

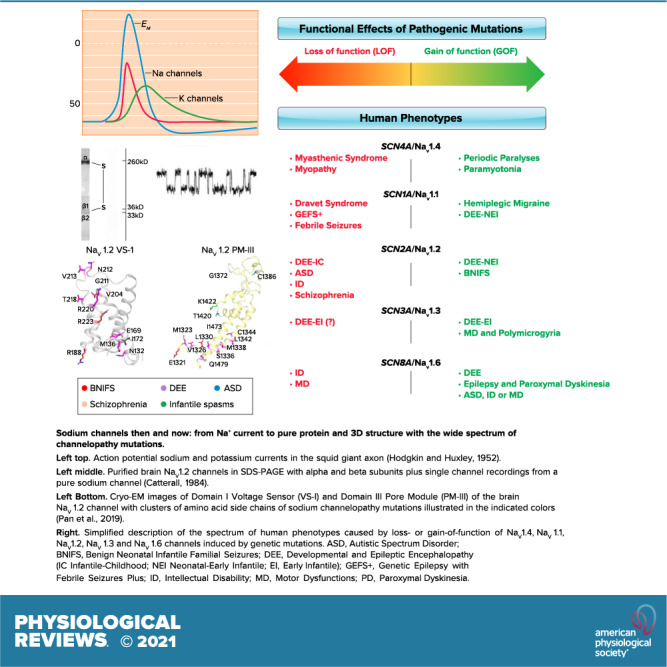
Keywords: autism, epilepsy, migraine, periodic paralysis, sodium channels
Abstract
Voltage-gated sodium channels initiate action potentials in nerve, skeletal muscle, and other electrically excitable cells. Mutations in them cause a wide range of diseases. These channelopathy mutations affect every aspect of sodium channel function, including voltage sensing, voltage-dependent activation, ion conductance, fast and slow inactivation, and both biosynthesis and assembly. Mutations that cause different forms of periodic paralysis in skeletal muscle were discovered first and have provided a template for understanding structure, function, and pathophysiology at the molecular level. More recent work has revealed multiple sodium channelopathies in the brain. Here we review the well-characterized genetics and pathophysiology of the periodic paralyses of skeletal muscle and then use this information as a foundation for advancing our understanding of mutations in the structurally homologous α-subunits of brain sodium channels that cause epilepsy, migraine, autism, and related comorbidities. We include studies based on molecular and structural biology, cell biology and physiology, pharmacology, and mouse genetics. Our review reveals unexpected connections among these different types of sodium channelopathies.
1. INTRODUCTION TO SODIUM CHANNELOPATHIES
Inherited sodium channelopathies are genetic diseases caused by mutations in sodium channels (1, 2), which are the primary focus of this review. In some diseases, alterations in sodium channel expression or function can occur through other means than genetic mutations. These acquired sodium channelopathies are of great interest, but they are not our focus here. The inherited periodic paralyses of skeletal muscle were the first recognized channelopathies (2, 3). They were mapped to the locus of the skeletal muscle sodium channel gene SCN4A that encodes the sodium channel NaV1.4, and both genetic and physiological studies confirmed a causal relationship between mutations in that gene and specific phenotypes of the periodic paralyses (2, 3). After these first studies, it was shown that mutations in the SCN1A gene encoding the brain sodium channel NaV1.1 cause genetic epilepsy (4), mutations in the SCN8A gene encoding the NaV1.6 channel cause paralysis in mice (4, 5), and mutations in the SCN9A gene encoding the NaV1.7 channel cause pain syndromes (6). Similarly, a large number of mutations in the SCN5A gene, encoding the cardiac sodium channel NaV1.5, cause inherited arrhythmias (7). In this article, we review the periodic paralyses as the foundation for studies of channelopathies, and we consider the ever-increasing number of mutations in brain sodium channels that cause genetic forms of epilepsy, autism, and cognitive dysfunction. Recent review articles have covered other aspects of this large field, including inherited pain syndromes (8), inherited cardiac arrhythmias (9–11), and channelopathies caused by mutations in the non-pore-forming auxiliary β-subunits of sodium channels (12, 13).
1.1. The Sodium Channel Protein Complex
Voltage-gated sodium channels initiate and conduct action potentials in nerve and muscle cells (14). Sodium channel proteins isolated from nerve and muscle are complexes of a large, pore-forming α-subunit of 250 kDa with one or two β-subunits of 30–40 kDa (FIGURE 1A; Ref. 16). The α-subunits are composed of 24 transmembrane segments organized in four homologous domains containing six transmembrane segments in each (FIGURE 1B; Ref. 16). The short intracellular linker between domains III and IV serves as the inactivation gate (FIGURE 1, B and C). The three-dimensional structure of the core functional unit of the sodium channel was first revealed by X-ray crystallographic studies of the homotetrameric ancestral bacterial sodium channel NaVAb (FIGURE 1, D–F; Ref. 17). As expected from structures of voltage-gated potassium channels, the pore is formed by the S5 and S6 segments in the center of a square array of four subunits, and the voltage sensor is a bundle of four transmembrane α-helices (S1–S4), connected to the pore by the S4–S5 linker (17, 19). The structures of eukaryotic nerve and skeletal muscle sodium channels have been determined by cryogenic electron microscopy (cryo-EM), including human sodium channels from nerve and skeletal muscle (FIGURE 1, G and H; Refs. 18, 20). The structure of the functional core of these channels is virtually identical to NaVAb (17), which was used as a search template to solve the initial structure (20). These detailed analyses of eukaryotic sodium channels reveal the structure of the complex of α- and β-subunits, the conformations of the selectivity filter and fast inactivation gate, and the structures of portions of the large intracellular and extracellular domains that are not present in the bacterial sodium channels.
FIGURE 1.
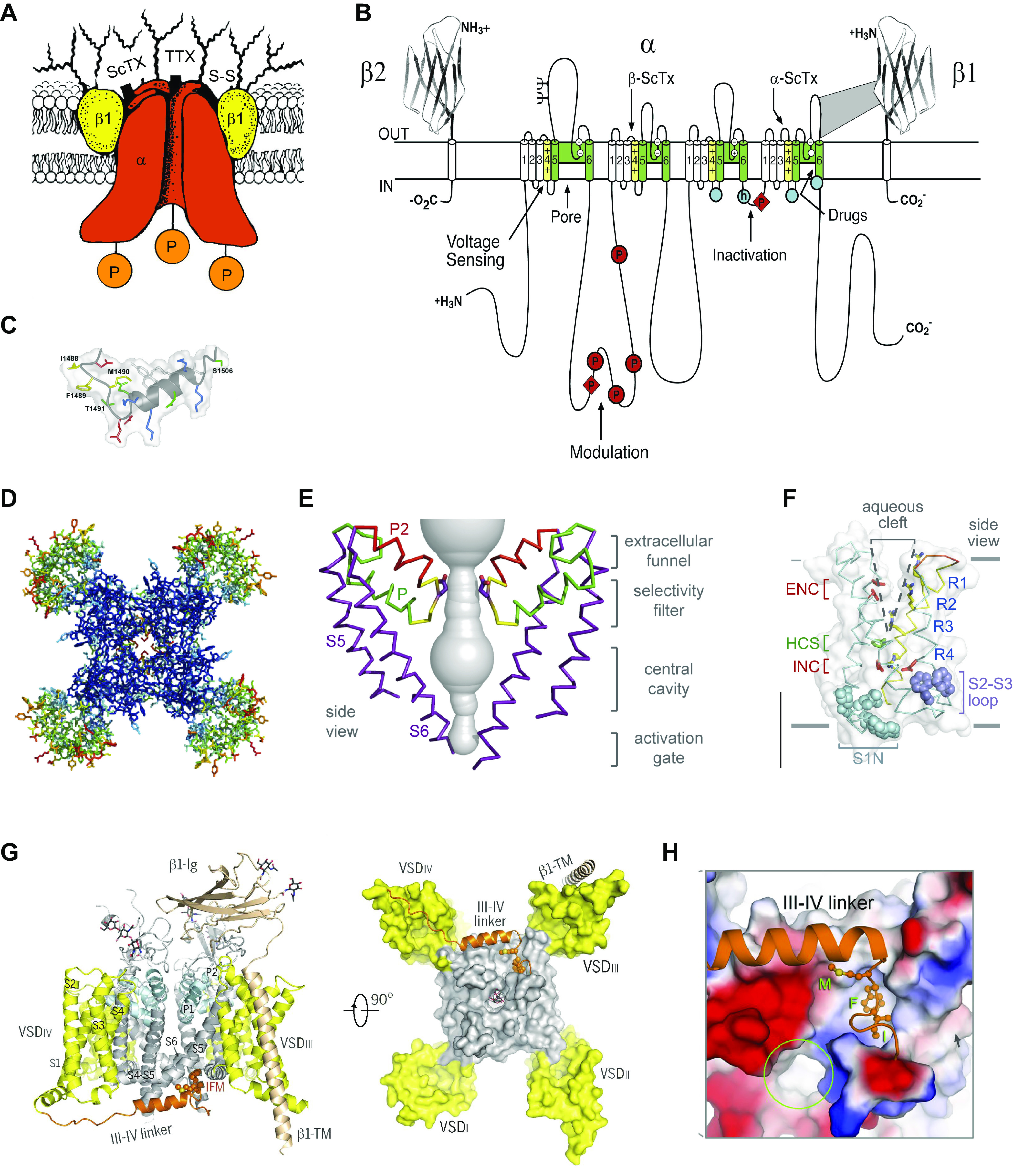
Structure of voltage-gated sodium channels revealed step by step over >30 yr. A: cartoon model of purified brain sodium channels with α- and β-subunits circa 1986. ScTx, scorpion toxin; TTX, tetrodotoxin; P, protein phosphorylation. B: transmembrane folding diagram of the sodium channel α-subunit with key functional domains indicated circa 2000. Blue circle with h, inactivation particle with isoleucine, phenylalanine, methionine (IFM) motif; empty blue circles, inactivation gate receptor. C: structure of the inactivation gate peptide of the brain NaV1.2 channel determined by NMR. D: top view of the structure of NaVAb determined by X-ray crystallography circa 2011. Blue, pore module; green, voltage sensors. E: structure of the pore of NaVAb. F: structure of the voltage sensor of NaVAb. G: structure of NaV1.4 with β1-subunit circa 2018. The domain III–IV linker (brown) serves as the fast inactivation gate. H: close-up of the IFM motif of the inactivation gate (brown) interacting with its receptor. A: adapted from Ref. 15 with permission from Annual Review of Biochemistry. B and C: adapted from Ref. 16 with permission from Neuron. D–F: adapted from Ref. 17 with permission from Nature. G and H: adapted from Ref. 18 with permission from Science.
Numerous proteins can interact with the core sodium channel complex formed by α- and β-subunits, leading to heterogeneous multimolecular complexes that can be specific of different cell types or different cell subcompartments (9, 21–24). These interactions play a role in trafficking and localization of the core channel complex, as well as in modulation of its functional properties, often by means of posttranslational modifications. They can also be implicated in rescuing folding/trafficking defective mutants, as we have outlined below in sect. 3.3. Interestingly, recent studies of cardiac NaV1.5 channels indicate that there can also be interactions of α-subunits: dimers of α-subunits can be isolated from cardiac tissue, and activities consistent with the existence of functional dimers are observed upon expression in noncardiac cells (25). The two channel proteins in these dimers are functionally coupled, and mutations in one channel protein can influence the biophysical and functional properties of its partner (25, 26). Biochemical studies revealed that NaV1.1 and NaV1.2 channels can also form dimers (25). These surprising findings could have implications for recessive NaV1.1 and NaV1.2 sodium channelopathies, in which wild-type (WT) and mutant sodium channels are coexpressed in neurons and other cell types in vivo. Although some of the complexity of functional phenotypes observed in sodium channelopathies may arise from changes in interaction with associated proteins and functional dimerization of α-subunits, we do not review them further here because they have been less studied in comparison with the direct effect of mutations of the core sodium channel complex, and some defective interactions implicated in channelopathies have been recently reviewed (22, 24).
1.2. Structural Basis for Sodium Channel Function
Detailed structure-function studies using mutagenesis, electrophysiology, and molecular modeling have given a two-dimensional map of the functional parts of sodium channels (FIGURE 1B; Refs. 16, 27). Channelopathy mutations affect all aspects of sodium channel function. The structural and molecular mechanisms for sodium channel function are introduced in the paragraphs below, and the impact of different classes of channelopathy mutations are considered in the subsequent sections.
1.2.1. Voltage-dependent activation.
Voltage-dependent activation of sodium channels is initiated by voltage-driven outward movement of the positive gating charges, usually arginine residues, in the S4 transmembrane segments of the voltage sensors (16, 28, 29). The voltage sensor is a four-helix bundle with a substantial aqueous cleft that faces the extracellular milieu (FIGURE 1F). The gating charges in the S4 segment are usually arginine residues spaced at three-residue intervals, which span the membrane (R1–R4; FIGURE 1F). Upon depolarization, the S4 segment moves outward, exchanging ion pair partners and transporting the arginine gating charges through the hydrophobic constriction site (HCS; FIGURE 1F), which serves to seal the voltage sensor and prevent transmembrane movement of water and ions. Changes in membrane potential drive the S4 segment inward and outward in response to hyperpolarization and depolarization, moving the gating charges through the HCS and across the complete transmembrane electric field. This “sliding-helix” mechanism of gating charge movement (15) has been confirmed for sodium channels by extensive mutagenesis, disulfide locking, molecular modeling, and most recently determination of the high-resolution structure of NaVAb by cryo-EM (19, 30–35). Similar experiments on potassium channels led to a consensus model for this voltage-sensing mechanism (36). This outward movement of the S4 segment initiates a conformational change in the voltage sensor, which is transmitted to the pore by a twisting motion of the S4–S5 linker (17, 34, 35). The intracellular ends of the S6 segments cross and interact closely to form the closed activation gate, which opens in an irislike motion in response to the voltage-dependent conformational changes in the voltage sensor (17, 19, 34, 37, 38). Structures of the sodium channel in closed and open states reveal a substantial movement of the amino acid side chains at the intracellular ends of the S6 segments, from a closed conformation with an orifice of <1 Å to an open conformation with an orifice of 8.5 Å (37) to 10.5 Å (38). Determination of the structure of the resting state of a voltage-gated sodium channel fits closely with the expectations of this gating model and provides a complete picture of voltage sensing, electromechanical coupling, and pore opening at the atomic level (34). Currents generated by the activation of sodium channels are studied with voltage-clamp experiments, in which the transmembrane potential is controlled and the current elicited at different potentials is recorded (FIGURE 2A).
FIGURE 2.
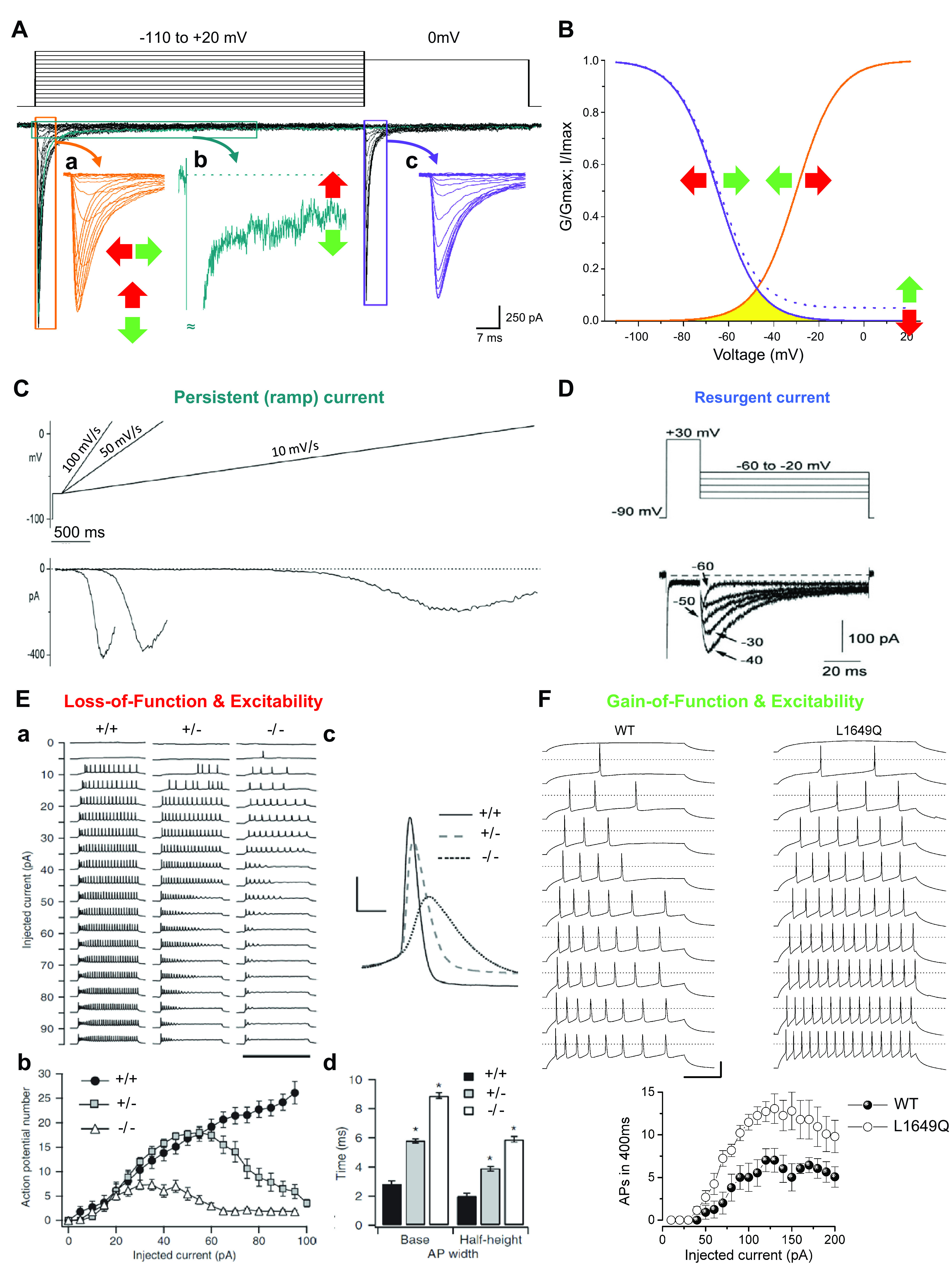
Functional properties of sodium channels. Functional properties and their modifications induced by mutations involved in channelopathies are studied by performing voltage-clamp experiments that follow the general methods introduced by Hodgkin and Huxley in their seminal work (39). In modern experiments aimed at identifying functional effects of mutations, sodium channels are often expressed in transfected cell lines that do not have endogenous channels of interest, and currents are evoked controlling the membrane potential in voltage-clamp mode by means of the whole cell configuration of the patch-clamp technique. A: representative recordings of families of NaV1.1 sodium currents with the 2-pulse voltage protocol shown at top. Inward sodium currents are a negative quantity by convention and therefore plotted downward. The first pulse is a step depolarization at different potentials (up to +20 mV) activating inward sodium currents (inset a), which then undergo fast inactivation during the 100-ms depolarization. These recordings determine the maximal current amplitude and the kinetics of fast inactivation from the open state within a few milliseconds. Current amplitude can be increased by gain-of-function mutations (depicted by green vertical arrow) or decreased by loss-of-function mutations (red vertical arrow). Sodium currents can be prolonged by gain-of-function mutations (green horizontal arrow) or shortened by loss-of-function mutations (red horizontal arrow). The small fraction of current with slower inactivation is called persistent sodium current (inset b, in which a single trace is shown). Gain-of-function mutations can increase its amplitude, whereas loss-of-function mutations can decrease it. The second pulse at 0 mV evokes sodium currents (inset c) whose amplitude decreases according to the amount of fast inactivation induced by the first pulse. Scale bars refer to the black traces. Similar voltage protocols with longer depolarizations (up to tens of seconds) can be used to study slow inactivation. B: the activation curve (orange) is obtained by plotting the normalized peak conductance (G/Gmax) of the currents in inset a as a function of the stimulus potential. The fast inactivation curve (violet) is obtained by plotting the normalized peak values of the currents (I/Imax) displayed in inset c as a function of the potential of the first pulse. Gain- and loss-of-function mutations can induce shifts of the curves (horizontal arrows). The current elicited at membrane potentials in which activation and inactivation curves overlap (yellow area) is called window current. When there is persistent current, the inactivation curve shows a baseline at positive voltages (solid line, 0% persistent current; dashed line, 5% persistent current). C: the persistent current can also be elicited with slow depolarizing voltage ramps that inactivate the fast transient current; of note, relatively fast voltage ramps can elicit a mixed persistent and fast current, whose amplitude depends on the kinetics of fast inactivation, and very slow voltage ramps can induce inactivation of persistent current. Modified from Ref. 40 with permission from Neuropharmacology. D: the resurgent sodium current is generated in some cell types during repolarizations from positive voltages to moderately negative potentials, and gain-of-function mutations can increase it, whereas loss-of-function mutations can decrease it. Modified from Ref. 41 with permission from Journal of Neuroscience. E: loss-of-function mutations of sodium channels decrease neuronal excitability. a: Loss of action potential firing in GABAergic neurons from Scn1a-knockout mice. Action potential discharges, recorded in current-clamp mode with the whole cell configuration of the patch-clamp technique, elicited by injecting depolarizing currents of increasing amplitude in control (+/+), heterozygote Scn1a+/− (+/−), and homozygote Scn1a−/− (−/−) GABAergic neurons. b: Input-output relationships of the number of action potentials vs. the injected current show a large reduction of excitability in Scn1a−/− neurons and less severe reduction in Scn1a+/− neurons. The rheobase (i.e., the minimum depolarizing current that elicits an action potential) is not modified in this model, but loss of function of some sodium channels can increase it. c: Representative single action potentials, recorded for each genotype of GABAergic neurons and elicited by the same injected current amplitude (35 pA), show reduced amplitude with NaV1.1 loss of function, as well as increased width and half-width, which are defined as the width at the base and at the half-maximum amplitude of the action potential (AP). d: Modified from Ref. 42 with permission from Nature Neuroscience. F: action potential discharges recorded in cultured GABAergic neurons transfected with wild-type (WT) NaV1.1 or the familial hemiplegic migraine gain-of-function L1649Q mutant show increased excitability when the mutant is expressed, as quantified in the input-output relationships (bottom). Modified from Ref. 43 with permission from Proceedings of the National Academy of Sciences of the United States of America. Sodium channel gain of function can also decrease rheobase and modify features of action potentials (e.g., amplitude, slope, width); not shown.
A surprising finding from structure-function studies of both sodium and potassium channels was the ability of the voltage sensor to serve as a proton-conducting (44) or cation-conducting (45, 46) pathway after mutation of the arginine gating charges. Neutralization of the gating charges of voltage-gated sodium and potassium channels causes a voltage-dependent leak current to flow through the mutant voltage sensors continuously. These gating pore currents (or omega currents) are voltage dependent because the leak occurs only when the mutant residue replaces the arginine gating charge in the HCS that seals the voltage sensor against transmembrane ion movement (45, 46). In sodium channels, mutation of the R1 and R2 gating charges causes gating pore current in the resting state, whereas mutation of R2 and R3 gives gating pore current in the activated and inactivated states (45). These characteristics are consistent with the sliding-helix model of voltage sensor function, which predicts that the gating charges slide through the HCS in sequence during activation and deactivation of the voltage sensor (45). Mutant gating pores can be highly selective for protons when histidine is substituted for arginine (44), but they are not highly selective among monovalent cations when other amino acid substitutions are present (45–47). However, mutant gating pores often have a high conductance to guanidine (46, 47), likely because it fits well in the space left by removal of the guanidine-containing arginine side chain.
1.2.2. Sodium conductance and selectivity.
Sodium conductance is mediated by the pore domain formed by the S5 and S6 segments and the P loop between them. As for potassium channels, sodium selectivity is mediated by the P loops in the four pore domains of sodium channels, which interact with Na+ as it approaches and enters the ion selectivity filter (17, 48–50). However, in sharp contrast to potassium channels, the outward-facing edge of the ion selectivity filter is composed of a square array of four glutamate residues in bacterial NaV channels (17) or a square array of four different amino acid residues, Asp-Glu-Lys-Ala, in vertebrate NaV channels (18). This high-field strength site partially dehydrates the approaching Na+ ion and allows only Na+ to pass efficiently (48). A dunking motion of the glutamate residues accompanies the partial dehydration of Na+ and its inward movement through the selectivity filter (48).
1.2.3. Fast inactivation.
Within 1–2 ms after opening, the fast inactivation gate formed by the intracellular linker connecting domains III and IV folds into the pore and inactivates it (FIGURE 1, B and C; Refs. 16, 27). In voltage-clamp experiments, some properties of fast inactivation can be studied by analyzing the current decay during step depolarizations and the decrease of the amplitude of the current elicited by the second depolarizing pulse in a two-pulse protocol (FIGURE 2, A and B). Fast inactivation does not have its own intrinsic voltage sensor. Structure-function studies indicate that outward movement of the gating charges in the S4 segment of the voltage sensor in domain IV plays a key role in coupling activation to fast inactivation (51–54). A series of key amino acid residues in this linker, Ile-Phe-Met, serves as the classically defined “inactivation particle,” which folds into the inner pore domain and blocks sodium conductance (55, 56). The structure of the fast inactivation gate contains an α-helical motif preceded by two turns that present the key interacting residues in the Ile-Phe-Met motif to the inner pore domain, where they are bound and close the pore (FIGURE 1, C and H; Refs. 18, 57). The “receptor” that binds the inactivation gate to the intracellular end of the pore is formed by amino acid residues in the S4–S5 linkers in domains III and IV and the intracellular end of the S6 segment in domain IV (18, 20, 58–62).
The intracellular COOH-terminal domain can modulate fast inactivation (63), possibly interacting with the fast inactivation gate (64). Consistently, this domain is implicated in modulations that modify inactivation properties, including G protein βγ-subunits that increase the persistent sodium current (65). The persistent current is a slowly inactivating fraction of the sodium current, which is generated because fast inactivation is not complete, even at very depolarized potentials (66) (FIGURE 2, A and C). Its kinetics of inactivation shows multiple time constants, and there is a residual component even after depolarizations of tens of seconds (67). Although it is often a small fraction of the peak transient sodium current, it can have important effects on cellular excitability. Distinct from the persistent current, the window current is a noninactivating current generated in the sharp window of membrane potentials where the curves of voltage dependence of fast inactivation and activation overlap, and it is important for boosting subthreshold depolarizations (FIGURE 2B). Another subthreshold sodium current is the resurgent current, an unusual transient sodium current elicited by repolarizations that follow strong depolarizations and can contribute to spontaneous and high-frequency firing (41, 68) (FIGURE 2D). It has been proposed that this current is produced by a putative intracellular blocking factor that would bind to open sodium channels and prevent fast inactivation.
1.2.4. Slow inactivation.
During long single depolarizations or long trains of depolarizations, sodium channels enter a slow inactivated state, from which recovery requires prolonged repolarization (69). Slow inactivation has a structural basis different from fast inactivation. Slow inactivation of bacterial sodium channels is caused by an asymmetric collapse of the pore involving amino acid residues in the ion selectivity filter and the full length of the pore-lining S6 segments (19, 70, 71). This mechanism is conserved in vertebrate sodium channels, in which analogous amino acid residues in the selectivity filter and the pore-lining S6 segments undergo conformational changes in the slow inactivation process (69, 72–74). The close structural similarity in the core transmembrane regions of bacterial and mammalian sodium channels (<2 Å root mean square deviation in backbone structure) supports close similarity in the underlying mechanisms of slow inactivation.
These structure-function models of sodium channels derived from a combination of mutagenesis, electrophysiological analysis, and structural studies have provided valuable templates for understanding the deleterious effects of mutations that cause sodium channelopathies. Impairments of voltage-dependent activation, generation of gating pore current through the voltage sensor, altered coupling of activation to pore opening, and defects in ion conductance, fast inactivation, and slow inactivation have all been associated with channelopathies of skeletal muscle and brain sodium channels. These defects can have a direct effect on generation of action potentials and their features (FIGURE 2, E and F).
2. NaV1.4 AND PERIODIC PARALYSIS
The periodic paralyses are rare genetic diseases with a dominant pattern of inheritance within families (2, 75, 76). They cause episodic flaccid paralyses, sometimes accompanied by episodic stiffening of skeletal muscles, which are associated with a wide range of physiological and environmental factors, including exercise, temperature, and changes in serum potassium levels that result from exercise and food intake (2, 76, 77). As outlined below, these diseases are generally caused by heterozygous mutations in the SCN4A gene encoding the skeletal muscle sodium channel NaV1.4 (2, 76) (FIGURE 3).
FIGURE 3.
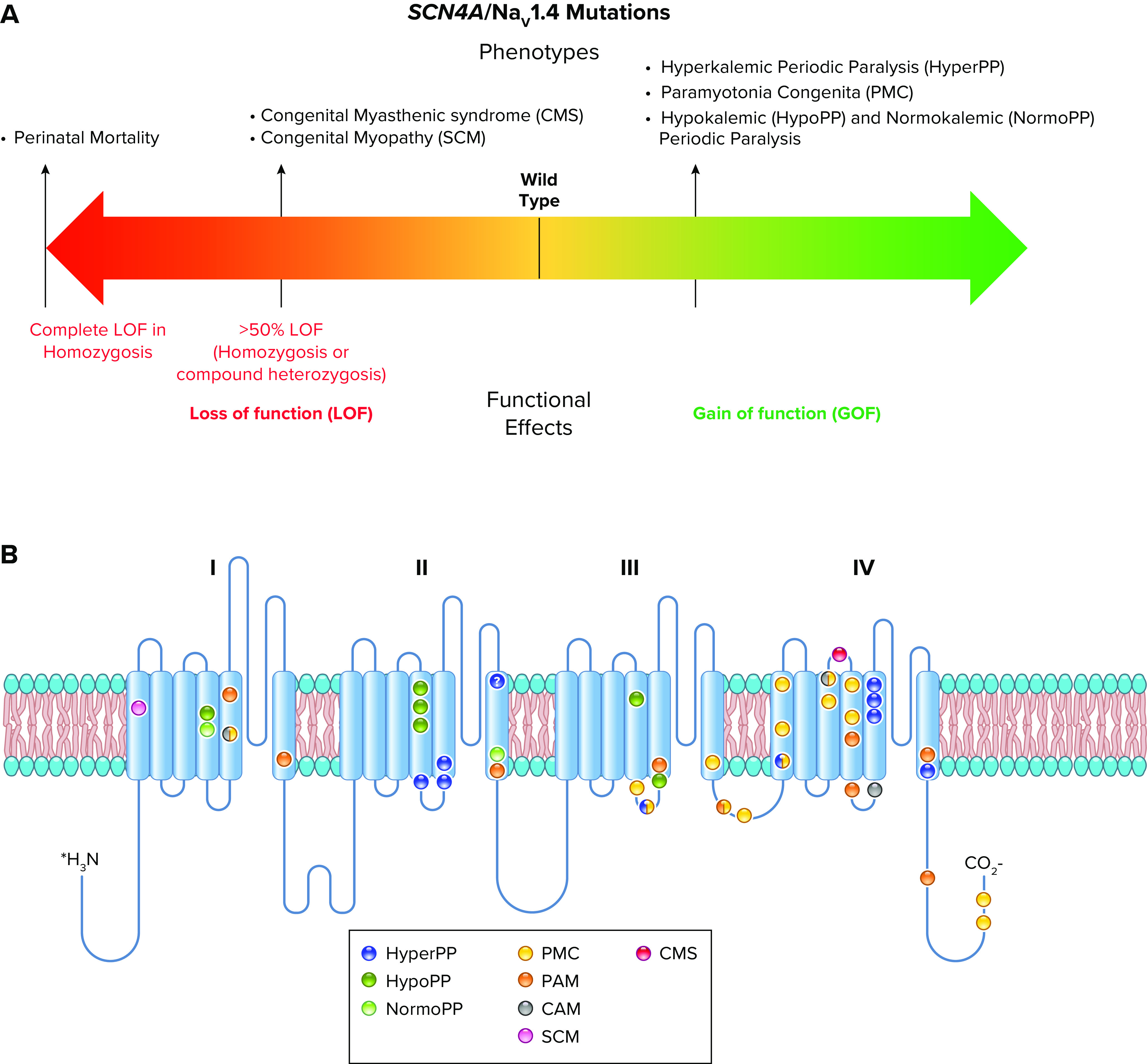
Spectrum of mutations and phenotypes for SCN4A/NaV1.4. A: phenotypic spectrum. Most SCN4A mutations cause NaV1.4 gain of function with different mechanisms, including induction of gating pore current, leading to specific clinical entities. A minority of mutations cause loss of function, inducing clinical symptoms when the loss is >50% (homozygosis or compound heterozygosis). The few patients with complete loss-of-function mutations in homozygosis showed perinatal lethality. B: molecular map of SCN4A mutations color-coded as indicated: hyperkalemic periodic paralysis (HyperPP), hypokalemic periodic paralysis (HypoPP), normokalemic periodic paralysis (NormoPP), paramyotonia congenita (PMC), potassium-aggravated myotonia (PAM), cold-aggravated myotonia (CAM), congenital myopathy (SCM), and congenital myasthenic syndrome (CMS).
2.1. Hyperkalemic Periodic Paralysis
In hyperkalemic periodic paralysis (HyperPP), affected individuals have episodes of flaccid paralysis associated with high levels of serum potassium (2, 76). Electrophysiological studies of skeletal muscle fibers from patients revealed impaired inactivation of the sodium current and increased persistent sodium current that caused depolarization, block of action potential generation, and flaccid paralysis (78). These studies implicated the skeletal muscle sodium channel as either a direct or indirect target of HyperPP mutations. Genetic mapping identified the SCN4A gene encoding the skeletal muscle NaV1.4 channel as a prime target of these mutations (79, 80). In landmark studies, Ptácek et al. (81) and Rojas et al (82) cloned and characterized mutations that cause HyperPP in the SCN4A gene encoding the skeletal muscle sodium channel NaV1.4, the first discovery of the genetic basis for an ion channelopathy. Sequencing of several mutations in this gene from individuals with HyperPP and their close relatives definitively identified SCN4A as the site of mutations that cause this disease (81). Mutations that cause HyperPP are concentrated in the S4–S5 linkers in domains II and III and in the S6 segments near the intracellular end of the pore in domain IV (FIGURE 4, A and B, blue; Ref. 83). These segments are crucial for coupling of voltage sensor movement to pore opening and for forming the receptor for the fast inactivation gate as it folds in and blocks the pore (18, 20, 58–62). Structure-function studies reveal that disruption of the protein interactions that keep the fast inactivation gate closed impairs fast inactivation, allows reopening of sodium channels, and causes persistent sodium currents (55, 56). Therefore, as expected from these structure-function studies, many HyperPP mutations impair fast inactivation (e.g., Refs. 84, 85). HyperPP mutations also impair slow inactivation (69, 86, 87). Mutations in distinct locations cause a variable mixture of impairment of fast and slow inactivation (86). These dual functional deficits may work together by first producing prolonged depolarization following action potentials due to impaired fast inactivation, which leads to persistent depolarization in the absence of effective slow inactivation.
FIGURE 4.

Structural features of Nav1.4 mutations. A–D: structural location of periodic paralysis mutations. CMS, congenital myasthenic syndrome; GEFS + 1, generalized epilepsy with febrile seizures plus 1; HOKPP2, hypokalemic periodic paralysis; HYPP, hyperkalemic periodic paralysis; MYOSCN4A, myotonia caused by SCN4A mutations; NKPP, normokalemic periodic paralysis; PMC, paramyotonia congenita; VSD, voltage-sensing domain. Adapted from Ref. 18 with permission from Science.
2.2. Paramyotonia Congenita and Potassium Aggravated Myotonia
Paramyotonia congenita (PMC) causes episodic paralysis of skeletal muscle, which initially presents as stiffness and rigidity triggered or aggravated by low temperature and is often followed by flaccid paralysis and muscle weakness (88–90). PMC mutations mapped to the same gene as HyperPP (80) and cDNA cloning and sequencing of mutations from affected and unaffected members of the same families demonstrated that PMC is caused by mutations in the SCN4A gene encoding NaV1.4 like HyperPP (91, 92). PMC mutations are clustered in the inactivation gate and nearby transmembrane segments and in the S4–S5 linker in domain III (FIGURE 4, A and B; Ref. 83). They are also clustered in the S4 segment in domain IV, which is thought to trigger fast inactivation (91, 93). Physiological studies of muscle fibers dissected from patients exhibited an unusual persistent depolarization of myofibers, which was blocked by tetrodotoxin and therefore required the activity of the mutant sodium channels (94). Single-channel recordings revealed many late openings following depolarization of muscle fibers from PMC patients but not in fibers from control individuals (94). These results pointed to gain-of-function effects of PMC mutations on sodium channels. Electrophysiological analysis of the functional changes in the mutant NaV1.4 channel expressed from cDNA in cultured nonmuscle cells revealed impaired inactivation of the sodium currents, caused by a combination of slowed and less steeply voltage-dependent fast inactivation and accelerated recovery from fast inactivation (95–99). These functional changes would cause persistent sodium current and reopening of single sodium channels in cells expressing these mutants. These gain-of-function effects lead to generation of inappropriate trains of action potentials and to uncoordinated twitching or myotonia. Although these mutations impair inactivation and increase persistent sodium current, they do not cause an increase in resting sodium levels in muscle fibers; however, cold challenge during exercise does cause an increase in intracellular sodium levels (100). Increased intracellular sodium during the bursts of action potentials that drive forceful muscle contractions would reduce the driving force for sodium current, depolarize muscle fibers, and cause failure of action potential generation. These cellular events may lead to the flaccid paralysis that follows episodes of myotonia and muscle stiffening in PMC.
Potassium-aggravated myotonia (PAM) is a milder disease than PMC. Stiffness and myotonia are initiated during exercise, and they may persist for hours, but they are not followed by flaccid paralysis (83). Muscle stiffness is aggravated by elevated serum potassium but is unaffected by cold (83). Mutations that cause this disease are scattered along the intracellular surface of the sodium channel, especially in domain IV, and they include amino acid residues identified as important components of the fast inactivation gate and its receptor region (FIGURE 4, A and B; Ref. 83). The functional effects are milder than those that cause PMC, but there is one mutation in the fast inactivation gate that causes both diseases, depending on the family and the genetic backgrounds of the patients (101).
2.3. Hypokalemic and Normokalemic Periodic Paralysis
Hypokalemic periodic paralysis (HypoPP) causes episodic flaccid paralysis associated with low potassium levels in serum and high potassium levels in skeletal muscle (2, 76, 102). Muscle fibers are depolarized and have a high internal sodium concentration, which leads to loss of excitability (2, 76, 102). Mutations in the CACNAL1 gene encoding the skeletal muscle calcium channel CaV1.1 are the most frequent cause of HypoPP (103). In contrast to the broad range of mutations that cause HyperPP and PMC, mutations that cause HypoPP always change the first or second arginine gating charge in an S4 segment to a neutral amino acid residue (83, 104, 105). Introduction of a HypoPP mutation into mice reproduced the potassium-sensitive paralysis and weakness characteristic of HypoPP, further confirming the causative role of these gating charge mutations in the disease (106). Surprisingly, analogous mutations in the first or second gating charges (R1 and R2) in the S4 segment of the NaV1.4 skeletal muscle sodium channel also cause this disease in humans and mice (FIGURE 4, A and B; Refs. 107–109), further highlighting the pathogenic role of these gating charge mutations in HypoPP. The rare related disease normokalemic periodic paralysis (NormoPP) causes similar flaccid paralysis at normal levels of serum potassium (110). Remarkably, it is caused by mutations of the gating charge arginine in the third (R3) position in S4 segment of NaV1.4 (110). This characteristic genetic feature that is shared by HypoPP and NormoPP mutations suggests a unique common mechanism of action of these mutations that are exclusively in gating charges. However, even though HypoPP mutations have a variety of effects on fast and slow inactivation of NaV1.4 channels, none of these effects is potent enough to be the primary cause of the symptoms of HypoPP or NormoPP (108, 111, 112).
