpiperazine-1-carbonyl)pyrrolidin-1-yl)-sulfonyl)phenyl)-3-(4-(trifluoromethyl)phenyl)urea (47).
Compound 47 (76 mg, 60% for two steps) was obtained in a yield of 60% for two steps as a white solid following the procedure of 11. HPLC purity 95% (tR = 17.10 min). 1H NMR (300 MHz, MeOD) Δ 7.82 (d, J = 8.7 Hz, 2H), 7.69 (dd, J = 11.7, 8.9 Hz, 4H), 7.59 (d, J = 8.5 Hz, 2H), 4.70 (dd, J = 7.9, 4.7 Hz, 1H), 3.79–3.41 (m, 6H), 2.47 (d, J = 20.7 Hz, 4H), 2.32 (s, 3H), 2.05 (m, 1H), 1.94 (m, 1H), 1.88–1.77 (m, 1H), 1.64 (dt, J = 12.3, 6.2 Hz, 1H). 13C NMR (75 MHz, MeOD) Δ 171.1, 152.6, 143.8, 142.6, 131.0, 128.6, 125.7 (q, J = 3.75 Hz), 124.0 (q, J = 32.25 Hz),118.4, 118.2, 58.4, 54.5, 54.0, 48.5, 44.9, 44.6, 41.7, 30.6, 24.4. HRMS (ESI+): m/z calcd for C24H29F3N5O4S [M + H]+, 540.1892; found, 540.1882.
(S)-N-(Pyridin-2-yl)-1-((4-(3-(4-(trifluoromethyl)phenyl)ureido)-phenyl)sulfonyl) pyrrolidine-2-carboxamide (48).
Compound 48 (20 mg, 39% for two steps) was obtained in a yield of 39% for two steps as a white solid following the procedure of 11. HPLC purity 96.5% (tR = 19.39 min). 1H NMR (300 MHz, MeOD) Δ 8.32 (d, J = 4.2 Hz, 1H), 8.15 (d, J = 8.3 Hz, 1H), 7.87 (d, J = 8.8 Hz, 2H), 7.84–7.77 (m, 1H), 7.74 (d, J = 8.8 Hz, 2H), 7.68 (d, J = 8.7 Hz, 2H), 7.60 (d, J = 8.8 Hz, 2H), 7.15 (dd, J = 7.0, 5.1 Hz, 1H), 4.32 (dd, J = 7.9, 4.0 Hz, 1H), 3.68–3.58 (m, 1H), 3.39–3.33 (m, 1H), 2.15–2.03 (m, 1H), 1.98–1.84 (m, 2H), 1.68 (m, 1H). 13C NMR (75 MHz, MeOD) Δ 171.5, 152.6, 151.0, 147.7, 144.3, 142.5, 138.4, 129.3, 128.9, 126.2, 125.7 (q, J = 3.75 Hz), 124.1 (q, J = 32.25 Hz)122.7, 120.0, 118.4, 118.2, 114.1, 62.7, 49.4, 30.5, 24.3. HRMS (ESI+): m/z calcd for C24H23F3N5O4S [M + H]+, 534.1423; found, 534.1414.
Methyl ((4-(3-(4-(Trifluoromethyl)phenyl)ureido)phenyl)-sulfonyl)-l-prolinate (49).
To a solution of 11 (325 mg, 0.69 mmol) in 6 mL of CH3OH and LiOH·H2O (30 mg, 6.9 mmol) in 2 mL of H2O was added. The solution was stirred at room temperature for 4 h. Then the solution was acidified by 10% HCl to pH = 1 and extracted by EtOAc. The organic phase was dried over anhydrous Na2SO4 and concentrated to get 49 (320 mg, 98%) as a light-yellow foam. HPLC purity 95% (tR = 16.36 min). 1H NMR (300 MHz, MeOD) Δ 8.82 (d, J = 25.3 Hz, 1H), 7.82 (d, J = 8.8 Hz, 2H), 7.74–7.64 (m, 4H), 7.59 (d, J = 8.7 Hz, 2H), 4.22 (dd, J = 7.8, 4.8 Hz, 1H), 3.48 (m, 1H), 3.32–3.22 (m, 1H), 2.09–2.01 (m, 1H), 2.00–1.87 (m, 2H), 1.75–1.63 (m, 1H). 13C NMR (75 MHz, MeOD) Δ 174.5, 152.7, 143.8, 142.5, 130.6, 128.5, 126.2, 125.7 (q, J = 3.75 Hz), 124.7, 124.0 (q, J = 32.25 Hz), 123.8, 123.4, 122.6, 118.3, 118.2, 60.6, 48.5, 30.6, 24.3. HRMS (ESI+): m/z calcd for C19H19F3N3O5S [M + H]+, 458.0998; found, 458.1006.
(S)-1-(4-((2-(Hydroxymethyl)pyrrolidin-1-yl)sulfonyl)phenyl)-3-(4 (trifluoromethyl)phenyl)urea (50).
To a solution of 11 (50 mg, 0.1 mmol) in 4 mL of THF, LiAlH4 (11 mg, 0.3 mmol) was added under N2. After 30 min, the mixture was poured into H2O, and extracted with CH2Cl2. After drying over anhydrous Na2SO4, the solution was concentrated and purified with a silica gel column (CH2Cl2/CH3OH = 20/1) to obtain the desired product 50 (46 mg, quant) as a white solid. HPLC purity 99.6% (tR = 18.29 min). 1H NMR (300 MHz, CDCl3) Δ 7.86 (s, 1H), 7.80 (s, 1H), 7.74 (s, 1H), 7.72 (s, 1H), 7.57 (s, 1H), 7.55 (s, 4H), 3.70 (d, J = 3.3 Hz, 3H), 3.47 (dt, J = 12.0, 6.2 Hz, 1H), 3.36–3.16 (m, 2H), 1.90–1.77 (m, 1H), 1.74 (m, 1H), 1.62–1.48 (m, 1H). 13C NMR (75 MHz, CDCl3) Δ 152.1, 143.6, 141.3, 129.8, 128.8, 126.3 (q, J = 3.75 Hz), 119.1, 119.0, 65.7, 61.9, 50.1, 28.7, 24.2. HRMS (ESI+): m/z calcd for C19H21F3N3O4S [M + H]+, 444.1205; found, 444.1218.
(S)-(1-((4-(3-(4-(Trifluoromethyl)phenyl)ureido)phenyl)sulfonyl)-pyrrolidin-2-yl)methyl 4-methylbenzenesulfonate (51).
