

Abstract
Purpose:
Activation of STING (stimulator of interferon genes) can trigger a robust, innate antitumor immune response in immunologically “cold” tumors such as glioblastoma.
Patients and Methods:
A small-molecule STING agonist, IACS-8779, was stereotactically administered using intraoperative navigation intratumorally in dogs with spontaneously arising glioblastoma. The phase I trial used an escalating dose design, ascending through four dose levels (5–20 μg). Treatment was repeated every 4–6 weeks for a minimum of two cycles. Radiographic response to treatment was determined by response assessment in neuro-oncology (RANO) criteria applied to isovoxel postcontrast T1-weighted MR images obtained on a single 3T magnet.
Results:
Six dogs were enrolled and completed ≥1 cycle of treatment. One dog was determined to have an abscess and was removed from further analysis. One procedure-related fatality was observed. Radiographic responses were dose dependent after the first cycle. The first subject had progressive disease, whereas there was 25% volumetric reduction in one subject and greater than 50% in the remaining surviving subjects. The median progression-free survival time was 14 weeks (range: 0–22 weeks), and the median overall survival time was 32 weeks (range: 11–39 weeks).
Conclusions:
Intratumoral STING agonist (IACS-8779) administration was well tolerated in dogs with glioblastoma to a dose of 15 μg. Higher doses of IACS-8779 were associated with radiographic responses.
Translational Relevance.
Preclinical murine models of gliomas have not been predictive of responses to immunotherapy in human subjects. As one of the few species that develop spontaneous intracranial gliomas with a frequency similar to that seen in humans, and that recapitulate the size, genetic, molecular, and immunologic tumor landscape of human tumors, dogs are an especially relevant large animal clinical model for testing novel glioma therapies. Dogs with intracranial glioblastoma are able to tolerate repeated intratumoral administration of IACS-8779 at doses that produce radiographic response, thereby extending the results from orthotopic rodent models to a more rigorous species for vetting therapeutic potential.
Introduction
STING (stimulator of interferon genes) agonists can increase T-cell infiltration into immunologically “cold” tumors, such as gliomas, through proinflammatory activation of suppressive tumor stroma, and can reverse the suppressive phenotype of myeloid-derived suppressor cells (1). STING is a widely expressed sensor of cellular stress, specifically the presence of DNA in the cytoplasm, that bridges the innate and adaptive immune systems by triggering interferon (IFN) release and through cis-activation of myeloid cells. STING is activated by cyclic dinucleotides, either originating directly from invading bacteria or generated by the protein cGAS upon binding to cytoplasmic DNA—a hallmark of viral infection. Tumor-cell DNA in host antigen-presenting cells has been shown to correlate with STING activation (2). Activation of STING culminates in increased production of IFNβ and other pro-inflammatory cytokines. STING activation can reeducate tumor-supportive M2 macrophages toward a proinflammatory M1 phenotype (3), and is also required for optimal priming of cytotoxic T cells against tumor antigens (4). Even in malignancies that lack STING expression, in vivo antitumor response can be mediated by the myeloid and endothelial stroma (5), which is particularly significant in glioblastoma.
Orthotopic gliomas grow faster in STING knockout mice, demonstrating the critical role of this pathway in limiting tumor progression (6). Such mice also lack effective spontaneous CD8+ T-cell priming against tumors (2). Prior studies have demonstrated a remarkable capacity for intratumorally injected STING agonists to eliminate not only the treated tumor, but also distant, untreated sites of disease (1, 7). STING agonists activate many of the same innate pathways as oncolytic viruses but in a vastly more potent and focused fashion. Early clinical trials of viral therapy have achieved sporadic clinical responses in glioblastoma patients, but these therapeutics are complex to manufacture, challenging to administer, and are often limited to a single, direct intratumoral (i.e., surgical) treatment. In contrast, STING agonists are free from the complexities and high costs of viral therapy and are easy to generate in accordance with good manufacturing practice standards.
Preclinical murine glioma models have not predicted immunotherapy responses in human glioma subjects, in part because of poor correlative immunobiology, but recent “omic” profiling has demonstrated a marked association of spontaneously arising canine gliomas with those in human subjects (8). Dogs experience a high rate of spontaneous gliomas, and their more comparable size and shared environment with humans make them ideal for translational clinical trials involving intratumoral treatment. Using a previously developed small-molecule STING agonist shown to be effective in murine glioma models, IACS-8779 (9), we treated dogs with spontaneous glioblastoma with at least two intratumoral injections and measured radiographic response at 4–6 weeks after each treatment.
Patients and Methods
All procedures were performed in accordance with an approved animal use protocol under the auspices of the Texas A&M Institutional Animal Care and Use Committee. The study drug was manufactured as we have described previously (9). The purity and identity of the compounds are validated by LC/MS and nuclear magnetic resonance and their biologic activity is validated using the InvivoGen THP1-Dual KO-STING Cells. Prior to administration, the agents were assessed as ≥99% pure and were endotoxin free.