The enigma of the mechanism of HypoPP and NormoPP was resolved by discovery of gating pore currents that are induced by HypoPP and NormoPP mutations (47, 113–116). As described above in sect. 1.2, mutation of gating charge arginine residues to neutral amino acid residues can cause gating pore current through the mutant voltage sensor. On the basis of these earlier findings, it was natural to hypothesize that HypoPP mutations cause pathogenic gating pore current. As expected from this hypothesis, pathogenic gating pore currents could be detected for HypoPP mutations in NaV1.4 (114, 115). The inward currents conducted by the pathogenic gating pores are in the range of 1% of the peak sodium current, and they are nonselective among monovalent cations (47, 114–116). In contrast to the nonselectivity among monovalent cations, mutants having His substituted for Arg often conduct protons selectively (115, 116). HypoPP mutations in NaV1.4 channels are located in the R1 and R2 gating charges, and they cause gating pore current in the resting state (114). Even though the gating pore currents are small, they are constant, and calculations indicate a large effect on sodium entry into muscle fibers, consistent with the increased intracellular sodium levels and depolarized membrane potentials in HypoPP muscle (114, 115).
In contrast to HypoPP, mutations that cause NormoPP specifically neutralize the R3 gating charge (110). They cause gating pore current in the activated and inactivated states, because the R3 gating charge is located in or near the HCS in the activated state of the voltage sensor (113). The voltage dependence of pathogenic gating pore current is complex: it is activated at strongly depolarized potentials but deactivated only at much more negative potentials resulting in a hysteresis loop (113). The low conductance of the mutant gating pore would not be pathogenic if the gating pore were only open briefly during an action potential. However, voltage-clamp analysis showed that the mutant gating pore is open in both activated and slow-inactivated NaV1.4 channels (113). Because slow inactivation accumulates during the long trains of action potentials that induce forceful muscle contraction, the mutant gating pore is open for sufficiently long intervals to cause periodic paralysis and degeneration of muscle fibers.
HypoPP and NormoPP mutations have been introduced into the bacterial sodium channel NaVAb to study the structural basis for gating pore current (117). The central pore currents and gating pore currents generated by these mutants were analogous to those observed in pathogenic mutations in NaV1.4 channels (FIGURE 5, A and B; Ref. 117). The central pore currents were typical for sodium channels (FIGURE 5, A and B, left). However, the mutants exhibited marked gating pore currents (FIGURE 5, A and B, center and right). The HypoPP mutant R2S conducted gating pore leak currents at negative membrane potentials in the resting state (FIGURE 5A, center and right, blue), whereas the R3G mutant conducted gating pore leak currents at positive potentials in the activated state (FIGURE 5B, center and right, red). As expected from physiological studies, these mutations cause a voltage-dependent transmembrane conductance pathway through the voltage sensor itself that is clearly observed by X-ray crystallography (FIGURE 5C). The diameter of the pathogenic gating pore is 2–3 Å, consistent with conductance of dehydrated sodium ions at the low rate characteristic of gating pore current. The complete water-filled pathway through which the sodium ion moves is highlighted in magenta in FIGURE 4C, right, as revealed by analysis with the program MOLE (117).
FIGURE 5.
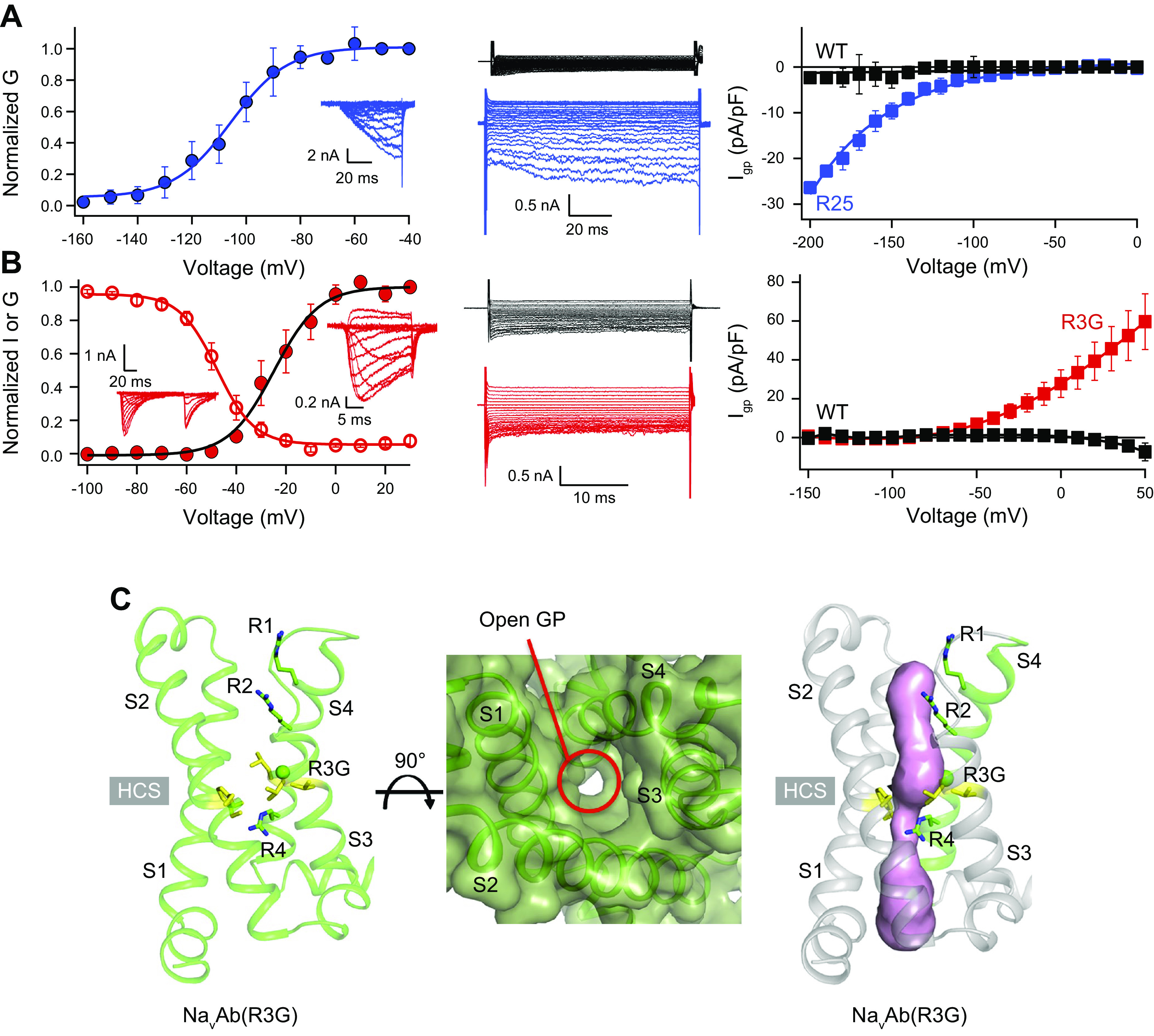
Pathogenic gating pore current and structure. A, left: central pore Na+ currents (inset) and conductance (G)/voltage (V) curve for NavAb/R2S recorded during 200-ms depolarizations from −200 mV to the indicated potentials. Center: leak Na+ currents for wild-type (WT; black) and NavAb/R2S (blue). Note the larger negative leak currents in NavAb/R2S due to gating pore current. Right: current (Igp)/V curves for nonlinear leak currents for NavAb/R2S (blue) or NavAb/WT (black) elicited by depolarization from −100 mV to the indicated potentials. B, left: central pore Na+ currents (inset) and G/V curve for NavAb/R3G from a holding potential of −160 mV (filled circles). Voltage dependence of steady-state inactivation (open circles) for NavAb/R3G. Center: leak Na+ currents for NaVAb/R3G (red) or NaVAb/WT (black) for voltage steps from 0 mV to the indicated potentials. Right: Igp/V curves for nonlinear leak currents for NavAb/R3G (red) or NaVAb/WT (black, n = 11). Note the larger positive leak currents in NavAb/R3G due to gating pore current. C: structure of a pathogenic gating pore (GP) in a normokalemic periodic paralysis (NormoPP) mutation. Magenta shading, water-accessible space determined by MOLE2. HCS, hydrophobic constriction site; R1–R4, gating charges; S1–S4, transmembrane segments. Adapted from Ref. 117 with permission from Nature.
Although gating pore current is nonselective, its primary pathogenic effects are caused by inward leak of Na+. Quantitative estimates indicate that entry of Na+ through the open gating pore would increase the resting leak of Na+ >2-fold and possibly as much as 10-fold (47, 114–116). This increase in inward leak of Na+ causes elevation of intracellular Na+ from ∼15 mM in control subjects to a range of 19–25 mM in patients with different mutations (118). Muscle fibers are also depolarized from −87 mV in control subjects to a range of −74 mV to −77 mV in patients (118). These deficits in patients were significantly worsened by local cooling of muscle fibers, which had no significant effect on control subjects. The alterations in ion gradients and membrane potential also exacerbate a bistable state of excitability, in which the membrane potential jumps from hyperpolarized to depolarized levels in muscle fibers from patients much more than in control subjects (118). Similar effects were observed in a mouse model of NaV1.4 HypoPP, including gating pore current, persistent depolarization, impaired action potential generation, and episodic weakness without myotonia (109). Although mutations in HypoPP are heterozygous, the gating pore current of the defective sodium channel causes dominant-negative physiological impairment of action potential firing by the wild-type sodium channels through reduction of the sodium gradient and inactivation by chronic depolarization. Low extracellular potassium levels contribute to episodes of flaccid paralysis in two parallel ways. First, low potassium reduces the effectiveness of sodium efflux mediated by the Na+-K+-ATPase, which exchanges extracellular potassium for intracellular sodium. This effect exacerbates the increase in intracellular sodium and membrane depolarization. Second, the reduced extracellular potassium increases the shift to the depolarized state by changing the ratio of the bistable hyperpolarized and depolarized states of membrane potential (118).
2.4. Congenital Myasthenic Syndrome and Congenital Myopathy
Myasthenias are characterized by development of weakness and fatigue during continuous use of skeletal muscles (119, 120). Most of the myasthenia syndromes are caused by failure of synaptic transmission, similar to myasthenia gravis (119, 120). However, rare cases of failure of action potential firing downstream of synaptic transmission have been discovered (119, 120). The congenital myasthenic syndrome (CMS) is a recessively inherited disease most often caused by homozygous mutations that neutralize a gating charge Arg in the S4 segment in domain IV of NaV1.4 (121, 122). Similar symptoms have been observed in patients with heterozygous mutations in the S4–S5 linker in domain I and the S4 segment and S3–S4 linker in domain IV (123, 124). These mutations cause a large negative shift in the voltage dependence of fast inactivation, slower recovery from inactivation, and, in some cases, dramatically accelerated slow inactivation (121, 122). Altogether, these mutations greatly reduce the sodium current in transfected cells and impair action potential firing in skeletal muscle fibers (77). Their effects are most pronounced during sustained trains of action potentials, which trigger forceful muscle contraction. Early in an action potential train, generation of action potentials is nearly normal; however, as the train continues, increasing numbers of the mutant and wild-type sodium channels enter the fast- and slow-inactivated states, and eventually action potential generation fails, causing fatigue (77).
Congenital myopathy is characterized by neonatal or early-onset weakness (125). It is caused by mutations in many genes. Fewer than 20 families worldwide have been identified with congenital myopathy caused by recessive SCN4A mutations (126). The mutations that have been analyzed have loss-of-function phenotypes, including complete or nearly complete loss of functional expression and large positive shifts in the voltage dependence of channel activation (126). The degree of impairment of muscle function correlates with the extent of loss of function of the mutant NaV1.4 channels. The inheritance of full loss-of-function mutations on both SCN4A alleles causes a particularly severe phenotype resulting in early lethality.
2.5. Genotype-Phenotype Correlations in Periodic Paralysis
The periodic paralyses have characteristic differences in their effects on action potential sodium currents and on sodium leak, which determine the pathology of these muscle diseases (77). Gain-of-function mutations cause HyperPP and PMC by producing NaV1.4 channels that are hyperactive, conduct excess persistent sodium current, and/or reopen frequently after inactivation. In HyperPP, these functional effects at the cellular level cause repetitive action potential firing leading to sustained depolarization, inactivation of both wild-type and mutant NaV1.4 channels, and episodes of weakness or flaccid paralysis (127). In PMC, changes in the kinetics of fast inactivation slow its onset and accelerate its recovery, thereby producing mutant sodium channels that are ready to reactivate immediately upon repolarization from the preceding action potential. These changes lead to inappropriate repetitive firing of action potentials that cause stiffness, the clinical hallmark of myotonia (77).
The pathogenesis of HypoPP and NormoPP is remarkably different from HyperPP and PMC (77, 106, 109, 113–115). Mutations in the R1, R2, and R3 gating charges cause pathogenic gating pore currents, which leak protons and/or sodium into the cell, reduce the sodium gradient, depolarize the cell, and inactivate both wild-type and mutant NaV1.4 channels because of depolarization. The constant inward gating pore current destabilizes the bistable resting membrane potential in skeletal muscle and causes oscillating changes that impair the electrophysiological stability of the muscle fiber (77). Gating pore current is a gain-of-function effect at the molecular level, but it causes loss of contractile function at the cellular level by these diverse mechanisms. Adult patients with HypoPP and NormoPP experience pathological changes in their skeletal muscle fibers with advancing age, and a similar vacuolar pathology with disruption of transverse tubules and triad junctions is observed in mice bearing a HypoPP mutation in CaV1.1 (106), but the long-term outcomes of sodium channel HypoPP mutations in skeletal muscle of mutant mice have not yet been studied in detail. In humans, it is likely that the prolonged elevation of intracellular sodium and prolonged depolarization in HypoPP are responsible for local edema and changes of intracellular calcium signaling that cause degenerative cellular effects over time. Swelling and degeneration of muscle fibers increases progressively during the disease in response to altered ionic homeostasis. Repair mechanisms are not sufficient to counteract these cellular pathologies, leading to progressive failure of muscle function in addition to periodic paralysis.
3. BRAIN SODIUM CHANNELS: NaV1.1 AND EPILEPSY
Epilepsy is caused by excess synchronized action potential generation in the brain, which in turn depends directly on sodium channels (128). Sodium channels are the molecular targets for major antiepileptic drugs, including phenytoin, carbamazepine, and lamotrigine (129). Therefore, it is not surprising that sodium channels are important molecular targets for mutations that cause epilepsy. Four sodium channel genes are expressed in the brain: NaV1.3, which is expressed primarily in embryonic and early postnatal brain, plus NaV1.1, NaV1.2, and NaV1.6, which are expressed increasingly in development and remain highly expressed in the mature brain (130–134). These three adult sodium channel types are all broadly expressed in the brain, but they differ in expression in different types of neurons and in localization in specific subcellular compartments of neurons. Early studies revealed differential expression of sodium channel types in hippocampal neurons, with NaV1.1 primarily in cell bodies and NaV1.2 primarily in dendrites and unmyelinated axons (135). NaV1.1 is the dominant sodium channel in interneurons (42), whereas NaV1.6 and NaV1.2 are the dominant sodium channel types in excitatory neurons and their myelinated axons (134, 136, 137). It is likely that the differential expression and subcellular localization of these sodium channel subtypes prevents effective functional compensation when one of these channel types is mutated, even though their functional properties are similar.
All sodium channel types that are highly expressed in the brain are targets for epilepsy mutations, which cause epileptic syndromes with a wide range of severity and comorbidities (128, 138, 139). The SCN1A gene that encodes the NaV1.1 channel is the most frequently mutated gene in sodium channel epilepsies, and these mutations cause a broad spectrum of epilepsy syndromes from mild febrile seizures to intractable, drug-resistant developmental and epileptic encephalopathies (140–142) (FIGURE 6).
FIGURE 6.
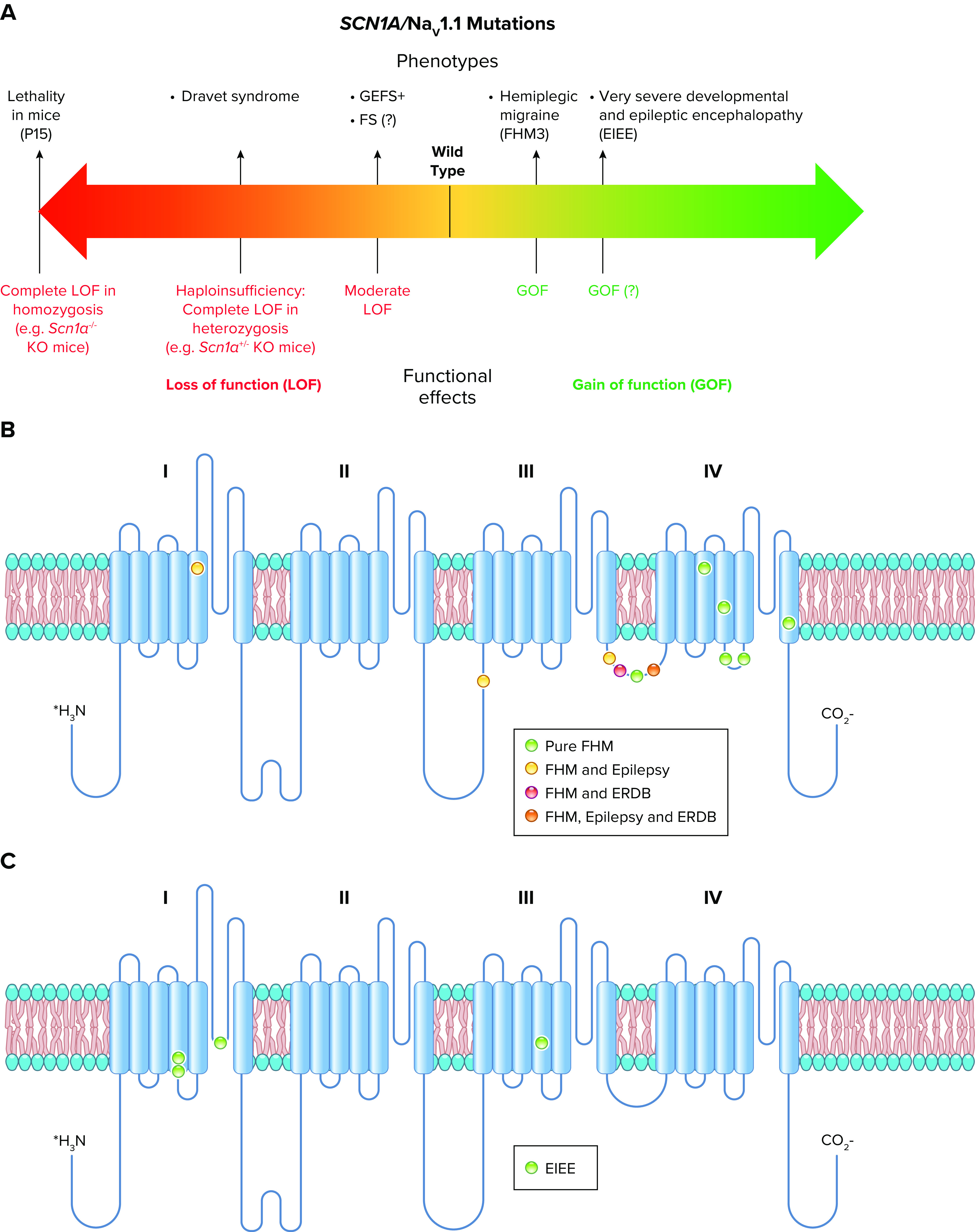
Spectrum of mutations and phenotypes for SCN1A/NaV1.1. A: phenotypic spectrum of SCN1A mutations. Most SCN1A mutations cause epileptic phenotypes, including the severe developmental and epileptic encephalopathy Dravet syndrome and the milder form genetic epilepsy with febrile seizures plus (GEFS+), which, however, shows large phenotypic variability and can include severe cases. SCN1A variants may be also involved in febrile seizure phenotypes (FS) that can include development of temporal lobe epilepsy with hippocampal sclerosis. These forms are caused by loss of function of NaV1.1 in heterozygosis, with often complete loss of function for Dravet syndrome, which is modeled by Scn1a+/− knockout mice. Gain of function of NaV1.1 has been identified for hemiplegic migraine (FHM3) mutations and proposed for mutations that cause an extremely severe early infantile epileptic encephalopathy (EIEE), although functional studies for this form have been performed for a single mutation. P15, postnatal day 15. B: molecular map of the NaV1.1 sodium channel with the location of hemiplegic migraine mutations color-coded as indicated (ERDB, elicited repetitive daily blindness). C: molecular map of the NaV1.1 sodium channel with the location of SCN1A EIEE mutations. Mutations causing other phenotypes are not shown because there are too many for a graphical representation.
3.1. Dravet Syndrome
Dravet syndrome is a developmental and epileptic encephalopathy that was first described clinically by Dr. Charlotte Dravet in Marseille, France in the late 1970s in an exceptional clinical investigation for its time (143, 144). Children with this disease develop normally for 6–9 mo. The first symptom of the disease is usually febrile seizures, which evolve over a few months to frequent spontaneous seizures that become drug resistant. Affected children lose developmental milestones that had previously been achieved, and they have severe cognitive impairment, autistic-like behaviors, ataxia, and disruption of circadian rhythms and sleep quality. Before the pathophysiology of the disease was well understood, >30% of children died prematurely from sudden unexpected death in epilepsy (SUDEP) or from accidents related to their seizures and impaired cognition. Even with modern care, >15% of affected individuals die prematurely (145), and most of those who survive to their teenage years have IQs in the range of 50 and require lifelong care (143, 144).
Dravet syndrome is caused by genetically dominant loss-of-function mutations in NaV1.1, which lead to deletions, truncations, nonsense codons, and single amino acid substitutions located throughout the coding exons of the gene (146–149). Mutations that cause the truncation of NaV1.1 lead to pure haploinsufficiency (150). More than 80% of patients diagnosed with Dravet syndrome have a mutation in NaV1.1 (145). Moreover, because only the 6-kb exons of the gene that code for the NaV1.1 protein are normally sequenced, it is likely that many of the other patients diagnosed with Dravet syndrome have mutations in the remaining 94% of the 100-kb SCN1A gene that cause loss of expression of the NaV1.1 protein and result in Dravet syndrome.
3.1.1. Loss of action potential firing in inhibitory interneurons in mouse models of Dravet syndrome.
It was a paradox that loss-of-function mutations in a sodium channel would cause epilepsy. This paradox was resolved through studies of mouse models of Dravet syndrome, which have spontaneous generalized tonic-clonic seizures that are easily observed by video recording or by electroencephalography (FIGURE 7A; Refs. 42, 153, 154). Electrophysiological analysis of these mice revealed a selective loss of sodium currents and action potential firing in GABAergic interneurons in the hippocampus (42, 140, 153, 154). Subsequent studies have shown a similar selective loss of electrical excitability in the GABAergic Purkinje neurons in the cerebellum, in the GABAergic neurons of the reticular nucleus of the thalamus, and in both parvalbumin-expressing and somatostatin-expressing interneurons in layer V of the cerebral cortex (FIGURE 7B; Refs. 151, 155–157). In each case, GABAergic inhibitory neurons were unable to sustain long trains of action potentials, which is required for effective control of the firing of excitatory neurons in neural circuits.
FIGURE 7.
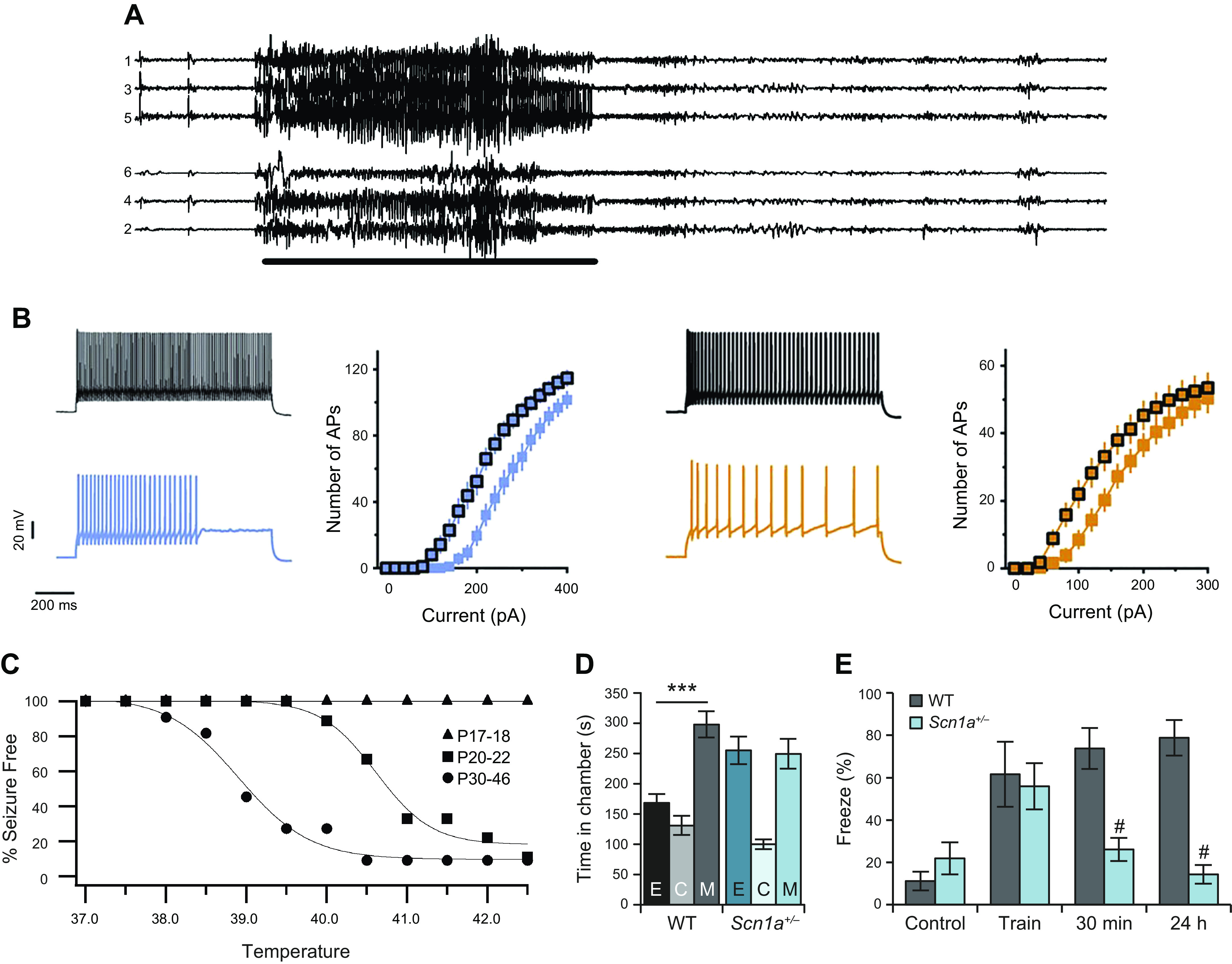
Cellular and systems phenotypes of Dravet syndrome (DS) in mice. A: electroencephalogram of a generalized tonic-clonic seizure in a DS mouse. B: loss of firing in interneurons. APs, action potentials; pA, picoamperes of stimulating current. Left: parvalbumin-expressing interneurons in layer V of the cerebral cortex. Black, wild type; blue, DS. Right, somatostatin-expressing interneurons in layer V of the cerebral cortex. Black, wild type (WT); gold, DS. Adapted from Ref. 151 with permission from Proceedings of the National Academy of Sciences of the United States of America. C: thermal induction of seizures. P, postnatal day. D: autistic-like social interaction behavior. E, empty; C, center; M, mouse. E: context-dependent fear conditioning. #P < 0.05. Adapted from Ref. 152 with permission from Nature.
Studies of local synaptic circuits are also consistent with this phenotype. Recordings of spontaneous, action potential-driven excitatory postsynaptic currents and inhibitory postsynaptic currents in the CA1 neurons in hippocampal slices showed that the frequency of inhibitory postsynaptic currents was decreased (152, 158). In response to that decrease, the frequency of excitatory postsynaptic currents was increased, and the hippocampal network became hyperexcitable (152, 158, 159). In recordings of hippocampal circuit function in vivo, the frequency of occurrence of sharp waves and sharp-wave ripples was reduced, and the frequency of individual ripples within a single sharp-wave ripple was also significantly reduced (160). These findings are consistent with impairment of action potential firing by parvalbumin-sensitive inhibitory basket cells in the hippocampus, whose firing provides critical timing information for sharp-wave ripples (161). Thus, action potential firing in interneurons is impaired, the ratio of excitatory to inhibitory synaptic activity in neural circuits is consistently increased, and the timing of circuit function is altered in the hippocampus in the early stages of Dravet syndrome in mice. Moreover, the hippocampus is directly implicated in the generation of seizures (159), and selective heterozygous deletion of NaV1.1 in the hippocampus is sufficient to cause thermal seizures and cognitive deficit characteristic of Dravet syndrome (162).
In the cerebral cortex, disynaptic inhibition between neighboring pyramidal neurons in layer V, which is mediated by somatostatin-positive frequency-accommodating interneurons (163), was strikingly impaired in Dravet syndrome (DS) mice (151). In brain slices of cerebral cortex ex vivo, the excitability of parvalbumin-positive and somatostatin-positive interneurons was reduced in DS mice, and optogenetic silencing of those neurons led to circuit hyperexcitability (155). However, studies of network function in vivo did not reveal consistent changes in the cerebral cortex, suggesting that compensatory mechanisms may be engaged in the in vivo experimental setting (155). Of note, dysfunctions might be more visible when the circuit is challenged and involved in high-frequency activity, as observed in the hippocampus (159).
3.1.2. Febrile seizures in a mouse model of Dravet syndrome.
Dravet syndrome usually begins with febrile seizures during a fever, a hot day, or a hot bath (143, 164). This characteristic of Dravet syndrome is mimicked in genetically modified mice by thermally induced seizures (165). Slow increase in the core body temperature of DS mice into the fever range of 38–41°C generates myoclonic seizures followed by generalized tonic-clonic seizures (FIGURE 7C). As in children with Dravet syndrome, susceptibility to thermally induced seizures is age dependent, with onset at a similar developmental time near the age of weaning, followed by more severe seizures and premature death (FIGURE 7C; Refs. 143, 165, 166).
3.1.3. Genetic evidence for a primary role of interneurons in Dravet syndrome.
Genetic evidence for a primary role of inhibitory interneurons in Dravet syndrome has come from specific gene deletion with the Cre-Lox method (167, 168). Deletion in forebrain interneurons using the Dlx promoter/enhancer, which is specifically expressed in developing GABAergic interneurons that arise in the medial ganglionic eminence and migrate to cerebral cortex, hippocampus, thalamus, and other forebrain structures (169), recapitulates the thermally induced seizures, spontaneous seizures, and premature death of mice observed with global deletion of NaV1.1 channels (167). In contrast, specific deletion of NaV1.1 in excitatory neurons ameliorates the phenotypes of Dravet syndrome (168). These results clearly demonstrate that the primary cause of the core symptoms of Dravet syndrome is failure of normal action potential firing by GABAergic interneurons. Consistently, single-cell transcriptomic data from humans and mice have confirmed that SCN1A is predominantly expressed in inhibitory neurons (170), and transcranial magnetic stimulation paradigms applied to Dravet syndrome patients have disclosed reduced intracortical inhibition in vivo (171).
3.1.4. Comorbidities in a mouse model of Dravet syndrome.
The major comorbidities of Dravet syndrome are also caused by failure of firing of GABAergic interneurons (172). 1) Ataxia is correlated with failure of action potential firing of the GABAergic Purkinje neurons of the cerebellum, which are crucial for coordination of movement (157). 2) Cognitive deficit in context-dependent fear conditioning (FIGURE 7E) and spatial learning and autistic-like behaviors in social interaction tests (FIGURE 7D) are all observed in mice in which NaV1.1 channels have been specifically deleted in forebrain inhibitory interneurons (152, 173). 3) Impaired sleep quality is also observed in mice with specific deletion of NaV1.1 channels in forebrain interneurons, and it is correlated with failure of firing of the GABAergic interneurons of the reticular nucleus of the thalamus, which set sleep rhythms by driving sleep spindles (156). Failure of firing of these neurons has also been observed in different epileptic Scn1a models (158); however, one study puzzlingly reported hyperexcitability of these neurons in brain slices from Scn1aR1407X/+ mice (174). 4) The circadian rhythm defect is also correlated with failure of firing of the GABAergic neurons of the suprachiasmatic nucleus of the hypothalamus, and it is rescued by enhancement of GABAergic neurotransmission with the benzodiazepine clonazepam, implicating failure of firing of interneurons as the primary cause of this defect (175). Thus, Dravet syndrome is an interneuronopathy in which an increase in excitation:inhibition balance in neural circuits throughout the brain causes the multifaceted comorbid disease phenotypes.
The Cre-Lox method has also been used to probe the functional roles of different classes of interneurons in the multifaceted phenotypes of Dravet syndrome (TABLE 1; Ref. 176). Nearly all of the interneurons in the cerebral cortex can be divided into three classes identified by their expression of parvalbumin, somatostatin, or the 5-HT3a receptor (177, 178). Heterozygous deletion of NaV1.1 channels in parvalbumin-expressing interneurons causes proepileptic effects and autistic-like behavior (176). Deletion in somatostatin-expressing interneurons causes proepileptic effects and hyperactivity (176). Deletion in these two classes of interneurons together gives synergistic effects on epilepsy, premature death, and cognitive deficit (176). In contrast, deletion in 5-HT3a receptor-expressing interneurons has much milder effects (TABLE 1; Ref. 179). It has been suggested that 5-HT3a receptor-expressing interneurons do not express NaV1.1 (180). However, single-cell RNA sequencing investigations suggest expression of NaV1.1 in these neurons (181, 182), and recent studies have shown reduced excitability of a subset of 5-HT3a receptor-expressing interneurons [irregular spiking vasoactive intestinal peptide (VIP)-positive interneurons] in layer II–III of the primary somatosensory and visual cortex (179, 183). Altogether, these studies indicate that altered excitation:inhibition balance in brain circuits, induced mainly by failure of action potential firing by parvalbumin-expressing interneurons, somatostatin-expressing interneurons, or both, induces both the core symptoms and the comorbidities of Dravet syndrome.
Table 1.
Functional impacts of deletion of NaV1.1 channels in specific classes of interneurons
| Interneuron | Epilepsy | SUDEP | Hyperactivity | Autistic Behavior | Cognitive Deficit |
|---|---|---|---|---|---|
| PV | ++ | +/− | − | + | − |
| SST | + | − | + | − | − |
| PV + SST | +++ | ++ | + | + | + |
| 5-HT3R | − | − | − | +/− | − |
| All: Dlx-Cre | ++++ | ++++ | + | + | ++ |
PV, parvalbumin; SST, somatostatin; SUDEP, sudden unexpected death in epilepsy; 5-HT3R, 5-HT3 receptor.
3.1.5. SUDEP in a mouse model of Dravet syndrome.
SUDEP is the most serious result of Dravet syndrome, and parents and caregivers live in fear of this devastating outcome (144, 184, 185). In C57BL/6J mice, Dravet syndrome mutations cause a wave of high incidence of SUDEP on postnatal day (P)21–P28 (42, 154, 186). Specific deletion of NaV1.1 channels in forebrain interneurons is sufficient to cause the primary wave of SUDEP during P21–P28, indicating that epilepsy itself causes premature death at this time rather than other parallel effects of deletion of NaV1.1 channels in heart or other peripheral tissues (186). Consistent with this conclusion, specific heterozygous deletion in the heart does not cause SUDEP (186). SUDEP in DS mice is correlated with dramatic bradycardia (186), respiratory disturbances (187), and sudden cardiac arrest following generalized tonic-clonic seizures (186). Sudden death is prevented by peripheral administration of N-methylscopolamine, a peripherally restricted inhibitor of muscarinic acetylcholine receptors (186). These results point to overactive parasympathetic cholinergic outflow from the central nervous system (CNS) during and after generalized tonic-clonic seizures, resulting in hyperactivation of cardiac muscarinic acetylcholine receptors, as the primary mechanism underlying SUDEP during P21–P28 in this model of Dravet syndrome (186). Abnormal parasympathetic regulation may disturb both respiration and cardiac function; however, prevention of sudden death by treatment with the non-CNS-penetrant muscarinic antagonist N-methylscopolamine administered in the peripheral circulation supports the conclusion that hyperactivation of muscarinic acetylcholine receptors in the heart, resulting in severe bradycardia and ventricular arrhythmia, is the proximate cause of death in the Dravet syndrome model in C57BL/6J mice during P21–P28 (186).
3.1.6. Genetic background effects in Dravet syndrome.
Genetic background effects are important in Dravet syndrome. Complete loss-of-function truncation mutations cause differing degrees of disease severity in Dravet syndrome patients with different genetic backgrounds (143), and dramatic genetic background effects are observed for identical mutations in different strains of mice (42, 188). In the first study of Dravet syndrome in mice, the C57BL/6J strain was found to be much more susceptible to the disease mutation compared with the 129SvJ strain (42). For that reason, most subsequent studies have been carried out on mice bred for >10 generations into C57BL/6J. In this genetic background, Dravet syndrome mice recapitulate all of the complex symptoms and comorbidities of the human disease, as reviewed above. Physiological studies comparing Dravet syndrome mice in the C57BL/6 genetic background and the 129SvJ genetic background revealed less severely impaired action potential firing and less severely impaired sodium channel-dependent boosting of excitatory postsynaptic currents recorded in GABAergic inhibitory neurons in 129SvJ mice (188). These milder effects of the DS mutation on action potential firing in 129SvJ mice may contribute to the milder phenotype of DS in these mice. Gene mapping has identified a subunit of GABAA receptors as a contributing molecular factor to this difference in disease severity, again pointing to defects in inhibitory neurotransmission as fundamental in Dravet syndrome (189).
Dravet syndrome has also been studied in Scn1a+/− mice of mixed genetic backgrounds generated by crossing C57BL/6J with 129S6.SvEvTac (190, 191). The resulting 50:50 F1 generation mice have a milder epileptic phenotype than pure-bred C57BL/6J, and they require temperatures in the range of 42.5°C for thermal induction of seizures (190). This temperature range is characteristic of heat stroke, so the seizures induced at that temperature may not be epileptic in origin. These mice have not been analyzed for comorbidities of Dravet syndrome to date. Thus, at this stage, they are a less complete model for studies of Dravet syndrome than pure-bred C57BL/6J.
3.1.7. Compensatory effects in Dravet syndrome.
In addition to SCN1A, three other sodium channel genes are broadly expressed in the brain (133), but they do not effectively compensate for loss of NaV1.1 channels in Dravet syndrome. Early studies revealed increased expression of NaV1.3 channels in the hippocampus (42) and increased activity of NaV1.6 channels in the Purkinje cells of the cerebellum (157), but neither of these compensatory changes is sufficient to prevent the symptoms of Dravet syndrome (42, 157). In mice with mixed 50:50 C57BL/6J:129S6.SvEvTac genetic background, upregulation of sodium current was observed in dissociated hippocampal pyramidal neurons from P21 mice (191). Increased sodium channel activity at P21 may be important for generation of the first spontaneous seizures, which occur at this age in Dravet syndrome mice. In parvalbumin-expressing layer II–III interneurons in brain slices from the primary somatosensory cortex of these mixed-breed mice, action potential firing was diminished at P18–P21 but returned to normal at P35–P56 (192). These results open the possibility that normalization of action potential firing in these neurons takes place with increasing age, which would correspond to late childhood and early teenage years in humans, when seizures in Dravet syndrome become less severe and are more effectively treated with antiepileptic drugs (144). However, the reduction of sodium current and the impairment of action potential firing in cortical neurons is less prominent than in hippocampal neurons in C57BL/6 mice (42, 151, 155, 159). Therefore, it will be important to determine whether the excitability of these highly sensitive hippocampal neurons, as well as of cortical interneurons of other layers and cortical regions, also returns toward wild type in older mice or remains low and continues to create hyperexcitability in neuronal circuits in the adult.