To a solution of 50 (140 mg, 0.32 mmol) in 4 mL of CH2Cl2, Et3N (65 mg, 0.64 mmol) and TsCl (76 mg, 0.4 mmol) were added under N2. After overnight, the mixture was poured into H2O, and extracted with CH2Cl2. After drying over anhydrous Na2SO4, the solution was concentrated and purified with silica gel column (CH2Cl2/CH3OH = 20/1) to obtain the desired product 51 (157 mg, 82%) as a white foam. HPLC purity 97.9% (tR = 20.97 min). 1H NMR (300 MHz, CDCl3) Δ 7.81 (d, J = 8.2 Hz, 2H), 7.75–7.63 (m, 4H), 7.62–7.51 (m, 6H), 7.37 (d, J = 8.1 Hz, 2H), 4.28 (dd, J = 10.1, 3.6 Hz, 1H), 4.07–3.97 (m, 1H), 3.82 (s, 1H), 3.39 (m, 1H), 3.10 (dd, J = 16.1, 6.7 Hz, 1H), 2.46 (s, 3H), 1.81 (m, 2H), 1.65 (m, 2H). 13C NMR (75 MHz, CDCl3) Δ 152.2, 145.6, 143.8, 141.6, 132.1, 130.1, 129.1, 128.8, 127.8, 126.2 (q, J = 3.75 Hz), 119.0, 118.9, 71.7, 58.0, 49.5, 28.5, 23.8, 21.6. HRMS (ESI+): m/z calcd for C26H27F3N3O6S2 [M + H]+, 598.1293; found, 598.1280.
(S)-1-(4-((2-(Morpholinomethyl)pyrrolidin-1-yl)sulfonyl)phenyl)-3-(4-(trifluoromethyl)phenyl)urea (52).
To a solution of 51 (50 mg, 0.1 mmol) in 5 mL of CH3CN, K2CO3 (41 mg, 0.3 mmol) was added. The mixture was allowed to stir at 50 °C for overnight. Then the mixture was poured into H2O and extracted by CH2Cl2. The organic layer was washed with saturated NaHCO3 (aq) and brine and dried over anhydrous Na2SO4. The resulting solution was evaporated, and the residue was purified by PTLC (CH2Cl2/MeOH = 50:1) to give the desired product 52 (29 mg, 57%) as a white solid. HPLC purity 97.9% (tR = 17.97 min). 1H NMR (300 MHz, MeOD) Δ 7.88 (d, J = 8.5 Hz, 2H), 7.79 (d, J = 8.5 Hz, 2H), 7.70 (d, J = 8.5 Hz, 2H), 7.59 (d, J = 8.5 Hz, 2H), 4.29 (m, 1H), 4.18–4.05 (m, 2H), 4.03–3.77 (m, 4H), 3.61 (d, J = 12.5 Hz, 1H), 3.52–3.34 (m, 3H), 3.25 (m, 2H), 1.92–1.79 (m, 1H), 1.63 (q, J = 6.4 Hz, 2H), 1.51 (m, 1H).13C NMR (75 MHz, MeOD) Δ 152.7, 143.7, 142.6, 130.4, 128.5, 126.3, 125.7 (q, J = 3.75 Hz), 124.0 (q, J = 32.25 Hz), 122.7, 118.3, 118.2, 66.5, 63.4, 57.7, 54.1, 48.8, 29.5, 23.5. HRMS (ESI+): m/z calcd for C23H28F3N4O4S [M + H]+, 513.1783; found, 513.1777.
(S)-1-(4-((2-((4-Hydroxypiperidin-1-yl)methyl)pyrrolidin-1-yl)-sulfonyl)phenyl)-3-(4-(trifluoromethyl)phenyl)urea (53).
Compound 53 was synthesized in a yield of 44% as a white solid following the procedure of 52. HPLC purity 98.8% (tR = 17.97 min). 1H NMR (300 MHz, MeOD) Δ 7.81 (d, J = 8.7 Hz, 2H), 7.70 (m,, 4H), 7.60 (d, J = 8.7 Hz, 2H), 3.78 (s, 1H), 3.70–3.58 (m, 1H), 3.37 (m, 1H), 3.25–3.14 (m, 1H), 3.02–2.83 (m, 2H), 2.69 (dd, J = 12.6, 3.8 Hz, 1H), 2.39 (m, 3H), 1.94–1.76 (m, 4H), 1.60 (m, 4H). 13C NMR (75 MHz, MeOD) Δ 152.7, 143.8, 142.6, 130.3, 128.5, 126.3, 125.7 (q, J = 3.75 Hz), 124.1 (q, J = 32.3 Hz), 122.7, 118.3, 118.2, 66.8, 62.9, 58.0, 52.2, 51.2, 48.7, 33.5, 33.4, 29.7, 23.4. HRMS (ESI+): m/z calcd for C24H30F3N4O4S [M + H]+, 527.1940; found, 527.1931.
Protein Expression and Purification.
Plasmid DNA encoding the canonical human BRD4 bromodomain (42–168) protein was purchased from Addgene. The recombinant protein was expressed in Escherichia coli BL21 (DE3) grown in Terrific Broth. E. coli cultures were allowed to grow to an optical density at 600 nm of 1.0, prior to lowering the temperature to 18 °C and the addtion of 0.5 mM IPTG. After harvesting the cells by centrifugation, cell pellets were frozen and stored at −80 °C until required. The protein purification was performed at 4 °C using the following procedure. The E. coli cell pellets were thawed and resuspended in 50 mM HEPES, 300 mM NaCl, 5% (v/v) glycerol, 0.5 mM TCEP, complete protease inhibitors (Roche) and 20 μg/mL DNase at pH 8.0 and lysed by sonication. The lysate was centrifuged at 12 000 rpm for 40 min to remove insoluble material. The BRD442–168 protein was purified from the clarified lysate using Ni-NTA resin in 50 mM HEPES, 300 mM NaCl, 5% glycerol, and 0.5 mM TCEP at pH 8.0. Contaminant proteins were washed from the resin using the same buffer supplemented with 20 mM imidazole before eluting the recombinant protein with buffer supplemented with 500 mM imidazole. His6-TEV protease was added to cleave the N-terminal His-tag during dialysis against 50 mM HEPES, 300 mM NaCl, 5% glycerol, and 0.5 mM TCEP at pH 7.5. The protein solution was passed through the Ni-NTA column to remove the TEV protease and the cleaved His-tags. As a final purification step, size exclusion chromatography was performed using a Sepax STR-10 S300 column pre-equilibrated with 10 mM HEPES, 100 mM NaCl, and 10 mM DTT at pH 7.5. All proteins were >95% pure based on SDS PAGE analysis.
Crystallization.
Crystals of human BRD4 BD1 in complex with compound 52 were prepared via hanging drop vapor diffusion at 20 °C. Then 10 mg/mL human BRD4 BD1 protein solution with 2 mM of 52 was preincubated on ice for 10 min prior to being mixed in 1:1 ratio (protein:reservoir solution) with 100 mM HEPES and 15% (w/v) PEG 3350 at pH 7.5. Orthorhombic crystals grew within 3 days and were subsequently cryoprotected with 100 mM HEPES and 30% (w/v) PEG 3350 at pH 7.5 containing 2 mM of 52. X-ray diffraction data were collected at beamline 22-ID at the Advanced Photon Source (Argonne National Laboratories). The diffraction data was processed and intergrated using the iMOSFLM v7.2.2,71 AIMLESS, and CTRUNCATE72 programs were used for scaling and anisotropy correction, respectively. The phase information was obtained by molecular replacement using a single chain of human BRD4 bromodomain structure (PDB 5KU3) as a search model. Iterative cycles of manual model building and refinement were performed within Phenix v1.16–354973 and COOT v0.8.9.174 software.
