

Abstract
The pharmacokinetics of gentamicin C1, C2, and C1a were studied in six beagles after administration of gentamicin at 4 mg/kg of body weight as a single intravenous bolus dose. Plasma concentrations of the gentamicin components were analyzed with a novel high-performance liquid chromatography method capable of identifying and quantifying each of the components. The pharmacokinetic analysis of the plasma concentration-versus-time data was performed using the noncompartmental approach. The results indicated significant differences in the pharmacokinetic characteristics between the gentamicin components C1, C1a, and C2. The mean residence times of gentamicin C1, C1a, and C2 were 81 ± 13, 84 ± 12, and 79 ± 13 min (mean ± standard deviation), respectively. The half-lives of the respective components were 64 ± 12, 66 ± 12 and 63 ± 12 min. Clearance (CL) of gentamicin C1, 4.62 ± 0.71 ml min−1 kg−1, was significantly higher (P = 0.0156) than CL of gentamicin C1a, 1.81 ± 0.26 ml min−1 kg−1, and C2, 1.82 ± 0.25 ml min−1 kg−1. Similarly, the volume of distribution at steady state (Vss) of gentamicin C1, 0.36 ± 0.04 liter kg−1, was significantly higher (P = 0.0156) than the Vss of gentamicin C1a, 0.14 ± 0.01 liter kg−1, and C2, 0.15 ± 0.02 liter kg−1. Tissue binding was considered the most likely cause for the difference. The difference may have clinical and toxicological significance.
Gentamicin is an aminoglycoside antibiotic used in treatment of serious infections caused by gram-negative aerobic bacteria. Gentamicin is not a single compound but a mixture of three major components, gentamicin C1, C1a, and C2, and a number of minor components. The major components differ in the degree of methylation in the 2-amino-hexose (purpurosamine) ring. Gentamicin C1a lacks methyl groups in this ring, while C1 and C2 have a methyl group in the 6′ position (Fig. 1). Gentamicin C1 is also N methylated in this position, while C1a and C2 have free amines instead. The C2 component consists of two stereoisomers (C2 and C2a). It should be emphasized that the difference in the chemical structure between the gentamicin components is essentially similar to the difference between gentamicins and some other aminoglycosides, such as tobramycin and netilmicin. It has also been recognized that there is a wide variation in the component ratio between different pharmaceutical gentamicin preparations (4, 10, 22). Gentamicin, like all aminoglycoside antibiotics, is nephrotoxic and ototoxic. Nephrotoxicity occurred in 17% and ototoxicity in 8% of patients treated with gentamicin (16), but in some populations the numbers could be higher (27). There appears to be a difference in the incidence of toxicity as a result of once- or multiple-daily administration protocols (15), suggesting a correlation between the toxicity and pharmacokinetics of gentamicin. Furthermore, the components were reported to possess different nephrotoxicity in animals (10), but the human data were inconclusive (7, 13). Strong tissue binding of gentamicin was reported (21).
FIG. 1.

Structures of gentamicin C1a, C2, and C1.
Radioimmunoassays, fluorescence polarization immunoassays, or microbiological assays have been used for quantitative determination of gentamicin in serum and/or plasma in pharmacokinetic studies (23). The limits of quantification of total gentamicin of these methods vary considerably but were generally in the range of 0.2 to 0.5 μg/ml. These methods lack the ability to identify and measure separately the three components. It is also not clear whether the performance characteristics of these methods were equal for the individual components, hence causing a potential bias in the accuracy of total gentamicin concentration. Consequently, the composition of the analytical standard, against which the concentrations are measured, and the different ratios of the three components in pharmaceutical gentamicin preparations are also bound to increase the analytical bias. Therefore, comprehensive understanding of gentamicin pharmacokinetics is profoundly dependent on an analytical method capable of analyzing the different components separately.