3.1.8. Antiepileptic therapies tested in animal models of Dravet syndrome.
Mouse models of Dravet syndrome have been used to test both pharmacological treatments and gene therapy approaches. The first studies tested the effect of drugs and treatments already used in the clinic on seizures induced with hyperthermia or the convulsant flurothyl. The ketogenic diet (a high-fat/low-carbohydrate and protein diet used as an alternative treatment for refractory epilepsy) was effective in reducing the sensitivity to flurothyl-induced seizures (193). Stiripentol (an antiepileptic approved specifically for Dravet syndrome) and combinatorial therapy with stiripentol plus clobazam (a positive allosteric modulator of GABAA receptors) were effective in reducing convulsant-induced seizures (194). Moreover, clonazepam (a positive allosteric modulator of GABAA receptors) and tiagabine (a presynaptic GABA reuptake inhibitor) were effective individually in increasing the threshold for thermal induction of seizures and had synergistic beneficial effects when given together (195). Overall, these results established the Dravet syndrome mouse model as a potentially valuable asset in testing new drugs and drug combinations in Dravet syndrome.
More recent studies have tested drugs that are not yet used extensively in the clinic. The synthetic neuroactive steroid SGE-516 is a potent positive allosteric modulator of both synaptic and extrasynaptic GABAA receptors. It increased threshold for hyperthermia-induced seizures, decreased frequency of spontaneous convulsive seizures, and prolonged survivals (196), consistent with other GABAA receptor-enhancing treatments. GS967, a nonconventional sodium channel blocker that preferentially inhibits persistent sodium current, was unexpectedly effective in reducing frequency of spontaneous seizures and mortality (197). The effect was probably related to a reduction of the excitability of excitatory neurons, which was enhanced in chronic administration because it reduced expression of NaV1.6 channels. A recent study reported reduction of seizures with chronic administrations of the peptidic toxin Hm1a, which can act as a specific NaV1.1 enhancer at nanomolar concentrations (198). However, delivery and dosage in clinical settings are challenging problems for this approach, in particular considering that at slightly higher concentrations Hm1a can hit numerous other targets (199).
Cannabidiol (CBD), a nonpsychotropic cannabinoid extracted from Cannabis sativa, is increasingly used to treat refractory epilepsy, and a small clinical trial showed a beneficial effect on Dravet syndrome patients (200). Clinical trials in Dravet syndrome are challenging because of the small number of patients, the wide age range of patients, and the differences in background standard-of-care medications that they receive concomitantly. Nevertheless, consistent with these clinical trial results, treatment with CBD reduced the duration and severity of hyperthermia-induced seizures and the frequency of spontaneous convulsive seizures of Scn1a+/− mice under carefully controlled laboratory conditions without other drugs (201), further supporting its therapeutic efficacy. When tested in brain slices in vitro, CBD increased inhibitory neurotransmission and decreased excitatory neurotransmission in the granule cell neurons of the dentate gyrus of the hippocampus, as measured by the frequency of spontaneous inhibitory and excitatory postsynaptic currents. These effects were caused by direct increase of the frequency of action potentials in GABAergic neurons and a resulting decrease in dentate granule cell neurons. This latter effect may be due in part to the reduction of persistent current generated by NaV1.6 channels, which can be inhibited by CBD in transfected nonneuronal cells expressing NaV1.6 (202, 203). CBD binds in the pore of an ancestral bacterial sodium channel, providing a molecular mechanism for this inhibitory effect (204). Notably, CBD’s actions on mouse dentate granule cells from Scn1a+/− mice did not depend on activation of CB1 cannabinoid receptor but were potentially mediated by antagonism of the lipid-activated G protein-coupled receptor GPR55 (201).
Surprisingly, Scn1Lab−/− mutant zebrafish have provided a model of Dravet syndrome that can be efficiently used in drug screens (205, 206). These screens have identified serotoninergic drugs that in some cases were effective when tested in Dravet syndrome patients, and more detailed studies identified 5-HT2B receptors as the potential drug target (207, 208). Clinical trials are in progress to test the efficacy of previously developed drugs that target serotonin receptors for prevention of seizures in Dravet syndrome (https://www.epygenix.com/pipelines/). Although the motor dysfunction induced in zebrafish by Scn1a mutations is not similar to seizures in mammalian brain and the concentrations of drug candidates used in this system are much higher than typical for human pharmacology, the successes to date indicate that phenotypic screens in this novel experimental system have promise for discovery of unexpected classes of drug candidates for treatment of intractable epilepsy.
Gene therapy approaches have been recently developed for upregulating the wild-type Scn1a allele in Scn1a+/− mice. One study identified a novel antisense noncoding RNA (SCN1ANAT) implicated in a physiological mechanism that downregulates the expression of Scn1a (209). Antisense oligonucleotides (AntagoNATs) targeting and degrading SCN1ANAT were able to specifically upregulate Scn1a expression in vitro and in vivo after intrathecal administration in the brain of a Dravet syndrome mouse model and a wild-type nonhuman primate (209). Four weekly injections of AntagoNATs decreased frequency of spontaneous convulsive seizures and increased the threshold for hyperthermic seizures. Moreover, the treatment partially rescued the excitability of parvalbumin-positive hippocampal GABAergic neurons. A more recent study used antisense oligonucleotides targeted to Scn8a for reducing expression of NaV1.6,, which is the primary sodium channel driving firing of excitatory neurons. A single treatment of Dravet syndrome mice with Scn8a antisense oligonucleotides at P2 reduced spontaneous convulsive seizures and delayed mortality onset from 3 wk to beyond 5 mo of age (210), a surprisingly long-lasting beneficial effect that is consistent with the therapeutic benefits obtained by heterozygous gene deletion of NaV1.6 in another study (211). An additional approach used the CRISPR-ON system with a catalytically inactive Cas9 (dCas9) to upregulate Scn1a expression (212). A specific single guide RNA (sgRNA) that increases Scn1a gene expression levels in cell lines and primary neurons with high specificity was identified. Treatment with this CRISPR-ON strategy was able to increase NaV1.1 protein levels and rescue excitability of GABAergic neurons from Scn1a+/− mice in primary culture. Delivery of the Scn1a-dCas9 activation system to Scn1a+/− neonates (before disease onset) using adeno-associated viruses enhanced excitability of parvalbumin-positive interneurons in brain slices and increased threshold for thermally induced seizures (212). Most recently, Han et al. (213) used a targeted augmentation of nuclear gene output (TANGO) approach to increase the expression of functional NaV1.1 channels in Dravet syndrome mice. They observed a substantial decrease in spontaneous seizures and SUDEP when the specific antisense oligonucleotides were injected in newborn mice. These methods hold great promise for treatment of Dravet syndrome in children who can be identified by gene sequencing early in life before their symptoms arise. Altogether, these genetic approaches have a high level of specificity for NaV1.1 channels and reverse the effects of loss of function of this channel, but the timing of treatment, method of delivery, and half-life of therapeutic agents are still challenging problems that need to be solved.
3.2. Genetic Epilepsy with Febrile Seizures Plus
The first pathogenic NaV1.1 mutations were identified in genetic epilepsy with febrile seizure plus (GEFS+) (214), which is characterized by a large phenotypic spectrum. Disease onset is between 3 mo and 6 yr, with febrile/hyperthermic seizures that continue to occur into adulthood. Spontaneous afebrile seizures develop later, and the most severe phenotypes in the GEFS+ spectrum are similar to Dravet syndrome (215). GEFS+ is genetically heterogeneous. Mutations are often identified in large families with autosomal dominant segregation and incomplete penetrance. Mutations of NaV1.1 have been identified in ∼20% of GEFS+ families, and they are all missense mutations.
GEFS+ NaV1.1 mutations were also the first to be functionally investigated in vitro (216). For this study, the cDNA of the long human splice variant was used, and the observed effect on NaV1.1 current was reduction of inactivation leading to increased persistent sodium current. This gain-of-function effect would be consistent with enhanced neuronal excitability, but that was opposite to the predicted effect of truncating DS mutations that had been previously identified (147). However, the same group reported loss of function for other GEFS+ mutations, in some causing complete loss of function, as for a DS missense mutation that was studied in parallel (217). Although a net gain-of-function effect was reported for a few other GEFS+ mutations in early work, studies performed thereafter have consistently shown that the general functional effect identified in transfected cells of both DS and GEFS+ NaV1.1 mutations is loss of function (153). The initial variability of functional effects was probably generated by the smaller functional effect of GEFS+ mutations compared to Dravet syndrome mutations, and by the experimental conditions used for the functional studies; in particular, the cellular background and the type of cDNA (including the specific splice variants) can have an important modulatory effect. Notably, the R1648H NaV1.1 GEFS+ mutation, which has been reported as a gain of function in an investigation that used heterologous expression systems (216), has been expressed in transgenic mice, and its functional effects have been studied in dissociated neocortical neurons (218). This study showed that in a neuronal cell background it induces loss of function but with modifications that are neuron subtype specific: slower recovery from inactivation and increased use-dependent inactivation in bipolar GABAergic interneurons but negative shift of voltage dependence of inactivation in pyramidal neurons.
Clinical phenotypes observed in patients carrying the R1648H mutation show large variability, ranging from mild GEFS+ to severity approaching that of Dravet syndrome (214, 219). A gene-targeted knockin mouse model of the R1648H mutation was subsequently generated (Scn1aR1648H/+) to correlate neuronal dysfunctions with phenotypic features (219). In the first studies, it was reported that the phenotype of this model is characterized by hyperthermic and spontaneous seizures (219) and sleep dysfunctions (220), which were milder in comparison with mice carrying truncating mutations that model Dravet syndrome.
Experiments performed in brain slices obtained from Scn1aR1648H/+ mice (158) have shown hypoexcitability of GABAergic interneurons in cerebral cortex, hippocampus, and thalamus, as quantified by the reduced number of action potentials in input-output relationships, without detectable modifications in excitatory neurons. Reduced posthyperpolarization firing in GABAergic neurons of the reticular nucleus of the thalamus was also observed (158), as reported in global-knockout Scn1a+/− mice (156). Hypoexcitability of GABAergic neurons caused a reduction in action potential-induced postsynaptic GABAergic currents and tonic GABAergic current, as well as abnormal thalamo-cortical and hippocampal spontaneous network activities, which showed pathological high-frequency oscillations that were not observed in control. Thus, the basic pathological mechanisms observed in Scn1aR1648H/+ mice are similar to those observed in models of Dravet syndrome truncating mutations, supporting the conclusion that loss of function is responsible for the range of GEFS+ phenotypes of this mutation. However, the observed phenotype is milder, recapitulating the mildest phenotypes in the GEFS+ spectrum rather than the more severe phenotype approaching that of Dravet syndrome observed in some R1648H patients. The spectrum of phenotypes observed in these patients is consistent with the hypothesis that loss-of-function mutations in SCN1A cause a range of phenotypes, of which Dravet syndrome is the most severe (140).
Interestingly, a recent study has shown that synergistic interactions between the genetic mutation and seizures can cause pathological remodeling in Scn1aR1648H/+ mice that leads to a severe Dravet syndrome-like phenotype (221) (FIGURE 8).To exclude the contribution of spontaneous seizures, this study used Scn1aR1648H/+ mice in a 50%-50% 129:C57Bl/6 F1 genetic background, which are asymptomatic and do not show spontaneous seizures but have hyperthermia-triggered seizures. Short seizures (<1 min) were induced experimentally with hyperthermia or with the convulsant flurothyl (1 seizure/day for 10 days, starting at P21). Hyperthermia triggered seizures only in Scn1aR1648H/+ mice, generating a chronic severe epileptic phenotype and cognitive/behavioral defects of hyperactivity, reduced social interaction, and deficits in learning and memory in adulthood, without major modifications in cytoarchitecture or neuronal death (221). Increased excitability was observed in hippocampal dentate granule cells but not CA1 pyramidal neurons, consistent with pathological remodeling that is not directly related to the effect of the mutation on GABAergic neurons. Flurothyl triggered similar seizures in both Scn1aR1648H/+ mice and wild-type littermates but led to chronic epilepsy, behavioral/cognitive dysfunctions, and hyperexcitability of hippocampal granule cells in Scn1aR1648H/+ mice, which showed a phenotype similar to that observed after induction of hyperthermic seizures. Thus, these results show that Scn1a genetic mutations/variants, even if mild per se, can make the brain sensitive to the deleterious effect of seizures, even short ones, and increase the risk of second hits to develop severe phenotypes. Strikingly, the interaction between the Scn1a mutation and seizures is necessary for generating pathological remodeling leading to a severe phenotype. This is relevant for both GEFS+ and Dravet syndrome, suggesting that seizures should be controlled to avoid phenotypic worsening, and also for other common epilepsies in which SCN1A genetic variants have been identified as risk factors (222–225).
FIGURE 8.
Synergistic interactions between SCN1A mutations and seizures leading to pathological remodeling and development of severe phenotypes. Top: protocol timeline. Bottom: outcomes of the experiments that showed that the Scn1aR1648H/+ knockin mice in the 129-C57BL/6 genetic background have an asymptomatic phenotype. The induction of short repeated seizures had no effect on wild-type (WT) mice, but it transformed the asymptomatic phenotype of Scn1aR1648H/+ mice into a severe Dravet syndrome-like phenotype, including frequent spontaneous seizures and cognitive/behavioral deficits. In these mice, there were no major modifications in cytoarchitecture or neuronal death but increased excitability of hippocampal dentate gyrus granule cells and increased expression of calbindin, consistent with a pathological remodeling of neuronal functions. Thus, an SCN1A mutation is a prerequisite for a long-term deleterious effect of seizures on the brain, indicating a clear interaction between seizures and the mutation (2-hit) for the development of a severe phenotype generated by pathological remodeling. ECoG, electrocorticogram. Adapted from Ref. 221 with permission from Neurobiology of Disease.
These results, emphasizing the importance of seizures in the severity of SCN1A phenotypes, contrast with data obtained by downregulating NaV1.1 expression in the hippocampus of wild-type rats by means of RNA interference, which showed cognitive deficits without seizures (226, 227). However, the phenotypic effects obtained with RNA interference experiments could be enhanced by the large (>70%) reduction of NaV1.1 expression, which is larger than the 50% reduction expected from haploinsufficiency (150).
3.3. Rescuable Folding-Defective Epileptogenic NaV1.1 Missense Mutants
The severity spectrum of NaV1.1-related epilepsies could be a continuum and depend on the amount of loss of function of the mutant: a mild impairment of NaV1.1 function would cause mild phenotypes, whereas a more complete loss of function would cause severe phenotypes (140). Interestingly, some NaV1.1 missense mutations cause loss of function because of folding/trafficking defects that lead to channel degradation in intracellular compartments (24). These mutants can often be rescued by incubation of the cells at low temperature or by interacting proteins that probably stabilize the correct folding conformation in the endoplasmic reticulum (ER) (228–232) (FIGURE 9A). Similar interactions may rescue the mutants in vivo, regulate the extent of loss of function, and modulate the phenotype in this way. Possibly, the complete loss of function observed for some GEFS+ mutants in functional studies in vitro may be caused by lack of rescue in the experimental conditions used, and, conversely, lack of rescue in vivo could induce complete loss of function of Dravet syndrome mutants that show only mild loss of function in some in vitro expression systems. Notably, folding-defective NaV1.1 missense mutants can also be partially rescued by interactions with small drugs (pharmacological chaperones), which bind to them in the ER and probably stabilize the correct folding conformation. Similar to interacting proteins, they can bind to rescued channels targeted to the plasma membrane and modify their properties (FIGURE 9A).
FIGURE 9.
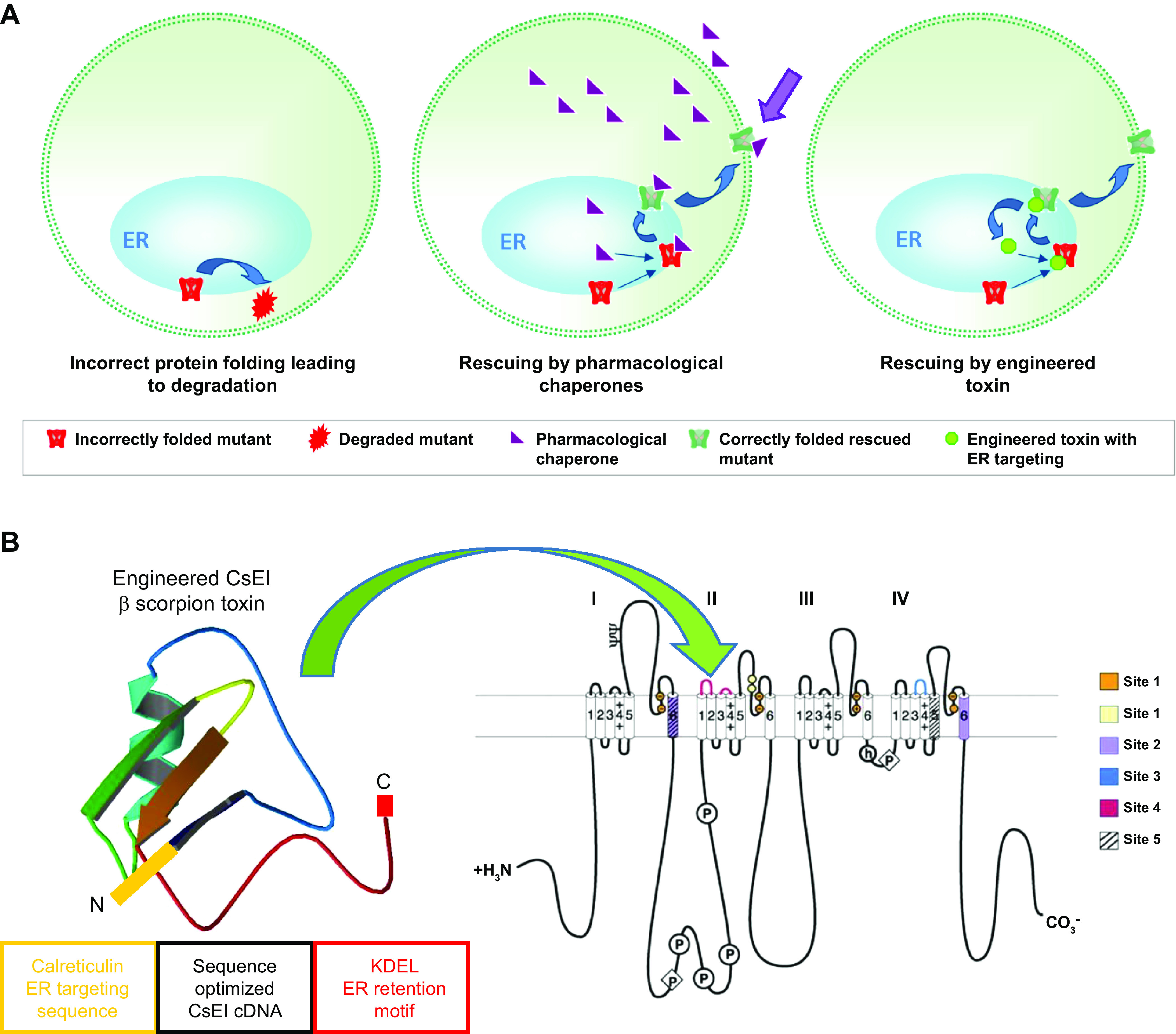
Rescue of expression and function of SCN1A/NaV1.1 mutants. A: mutations of NaV1.1 and protein folding. Left: folding-defective mutants are recognized as incorrectly folded proteins by the quality control system of the endoplasmic reticulum (ER) and degraded. Center: folding defects can be rescued by molecular interactions with interacting proteins or pharmacological chaperones, which probably act by stabilizing a correct folding conformation and preventing degradation. They also interact with rescued proteins by modifying their functional properties (purple arrow), which can be a major drawback for therapeutic applications of pharmacological chaperones. Rescue of epileptogenic NaV1.1 folding-defective mutants attenuates the loss of function but, different than for FHM mutations, never induces gain of function. Right: engineered peptide toxins targeted to the ER can rescue NaV1.1 folding-defective mutants but, unlike pharmacological chaperones, do not interact with rescued proteins at the plasma membrane. B, left: the cDNA of Centruroides sculpturatus Ewing (CsEI) β-scorpion toxin, sequence-optimized for expression in human cells, was engineered to target the peptide to the ER by insertion of an NH2-terminal calreticulin ER targeting sequence (orange) and a COOH-terminal KDEL ER retention motif (red); cells transfected with a plasmid containing the engineered cDNA expressed the engineered ER-resident toxin that rescued some NaV1.1 mutants and was not released extracellularly, as tested with immunoblots of the extracellular medium and with functional evaluations (228). Right: interaction of the toxin with sodium channels at neurotoxin receptor site 4 in domain II; toxin binding sites are color coded as indicated. Adapted from Ref. 228 with permission of Neurobiology of Disease and from Ref. 233 with permission from Biochimie.
Interestingly, a peptide toxin selective for sodium channels, whose binding site is in the S1–S2 and S3–S4 loops of domain II (233), has been engineered to function as ER-resident pharmacological chaperone (228) (FIGURE 9). In fact, the cDNA of the β-toxin from the venom of the scorpion Centruroides sculpturatus Ewing (CsEI) was synthetized, optimizing the sequence for expression in human cells and adding a calreticulin ER targeting sequence and a KDEL ER retention motif. Its expression in cells transfected with a plasmid containing the synthetic DNA was able to rescue some folding-defective mutants, without being released in the extracellular medium and not modifying functional channels in the plasma membrane. These results showed that interaction in the ER can be sufficient for rescuing folding-defective mutants, circumventing potential unwanted side effects of classical pharmacological chaperones, and these rescuing approaches may be used for developing therapeutic strategies (24). Importantly, epileptogenic NaV1.1 mutants do not show gain-of-function changes in functional properties upon rescue (228–232). Folding-defective mutants have also been identified in familial hemiplegic migraine (43, 234) (see below). These mutants show gain of function even when partially rescued, because of the modifications of their intrinsic gating properties.
3.4. Febrile Seizures
Febrile seizures (FS) affect 2–4% of children and are defined as seizure events associated with fever, with onset after the age of 6 mo and spontaneous remission by the age of 6 yr, but without pathological or traumatic cause (235). They are classified as a distinct pathological entity rather than epilepsy. Approximately 3–8% of children with FS develop epilepsy later in life, in particular idiopathic generalized epilepsy or temporal lobe epilepsy with hippocampal sclerosis (TLE-HS), although the causal link between FS and the subsequent development of these epilepsies is still debated (235). In contrast, children in GEFS+ families present with febrile/hyperthermic seizures early in life that can continue to occur in adulthood, but they do not develop TLE-HS (215). Interestingly, these features are shared by Dravet syndrome patients, who do not develop hippocampal sclerosis, even if they have frequent febrile status epilepticus (236, 237), and by animal models carrying Scn1a epileptogenic mutations, in which there is no evident neuronal death (42, 154, 221).
A genetic predisposition has been proposed for FS, and in large, multigeneration families with presumed autosomal dominant inheritance several chromosomal loci for FS have been identified (238). The FEB3 locus contains one of the few genes for which a causal role has been proposed: SCN1A. Consistently, the SCN1A M145T missense mutation was identified in all 14 affected members of a large family showing pure FS that always ceased by the age of 6 yr, and 3 of them subsequently developed TLE-HS (239, 240). Functional studies of sodium currents in transfected tsA-201 cells have shown that M145T causes a partial loss of function, inducing 60% reduction of current density and a 10-mV positive shift of the voltage dependence of activation, consistent with hypoexcitability (240). Although it has been proposed that this family could be included in the GEFS+ spectrum (241), it fits better with the clinical features of typical FS (239), including the subsequent development of TLE-HS, which is not observed in GEFS+. Moreover, a genomewide association study (GWAS) has subsequently identified significant association between the SCN1A gene and TLE-HS that develops after a history of FS (225). However, it is not clear why SCN1A mutations that induce quantitatively similar loss of function of NaV1.1 can cause either GEFS+ or typical FS with possible development of TLE-HS.
3.5. NaV1.1 Mutations in an Extremely Severe Early Infantile Epileptic Encephalopathy
Recently, heterozygous de novo SCN1A mutations have been implicated in an additional epileptic phenotype, developmental and early infantile epileptic encephalopathy (EIEE), that is even more severe than typical SCN1A Dravet syndrome, with seizure onset between 6 and 12 wk, profound cognitive and motor impairments, and hyperkinetic movement disorder. Sequencing of SCN1A in the nine patients that were initially identified showed that eight of them had a recurrent missense mutation, T226M, and one child had the missense mutation P1345S (242). One more case, which was retrospectively thought to have a phenotype consistent with SCN1A EIEE, carried the V422L variant (243). Moreover, a subsequent study identified the S228P SCN1A mutation in a patient showing overall features that are consistent with SCN1A EIEE, but disease onset occurred in the first hours of life (244), which is different from the other SCN1A EIEE patients described. This feature is puzzling because SCN1A is thought to be expressed primarily later in postnatal development. Pathogenicity of S228P was supported by the exclusion of additional genetic variants by performing whole exome sequencing and analyses of deletions/duplication and copy number variants targeting all protein-coding exons, exon-intron boundaries, and selected noncoding clinically relevant variants (244), although pathogenic contribution by other noncoding regions or by other yet undetectable epigenetic factors cannot be excluded.
Functional studies were performed for the T226M mutation expressed in transfected Chinese hamster ovary (CHO) cells using a semisynthetic cDNA of the NaV1.1 long splicing variant, modified for reducing spontaneous mutagenesis in bacteria (245). T226M showed mixed effects on functional properties that could lead to either gain of function, because of a hyperpolarizing shift of the activation curve, or loss of function, because of a hyperpolarizing shift of the inactivation curve and faster time course of inactivation. To shed some light on the overall functional effect of T226M, which could not be directly predicted from these mixed dysfunctions, the dynamic action potential clamp method was used (246, 247). Simulations of action potential firing were generated with a “hybrid cell” based on a computational model in which the sodium conductance was obtained in real time from the whole cell recording of a CHO cell expressing either wild-type or T226M channels. Model hybrid cells with the T226M conductance displayed a negative shift in rheobase and depolarization block with cessation of action potential firing upon injection of depolarizing current stimuli that instead generated repetitive action potential firing in control cells. Similar results were obtained with traditional fully computational model neurons. The interpretation of these results was that the overall effect of T226M on NaV1.1 is consistent with gain of function at the molecular level, which causes pathological facilitation of action potential firing and depolarization block in GABAergic interneurons, leading to a dominant-negative effect at the cellular and in vivo levels. This form of depolarization block was hypothesized to produce a larger decrease of inhibition in neuronal networks than the haploinsufficiency that is typical of Dravet syndrome.
The results of these functional and cell modeling studies are controversial, because gain of function is a common effect observed for migraine NaV1.1 mutations that do not cause epilepsy (248) (see below). Moreover, the effects of T226M on NaV1.1 are very similar to those observed for other epileptogenic mutations, such as W1204R (228, 249), which causes mild epilepsy. This complicates interpretation of the data obtained with T226M. To resolve this discrepancy, functional investigation of the mutation T226M should be completed with studies in real neurons, and results should be supported by functional studies of other NaV1.1 EIEE mutations.
4. NaV1.1 AND MIGRAINE
Migraine is a highly prevalent debilitating, episodic disorder characterized by a severe and long-lasting headache that can coincide with nausea and neurological signs (250, 251). In ∼30% of the patients, headaches are preceded by transient symptoms, including visual effects, called aura. The pathophysiology of migraine is complex, and some understanding has emerged from the studies of a rare monogenic form of migraine with aura, familial hemiplegic migraine (FHM), in which the attacks are associated with hemiparesis (i.e., transient, one-sided motor weakness). It is noteworthy that the features of migraine attacks in FHM, except for the motor symptoms, are identical to common migraine with aura, and most FHM patients also have attacks of migraine without motor symptoms, suggesting similar pathophysiological mechanisms for FHM and common migraine. (250, 251). Mutations in three genes have been identified as the causes of FHM: CACNA1A, encoding the α1a-subunit of the Cav2.1 neuronal calcium channel (FHM1) (252); ATP1A2, encoding the α2-subunit of the astrocytic sodium/potassium pump (FHM2) (253); and SCN1A, encoding the Nav1.1 sodium channel (FHM3) (254). As reviewed above, SCN1A is the most frequently mutated gene in epilepsy, with hundreds of mutations identified in a range of epilepsy syndromes with different phenotypes (141). Thus far, 11 mutations in the SCN1A gene have been shown to cause FHM3, including sporadic de novo mutations in nonfamilial cases (248). They are all missense mutations, mainly clustered in domain IV of the NaV1.1 channel (FIGURE 6B).
The different types of FHM can show distinct clinical phenotypes. FHM1 and 2 are often characterized by complex phenotypes, comprising permanent cerebellar signs/ataxia and intellectual disability, as well as severe hemiplegic attacks lasting days, which are sometimes followed by loss of consciousness, coma, and in some cases even death (251). Interestingly, thus far there are no FHM3 patients with these features (248). Moreover, epilepsy has been reported as a comorbidity in FHM. However, in contrast to CACNA1A and ATP1A2 mutations, in the few FHM patients carrying SCN1A mutations who also present with seizures, hemiplegic migraine attacks are always independent from seizures, and in general the two phenotypes do not temporally overlap (248). A few patients show a further FHM3-specific comorbidity: elicited repetitive daily blindness (ERDB), which is independent from hemiplegic migraine attacks and has been reported in one of the four patients carrying the F1499L mutation in one family and in four of the five patients carrying the Q1489H mutation in a different family (255). Notably, in two additional independent FHM3 families carrying the F1499L mutation, the phenotype was pure hemiplegic migraine without ERDB (256).
Functional studies of seven mutations causing pure hemiplegic migraine or both hemiplegic migraine and epilepsy have been undertaken and have provided major insights into pathophysiological mechanisms as described below. Thus far, there are no functional studies of mutations that cause hemiplegic migraine and ERDB.
4.1. Folding-Defective FHM3 Mutants Leading to Gain of Function upon Rescue
The initial functional studies of FHM3 mutations engineered in human NaV1.1 cDNAs generated controversial results, with gain of function identified for some mutations and loss of function for others (257, 258). In particular, the mutation L1649Q, which causes pure hemiplegic migraine in a family with seven carriers, was engineered into the long NaV1.1 splice variant cDNA and did not generate quantifiable sodium current in a transfected human embryonic kidney cell line, suggesting a complete loss of function (258). In the same study, using cell surface biotinylation and immunoblot analysis, it was shown that WT and L1649Q channels had similar levels of total protein expression, but L1649Q exhibited a 10-fold lower level of cell surface expression compared with WT (258), reflecting impaired trafficking to the plasma membrane. Puzzlingly, this complete loss of function is similar to the effects of most mutations causing Dravet syndrome. A subsequent study (43) investigated L1649Q using the short Nav1.1 human splice variant, which is the predominant variant expressed in the brain (259, 260). Results under control conditions were similar to those of Ref. 258; however, this study showed that the L1649Q mutant is folding defective, as it can be rescued by incubation of the cells at 30°C, a typical feature of folding-defective mutants (261), or by coexpression with interacting proteins (43). Strikingly, L1649Q was partially functional when expressed in transfected cortical GABAergic neurons in primary culture at physiological temperature. Evidently, the neuronal cell background was sufficient to induce a partial rescue, probably because of interactions with endogenous proteins. Analysis of the gating properties of L1649Q channels showed both gain- and loss-of-function modifications that were similar in a human cell line and in neurons. Investigation of the overall effect of the mutant by means of voltage stimuli mimicking action potential discharges showed that frequency-dependent inactivation was reduced, consistent with better capacity to sustain high-frequency firing than WT. In fact, current-clamp recordings in transfected neurons showed that expression of L1649Q NaV1.1 induces hyperexcitability that is greater than with expression of WT NaV1.1. A computational model in which the effects of this mutation were implemented showed that a limited partial rescue (30% in the model) is sufficient for transforming the apparently nonfunctional L1649Q mutant into a gain-of-function mutant (43).
Interestingly, loss-of-function mutations that lead to overall gain of function upon rescue are recurrent in FHM3. The mutation L1670W, causing pure hemiplegic migraine in two unrelated families, was studied in a human cell line and in cortical neurons in culture using the short NaV1.1 human splice variant (234). The results showed that L1670W is characterized by nearly complete loss of function in transfected nonneuronal cells maintained at physiological temperature, similar to L1649Q. Incubation of the transfected cells at 30°C or transfection into cortical neurons was able to partially rescue the function of the mutant. Therefore, L1670W, similar to L1649Q, is a folding-defective mutant. Analysis of the functional effects of rescued L1670W revealed numerous modifications, in most cases consistent with destabilization of the inactivated states. In fact, the study of the overall effect of the mutation using voltage stimuli mimicking action potential discharges indicated that L1670W induces a gain of function both in a human cell line and in cortical neurons (234). The mutation L1670W was studied by another group with a synthetic NaV1.1 clone, expressed for functional analysis in a human cell line (262). Similar to Ref. 245 (see above), the synthetic clone was generated to reduce spontaneous mutagenesis in bacteria expressing plasmids that contain human NaV1.1 cDNA. In this case, it was formed with a NaV1.5 cDNA backbone in which codons were modified to obtain the NaV1.1 amino acid sequence. With this strategy, the NaV1.1 protein should be produced, but RNA processing could be different compared with that of the native NaV1.1 gene sequence. The functional effects observed were consistent with destabilization of inactivation and were similar to those observed after rescue in Ref. 234. However, in the experimental conditions of Ref. 262, L1670W was evidently already partially rescued in the cells used for functional studies, because current density was reduced less than in Ref. 234.
Overall, these mutants show nearly complete loss of function in some conditions, but they are transformed into gain of function upon rescue of folding defects. This feature is specific to FHM mutants, because epileptogenic NaV1.1 mutants can be partially rescued but do not become gain of function (see above).
4.2. Functional Studies of Other Mutations Causing FHM
Two other SCN1A mutations causing pure FHM3 have been functionally studied. The mutation L1624P was identified in a three-generation family containing five patients with FHM attacks, showing high individual clinical variability with respect to attack duration, severity, and frequency but without comorbidities. Functional studies of L1624P revealed numerous dysfunctions in general consistent with a severe destabilization of fast inactivation, leading to gain of function and neuronal hyperexcitability (263). A further mutation, F1174S, was engineered into the synthetic NaV1.1 clone outlined above, and the data obtained from expression and electrophysiological analysis in a human cell line showed results that are also consistent with destabilization of fast inactivation leading to gain of function (262).
Other functional studies have investigated mutations identified in families with FHM and epilepsy. The mutation Q1489K was the first FHM3 mutation reported and was identified in 22 carriers from three families (254). One carrier had benign focal epilepsy of childhood that remitted before the onset of FHM, and a single febrile seizure was reported in two carriers before the onset of FHM (254). The functional study of this mutation was initially performed using the cardiac NaV1.5 isoform to bypass the difficulties encountered to manipulate and express the human NaV1.1 clone. In this study, the only altered gating property was the recovery from fast inactivation, which was accelerated by the mutation (254). Two further studies analyzed the effect of this mutation by introducing it into the human NaV1.1 cDNA. One study introduced the mutation in the short human NaV1.1 splice variant and analyzed its effects in a human cell line and in neurons in primary culture (257). The other study used the long splice variant expressed in a human cell line (258). Both studies found that Q1489K exhibited numerous contrasting functional defects in voltage-clamp recordings, interpreted as preponderant loss of function in Ref. 258 and preponderant gain of function in Ref. 257. Current-clamp recordings reported in Ref. 257 showed that neurons transfected with Q1489K generated more action potentials than neurons transfected with WT NaV1.1, consistent with gain of function and hyperexcitability.
The FHM mutation L263V was identified in the only reported family in which hemiplegic migraine cosegregated with epilepsy in one branch of the family (264) but another branch showed pure hemiplegic migraine. Moreover, the same group identified a second family carrying the same mutation, in which the phenotype was pure hemiplegic migraine, consistent with polygenic inheritance in the first family (265). Functional analysis of L263V using the long Nav1.1 splice variant indicated that the mutation induces gain of function because of destabilization of both fast and slow inactivation (258). Recently, L263V has been engineered in the first FHM3 gene-targeted mouse model (266), which shows a phenotype that is consistent with migraine but does not show seizures (see below).
The heterozygous T1174S missense mutation was found in a three-generation family in which three affected individuals in a branch had febrile seizures and/or focal epilepsy whereas two additional relatives in another branch had only typical FHM3. To investigate the pathophysiological mechanisms leading to these different phenotypes in the same family, both functional studies and simulations with a computational model were performed (267). The study showed that T1174S induces divergent functional effects that can be consistent with both gain and loss of function: positive shift of the activation curve (loss of function), deceleration of the recovery from fast inactivation (loss of function), and increase of persistent sodium current (gain of function). Notably, the increase in persistent current was a labile property for this mutant in long-lasting whole cell recordings, indicating that it is most likely caused by sensitization of the mutant channel to neuromodulation by intracellular signaling pathways. Both functional and modeling data suggest that the overall functional effect of the mutation can be regulated. The increase of the persistent current may be dependent on the action of modulators that switch the effect of the mutation from loss of function (epilepsy) to gain of function (FHM). Interestingly, this mutation has been identified in another family with migraine and motor impairments (268), but it has also been identified in patients with different epileptic phenotypes, although with incomplete penetrance (269). Thus, T1174S could be a variant that needs modifiers or cofactors to be pathogenic, which could also switch its effect from mainly proepileptic to mainly promigraine.
4.3. Overall Pathophysiological Mechanisms in FHM3
The functional studies outlined above have shown that FHM3 mutations induce overall gain of function of NaV1.1 and neuronal hyperexcitability. This has allowed us to infer a possible pathophysiological mechanism. In fact, as illustrated in previous sections, NaV1.1 is expressed mainly in GABAergic inhibitory neurons, in which it has a key role in the generation of action potentials in the axon initial segment (42, 154, 158, 270). The hyperexcitability of GABAergic neurons could cause increased extracellular K+, neuronal depolarization, and initiation of cortical spreading depression (CSD), a wave of transient network hyperexcitability leading to a long-lasting depolarization block of neuronal firing that is the electrophysiological correlate of migraine aura and a proposed pathological mechanism of migraine headache (248, 257, 271) (FIGURE 10). In fact, we have shown that hyperexcitability of GABAergic neurons induced with the selective NaV1.1 enhancer Hm1a or with optogenetic stimulation triggers CSD specifically in the neocortex, because of extracellular accumulation of K+ initially generated by GABAergic neurons’ spiking, which eventually also engages glutamatergic neurons (272). Consistently, we have shown in a recent computational model that the increase of extracellular K+ generated by the spiking of a hyperexcitable GABAergic neuron can be sufficient to induce depolarizing block, the cellular correlate of CSD, of a connected pyramidal excitatory neuron (273).
FIGURE 10.