Determination of Solubility.
Solubility in water for 52 was determined by HPLC analysis according to a previously published protocol.75,76 First, 1–2 mg of 52 was weighed and added to 1 mL of water, respectively. Then, 10–15 mg of 52 was weighed and added to 0.1–0.6 mL of water. The suspensions were shaken at 25 °C for 24 h and then centrifuged, and the supernatants were filtered. Aliquots (5 μL) of the supernatants were injected into the HPLC system equipped with a C18 reverse-phase column under the same condition which was described in the general Experimental Section. One-point calibration77 was done by injecting 5 μL aliquots of the corresponding buffer solutions of 52 with known concentrations.
Cell Culture.
Immortalized human small airway epithelial cells (hSAECs) were previously described.9 hSAECs were grown in small airway epithelial cell growth medium (Lonza, Walkersville, MD) in a humidified atmosphere of 5% CO2. Poly(I:C) was obtained from Sigma (St. Louis, MO) and used at 10 μg/mL in cell culture. Compound 1 was purchased from Tocris, and compound 3 was purchased from Cayman Chemical (Ann Arbor, Michigan). Compounds were solubilized in DMSO and added at the indicated concentrations.
Quantitative Real-time PCR (qRT-PCR) Analysis.
For gene expression analyses, 1 μg of RNA was reverse transcribed using Super Script III as previously described.9 Then 1 μL of cDNA product was amplified using SYBR Green Supermix (Bio-Rad) and indicated gene-specific primers. The reaction mixtures were subjected to 40 cycles of 15 s at 94 °C, 60 s at 60 °C, and 1 min at 72 °C in an iCycler (BioRad). Quantification of relative changes in gene expression was calculated using the ΔΔCt method, and expression as the fold change between experimental and control samples was normalized to internal control cyclophilin (PPIA).9
In Vitro Efficacy of BRD4 Inhibitors on Poly(I:C)-Induced Innate Immune Response.
hSAECs were first pretreated with a series of final concentrations of BRD4 inhibitors from 0.01 nM to 100 μM for 24 h and were then added poly(I:C) at 10 μg/mL for another 4 h prior to harvesting the cells. The harvested cells were first washed with PBS twice, and then the total RNA was extracted using acid guanidinium phenol extraction (Tri Reagent; Sigma). The total RNA was further reverse-transcribed for gene expression analysis by RT-qPCR. The inhibitory effect of BRD4 inhibitors on poly(I:C)-induced innate immune gene expression was compared with that of poly(I:C) alone and inhibitory percentage of each treatment was obtained. For compounds 1, 3, 11, 45, 50, 51, 52, and 53, in vitro efficacy of these BRD4 inhibitors on poly(I:C) induced innate immune response were presented as the IC50 values of these compounds. The IC50 values are calculated out using the Four Parameters Regression method (https://www.aatbio.com/tools/ic50-calculator/). Compounds were dissolved in DMSO and further diluted at cell culture medium to appropriate concentrations.
Time-resolved Fluorescence Energy Transfer (TR-FRET) Assays.
The 384-well plate-based commercial TR-FRET assay kits (Cayman Chemical, Ann Arbor, Michigan) were used to determine the binding ability of tested BRD4 inhibitors to the BRD4 and BRD2 bromodomains (BD) using the two recombinant BRD4 BDs or BRD2 BDs by TR-FRET assays. A series of concentrations of BRD4 inhibitors from 0.01 nM to 100 μM were added into a 384 well test plate and mixed with other reaction components based on the instructions from vendor followed by incubation 1 h at room temperature. The commercially available BRD inhibitors compounds 1 and 3 were used as the controls. The plates were read in time-resolved format by exciting the sample at 340 nm and reading emissions at 620 and 670 nm, using a 100 μs delay and a 500 μs window at a Tecan M1000 pro reader. A plot of the TR-FRET ratio (670 nm emission/620 nm emission) versus inhibitor concentration on semilog axes results in a sigmoidal dose–response curve typical of competitive assays. These data were further calculated out with the IC50 values of tested BRD4 inhibitors to the bromodomains of BRD2 and BRD4 as well as other relevant target proteins, respectively. The IC50 values are calculated out using the Four Parameters Regression method (https://www.aatbio.com/tools/ic50-calculator/).
Plasmid Construction.
Human BRD4-BD1 (aa 44–168) and BRD4-BD2 (aa 349–461) coding sequences were amplified by PCR from pF:hBRD4(1–722)-11d DNA template78 using Phusion polymerase (NEB, M0530L) and primer pairs 5′-CGCGGA-TCCAACCCCCCGCCCCCAGAGACCTCCAAC-3′ and 5′-CCGCTCGAGTCATTCTTCTGTGGGTAGCTCATTTATTTT-3′ for BD1 and 5′-CGCGGATCCAAGGTCTCGGAGCAGCTCA-3′ and 5′-TTCCCGGGTCTAGATCAAGGCTCGTCCGGC-ATCTT-3′ for BD2. The PCR fragments were then digested with BamHI and XhoI sites (for BD1) or BamHI and SmaI sites (for BD2) and inserted into a linearized pGEX-6P-1 (Sigma-Aldrich GE28–9546-48) with the corresponding enzymes to generate pGEX-6P-1-hBRD4-BD1 and pGEX-6P-1-hBRD4-BD2, respectively. Generation of BD1 E151A mutation was done by site-directed mutagenesis using primer pair 5′-GTCTTAATGGCAGCAGCTCTGGAAAAG-3′ and 5′-CTTTTCCAGAGCTGCTGCCATTAAGAC and pGEX-6P-1-hBRD4-BD1 template for PCR amplification at 98°C for 1 min, followed by 25 cycles of 98°C for 45 s, 55°C for 45 s, and 68°C for 12 min. The PCR products were then digested with DpnI at 37°C for 2 h to degrade the template plasmid with the mutated construct named pGEX-6P-1-hBRD4-BD1-E151A.
Protein Purification.
Plasmids containing GST-tagged BD1/2-coding sequences were transformed respectively into OverExpress C43(DE3) bacterial competent cells (Lucigen). Then 12 L of bacterial cultures were induced at 37°C for 4 h upon the addition of IPTG (final concentration 1 mM) when cell density reaches OD600 between 0.6 and 0.8. Bacteria cultures were spun down and pellets were resuspended in 180 mL of Bacterial Lysis Buffer (BLB) containing 500 mM NaCl, 20 mM Tris-HCl pH 7.9 at 4°C, 20% glycerol, 1 mM EDTA, 0.2 mM DTT, and protease inhibitors, followed by sonication. The supernatant was then collected after centrifugation at 20 000 rpm for 30 min at 4°C using the JA-25.50 rotor (Beckman). Glutathione (GSH) agarose beads (12 mL, GOLDBIO, catalogue no. G-250–100) were added for an overnight rotation at 4°C. The GSH beads with bound GST-BRD4-BD1 or -BD2 were spun down and washed three times with 120 mL of BLB and digested with human rhinovirus 3C protease (HRV-P3C; Thermal Fisher 88946) at 4°C for 4 h of rotation to release BD1/2 proteins without GST tags. To remove HRV-P3C, untagged BRD4-BD1 or -BD2 eluates were passed through a HiTrap Q HP anion exchange chromatography column (GE Healthcare, SKU no. GE17–1154-01) using the ÄKTA FPLC system in 10 mM HEPES pH 7.5 and eluted by a salt gradient from 100 mM to 1 M NaCl. FPLC fractions containing BD1/2 were combined and concentrated using Amicon ultracentrifugal filter 3000 Da MWCO (Millipore, UFC900324) with the final buffer 10 mM HEPES pH 7.5 and 150 mM NaCl.