Preclinical safety testing
Three purpose-bred Beagle dogs were acquired from a commercial vendor. The dogs were female, intact, and healthy. All three dogs weighed between 6 and 10 kg. Each dog was administered two doses of 100 μg IACS-8803, a compound closely related to IACS-8779 (9), in a volume of 500 μL, separated by 14 days, subcutaneously over the lateral aspect of each pelvic limb. Dogs were observed every 4 hours for the first 12 hours after injection, and at least twice daily for the next 7 days. Within 1 hour before each injection, and 48 hours after each injection, complete blood counts and routine chemistry panels with electrolytes were collected from each dog. Photographs of the injection site were taken at 24 and 48 hours after each injection. Adverse events (AE) were scored according to the Veterinary Comparative Oncology Group Common Terminology Criteria for Adverse Events (VCOG-CTCAE v1.1) rubric (10), and were reported to the principal investigators within 12 hours of first observation.
Study design and participants
Six dogs with suspected spontaneously arising glioblastoma were recruited from The Texas A&M University Veterinary Medical Teaching Hospital (TAMU-VMTH). All subjects were enrolled with fully informed and written consent of their owners. This was an open-label, single-arm phase I dose-escalation trial in dogs with newly diagnosed glioblastoma. All enrollees were required to be at least 1 year of age and weigh over 8.5 kg. Dogs were permitted to have mild neurologic impairments, similar to human subjects, according to the modified Glasgow Coma Scale score (11). All dogs were screened for metabolic or bone marrow dysfunction using a biochemistry panel and complete blood count before each treatment cycle. Dogs that had previously received definitive treatment (surgical resection or radiotherapy), or had imaging findings inconsistent with a high grade were excluded from enrollment. Following injection and a minimum of 24 hours observation for AEs, dogs were discharged under their owners' care, with instructions to return to the hospital for general anesthesia and MRI at 4–6 weeks after injection. At the recheck visit, dogs were assessed for general health and neurologic status. A venous blood sample was drawn for evaluation as indicated above. Following MRI, a second image-guided injection of IACS-8779 was given as described above. The skin, subcutaneous tissues, and muscle were reopened and the second injection was administered through the same craniotomy site as the first injection. Dogs returned again for evaluation, general anesthesia, and brain MRI at approximately 15 weeks after the first injection (∼10 weeks after the second injection). At that time, the dogs were also evaluated for overall health and neurologic function. No dog received a third injection of IACS-8779.
Neurosurgical procedures
Following presumptive diagnosis of a high-grade, gadolinium-enhancing space-occupying glial brain tumor by MRI criteria (12), and confirmation of normal organ and bone marrow function, dogs were placed under general anesthesia and positioned in a custom-made MRI-compatible bite plate in sternal recumbency and secured with dental putty conformed to the maxillary dentition. A minimum of six gadolinium-impregnated fiducial rings were fixed in place as close to the head as possible with offsets in three planes. Dogs were imaged on a 3T Siemens Verio MR system (see Imaging Protocol below). The postcontrast T1-weighted images were used for reconstruction and trajectory planning using the Brainsight system (13). A single, approximately 5-mm-diameter burr hole craniectomy was made at an entry point determined using the images obtained as described above. The dura was directly visualized within the craniectomy and opened using a #12 scalpel blade. Prophylactic administration of 0.5 g/kg of mannitol, through a filter needle on a constant-rate infusion over 15 minutes, was initiated at the time of the dural opening. A 100 μL Hamilton syringe secured in a calibrated holder on an x-y stage, containing 50 μL of IACS-8779 at varying concentrations, was positioned along the selected trajectory and lowered to a depth calculated using the MR image reconstruction (Fig. 1). IACS-8779 was injected at a rate of 2 μL/minute. After completion of the entire 50 μL injection, the syringe was withdrawn, and the craniectomy and durotomy were examined for evidence of hemorrhage or swelling. Minor dural hemorrhage was controlled with a hemostatic sponge, and hemostasis was confirmed before replacement with a dry piece of hemostatic sponge, over which the tissues were then closed. Dogs were recovered from anesthesia and maintained in the TAMU-VMTH ICU for a minimum of 24 hours following each injection. Medications (corticosteroids and anticonvulsants) previously prescribed for control of clinical signs related to their intracranial disease were continued without interruption during hospitalization.
Figure 1.