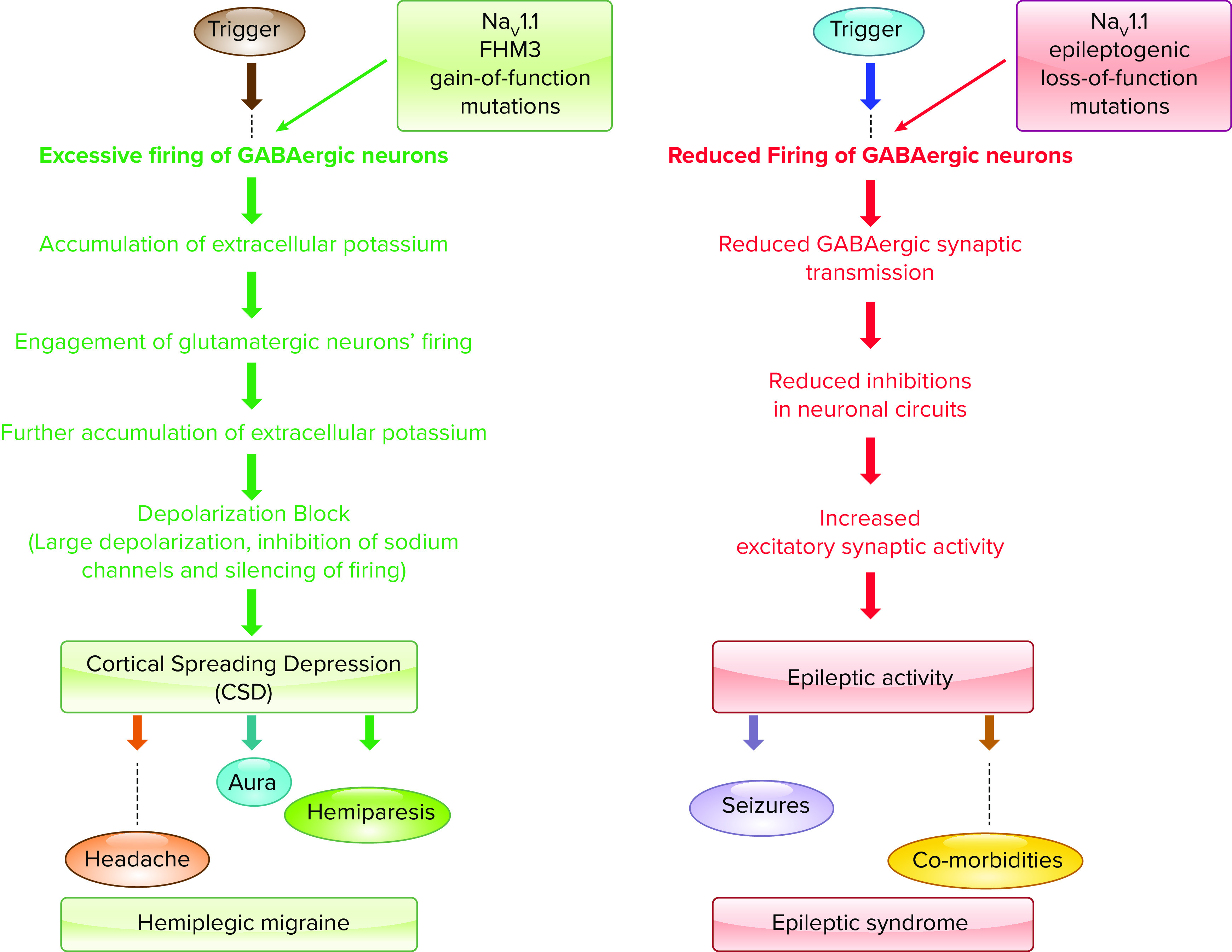
Comparison of proposed mechanisms for generation of migraine attacks by gain-of-function NaV1.1 FHM3 mutations and seizure generation by NaV1.1 epileptogenic mutations. Left: in migraine the genetic background is important for determining an intrinsic threshold for migraine attacks, which is modulated by internal and external factors (trigger stimuli). Emotional stress and minor head trauma are among the most common triggers of hemiplegic migraine attacks (250, 251). Some triggers are thought to induce excessive neuronal firing and consequently lead to extracellular K+ accumulation, which can eventually lead to long-lasting neuronal depolarization and silencing of firing caused by inactivation of sodium channels (depolarization block). These events produce cortical spreading depolarization (CSD), a wave of transient network hyperexcitability leading to a long-lasting depolarization block of neuronal firing. CSD directly causes aura, and it could also induce headache by activating trigeminal nociceptors and hemiparesis (248, 271). FHM3 NaV1.1 gain-of-function mutations can lower the triggering threshold of migraine attacks by increasing excitability of GABAergic neurons, which can induce spike-dependent accumulation of extracellular potassium that engages the spiking of glutamatergic neurons, eventually leading to depolarizing block and CSD initiation. Extracellular accumulation of the excitatory neurotransmitter glutamate plays a minor role in this proposed GABAergic neuron-dependent mechanism (272). Right: similar to migraine, it is hypothesized that epileptogenic mutations lower the threshold for seizure generation, although seizure triggers are less clearly identified than migraine triggers (248). Epileptogenic NaV1.1 loss-of-function mutations lower seizure threshold by reducing firing of GABAergic neurons, which leads to reduced GABAergic synaptic transmission and reduced inhibition in neuronal circuits, which can lead to generation of epileptic activity. Comorbidities could be generated by both seizures and the direct effect of the mutation (140, 141, 172).
It is worthwhile to emphasize that pathophysiological mechanisms of epilepsy and migraine can share some similarities, because hyperexcitability of cortical neuronal networks can be involved in both these diseases. In particular, as reported above, it has been proposed that some FHM3 mutations, such as L263V, can cause both hemiplegic migraine and epilepsy in the same family, although there is no evidence of association between migraine attacks and seizures (248). The first FHM3 mouse model, a knockin that expresses the L263V Scn1a mutation, has been recently reported (266). Interestingly, these mice show spontaneous CSD events as well as facilitation of CSD induction with electrical stimulations, recapitulating what has been shown in patients having migraine with aura. No seizure-related or other abnormal behaviors were observed, and electrographic recordings did not reveal epileptic activity during a total recording time of 2,228 h in 24 animals. Thus, the model replicates a migraine phenotype without epileptic comorbidity, and the pathophysiological mechanism is consistent with facilitation of CSD initiation. Notably, this is different in comparison with other FHM models; for example, knockin mice carrying the S218L FHM1 mutation of CaV2.1 calcium channels, which exhibit facilitation of CSD induction and spontaneous generalized tonic-clonic seizures (274). A feature of L263V Scn1a mice that is not consistent with the human FHM3 phenotype is the high mortality rate, with only ∼10% of heterozygous mutant mice surviving to the age of P60. Different from epileptic Scn1a models, maintaining the mutation in either the genetic background of pure C57BL/6J or mixed 50:50 C57BL/6J-129/SvJ had no significant impact on survival of L263V Scn1a mice.
5. NaV1.1 AND AUTISM SPECTRUM DISORDER
Autism spectrum disorders (ASD) are a group of neurodevelopmental disorders that are defined by impairments in social interactions along with restricted and repetitive behaviors. High-throughput sequencing has implicated numerous genes in the autism spectrum (275–277), including SCN1A discussed here and SCN2A discussed below. These studies included sequencing of “trios,” that is, a set of DNAs from two parents, an unaffected sibling, and an affected individual. This approach identified de novo mutations in NaV1.1 in the affected individuals that are not present in parents or siblings and therefore are causative for autism. Consistent with these genetic results, behavioral studies of Dravet syndrome patients with SCN1A loss-of-function mutations (278–280) revealed autistic features in a large fraction of affected individuals. Moreover, autistic-like behaviors, including social interaction deficit and repetitive behaviors, were observed in Dravet syndrome Scn1a+/− mice analyzed under carefully controlled laboratory conditions (152, 173). Because Dravet syndrome is caused by loss-of-function mutations in NaV1.1 channels, which cause specific loss of excitability in GABAergic interneurons, it is likely that these autistic behaviors result from impaired firing of interneurons. Direct evidence for this conclusion came from studies of mice with NaV1.1 channels specifically deleted in forebrain GABAergic inhibitory neurons (152), which exhibited both impaired social interactions and repetitive behaviors that could be rescued by treatment with low-dose benzodiazepines that enhance the postsynaptic response to GABA.
6. NaV1.2 AND EPILEPSY
6.1. Functions of NaV1.2 Channels in Neurons
Early studies showed that NaV1.1 and NaV1.2 channels are differentially localized in neurons in the hippocampus: NaV1.1 in the cell bodies of excitatory and inhibitory neurons versus NaV1.2 channels in unmyelinated axons and dendrites of excitatory neurons (133, 135). The Nav1.2 sodium channel is widely expressed in the central nervous system, particularly in cortical and hippocampal glutamatergic pyramidal cells (281, 282). In myelinated axons, Na+ channels are clustered at high density at the nodes of Ranvier to allow saltatory conduction. NaV1.2 channels are the main channels in nodes of Ranvier during early postnatal development and are largely replaced by Nav1.6 channels during development of maturing nodes of Ranvier (283, 284). Moreover, a high density of Na+ channels is present at the axonal initial segment (AIS), which is, for this reason, the primary site for generation of action potentials in neurons (136, 285, 286). Similar to nodes of Ranvier, in the first days of rodents’ postnatal development, Nav1.2 is the main sodium channel of the AIS; after the 10th day of postnatal life, it is partially replaced by Nav1.6, which becomes the main sodium channel in the distal part of the AIS, whereas Nav1.2 is segregated in the proximal part (136, 282, 287). This separation can be functionally important, as it has been proposed that Nav1.6 channels in the distal AIS promote action potential initiation whereas Nav1.2 channels in the proximal AIS promote backpropagation to the soma (281). In adult rodents, NaV1.2 is still present in thin processes, presumably distal unmyelinated portions of preterminal axons (288).
In these cellular contexts, NaV1.2 channels are in position to drive action potential generation and backpropagation and possibly contribute to epilepsy. It was surprising to discover NaV1.2 channels in high density in dendrites of hippocampal neurons in early immunocytochemical studies (135). Later work revealed backpropagating action potentials moving from cell bodies into many types of dendrites (e.g., Ref. 289). Moreover, recent work with heterozygous NaV1.2-knockout mice has shown that these channels are crucial for backpropagating action potentials in dendrites of prefrontal cortex pyramidal neurons (290). Backpropagating action potentials are thought to distribute and integrate action potential generation and calcium signaling in the large dendrites of cortical pyramidal neurons (291). In this cellular context, recent work has shown that NaV1.2 channels are crucial in a spectrum of diseases, including epilepsy, autism, and other neuropsychiatric disorders (FIGURE 11A).
FIGURE 11.
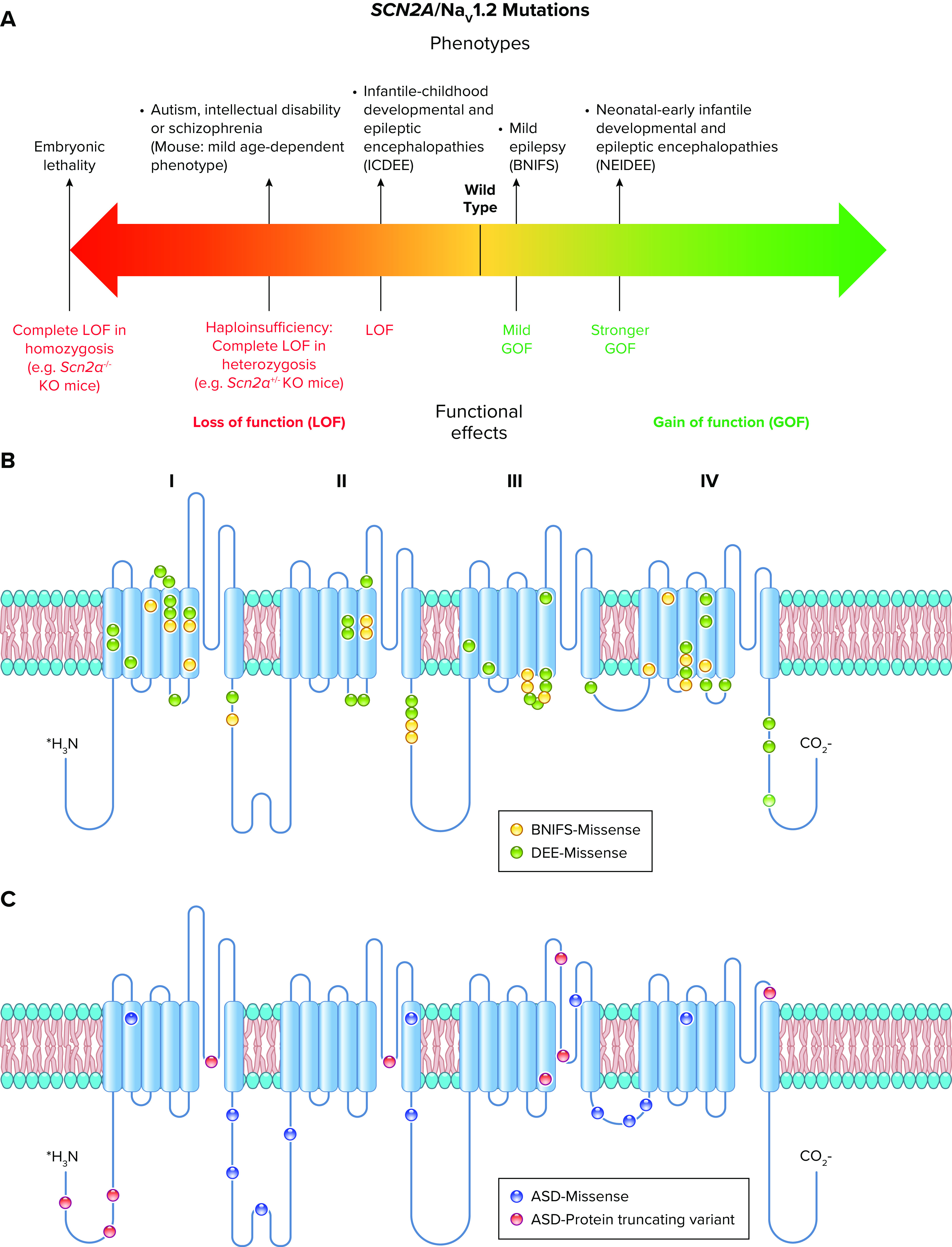
Spectrum of mutations and phenotypes for SCN2A/NaV1.2. A: SCN2A/NaV1.2 mutations inducing mild gain of function cause benign neonatal-infantile familial seizures (BNIFS) with onset between 3 and 6 mo of age. Neonatal-early infantile developmental and epileptic encephalopathies (NEIDEE) with onset before 3 mo are in general caused by mutations that induce larger gain of function. Mutations inducing loss of function cause infantile-childhood developmental and epileptic encephalopathies (ICDEE), with onset after 3 mo. Complete loss of function in heterozygosis (haploinsufficiency) can lead to behavioral/cognitive phenotypes without epilepsy: autism spectrum disorders (ASD), intellectual disability, or schizophrenia. Haploinsufficiency in Scn2a+/− mice causes a relatively mild and age-dependent phenotype including short absence-like seizures, autistic/schizophrenic traits, and memory dysfunctions. Homozygote Scn2a−/− knockout (KO) mice show embryonic mortality. B: molecular map of the NaV1.2 sodium channel with the location of 19 missense SCN2A mutations in BNIFS (yellow) and 49 missense SCN2A mutations in developmental and epileptic encephalopathy (DEE: both ICDEE and NEIDEE) (green). C: molecular map of the NaV1.2 sodium channel with the location of 12 missense (red) and 9 protein-truncating (blue) SCN2A variants identified in ASD cases.
The possible involvement of NaV1.2 mutations in epilepsy was initially proposed in 2001, with the identification of the variant R187W in a family in which the proband developed complex partial seizures after an initial diagnosis of FS plus (FS+) (292). However, the small pedigree did not show clear cosegregation with the disease phenotype, and there was bilineal inheritance of epilepsy in the family, so it is possible that there are other genes implicated in the phenotype of this patient (293).
6.2. Nav1.2 Channels in Benign Familial Neonatal/Infantile Seizures
The first epileptic syndrome clearly associated with mutations of SCN2A was benign familial neonatal/infantile seizures (BFNIS) (294, 295). This autosomal dominant disorder is caused by missense mutations with high penetrance and is characterized by afebrile seizures with onset between 3 and 6 mo of age and spontaneous remission within the first year of life, without subsequent neurological deficits. BFNIS is a relatively mild syndrome within the spectrum of NaV1.2 channelopathies (FIGURE 11, A and B). After the initial studies, several other missense mutations that map broadly across the channel structure have been identified in SCN2A that are responsible for BFNIS, further confirming BFNIS as a NaV1.2 channelopathy (FIGURE 11B) (296–298).
The first functional analysis of NaV1.2 BFNIS mutations was performed by Scalmani et al. in 2006 (299). Four BFNIS mutations, L1330F, L1563V, R223Q, and R1319Q, were studied with the rat Nav1.2 isoform and transfected pyramidal and bipolar-shaped (GABAergic) neocortical neurons in primary culture. Classical voltage-clamp experiments showed gain-of-function effects for L1330F and L1563V, with mild modifications of gating properties, whereas mixed gain- and loss-of-function effects were observed for R223Q and R1319Q. Action potential-clamp experiments showed that R223Q and R1319Q induce an increase of both subthreshold and action sodium currents. Thus, the overall functional effect was consistent with neuronal hyperexcitability for all four mutations studied.
A subsequent study challenged these results, investigating three of these mutations (R1319Q, L1330F, and L1563V) using the human adult NaV1.2 isoform expressed in a human cell line (300). This study reported a reduction in current density and cell surface expression (quantified with biotinylation), together with mild and contrasting alterations of gating properties. Overall, the functional outcome was consistent with mild loss of function, opposite to the first study of these mutations.
However, further studies confirmed that BFNIS mutations cause gain of function of NaV1.2. These investigations have often been performed comparing effects in “neonatal” and “adult” human NaV1.2 splice variants (301, 302), because BFNIS onset is in the first months of life. Gain of function has been observed for the L1563V BFNIS mutation (303), studied with both neonatal and adult splice variants expressed in a human cell line. This study showed that the adult WT variant can sustain increased excitability compared with the neonatal WT variant, because of mild modifications of gating properties. Notably, L1563V did not induce significant functional effects in the adult isoform, but it caused the neonatal isoform to behave like the adult isoform, consistent with gain of function and increased neuronal excitability. However, a subsequent study from the same group of the L1563V mutant expressed in Chinese hamster ovary (CHO) cells (246) reported effects in the adult variant that were similar to those previously observed with the neonatal isoform (303). In this study, simulations obtained with the dynamic action potential clamp technique (see above) predicted that L1563V would induce mild neuronal hyperexcitability also when inserted into the adult variant. Moreover, a further recent study confirmed a gain of function for L1563V engineered in the adult variant expressed in a human cell line (304). Thus, L1563V can induce mild gain of function in both neonatal and adult splice variants.
Another study compared the effect of two BFNIS mutations, M252V and V261M, on human neonatal and adult NaV1.2 variants transiently expressed in a human cell line (282), showing that both mutations induce gain of function by altering different gating properties. Functional effects were observed in both neonatal and adult variants for V261M but only in the neonatal variant for M252V. Further studies investigated the BFNIS mutations Y1589C (305), V280E, and K908E (306) using the human adult NaV1.2 variant transiently expressed in a human cell line, showing an overall gain of function for all mutations due to modifications of several gating properties.
Therefore, the common functional effect of BFNIS mutations is a mild gain of function consistent with hyperexcitability of excitatory neurons. Functional studies have shown that the major factor for the transient generation of seizures limited to the first months of life is probably the age-dependent differential expression and localization of NaV1.2 channels in neuronal subcompartments, rather than specific effects of the mutations on the NaV1.2 neonatal splice variant.
6.3. Nav1.2 Channels in Developmental and Epileptic Encephalopathies
Since the initial identification of SCN2A mutations in BFNIS (295), the phenotypic spectrum of epilepsies caused by SCN2A mutations has expanded considerably, in particular including severe developmental and epileptic encephalopathies (DEE) (307–309) (FIGURE 11A). Similar to BFNIS, these mutations map broadly across the NaV1.2 channel structure (FIGURE 11B)
The first NaV1.2 mutation reported in a severe epilepsy was the de novo heterozygous truncation R102X, identified in a patient showing pharmacoresistant seizures with onset at 1 yr and 7 mo of age, severe intellectual and psychomotor retardation, and autistic traits (304, 310). R102X causes a large truncation, sparing just a cytoplasmic NH2-terminal fragment of the NaV1.2 protein and inducing complete loss of function, as confirmed by more recent work of the same group that compared features of knockin mice expressing the R102X mutation to Scn2a+/− knockout mice (288).
After the identification of R102X, other SCN2A mutations have been identified that cause infantile and childhood DEE (ICDEE), representing ∼40% of the total SCN2A DEE cases (308, 309) (FIGURE 11A). Phenotypes of SCN2A ICDEE patients can be correlated to the age of onset. Patients with onset between 3 mo and 1 yr of age in general show a West syndrome phenotype (infantile spasms as seizure type, EEG with hypsarrhythmia, and developmental regression), with possible evolution into a Lennox–Gastaut phenotype (multiple seizure types, EEG with diffuse spike-and-wave and paroxysmal fast activity, and intellectual disability). SCN2A ICDEE patients with onset at >1 yr of age often show variable seizure phenotypes that cannot be classified as an established epileptic syndrome, including tonic-clonic, myoclonic, and absence seizures, with developmental/cognitive delay and autistic traits that can appear before seizure onset.
Notably, ∼60% of SCN2A DEE have onset in the first 3 mo of life, mostly in the neonatal period (308, 309). These neonatal-early infantile DEE (NEIDEE) patients show intellectual disability in all the cases. About 50% of them have unclassifiable epileptic phenotypes with variable seizure types, whereas the others have phenotypes that fit the features of two epileptic syndromes: Ohtahara syndrome (neonatal-onset spasms or tonic seizures and EEG with burst suppression pattern, sometimes evolving into West syndrome and/or Lennox–Gastaut phenotypes) or epilepsy with early infantile migrating focal seizures (EIMFS; multiple types of focal seizures that migrate from one hemisphere to the other). SCN2A DEE mutations generally arise de novo, and ∼80% are missense (FIGURE 11B). However, a few mutations are recurrent in different patients, for example, A263V, R853Q, and L1342P, which cause ICDEE, and E999K and R1882Q, which cause NEIDEE (309, 311).
The first functional study of a SCN2A NEIDEE mutation (I1473M) appeared controversial because it showed a net gain of function (312), which is opposite to the effect of the original R102X ICDEE mutation (310). However, more recent studies have delineated a clearer correlation between gain-of-function effects and clinical phenotypes (313). Functional studies of the A263V mutation, identified in a patient with neonatal-onset seizures and childhood-onset episodic ataxia, provided additional evidence that SCN2A mutations involved in neonatal/early infantile seizures induce gain of function (314). In fact, A263V expressed in a human cell line induced a prominent gain of function in both neonatal and adult NaV1.2 variants because of several modifications of gating properties that are consistent with destabilized inactivation. A subsequent report confirmed these results, identifying SCN2A mutations in other patients with neonatal-onset seizures and ataxia: the same mutation reported before, A263V, in one patient; G1522A in a second patient; and the combination of G1522A and R1882G in the third patient. All of these mutations induced gain of function with increased persistent current, negative shifts of the voltage dependence of activation, or positive shifts of inactivation (315). The clinical spectrum of these patients, and of others subsequently identified, is consistent with a milder NEIDEE characterized by childhood ataxia (316). Moreover, a report including data from 201 patients shed further light on genotype-phenotype relationships of DEE SCN2A mutations and suggested therapeutic implications (309). This work showed that truncating mutations, which are likely to cause loss of function, are found only in ICDEE, in general when onset is after 2 yr of life. Consistent with this, antiepileptic sodium channel blockers are often effective in NEIDEE but not in ICDEE, in which they often induce phenotype worsening. These findings are consistent with NaV1.2 gain of function in NEIDEE and loss of function in ICDEE, a dichotomy that was supported in this study by the functional analysis of four mutations. In fact, two NEIDEE mutations showed gain of function because of large increase of persistent current (V423L) or accelerated recovery from fast inactivation (F1597L), whereas two ICFEE mutations showed loss of function because of a large negative shift of the voltage dependence of inactivation and faster current decay (P1622S) or a positive shift of the voltage dependence of activation (G899S).
Two further studies (246, 317) confirmed these results, comparing two recurrent mutations and showing gain of function for R1882Q, implicated in NEIDEE, and loss of function for R853Q, implicated in ICDEE. R1882Q impaired fast inactivation and increased persistent sodium current (317). In contrast, R853Q reduced peak sodium current, enhanced fast inactivation, and reduced persistent sodium current. In Xenopus oocytes, the mutation R853Q, which is located in the S4 segment of domain II, reduced functional expression and induced a substantial inward gating-pore current at membrane potentials negative to –30 mV. This is an unusual functional effect, but it cannot be generalized to other ICDEE mutations, and its influence on the overall effect of the R853Q mutation is not clear. The second study used dynamic action potential clamp methods (246), predicting a dramatic increase in firing for R1882Q and a marked reduction in firing for R853Q. Altogether, these results strongly support the conclusion that loss-of-function mutations primarily cause ICDEE whereas gain-of-function mutations primarily cause NEIDEE.
Separation of mutations that induce the same type of functional effect (gain vs. loss of function) but cause phenotypes of different severity can have substantial clinical relevance, in particular for genetic counseling and for orienting therapies. Toward that goal, an “electrophysiological index” was developed for correlating functional effects and clinical severity based on parameters obtained from classic voltage-clamp experiments and was used to evaluate 3 novel and 21 previously characterized gain-of-function NaV1.2 mutations (306). This electrophysiological index correlated well on average with clinical severity, but there were overlaps between some single mutations that did not allow consistent predictions of phenotypic severity based only on their functional properties analyzed by voltage-clamp recording in vitro. These potentially important tools should be further developed and refined in future studies.
A recent study extended the comparison of the functional effects observed in the neonatal and adult splicing variants to five NEIDEE mutations (318). This comparison had already been attempted for BFNIS mutations with controversial results and for the A263V NEIDEE mutation without observing differential effects (314) (see above). The five mutations (T236S, E999K, S1336Y, T1623N, and R1882Q) were identified in patients that had seizure onset in the first days of life. All mutations, expressed in a human cell line, exhibited gain-of-function modifications consistent with enhanced neuronal excitability. Only three of the five mutations (T236S, E999K, and S1336Y) exhibited greater dysfunctions in the neonatal variant compared with those observed in the adult variant. Therefore, these studies showed that a larger effect in the neonatal variant is not a general property of mutations involved in NEIDEE with neonatal onset, similar to BFNIS mutations.
Overall, the consensus based on these extensive functional studies is that ICDEE mutations cause loss of function and that NEIDEE mutations cause gain of function, which is in general more pronounced than for BFNIS mutations.
7. NaV1.2: AUTISM SPECTRUM DISORDER, INTELLECTUAL DISABILITY, AND SCHIZOPHRENIA
Interestingly, SCN2A mutations have also been identified in patients with neurodevelopmental dysfunctions, whose phenotypes include autism spectrum disorders (ASD) and/or intellectual disability (ID), as well as other neuropsychiatric phenotypes such as schizophrenia (307–309) (FIGURE 11A). These mutations map broadly across the structure of NaV1.2 (FIGURE 11C). Notably, SCN2A mutations have been reported as among the most frequent de novo mutations identified in ASD by genome sequencing of trios (275–277, 307), even though their frequency is probably underestimated because many more epileptic patients are screened for SCN2A mutations compared with ASD/ID patients (307). Although seizures have been observed in some of these patients after the onset of neurodevelopmental dysfunction, epilepsy is not a major feature of their phenotype.
7.1. Nav1.2 Channels and Autism Spectrum Disorder
Functional analysis of 10 SCN2A mutations identified in ASD patients without seizure was performed using the adult splicing variant of NaV1.2 expressed in a human cell line (311). Two nonsense mutants (C959X and G1013X) and one frameshift mutant (S686fs), which generate truncated NaV1.2 proteins, as well as four missense mutants (R379H, R937H, R937C, and C1386R) caused complete loss of NaV1.2 activity. Three additional missense mutants (D12N, D82G, and T1420M) induced partial loss of function because of reduction of current amplitude and/or modifications of gating properties. Altogether, the data demonstrated that the most common effect of ASD-related missense and frameshift mutations was loss of channel function. The missense mutants often caused complete block of ion conduction, although three of these mutants were conductive and showed only partial loss of function. A computational model predicted that all of the mutations studied would induce a deficit of neuronal excitability. Considering the expression pattern and distribution of NaV1.2 channels during development, the authors suggested that these defects in neuronal excitability would result in a persistent change in neuronal circuit function, which together with neurodevelopment changes could result in induction of permanent ASD behaviors. Mouse models have been used to test this hypothesis (see below), although those available thus far are knockouts that do not carry a mutation identified in patients.
7.2. Nav1.2 Channels and Intellectual Disability
De novo heterozygous mutations of SCN2A, identified by exome sequencing studies in patients with sporadic ID without seizures, were functionally evaluated in a recent study using the human adult NaV1.2 variant transiently expressed in a human cell line. The three ID mutations were the frameshift L611Vfs*35 (319, 320) and the stop codon W1716X (309), which both induce a large truncation, and the missense R937C (320). Their effects were compared with those of the NEIDEE mutation E1803G (321), the ICDEE mutation L1342P (322), and the BFNIS mutation L1563V. ID mutations showed complete loss of function because they did not produce measurable currents, similarly to most of the ASD mutants investigated in Ref. 309 (see above). The NEIDEE mutant E1803G had a shallower slope of the voltage dependence of inactivation, which induced a partial positive shift and an increase of the window current, both gains of function consistent with the effect of other NEIDEE mutants. The ICDEE mutation L1342P induced contrasting modifications of gating properties, which can lead to an overall loss of function, similar to most of the ICDEE mutations studied. A mild gain of function was confirmed for the BFNIS L1563V mutation. These findings confirm the results obtained in other functional studies of NaV1.2 mutations involved in epilepsy and support the hypothesis that complete loss of function usually leads to ID or ASD without epilepsy; however, it is not clear yet why some complete loss-of-function mutations cause ICDEE and not ID or ASD without epilepsy (308, 309).
7.3. NaV1.2 Channels and Schizophrenia
Schizophrenia is a neurodevelopmental disorder whose pathophysiology is largely unknown, characterized by incoherent or illogical thoughts, abnormal behavior, and altered perception, with abnormalities of brain function. The first report that showed possible involvement of SCN2A variants in schizophrenia was a genomewide association study (GWAS) of 860 schizophrenic patients and 510 control subjects that revealed a significant association of general cognitive ability with polymorphisms in SCN2A and thereby implicated the NaV1.2 channel in the pathophysiology of cognitive impairments in schizophrenia (323). The same study showed reduced expression of SCN2A mRNA in postmortem prefrontal cortex tissue samples from schizophrenic patients compared with control subjects. Subsequent gene-sequencing studies have identified rare SCN2A variants in schizophrenic patients, including the missense mutation R850P discovered by exome sequencing of 623 schizophrenia trios (324), the missense mutation V1282F discovered in two cases, and the truncating mutation E169X discovered by sequencing SCN2A in 980 schizophrenic patients (325). The two missense mutations have uncertain functional effects, whereas the large truncation is predicted to cause complete loss of function. Functional studies are warranted in order to clarify better the genotype-phenotype relationship of these mutations and to gain insight into the specific pathophysiological mechanism underlying this neurodevelopmental disorder compared with ASD and ID.
8. MOUSE GENETIC MODELS OF NaV1.2 CHANNELOPATHIES
Although studies using heterologous expression systems are essential for screening the functional effects of numerous mutations and have given insights into the alterations of NaV1.2 channel function caused by SCN2A mutations, gene-targeted mouse models carrying human variants are important tools for investigating the mechanisms of SCN2A mutations in different subtypes of neurons, disclosing their effects on neuronal networks, and linking them to behavioral dysfunctions. For instance, as outlined above, the neuropathogenic mechanisms of mutations of NaV1.1 have emerged from studies of a gene-targeted mouse model, showing that NaV1.1 is the major sodium channel in inhibitory neurons and thus providing an explanation for the network disinhibition in patients with loss-of-function NaV1.1 mutations in Dravet syndrome and other diseases (42, 172). The mouse models of SCN2A mutations studied extensively thus far either delete Scn2a or express a truncated nonfunctional form of NaV1.2.
The first NaV1.2 mouse model was generated >20 yr ago, before the identification of the first human pathogenic SCN2A mutation, in order to obtain insights into the physiological functions of NaV1.2 (326). It is a gene knockout that completely blocks NaV1.2 expression by deletion of the first Scn2a exon. These global homozygous knockout mice (Scn2a−/−) showed perinatal death and therefore suggested a crucial role of NaV1.2 for the nervous system, despite possible compensatory effects of other sodium channels. In contrast, heterozygous Scn2a+/− mice did not show an overt phenotype, even though hippocampal pyramidal neurons dissociated from P5–P9 Scn2a+/− mice had 50% reduction of sodium current. Later follow-up studies did not detect convulsive seizures in Scn2a+/− mice (327). However, several papers based on this original mouse line and other lines more recently developed do indeed show epileptic features. A recent study (288) compared the Scn2a+/− global knockout mouse developed in Ref. 326 with two other Scn2a mouse lines: a knockin mouse model carrying the nonsense R102X SCN2A mutation that causes ICDEE (310) (see above) and conditional mouse lines with a Scn2a deletion in specific brain areas and cell types. The results showed that adult mice with both Scn2a+/− global knockout and Scn2a+/R102X genotypes exhibit a mild epileptic phenotype of short absence-like seizures (FIGURE 12A), which are associa ted with spike-and-wave discharges (SWDs) in EEG recordings and are sensitive to the anti-absence seizure drug ethosuximide. This phenotype was more prominent in heterozygous conditional mice in which Scn2a was deleted specifically in dorsal-telencephalic excitatory neurons, whereas seizures were not detected in mice with selective Scn2a deletion in inhibitory neurons. A follow-up study showed that heterozygous Scn2a deletion in corticostriatal but not corticothalamic neurons was sufficient for the generation of SWDs, suggesting that impaired cortico-striatal excitatory transmission is a possible mechanism for absence-like seizures in this model (330).
FIGURE 12.
Phenotypes of heterozygous Scn2a knockout (Scn2a+/−) mice. A: absence-like seizures with spike and wave discharges (SWDs) associated with electromyogram (EMG) suppression are observed in 10- to 27-wk-old Scn2a+/− mice, as shown by electrocorticogram and multisite local field potential (ECoG-LFP) recordings, which reveal the predominant appearance of LFP epileptiform discharges in medial prefrontal cortex (mPFC) and caudate putamen (CPu). Black arrowheads indicate the onset of SWD. Scale bars: vertical, 500 µV; horizontal, 1 s. Modified from Ref. 288 with permission from Communications Biology. B: stereotyped behaviors consistent with autistic-like traits are present in young (a–c) but not adult (d–f) Scn2a+/− (HZ) mice. WT, wild type. a and d: Time spent in self-grooming. b and e: Number of marbles buried. c and f: Persistence in repetitive rotarod trials, which evaluate repetitive motor behaviors. These stereotyped behaviors are not observed in adult [postnatal day (P)60–P95] mice. Modified from Ref. 328 with permission from Scientific Reports. C: impaired somatic excitability. a: Severe impairment in developing cortical pyramidal neurons from P4–P7 Scn2a+/− mice. Morphology of a developing pyramidal cell (left) and action potential discharges (right). b: Moderate impairment in mature neurons from adult Scn2a+/− mice (>P60). Morphology of a mature, thick-tufted pyramidal cell (left) and action potential discharges (right). Modified from Ref. 290 with permission from Neuron. D: backpropagating dendritic excitability is impaired in mature cortical layer V pyramidal neurons of Scn2a+/− mice. Calcium transients evoked by trains of action potential duplets were recorded at various locations throughout the apical dendrite. In WT neurons duplets reliably evoke calcium transients throughout the apical dendrite, whereas calcium transients in Scn2a+/− neurons rapidly diminish in amplitude with increasing distance from the soma, becoming virtually absent in the most distal dendritic branches. G/Gsat, relative fluorescence intensity; Vm, membrane voltage; Iinj, injected current. Modified from Ref. 290 with permission from Neuron. E: decreased long-term potentiation (LTP) induced by theta burst stimulation (10 trains of 4 pulses at 100 Hz) at hippocampal Schaffer collaterals-CA1 synapses of 3-mo-old floxed Scn2a+/− mice (HT; red) compared with WT littermates. fEPSP, field excitatory postsynaptic potential. Modified from Ref. 329 with permission from Frontiers in Molecular Neuroscience.
Other recent studies of Scn2a mouse lines focused on behavioral and cognitive alterations as well as cellular and local network defects. Behavioral and cognitive alterations identified in adult (>P60) Scn2a+/− global knockout mice are quite mild. One study showed novelty-induced exploratory hyperactivity, decreased anxiety, and quite mild dysfunctions of social behavior (330), which may be interpreted as autistic- and/or schizophrenic-like features, whereas another study showed only trends toward dysfunctions in social behavior (290). Similarly, social behavior was not modified in a novel global heterozygous conditional floxed Scn2a-knockout mouse line (329), in which social approach, social communication, and repetitive behaviors were investigated. Tests of cognitive features showed enhanced fear memory in two studies (329, 330), one of which also showed mildly impaired spatial learning and memory in the Morris water maze (329), and a third study showed only trends toward dysfunctions in a few other learning tasks (290). Spatial learning dysfunctions were studied in detail in another study (331). Slowed hippocampus-related spatial learning and abnormalities in hippocampal replay were observed, including decreased cell assembly reactivation strengths and truncated hippocampal replay sequences, which may be involved in consolidation of spatial memories. In contrast, no changes to single place cells or cell assemblies were observed during encoding of spatial information.
Two studies analyzed autistic features in newborn and juvenile (up to P21) mice, including ultrasonic vocalizations, but these investigations did not show abnormalities (329, 330). In contrast with the very mild behavioral dysfunctions observed in adult mice and the absence of them in newborn/juvenile mice, a recent study (328) identified more evident autistic features in young (P22–P45) global Scn2a+/− mice. Social communication quantified with ultrasonic vocalizations was strongly reduced, impairment in social interactions was very mild, and several motor stereotypies were observed (FIGURE 12B). These young Scn2a+/− mice also showed decreased memory performance in the novel object recognition test and a trend in the Y maze test. Moreover, they were less anxious and resigned than WT littermates. Consistent with the other studies, adult Scn2a+/− mice (>P60) showed a much milder phenotype, because only social communication was significantly reduced. Thus, behavioral-cognitive phenotypes are more prominent in young global Scn2a+/− mice than in adults.
Two studies that performed behavioral tests also investigated cellular/local network defects in the prefrontal cortex (290), which may be particularly important for autistic and schizophrenic features, and in the CA1 area of the hippocampus (329), which may be particularly important for memory dysfunction. Investigations in prefrontal cortex brain slices of global Scn2a+/− mice (290) showed, as expected, reduced excitability of cortical layer V pyramidal neurons across developmental periods, including reduced action potential threshold and maximal firing frequency in neonatal neurons and reduced action potential slope after P20 (FIGURE 12C). In contrast, excitability of cortical parvalbumin- and somatostatin-positive GABAergic neurons, as well as excitability of pyramidal neurons in the CA1 area of the hippocampus, were not altered in neurons from P34–P40 animals. Surprisingly, backpropagating action potentials in mature neurons were more attenuated in distal dendrites than in the soma, because NaV1.2 haploinsufficiency severely impaired dendritic excitability (FIGURE 12D). Moreover, in juvenile (P27) mice, but not in neonates, both the frequency of miniature excitatory postsynaptic currents (mEPSCs) and the AMPA-to-NMDA ratio of stimulated synaptic currents were reduced and dendritic spine morphology was altered, consistent with a pathological remodeling of synaptic properties. In contrast, no modifications of miniature inhibitory postsynaptic currents (mIPSCs) were observed.
To determine whether these defects are caused by neurodevelopmental dysfunctions induced in the neonatal period or directly by NaV1.2 haploinsufficiency, a conditional floxed Scn2a-knockout line was crossed with a (CaMKIIa)-Cre line, which expresses Cre recombinase in neocortical pyramidal cells only after P10 (290). These mice, which are haploinsufficient for NaV1.2 after P10, show the same dysfunctions observed in global Scn2a+/− mice, ruling out the involvement of early neurodevelopmental dysfunctions in causing phenotypes later in life. Moreover, injections of low-titer Cre-expressing adeno-associated virus at P28 were used to obtain sparse NaV1.2-haploinsufficient cortical neurons. These neurons showed reduced action potential slope, AMPA-to-NMDA ratio, and long-term potentiation (LTP), as well as small alterations of spine morphology. Thus, network effects were not necessary for generating the dysfunctions observed in these experiments.
Recordings in the CA1 area of the hippocampus of brain slices obtained from 3-wk-old floxed global Scn2a+/− mice (329) showed some differences in comparison with the data obtained in the prefrontal cortex (290). In fact, AMPA-to-NMDA ratio and basal evoked synaptic transmission at Schaffer collateral-CA1 synapses were not modified, and there was a reduction of spontaneous excitatory postsynaptic currents (sEPSCs), which depend on network excitability, but not of mEPSCs, which depend on synaptic properties. This is consistent with lack of modification of functional properties or number of synapses. Nevertheless, LTP induced by high-frequency stimulation was reduced (FIGURE 12E), although long-term depression (LTD) was not modified. Excitability of CA1 pyramidal neurons showed quite mild, if any, impairment, and inhibitory synaptic transmission was not modified.