Thermal Shift Assay (TSA).
TSA experiments were performed in triplicate in a 384-well plate (Hard-Shell 384-well thin-wall PCR plates from Bio-Rad) using the CFX384 Real-Time C1000 touch thermal cycler (Bio-Rad). In each 10-μL reaction well, 2 μL of 25X SyproOrange dye (diluted from 5000× stock solution, Invitrogen, ref no. S6650, in sterile ddH2O) was mixed with 6 μL of BD1/2 proteins (in 20 mM HEPES pH 6.5, 100 mM NaCl, 2.5% glycerol) and 2 μL of 52 or JQ1 in 5% DMSO (final 0–126.5 μM). The protein concentrations (i.e., 5 μM of wild-type and mutant BD1 and 25 μM of BD2) used for the experiments were determined by comparable fluorescence signals between dye-bound BD1 and BD2 as well as JQ1-induced thermal stability for BD1 and BD2 binding. After combining dye/protein/compd, the 384-well plate was sealed with Microseal “B” seal adhesive film (Bio-Rad, catalogue no. MSB1001) and centrifuged at 1000 rpm for 10 s in a Beckman Coulter Allegra X-22 centrifuge to remove air bubbles. Fluorescence signals generated by dye binding to unfolded proteins were monitored by using the melt curve protocol at the rate of 1°C temperature increment in 30 s from 25 °C to 95 °C with signal capture every 1 °C. The melt curves obtained by fluorescence signal (F) versus temperature (T) were converted to melting peaks by plotting the negative derivative of the fluorescence with respect to temperature against temperature (−ΔF/ΔT vs T)63,79 using Bio-Rad CFX Manager v3.1 software. Tm (temperature with 50% of protein unfolded and bound by the dye) represents the lowest part of the melting peaks.79
In Vivo Efficacy of BRD4 Inhibitors on Poly(I:C)-Induced Acute Airway Inflammation.
Animal experiments were performed according to the NIH Guide for Care and Use of Experimental Animals and approved by the University of Texas Medical Branch (UTMB) Animal Care and Use Committee (approval no. 1312058B). Male C57BL/6J mice (12 weeks old) were purchased from The Jackson Laboratory (Bar Harbor, ME) and housed under pathogen-free conditions with food and water ad libitum. C57BL/6J mice were pretreated in the absence or presence of the indicated BRD4 inhibitors (10 mg/kg body weight, via po route) 1 day prior to poly(I:C) stimulation. The next day, animals were given another dose of BRD4 inhibitor immediately followed by intranasal (i.n.) administration of phosphate-buffered saline (PBS, 50 μL) or poly(I:C) (300 μg dissolved in 50 μL of PBS). One day later, the mice were euthanized. The BALF and lung tissues of treated mice were collected for further analysis. Compounds were first dissolved in DMSO and further diluted in 10% hydroxypropyl β-cyclodextrin in PBS to appropriate concentration prior to intraperitoneal administration.
Evaluation of Airway Inflammation.
Cellular recruitment into the airway lumen was assessed in the BALF. Lungs were perfused twice with 1 mL of sterile PBS (pH 7.4) to obtain the BALF. Total cell counts were determined by trypan blue staining of 50 μL of BALF and counting viable cells using a hemocytometer. Differential cell counts were performed on cytocentrifuge preparations (Cytospin 3; Thermo Shandon, Pittsburgh, Pa) stained with Wright-Giemsa. A total of 300 cells were counted per sample using light microscopy. Formalin-fixed lungs were embedded in paraffin, sectioned at a 4 μm thickness, and stained with hematoxylin and eosin or Masson’s trichrome. Microscopy was performed on a NIKON Eclipse Ti System.
Cytokine Bio-Plex Assay.
BALF samples were centrifuged (800g for 5 min at 4 °C) and the cytokines quantitated in the supernatant using the Bio-Plex Pro mouse cytokine assay (Bio-Rad, Hercules, CA) with recombinant cytokine standards (in triplicate). Readings were performed on a Bioplex 200 system (Bio-Rad). Data was analyzed using Bio-Plex Manager Software Version 6.0 Build 617 (Bio-Rad).
Supplementary Material
ACKNOWLEDGMENTS
This work was partially supported by the Crohn’s & Colitis Foundation Entrepreneurial Investing (EI) Initiative award (J.Z., A.R.B., and B.T.), Litwin IBD Pioneers Program Award (B.T. and J.Z.), and a postdoctoral research fellowship award (Z.L.) from the Crohn’s & Colitis Foundation of America, UTMB Technology Commercialization Program, and Sanofi Innovation Awards (iAwards) (A.R.B., J.Z., and B.T.), the John D. Stobo, M.D. Distinguished Chair Endowment Fund (J.Z.), and the NIH grants AI157852 (H.H. and J.Z.), AI062885 (A.R.B.), CA251698 (C.M.C.), and CPRIT grants RP180349 and RP190077 (C.M.C.). Core laboratory support was provided by the UTMB Histopathology facility core. We thank Drs. Lawrence C. Sowers and Jason Herring at the Department of Pharmacology as well as Dr. Tianzhi Wang at the NMR core facility of UTMB for the NMR spectroscopy assistance, and Dr. Xuemei Luo at the UTMB mass spectrometry core with funding support from UT system proteomics network for the HRMS analysis.
ABBREVIATIONS USED
- Ala
alanine
- AUC
area under the curve
- BALF
bronchoalveolar lavage fluid
- BET
bromodomain and extra-terminal domain
- BRD4
bromodomain-containing protein 4
- BD
bromodomain
- BD1
bromodomain 1
- BD2
bromodomain 2
- BRDT
bromodomain testis-specific protein
- BID
basic residue-enriched interaction domain
- COPD
chronic obstructive pulmonary disease
- CTM
C-terminal motif
- C max
maximum concentration
- CL
clearance
- DMPK
drug metabolism and pharmacokinetics
- DIPEA
N,N-diisopropylethylamine
- ET
extra-terminal domain
- E151A
glutamic acid 151 to alanine mutation
- F
oral bioavailability
- Glu
glutamic acid
- hSAECs
human small airway epithelial cells
- HBTU
N,N,N′,N′-tetramethyl-O-(1H-benzotriazol-1-yl)uranium hexafluorophosphate
- ip
intraperitoneal injection
- i.n.
intranasal administration
- KAc
lysine acetylation
- NPS
N-terminal cluster of CK2 phosphorylation sites
- PROTAC
proteolysis targeted chimeras
- poly(I:C)
polyinosinic:polycytidylic acid
- po. per os
oral administration
- qRT-PCR
quantitative real-time PCR
- SAR
structure–activity relationship
- TR-FRET
time-resolved fluorescence energy transfer
- TsCl
4-toluene-sulfonyl chloride
- t 1/2
half-life
- TSA
thermal shift assay
- ΔTm
change of the melting temperature
- V ss
volume of distribution at steady state
- WT
wild-type
Footnotes
Accession Codes
PDB code for BRD4 with bound 52 is 6U0D. Authors will release the atomic coordinates and experimental data upon article publication.