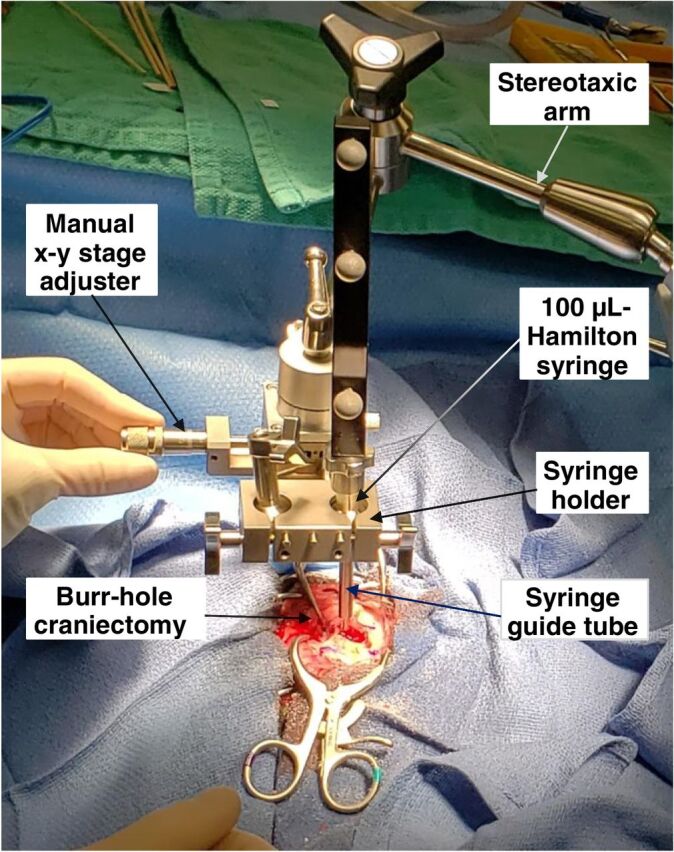
Intraoperative image of direct intratumoral administration of IACS-8779 using navigational guidance.
Outcomes
One dog was determined to have had an abscess rather than a glioblastoma postmortem and was removed from further analysis. One dog did not survive the initial post-injection period, developing clinical signs of increased intracranial pressure that was unresponsive to medical management within the first 24 hours after the procedure. This dog was ultimately euthanized within 48 hours of the procedure. In the surviving dogs, responses were assessed by clinical examinations and MRI scans every 4–6 weeks to coincide with the next intratumoral injection. The brain MRI used for initial image-guidance planning was considered the baseline MRI for evaluation of treatment response. Toxic effects were assessed at baseline and before each cycle. AEs were measured according to the VCOG CTCAE v1.1. Treatment was continued for at least two cycles and until objective disease progression (PD) occurred, intercurrent illness prevented further drug administration, unacceptable AEs occurred, or consent was withdrawn. A toxicity evaluation endpoint was defined as treatment-related unmanageable toxicities, including grade 3 or 4 AEs that require termination of the treatment during the first cycle. Overall survival (OS) was defined as the time from onset of clinical signs related to intracranial disease to the time of natural death or euthanasia for any reason. Subjects who were alive were censored at the time of last contact.
Imaging protocol and imaging response-assessment criteria
MRI scans were acquired on a 3.0 T Siemens Verio MRI scanner using the standard protocol: for each subject, 3-plane T2-weighted, transverse T2-FLAIR, transverse T2*, and isovoxel T1 (RAGE) pre- and post-gadolinium were obtained (Supplementary Table S1). The postcontrast T1-weighted sequences were used to assess response to therapy via the response assessment in neuro-oncology criterion (RANO) criteria (12). Contrast-enhancing lesions with volume measurements of >0.5 cm3 were considered as measurable lesions, whereas smaller lesions and those with nonenhancing T2/FLAIR hyperintensity were considered to be nonmeasurable lesions. Subjects were categorized based on best response as having: (i) progressive disease (PD) as defined by a ≥25% increase in the sum of the products of the perpendicular diameters of contrast enhancement; (ii) stable disease (SD) as defined as no substantial change; (iii) partial response (PR) as defined by ≥50% decrease in the baseline of the sum of products of the perpendicular diameters of contrast enhancement; or (iv) complete response (CR) as defined as complete disappearance of all enhancing measurable disease.
Histopathology
Formalin-fixed, paraffin-embedded sections of canine central nervous system were cut at 4 μm and mounted on charged slides. The sections were deparaffinized in xylene and rehydrated through graded alcohols.
Myeloperoxidase
Antigen retrieval was performed by immersing the slides in a citrate buffer and heating them in a pressure cooker (Decloaking Chamber, Biocare Medical). After retrieval, the slides were washed with Tris buffer prior to beginning the immunostaining procedure. The IHC procedure was run on an automated platform (intelliPATH FLX, Biocare Medical). All incubations were carried out at room temperature. Endogenous peroxidase activity was blocked by incubating the slides with 3% hydrogen peroxide for 10 minutes. A non-serum reagent (Background Punisher, Biocare Medical) was used to block non-specific protein binding. The sections were then incubated with the primary antibody (rabbit anti-human myeloperoxidase, dilution 1:1,000, Agilent Technologies) for 30 minutes. Next, the slides were incubated with a polymer detection reagent (Mach 2 Rabbit HRP-Polymer, Biocare Medical) for 25 minutes. Sites of antibody–antigen interaction were visualized by incubating with a DAB chromogen (ImmPACT DAB Substrate kit, peroxidase, Vector Laboratories) for 5 minutes.