Overall, these studies show that Scn2a+/− mice have a relatively mild phenotype, including short absence-like seizures, spatial memory deficits but enhanced fear memory, plus autistic and possibly schizophrenic traits. Some features are age dependent; in particular, autistic traits are more prominent in young mice and tend to remit in adults. Moreover, recordings in brain slices of these models have shown that NaV1.2, besides its established axonal role, has important dendritic functions in pyramidal neurons of the prefrontal cortex, where its haploinsufficiency impairs synaptic plasticity and synaptic strength, even when NaV1.2 expression is reduced in single neurons late in postnatal development. Notably, NaV1.2 haploinsufficiency may lead to differential synaptic remodeling in the hippocampus compared with the prefrontal cortex. It would be interesting to investigate whether developmental reversal of cellular/network dysfunctions may be related to the strong attenuation of autistic-like behaviors in adult mice.
In summary, it is remarkable that studies to date reveal mutations of SCN2A/Nav1.2 that cause a wide range of neurodevelopmental disorders, including epilepsy of varying severity with neonatal, infantile, or childhood onset, ASD, ID, and possibly schizophrenia. The first line of functional studies gives some explanations about genotype-phenotype relationships: gain-of-function mutations are related to mild benign neonatal/infantile epilepsy and to neonatal/early infantile epileptic encephalopathy, whereas loss-of-function mutations are linked to infantile/childhood epileptic encephalopathy or neurodevelopmental disorders like autism and intellectual disability without seizures. However, it is more difficult to establish genotype-phenotype relationships within categories of these gain-of-function or loss-of-function mutants, because functional effects of mutations causing different phenotypes often overlap. Different from Scn1a models, the available Scn2a mouse models show much milder phenotypes than patients, who have more prominent autistic traits, cognitive deficits, and epilepsy. In particular, haploinsufficient knockout models show only mild absence seizures, whereas most patients have more severe epilepsy and there are only a few patients who show absence seizures alone. More studies are now required to shed light on detailed pathophysiological mechanisms specific for each disorder within the SCN2A spectrum. The development of further Scn2a mouse models carrying different types of human mutations may help to unravel in more detail the pathophysiological mechanisms of patients.
Thus far, the only mouse model of a specific human pathogenic mutation is the knockin of A263V (332), a mutation that induces neonatal-onset seizures and late-onset episodic ataxia in patients and causes NaV1.2 gain of function because of gating modifications in cellular expression systems (314). However, these mice have been studied in much less detail than the knockout models presented above.
9. NaV1.6: EPILEPSY AND MOVEMENT DISORDERS
As outlined above, NaV1.6 is the main sodium channel in AIS and nodes of Ranvier of myelinated axons of excitatory neurons and is now implicated in epilepsy and movement disorders (FIGURE 13). The first pathogenic mutations of NaV1.6 were identified in the “motor endplate disease” (now termed Scn8amed; Ref. 333) spontaneous mutant mouse line, which is characterized by severe motor dysfunction, including dystonia, ataxia, tremor, and progressive paralysis of the hindlimbs, and by juvenile lethality. This mouse line carries a recessive Scn8a mutation, which truncates the NaV1.6 channel protein, leading to complete loss of function (333–335). The closely related mutant mouse line Scn8amedJ has a nearly complete loss of function and similar phenotype (333, 335). A milder phenotype in the jolting mouse (now termed Scn8ajo) (333) is caused by a missense mutation in the S4–S5 linker in domain III, which positively shifts the voltage dependence of activation and thereby impairs channel function (132). In-frame deletion of three residues in the S6 segment in domain IV in the Scn8a9J mouse line (336) leads to partial loss of function of NaV1.6 and less severe phenotypes. Different from the phenotypes observed in homozygous mice, anxiety-like behavioral dysfunction was initially identified as the main phenotype of heterozygous mice carrying these mutations (333). These mouse lines do not show spontaneous convulsive seizures, but spike-wave discharges (SWDs) during periods of behavioral arrest, the hallmark of absence epilepsy, are observed in heterozygous Scn8amed and Scn8ajo mice, as well as in a line carrying a chemically induced Scn8a missense mutation (337).
FIGURE 13.
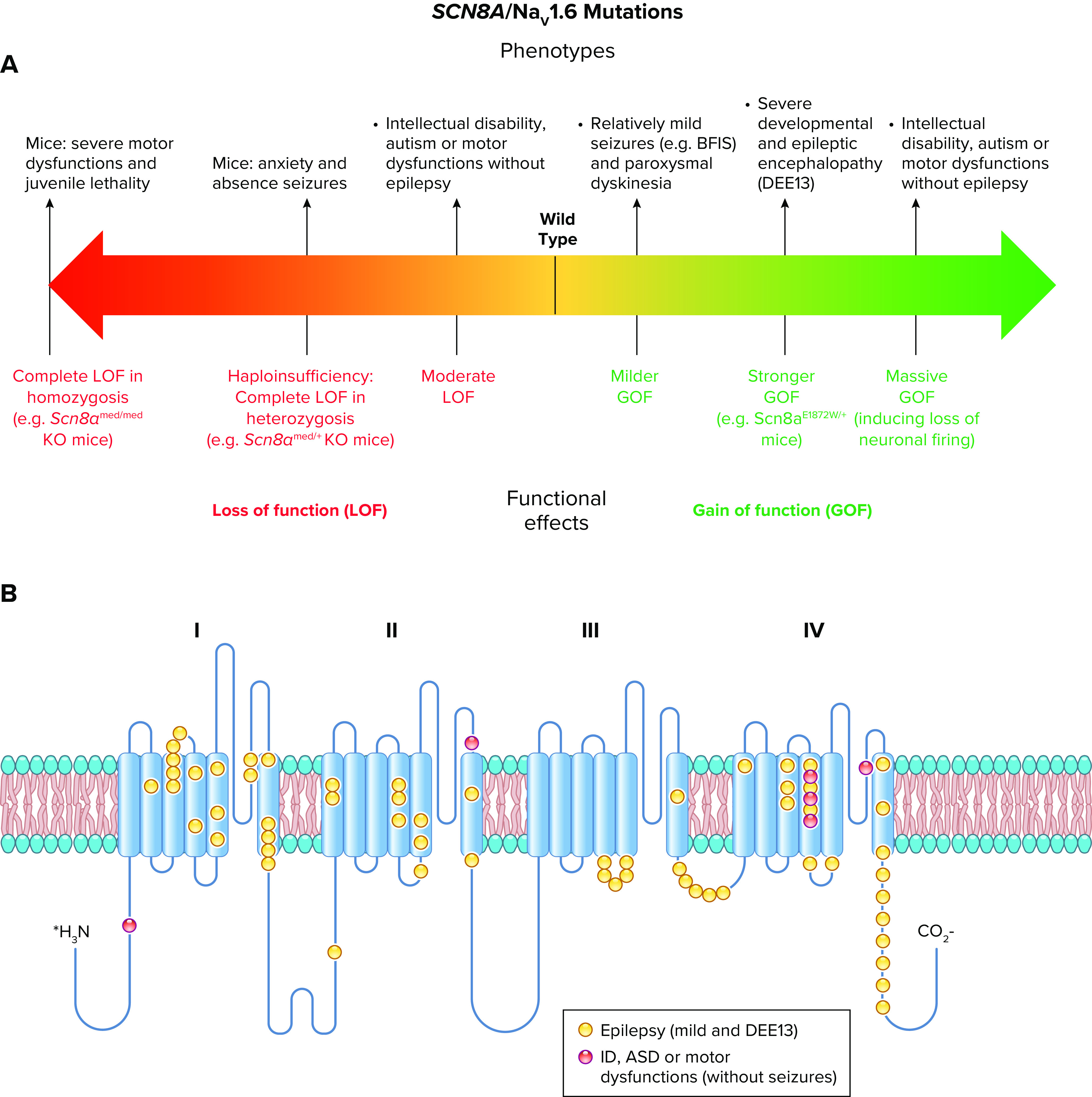
Spectrum of mutations and phenotypes for SCN8A/NaV1.6. A: phenotypic spectrum. Gain-of-function SCN8A/NaV1.6 mutations can cause relatively mild epilepsy [e.g., benign familial infantile seizures (BFIS)], paroxysmal dyskinesia when the functional effect is moderate, or severe developmental and epileptic encephalopathy (DEE13) when the functional effect is stronger. Loss-of-function mutations can cause intellectual disability (ID), autism [autism spectrum disorders (ASD)], or motor dysfunctions without epilepsy, which can be caused also by mutations that cause a massive gain of function inducing loss of neuronal firing by depolarizing block. Knockin mice carrying gain-of-function mutations show hyperexcitability of excitatory neurons and phenotype consistent with DEE13. Knockout (KO) mice reproducing complete loss of function in heterozygosis (haploinsufficiency) show anxiety and absence seizures. Mice with complete loss of function in homozygosis show severe motor dysfunctions and juvenile lethality. B: molecular map of the NaV1.6 sodium channel with the location of SCN8A mutations color-coded as indicated.
Recordings from neurons obtained from mouse lines carrying Scn8a loss-of-function mutations have shown that firing is reduced in cerebellar Purkinje cells (41, 338) and granule neurons (339), consistent with motor dysfunction phenotypes, but also in numerous other types of neurons, including hippocampal CA1 pyramidal neurons (340), subthalamic neurons (341), globus pallidus neurons (342), trigeminal mesencephalic neurons (343), retinal ganglion cells (344), and dorsal root ganglion (DRG) sensory neurons (345). A reduction of transient, persistent, and resurgent sodium currents has been observed in several of these neurons (41, 339–341, 343, 346, 347).
A recent investigation, which used conditional cre-lox lines that allow specific NaV1.6 knockout in different neuron subtypes, proposed that SWDs are generated by reduced tonic firing of GABAergic neurons of the thalamic reticular nucleus, leading to an impairment of desynchronizing recurrent synaptic inhibition within the reticular nucleus and thus to hyperexcitation of thalamo-cortical circuits (348). Notably, in this study, specific decrease of NaV1.6 expression in cortical excitatory neurons was antiepileptic, reducing convulsive seizures induced with flurothyl. Reduced epileptic activity was also observed previously in double heterozygous mice carrying a loss-of-function mutation in Scn1a that causes the Dravet syndrome phenotype plus a loss-of-function mutation for NaV1.6 that reduces action potential generation in excitatory neurons (211). These observations suggest that reduced expression of Scn8a protects against convulsive seizures by decreasing excitability of excitatory glutamatergic neurons.
The first human SCN8A mutation, introducing a premature stop codon that truncates NaV1.6, causing complete loss of function, was identified in a heterozygous proband with cognitive impairment and cerebellar ataxia without seizures (349). However, this study was limited by lack of segregation in the small pedigree and incomplete information about family members. The second human mutation was identified by whole genome sequencing in a proband presenting with an infantile epileptic encephalopathy (early-onset seizures, intellectual disability, and ataxia), who died of sudden unexplained death in epilepsy (SUDEP) (350). The functional study of the missense mutation (N1768D) was carried out in neuroblastoma ND7/23 cells because NaV1.6 does not express well in other cell lines normally used for functional studies of ion channels. The results showed a large increase in persistent current and incomplete sodium channel inactivation, consistent with a gain of function of NaV1.6, leading to increased action potential firing in transfected neurons (350).
At present, a few hundred patients with SCN8A mutations have been reported with a wide phenotypic spectrum. The vast majority of the published SCN8A patients suffer from severe developmental and epileptic encephalopathy (DEE13), characterized by early-onset epilepsy with multiple seizure types, infrequent febrile seizures, EEG abnormalities mainly in the temporo-occipital regions, severe intellectual disability, and movement disorders (351). Other patients show milder phenotypes, including benign familial infantile seizures (BFIS) with paroxysmal dyskinesia (352), and epilepsies with intermediate phenotypes (353). Furthermore, some patients carrying SCN8A mutations and showing intellectual disability, autism, or movement disorders without epilepsy have been reported (354, 355). Their phenotype is similar to that induced by the first human SCN8A mutation identified (349).
Although the first SCN8A disease mutation identified was truncating (349), thus leading to complete loss of function, those subsequently identified cause missense substitutions of evolutionarily conserved amino acid residues that alter channel function. In contrast to pathologies caused by mutations in other sodium channels, >20% of patients have recurrent mutations, in particular at residues R1617 or R1872 (356). Functional studies of 16 mutations have been carried out by expression in vitro in transfected ND7/23 cells and, in some studies, in transfected neurons in primary culture to investigate effects on excitability (350, 354–358). Functional effects of mutations that cause DEE13 or milder epilepsy induce gain of function and include negative shifts of voltage dependence of activation, positive shifts of voltage dependence of inactivation, slowed channel inactivation, or increased persistent or resurgent current. These functional changes are all consistent with neuronal hyperexcitability, which has been observed in the studies that evaluated the effect on action potential discharges of transfected cultured neurons (350, 354, 357). Other mutations causing intellectual disability, autism, or movement disorders without epilepsy induce loss of function, which can be complete (349, 354, 355). Interestingly, a recent study has shown that functional analysis in transfected neurons is crucial for correctly disclosing the overall effect of some mutations and obtaining clear genotype-phenotype relationships (354). In fact, the mutation A1622D, identified in a patient with severe developmental delay and intellectual disability without epilepsy, caused a profound destabilization of the inactivated state leading to massive slowing of inactivation, positive shift of voltage dependence of inactivation, and increased persistent current, all effects causing gain of function at the channel level. However, expression in neurons showed that the large effects of this mutation lead to reduced excitability because of induction of depolarizing block of action potential firing. Moreover, the mutation E1483K causing mild epilepsy induced small and statistically insignificant modifications of the functional properties of the channel in ND7/23 cells but increased firing in transfected neurons. Thus, it is important to include studies performed in neurons in future functional evaluations, as has been already done for NaV1.1 (43, 234).
Two human SCN8A mutations causing DEE13 have been studied by engineering them in knockin mouse models: the first mutation identified, N1768D, which was engineered in a standard knockin (359), and the recurrent mutation R1872W, which was engineered in a conditional floxed knockin (360). Both mice recapitulate in heterozygosis the basic features of SCN8A encephalopathy with spontaneous seizures, SUDEP, and mild impairment of motor coordination. Heterozygous N1768D mice show gain of function of sodium currents (increased persistent and resurgent current; positive shift of inactivation) and hyperexcitability in pyramidal neurons of the CA1 area of the hippocampus, layer II stellate neurons of the medial entorhinal cortex, and subiculum neurons of the hippocampus (361–363). In contrast, no changes were observed in GABAergic bipolar neurons from the CA1 area or pyramidal neurons from the cortex and the CA3 area of the hippocampus (361–363). Conditional R1872W mice confirmed that excitatory neurons are the target of the functional effect, because restriction of R1872W expression to excitatory neurons induced seizure and SUDEP phenotypes that were similar to those of global expression, whereas restriction of R1872W expression to inhibitory neurons did not induce an overt phenotype (360). Notably, expression of R1872W in adulthood was sufficient to generate seizures and SUDEP, suggesting that successful therapies would probably require lifelong treatment (360). Cardiac features have been studied in heterozygous N1768D mice, showing that they have hyperexcitability of cardiac myocytes and increased parasympathetic tone leading to cardiac arrhythmia, which might be implicated in SUDEP (364). Thus, knockin mice have confirmed that gain-of-‐function mutations of SCN8A are sufficient to induce hyperexcitability of some subtypes of excitatory neurons, generating severe seizures and a lethal phenotype, similar to the clinical features of DEE13. In contrast, spontaneous mouse models carrying loss-of-function NaV1.6 mutations (333) show a phenotype that is similar to that of patients with intellectual disability and/or movement disorders without epilepsy. Future genetic studies of human pathologies characterized by motor dysfunctions may reveal that NaV1.6 is more widely implicated in these pathologies.
Patients with SCN8A mutations are in general considered drug resistant. Knockin Scn8a mice and transfected cells have been also used to identify potential new drug candidates for treatment of epilepsies caused by SCN8A mutations, showing that both new and old sodium channel blockers can be effective in reducing the functional effects of gain-of-function mutations (203, 360, 363, 365–367). Interestingly, it has been shown that DEE13 patients, who carry gain-of-function mutations, can respond to high-dose treatment with the classical antiepileptic sodium channel blocker phenytoin (368), although another study cautioned regarding the long-lasting adverse effects of high-dose phenytoin treatment (369). Specific NaV1.6 blockers are under development (e.g., https://www.neurocrine.com/pipeline/epilepsy/nbi-921352-xen901/), and they might be more effective for long-lasting treatments than the classical drugs, which are not isoform specific. Recently, antisense oligonucleotides that reduce Scn8a expression by 25–50% have been shown to delay seizure onset and lethality in both Scn8aR1872W/+ and Scn1a+/− (Dravet syndrome model) mice (210). Such genetic approaches offer a high level of specificity for inhibition of NaV1.6 expression, if the challenges of drug delivery and drug half-life can be addressed.
10. NaV1.3: EPILEPSY AND IMPAIRED NEURODEVELOPMENT
NaV1.3, encoded by the SCN3A gene, is a still mysterious member of the sodium channel family, because its functions have not been completely determined and, until recently, it had a marginal role in channelopathies. NaV1.3 is broadly expressed in the brain at high levels during embryonic development, but postnatal expression is very low in both rodents and humans (130, 131, 166, 370). In the past, the implication of NaV1.3 in pathologies was limited to postnatal upregulation of its expression, observed in pain-related pathological remodeling (371) or in homeostatic responses of epileptic rodent models, for example in Scn1a models (42). More recently, mutations of SCN3A have been identified in patients with different neurological phenotypes (FIGURE 14).
FIGURE 14.
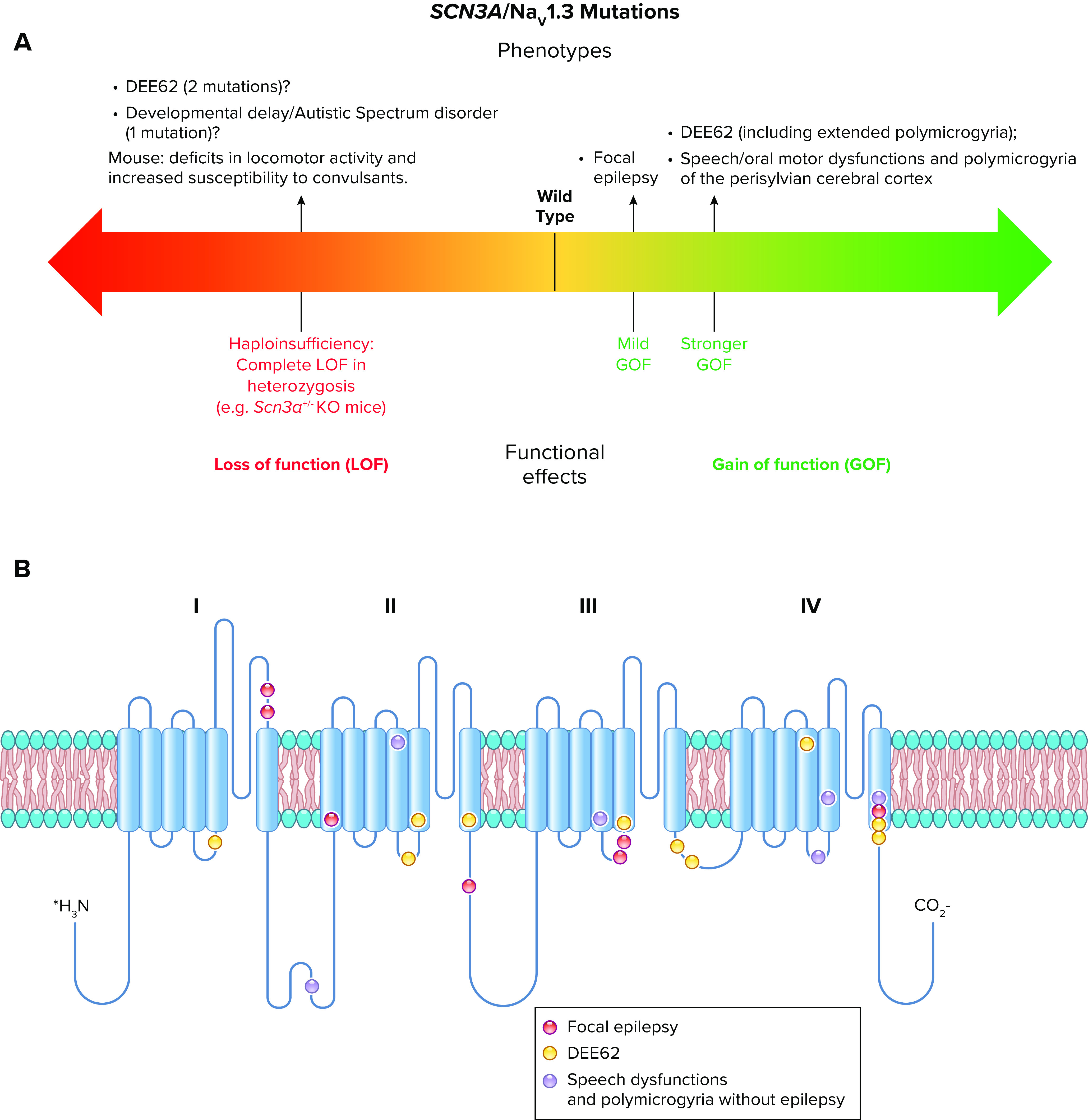
Spectrum of mutations and phenotypes in SCN3A/NaV1.3. A: phenotypic spectrum of NaV1.3 mutations. Most SCN3A/NaV1.3 pathogenic mutations cause gain of function, with milder ones identified in patients with focal epilepsy and stronger ones in patients with early infantile epileptic encephalopathy often including extended polymicrogyria (DEE62) or speech/oral motor dysfunctions and polymicrogyria limited to the perisylvian cerebral cortex without epilepsy. Loss-of-function mutations have been identified in 2 patients with DEE62 and 1 patient with developmental delay/autism spectrum disorders (ASD), but it is not clear how they can give rise to phenotypes that are similar to those of the gain-of-function mutations, which are the large majority. Haploinsufficiency in Scn3a+/- knockout (KO) mice causes deficits in locomotor activity and increased susceptibility to convulsants but not an overt epileptic phenotype or behavioral dysfunctions. B: molecular map of the NaV1.3 sodium channel with the location of SCN3A mutations color-coded as indicated.
The first mutation, the missense mutation K345Q, was identified in one patient in a study that screened a cohort of 18 children with pharmacoresistant childhood focal epilepsy. Functional analysis of this mutation, obtained by inserting the NaV1.3 mutation into the cDNA of the NaV1.5 cardiac channel, showed an increase in persistent current, consistent with gain of function (372). Functional analysis of this mutation performed subsequently with the human NaV1.3 cDNA confirmed that it increases persistent sodium current in a transfected human cell line and induces hyperexcitability in transfected neurons (373). A genetic screen of pediatric patients with neonatal-childhood focal epilepsy identified four other SCN3A missense variants causing relatively mild phenotypes (374). All the variants identified in this study showed increased sodium current elicited with slow depolarizing ramps, although only one variant induced an increase of persistent sodium current elicited with step depolarizations, consistent with a preponderant effect on kinetics of fast inactivation for the three others. Thus, the results of the functional studies are consistent with neuronal hyperexcitability as the pathological mechanism of these NaV1.3 mutations. Other variants have been identified in individuals with mild epileptic phenotypes but with uncertain pathogenicity and without functional analysis (375, 376).
More recently, three de novo SCN3A heterozygous missense mutations (I875T in 2 cases, P1333L, and V1769A) have been identified in four unrelated early infantile developmental and epileptic encephalopathy (DEE62) patients, characterized by multifocal seizures, severe intellectual disability and, for the two cases with the I875T mutation, polymicrogyria (multiple small gyri creating excessive folding of the cerebral cortex) (377). The functional effect of the three mutations was studied in a human cell line transfected with a human NaV1.3 cDNA, showing various mechanisms of gain of function consistent with neuronal hyperexcitability, including robust increases of persistent sodium current, negative shifts of voltage dependence of activation, and positive shifts of voltage dependence of inactivation (377). After the first report, the recurrent I875T mutation was identified in two other unrelated patients with DEE62 and polymicrogyria (378), and two further de novo mutations were identified (without performing functional analysis) in two patients from another cohort (379), the M1765I mutation in one patient with DEE62 and severe polymicrogyria and the L984del mutation in one patient showing intellectual disability with autistic features and pharmacoresponsive epilepsy but no polymicrogyria.
Interestingly, five SCN3A heterozygous missense mutations have been recently identified in six unrelated families presenting with speech and oral motor dysfunctions associated with polymicrogyria of the perisylvian cerebral cortex but that did not typically exhibit epilepsy (370). Disrupted cerebral cortical folding and neuronal migration were observed in ferrets expressing mutant Nav1.3, which were chosen for study because they have cortical gyri like humans, whereas rats and mice do not (370). These studies in ferrets disclose a possible unexpected role of SCN3A in progenitor cells and migrating neurons involved in prenatal development of human cortical language areas. Interestingly, one of the mutations identified in this study (I875T) was also identified in a DEE62 patient with polymicrogyria (377). Functional analysis in a transfected human cell line using a human NaV1.3 cDNA showed that these mutations induce gain of function, including increased persistent current and positive shift of voltage dependence of inactivation, similar to the effects reported in Ref. 377.
A further recent study (380), performed with a cohort of 22 patients, confirmed that SCN3A-related clinical phenotypes show a wide spectrum, including mild epilepsy with intellectual dysfunction, early infantile developmental and epileptic encephalopathy often associated with polymicrogyria, and speech and oral motor dysfunctions associated with polymicrogyria without epilepsy; ictal and nonictal autonomic dysfunction or microcephaly can be additional clinical features. Relatively mild neonatal-childhood focal epilepsy, caused by the first SCN3A identified mutations (372, 374), would be the mildest phenotype in the spectrum. Onset is scattered between the neonatal and the early childhood period for all the phenotypes.
Functional studies in transfected human cell lines have shown that most of the SCN3A mutations identified in patients with severe phenotypes exhibit prominent gain of function, inducing in particular large increases of persistent sodium current, whereas variants identified in patients with milder phenotypes exhibit less pronounced gain of function (370, 373, 374, 377, 380). A puzzling finding is that loss of function caused by markedly reduced current density has been reported for three SCN3A variants: L247P in a child with focal epilepsy, developmental delay, microcephaly, and autonomic nervous system dysfunctions (381); Y1669C in a child with mild intellectual disability and autism spectrum disorder with no history of seizures or epilepsy (380); and K1506Nfs*18 in a DEE62 infant with profound developmental delay and without polymicrogyria (380). Heterozygous adult Scn3a-knockout mice were investigated as a model of SCN3A loss-of-function mutations, but they do not show features of developmental and epileptic encephalopathy. In fact, they have increased susceptibility to induced seizures and deficits in locomotor activity but no spontaneous seizures or abnormalities in other behavioral features (381). Interestingly, lack of plasma membrane targeting was reported for the L247P mutant, consistent with folding/trafficking defects (381). It would be interesting to test whether SCN3A folding/trafficking mutants can be functionally rescued by expression in a neuronal cell background leading to gain of function, like some NaV1.1 mutations (43, 234) (see above).
Most SCN3A-positive patients are drug resistant, and patients who carry gain-of-function mutations are also reported as resistant to antiepileptic treatment with sodium channel blockers. Perhaps earlier initiation of therapy or use of higher doses might be more efficacious, although we do not know yet whether epilepsy is directly caused by the NaV1.3 dysfunction, the brain malformations, or both.
Notably, patients carrying chromosomal deletions encompassing SCN3A, which should induce loss of function of NaV1.3, show microcephaly, autistic features, and language delay but no seizures (382), and patients carrying chromosomal duplications encompassing SCN3A, which may induce gain of function of NaV1.3 because of increased expression (but without modifications of gating properties), show neonatal seizures with normal intellectual development (383). Although disruption or duplication of neighboring genes is implicated in these chromosomal defects and may also contribute to these complex phenotypes, these findings are not consistent with some of the results presented above. These complex and variable phenotypes of SCN3A mutations may reflect an important role of SCN3A in neurodevelopment, which causes variable phenotypic outcomes through individual differences in neurodevelopmental processes and genetic background of affected individuals.
11. LOOKING AHEAD
In addition to presentation of their physiology and pathophysiology, our review of sodium channelopathies of skeletal muscle and brain leads to several overarching themes with significance for the future.
Small effects matter. Both skeletal muscle and brain sodium channels are involved in numerous human genetic diseases, in which their function can be either increased or decreased. Lessons from classic studies of skeletal muscle channelopathies taught us that small effects, such as impairment of sodium channel inactivation by only a few percent, are sufficient to be pathogenic. Mutations of brain sodium channels often follow this pattern, with relatively small effects on the voltage dependence and kinetics of sodium channel function leading to epilepsy, autism, and other neuropsychiatric disorders.
Functional analysis in detail is crucial. Functional analysis in transfected cells is an important first step for performing screens of numerous mutants and may also be exploited for the development of diagnostic tests that could be included in clinical evaluations. However, specific experimental conditions for each gene should be better identified and standardized, in order to obtain robust functional evaluations. It will be important to use the correct cDNA clones, to express mutants in relevant subtypes of neurons, to study all the functional properties of sodium channels (including slow inactivation), and to perform experiments for investigating overall effects, including prediction of the effect on neuronal excitability (action potential clamp, dynamic clamp, computational modeling). These predictions and expression in neurons are particularly important for functional analysis of mutations causing loss of function that can be rescued or transformed into gain of function in neuronal cell backgrounds, as well as for mutations causing large gain-of-function effects, which may be equivalent at the cellular level to those that cause large loss of function, because they can induce depolarizing block of excitability.
Homeostasis often cannot compensate. There are nine genes that encode voltage-gated sodium channels, and four of them are widely expressed in the brain. Nevertheless, even when homeostatic responses have been identified, in most of the cases they are not sufficient to rescue the effect of pathogenic mutations, especially those causing severe loss of function. Moreover, pathological remodeling can often amplify the pathological effects of mutations. These complex responses to the initial direct functional effect of a mutation, which can be modulated both qualitatively and quantitatively by individual genetic background, are implicated in the substantial phenotypic variability that is observed in sodium channelopathies.
The genetic background of human nerve and muscle can determine the overall effects of mutations. The use of human neurons, in particular neurons obtained with induced pluripotent stem cell (iPSC) technologies, will complement phenotypic studies in the future. Thus far, only a few papers have been published on iPSC models of brain sodium channelopathies, and there are none for skeletal muscle sodium channelopathies. Most of these studies have investigated NaV1.1 mutations, and some technical problems related to variability and incomplete cellular maturation have limited their effectiveness, as recently reviewed (141). These technical problems may be solved in the future, but iPSC technologies are too time consuming and expensive to be exploited for routine drug screening and clinical tests at this stage.
Animal models are essential. Analysis of pathogenesis of sodium channel mutations in vitro is insufficient, although it can provide information on the initial direct functional effect of a mutation. Mutations should be introduced into animal models for testing therapeutic approaches in vivo and for identifying detailed mechanisms at the systems level, including homeostatic responses and pathological remodeling. However, careful comparisons and critical evaluations of human and animal phenotypes are necessary to integrate phenotypic features and age-related specificities of diseases in different species, whose time course of development and complexity of brain and muscle function are so different.
Structure is coming. Though we have not focused on this point here, the tidal wave of sodium channel structures that have emerged in recent years provides exceptional opportunities for defining common molecular and structural mechanisms of pathogenesis and using high-resolution structural data in structure-based drug design for seemingly intractable diseases.
GRANTS
Research in the authors’ laboratories presented here was supported by research grants from the Laboratory of Excellence “Ion Channel Science and Therapeutics” (LabEx ICST, ANR-11-LABX-0015-01, France), the ComputaBrain project (UCA-Jedi, ANR-15-IDEX-01, France), the European Commission (FP7 project 602531-DESIRE), the Foundation Famiglie Dravet Onlus (Italy), the Foundation Jérôme-Lejeune (France), the Interdisciplinary Institute for Modeling in Neuroscience and Cognition (NeuroMod) of the Université Côte d'Azur (France) (M.M. and S.C.), and the National Institutes of Health (W.A.C.: R01 NS-25704, R01 NS-15751, and R35 NS-111573). M.M. and S.C. are members of the “Fédération Hospitalo-Universitaire” InovPain (FHU-InovPain, France). W.A.C. sincerely thanks the Institut de Pharmacologie Moléculaire et Cellulaire, Université Cote d’Azur (Valbonne-Sophia Antipolis, France) and especially Directeur Professeur Jean-Louis Nahon, Directeur Adjoint Professeur Florian Lesage, and Directeur Emeritus Professeur Michel Lazdunski for their hospitality during his appointment as a Visiting Scientist in autumn 2018, when the concept, outline, and initial drafts for this review article were developed. The visit was sponsored in part by the Académie 4-Université Cote d’Azur (UCA-Jedi, ANR-15-IDEX-01).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
M.M., S.C., and W.A.C. prepared figures; M.M., S.C. and W.A.C. drafted manuscript; M.M. and W.A.C. edited and revised manuscript; M.M., S.C., and W.A.C. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank all the members of our laboratories who have contributed to obtain the results presented in this review.