The authors declare no competing financial interest.
Contributor Information
Zhiqing Liu, Chemical Biology Program, Department of Pharmacology and Toxicology, University of Texas Medical Branch (UTMB), Galveston, Texas 77555, United States.
Yi Li, Chemical Biology Program, Department of Pharmacology and Toxicology, University of Texas Medical Branch (UTMB), Galveston, Texas 77555, United States.
Haiying Chen, Chemical Biology Program, Department of Pharmacology and Toxicology, University of Texas Medical Branch (UTMB), Galveston, Texas 77555, United States.
Hsien-Tsung Lai, Simmons Comprehensive Cancer Center, University of Texas Southwestern Medical Center, Dallas, Texas 75390, United States.
Pingyuan Wang, Chemical Biology Program, Department of Pharmacology and Toxicology, University of Texas Medical Branch (UTMB), Galveston, Texas 77555, United States.
Shwu-Yuan Wu, Simmons Comprehensive Cancer Center and Department of Biochemistry, University of Texas Southwestern Medical Center, Dallas, Texas 75390, United States.
Eric A. Wold, Chemical Biology Program, Department of Pharmacology and Toxicology, University of Texas Medical Branch (UTMB), Galveston, Texas 77555, United States.
Paul G. Leonard, Core for Biomolecular Structure and Function, MD Anderson Cancer Center, Houston, Texas 77054, United States
Sarah Joseph, Core for Biomolecular Structure and Function, MD Anderson Cancer Center, Houston, Texas 77054, United States.
Haitao Hu, Department of Microbiology and Immunology, University of Texas Medical Branch (UTMB), Galveston, Texas 77555, United States.
Cheng-Ming Chiang, Simmons Comprehensive Cancer Center, Department of Biochemistry, and Department of Pharmacology, University of Texas Southwestern Medical Center, Dallas, Texas 75390, United States.
Allan R. Brasier, Institute for Clinical and Translational Research (ICTR), University of Wisconsin-Madison School of Medicine and Public Health, Madison, Wisconsin 53705, United States.
Bing Tian, Department of Internal Medicine and Sealy Center for Molecular Medicine, University of Texas Medical Branch (UTMB), Galveston, Texas 77555, United States.
Jia Zhou, Chemical Biology Program, Department of Pharmacology and Toxicology, Sealy Center for Molecular Medicine, and Institute for Translational Sciences, University of Texas Medical Branch (UTMB), Galveston, Texas 77555, United States.
REFERENCES
- (1).Barnes PJ; Burney PG; Silverman EK; Celli BR; Vestbo J; Wedzicha JA; Wouters EFM Chronic obstructive pulmonary disease. Nat. Rev. Dis. Primers 2015, 1, 15076. [DOI] [PubMed] [Google Scholar]
- (2).Chronic Obstructive Pulmonary Disease (COPD); World Health Organization, 2021; https://www.who.int/news-room/fact-sheets/detail/chronic-obstructive-pulmonary-disease-(COPD) (accessed 2017-12-01). [DOI] [PMC free article] [PubMed]
- (3).Wedzicha JA Role of viruses in exacerbations of chronic obstructive pulmonary disease. Proc. Am. Thorac. Soc 2004, 1, 115– 120. [DOI] [PubMed] [Google Scholar]
- (4).Tian B; Yang J; Zhao Y; Ivanciuc T; Sun H; Wakamiya M; Garofalo RP; Brasier AR Central role of the NF-kappaB pathway in the scgb1a1-expressing epithelium in mediating respiratory syncytial virus-induced airway inflammation. J. Virol 2018, 92, e00441-1B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Tian B; Yang J; Zhao Y; Ivanciuc T; Sun H; Garofalo RP; Brasier AR BRD4 couples NF-kappaB/RelA with airway inflammation and the IRF-RIG-I amplification loop in respiratory syncytial virus infection. J. Virol 2017, 91, e00007–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Yang J; Tian B; Sun H; Garofalo RP; Brasier AR Epigenetic silencing of IRF1 dysregulates type III interferon responses to respiratory virus infection in epithelial to mesenchymal transition. Nat. Microbiol 2017, 2, 17086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Tian B; Patrikeev I; Ochoa L; Vargas G; Belanger KK; Litvinov J; Boldogh I; Ameredes BT; Motamedi M; Brasier AR NF-kappaB mediates mesenchymal transition, remodeling, and pulmonary fibrosis in response to chronic inflammation by viral rna patterns. Am. J. Respir. Cell Mol. Biol 2017, 56, 506–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Tian B; Hosoki K; Liu Z; Yang J; Zhao Y; Sun H; Zhou J; Rytting E; Kaphalia L; Calhoun WJ; Sur S; Brasier AR Mucosal bromodomain-containing protein 4 mediates aeroallergen-induced inflammation and remodeling. J. Allergy Clin. Immunol 2019, 143, 1380–1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Tian B; Liu Z; Yang J; Sun H; Zhao Y; Wakamiya M; Chen H; Rytting E; Zhou J; Brasier AR Selective antagonists of the bronchiolar epithelial NFkB-bromodomain-containing protein 4 pathway in viral-induced airway inflammation. Cell Rep 2018, 23, 1138–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Zhao Y; Tian B; Sun H; Zhang J; Zhang Y; Ivannikov M; Motamedi M; Liu Z; Zhou J; Kaphalia L; Calhoun WJ; Maroto R; Brasier AR Pharmacoproteomics reveal novel protective activity of bromodomain containing 4 inhibitors on vascular homeostasis in TLR3-mediated airway remodeling. J. Proteomics 2019, 205, 103415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Brasier AR; Zhou J Validation of the epigenetic reader bromodomain-containing protein 4 (BRD4) as a therapeutic target for treatment of airway remodeling. Drug Discovery Today 2020, 25, 126–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Filippakopoulos P; Knapp S Targeting bromodomains: Epigenetic readers of lysine acetylation. Nat. Rev. Drug Discovery 2014, 13, 337–356. [DOI] [PubMed] [Google Scholar]
- (13).Wu SY; Chiang CM The double bromodomain-containing chromatin adaptor BRD4 and transcriptional regulation. J. Biol. Chem 2007, 282, 13141–13145. [DOI] [PubMed] [Google Scholar]