The presence of a deep-compartment and a three-compartment model of gentamicin disposition was suggested (3, 24, 27), and in some studies a terminal half-life (t1/2) of more than 100 h was observed (3, 20, 21). However, other reports concluded that a two-compartment model best described gentamicin pharmacokinetics and were unable to observe the deep compartment (6, 17, 18). At this point there seems to be no general agreement on a specific compartmental model best describing gentamicin pharmacokinetics. The basic problem in these studies is that the reported pharmacokinetics of gentamicin relate to an unknown combination of chemically related but different compounds addressed as gentamicin. Therefore, the variation in gentamicin pharmacokinetics and nephrotoxicity, reported in the different studies, could have resulted from the different pharmacokinetic characteristics of the components. Because the analytical uncertainties have not been clarified, studies concerning gentamicin pharmacokinetics may be indicative of the pharmaceutical preparation used in the study. However, meaningful pharmacokinetics can be determined for a single compound only. Gentamicin C1 pharmacokinetics were reported to differ from total gentamicin pharmacokinetics (13). To our knowledge the pharmacokinetics of the three major gentamicin components have not been investigated.
This paper describes the pharmacokinetics of the three major gentamicin components in six beagles, using a reversed-phase high-performance liquid chromatography (HPLC) method for analysis of plasma concentrations.
MATERIALS AND METHODS
Gentamicin (gentamicin base as sulphate [80 mg/2 ml], RAFA Laboratories Ltd., Jerusalem, Israel) containing 19.1 mg of gentamicin C1a, 31.3 mg of gentamicin C1, and 49.6 mg of gentamicin C2 in 100.0 mg total gentamicin was administered as a single intravenous (i.v.) bolus into the saphenic vein at 4 mg of total gentamicin/kg of body weight to six beagles (four males and two females) weighing 16 to 20 kg. Venous blood samples (5 ml) were collected at 0, 10, 20, 30, 45, 60, 90, 120, 180, 240, 360, 480, 600, and 720 min and 24, 48, and 72 h after drug administration in heparinized tubes via an indwelling jugular vein catheter. Plasma samples were stored at −30°C until analysis. The analytical work was completed within 2 months after sample collection.
The gentamicin C1, C1a, and C2 components were separated in a silica column according to the method described by Claes et al. (4) and used as analytical standards. Concentrations of the gentamicin components in plasma were assayed according to the method of Isoherranen and Soback (9). A polymer solid-phase extraction cartridge (Oasis, 30 mg; Waters Associates, Milford, Mass.) was conditioned with 1 ml of methanol followed by 1 ml of 0.17 M Tris buffer at pH 10.0. One milliliter of plasma sample, mixed thoroughly with 5 ml of 0.17 M Tris buffer at pH 12.0, was loaded on the cartridge. The column was washed with 2 ml of 0.17 M Tris buffer, pH 10.0, and dried. An aliquot of 300 μl of derivatization reagent (0.5 ml of 0.17 M Tris at pH 12.0, 0.5 ml of water, and 50 mg of 1-fluoro-2,4,dinitrobenzene in 2.2 ml of acetonitrile) was applied to the solid-phase cartridge, and the cartridge was placed into an oven at 100°C for 1 h. The derivatized gentamicin was then eluted with 5 ml of acetonitrile and evaporated to dryness. The residue was dissolved into 300 μl of acetonitrile and transferred to an autosampler vial for HPLC analysis. A 20-μl aliquot was then injected to the chromatography, consisting of a low-pressure mixing gradient HPLC system, a diode array detector, and an autosampler (model H-P 1100; Hewlett-Packard, Waldbron, Germany). The separation was performed using a reversed-phase column (Symmetry C18; 100 by 4.6 mm; 3.5-μm particle size; Waters Associates) with a C18 precolumn and acetonitrile-Tris buffer (8.3 mM) at pH 7.0 (68:32, vol/vol) in the mobile phase at a flow rate of 1.2 ml/min. The 2,4-dinitrophenyl derivatives of gentamicin components were detected by UV absorption at 365 nm. The limits of quantification (LOQ) of the components, defined as nine times noise, were 0.07 μg/ml (C1) and 0.1 μg/ml (C1a and C2), and the recovery was 72%. The linear range was from 0.07 μg/ml or 0.1 μg/ml (C1a and C2) to 20 μg/ml for the components. The intraday coefficients of variation of the assay were 7.7, 10, and 11 and 2.1, 4.2, and 1.1 for gentamicin C1a, C2, and C1 at 0.1 and 20 μg/ml, respectively. The interday coefficients of variation of the assay were 13, 12, and 7.7 and 4.8, 6.3, and 2.0 for gentamicin C1a, C2, and C1 at 0.1 and 20 μg/ml, respectively.