REFERENCES
- 1.Ashcroft FM. From molecule to malady. Nature 440: 440–447, 2006. doi: 10.1038/nature04707. [DOI] [PubMed] [Google Scholar]
- 2.Venance SL, Cannon SC, Fialho D, Fontaine B, Hanna MG, Ptacek LJ, Tristani-Firouzi M, Tawil R, Griggs RC; CINCH investigators. The primary periodic paralyses: diagnosis, pathogenesis and treatment. Brain 129: 8–17, 2006. doi: 10.1093/brain/awh639. [DOI] [PubMed] [Google Scholar]
- 3.Ptácek LJ. Channelopathies: ion channel disorders of muscle as a paradigm for paroxysmal disorders of the nervous system. Neuromuscul Disord 7: 250–255, 1997. doi: 10.1016/S0960-8966(97)00046-1. [DOI] [PubMed] [Google Scholar]
- 4.Meisler MH, Kearney J, Ottman R, Escayg A. Identification of epilepsy genes in human and mouse. Annu Rev Genet 35: 567–588, 2001. doi: 10.1146/annurev.genet.35.102401.091142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Meisler MH, Sprunger LK, Plummer NW, Escayg A, Jones JM. Ion channel mutations in mouse models of inherited neurological disease. Ann Med 29: 569–574, 1997. doi: 10.3109/07853899709007484. [DOI] [PubMed] [Google Scholar]
- 6.Dib-Hajj SD, Cummins TR, Black JA, Waxman SG. From genes to pain: NaV1.7 and human pain disorders. Trends Neurosci 30: 555–563, 2007. doi: 10.1016/j.tins.2007.08.004. [DOI] [PubMed] [Google Scholar]
- 7.Keating MT, Sanguinetti MC. Molecular and cellular mechanisms of cardiac arrhythmias. Cell 104: 569–580, 2001. doi: 10.1016/s0092-8674(01)00243-4. [DOI] [PubMed] [Google Scholar]
- 8.Bennett DL, Clark AJ, Huang J, Waxman SG, Dib-Hajj SD. The role of voltage-gated sodium channels in pain signaling. Physiol Rev 99: 1079–1151, 2019. doi: 10.1152/physrev.00052.2017. [DOI] [PubMed] [Google Scholar]
- 9.Abriel H, Rougier JS, Jalife J. Ion channel macromolecular complexes in cardiomyocytes: roles in sudden cardiac death. Circ Res 116: 1971–1988, 2015. doi: 10.1161/CIRCRESAHA.116.305017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bohnen MS, Peng G, Robey SH, Terrenoire C, Iyer V, Sampson KJ, Kass RS. Molecular pathophysiology of congenital long QT syndrome. Physiol Rev 97: 89–134, 2017. doi: 10.1152/physrev.00008.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Moreau A, Chahine M. A new cardiac channelopathy: from clinical phenotypes to molecular mechanisms associated with NaV1.5 gating pores. Front Cardiovasc Med 5: 139, 2018. doi: 10.3389/fcvm.2018.00139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bouza AA, Isom LL. Voltage-gated sodium channel beta subunits and their related diseases. Handb Exp Pharmacol 246: 423–450, 2018. doi: 10.1007/164_2017_48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Edokobi N, Isom LL. Voltage-gated sodium channel beta1/beta1B subunits regulate cardiac physiology and pathophysiology. Front Physiol 9: 351, 2018. doi: 10.3389/fphys.2018.00351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hille B. Ionic Channels of Excitable Membranes (3rd ed.). Sunderland, MA: Sinauer Associates Inc., 2001. [Google Scholar]
- 15.Catterall WA. Molecular properties of voltage-sensitive sodium channels. Annu Rev Biochem 55: 953–985, 1986. doi: 10.1146/annurev.bi.55.070186.004513. [DOI] [PubMed] [Google Scholar]
- 16.Catterall WA. From ionic currents to molecular mechanisms: the structure and function of voltage-gated sodium channels. Neuron 26: 13–25, 2000. doi: 10.1016/S0896-6273(00)81133-2. [DOI] [PubMed] [Google Scholar]
- 17.Payandeh J, Scheuer T, Zheng N, Catterall WA. The crystal structure of a voltage-gated sodium channel. Nature 475: 353–358, 2011. doi: 10.1038/nature10238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pan X, Li Z, Zhou Q, Shen H, Wu K, Huang X, Chen J, Zhang J, Zhu X, Lei J, Xiong W, Gong H, Xiao B, Yan N. Structure of the human voltage-gated sodium channel NaV1.4 in complex with beta1. Science 362: eaau2486, 2018. doi: 10.1126/science.aau2486. [DOI] [PubMed] [Google Scholar]
- 19.Catterall WA, Zheng N. Deciphering voltage-gated Na+ and Ca2+ channels by studying prokaryotic ancestors. Trends Biochem Sci 40: 526–534, 2015. doi: 10.1016/j.tibs.2015.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shen H, Zhou Q, Pan X, Li Z, Wu J, Yan N. Structure of a eukaryotic voltage-gated sodium channel at near-atomic resolution. Science 355: eaal4326, 2017. doi: 10.1126/science.aal4326. [DOI] [PubMed] [Google Scholar]
- 21.Catterall WA. Signaling complexes of voltage-gated sodium and calcium channels. Neurosci Lett 486: 107–116, 2010. doi: 10.1016/j.neulet.2010.08.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Curran J, Mohler PJ. Alternative paradigms for ion channelopathies: disorders of ion channel membrane trafficking and posttranslational modification. Annu Rev Physiol 77: 505–524, 2015. doi: 10.1146/annurev-physiol-021014-071838. [DOI] [PubMed] [Google Scholar]
- 23.Dib-Hajj SD, Waxman SG. Isoform-specific and pan-channel partners regulate trafficking and plasma membrane stability; and alter sodium channel gating properties. Neurosci Lett 486: 84–91, 2010. doi: 10.1016/j.neulet.2010.08.077. [DOI] [PubMed] [Google Scholar]
- 24.Terragni B, Scalmani P, Franceschetti S, Cestèle S, Mantegazza M. Post-translational dysfunctions in channelopathies of the nervous system. Neuropharmacology 132: 31–42, 2018. doi: 10.1016/j.neuropharm.2017.05.028. [DOI] [PubMed] [Google Scholar]
- 25.Clatot J, Hoshi M, Wan X, Liu H, Jain A, Shinlapawittayatorn K, Marionneau C, Ficker E, Ha T, Deschênes I. Voltage-gated sodium channels assemble and gate as dimers. Nat Commun 8: 2077, 2017. doi: 10.1038/s41467-017-02262-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Clatot J, Zheng Y, Girardeau A, Liu H, Laurita KR, Marionneau C, Deschênes I. Mutant voltage-gated Na+ channels can exert a dominant negative effect through coupled gating. Am J Physiol Heart Circ Physiol 315: H1250–H1257, 2018. doi: 10.1152/ajpheart.00721.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mantegazza M, Catterall WA. Voltage-gated Na+ channels: structure, function, and pathophysiology. In: Jasper’s Basic Mechanisms of the Epilepsies, edited by Noebels JL, Avoli M, Rogawski MA, Olsen RW, Delgado-Escueta AV.. Bethesda, MD: National Center for Biotechnology Information, 2012. [PubMed] [Google Scholar]
- 28.Bezanilla F. The voltage sensor in voltage-dependent ion channels. Physiol Rev 80: 555–592, 2000. doi: 10.1152/physrev.2000.80.2.555. [DOI] [PubMed] [Google Scholar]
- 29.Horn R. Molecular basis for function in sodium channels. Novartis Found Symp 241: 21–26, 2002. [PubMed] [Google Scholar]
- 30.DeCaen PG, Yarov-Yarovoy V, Scheuer T, Catterall WA. Gating charge interactions with the S1 segment during activation of a Na+ channel voltage sensor. Proc Natl Acad Sci USA 108: 18825–18830, 2011. doi: 10.1073/pnas.1116449108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.DeCaen PG, Yarov-Yarovoy V, Sharp EM, Scheuer T, Catterall WA. Sequential formation of ion pairs during activation of a sodium channel voltage sensor. Proc Natl Acad Sci USA 106: 22498–22503, 2009. doi: 10.1073/pnas.0912307106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.DeCaen PG, Yarov-Yarovoy V, Zhao Y, Scheuer T, Catterall WA. Disulfide locking a sodium channel voltage sensor reveals ion pair formation during activation. Proc Natl Acad Sci USA 105: 15142–15147, 2008. doi: 10.1073/pnas.0806486105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mantegazza M, Cestèle S. Beta-scorpion toxin effects suggest electrostatic interactions in domain II of voltage-dependent sodium channels. J Physiol 568: 13–30, 2005. doi: 10.1113/jphysiol.2005.093484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wisedchaisri G, Tonggu L, McCord E, Gamal El-Din TM, Wang L, Zheng N, Catterall WA. Resting-state structure and gating mechanism of a voltage-gated sodium channel. Cell 178: 993–1003.e12, 2019. doi: 10.1016/j.cell.2019.06.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yarov-Yarovoy V, Decaen PG, Westenbroek RE, Pan CY, Scheuer T, Baker D, Catterall WA. Structural basis for gating charge movement in the voltage sensor of a sodium channel. Proc Natl Acad Sci USA 109: E93–E102, 2012. doi: 10.1073/pnas.1118434109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vargas E, Yarov-Yarovoy V, Khalili-Araghi F, Catterall WA, Klein ML, Tarek M, Lindahl E, Schulten K, Perozo E, Bezanilla F, Roux B. An emerging consensus on voltage-dependent gating from computational modeling and molecular dynamics simulations. J Gen Physiol 140: 587–594, 2012. doi: 10.1085/jgp.201210873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bagnéris C, Naylor CE, McCusker EC, Wallace BA. Structural model of the open-closed-inactivated cycle of prokaryotic voltage-gated sodium channels. J Gen Physiol 145: 5–16, 2015. doi: 10.1085/jgp.201411242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lenaeus MJ, Gamal El-Din TM, Ing C, Ramanadane K, Pomes R, Zheng N, Catterall WA. Structures of closed and open states of a voltage-gated sodium channel. Proc Natl Acad Sci USA 114: E3051–E3060, 2017. doi: 10.1073/pnas.1700761114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hodgkin AL, Huxley AF. A quantitative description of membrane current and its application to conduction and excitation in nerve. J Physiol 117: 500–544, 1952. doi: 10.1113/jphysiol.1952.sp004764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Terragni B, Scalmani P, Colombo E, Franceschetti S, Mantegazza M. Ranolazine vs. phenytoin: greater effect of ranolazine on the transient Na+ current than on the persistent Na+ current in central neurons. Neuropharmacology 110: 223–236, 2016. doi: 10.1016/j.neuropharm.2016.06.029. [DOI] [PubMed] [Google Scholar]
- 41.Raman IM, Bean BP. Resurgent sodium current and action potential formation in dissociated cerebellar Purkinje neurons. J Neurosci 17: 4517–4526, 1997. doi: 10.1523/JNEUROSCI.17-12-04517.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yu FH, Mantegazza M, Westenbroek RE, Robbins CA, Kalume F, Burton KA, Spain WJ, McKnight GS, Scheuer T, Catterall WA. Reduced sodium current in GABAergic interneurons in a mouse model of severe myoclonic epilepsy in infancy. Nat Neurosci 9: 1142–1149, 2006. doi: 10.1038/nn1754. [DOI] [PubMed] [Google Scholar]
- 43.Cestèle S, Schiavon E, Rusconi R, Franceschetti S, Mantegazza M. Nonfunctional NaV1.1 familial hemiplegic migraine mutant transformed into gain of function by partial rescue of folding defects. Proc Natl Acad Sci USA 110: 17546–17551, 2013. doi: 10.1073/pnas.1309827110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Starace DM, Bezanilla F. A proton pore in a potassium channel voltage sensor reveals a focused electric field. Nature 427: 548–553, 2004. doi: 10.1038/nature02270. [DOI] [PubMed] [Google Scholar]
- 45.Sokolov S, Scheuer T, Catterall WA. Ion permeation through a voltage- sensitive gating pore in brain sodium channels having voltage sensor mutations. Neuron 47: 183–189, 2005. doi: 10.1016/j.neuron.2005.06.012. [DOI] [PubMed] [Google Scholar]
- 46.Tombola F, Pathak MM, Isacoff EY. Voltage-sensing arginines in a potassium channel permeate and occlude cation-selective pores. Neuron 45: 379–388, 2005. doi: 10.1016/j.neuron.2004.12.047. [DOI] [PubMed] [Google Scholar]
- 47.Sokolov S, Scheuer T, Catterall WA. Ion permeation and block of the gating pore in the voltage sensor of NaV1.4 channels with hypokalemic periodic paralysis mutations. J Gen Physiol 136: 225–236, 2010. doi: 10.1085/jgp.201010414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chakrabarti N, Ing C, Payandeh J, Zheng N, Catterall WA, Pomès R. Catalysis of Na+ permeation in the bacterial sodium channel NaVAb. Proc Natl Acad Sci USA 110: 11331–11336, 2013. doi: 10.1073/pnas.1309452110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Naylor CE, Bagnéris C, DeCaen PG, Sula A, Scaglione A, Clapham DE, Wallace BA. Molecular basis of ion permeability in a voltage-gated sodium channel. EMBO J 35: 820–830, 2016. doi: 10.15252/embj.201593285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ulmschneider MB, Bagnéris C, McCusker EC, DeCaen PG, Delling M, Clapham DE, Ulmschneider JP, Wallace BA. Molecular dynamics of ion transport through the open conformation of a bacterial voltage-gated sodium channel. Proc Natl Acad Sci USA 110: 6364–6369, 2013. doi: 10.1073/pnas.1214667110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Capes DL, Goldschen-Ohm MP, Arcisio-Miranda M, Bezanilla F, Chanda B. Domain IV voltage-sensor movement is both sufficient and rate limiting for fast inactivation in sodium channels. J Gen Physiol 142: 101–112, 2013. doi: 10.1085/jgp.201310998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chanda B, Bezanilla F. Tracking voltage-dependent conformational changes in skeletal muscle sodium channel during activation. J Gen Physiol 120: 629–645, 2002. doi: 10.1085/jgp.20028679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rogers JC, Qu Y, Tanada TN, Scheuer T, Catterall WA. Molecular determinants of high affinity binding of alpha-scorpion toxin and sea anemone toxin in the S3-S4 extracellular loop in domain IV of the Na+ channel alpha subunit. J Biol Chem 271: 15950–15962, 1996. doi: 10.1074/jbc.271.27.15950. [DOI] [PubMed] [Google Scholar]
- 54.Sheets MF, Kyle JW, Kallen RG, Hanck DA. The Na channel voltage sensor associated with inactivation is localized to the external charged residues of domain IV, S4. Biophys J 77: 747–757, 1999. doi: 10.1016/S0006-3495(99)76929-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kellenberger S, West JW, Scheuer T, Catterall WA. Molecular analysis of the putative inactivation particle in the inactivation gate of brain type IIA Na+ channels. J Gen Physiol 109: 589–605, 1997. doi: 10.1085/jgp.109.5.589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.West JW, Patton DE, Scheuer T, Wang Y, Goldin AL, Catterall WA. A cluster of hydrophobic amino acid residues required for fast Na+ channel inactivation. Proc Natl Acad Sci USA 89: 10910–10914, 1992. doi: 10.1073/pnas.89.22.10910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rohl CA, Boeckman FA, Baker C, Scheuer T, Catterall WA, Klevit RE. Solution structure of the sodium channel inactivation gate. Biochemistry 38: 855–861, 1999. doi: 10.1021/bi9823380. [DOI] [PubMed] [Google Scholar]
- 58.McPhee JC, Ragsdale D, Scheuer T, Catterall WA. A critical role for the S4-S5 intracellular loop in domain IV of the sodium channel alpha subunit in fast inactivation. J Biol Chem 273: 1121–1129, 1998. doi: 10.1074/jbc.273.2.1121. [DOI] [PubMed] [Google Scholar]
- 59.McPhee JC, Ragsdale DS, Scheuer T, Catterall WA. A critical role for transmembrane segment IVS6 of the sodium channel alpha subunit in fast inactivation. J Biol Chem 270: 12025–12034, 1995. doi: 10.1074/jbc.270.20.12025. [DOI] [PubMed] [Google Scholar]
- 60.McPhee JC, Ragsdale DS, Scheuer T, Catterall WA. A mutation in segment IVS6 disrupts fast inactivation of sodium channels. Proc Natl Acad Sci USA 91: 12346–12350, 1994. doi: 10.1073/pnas.91.25.12346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Smith MR, Goldin AL. Interaction between the sodium channel inactivation linker and domain III S4-S5. Biophys J 73: 1885–1895, 1997. doi: 10.1016/S0006-3495(97)78219-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tang LH, Kallen RG, Horn R. Role of an S4-S5 linker in sodium channel inactivation probed by mutagenesis and a peptide blocker. J Gen Physiol 108: 89–104, 1996. doi: 10.1085/jgp.108.2.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mantegazza M, Yu FH, Catterall WA, Scheuer T. Role of the C-terminal domain in inactivation of brain and cardiac sodium channels. Proc Natl Acad Sci USA 98: 15348–15353, 2001. doi: 10.1073/pnas.211563298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Motoike HK, Liu H, Glaaser IW, Yang AS, Tateyama M, Kass RS. The Na+ channel inactivation gate is a molecular complex: a novel role of the COOH-terminal domain. J Gen Physiol 123: 155–165, 2004. doi: 10.1085/jgp.200308929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mantegazza M, Yu FH, Powell AJ, Clare JJ, Catterall WA, Scheuer T. Molecular determinants for modulation of persistent sodium current by G-protein betagamma subunits. J Neurosci 25: 3341–3349, 2005. doi: 10.1523/JNEUROSCI.0104-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Crill WE. Persistent sodium current in mammalian central neurons. Annu Rev Physiol 58: 349–362, 1996. doi: 10.1146/annurev.ph.58.030196.002025. [DOI] [PubMed] [Google Scholar]
- 67.Colombo E, Franceschetti S, Avanzini G, Mantegazza M. Phenytoin inhibits the persistent sodium current in neocortical neurons by modifying its inactivation properties. PLoS One 8: e55329, 2013. doi: 10.1371/journal.pone.0055329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lewis AH, Raman IM. Resurgent current of voltage-gated Na+ channels. J Physiol 592: 4825–4838, 2014. doi: 10.1113/jphysiol.2014.277582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Vilin YY, Ruben PC. Slow inactivation in voltage-gated sodium channels: molecular substrates and contributions to channelopathies. Cell Biochem Biophys 35: 171–190, 2001. doi: 10.1385/CBB:35:2:171. [DOI] [PubMed] [Google Scholar]
- 70.Payandeh J, Gamal El-Din TM, Scheuer T, Zheng N, Catterall WA. Crystal structure of a voltage-gated sodium channel in two potentially inactivated states. Nature 486: 135–139, 2012. doi: 10.1038/nature11077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhang X, Ren W, DeCaen P, Yan C, Tao X, Tang L, Wang J, Hasegawa K, Kumasaka T, He J, Wang J, Clapham DE, Yan N. Crystal structure of an orthologue of the NaChBac voltage-gated sodium channel. Nature 486: 130–134, 2012. doi: 10.1038/nature11054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Balser JR, Nuss HB, Chiamvimonvat N, Pérez-García MT, Marban E, Tomaselli GF. External pore residue mediates slow inactivation in mu-1 rat skeletal muscle sodium channels. J Physiol 494: 431–442, 1996. doi: 10.1113/jphysiol.1996.sp021503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Gamal El-Din TM, Lenaeus MJ, Ramanadane K, Zheng N, Catterall WA. Molecular dissection of multiphase inactivation of the bacterial sodium channel NaVAb. J Gen Physiol 151: 174–185, 2019. doi: 10.1085/jgp.201711884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.O’Reilly JP, Wang SY, Wang GK. Residue-specific effects on slow inactivation at V787 in D2-S6 of NaV1.4 sodium channels. Biophys J 81: 2100–2111, 2001. doi: 10.1016/S0006-3495(01)75858-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Fontaine B. Periodic paralysis. Adv Genet 63: 3–23, 2008. doi: 10.1016/S0065-2660(08)01001-8. [DOI] [PubMed] [Google Scholar]
- 76.Jurkat-Rott K, Lehmann-Horn F. Paroxysmal muscle weakness: the familial periodic paralyses. J Neurol 253: 1391–1398, 2006. doi: 10.1007/s00415-006-0339-0. [DOI] [PubMed] [Google Scholar]
- 77.Cannon SC. Sodium channelopathies of skeletal muscle. Handb Exp Pharmacol 246: 309–330, 2018. doi: 10.1007/164_2017_52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lehmann-Horn F, Iaizzo PA, Hatt H, Franke C. Altered gating and conductance of Na+ channels in hyperkalemic periodic paralysis. Pflugers Arch 418: 297–299, 1991. doi: 10.1007/BF00370530. [DOI] [PubMed] [Google Scholar]
- 79.Fontaine B, Khurana TS, Hoffman EP, Bruns GA, Haines JL, Trofatter JA, Hanson MP, Rich J, McFarlane H, Yasek DM, Romano D, Gusella JF, Brown RH Jr.. Hyperkalemic periodic paralysis and the adult muscle sodium channel alpha-subunit gene. Science 250: 1000–1002, 1990. doi: 10.1126/science.2173143. [DOI] [PubMed] [Google Scholar]
- 80.Ptacek LJ, Tyler F, Trimmer JS, Agnew WS, Leppert M. Analysis in a large hyperkalemic periodic paralysis pedigree supports tight linkage to a sodium channel locus. Am J Hum Genet 49: 378–382, 1991. [PMC free article] [PubMed] [Google Scholar]
- 81.Ptácek LJ, George AL Jr, Griggs RC, Tawil R, Kallen RG, Barchi RL, Robertson M, Leppert MF. Identification of a mutation in the gene causing hyperkalemic periodic paralysis. Cell 67: 1021–1027, 1991. doi: 10.1016/0092-8674(91)90374-8. [DOI] [PubMed] [Google Scholar]
- 82.Rojas CV, Wang JZ, Schwartz LS, Hoffman EP, Powell BR, Brown RH. A Met-to-Val mutation in the skeletal muscle Na+ channel alpha-subunit in hyperkalemic periodic paralysis. Nature 354: 387–389, 1991. doi: 10.1038/354387a0. [DOI] [PubMed] [Google Scholar]
- 83.Simkin D, Bendahhou S. Skeletal muscle Na channel disorders. Front Pharmacol 2: 63, 2011. doi: 10.3389/fphar.2011.00063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Cannon SC, Brown RH Jr, Corey DP. A sodium channel defect in hyperkalemic periodic paralysis: potassium-induced failure of inactivation. Neuron 6: 619–626, 1991. doi: 10.1016/0896-6273(91)90064-7. [DOI] [PubMed] [Google Scholar]
- 85.Cannon SC, Hayward LJ, Beech J, Brown RH Jr.. Sodium channel inactivation is impaired in equine hyperkalemic periodic paralysis. J Neurophysiol 73: 1892–1899, 1995. doi: 10.1152/jn.1995.73.5.1892. [DOI] [PubMed] [Google Scholar]
- 86.Bendahhou S, Cummins TR, Kula RW, Fu YH, Ptácek LJ. Impairment of slow inactivation as a common mechanism for periodic paralysis in DIIS4-S5. Neurology 58: 1266–1272, 2002. doi: 10.1212/wnl.58.8.1266. [DOI] [PubMed] [Google Scholar]
- 87.Hayward LJ, Brown RH Jr, Cannon SC. Slow inactivation differs among mutant Na channels associated with myotonia and periodic paralysis. Biophys J 72: 1204–1219, 1997. doi: 10.1016/S0006-3495(97)78768-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Haass A, Ricker K, Rüdel R, Lehmann-Horn F, Böhlen R, Dengler R, Mertens HG. Clinical study of paramyotonia congenita with and without myotonia in a warm environment. Muscle Nerve 4: 388–395, 1981. doi: 10.1002/mus.880040507. [DOI] [PubMed] [Google Scholar]
- 89.Ricker K, Rüdel R, Lehmann-Horn F, Küther G. Muscle stiffness and electrical activity in paramyotonia congenita. Muscle Nerve 9: 299–305, 1986. doi: 10.1002/mus.880090403. [DOI] [PubMed] [Google Scholar]
- 90.Rüdel R, Ricker K, Lehmann-Horn F. Genotype-phenotype correlations in human skeletal muscle sodium channel diseases. Arch Neurol 50: 1241–1248, 1993. doi: 10.1001/archneur.1993.00540110113011. [DOI] [PubMed] [Google Scholar]
- 91.Ptácek LJ, George AL Jr, Barchi RL, Griggs RC, Riggs JE, Robertson M, Leppert MF. Mutations in an S4 segment of the adult skeletal muscle sodium channel cause paramyotonia congenita. Neuron 8: 891–897, 1992. doi: 10.1016/0896-6273(92)90203-P. [DOI] [PubMed] [Google Scholar]
- 92.Ptacek LJ, Gouw L, Kwieciński H, McManis P, Mendell JR, Barohn RJ, George AL Jr, Barchi RL, Robertson M, Leppert MF. Sodium channel mutations in paramyotonia congenita and hyperkalemic periodic paralysis. Ann Neurol 33: 300–307, 1993. doi: 10.1002/ana.410330312. [DOI] [PubMed] [Google Scholar]
- 93.Bendahhou S, Cummins TR, Kwiecinski H, Waxman SG, Ptácek LJ. Characterization of a new sodium channel mutation at arginine 1448 associated with moderate paramyotonia congenita in humans. J Physiol 518: 337–344, 1999. doi: 10.1111/j.1469-7793.1999.0337p.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Lerche H, Mitrovic N, Dubowitz V, Lehmann-Horn F. Paramyotonia congenita: the R1448P Na+ channel mutation in adult human skeletal muscle. Ann Neurol 39: 599–608, 1996. doi: 10.1002/ana.410390509. [DOI] [PubMed] [Google Scholar]
- 95.Bouhours M, Luce S, Sternberg D, Willer JC, Fontaine B, Tabti N. A1152D mutation of the Na+ channel causes paramyotonia congenita and emphasizes the role of DIII/S4-S5 linker in fast inactivation. J Physiol 565: 415–427, 2005. doi: 10.1113/jphysiol.2004.081018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Bouhours M, Sternberg D, Davoine CS, Ferrer X, Willer JC, Fontaine B, Tabti N. Functional characterization and cold sensitivity of T1313A, a new mutation of the skeletal muscle sodium channel causing paramyotonia congenita in humans. J Physiol 554: 635–647, 2004. doi: 10.1113/jphysiol.2003.053082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Fleischhauer R, Mitrovic N, Deymeer F, Lehmann-Horn F, Lerche H. Effects of temperature and mexiletine on the F1473S Na+ channel mutation causing paramyotonia congenita. Pflugers Arch 436: 757–765, 1998. doi: 10.1007/s004240050699. [DOI] [PubMed] [Google Scholar]
- 98.Mitrovic N, Lerche H, Heine R, Fleischhauer R, Pika-Hartlaub U, Hartlaub U, George AL Jr, Lehmann-Horn F. Role in fast inactivation of conserved amino acids in the IV/S4-S5 loop of the human muscle Na+ channel. Neurosci Lett 214: 9–12, 1996. doi: 10.1016/0304-3940(96)12866-4. [DOI] [PubMed] [Google Scholar]
- 99.Yang N, Ji S, Zhou M, Ptácek LJ, Barchi RL, Horn R, George AL Jr.. Sodium channel mutations in paramyotonia congenita exhibit similar biophysical phenotypes in vitro. Proc Natl Acad Sci USA 91: 12785–12789, 1994. doi: 10.1073/pnas.91.26.12785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Weber MA, Nielles-Vallespin S, Huttner HB, Wöhrle JC, Jurkat-Rott K, Lehmann-Horn F, Schad LR, Kauczor HU, Essig M, Meinck HM. Evaluation of patients with paramyotonia at 23Na MR imaging during cold-induced weakness. Radiology 240: 489–500, 2006. doi: 10.1148/radiol.2401050737. [DOI] [PubMed] [Google Scholar]
- 101.Mitrovic N, George AL Jr, Lerche H, Wagner S, Fahlke C, Lehmann-Horn F. Different effects on gating of three myotonia-causing mutations in the inactivation gate of the human muscle sodium channel. J Physiol 487: 107–114, 1995. doi: 10.1113/jphysiol.1995.sp020864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Rüdel R, Lehmann-Horn F, Ricker K, Küther G. Hypokalemic periodic paralysis: in vitro investigation of muscle fiber membrane parameters. Muscle Nerve 7: 110–120, 1984. doi: 10.1002/mus.880070205. [DOI] [PubMed] [Google Scholar]
- 103.Jurkat-Rott K, Lehmann-Horn F, Elbaz A, Heine R, Gregg RG, Hogan K, Powers PA, Lapie P, Vale-Santos JE, Weissenbach J, Fontaine B. A calcium channel mutation causing hypokalemic periodic paralysis. Hum Mol Genet 3: 1415–1419, 1994. doi: 10.1093/hmg/3.8.1415. [DOI] [PubMed] [Google Scholar]
- 104.Jurkat-Rott K, Uetz U, Pika-Hartlaub U, Powell J, Fontaine B, Melzer W, Lehmann-Horn F. Calcium currents and transients of native and heterologously expressed mutant skeletal muscle DHP receptor alpha1 subunits (R528H). FEBS Lett 423: 198–204, 1998. doi: 10.1016/s0014-5793(98)00090-8. [DOI] [PubMed] [Google Scholar]
- 105.Matthews E, Labrum R, Sweeney MG, Sud R, Haworth A, Chinnery PF, Meola G, Schorge S, Kullmann DM, Davis MB, Hanna MG. Voltage sensor charge loss accounts for most cases of hypokalemic periodic paralysis. Neurology 72: 1544–1547, 2009. doi: 10.1212/01.wnl.0000342387.65477.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Wu F, Mi W, Hernández-Ochoa EO, Burns DK, Fu Y, Gray HF, Struyk AF, Schneider MF, Cannon SC. A calcium channel mutant mouse model of hypokalemic periodic paralysis. J Clin Invest 122: 4580–4591, 2012. doi: 10.1172/JCI66091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Bulman DE, Scoggan KA, van Oene MD, Nicolle MW, Hahn AF, Tollar LL, Ebers GC. A novel sodium channel mutation in a family with hypokalemic periodic paralysis. Neurology 53: 1932–1936, 1999. doi: 10.1212/wnl.53.9.1932. [DOI] [PubMed] [Google Scholar]
- 108.Sternberg D, Maisonobe T, Jurkat-Rott K, Nicole S, Launay E, Chauveau D, Tabti N, Lehmann-Horn F, Hainque B, Fontaine B. Hypokalaemic periodic paralysis type 2 caused by mutations at codon 672 in the muscle sodium channel gene SCN4A. Brain 124: 1091–1099, 2001. doi: 10.1093/brain/124.6.1091. [DOI] [PubMed] [Google Scholar]
- 109.Wu F, Mi W, Burns DK, Fu Y, Gray HF, Struyk AF, Cannon SC. A sodium channel knockin mutant (NaV1.4-R669H) mouse model of hypokalemic periodic paralysis. J Clin Invest 121: 4082–4094, 2011. doi: 10.1172/JCI57398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Vicart S, Sternberg D, Fournier E, Ochsner F, Laforet P, Kuntzer T, Eymard B, Hainque B, Fontaine B. New mutations of SCN4A cause a potassium-sensitive normokalemic periodic paralysis. Neurology 63: 2120–2127, 2004. doi: 10.1212/01.wnl.0000145768.09934.ec. [DOI] [PubMed] [Google Scholar]
- 111.Jurkat-Rott K, Mitrovic N, Hang C, Kouzmekine A, Iaizzo P, Herzog J, Lerche H, Nicole S, Vale-Santos J, Chauveau D, Fontaine B, Lehmann-Horn F. Voltage-sensor sodium channel mutations cause hypokalemic periodic paralysis type 2 by enhanced inactivation and reduced current. Proc Natl Acad Sci USA 97: 9549–9554, 2000. doi: 10.1073/pnas.97.17.9549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Kuzmenkin A, Muncan V, Jurkat-Rott K, Hang C, Lerche H, Lehmann-Horn F, Mitrovic N. Enhanced inactivation and pH sensitivity of Na+ channel mutations causing hypokalaemic periodic paralysis type II. Brain 125: 835–843, 2002. doi: 10.1093/brain/awf071. [DOI] [PubMed] [Google Scholar]
- 113.Sokolov S, Scheuer T, Catterall WA. Depolarization-activated gating pore current conducted by mutant sodium channels in potassium-sensitive normokalemic periodic paralysis. Proc Natl Acad Sci USA 105: 19980–19985, 2008. doi: 10.1073/pnas.0810562105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Sokolov S, Scheuer T, Catterall WA. Gating pore current in an inherited ion channelopathy. Nature 446: 76–78, 2007. doi: 10.1038/nature05598. [DOI] [PubMed] [Google Scholar]
- 115.Struyk AF, Cannon SC. A Na+ channel mutation linked to hypokalemic periodic paralysis exposes a proton-selective gating pore. J Gen Physiol 130: 11–20, 2007. doi: 10.1085/jgp.200709755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Struyk AF, Markin VS, Francis D, Cannon SC. Gating pore currents in DIIS4 mutations of NaV1.4 associated with periodic paralysis: saturation of ion flux and implications for disease pathogenesis. J Gen Physiol 132: 447–464, 2008. doi: 10.1085/jgp.200809967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Jiang D, Gamal El-Din TM, Ing C, Lu P, Pomès R, Zheng N, Catterall WA. Structural basis for gating pore current in periodic paralysis. Nature 557: 590–594, 2018. doi: 10.1038/s41586-018-0120-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Jurkat-Rott K, Weber MA, Fauler M, Guo XH, Holzherr BD, Paczulla A, Nordsborg N, Joechle W, Lehmann-Horn F. K+-dependent paradoxical membrane depolarization and Na+ overload, major and reversible contributors to weakness by ion channel leaks. Proc Natl Acad Sci USA 106: 4036–4041, 2009. doi: 10.1073/pnas.0811277106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Engel AG. Genetic basis and phenotypic features of congenital myasthenic syndromes. Handb Clin Neurol 148: 565–589, 2018. doi: 10.1016/B978-0-444-64076-5.00037-5. [DOI] [PubMed] [Google Scholar]
- 120.Finsterer J. Congenital myasthenic syndromes. Orphanet J Rare Dis 14: 57, 2019. doi: 10.1186/s13023-019-1025-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Arnold WD, Feldman DH, Ramirez S, He L, Kassar D, Quick A, Klassen TL, Lara M, Nguyen J, Kissel JT, Lossin C, Maselli RA. Defective fast inactivation recovery of NaV1.4 in congenital myasthenic syndrome. Ann Neurol 77: 840–850, 2015. doi: 10.1002/ana.24389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Habbout K, Poulin H, Rivier F, Giuliano S, Sternberg D, Fontaine B, Eymard B, Morales RJ, Echenne B, King L, Hanna MG, Mannikko R, Chahine M, Nicole S, Bendahhou S. A recessive NaV1.4 mutation underlies congenital myasthenic syndrome with periodic paralysis. Neurology 86: 161–169, 2016. doi: 10.1212/WNL.0000000000002264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Elia N, Palmio J, Castañeda MS, Shieh PB, Quinonez M, Suominen T, Hanna MG, Männikkö R, Udd B, Cannon SC. Myasthenic congenital myopathy from recessive mutations at a single residue in NaV1.4. Neurology 92: e1405–e1415, 2019. doi: 10.1212/WNL.0000000000007185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Tsujino A, Maertens C, Ohno K, Shen XM, Fukuda T, Harper CM, Cannon SC, Engel AG. Myasthenic syndrome caused by mutation of the SCN4A sodium channel. Proc Natl Acad Sci USA 100: 7377–7382, 2003. doi: 10.1073/pnas.1230273100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Ravenscroft G, Sollis E, Charles AK, North KN, Baynam G, Laing NG. Fetal akinesia: review of the genetics of the neuromuscular causes. J Med Genet 48: 793–801, 2011. doi: 10.1136/jmedgenet-2011-100211. [DOI] [PubMed] [Google Scholar]
- 126.Zaharieva IT, Thor MG, Oates EC, van Karnebeek C, Hendson G, Blom E, et al. Loss-of-function mutations in SCN4A cause severe foetal hypokinesia or ‘classical’ congenital myopathy. Brain 139: 674–691, 2016. doi: 10.1093/brain/awv352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Hayward LJ, Kim JS, Lee MY, Zhou H, Kim JW, Misra K, Salajegheh M, Wu FF, Matsuda C, Reid V, Cros D, Hoffman EP, Renaud JM, Cannon SC, Brown RH Jr.. Targeted mutation of mouse skeletal muscle sodium channel produces myotonia and potassium-sensitive weakness. J Clin Invest 118: 1437–1449, 2008. doi: 10.1172/JCI32638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Guerrini R, Marini C, Mantegazza M. Genetic epilepsy syndromes without structural brain abnormalities: clinical features and experimental models. Neurotherapeutics 11: 269–285, 2014. doi: 10.1007/s13311-014-0267-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Mantegazza M, Curia G, Biagini G, Ragsdale DS, Avoli M. Voltage-gated sodium channels as therapeutic targets in epilepsy and other neurological disorders. Lancet Neurol 9: 413–424, 2010. doi: 10.1016/S1474-4422(10)70059-4. [DOI] [PubMed] [Google Scholar]
- 130.Beckh S, Noda M, Lübbert H, Numa S. Differential regulation of three sodium channel messenger RNAs in the rat central nervous system during development. EMBO J 8: 3611–3616, 1989. doi: 10.1002/j.1460-2075.1989.tb08534.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Gordon D, Merrick D, Auld V, Dunn R, Goldin AL, Davidson N, Catterall WA. Tissue-specific expression of the RI and RII sodium channel subtypes. Proc Natl Acad Sci USA 84: 8682–8686, 1987. doi: 10.1073/pnas.84.23.8682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Kohrman DC, Smith MR, Goldin AL, Harris J, Meisler MH. A missense mutation in the sodium channel Scn8a is responsible for cerebellar ataxia in the mouse mutant jolting. J Neurosci 16: 5993–5999, 1996. doi: 10.1523/JNEUROSCI.16-19-05993.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Trimmer JS, Rhodes KJ. Localization of voltage-gated ion channels in mammalian brain. Annu Rev Physiol 66: 477–519, 2004. doi: 10.1146/annurev.physiol.66.032102.113328. [DOI] [PubMed] [Google Scholar]
- 134.Tzoumaka E, Tischler AC, Sangameswaran L, Eglen RM, Hunter JC, Novakovic SD. Differential distribution of the tetrodotoxin-sensitive rPN4/NaCh6/Scn8a sodium channel in the nervous system. J Neurosci Res 60: 37–44, 2000. doi:. [DOI] [PubMed] [Google Scholar]
- 135.Westenbroek RE, Merrick DK, Catterall WA. Differential subcellular localization of the RI and RII Na+ channel subtypes in central neurons. Neuron 3: 695–704, 1989. doi: 10.1016/0896-6273(89)90238-9. [DOI] [PubMed] [Google Scholar]
- 136.Boiko T, Van Wart A, Caldwell JH, Levinson SR, Trimmer JS, Matthews G. Functional specialization of the axon initial segment by isoform-specific sodium channel targeting. J Neurosci 23: 2306–2313, 2003. doi: 10.1523/JNEUROSCI.23-06-02306.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Vacher H, Mohapatra DP, Trimmer JS. Localization and targeting of voltage-dependent ion channels in mammalian central neurons. Physiol Rev 88: 1407–1447, 2008. doi: 10.1152/physrev.00002.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Brunklaus A, Du J, Steckler F, Ghanty II, Johannesen KM, Fenger CD, et al. Biological concepts in human sodium channel epilepsies and their relevance in clinical practice. Epilepsia 61: 387–399, 2020. doi: 10.1111/epi.16438. [DOI] [PubMed] [Google Scholar]
- 139.Meisler MH, O’Brien JE, Sharkey LM. Sodium channel gene family: epilepsy mutations, gene interactions and modifier effects. J Physiol 588: 1841–1848, 2010. doi: 10.1113/jphysiol.2010.188482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Catterall WA, Kalume F, Oakley JC. NaV1.1 channels and epilepsy. J Physiol 588: 1849–1859, 2010. doi: 10.1113/jphysiol.2010.187484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Mantegazza M, Broccoli V. SCN1A/NaV1.1 channelopathies: Mechanisms in expression systems, animal models, and human iPSC models. Epilepsia 60: S25–S38, 2019. doi: 10.1111/epi.14700. [DOI] [PubMed] [Google Scholar]