- (14).Chiang CM Phospho-BRD4: transcription plasticity and drug targeting. Drug Discov. Today Technol 2016, 19, 17–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Sun C; Yin J; Fang Y; Chen J; Jeong KJ; Chen X; Vellano CP; Ju Z; Zhao W; Zhang D; Lu Y; Meric-Bernstam F; Yap TA; Hattersley M; O’Connor MJ; Chen H; Fawell S; Lin SY; Peng G; Mills GB BRD4 inhibition is synthetic lethal with PARP inhibitors through the induction of homologous recombination deficiency. Cancer Cell 2018, 33, 401–416. e408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Belkina AC; Denis GV BET domain co-regulators in obesity, inflammation and cancer. Nat. Rev. Cancer 2012, 12, 465–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Wu SY; Lee CF; Lai HT; Yu CT; Lee JE; Zuo H; Tsai SY; Tsai MJ; Ge K; Wan Y; Chiang CM Opposing functions of BRD4 isoforms in breast cancer. Mol. Cell 2020, 78, 1114–1132. e1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Duan Q; McMahon S; Anand P; Shah H; Thomas S; Salunga HT; Huang Y; Zhang R; Sahadevan A; Lemieux ME; Brown JD; Srivastava D; Bradner JE; McKinsey TA; Haldar SM BET bromodomain inhibition suppresses innate inflammatory and profibrotic transcriptional networks in heart failure. Sci. Transl. Med 2017, 9, eaah5084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Bao Y; Wu X; Chen J; Hu X; Zeng F; Cheng J; Jin H; Lin X; Chen LF BRD4 modulates the innate immune response through Mnk2-eIF4E pathway-dependent translational control of IκBα. Proc. Natl. Acad. Sci. U. S. A 2017, 114, E3993–E4001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Zhang G; Liu R; Zhong Y; Plotnikov AN; Zhang W; Zeng L; Rusinova E; Gerona-Nevarro G; Moshkina N; Joshua J; Chuang PY; Ohlmeyer M; He JC; Zhou MM Down-regulation of NF-kappaB transcriptional activity in hiv-associated kidney disease by BRD4 inhibition. J. Biol. Chem 2012, 287, 28840–28851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Li Y; Chen J; Bolinger AA; Chen H; Liu Z; Cong Y; Brasier AR; Pinchuk IV; Tian B; Zhou J Target-based small molecule drug discovery towards novel therapeutics for inflammatory bowel diseases. Inflamm. Bowel Dis 2021, 27, S38–S62. [DOI] [PubMed] [Google Scholar]
- (22).Zhu J; Gaiha GD; John SP; Pertel T; Chin CR; Gao G; Qu H; Walker BD; Elledge SJ; Brass AL Reactivation of latent HIV-1 by inhibition of BRD4. Cell Rep 2012, 2, 807–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Wu SY; Nin DS; Lee AY; Simanski S; Kodadek T; Chiang CM BRD4 phosphorylation regulates HPVE2-mediated viral transcription, origin replication, and cellular MMP-9 expression. Cell Rep 2016, 16, 1733–1748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Alamer E; Zhong C; Liu Z; Niu Q; Long F; Guo L; Gelman BB; Soong L; Zhou J; Hu H Epigenetic suppression of HIV in myeloid cells by the BRD4-selective small molecule modulator ZL0580. J. Virol 2020, 94, e01880–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Mann M; Roberts DS; Zhu Y; Li Y; Zhou J; Ge Y; Brasier AR Discovery of RSV-induced BRD4 protein interactions using native immunoprecipitation and parallel accumulation-serial fragmentation (PASEF) mass spectrometry. Viruses 2021, 13, 454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Xu X; Mann M; Qiao D; Li Y; Zhou J; Brasier AR Bromodomain containing protein 4 (BRD4) regulates expression of its interacting coactivators in the innate response to respiratory syncytial virus. Front. Mol. Biosci 2021, 8, 728661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Korb E; Herre M; Zucker-Scharff I; Darnell RB; Allis CD Bet protein BRD4 activates transcription in neurons and BET inhibitor JQ1 blocks memory in mice. Nat. Neurosci 2015, 18, 1464–1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Sartor GC; Powell SK; Brothers SP; Wahlestedt C Epigenetic readers of lysine acetylation regulate cocaine-induced plasticity. J. Neurosci 2015, 35, 15062–15072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Liu Z; Wang P; Chen H; Wold EA; Tian B; Brasier AR; Zhou J Drug discovery targeting bromodomain-containing protein 4. J. Med. Chem 2017, 60, 4533–4558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Zhou Z; Li X; Liu Z; Huang L; Yao Y; Li L; Chen J; Zhang R; Zhou J; Wang L; Zhang QQ A bromodomain-containing protein 4 (BRD4) inhibitor suppresses angiogenesis by regulating AP-1 expression. Front. Pharmacol 2020, 11, 1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Picaud S; Wells C; Felletar I; Brotherton D; Martin S; Savitsky P; Diez-Dacal B; Philpott M; Bountra C; Lingard H; Fedorov O; Muller S; Brennan PE; Knapp S; Filippakopoulos P RVX-208, an inhibitor of bet transcriptional regulators with selectivity for the second bromodomain. Proc. Natl. Acad. Sci. U. S. A 2013, 110, 19754–19759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Gavai AV; Norris D; Tortolani D; O’Malley D; Zhao Y; Quesnelle C; Gill P; Vaccaro W; Huynh T; Ahuja V; Dodd D; Mussari C; Harikrishnan L; Kamau M; Tokarski JS; Sheriff S; Rampulla R; Wu D-R; Li J; Zhang H; Li P; Sun D; Yip H; Zhang Y; Mathur A; Zhang H; Huang C; Yang Z; Ranasinghe A; D’Arienzo C; Su C; Everlof G; Zhang L; Raghavan N; Hunt JT; Poss M; Vite GD; Westhouse RA; Wee S Abstract 5789: Discovery of clinical candidate BMS-986158, an oral bet inhibitor, for the treatment of cancer. Cancer Res 2018, 78, 5789. [Google Scholar]
- (33).Filippakopoulos P; Qi J; Picaud S; Shen Y; Smith WB; Fedorov O; Morse EM; Keates T; Hickman TT; Felletar I; Philpott M; Munro S; McKeown MR; Wang Y; Christie AL; West N; Cameron MJ; Schwartz B; Heightman TD; La Thangue N; French CA; Wiest O; Kung AL; Knapp S; Bradner JE Selective inhibition of BET bromodomains. Nature 2010, 468, 1067–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Albrecht BK; Gehling VS; Hewitt MC; Vaswani RG; Cote A; Leblanc Y; Nasveschuk CG; Bellon S; Bergeron L; Campbell R; Cantone N; Cooper MR; Cummings RT; Jayaram H; Joshi S; Mertz JA; Neiss A; Normant E; O’Meara M; Pardo E; Poy F; Sandy P; Supko J; Sims RJ 3rd; Harmange JC; Taylor AM; Audia JE Identification of a benzoisoxazoloazepine inhibitor (CPI-0610) of the bromodomain and extra-terminal (BET) family as a candidate for human clinical trials. J. Med. Chem 2016, 59, 1330–1339. [DOI] [PubMed] [Google Scholar]
- (35).Faivre EJ; McDaniel KF; Albert DH; Mantena SR; Plotnik JP; Wilcox D; Zhang L; Bui MH; Sheppard GS; Wang L; Sehgal V; Lin X; Huang X; Lu X; Uziel T; Hessler P; Lam LT; Bellin RJ; Mehta G; Fidanze S; Pratt JK; Liu D; Hasvold LA; Sun C; Panchal SC; Nicolette JJ; Fossey SL; Park CH; Longenecker K; Bigelow L; Torrent M; Rosenberg SH; Kati WM; Shen Y Selective inhibition of the BD2 bromodomain of bet proteins in prostate cancer. Nature 2020, 578, 306–310. [DOI] [PubMed] [Google Scholar]