Pharmacokinetic analysis.
The pharmacokinetics of the gentamicin components were determined by use of a noncompartmental approach based on the statistical moment theory (26) and utilizing a computer program (25). The linear terminal slope (β) was calculated by a linear, least-squares regression analysis, using the last five to six plasma concentration-versus-time points. The t1/2 was calculated according to the following equation: t1/2 = 1n2/β. (8). The mean residence time (MRT) was determined by the equation MRT = AUMC/AUC, where AUMC is the area under the first moment curve and AUC is the area under the plasma drug concentration-time (zero moment) curve (26). The AUMC and AUC were calculated by the trapezoidal method and extrapolated to infinity (25). The volume of distribution at steady state (Vss) was estimated as follows:
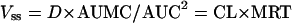 |
1 |
where D is the dose and CL is the total body clearance (2). The volume of distribution in the elimination phase (Vβ) was calculated according to the following equation: Vβ = D/(AUC × β). Total body clearance (CL) was calculated by use of the following equation (8).
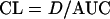 |
2 |
Statistical analysis.
Friedman's nonparametric repeated-measures test and the Wilcoxon signed-rank test were used to analyze statistical differences (P < 0.05) in the determined pharmacokinetic parameters.
RESULTS
Figure 2 illustrates a chromatogram of the separation of the gentamicin components in dog plasma. The pharmacokinetic parameters for each gentamicin component and for the total gentamicin in the six dogs are presented in Table 1. The total gentamicin pharmacokinetics, determined using the sum of the concentrations of the components, are given for comparison only with full knowledge that pharmacokinetics of a mixture of compounds cannot be determined unequivocally. Figure 3 depicts the plasma concentration-versus-time curves for the gentamicin components and total gentamicin. The plasma gentamicin component concentrations were below the LOQ in all samples collected at 480 min and thereafter.
FIG. 2.

Representative chromatogram of gentamicin analysis in the plasma of dog 3 at 120 min after administration, representing 0.94-μg/ml gentamicin C1a, 1.68-μg/ml gentamicin C2, and 0.92-μg/ml gentamicin C1.
TABLE 1.
Pharmacokinetic parameters determined for the total gentamicin and components after i.v. administration of gentamicin (4 mg/kg) to six beagles
| Gentamicin | AUC (μg ml−1 min) | CL (ml min−1 kg−1) | Vss (liter/kg) | MRT (min) | t1/2 (min) | β (min−1) | Vβ (liter/kg) |
|---|---|---|---|---|---|---|---|
| Total | 1,553 ± 227 | 2.63 ± 0.42 | 0.21 ± 0.02 | 82 ± 10 | 65 ± 10 | 0.011 ± 0.002 | 0.24 ± 0.02 |
| C1a | 429 ± 55 | 1.81 ± 0.26 | 0.15 ± 0.02 | 81 ± 13 | 64 ± 12 | 0.012 ± 0.003 | 0.16 ± 0.02 |
| C2 | 697 ± 90 | 1.82 ± 0.26 | 0.15 ± 0.02 | 84 ± 12 | 66 ± 12 | 0.011 ± 0.003 | 0.17 ± 0.02 |
| C1 | 437 ± 64 | 4.62a ± 0.71 | 0.36a ± 0.04 | 79 ± 13 | 63 ± 12 | 0.011 ± 0.003 | 0.41a ± 0.05 |
Significantly different (P < 0.05) from other values in the column (excluding total gentamicin).
FIG. 3.

Plasma concentration (mean ± standard deviation [error bars]) versus-time curves of gentamicin C1a, C2, and C1 after i.v. administration of 0.8-, 2-, and 1.2-mg/kg concentrations of the respective gentamicin components to six beagles.
No difference was observed in the MRT and t1/2 of the three gentamicin components. The CL of gentamicin C1 was 150% higher than the CL of gentamicin C1a and C2. The Vss of gentamicin C1 was 145% higher than the Vss of C1a and C2. There were significant differences in the Vss (P = 0.0055), Vβ (P = 0.0017), and CL (P = 0.0081) values between the three components. The volumes of distribution and CL clearance of gentamicin C1 were significantly higher than those of gentamicins C1a and C2 (P = 0.0156). There were no differences in Vss, Vβ, and CL values between C1a and C2.