- 142.Scheffer IE, Berkovic S, Capovilla G, Connolly MB, French J, Guilhoto L, Hirsch E, Jain S, Mathern GW, Moshé SL, Nordli DR, Perucca E, Tomson T, Wiebe S, Zhang YH, Zuberi SM. ILAE classification of the epilepsies: position paper of the ILAE Commission for Classification and Terminology. Epilepsia 58: 512–521, 2017. doi: 10.1111/epi.13709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Dravet C. The core Dravet syndrome phenotype. Epilepsia 52, Suppl 2: 3–9, 2011. doi: 10.1111/j.1528-1167.2011.02994.x. [DOI] [PubMed] [Google Scholar]
- 144.Genton P, Velizarova R, Dravet C. Dravet syndrome: the long-term outcome. Epilepsia 52: 44–49, 2011. doi: 10.1111/j.1528-1167.2011.03001.x. [DOI] [PubMed] [Google Scholar]
- 145.Cooper MS, Mcintosh A, Crompton DE, McMahon JM, Schneider A, Farrell K, Ganesan V, Gill D, Kivity S, Lerman-Sagie T, McLellan A, Pelekanos J, Ramesh V, Sadleir L, Wirrell E, Scheffer IE. Mortality in Dravet syndrome. Epilepsy Res 128: 43–47, 2016. doi: 10.1016/j.eplepsyres.2016.10.006. [DOI] [PubMed] [Google Scholar]
- 146.Claes L, Ceulemans B, Audenaert D, Smets K, Löfgren A, Del-Favero J, Ala-Mello S, Basel-Vanagaite L, Plecko B, Raskin S, Thiry P, Wolf NI, Van Broeckhoven C, De Jonghe P. De novo SCN1A mutations are a major cause of severe myoclonic epilepsy of infancy. Hum Mutat 21: 615–621, 2003. doi: 10.1002/humu.10217. [DOI] [PubMed] [Google Scholar]
- 147.Claes L, Del-Favero J, Ceulemans B, Lagae L, Van Broeckhoven C, De Jonghe P. De novo mutations in the sodium-channel gene SCN1A cause severe myoclonic epilepsy of infancy. Am J Hum Genet 68: 1327–1332, 2001. doi: 10.1086/320609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Fujiwara T, Sugawara T, Mazaki-Miyazaki E, Takahashi Y, Fukushima K, Watanabe M, Hara K, Morikawa T, Yagi K, Yamakawa K, Inoue Y. Mutations of sodium channel alpha subunit type 1 (SCN1A) in intractable childhood epilepsies with frequent generalized tonic-clonic seizures. Brain 126: 531–546, 2003. doi: 10.1093/brain/awg053. [DOI] [PubMed] [Google Scholar]
- 149.Sugawara T, Mazaki-Miyazaki E, Fukushima K, Shimomura J, Fujiwara T, Hamano S, Inoue Y, Yamakawa K. Frequent mutations of SCN1A in severe myoclonic epilepsy in infancy. Neurology 58: 1122–1124, 2002. doi: 10.1212/wnl.58.7.1122. [DOI] [PubMed] [Google Scholar]
- 150.Bechi G, Scalmani P, Schiavon E, Rusconi R, Franceschetti S, Mantegazza M. Pure haploinsufficiency for Dravet syndrome NaV1.1 (SCN1A) sodium channel truncating mutations. Epilepsia 53: 87–100, 2012. doi: 10.1111/j.1528-1167.2011.03346.x. [DOI] [PubMed] [Google Scholar]
- 151.Tai C, Abe Y, Westenbroek RE, Scheuer T, Catterall WA. Impaired excitability of somatostatin- and parvalbumin-expressing cortical interneurons in a mouse model of Dravet syndrome. Proc Natl Acad Sci USA 111: E3139–E3148, 2014. doi: 10.1073/pnas.1411131111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Han S, Tai C, Westenbroek RE, Yu FH, Cheah CS, Potter GB, Rubenstein JL, Scheuer T, de la Iglesia HO, Catterall WA. Autistic-like behaviour in Scn1a+/- mice and rescue by enhanced GABA-mediated neurotransmission. Nature 489: 385–390, 2012. doi: 10.1038/nature11356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Mantegazza M. Dravet syndrome: insights from in vitro experimental models. Epilepsia 52: 62–69, 2011. doi: 10.1111/j.1528-1167.2011.03005.x. [DOI] [PubMed] [Google Scholar]
- 154.Ogiwara I, Miyamoto H, Morita N, Atapour N, Mazaki E, Inoue I, Takeuchi T, Itohara S, Yanagawa Y, Obata K, Furuichi T, Hensch TK, Yamakawa K. NaV1.1 localizes to axons of parvalbumin-positive inhibitory interneurons: a circuit basis for epileptic seizures in mice carrying an Scn1a gene mutation. J Neurosci 27: 5903–5914, 2007. doi: 10.1523/JNEUROSCI.5270-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.De Stasi AM, Farisello P, Marcon I, Cavallari S, Forli A, Vecchia D, Losi G, Mantegazza M, Panzeri S, Carmignoto G, Bacci A, Fellin T. Unaltered network activity and interneuronal firing during spontaneous cortical dynamics in vivo in a mouse model of severe myoclonic epilepsy of infancy. Cereb Cortex 26: 1778–1794, 2016. doi: 10.1093/cercor/bhw002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Kalume F, Oakley JC, Westenbroek RE, Gile J, de la Iglesia HO, Scheuer T, Catterall WA. Sleep impairment and reduced interneuron excitability in a mouse model of Dravet Syndrome. Neurobiol Dis 77: 141–154, 2015. doi: 10.1016/j.nbd.2015.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Kalume F, Yu FH, Westenbroek RE, Scheuer T, Catterall WA. Reduced sodium current in Purkinje neurons from NaV1.1 mutant mice: implications for ataxia in severe myoclonic epilepsy in infancy. J Neurosci 27: 11065–11074, 2007. doi: 10.1523/JNEUROSCI.2162-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Hedrich UB, Liautard C, Kirschenbaum D, Pofahl M, Lavigne J, Liu Y, Theiss S, Slotta J, Escayg A, Dihné M, Beck H, Mantegazza M, Lerche H. Impaired action potential initiation in GABAergic interneurons causes hyperexcitable networks in an epileptic mouse model carrying a human NaV1.1 mutation. J Neurosci 34: 14874–14889, 2014. doi: 10.1523/JNEUROSCI.0721-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Liautard C, Scalmani P, Carriero G, de Curtis M, Franceschetti S, Mantegazza M. Hippocampal hyperexcitability and specific epileptiform activity in a mouse model of Dravet syndrome. Epilepsia 54: 1251–1261, 2013. doi: 10.1111/epi.12213. [DOI] [PubMed] [Google Scholar]
- 160.Cheah CS, Lundstrom BN, Catterall WA, Oakley JC. Impairment of sharp-wave ripples in a murine model of Dravet Syndrome. J Neurosci 39: 9251–9260, 2019. doi: 10.1523/JNEUROSCI.0890-19.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Buzsáki G. Hippocampal sharp wave-ripple: a cognitive biomarker for episodic memory and planning. Hippocampus 25: 1073–1188, 2015. doi: 10.1002/hipo.22488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Stein RE, Kaplan JS, Li J, Catterall WA. Hippocampal deletion of NaV1.1 channels in mice causes thermal seizures and cognitive deficit characteristic of Dravet Syndrome. Proc Natl Acad Sci USA 116: 16571–16576, 2019. doi: 10.1073/pnas.1906833116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Silberberg G, Markram H. Disynaptic inhibition between neocortical pyramidal cells mediated by Martinotti cells. Neuron 53: 735–746, 2007. doi: 10.1016/j.neuron.2007.02.012. [DOI] [PubMed] [Google Scholar]
- 164.Oguni H, Hayashi K, Awaya Y, Fukuyama Y, Osawa M. Severe myoclonic epilepsy in infants—a review based on the Tokyo Women’s Medical University series of 84 cases. Brain Dev 23: 736–748, 2001. doi: 10.1016/S0387-7604(01)00276-5. [DOI] [PubMed] [Google Scholar]
- 165.Oakley JC, Kalume F, Yu FH, Scheuer T, Catterall WA. Temperature- and age-dependent seizures in a mouse model of severe myoclonic epilepsy in infancy. Proc Natl Acad Sci USA 106: 3994–3999, 2009. doi: 10.1073/pnas.0813330106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Cheah CS, Westenbroek RE, Roden WH, Kalume F, Oakley JC, Jansen LA, Catterall WA. Correlations in timing of sodium channel expression, epilepsy, and sudden death in Dravet syndrome. Channels (Austin) 7: 468–472, 2013. doi: 10.4161/chan.26023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Cheah CS, Yu FH, Westenbroek RE, Kalume FK, Oakley JC, Potter GB, Rubenstein JL, Catterall WA. Specific deletion of NaV1.1 sodium channels in inhibitory interneurons causes seizures and premature death in a mouse model of Dravet syndrome. Proc Natl Acad Sci USA 109: 14646–14651, 2012. doi: 10.1073/pnas.1211591109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Ogiwara I, Iwasato T, Miyamoto H, Iwata R, Yamagata T, Mazaki E, Yanagawa Y, Tamamaki N, Hensch TK, Itohara S, Yamakawa K. NaV1.1 haploinsufficiency in excitatory neurons ameliorates seizure-associated sudden death in a mouse model of Dravet syndrome. Hum Mol Genet 22: 4784–4804, 2013. doi: 10.1093/hmg/ddt331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Potter GB, Petryniak MA, Shevchenko E, McKinsey GL, Ekker M, Rubenstein JL. Generation of Cre-transgenic mice using Dlx1/Dlx2 enhancers and their characterization in GABAergic interneurons. Mol Cell Neurosci 40: 167–186, 2009. doi: 10.1016/j.mcn.2008.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Du J, Simmons S, Brunklaus A, Adiconis X, Hession CC, Fu Z, Li Y, Shema R, Moller RS, Barak B, Feng G, Meisler M, Sanders S, Lerche H, Campbell AJ, McCarroll S, Levin JZ, Lal D. Differential excitatory vs inhibitory SCN expression at single cell level regulates brain sodium channel function in neurodevelopmental disorders. Eur J Paediatr Neurol 24: 129–133, 2020. doi: 10.1016/j.ejpn.2019.12.019. [DOI] [PubMed] [Google Scholar]
- 171.Stern WM, Sander JW, Rothwell JC, Sisodiya SM. Impaired intracortical inhibition demonstrated in vivo in people with Dravet syndrome. Neurology 88: 1659–1665, 2017. doi: 10.1212/WNL.0000000000003868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Catterall WA. Dravet Syndrome: a sodium channel interneuronopathy. Curr Opin Physiol 2: 42–50, 2018. doi: 10.1016/j.cophys.2017.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Tatsukawa T, Ogiwara I, Mazaki E, Shimohata A, Yamakawa K. Impairments in social novelty recognition and spatial memory in mice with conditional deletion of Scn1a in parvalbumin-expressing cells. Neurobiol Dis 112: 24–34, 2018. doi: 10.1016/j.nbd.2018.01.009. [DOI] [PubMed] [Google Scholar]
- 174.Ritter-Makinson S, Clemente-Perez A, Higashikubo B, Cho FS, Holden SS, Bennett E, Chkhaidze A, Eelkman Rooda OH, Cornet MC, Hoebeek FE, Yamakawa K, Cilio MR, Delord B, Paz JT. Augmented reticular thalamic bursting and seizures in Scn1a-Dravet Syndrome. Cell Rep 26: 1071, 2019. doi: 10.1016/j.celrep.2019.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Han S, Yu FH, Schwartz MD, Linton JD, Bosma MM, Hurley JB, Catterall WA, de la Iglesia HO. NaV1.1 channels are critical for intercellular communication in the suprachiasmatic nucleus and for normal circadian rhythms. Proc Natl Acad Sci USA 109: E368–E377, 2012. doi: 10.1073/pnas.1115729109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Rubinstein M, Han S, Tai C, Westenbroek RE, Hunker A, Scheuer T, Catterall WA. Dissecting the phenotypes of Dravet syndrome by gene deletion. Brain 138: 2219–2233, 2015. doi: 10.1093/brain/awv142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Rudy B, Fishell G, Lee S, Hjerling-Leffler J. Three groups of interneurons account for nearly 100% of neocortical GABAergic neurons. Dev Neurobiol 71: 45–61, 2011. doi: 10.1002/dneu.20853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Tremblay R, Lee S, Rudy B. GABAergic interneurons in the neocortex: from cellular properties to circuits. Neuron 91: 260–292, 2016. doi: 10.1016/j.neuron.2016.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Williams AD, Cheah CS, Catterall WA, Oakley JC. Diminished excitability of 5-HT3aR-expressing GABAergic interneurons but no pro-epileptic effects caused by selective deletion of NaV1.1 channels in a mouse model of Dravet Syndrome ( Abstract). In: 2017 Neuroscience Meeting Planner. Washington, DC: Society for Neuroscience, 2017, Program No. 203.06. [Google Scholar]
- 180.Yamagata T, Ogiwara I, Mazaki E, Yanagawa Y, Yamakawa K. NaV1.2 is expressed in caudal ganglionic eminence-derived disinhibitory interneurons: mutually exclusive distributions of NaV1.1 and NaV1.2. Biochem Biophys Res Commun 491: 1070–1076, 2017. [DOI] [PubMed] [Google Scholar]
- 181.Paul A, Crow M, Raudales R, He M, Gillis J, Huang ZJ. Transcriptional architecture of synaptic communication delineates GABAergic neuron identity. Cell 171: 522–539.e20, 2017. doi: 10.1016/j.cell.2017.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Tasic B, Menon V, Nguyen TN, Kim TK, Jarsky T, Yao Z, Levi B, Gray LT, Sorensen SA, Dolbeare T, Bertagnolli D, Goldy J, Shapovalova N, Parry S, Lee C, Smith K, Bernard A, Madisen L, Sunkin SM, Hawrylycz M, Koch C, Zeng H. Adult mouse cortical cell taxonomy revealed by single cell transcriptomics. Nat Neurosci 19: 335–346, 2016. doi: 10.1038/nn.4216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Goff KM, Goldberg EM. Vasoactive intestinal peptide-expressing interneurons are impaired in a mouse model of Dravet syndrome. Elife 8: e46846, 2019. doi: 10.7554/eLife.46846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Nolan K, Camfield CS, Camfield PR. Coping with a child with Dravet syndrome: insights from families. J Child Neurol 23: 690–694, 2008. doi: 10.1177/0883073808314162. [DOI] [PubMed] [Google Scholar]
- 185.Skluzacek JV, Watts KP, Parsy O, Wical B, Camfield P. Dravet syndrome and parent associations: the IDEA League experience with comorbid conditions, mortality, management, adaptation, and grief. Epilepsia 52: 95–101, 2011. doi: 10.1111/j.1528-1167.2011.03012.x. [DOI] [PubMed] [Google Scholar]
- 186.Kalume F, Westenbroek RE, Cheah CS, Yu FH, Oakley JC, Scheuer T, Catterall WA. Sudden unexpected death in a mouse model of Dravet syndrome. J Clin Invest 123: 1798–1808, 2013. doi: 10.1172/JCI66220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Kim Y, Bravo E, Thirnbeck CK, Smith-Mellecker LA, Kim SH, Gehlbach BK, Laux LC, Zhou X, Nordli DR Jr, Richerson GB. Severe peri-ictal respiratory dysfunction is common in Dravet syndrome. J Clin Invest 128: 1141–1153, 2018. doi: 10.1172/JCI94999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Rubinstein M, Westenbroek RE, Yu FH, Jones CJ, Scheuer T, Catterall WA. Genetic background modulates impaired excitability of inhibitory neurons in a mouse model of Dravet syndrome. Neurobiol Dis 73: 106–117, 2015. doi: 10.1016/j.nbd.2014.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Hawkins NA, Zachwieja NJ, Miller AR, Anderson LL, Kearney JA. Fine mapping of a Dravet Syndrome modifier locus on mouse chromosome 5 and candidate gene analysis by RNA-Seq. PLoS Genet 12: e1006398, 2016. doi: 10.1371/journal.pgen.1006398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Hawkins NA, Anderson LL, Gertler TS, Laux L, George AL Jr, Kearney JA. Screening of conventional anticonvulsants in a genetic mouse model of epilepsy. Ann Clin Transl Neurol 4: 326–339, 2017. doi: 10.1002/acn3.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Mistry AM, Thompson CH, Miller AR, Vanoye CG, George AL Jr, Kearney JA. Strain- and age-dependent hippocampal neuron sodium currents correlate with epilepsy severity in Dravet syndrome mice. Neurobiol Dis 65: 1–11, 2014. doi: 10.1016/j.nbd.2014.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Favero M, Sotuyo NP, Lopez E, Kearney JA, Goldberg EM. A transient developmental window of fast-spiking interneuron dysfunction in a mouse model of Dravet Syndrome. J Neurosci 38: 7912–7927, 2018. doi: 10.1523/JNEUROSCI.0193-18.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Dutton SB, Sawyer NT, Kalume F, Jumbo-Lucioni P, Borges K, Catterall WA, Escayg A. Protective effect of the ketogenic diet in Scn1a mutant mice. Epilepsia 52: 2050–2056, 2011. doi: 10.1111/j.1528-1167.2011.03211.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Cao D, Ohtani H, Ogiwara I, Ohtani S, Takahashi Y, Yamakawa K, Inoue Y. Efficacy of stiripentol in hyperthermia-induced seizures in a mouse model of Dravet syndrome. Epilepsia 53: 1140–1145, 2012. doi: 10.1111/j.1528-1167.2012.03497.x. [DOI] [PubMed] [Google Scholar]
- 195.Oakley JC, Cho AR, Cheah CS, Scheuer T, Catterall WA. Synergistic GABA-enhancing therapy against seizures in a mouse model of Dravet syndrome. J Pharmacol Exp Ther 345: 215–224, 2013. doi: 10.1124/jpet.113.203331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Hawkins NA, Lewis M, Hammond RS, Doherty JJ, Kearney JA. The synthetic neuroactive steroid SGE-516 reduces seizure burden and improves survival in a Dravet syndrome mouse model. Sci Rep 7: 15327, 2017. doi: 10.1038/s41598-017-15609-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Anderson LL, Hawkins NA, Thompson CH, Kearney JA, George AL Jr.. Unexpected efficacy of a novel sodium channel modulator in Dravet Syndrome. Sci Rep 7: 1682, 2017. doi: 10.1038/s41598-017-01851-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Richards KL, Milligan CJ, Richardson RJ, Jancovski N, Grunnet M, Jacobson LH, Undheim EA, Mobli M, Chow CY, Herzig V, Csoti A, Panyi G, Reid CA, King GF, Petrou S. Selective NaV1.1 activation rescues Dravet syndrome mice from seizures and premature death. Proc Natl Acad Sci USA 115: E8077–E8085, 2018. doi: 10.1073/pnas.1804764115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Escoubas P, Diochot S, Célérier ML, Nakajima T, Lazdunski M. Novel tarantula toxins for subtypes of voltage-dependent potassium channels in the KV2 and KV4 subfamilies. Mol Pharmacol 62: 48–57, 2002. doi: 10.1124/mol.62.1.48. [DOI] [PubMed] [Google Scholar]
- 200.Devinsky O, Cross JH, Laux L, Marsh E, Miller I, Nabbout R, Scheffer IE, Thiele EA, Wright S; Cannabidiol in Dravet Syndrome Study Group. Trial of cannabidiol for drug-resistant seizures in Dravet Syndrome. N Engl J Med 376: 2011–2020, 2017. doi: 10.1056/NEJMoa1611618. [DOI] [PubMed] [Google Scholar]
- 201.Kaplan JS, Stella N, Catterall WA, Westenbroek RE. Cannabidiol attenuates seizures and social deficits in a mouse model of Dravet syndrome. Proc Natl Acad Sci USA 114: 11229–11234, 2017. doi: 10.1073/pnas.1711351114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Ghovanloo MR, Shuart NG, Mezeyova J, Dean RA, Ruben PC, Goodchild SJ. Inhibitory effects of cannabidiol on voltage-dependent sodium currents. J Biol Chem 293: 16546–16558, 2018. doi: 10.1074/jbc.RA118.004929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Patel RR, Barbosa C, Brustovetsky T, Brustovetsky N, Cummins TR. Aberrant epilepsy-associated mutant NaV1.6 sodium channel activity can be targeted with cannabidiol. Brain 139: 2164–2181, 2016. doi: 10.1093/brain/aww129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Sait LG, Sula A, Ghovanloo MR, Hollingworth D, Ruben PC, Wallace BA. Cannabidiol interactions with voltage-gated sodium channels. Elife 9: e58593, 2020. doi: 10.7554/eLife.58593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Baraban SC, Dinday MT, Hortopan GA. Drug screening in Scn1a zebrafish mutant identifies clemizole as a potential Dravet syndrome treatment. Nat Commun 4: 2410, 2013. doi: 10.1038/ncomms3410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Dinday MT, Baraban SC. Large-scale phenotype-based antiepileptic drug screening in a zebrafish model of Dravet Syndrome. eNeuro 2: ENEURO.0068-15.2015, 2015. doi: 10.1523/ENEURO.0068-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Griffin A, Hamling KR, Hong S, Anvar M, Lee LP, Baraban SC. Preclinical animal models for Dravet Syndrome: seizure phenotypes, comorbidities and drug screening. Front Pharmacol 9: 573, 2018. doi: 10.3389/fphar.2018.00573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Sourbron J, Partoens M, Scheldeman C, Zhang Y, Lagae L, de Witte P. Drug repurposing for Dravet syndrome in scn1Lab-/- mutant zebrafish. Epilepsia 60: e8–e13, 2019. doi: 10.1111/epi.14647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Hsiao J, Yuan TY, Tsai MS, Lu CY, Lin YC, Lee ML, Lin SW, Chang FC, Liu Pimentel H, Olive C, Coito C, Shen G, Young M, Thorne T, Lawrence M, Magistri M, Faghihi MA, Khorkova O, Wahlestedt C. Upregulation of haploinsufficient gene expression in the brain by targeting a long non-coding RNA improves seizure phenotype in a model of Dravet Syndrome. EBioMedicine 9: 257–277, 2016. doi: 10.1016/j.ebiom.2016.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Lenk GM, Jafar-Nejad P, Hill SF, Huffman LD, Smolen CE, Wagnon JL, Petit H, Yu W, Ziobro J, Bhatia K, Parent J, Giger RJ, Rigo F, Meisler MH. Scn8a antisense oligonucleotide is protective in mouse models of SCN8A encephalopathy and Dravet syndrome. Ann Neurol 87: 339–346, 2020. doi: 10.1002/ana.25676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Martin MS, Tang B, Papale LA, Yu FH, Catterall WA, Escayg A. The voltage-gated sodium channel Scn8a is a genetic modifier of severe myoclonic epilepsy of infancy. Hum Mol Genet 16: 2892–2899, 2007. doi: 10.1093/hmg/ddm248. [DOI] [PubMed] [Google Scholar]
- 212.Colasante G, Lignani G, Brusco S, Di Berardino C, Carpenter J, Giannelli S, Valassina N, Bido S, Ricci R, Castoldi V, Marenna S, Church T, Massimino L, Morabito G, Benfenati F, Schorge S, Leocani L, Kullmann DM, Broccoli V. dCas9-based Scn1a gene activation restores inhibitory interneuron excitability and attenuates seizures in Dravet Syndrome mice. Mol Ther 28: 235–253, 2020. doi: 10.1016/j.ymthe.2019.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Han Z, Chen C, Christiansen A, Jin S, Lin Q, Anumonwo C, Liu C, Leiser SC, Meena, Aznarez I, Liau G, Isom LL. Antisense oligonucleotides increase Scn1a expression and reduce seizures and SUDEP incidence in a mouse model of Dravet syndrome. Sci Transl Med 12: eaaz6100, 2020. doi: 10.1126/scitranslmed.aaz6100. [DOI] [PubMed] [Google Scholar]
- 214.Escayg A, MacDonald BT, Meisler MH, Baulac S, Huberfeld G, An-Gourfinkel I, Brice A, Leguern E, Moulard B, Chaigne D, Buresi C, Malafosse A. Mutations of SCN1A, encoding a neuronal sodium channel, in two families with GEFS+2. Nat Genet 24: 343–345, 2000. doi: 10.1038/74159. [DOI] [PubMed] [Google Scholar]
- 215.Zhang YH, Burgess R, Malone JP, Glubb GC, Helbig KL, Vadlamudi L, Kivity S, Afawi Z, Bleasel A, Grattan-Smith P, Grinton BE, Bellows ST, Vears DF, Damiano JA, Goldberg-Stern H, Korczyn AD, Dibbens LM, Ruzzo EK, Hildebrand MS, Berkovic SF, Scheffer IE. Genetic epilepsy with febrile seizures plus: refining the spectrum. Neurology 89: 1210–1219, 2017. doi: 10.1212/WNL.0000000000004384. [DOI] [PubMed] [Google Scholar]
- 216.Lossin C, Wang DW, Rhodes TH, Vanoye CG, George AL Jr.. Molecular basis of an inherited epilepsy. Neuron 34: 877–884, 2002. doi: 10.1016/S0896-6273(02)00714-6. [DOI] [PubMed] [Google Scholar]
- 217.Lossin C, Rhodes TH, Desai RR, Vanoye CG, Wang D, Carniciu S, Devinsky O, George AL Jr.. Epilepsy-associated dysfunction in the voltage-gated neuronal sodium channel SCN1A. J Neurosci 23: 11289–11295, 2003. doi: 10.1523/JNEUROSCI.23-36-11289.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Tang B, Dutt K, Papale L, Rusconi R, Shankar A, Hunter J, Tufik S, Yu FH, Catterall WA, Mantegazza M, Goldin AL, Escayg A. A BAC transgenic mouse model reveals neuron subtype-specific effects of a generalized epilepsy with febrile seizures plus (GEFS+) mutation. Neurobiol Dis 35: 91–102, 2009. doi: 10.1016/j.nbd.2009.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Depienne C, Trouillard O, Gourfinkel-An I, Saint-Martin C, Bouteiller D, Graber D, Barthez-Carpentier MA, Gautier A, Villeneuve N, Dravet C, Livet MO, Rivier-Ringenbach C, Adam C, Dupont S, Baulac S, Héron D, Nabbout R, Leguern E. Mechanisms for variable expressivity of inherited SCN1A mutations causing Dravet syndrome. J Med Genet 47: 404–410, 2010. doi: 10.1136/jmg.2009.074328. [DOI] [PubMed] [Google Scholar]
- 220.Papale LA, Makinson CD, Christopher Ehlen J, Tufik S, Decker MJ, Paul KN, Escayg A. Altered sleep regulation in a mouse model of SCN1A-derived genetic epilepsy with febrile seizures plus (GEFS+). Epilepsia 54: 625–634, 2013. doi: 10.1111/epi.12060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Salgueiro-Pereira AR, Duprat F, Pousinha PA, Loucif A, Douchamps V, Regondi C, Ayrault M, Eugie M, Stunault MI, Escayg A, Goutagny R, Gnatkovsky V, Frassoni C, Marie H, Bethus I, Mantegazza M. A two-hit story: seizures and genetic mutation interaction sets phenotype severity in SCN1A epilepsies. Neurobiol Dis 125: 31–44, 2019. doi: 10.1016/j.nbd.2019.01.006. [DOI] [PubMed] [Google Scholar]
- 222.EPICURE Consortium, EMINet Consortium, Steffens M, Leu C, Ruppert AK, Zara F, et al. Genome-wide association analysis of genetic generalized epilepsies implicates susceptibility loci at 1q43, 2p16.1, 2q22.3 and 17q21.32. Hum Mol Genet 21: 5359–5372, 2012. doi: 10.1093/hmg/dds373. [DOI] [PubMed] [Google Scholar]
- 223.Epi4K Consortium, Epilepsy Phenome/Genome Project. Ultra-rare genetic variation in common epilepsies: a case-control sequencing study. Lancet Neurol 16: 135–143, 2017. doi: 10.1016/S1474-4422(16)30359-3. [DOI] [PubMed] [Google Scholar]
- 224.International League Against Epilepsy Consortium on Complex Epilepsies. Genetic determinants of common epilepsies: a meta-analysis of genome-wide association studies. Lancet Neurol 13: 893–903, 2014. doi: 10.1016/S1474-4422(14)70171-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Kasperaviciute D, Catarino CB, Matarin M, Leu C, Novy J, Tostevin A, et al. Epilepsy, hippocampal sclerosis and febrile seizures linked by common genetic variation around SCN1A. Brain 136: 3140–3150, 2013. doi: 10.1093/brain/awt233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Bender AC, Natola H, Ndong C, Holmes GL, Scott RC, Lenck-Santini PP. Focal Scn1a knockdown induces cognitive impairment without seizures. Neurobiol Dis 54: 297–307, 2013. doi: 10.1016/j.nbd.2012.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Sakkaki S, Barrière S, Bender AC, Scott RC, Lenck-Santini PP. Focal dorsal hippocampal NaV1.1 knock down alters place cell temporal coordination and spatial behavior. Cereb Cortex 30: 5049–5066, 2020. doi: 10.1093/cercor/bhaa101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Bechi G, Rusconi R, Cestèle S, Striano P, Franceschetti S, Mantegazza M. Rescuable folding defective NaV1.1 (SCN1A) mutants in epilepsy: properties, occurrence, and novel rescuing strategy with peptides targeted to the endoplasmic reticulum. Neurobiol Dis 75: 100–114, 2015. doi: 10.1016/j.nbd.2014.12.028. [DOI] [PubMed] [Google Scholar]
- 229.Rusconi R, Combi R, Cestèle S, Grioni D, Franceschetti S, Dalprà L, Mantegazza M. A rescuable folding defective NaV1.1 (SCN1A) sodium channel mutant causes GEFS+: common mechanism in NaV1.1 related epilepsies? Hum Mutat 30: E747–E760, 2009. doi: 10.1002/humu.21041. [DOI] [PubMed] [Google Scholar]
- 230.Rusconi R, Scalmani P, Cassulini RR, Giunti G, Gambardella A, Franceschetti S, Annesi G, Wanke E, Mantegazza M. Modulatory proteins can rescue a trafficking defective epileptogenic NaV1.1 Na+ channel mutant. J Neurosci 27: 11037–11046, 2007. doi: 10.1523/JNEUROSCI.3515-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Sugiura Y, Ogiwara I, Hoshi A, Yamakawa K, Ugawa Y. Different degrees of loss of function between GEFS+ and SMEI NaV1.1 missense mutants at the same residue induced by rescuable folding defects. Epilepsia 53: e111–e114, 2012. doi: 10.1111/j.1528-1167.2012.03467.x. [DOI] [PubMed] [Google Scholar]
- 232.Thompson CH, Porter JC, Kahlig KM, Daniels MA, George AL Jr.. Nontruncating SCN1A mutations associated with severe myoclonic epilepsy of infancy impair cell surface expression. J Biol Chem 287: 42001–42008, 2012. doi: 10.1074/jbc.M112.421883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Cestèle S, Catterall WA. Molecular mechanisms of neurotoxin action on voltage-gated sodium channels. Biochimie 82: 883–892, 2000. doi: 10.1016/s0300-9084(00)01174-3. [DOI] [PubMed] [Google Scholar]
- 234.Dhifallah S, Lancaster E, Merrill S, Leroudier N, Mantegazza M, Cestèle S. Gain of function for the SCN1A/hNaV1.1-L1670W mutation responsible for familial hemiplegic migraine. Front Mol Neurosci 11: 232, 2018. doi: 10.3389/fnmol.2018.00232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Patel N, Ram D, Swiderska N, Mewasingh LD, Newton RW, Offringa M. Febrile seizures. BMJ 351: h4240, 2015. doi: 10.1136/bmj.h4240. [DOI] [PubMed] [Google Scholar]
- 236.Catarino CB, Liu JY, Liagkouras I, Gibbons VS, Labrum RW, Ellis R, Woodward C, Davis MB, Smith SJ, Cross JH, Appleton RE, Yendle SC, McMahon JM, Bellows ST, Jacques TS, Zuberi SM, Koepp MJ, Martinian L, Scheffer IE, Thom M, Sisodiya SM. Dravet syndrome as epileptic encephalopathy: evidence from long-term course and neuropathology. Brain 134: 2982–3010, 2011. doi: 10.1093/brain/awr129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Guerrini R, Striano P, Catarino C, Sisodiya SM. Neuroimaging and neuropathology of Dravet syndrome. Epilepsia 52, Suppl 2: 30–34, 2011. doi: 10.1111/j.1528-1167.2011.02998.x. [DOI] [PubMed] [Google Scholar]
- 238.Baulac S, Gourfinkel-An I, Nabbout R, Huberfeld G, Serratosa J, Leguern E, Baulac M. Fever, genes, and epilepsy. Lancet Neurol 3: 421–430, 2004. doi: 10.1016/S1474-4422(04)00808-7. [DOI] [PubMed] [Google Scholar]
- 239.Colosimo E, Gambardella A, Mantegazza M, Labate A, Rusconi R, Schiavon E, Annesi F, Cassulini RR, Carrideo S, Chifari R, Canevini MP, Canger R, Franceschetti S, Annesi G, Wanke E, Quattrone A. Electroclinical features of a family with simple febrile seizures and temporal lobe epilepsy associated with SCN1A loss-of-function mutation. Epilepsia 48: 1691–1696, 2007. doi: 10.1111/j.1528-1167.2007.01153.x. [DOI] [PubMed] [Google Scholar]
- 240.Mantegazza M, Gambardella A, Rusconi R, Schiavon E, Annesi F, Cassulini RR, Labate A, Carrideo S, Chifari R, Canevini MP, Canger R, Franceschetti S, Annesi G, Wanke E, Quattrone A. Identification of an NaV1.1 sodium channel (SCN1A) loss-of-function mutation associated with familial simple febrile seizures. Proc Natl Acad Sci USA 102: 18177–18182, 2005. doi: 10.1073/pnas.0506818102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Mullen SA, Scheffer IE. Translational research in epilepsy genetics: sodium channels in man to interneuronopathy in mouse. Arch Neurol 66: 21–26, 2009. doi: 10.1001/archneurol.2008.559. [DOI] [PubMed] [Google Scholar]
- 242.Sadleir LG, Mountier EI, Gill D, Davis S, Joshi C, DeVile C, Kurian MA, DDD Study, Mandelstam S, Wirrell E, Nickels KC, Murali HR, Carvill G, Myers CT, Mefford HC, Scheffer IE. Not all SCN1A epileptic encephalopathies are Dravet syndrome: early profound Thr226Met phenotype. Neurology 89: 1035–1042, 2017. doi: 10.1212/WNL.0000000000004331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Ohashi T, Akasaka N, Kobayashi Y, Magara S, Kawashima H, Matsumoto N, Saitsu H, Tohyama J. Infantile epileptic encephalopathy with a hyperkinetic movement disorder and hand stereotypies associated with a novel SCN1A mutation. Epileptic Disord 16: 208–212, 2014. doi: 10.1684/epd.2014.0649. [DOI] [PubMed] [Google Scholar]
- 244.Spagnoli C, Frattini D, Rizzi S, Salerno GG, Fusco C. Early infantile SCN1A epileptic encephalopathy: expanding the genotype-phenotype correlations. Seizure 65: 62–64, 2019. doi: 10.1016/j.seizure.2019.01.002. [DOI] [PubMed] [Google Scholar]
- 245.Berecki G, Bryson A, Terhag J, Maljevic S, Gazina EV, Hill SL, Petrou S. SCN1A gain of function in early infantile encephalopathy. Ann Neurol 85: 514–525, 2019. doi: 10.1002/ana.25438. [DOI] [PubMed] [Google Scholar]
- 246.Berecki G, Howell KB, Deerasooriya YH, Cilio MR, Oliva MK, Kaplan D, Scheffer IE, Berkovic SF, Petrou S. Dynamic action potential clamp predicts functional separation in mild familial and severe de novo forms of SCN2A epilepsy. Proc Natl Acad Sci USA 115: E5516–E5525, 2018. doi: 10.1073/pnas.1800077115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Berecki G, Zegers JG, Verkerk AO, Bhuiyan ZA, de Jonge B, Veldkamp MW, Wilders R, van Ginneken AC. HERG channel (dys)function revealed by dynamic action potential clamp technique. Biophys J 88: 566–578, 2005. doi: 10.1529/biophysj.104.047290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Mantegazza M, Cestèle S. Pathophysiological mechanisms of migraine and epilepsy: similarities and differences. Neurosci Lett 667: 92–102, 2018. doi: 10.1016/j.neulet.2017.11.025. [DOI] [PubMed] [Google Scholar]
- 249.Spampanato J, Escayg A, Meisler MH, Goldin AL. Generalized epilepsy with febrile seizures plus type 2 mutation W1204R alters voltage-dependent gating of NaV1.1 sodium channels. Neuroscience 116: 37–48, 2003. doi: 10.1016/S0306-4522(02)00698-X. [DOI] [PubMed] [Google Scholar]
- 250.Ferrari MD, Klever RR, Terwindt GM, Ayata C, van den Maagdenberg AM. Migraine pathophysiology: lessons from mouse models and human genetics. Lancet Neurol 14: 65–80, 2015. doi: 10.1016/S1474-4422(14)70220-0. [DOI] [PubMed] [Google Scholar]
- 251.Pietrobon D, Moskowitz MA. Pathophysiology of migraine. Annu Rev Physiol 75: 365–391, 2013. doi: 10.1146/annurev-physiol-030212-183717. [DOI] [PubMed] [Google Scholar]
- 252.Ophoff RA, Terwindt GM, Vergouwe MN, van Eijk R, Oefner PJ, Hoffman SM, Lamerdin JE, Mohrenweiser HW, Bulman DE, Ferrari M, Haan J, Lindhout D, van Ommen GJ, Hofker MH, Ferrari MD, Frants RR. Familial hemiplegic migraine and episodic ataxia type-2 are caused by mutations in the Ca2+ channel gene CACNL1A4. Cell 87: 543–552, 1996. doi: 10.1016/s0092-8674(00)81373-2. [DOI] [PubMed] [Google Scholar]
- 253.De Fusco M, Marconi R, Silvestri L, Atorino L, Rampoldi L, Morgante L, Ballabio A, Aridon P, Casari G. Haploinsufficiency of ATP1A2 encoding the Na+/K+ pump alpha2 subunit associated with familial hemiplegic migraine type 2. Nat Genet 33: 192–196, 2003. doi: 10.1038/ng1081. [DOI] [PubMed] [Google Scholar]
- 254.Dichgans M, Freilinger T, Eckstein G, Babini E, Lorenz-Depiereux B, Biskup S, Ferrari MD, Herzog J, van den Maagdenberg AM, Pusch M, Strom TM. Mutation in the neuronal voltage-gated sodium channel SCN1A in familial hemiplegic migraine. Lancet 366: 371–377, 2005. doi: 10.1016/S0140-6736(05)66786-4. [DOI] [PubMed] [Google Scholar]
- 255.Vahedi K, Depienne C, Le Fort D, Riant F, Chaine P, Trouillard O, Gaudric A, Morris MA, Leguern E, Tournier-Lasserve E, Bousser MG. Elicited repetitive daily blindness: a new phenotype associated with hemiplegic migraine and SCN1A mutations. Neurology 72: 1178–1183, 2009. doi: 10.1212/01.wnl.0000345393.53132.8c. [DOI] [PubMed] [Google Scholar]
- 256.Schubert V, Auffenberg E, Biskup S, Jurkat-Rott K, Freilinger T. Two novel families with hemiplegic migraine caused by recurrent SCN1A mutation p.F1499L. Cephalalgia 38: 1503–1508, 2018. doi: 10.1177/0333102417742365. [DOI] [PubMed] [Google Scholar]
- 257.Cestèle S, Scalmani P, Rusconi R, Terragni B, Franceschetti S, Mantegazza M. Self-limited hyperexcitability: functional effect of a familial hemiplegic migraine mutation of the NaV1.1 (SCN1A) Na+ channel. J Neurosci 28: 7273–7283, 2008. doi: 10.1523/JNEUROSCI.4453-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Kahlig KM, Rhodes TH, Pusch M, Freilinger T, Pereira-Monteiro JM, Ferrari MD, van den Maagdenberg AM, Dichgans M, George AL Jr.. Divergent sodium channel defects in familial hemiplegic migraine. Proc Natl Acad Sci USA 105: 9799–9804, 2008. doi: 10.1073/pnas.0711717105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Lossin C. A catalog of SCN1A variants. Brain Dev 31: 114–130, 2009. doi: 10.1016/j.braindev.2008.07.011. [DOI] [PubMed] [Google Scholar]
- 260.Schaller KL, Krzemien DM, McKenna NM, Caldwell JH. Alternatively spliced sodium channel transcripts in brain and muscle. J Neurosci 12: 1370–1381, 1992. doi: 10.1523/JNEUROSCI.12-04-01370.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Bernier V, Lagacé M, Bichet DG, Bouvier M. Pharmacological chaperones: potential treatment for conformational diseases. Trends Endocrinol Metab 15: 222–228, 2004. doi: 10.1016/j.tem.2004.05.003. [DOI] [PubMed] [Google Scholar]
- 262.Bertelli S, Barbieri R, Pusch M, Gavazzo P. Gain of function of sporadic/familial hemiplegic migraine-causing SCN1A mutations: use of an optimized cDNA. Cephalalgia 39: 477–488, 2019. doi: 10.1177/0333102418788336. [DOI] [PubMed] [Google Scholar]
- 263.Fan C, Wolking S, Lehmann-Horn F, Hedrich UB, Freilinger T, Lerche H, Borck G, Kubisch C, Jurkat-Rott K. Early-onset familial hemiplegic migraine due to a novel SCN1A mutation. Cephalalgia 36: 1238–1247, 2016. doi: 10.1177/0333102415608360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Castro MJ, Stam AH, Lemos C, de Vries B, Vanmolkot KR, Barros J, Terwindt GM, Frants RR, Sequeiros J, Ferrari MD, Pereira-Monteiro JM, van den Maagdenberg AM. First mutation in the voltage-gated NaV1.1 subunit gene SCN1A with co-occurring familial hemiplegic migraine and epilepsy. Cephalalgia 29: 308–313, 2009. doi: 10.1111/j.1468-2982.2008.01721.x. [DOI] [PubMed] [Google Scholar]
- 265.Barros J, Ferreira A, Brandão AF, Lemos C, Correia F, Damásio J, Tuna A, Sequeiros J, Coutinho P, Alonso I, Pereira-Monteiro J. Familial hemiplegic migraine due to L263V SCN1A mutation: discordance for epilepsy between two kindreds from Douro Valley. Cephalalgia 34: 1015–1020, 2014. doi: 10.1177/0333102414527015. [DOI] [PubMed] [Google Scholar]