- (36).McDaniel KF; Wang L; Soltwedel T; Fidanze SD; Hasvold LA; Liu D; Mantei RA; Pratt JK; Sheppard GS; Bui MH; Faivre EJ; Huang X; Li L; Lin X; Wang R; Warder SE; Wilcox D; Albert DH; Magoc TJ; Rajaraman G; Park CH; Hutchins CW; Shen JJ; Edalji RP; Sun CC; Martin R; Gao W; Wong S; Fang G; Elmore SW; Shen Y; Kati WM Discovery of N-(4-(2,4-difluorophenoxy)-3-(6-methyl-7-oxo-6,7-dihydro-1H-pyrrolo[2,3-c]pyridin-4-yl)phenyl)ethanesulfonamide (ABBV-075/mivebresib), a potent and orally available bromodomain and extraterminal domain (BET) family bromodomain inhibitor. J. Med. Chem 2017, 60, 8369–8384. [DOI] [PubMed] [Google Scholar]
- (37).Liu Z; Ding Y; Ye N; Wild C; Chen H; Zhou J Direct activation of Bax protein for cancer therapy. Med. Res. Rev 2016, 36, 313–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Tam CS; Anderson MA; Pott C; Agarwal R; Handunnetti S; Hicks RJ; Burbury K; Turner G; Di Iulio J; Bressel M; Westerman D; Lade S; Dreyling M; Dawson SJ; Dawson MA; Seymour JF; Roberts AW Ibrutinib plus venetoclax for the treatment of mantle-cell lymphoma. N. Engl. J. Med 2018, 378, 1211–1223. [DOI] [PubMed] [Google Scholar]
- (39).Gilan O; Rioja I; Knezevic K; Bell MJ; Yeung MM; Harker NR; Lam EYN; Chung CW; Bamborough P; Petretich M; Urh M; Atkinson SJ; Bassil AK; Roberts EJ; Vassiliadis D; Burr ML; Preston AGS; Wellaway C; Werner T; Gray JR; Michon AM; Gobbetti T; Kumar V; Soden PE; Haynes A; Vappiani J; Tough DF; Taylor S; Dawson SJ; Bantscheff M; Lindon M; Drewes G; Demont EH; Daniels DL; Grandi P; Prinjha RK; Dawson MA Selective targeting of BD1 and BD2 of the BET proteins in cancer and immunoinflammation. Science 2020, 368, 387–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Zengerle M; Chan KH; Ciulli A Selective small molecule induced degradation of the BET bromodomain protein brd4. ACS Chem. Biol 2015, 10, 1770–1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Nowak RP; DeAngelo SL; Buckley D; He Z; Donovan KA; An J; Safaee N; Jedrychowski MP; Ponthier CM; Ishoey M; Zhang T; Mancias JD; Gray NS; Bradner JE; Fischer ES Plasticity in binding confers selectivity in ligand-induced protein degradation. Nat. Chem. Biol 2018, 14, 706–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Wootten D; Christopoulos A; Sexton PM Emerging paradigms in GPCR allostery: Implications for drug discovery. Nat. Rev. Drug Discovery 2013, 12, 630–644. [DOI] [PubMed] [Google Scholar]
- (43).Tang P; Zhang J; Liu J; Chiang CM; Ouyang L Targeting bromodomain and extraterminal proteins for drug discovery: From current progress to technological development. J. Med. Chem 2021, 64, 2419–2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Raux B; Voitovich Y; Derviaux C; Lugari A; Rebuffet E; Milhas S; Priet S; Roux T; Trinquet E; Guillemot JC; Knapp S; Brunel JM; Fedorov AY; Collette Y; Roche P; Betzi S; Combes S; Morelli X Exploring selective inhibition of the first bromodomain of the human bromodomain and extra-terminal domain (BET) proteins. J. Med. Chem 2016, 59, 1634–1641. [DOI] [PubMed] [Google Scholar]
- (45).Filippakopoulos P; Picaud S; Mangos M; Keates T; Lambert JP; Barsyte-Lovejoy D; Felletar I; Volkmer R; Muller S; Pawson T; Gingras AC; Arrowsmith CH; Knapp S Histone recognition and large-scale structural analysis of the human bromodomain family. Cell 2012, 149, 214–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Gashaw I; Ellinghaus P; Sommer A; Asadullah K What makes a good drug target? Drug Discovery Today 2012, 17, S24. [DOI] [PubMed] [Google Scholar]
- (47).Liu Z; Tian B; Chen H; Wang P; Brasier AR; Zhou J Discovery of potent and selective BRD4 inhibitors capable of blocking TLR3-induced acute airway inflammation. Eur. J. Med. Chem 2018, 151, 450–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Tian B; Liu Z; Litvinov J; Maroto R; Jamaluddin M; Rytting E; Patrikeev I; Ochoa L; Vargas G; Motamedi M; Ameredes BT; Zhou J; Brasier AR Efficacy of novel highly specific bromodomain-containing protein 4 inhibitors in innate inflammation-driven airway remodeling. Am. J. Respir. Cell Mol. Biol 2019, 60, 68–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Liu Z; Chen H; Wang P; Li Y; Wold EA; Leonard PG; Joseph S; Brasier AR; Tian B; Zhou J Discovery of orally bioavailable chromone derivatives as potent and selective BRD4 inhibitors: Scaffold hopping, optimization, and pharmacological evaluation. J. Med. Chem 2020, 63, 5242–5256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Saphier S; Haft A; Margel S Bacterial reduction as means for colonic drug delivery: Can other chemical groups provide an alternative to the azo bond? J. Med. Chem 2012, 55, 10781–10785. [DOI] [PubMed] [Google Scholar]
- (51).Liu Z; Zhu Y; Chen H; Wang P; Mei FC; Ye N; Cheng X; Zhou J Structure-activity relationships of 2-substituted phenyl-N-phenyl-2-oxoacetohydrazonoyl cyanides as novel antagonists of exchange proteins directly activated by cAMP (EPACs). Bioorg. Med. Chem. Lett 2017, 27, 5163–5166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Jung M; Philpott M; Muller S; Schulze J; Badock V; Eberspacher U; Moosmayer D; Bader B; Schmees N; Fernandez-Montalvan A; Haendler B Affinity map of bromodomain protein 4 (BRD4) interactions with the histone H4 tail and the small molecule inhibitor JQ1. J. Biol. Chem 2014, 289, 9304–9319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Wang P; Felsing DE; Chen H; Stutz SJ; Murphy RE; Cunningham KA; Allen JA; Zhou J Discovery of potent and brain-penetrant GPR52 agonist that suppresses psychostimulant behavior. J. Med. Chem 2020, 63, 13951–13972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Wold EA; Garcia EJ; Wild CT; Miszkiel JM; Soto CA; Chen J; Pazdrak K; Fox RG; Anastasio NC; Cunningham KA; Zhou J Discovery of 4-phenylpiperidine-2-carboxamide analogues as serotonin 5-HT2C receptor-positive allosteric modulators with enhanced drug-like properties. J. Med. Chem 2020, 63, 7529–7544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Lucas X; Wohlwend D; Hugle M; Schmidtkunz K; Gerhardt S; Schule R; Jung M; Einsle O; Gunther S 4-Acyl pyrroles: Mimicking acetylated lysines in histone code reading. Angew. Chem., Int. Ed 2013, 52, 14055–14059. [DOI] [PubMed] [Google Scholar]