DISCUSSION
Gentamicin pharmacokinetics has been a subject of considerable interest due to its clinical importance on the one hand and its toxicity on the other hand. Gentamicin is a very polar entity that does not undergo metabolism in the body and is excreted mainly by glomerular filtration (27). However, the fact that gentamicin is actually a combination of three major components, gentamicin C1, C1a, and C2, has gone practically unnoticed in pharmacokinetic studies. Furthermore, these components appear in pharmaceutical preparations in widely variable ratios (4, 10, 22). The analytical problems associated with determination of the combined concentration of the three compounds were identified already in the early studies (12, 21), but this aspect was widely overlooked in the subsequent research.
The plasma concentration-time curves of the gentamicin components generated in the present study represent disposition profiles of a multicompartment model. In one-compartment models Vβ is equal to Vss (8). These volume terms are exit site dependent, and the presence of a slowly equilibrating compartment could also cause the difference in these terms (14). The results of this study revealed that the difference between Vβ and Vss of the three components was small (7 to 14%). No sign of a long terminal phase could be distinguished from the plasma concentration-time curves, as the concentrations declined below the LOQ after 360 min, despite the limit of the detection in the present study that was equal to or less than those in most other studies describing gentamicin pharmacokinetics. The CL, Vss, and Vβ estimated in the present study for total gentamicin were lower than the respective values reported earlier (1, 17), which were closer to those determined here for gentamicin C1. In accordance with our results, gentamicin C1 was reported to have higher CL and volume of distribution than total gentamicin in humans (13).
The relationship of volume of distribution to the plasma and tissue volumes can be characterized as follows: V = VP + VT(fu/fuT), where VP is the volume of plasma, VT is the aqueous volume outside plasma into which the drug distributes, fu is the fraction unbound in plasma, and fuT is the fraction unbound in tissue (19). Because all the components were administered simultaneously to the dogs, the VP and VT were identical for all the components. Thus, the differences in Vss between the components indicated that C1 had either a larger fu or a smaller fuT than the two other components. The plasma protein binding of gentamicin (and aminoglycosides in general) is less than 10% (27), and it is inconceivable that it could contribute to the 145% difference in Vss. Therefore, gentamicins C1a and C2 appear to be less bound to tissue than gentamicin C1. This may be of clinical importance if gentamicin nephrotoxicity results from strong binding to the renal tissue. The binding between gentamicin and phospholipids was found to be ionic (12).
More than 95% of the total gentamicin dose is excreted unchanged in urine in dogs (24). The renal clearance (CLR) is defined as: CLR = fu × GFR + CLS − CLRa, where GFR is the glomerular filtration rate, CLS is the tubular secretion clearance, and CLRa is the tubular reabsorption clearance (11). Consequently, if the clearance of the unbound drug is less than the GFR, reabsorption occurs. A general estimate of the glomerular filtration rate of 6.13 ml min−1 kg−1 in dog was given (5), but GFR values of 3.8 and 4.0 ml min−1 kg−1 have also been described (11, 17). The CL of gentamicin C1 was similar to the lower GFR values reported for dogs. The significantly lower CL of gentamicin C1a and C2 compared to gentamicin C1 suggests that gentamicin C1a and C2 were reabsorbed in the kidney to a much higher extent than gentamicin C1. It is noteworthy that irreversible tissue binding would increase CL values by decreasing the AUC in equation 2.
A decrease in gentamicin CL as a function of increased dose was described (6). In the present study the components were given in different doses which, accordingly, could have caused the difference in their pharmacokinetics. However, this seems unlikely, because the lowest CL was determined for the component given at the lowest dose.
According to equation 1, Vss, CL, and MRT are interrelated. Because no difference was observed in the MRTs of the three components Vss and CL must change in the same direction and on the same order of magnitude. Analogously, the similar t1/2 values of the three components result from changes of Vβ and CL in the same direction and on the same order of magnitude according to the equation t1/2 = Vβ1n2/CL. The fact that both volume of distribution and CL of gentamicin C1a and C2 or gentamicin C1 are affected to a similar magnitude would support the hypothesis that the change results from the same physiological cause. Tissue binding appears the most likely reason to affect both parameters in a similar manner. Renal uptake by endocytosis of polybasic drugs, such as aminoglycosides, mediated by an epithelial glycoprotein was reported (12). Accordingly, the uptake of gentamicin C1 in the present study would be higher than those of gentamicin C1a and C2.