- 266.Jansen NA, Dehghani A, Linssen MM, Breukel C, Tolner EA, van den Maagdenberg A. First FHM3 mouse model shows spontaneous cortical spreading depolarizations. Ann Clin Transl Neurol 7: 132–138, 2020. doi: 10.1002/acn3.50971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Cestèle S, Labate A, Rusconi R, Tarantino P, Mumoli L, Franceschetti S, Annesi G, Mantegazza M, Gambardella A. Divergent effects of the T1174S SCN1A mutation associated with seizures and hemiplegic migraine. Epilepsia 54: 927–935, 2013. doi: 10.1111/epi.12123. [DOI] [PubMed] [Google Scholar]
- 268.Gargus JJ, Tournay A. Novel mutation confirms seizure locus SCN1A is also familial hemiplegic migraine locus FHM3. Pediatr Neurol 37: 407–410, 2007. doi: 10.1016/j.pediatrneurol.2007.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Lal D, Reinthaler EM, Dejanovic B, May P, Thiele H, Lehesjoki AE, Schwarz G, Riesch E, Ikram MA, van Duijn CM, Uitterlinden AG, Hofman A, Steinböck H, Gruber-Sedlmayr U, Neophytou B, Zara F, Hahn A, Genetic Commission of the Italian League against Epilepsy, EuroEPINOMICS CoGIE Consortium, Gormley P, Becker F, Weber YG, Cilio MR, Kunz WS, Krause R, Zimprich F, Lemke JR, Nürnberg P, Sander T, Lerche H, Neubauer BA. Evaluation of presumably disease causing SCN1A variants in a cohort of common epilepsy syndromes. PLoS One 11: e0150426, 2016. doi: 10.1371/journal.pone.0150426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Li T, Tian C, Scalmani P, Frassoni C, Mantegazza M, Wang Y, Yang M, Wu S, Shu Y. Action potential initiation in neocortical inhibitory interneurons. PLoS Biol 12: e1001944, 2014. doi: 10.1371/journal.pbio.1001944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Pietrobon D, Moskowitz MA. Chaos and commotion in the wake of cortical spreading depression and spreading depolarizations. Nat Rev Neurosci 15: 379–393, 2014. doi: 10.1038/nrn3770. [DOI] [PubMed] [Google Scholar]
- 272.Chever O, Zerimech S, Scalmani P, Lemaire L, Loucif A, Ayrault M, Krupa M, Desroches M, Duprat F, Cestèle S, Mantegazza M. GABAergic neurons and NaV1.1 channel hyperactivity: a novel neocortex-specific mechanism of cortical spreading depression. bioRxiv 991158, 2020. doi: 10.1101/2020.03.14.991158. [DOI]
- 273.Desroches M, Faugeras O, Krupa M, Mantegazza M. Modeling cortical spreading depression induced by the hyperactivity of interneurons. J Comput Neurosci 47: 125–140, 2019. doi: 10.1007/s10827-019-00730-8. [DOI] [PubMed] [Google Scholar]
- 274.van den Maagdenberg AM, Pizzorusso T, Kaja S, Terpolilli N, Shapovalova M, Hoebeek FE, Barrett CF, Gherardini L, van de Ven RC, Todorov B, Broos LA, Tottene A, Gao Z, Fodor M, De Zeeuw CI, Frants RR, Plesnila N, Plomp JJ, Pietrobon D, Ferrari MD. High cortical spreading depression susceptibility and migraine-associated symptoms in CaV2.1 S218L mice. Ann Neurol 67: 85–98, 2010. doi: 10.1002/ana.21815. [DOI] [PubMed] [Google Scholar]
- 275.O’Roak BJ, Vives L, Girirajan S, Karakoc E, Krumm N, Coe BP, Levy R, Ko A, Lee C, Smith JD, Turner EH, Stanaway IB, Vernot B, Malig M, Baker C, Reilly B, Akey JM, Borenstein E, Rieder MJ, Nickerson DA, Bernier R, Shendure J, Eichler EE. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature 485: 246–250, 2012. doi: 10.1038/nature10989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Sanders SJ, He X, Willsey AJ, Ercan-Sencicek AG, Samocha KE, Cicek AE, et al. Insights into autism spectrum disorder genomic architecture and biology from 71 risk loci. Neuron 87: 1215–1233, 2015. doi: 10.1016/j.neuron.2015.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Sanders SJ, Murtha MT, Gupta AR, Murdoch JD, Raubeson MJ, Willsey AJ, Ercan-Sencicek AG, DiLullo NM, Parikshak NN, Stein JL, Walker MF, Ober GT, Teran NA, Song Y, El-Fishawy P, Murtha RC, Choi M, Overton JD, Bjornson RD, Carriero NJ, Meyer KA, Bilguvar K, Mane SM, Sestan N, Lifton RP, Günel M, Roeder K, Geschwind DH, Devlin B, State MW. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature 485: 237–241, 2012. doi: 10.1038/nature10945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Berkvens JJ, Veugen I, Veendrick-Meekes MJ, Snoeijen-Schouwenaars FM, Schelhaas HJ, Willemsen MH, Tan IY, Aldenkamp AP. Autism and behavior in adult patients with Dravet syndrome (DS). Epilepsy Behav 47: 11–16, 2015. doi: 10.1016/j.yebeh.2015.04.057. [DOI] [PubMed] [Google Scholar]
- 279.Kwong AK, Fung CW, Chan SY, Wong VC. Identification of SCN1A and PCDH19 mutations in Chinese children with Dravet syndrome. PLoS One 7: e41802, 2012. doi: 10.1371/journal.pone.0041802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Li BM, Liu XR, Yi YH, Deng YH, Su T, Zou X, Liao WP. Autism in Dravet syndrome: prevalence, features, and relationship to the clinical characteristics of epilepsy and mental retardation. Epilepsy Behav 21: 291–295, 2011. doi: 10.1016/j.yebeh.2011.04.060. [DOI] [PubMed] [Google Scholar]
- 281.Hu W, Tian C, Li T, Yang M, Hou H, Shu Y. Distinct contributions of NaV1.6 and NaV1.2 in action potential initiation and backpropagation. Nat Neurosci 12: 996–1002, 2009. doi: 10.1038/nn.2359. [DOI] [PubMed] [Google Scholar]
- 282.Liao Y, Deprez L, Maljevic S, Pitsch J, Claes L, Hristova D, Jordanova A, Ala-Mello S, Bellan-Koch A, Blazevic D, Schubert S, Thomas EA, Petrou S, Becker AJ, De Jonghe P, Lerche H. Molecular correlates of age-dependent seizures in an inherited neonatal-infantile epilepsy. Brain 133: 1403–1414, 2010. doi: 10.1093/brain/awq057. [DOI] [PubMed] [Google Scholar]
- 283.Boiko T, Rasband MN, Levinson SR, Caldwell JH, Mandel G, Trimmer JS, Matthews G. Compact myelin dictates the differential targeting of two sodium channel isoforms in the same axon. Neuron 30: 91–104, 2001. doi: 10.1016/s0896-6273(01)00265-3. [DOI] [PubMed] [Google Scholar]
- 284.Kaplan MR, Cho MH, Ullian EM, Isom LL, Levinson SR, Barres BA. Differential control of clustering of the sodium channels NaV1.2 and NaV1.6 at developing CNS nodes of Ranvier. Neuron 30: 105–119, 2001. doi: 10.1016/S0896-6273(01)00266-5. [DOI] [PubMed] [Google Scholar]
- 285.Catterall WA. Localization of sodium channels in cultured neural cells. J Neurosci 1: 777–783, 1981. doi: 10.1523/JNEUROSCI.01-07-00777.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Wollner DA, Catterall WA. Localization of sodium channels in axon hillocks and initial segments of retinal ganglion cells. Proc Natl Acad Sci USA 83: 8424–8428, 1986. doi: 10.1073/pnas.83.21.8424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Rasband MN. The axon initial segment and the maintenance of neuronal polarity. Nat Rev Neurosci 11: 552–562, 2010. doi: 10.1038/nrn2852. [DOI] [PubMed] [Google Scholar]
- 288.Ogiwara I, Miyamoto H, Tatsukawa T, Yamagata T, Nakayama T, Atapour N, Miura E, Mazaki E, Ernst SJ, Cao D, Ohtani H, Itohara S, Yanagawa Y, Montal M, Yuzaki M, Inoue Y, Hensch TK, Noebels JL, Yamakawa K. NaV1.2 haplodeficiency in excitatory neurons causes absence-like seizures in mice. Commun Biol 1: 96, 2018. doi: 10.1038/s42003-018-0099-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Stuart GJ, Sakmann B. Active propagation of somatic action potentials into neocortical pyramidal cell dendrites. Nature 367: 69–72, 1994. doi: 10.1038/367069a0. [DOI] [PubMed] [Google Scholar]
- 290.Spratt PW, Ben-Shalom R, Keeshen CM, Burke KJ Jr, Clarkson RL, Sanders SJ, Bender KJ. The autism-associated gene Scn2a contributes to dendritic excitability and synaptic function in the prefrontal cortex. Neuron 103: 673–685.e5, 2019. doi: 10.1016/j.neuron.2019.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Larkum ME, Zhu JJ, Sakmann B. Dendritic mechanisms underlying the coupling of the dendritic with the axonal action potential initiation zone of adult rat layer 5 pyramidal neurons. J Physiol 533: 447–466, 2001. doi: 10.1111/j.1469-7793.2001.0447a.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Sugawara T, Tsurubuchi Y, Agarwala KL, Ito M, Fukuma G, Mazaki-Miyazaki E, Nagafuji H, Noda M, Imoto K, Wada K, Mitsudome A, Kaneko S, Montal M, Nagata K, Hirose S, Yamakawa K. A missense mutation of the Na+ channel alpha II subunit gene NaV1.2 in a patient with febrile and afebrile seizures causes channel dysfunction. Proc Natl Acad Sci USA 98: 6384–6389, 2001. doi: 10.1073/pnas.111065098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Ito M, Shirasaka Y, Hirose S, Sugawara T, Yamakawa K. Seizure phenotypes of a family with missense mutations in SCN2A. Pediatr Neurol 31: 150–152, 2004. doi: 10.1016/j.pediatrneurol.2004.02.013. [DOI] [PubMed] [Google Scholar]
- 294.Berkovic SF, Heron SE, Giordano L, Marini C, Guerrini R, Kaplan RE, Gambardella A, Steinlein OK, Grinton BE, Dean JT, Bordo L, Hodgson BL, Yamamoto T, Mulley JC, Zara F, Scheffer IE. Benign familial neonatal-infantile seizures: characterization of a new sodium channelopathy. Ann Neurol 55: 550–557, 2004. doi: 10.1002/ana.20029. [DOI] [PubMed] [Google Scholar]
- 295.Heron SE, Crossland KM, Andermann E, Phillips HA, Hall AJ, Bleasel A, Shevell M, Mercho S, Seni MH, Guiot MC, Mulley JC, Berkovic SF, Scheffer IE. Sodium-channel defects in benign familial neonatal-infantile seizures. Lancet 360: 851–852, 2002. doi: 10.1016/S0140-6736(02)09968-3. [DOI] [PubMed] [Google Scholar]
- 296.Herlenius E, Heron SE, Grinton BE, Keay D, Scheffer IE, Mulley JC, Berkovic SF. SCN2A mutations and benign familial neonatal-infantile seizures: the phenotypic spectrum. Epilepsia 48: 1138–1142, 2007. doi: 10.1111/j.1528-1167.2007.01049.x. [DOI] [PubMed] [Google Scholar]
- 297.Striano P, Bordo L, Lispi ML, Specchio N, Minetti C, Vigevano F, Zara F. A novel SCN2A mutation in family with benign familial infantile seizures. Epilepsia 47: 218–220, 2006. doi: 10.1111/j.1528-1167.2006.00392.x. [DOI] [PubMed] [Google Scholar]
- 298.Zara F, Specchio N, Striano P, Robbiano A, Gennaro E, Paravidino R, et al. Genetic testing in benign familial epilepsies of the first year of life: clinical and diagnostic significance. Epilepsia 54: 425–436, 2013. doi: 10.1111/epi.12089. [DOI] [PubMed] [Google Scholar]
- 299.Scalmani P, Rusconi R, Armatura E, Zara F, Avanzini G, Franceschetti S, Mantegazza M. Effects in neocortical neurons of mutations of the NaV1.2 Na+ channel causing benign familial neonatal-infantile seizures. J Neurosci 26: 10100–10109, 2006. doi: 10.1523/JNEUROSCI.2476-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Misra SN, Kahlig KM, George AL Jr.. Impaired NaV1.2 function and reduced cell surface expression in benign familial neonatal-infantile seizures. Epilepsia 49: 1535–1545, 2008. doi: 10.1111/j.1528-1167.2008.01619.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Kasai N, Fukushima K, Ueki Y, Prasad S, Nosakowski J, Sugata K, Sugata A, Nishizaki K, Meyer NC, Smith RJ. Genomic structures of SCN2A and SCN3A—candidate genes for deafness at the DFNA16 locus. Gene 264: 113–122, 2001. doi: 10.1016/s0378-1119(00)00594-1. [DOI] [PubMed] [Google Scholar]
- 302.Sarao R, Gupta SK, Auld VJ, Dunn RJ. Developmentally regulated alternative RNA splicing of rat brain sodium channel mRNAs. Nucleic Acids Res 19: 5673–5679, 1991. doi: 10.1093/nar/19.20.5673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.Xu R, Thomas EA, Jenkins M, Gazina EV, Chiu C, Heron SE, Mulley JC, Scheffer IE, Berkovic SF, Petrou S. A childhood epilepsy mutation reveals a role for developmentally regulated splicing of a sodium channel. Mol Cell Neurosci 35: 292–301, 2007. doi: 10.1016/j.mcn.2007.03.003. [DOI] [PubMed] [Google Scholar]
- 304.Begemann A, Acuña MA, Zweier M, Vincent M, Steindl K, Bachmann-Gagescu R, Hackenberg A, Abela L, Plecko B, Kroell-Seger J, Baumer A, Yamakawa K, Inoue Y, Asadollahi R, Sticht H, Zeilhofer HU, Rauch A. Further corroboration of distinct functional features in SCN2A variants causing intellectual disability or epileptic phenotypes. Mol Med 25: 6, 2019. doi: 10.1186/s10020-019-0073-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305.Lauxmann S, Boutry-Kryza N, Rivier C, Mueller S, Hedrich UB, Maljevic S, Szepetowski P, Lerche H, Lesca G. An SCN2A mutation in a family with infantile seizures from Madagascar reveals an increased subthreshold Na+ current. Epilepsia 54: e117–e121, 2013. doi: 10.1111/epi.12241. [DOI] [PubMed] [Google Scholar]
- 306.Lauxmann S, Verbeek NE, Liu Y, Zaichuk M, Müller S, Lemke JR, van Kempen MJ, Lerche H, Hedrich UB. Relationship of electrophysiological dysfunction and clinical severity in SCN2A-related epilepsies. Hum Mutat 39: 1942–1956, 2018. doi: 10.1002/humu.23619. [DOI] [PubMed] [Google Scholar]
- 307.Sanders SJ, Campbell AJ, Cottrell JR, Moller RS, Wagner FF, Auldridge AL, Bernier RA, Catterall WA, Chung WK, Empfield JR, George AL Jr, Hipp JF, Khwaja O, Kiskinis E, Lal D, Malhotra D, Millichap JJ, Otis TS, Petrou S, Pitt G, Schust LF, Taylor CM, Tjernagel J, Spiro JE, Bender KJ. Progress in understanding and treating SCN2A-mediated disorders. Trends Neurosci 41: 442–456, 2018. doi: 10.1016/j.tins.2018.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.Wolff M, Brunklaus A, Zuberi SM. Phenotypic spectrum and genetics of SCN2A-related disorders, treatment options, and outcomes in epilepsy and beyond. Epilepsia 60: S59–S67, 2019. doi: 10.1111/epi.14935. [DOI] [PubMed] [Google Scholar]
- 309.Wolff M, Johannesen KM, Hedrich UB, Masnada S, Rubboli G, Gardella E, et al. Genetic and phenotypic heterogeneity suggest therapeutic implications in SCN2A-related disorders. Brain 140: 1316–1336, 2017. doi: 10.1093/brain/awx054. [DOI] [PubMed] [Google Scholar]
- 310.Kamiya K, Kaneda M, Sugawara T, Mazaki E, Okamura N, Montal M, Makita N, Tanaka M, Fukushima K, Fujiwara T, Inoue Y, Yamakawa K. A nonsense mutation of the sodium channel gene SCN2A in a patient with intractable epilepsy and mental decline. J Neurosci 24: 2690–2698, 2004. doi: 10.1523/JNEUROSCI.3089-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311.Ben-Shalom R, Keeshen CM, Berrios KN, An JY, Sanders SJ, Bender KJ. Opposing effects on NaV1.2 function underlie differences between SCN2A variants observed in individuals with autism spectrum disorder or infantile seizures. Biol Psychiatry 82: 224–232, 2017. doi: 10.1016/j.biopsych.2017.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312.Ogiwara I, Ito K, Sawaishi Y, Osaka H, Mazaki E, Inoue I, Montal M, Hashikawa T, Shike T, Fujiwara T, Inoue Y, Kaneda M, Yamakawa K. De novo mutations of voltage-gated sodium channel alphaII gene SCN2A in intractable epilepsies. Neurology 73: 1046–1053, 2009. doi: 10.1212/WNL.0b013e3181b9cebc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313.Hedrich UB, Lauxmann S, Lerche H. SCN2A channelopathies: mechanisms and models. Epilepsia 60, Suppl 3: S68–S76, 2019. doi: 10.1111/epi.14731. [DOI] [PubMed] [Google Scholar]
- 314.Liao Y, Anttonen AK, Liukkonen E, Gaily E, Maljevic S, Schubert S, Bellan-Koch A, Petrou S, Ahonen VE, Lerche H, Lehesjoki AE. SCN2A mutation associated with neonatal epilepsy, late-onset episodic ataxia, myoclonus, and pain. Neurology 75: 1454–1458, 2010. doi: 10.1212/WNL.0b013e3181f8812e. [DOI] [PubMed] [Google Scholar]
- 315.Schwarz N, Hahn A, Bast T, Müller S, Löffler H, Maljevic S, Gaily E, Prehl I, Biskup S, Joensuu T, Lehesjoki AE, Neubauer BA, Lerche H, Hedrich UB. Mutations in the sodium channel gene SCN2A cause neonatal epilepsy with late-onset episodic ataxia. J Neurol 263: 334–343, 2016. doi: 10.1007/s00415-015-7984-0. [DOI] [PubMed] [Google Scholar]
- 316.Schwarz N, Bast T, Gaily E, Golla G, Gorman KM, Griffiths LR, Hahn A, Hukin J, King M, Korff C, Miranda MJ, Möller RS, Neubauer B, Smith RA, Smol T, Striano P, Stroud B, Vaccarezza M, Kluger G, Lerche H, Fazeli W. Clinical and genetic spectrum of SCN2A-associated episodic ataxia. Eur J Paediatr Neurol 23: 438–447, 2019. doi: 10.1016/j.ejpn.2019.03.001. [DOI] [PubMed] [Google Scholar]
- 317.Mason ER, Wu F, Patel RR, Xiao Y, Cannon SC, Cummins TR. Resurgent and gating pore currents induced by de novo SCN2A epilepsy mutations. eNeuro 6: ENEURO.0141-19.2019, 2019. doi: 10.1523/ENEURO.0141-19.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318.Thompson CH, Ben-Shalom R, Bender KJ, George AL. Alternative splicing potentiates dysfunction of early-onset epileptic encephalopathy SCN2A variants. J. Gen Physiol 152: e201912442, 2020. doi: 10.1085/jgp.201912442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319.Li J, Cai T, Jiang Y, Chen H, He X, Chen C, Li X, Shao Q, Ran X, Li Z, Xia K, Liu C, Sun ZS, Wu J. Genes with de novo mutations are shared by four neuropsychiatric disorders discovered from NPdenovo database. Mol Psychiatry 21: 298, 2016. doi: 10.1038/mp.2015.58. [DOI] [PubMed] [Google Scholar]
- 320.Rauch A, Wieczorek D, Graf E, Wieland T, Endele S, Schwarzmayr T, et al. Range of genetic mutations associated with severe non-syndromic sporadic intellectual disability: an exome sequencing study. Lancet 380: 1674–1682, 2012. doi: 10.1016/S0140-6736(12)61480-9. [DOI] [PubMed] [Google Scholar]
- 321.Papuc SM, Abela L, Steindl K, Begemann A, Simmons TL, Schmitt B, et al. The role of recessive inheritance in early-onset epileptic encephalopathies: a combined whole-exome sequencing and copy number study. Eur J Hum Genet 27: 408–421, 2019. doi: 10.1038/s41431-018-0299-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322.Hackenberg A, Baumer A, Sticht H, Schmitt B, Kroell-Seger J, Wille D, Joset P, Papuc S, Rauch A, Plecko B. Infantile epileptic encephalopathy, transient choreoathetotic movements, and hypersomnia due to a de novo missense mutation in the SCN2A gene. Neuropediatrics 45: 261–264, 2014. doi: 10.1055/s-0034-1372302. [DOI] [PubMed] [Google Scholar]
- 323.Dickinson D, Straub RE, Trampush JW, Gao Y, Feng N, Xie B, Shin JH, Lim HK, Ursini G, Bigos KL, Kolachana B, Hashimoto R, Takeda M, Baum GL, Rujescu D, Callicott JH, Hyde TM, Berman KF, Kleinman JE, Weinberger DR. Differential effects of common variants in SCN2A on general cognitive ability, brain physiology, and messenger RNA expression in schizophrenia cases and control individuals. JAMA Psychiatry 71: 647–656, 2014. doi: 10.1001/jamapsychiatry.2014.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324.Fromer M, Pocklington AJ, Kavanagh DH, Williams HJ, Dwyer S, Gormley P, et al. De novo mutations in schizophrenia implicate synaptic networks. Nature 506: 179–184, 2014. doi: 10.1038/nature12929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 325.Carroll LS, Woolf R, Ibrahim Y, Williams HJ, Dwyer S, Walters J, Kirov G, O'Donovan MC, Owen MJ. Mutation screening of SCN2A in schizophrenia and identification of a novel loss-of-function mutation. Psychiatr Genet 26: 60–65, 2016. doi: 10.1097/YPG.0000000000000110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 326.Planells-Cases R, Caprini M, Zhang J, Rockenstein EM, Rivera RR, Murre C, Masliah E, Montal M. Neuronal death and perinatal lethality in voltage-gated sodium channel alpha(II)-deficient mice. Biophys J 78: 2878–2891, 2000. doi: 10.1016/S0006-3495(00)76829-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327.Mishra V, Karumuri BK, Gautier NM, Liu R, Hutson TN, Vanhoof-Villalba SL, Vlachos I, Iasemidis L, Glasscock E. Scn2a deletion improves survival and brain-heart dynamics in the Kcna1-null mouse model of sudden unexpected death in epilepsy (SUDEP). Hum Mol Genet 26: 2091–2103, 2017. doi: 10.1093/hmg/ddx104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 328.Lena I, Mantegazza M. NaV1.2 haploinsufficiency in Scn2a knock-out mice causes an autistic-like phenotype attenuated with age. Sci Rep 9: 12886, 2019. doi: 10.1038/s41598-019-49392-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 329.Shin W, Kweon H, Kang R, Kim D, Kim K, Kang M, Kim SY, Hwang SN, Kim JY, Yang E, Kim H, Kim E. Scn2a haploinsufficiency in mice suppresses hippocampal neuronal excitability, excitatory synaptic drive, and long-term potentiation, and spatial learning and memory. Front Mol Neurosci 12: 145, 2019. doi: 10.3389/fnmol.2019.00145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 330.Miyamoto H, Tatsukawa T, Shimohata A, Yamagata T, Suzuki T, Amano K, Mazaki E, Raveau M, Ogiwara I, Oba-Asaka A, Hensch TK, Itohara S, Sakimura K, Kobayashi K, Kobayashi K, Yamakawa K. Impaired cortico-striatal excitatory transmission triggers epilepsy. Nat Commun 10: 1917, 2019. doi: 10.1038/s41467-019-09954-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 331.Middleton SJ, Kneller EM, Chen S, Ogiwara I, Montal M, Yamakawa K, McHugh TJ. Altered hippocampal replay is associated with memory impairment in mice heterozygous for the Scn2a gene. Nat Neurosci 21: 996–1003, 2018. doi: 10.1038/s41593-018-0163-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 332.Schattling B, Fazeli W, Engeland B, Liu Y, Lerche H, Isbrandt D, Friese MA. Activity of NaV1.2 promotes neurodegeneration in an animal model of multiple sclerosis. JCI Insight 1: e89810, 2016. doi: 10.1172/jci.insight.89810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 333.Meisler MH, Plummer NW, Burgess DL, Buchner DA, Sprunger LK. Allelic mutations of the sodium channel SCN8A reveal multiple cellular and physiological functions. Genetica 122: 37–45, 2004. doi: 10.1007/s10709-004-1441-9. [DOI] [PubMed] [Google Scholar]
- 334.Burgess DL, Kohrman DC, Galt J, Plummer NW, Jones JM, Spear B, Meisler MH. Mutation of a new sodium channel gene, Scn8a, in the mouse mutant ‘motor endplate disease’. Nat Genet 10: 461–465, 1995. doi: 10.1038/ng0895-461. [DOI] [PubMed] [Google Scholar]
- 335.Kohrman DC, Harris JB, Meisler MH. Mutation detection in the med and medJ alleles of the sodium channel Scn8a: unusual splicing due to a minor class AT-AC intron. J Biol Chem 271: 17576–17581, 1996. doi: 10.1074/jbc.271.29.17576. [DOI] [PubMed] [Google Scholar]
- 336.Jones JM, Dionne L, Dell’Orco J, Parent R, Krueger JN, Cheng X, Dib-Hajj SD, Bunton-Stasyshyn RK, Sharkey LM, Dowling JJ, Murphy GG, Shakkottai VG, Shrager P, Meisler MH. Single amino acid deletion in transmembrane segment D4S6 of sodium channel Scn8a (NaV1.6) in a mouse mutant with a chronic movement disorder. Neurobiol Dis 89: 36–45, 2016. doi: 10.1016/j.nbd.2016.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 337.Papale LA, Beyer B, Jones JM, Sharkey LM, Tufik S, Epstein M, Letts VA, Meisler MH, Frankel WN, Escayg A. Heterozygous mutations of the voltage-gated sodium channel SCN8A are associated with spike-wave discharges and absence epilepsy in mice. Hum Mol Genet 18: 1633–1641, 2009. doi: 10.1093/hmg/ddp081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 338.Raman IM, Sprunger LK, Meisler MH, Bean BP. Altered subthreshold sodium currents and disrupted firing patterns in Purkinje neurons of Scn8a mutant mice. Neuron 19: 881–891, 1997. doi: 10.1016/S0896-6273(00)80969-1. [DOI] [PubMed] [Google Scholar]
- 339.Osorio N, Cathala L, Meisler MH, Crest M, Magistretti J, Delmas P. Persistent NaV1.6 current at axon initial segments tunes spike timing of cerebellar granule cells. J Physiol 588: 651–670, 2010. doi: 10.1113/jphysiol.2010.183798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 340.Royeck M, Horstmann MT, Remy S, Reitze M, Yaari Y, Beck H. Role of axonal NaV1.6 sodium channels in action potential initiation of CA1 pyramidal neurons. J Neurophysiol 100: 2361–2380, 2008. doi: 10.1152/jn.90332.2008. [DOI] [PubMed] [Google Scholar]
- 341.Do MT, Bean BP. Sodium currents in subthalamic nucleus neurons from NaV1.6-null mice. J Neurophysiol 92: 726–733, 2004. doi: 10.1152/jn.00186.2004. [DOI] [PubMed] [Google Scholar]
- 342.Mercer JN, Chan CS, Tkatch T, Held J, Surmeier DJ. NaV1.6 sodium channels are critical to pacemaking and fast spiking in globus pallidus neurons. J Neurosci 27: 13552–13566, 2007. doi: 10.1523/JNEUROSCI.3430-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 343.Enomoto A, Han JM, Hsiao CF, Chandler SH. Sodium currents in mesencephalic trigeminal neurons from NaV1.6 null mice. J Neurophysiol 98: 710–719, 2007. doi: 10.1152/jn.00292.2007. [DOI] [PubMed] [Google Scholar]
- 344.Van Wart A, Matthews G. Impaired firing and cell-specific compensation in neurons lacking NaV1.6 sodium channels. J Neurosci 26: 7172–7180, 2006. doi: 10.1523/JNEUROSCI.1101-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 345.Chen L, Huang J, Zhao P, Persson AK, Dib-Hajj FB, Cheng X, Tan A, Waxman SG, Dib-Hajj SD. Conditional knockout of NaV1.6 in adult mice ameliorates neuropathic pain. Sci Rep 8: 3845, 2018. doi: 10.1038/s41598-018-22216-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 346.Cummins TR, Dib-Hajj SD, Herzog RI, Waxman SG. NaV1.6 channels generate resurgent sodium currents in spinal sensory neurons. FEBS Lett 579: 2166–2170, 2005. doi: 10.1016/j.febslet.2005.03.009. [DOI] [PubMed] [Google Scholar]
- 347.Maurice N, Tkatch T, Meisler M, Sprunger LK, Surmeier DJ. D1/D5 dopamine receptor activation differentially modulates rapidly inactivating and persistent sodium currents in prefrontal cortex pyramidal neurons. J Neurosci 21: 2268–2277, 2001. doi: 10.1523/JNEUROSCI.21-07-02268.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 348.Makinson CD, Tanaka BS, Sorokin JM, Wong JC, Christian CA, Goldin AL, Escayg A, Huguenard JR. Regulation of thalamic and cortical network synchrony by Scn8a. Neuron 93: 1165–1179, 2017. doi: 10.1016/j.neuron.2017.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 349.Trudeau MM, Dalton JC, Day JW, Ranum LP, Meisler MH. Heterozygosity for a protein truncation mutation of sodium channel SCN8A in a patient with cerebellar atrophy, ataxia, and mental retardation. J Med Genet 43: 527–530, 2006. doi: 10.1136/jmg.2005.035667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 350.Veeramah KR, O’Brien JE, Meisler MH, Cheng X, Dib-Hajj SD, Waxman SG, Talwar D, Girirajan S, Eichler EE, Restifo LL, Erickson RP, Hammer MF. De novo pathogenic SCN8A mutations identified by whole-genome sequencing of a family quartet affected by infantile epileptic encephalopathy and SUDEP. Am J. Hum Genet 90: 502–510, 2012. doi: 10.1016/j.ajhg.2012.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 351.Gardella E, Marini C, Trivisano M, Fitzgerald MP, Alber M, Howell KB, Darra F, Siliquini S, Bölsterli BK, Masnada S, Pichiecchio A, Johannesen KM, Jepsen B, Fontana E, Anibaldi G, Russo S, Cogliati F, Montomoli M, Specchio N, Rubboli G, Veggiotti P, Beniczky S, Wolff M, Helbig I, Vigevano F, Scheffer IE, Guerrini R, Møller RS. The phenotype of SCN8A developmental and epileptic encephalopathy. Neurology 91: e1112–e1124, 2018. doi: 10.1212/WNL.0000000000006199. [DOI] [PubMed] [Google Scholar]
- 352.Gardella E, Becker F, Møller RS, Schubert J, Lemke JR, Larsen LH, Eiberg H, Nothnagel M, Thiele H, Altmüller J, Syrbe S, Merkenschlager A, Bast T, Steinhoff B, Nürnberg P, Mang Y, Bakke Møller L, Gellert P, Heron SE, Dibbens LM, Weckhuysen S, Dahl HA, Biskup S, Tommerup N, Hjalgrim H, Lerche H, Beniczky S, Weber YG. Benign infantile seizures and paroxysmal dyskinesia caused by an SCN8A mutation. Ann Neurol 79: 428–436, 2016. doi: 10.1002/ana.24580. [DOI] [PubMed] [Google Scholar]
- 353.Johannesen KM, Gardella E, Encinas AC, Lehesjoki AE, Linnankivi T, Petersen MB, et al. The spectrum of intermediate SCN8A-related epilepsy. Epilepsia 60: 830–844, 2019. doi: 10.1111/epi.14705. [DOI] [PubMed] [Google Scholar]
- 354.Liu Y, Schubert J, Sonnenberg L, Helbig KL, Hoei-Hansen CE, Koko M, Rannap M, Lauxmann S, Huq M, Schneider MC, Johannesen KM, Kurlemann G, Gardella E, Becker F, Weber YG, Benda J, Møller RS, Lerche H. Neuronal mechanisms of mutations in SCN8A causing epilepsy or intellectual disability. Brain 142: 376–390, 2019. doi: 10.1093/brain/awy326. [DOI] [PubMed] [Google Scholar]
- 355.Wagnon JL, Barker BS, Ottolini M, Park Y, Volkheimer A, Valdez P, Swinkels ME, Patel MK, Meisler MH. Loss-of-function variants of SCN8A in intellectual disability without seizures. Neurol Genet 3: e170, 2017. doi: 10.1212/NXG.0000000000000170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 356.Wagnon JL, Barker BS, Hounshell JA, Haaxma CA, Shealy A, Moss T, Parikh S, Messer RD, Patel MK, Meisler MH. Pathogenic mechanism of recurrent mutations of SCN8A in epileptic encephalopathy. Ann Clin Transl Neurol 3: 114–123, 2016. doi: 10.1002/acn3.276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 357.de Kovel CG, Meisler MH, Brilstra EH, van Berkestijn FM, van’t Slot R, van Lieshout S, Nijman IJ, O'Brien JE, Hammer MF, Estacion M, Waxman SG, Dib-Hajj SD, Koeleman BP. Characterization of a de novo SCN8A mutation in a patient with epileptic encephalopathy. Epilepsy Res 108: 1511–1518, 2014. doi: 10.1016/j.eplepsyres.2014.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 358.Pan Y, Cummins TR. Distinct functional alterations in SCN8A epilepsy mutant channels. J Physiol 598: 381–401, 2020. doi: 10.1113/JP278952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 359.Wagnon JL, Korn MJ, Parent R, Tarpey TA, Jones JM, Hammer MF, Murphy GG, Parent JM, Meisler MH. Convulsive seizures and SUDEP in a mouse model of SCN8A epileptic encephalopathy. Hum Mol Genet 24: 506–515, 2015. doi: 10.1093/hmg/ddu470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 360.Bunton-Stasyshyn RK, Wagnon JL, Wengert ER, Barker BS, Faulkner A, Wagley PK, Bhatia K, Jones JM, Maniaci MR, Parent JM, Goodkin HP, Patel MK, Meisler MH. Prominent role of forebrain excitatory neurons in SCN8A encephalopathy. Brain 142: 362–375, 2019. doi: 10.1093/brain/awy324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 361.Lopez-Santiago LF, Yuan Y, Wagnon JL, Hull JM, Frasier CR, O'Malley HA, Meisler MH, Isom LL. Neuronal hyperexcitability in a mouse model of SCN8A epileptic encephalopathy. Proc Natl Acad Sci USA 114: 2383–2388, 2017. doi: 10.1073/pnas.1616821114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 362.Ottolini M, Barker BS, Gaykema RP, Meisler MH, Patel MK. Aberrant sodium channel currents and hyperexcitability of medial entorhinal cortex neurons in a mouse model of SCN8A encephalopathy. J Neurosci 37: 7643–7655, 2017. doi: 10.1523/JNEUROSCI.2709-16.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 363.Wengert ER, Saga AU, Panchal PS, Barker BS, Patel MK. Prax330 reduces persistent and resurgent sodium channel currents and neuronal hyperexcitability of subiculum neurons in a mouse model of SCN8A epileptic encephalopathy. Neuropharmacology 158: 107699, 2019. doi: 10.1016/j.neuropharm.2019.107699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 364.Frasier CR, Wagnon JL, Bao YO, McVeigh LG, Lopez-Santiago LF, Meisler MH, Isom LL. Cardiac arrhythmia in a mouse model of sodium channel SCN8A epileptic encephalopathy. Proc Natl Acad Sci USA 113: 12838–12843, 2016. doi: 10.1073/pnas.1612746113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 365.Atkin TA, Maher CM, Gerlach AC, Gay BC, Antonio BM, Santos SC, Padilla KM, Rader J, Krafte DS, Fox MA, Stewart GR, Petrovski S, Devinsky O, Might M, Petrou S, Goldstein DB. A comprehensive approach to identifying repurposed drugs to treat SCN8A epilepsy. Epilepsia 59: 802–813, 2018. doi: 10.1111/epi.14037. [DOI] [PubMed] [Google Scholar]
- 366.Baker EM, Thompson CH, Hawkins NA, Wagnon JL, Wengert ER, Patel MK, George AL Jr, Meisler MH, Kearney JA. The novel sodium channel modulator GS-458967 (GS967) is an effective treatment in a mouse model of SCN8A encephalopathy. Epilepsia 59: 1166–1176, 2018. doi: 10.1111/epi.14196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 367.Barker BS, Ottolini M, Wagnon JL, Hollander RM, Meisler MH, Patel MK. The SCN8A encephalopathy mutation p.Ile1327Val displays elevated sensitivity to the anticonvulsant phenytoin. Epilepsia 57: 1458–1466, 2016. doi: 10.1111/epi.13461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 368.Boerma RS, Braun KP, van den Broek MP, van Berkestijn FM, Swinkels ME, Hagebeuk EO, Lindhout D, van Kempen M, Boon M, Nicolai J, de Kovel CG, Brilstra EH, Koeleman BP. Remarkable phenytoin sensitivity in 4 children with SCN8A-related epilepsy: a molecular neuropharmacological approach. Neurotherapeutics 13: 192–197, 2016. doi: 10.1007/s13311-015-0372-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 369.Braakman HM, Verhoeven JS, Erasmus CE, Haaxma CA, Willemsen MH, Schelhaas HJ. Phenytoin as a last-resort treatment in SCN8A encephalopathy. Epilepsia Open 2: 343–344, 2017. doi: 10.1002/epi4.12059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 370.Smith RS, Kenny CJ, Ganesh V, Jang A, Borges-Monroy R, Partlow JN, Hill RS, Shin T, Chen AY, Doan RN, Anttonen AK, Ignatius J, Medne L, Bönnemann CG, Hecht JL, Salonen O, Barkovich AJ, Poduri A, Wilke M, de Wit MC, Mancini GM, Sztriha L, Im K, Amrom D, Andermann E, Paetau R, Lehesjoki AE, Walsh CA, Lehtinen MK. Sodium channel SCN3A (NaV1.3) regulation of human cerebral cortical folding and oral motor development. Neuron 99: 905–913.e7, 2018. doi: 10.1016/j.neuron.2018.07.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 371.Waxman SG. Mechanisms of disease: sodium channels and neuroprotection in multiple sclerosis-current status. Nat Clin Pract Neurol 4: 159–169, 2008. doi: 10.1038/ncpneuro0735. [DOI] [PubMed] [Google Scholar]
- 372.Holland KD, Kearney JA, Glauser TA, Buck G, Keddache M, Blankston JR, Glaaser IW, Kass RS, Meisler MH. Mutation of sodium channel SCN3A in a patient with cryptogenic pediatric partial epilepsy. Neurosci Lett 433: 65–70, 2008. doi: 10.1016/j.neulet.2007.12.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 373.Estacion M, Gasser A, Dib-Hajj SD, Waxman SG. A sodium channel mutation linked to epilepsy increases ramp and persistent current of NaV1.3 and induces hyperexcitability in hippocampal neurons. Exp Neurol 224: 362–368, 2010. doi: 10.1016/j.expneurol.2010.04.012. [DOI] [PubMed] [Google Scholar]
- 374.Vanoye CG, Gurnett CA, Holland KD, George AL Jr, Kearney JA. Novel SCN3A variants associated with focal epilepsy in children. Neurobiol Dis 62: 313–322, 2014. doi: 10.1016/j.nbd.2013.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 375.Trujillano D, Bertoli-Avella AM, Kumar Kandaswamy K, Weiss ME, Köster J, Marais A, Paknia O, Schröder R, Garcia-Aznar JM, Werber M, Brandau O, Calvo Del Castillo M, Baldi C, Wessel K, Kishore S, Nahavandi N, Eyaid W, Al Rifai MT, Al-Rumayyan A, Al-Twaijri W, Alothaim A, Alhashem A, Al-Sannaa N, Al-Balwi M, Alfadhel M, Rolfs A, Abou Jamra R. Clinical exome sequencing: results from 2819 samples reflecting 1000 families. Eur J Hum Genet 25: 176–182, 2017. doi: 10.1038/ejhg.2016.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 376.Wang Y, Du X, Bin R, Yu S, Xia Z, Zheng G, Zhong J, Zhang Y, Jiang YH, Wang Y. Genetic variants identified from epilepsy of unknown etiology in Chinese children by targeted exome sequencing. Sci Rep 7: 40319, 2017. doi: 10.1038/srep46520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 377.Zaman T, Helbig I, Božović IB, DeBrosse SD, Bergqvist AC, Wallis K, Medne L, Maver A, Peterlin B, Helbig KL, Zhang X, Goldberg EM. Mutations in SCN3A cause early infantile epileptic encephalopathy. Ann Neurol 83: 703–717, 2018. doi: 10.1002/ana.25188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 378.Miyatake S, Kato M, Sawaishi Y, Saito T, Nakashima M, Mizuguchi T, Mitsuhashi S, Takata A, Miyake N, Saitsu H, Matsumoto N. Recurrent SCN3A p.Ile875Thr variant in patients with polymicrogyria. Ann Neurol 84: 159–161, 2018. doi: 10.1002/ana.25256. [DOI] [PubMed] [Google Scholar]
- 379.Inuzuka LM, Macedo-Souza LI, Della-Ripa B, Cabral KS, Monteiro F, Kitajima JP, de Souza Godoy LF, de Souza Delgado D, Kok F, Garzon E. Neurodevelopmental disorder associated with de novo SCN3A pathogenic variants: two new cases and review of the literature. Brain Dev 42: 211–216, 2020. doi: 10.1016/j.braindev.2019.09.004. [DOI] [PubMed] [Google Scholar]
- 380.Zaman T, Helbig KL, Clatot J, Thompson CH, Kang SK, Stouffs K, et al. SCN3A-related neurodevelopmental disorder: a spectrum of epilepsy and brain malformation. Ann Neurol 88: 348–362, 2020. doi: 10.1002/ana.25809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 381.Lamar T, Vanoye CG, Calhoun J, Wong JC, Dutton SB, Jorge BS, Velinov M, Escayg A, Kearney JA. SCN3A deficiency associated with increased seizure susceptibility. Neurobiol Dis 102: 38–48, 2017. doi: 10.1016/j.nbd.2017.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 382.Celle ME, Cuoco C, Porta S, Gimelli G, Tassano E. Interstitial 2q24.3 deletion including SCN2A and SCN3A genes in a patient with autistic features, psychomotor delay, microcephaly and no history of seizures. Gene 532: 294–296, 2013. doi: 10.1016/j.gene.2013.09.073. [DOI] [PubMed] [Google Scholar]
- 383.Thuresson AC, Van Buggenhout G, Sheth F, Kamate M, Andrieux J, Clayton Smith J, Soussi Zander C. Whole gene duplication of SCN2A and SCN3A is associated with neonatal seizures and a normal intellectual development. Clin Genet 91: 106–110, 2017. doi: 10.1111/cge.12797. [DOI] [PubMed] [Google Scholar]