- (56).Zhang G; Smith SG; Zhou MM Discovery of chemical inhibitors of human bromodomains. Chem. Rev 2015, 115, 11625–11668. [DOI] [PubMed] [Google Scholar]
- (57).Ember SW; Zhu JY; Olesen SH; Martin MP; Becker A; Berndt N; Georg GI; Schonbrunn E Acetyl-lysine binding site of bromodomain-containing protein 4 (BRD4) interacts with diverse kinase inhibitors. ACS Chem. Biol 2014, 9, 1160–1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Olp MD; Sprague DJ; Goetz CJ; Kathman SG; Wynia-Smith SL; Shishodia S; Summers SB; Xu Z; Statsyuk AV; Smith BC Covalent-fragment screening of BRD4 identifies a ligandable site orthogonal to the acetyl-lysine binding sites. ACS Chem. Biol 2020, 15, 1036–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Ye F; Huang J; Wang H; Luo C; Zhao K Targeting epigenetic machinery: Emerging novel allosteric inhibitors. Pharmacol. Ther 2019, 204, 107406. [DOI] [PubMed] [Google Scholar]
- (60).Wold EA; Chen J; Cunningham KA; Zhou J Allosteric modulation of class a GPCRs: Targets, agents, and emerging concepts. J. Med. Chem 2019, 62, 88–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Conn PJ; Christopoulos A; Lindsley CW Allosteric modulators of GPCRs: A novel approach for the treatment of CNS disorders. Nat. Rev. Drug Discovery 2009, 8, 41–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Conn PJ; Lindsley CW; Meiler J; Niswender CM Opportunities and challenges in the discovery of allosteric modulators of GPCRs for treating cns disorders. Nat. Rev. Drug Discovery 2014, 13, 692–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Huynh K; Partch CL Analysis of protein stability and ligand interactions by thermal shift assay. Curr. Protoc. Protein Sci 2015, 79, 28.29.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Christopoulos A Allosteric binding sites on cell-surface receptors: Novel targets for drug discovery. Nat. Rev. Drug Discovery 2002, 1, 198–210. [DOI] [PubMed] [Google Scholar]
- (65).Wootten D; Christopoulos A; Sexton PM Emerging paradigms in GPCR allostery: Implications for drug discovery. Nat. Rev. Drug Discovery 2013, 12, 630–644. [DOI] [PubMed] [Google Scholar]
- (66).Wold EA; Zhou J GPCR allosteric modulators: Mechanistic advantages and therapeutic applications. Curr. Top. Med. Chem 2019, 18, 2002–2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Wild CT; Miszkiel JM; Wold EA; Soto CA; Ding C; Hartley RM; White MA; Anastasio NC; Cunningham KA; Zhou J Design, synthesis, and characterization of 4-undecylpiper-idine-2-carboxamides as positive allosteric modulators of the serotonin (5-HT) 5-HT(2C) receptor. J. Med. Chem 2019, 62, 288–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Vogel GH Determination of solubility by hyphenated HPLC methods. In Drug Discovery and Evaluation: Safety and Pharmacokinetics Assay; Springer: New York, 2006; pp 400–402. [Google Scholar]
- (69).Niu Q; Liu Z; Alamer E; Fan X; Chen H; Endsley J; Gelman BB; Tian B; Kim JH; Michael NL; Robb ML; Ananworanich J; Zhou J; Hu H Structure-guided drug design identifies a BRD4-selective small molecule that suppresses hiv. J. Clin. Invest 2019, 129, 3361–3373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Li Z; Guo J; Wu Y; Zhou Q The BET bromodomain inhibitor JQ1 activates hiv latency through antagonizing BRD4 inhibition of TAT-transactivation. Nucleic Acids Res 2013, 41, 277–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Battye TG; Kontogiannis L; Johnson O; Powell HR; Leslie AG Imosflm: A new graphical interface for diffraction-image processing with mosflm. Acta Crystallogr. D Biol. Crystallogr 2011, 67, 271–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Evans PR; Murshudov GN How good are my data and what is the resolution? Acta Crystallogr. D Biol. Crystallogr 2013, 69, 1204–1214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Adams PD; Afonine PV; Bunkoczi G; Chen VB; Davis IW; Echols N; Headd JJ; Hung LW; Kapral GJ; Grosse-Kunstleve RW; McCoy AJ; Moriarty NW; Oeffner R; Read RJ; Richardson DC; Richardson JS; Terwilliger TC; Zwart PH Phenix: A comprehensive python-based system for macro-molecular structure solution. Acta Crystallogr. D Biol. Crystallogr 2010, 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Emsley P; Lohkamp B; Scott WG; Cowtan K Features and development of coot. Acta Crystallogr. D Biol. Crystallogr 2010, 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Ding C; Zhang Y; Chen H; Yang Z; Wild C; Chu L; Liu H; Shen Q; Zhou J Novel nitrogen-enriched oridonin analogues with thiazole-fused A-ring: Protecting group-free synthesis, enhanced anticancer profile, and improved aqueous solubility. J. Med. Chem 2013, 56, 5048–5058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Chen H; Yang Z; Ding C; Chu L; Zhang Y; Terry K; Liu H; Shen Q; Zhou J Discovery of O-alkylamino tethered niclosamide derivatives as potent and orally bioavailable anticancer agents. ACS Med. Chem. Lett 2013, 4, 180–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (77).Kiselev E; DeGuire S; Morrell A; Agama K; Dexheimer TS; Pommier Y; Cushman M 7-Azaindenoisoquinolines as topoisomerase i inhibitors and potential anticancer agents. J. Med. Chem 2011, 54, 6106–6116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (78).Wu SY; Lee AY; Lai HT; Zhang H; Chiang CM Phospho switch triggers BRD4 chromatin binding and activator recruitment for gene-specific targeting. Mol. Cell 2013, 49, 843–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (79).Toyota T; Watanabe A; Shibuya H; Nankai M; Hattori E; Yamada K; Kurumaji A; Karkera JD; Detera-Wadleigh SD; Yoshikawa T Association study on the DUSP6 gene, an affective disorder candidate gene on 12q23, performed by using fluorescence resonance energy transfer-based melting curve analysis on the lightcycler. Mol. Psychiatry 2000, 5, 489–494. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.