In commercial preparations, gentamicin C1 consists of 25 to 50% of the total gentamicin. Equation 2 can be rearranged to AUC = D/CL, and the extreme values of the component ratio can be used as the dose and the values obtained in this study as the CL. Consequently, the total gentamicin AUC, calculated as the sum of the AUC values for each component, may vary up to 20%. This simulation emphasizes the importance of determining the pharmacokinetics of each gentamicin component separately, including the quantitative assessment of each component in the administered dose. Furthermore, the different pharmacokinetics of the components may warrant reevaluation of the use of single-component gentamicin preparations in clinical situations.
ACKNOWLEDGMENTS
We thank Rica Benita and Dana Levin for their skillful technical assistance.
REFERENCES
- 1.Batra V K, Morrison J A, Hoffman T R. Pharmacokinetics of piperacillin and gentamicin following intravenous administration to dogs. J Pharm Sci. 1983;72:894–898. doi: 10.1002/jps.2600720813. [DOI] [PubMed] [Google Scholar]
- 2.Benet L Z, Galeazzi R. Noncompartmental determination of the steady-state volume of distribution. J Pharm Sci. 1979;68:1071–1074. doi: 10.1002/jps.2600680845. [DOI] [PubMed] [Google Scholar]
- 3.Brown S A, Nelson R W, Scott-Moncrieff C. Gentamicin pharmacokinetics in diabetic dogs. J Vet Pharmacol Ther. 1991;14:90–95. doi: 10.1111/j.1365-2885.1991.tb00808.x. [DOI] [PubMed] [Google Scholar]
- 4.Claes P J, Busson R, Vanderhaeghe H. Determination of the component ratio of commercial gentamicins by high-performance liquid chromatography using pre-column derivatization. J Chromatogr. 1984;298:445–457. doi: 10.1016/s0021-9673(01)92742-6. [DOI] [PubMed] [Google Scholar]
- 5.Davies B, Morris T. Physiological parameters in laboratory animals and humans. Pharm Res. 1993;10:1093–1096. doi: 10.1023/a:1018943613122. [DOI] [PubMed] [Google Scholar]
- 6.Demczar D J, Nafziger A N, Bertino J S., Jr Pharmacokinetics of gentamicin at traditional versus high doses: implications for once-daily aminoglycoside dosing. Antimicrob Agents Chemother. 1997;41:1115–1119. doi: 10.1128/aac.41.5.1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Forrey A W, Meijsen-Ludwick B T, O'Neill M A, Maxwell B M, Blair A D, Cutler R E. Nephrotoxicity: a comparison in humans of gentamicin and gentamicin C1 administration. Toxicol Appl Pharmacol. 1978;44:453–462. doi: 10.1016/0041-008x(78)90253-3. [DOI] [PubMed] [Google Scholar]
- 8.Gibaldi M, Perrier D. Pharmacokinetics. 2nd ed. New York, N.Y: Marcel Dekker; 1982. pp. 199–219. [Google Scholar]
- 9.Isoherranen, N., and S. Soback. Determination of gentamicin C1, C1a and C2 in plasma and urine by use of high performance liquid chromatography. Clin. Chem., in press.
- 10.Kohlhepp S J, Loveless M O, Kohnen P W, Houghton D C, Bennett W M, Gilbert D N. Nephrotoxicity of the constituents of gentamicin complex. J Infect Dis. 1984;149:605–614. doi: 10.1093/infdis/149.4.605. [DOI] [PubMed] [Google Scholar]
- 11.Lin J H. Species similarities and differences in pharmacokinetics. Drug Metab Disp. 1995;24:1008–1021. [PubMed] [Google Scholar]
- 12.Moerstrup S K, Cui S, Vorum H, Bregengard C, Bjorn S E, Norris K, Gliemann J, Christenssen E I. Evidence that epithelial glycoprotein 330/megalin mediates uptake of polybasic drugs. J Clin Investig. 1995;96:1404–1413. doi: 10.1172/JCI118176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mosegaard A, Welling P G, Madsen P O. Gentamicin and gentamicin C1 in the treatment of complicated urinary tract infections: comparative study of efficacy, tolerance, and pharmacokinetics. Antimicrob Agents Chemother. 1975;7:328–332. doi: 10.1128/aac.7.3.328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nakashima E, Benet L Z. General treatment of mean residence time, clearance and volume parameters in linear mammillary models with elimination from any compartment. J Pharmacokin Biopharm. 1988;16:475–493. doi: 10.1007/BF01062381. [DOI] [PubMed] [Google Scholar]
- 15.Nicolau D P, Freeman C D, Belliveau P P, Nightingale C H, Ross J W, Quintiliani R. Experience with a once-daily aminoglycoside program administered to 2,184 adult patients. Antimicrob Agents Chemother. 1995;39:650–655. doi: 10.1128/AAC.39.3.650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Prins J M, Buller H R, Kuijper E J, Tange R A, Speelman P. Once versus thrice daily gentamicin in patients with serious infections. Lancet. 1993;341:335–339. doi: 10.1016/0140-6736(93)90137-6. [DOI] [PubMed] [Google Scholar]
- 17.Riviere J E, Coppoc G L. Pharmacokinetics of gentamicin in juvenile dog. Am J Vet Res. 1981;42:1621–1623. [PubMed] [Google Scholar]
- 18.Riviere J E, Carver M P, Coppoc G L, Carlton W W, Lantz G C, Shy-Modjeska J. Pharmacokinetics and comparative nephrotoxicity of fixed-dose versus fixed-interval reduction of gentamicin dosage in subtotal nephrectomized dogs. Toxicol Appl Pharmacol. 1984;75:496–509. doi: 10.1016/0041-008x(84)90186-8. [DOI] [PubMed] [Google Scholar]
- 19.Rowland M, Tozer T. Clinical pharmacokinetics: concepts and applications. 3rd ed. Philadelphia, Pa: Lea & Febiger; 1995. p. 148. [Google Scholar]
- 20.Schentag J J, Jusko W J. Renal clearance and tissue accumulation of gentamicin. Clin Pharmacol Ther. 1977;22:364–370. doi: 10.1002/cpt1977223364. [DOI] [PubMed] [Google Scholar]
- 21.Schentag J J, Jusko W J, Vance J W, Cumbo T J, Abrutyn E, DeLattre M, Gerbracht L M. Gentamicin disposition and tissue accumulation on multiple dosing. J Pharmacokin Biopharm. 1977;5:559–577. doi: 10.1007/BF01059684. [DOI] [PubMed] [Google Scholar]
- 22.White L O, Lovering A M, Reeves D S. Variations in gentamicin C1, C1a, C2 and C2a content of some preparations of gentamicin sulfate used clinically as determined by high-performance liquid chromatography. Ther Drug Monit. 1983;5:123–126. doi: 10.1097/00007691-198303000-00014. [DOI] [PubMed] [Google Scholar]
- 23.White L O. Assays for therapeutic monitoring and pharmacokinetic investigations of aminoglycosides: quality aspects. Ther Drug Monit. 1998;20:464–468. doi: 10.1097/00007691-199810000-00003. [DOI] [PubMed] [Google Scholar]
- 24.Whittem T, Parton K, Turner K. Effect of polyaspartic acid on pharmacokinetics of gentamicin after single intravenous dose in the dog. Antimicrob Agents Chemother. 1996;40:1237–1241. doi: 10.1128/aac.40.5.1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yamaoka K. Methods for pharmacokinetic analysis by personal computer. 2nd edn. Tokyo, Japan: Nanko-do Ltd.; 1986. pp. 145–162. [Google Scholar]
- 26.Yamaoka K, Nakagawa T, Uno T. Statistical moments in pharmacokinetics. J Pharmacokin Biopharm. 1978;6:547–558. doi: 10.1007/BF01062109. [DOI] [PubMed] [Google Scholar]
- 27.Zaske D E. Aminoglycosides. In: Ewans W E, Schentag J J, Jusko W J, Relling M V, editors. Applied pharmacokinetics. 3rd ed. Vancouver, Wash: Applied Therapeutics Inc.; 1992. pp. 14-1–14-47. [Google Scholar]