Hematoxylin and eosin
The staining procedure was run on a Leica automated stainer. Slides were heated to 65°C for 9 minutes, then cleared [with Pro-Par xylene substitute (Anatech Ltd)], and rehydrated before treatment with SPECTRA Hemalast (Leica) and sequential staining with hematoxylin for 4 minutes, bluing for 30 seconds, and eosin for 1 minute following a 1-minute 80% ethyl alcohol wash. Slides were then dehydrated, recleared with Pro-Par, and cover slipped using Permount.
Statistical analysis
Median progression-free survival (PFS) and OS times from onset of signs were estimated by Kaplan–Meier analyses. AEs were recorded and tabulated according to type and grade. Data were analyzed using Matlab for Windows version R2020a (Mathworks). Tumor volume measurements were made using OsiriX MD version 12.0.0 (Pixmeo).
Results
Preclinical safety study
Three intact female beagle dogs between 6 months and 1 year of age were treated as described above. Mild (VCOG CTCAE v1.1 grade 1) local erythema was identified at all injection sites between 4 and 24 hours after injection. All injection site reactions were transient and resolved without intervention. No dog showed changes in complete blood count or serum chemistry panel values, relative to baseline, after either the first or the second injection. One dog experienced grade 1 diarrhea of <48 hours duration that was responsive to oral probiotics during the trial.
Clinical trial
We screened and obtained consent from the owners of six dogs between February 2020 and November 2020. The cut-off date for data analysis was December 2020. There were no treatment-related deaths. Two dogs experienced post-procedure complications that resulted in euthanasia prior to discharge; one of these was determined to have an abscess, rather than a glioblastoma postmortem. One additional dog (Dog 4) experienced an AE possibly related to treatment that consisted of grade 2 regurgitation within 24 hours after each injection. This AE was managed symptomatically with prokinetics, proton-pump inhibitors, and acid buffers. These symptoms resolved within 48 hours on both occasions. There were no metabolic or hematologic adverse effects identified in any dog as assessed by complete blood count or serum chemistry panel values on samples drawn as indicated in the Patients and Methods. No subjects that survived to discharge discontinued the study because of drug-related toxic effects.
Table 1 shows the baseline demographic and outcome data. Table 2 shows detailed outcome and radiographic response data. All enrollees were followed until date of death. The longest PFS was 22 weeks. The first subject treated at the smallest dose of 5 μg was euthanized because of clinical disease progression. The second dog died from seizure-related complications (aspiration pneumonia). The median PFS time for enrolled subjects that survived 48 hours after injection was 14 weeks (range: 0–22 weeks) and the median OS time was 32 weeks (range: 11–39 weeks).
Table 1.
Clinical demographics and outcomes of dogs enrolled in the STING clinical trial.
| Dog identifier | 1 | 2 | 3 | 4 | 5 |
| Breed | French bulldog | French bulldog | Cane Corso mastiff | American pit bull terrier | French bulldog |
| Gender/neuter status | MN | MN | FI | FN | MN |
| Age at onset of signs (years) | 9 | 9 | 4 | 10 | 5 |
| Presenting sign | Seizures | Seizures | Seizures | Seizures | Seizures |
| Duration of signs at first treatment | 4 months | 1 month | 6 months | 2 months | 1 month |
| Anatomic location of tumor | Left forebrain | Left forebrain | Right forebrain | Right forebrain | Right forebrain |
| Tumor type/grade | GBM/grade 4 | Small-cell GBM/grade 4 | No tissue | Small-cell GBM/grade 4 | GBM/grade 4 |
| Outcome | Euthanized (progressive disease) | Deceased (aspiration pneumonia) | Euthanized (progressive disease) | Euthanized (progressive disease) | Deceased (loss of cerebral perfusion) |
Abbreviations: F, female; GBM, glioblastoma; I = intact; M, male; N, neutered.
Table 2.
Detailed outcome and radiographic response data for STING clinical trial.
| Dog identifier | 1 | 2 | 3 | 4 | 5 |
| Dose—first injection | 5 μg | 5 μg | 15 μg | 20 μg | 15 μg |
| Dose—second injection | 5 μg | 10 μg | 20 μg | 20 μg | — |
| PFS (weeks) | 0 | 11 | 16 | 22 | 0 |
| OS (weeks) | 33 | 11 | 39 | 31 | 0 |
| Contrast-enhancing tumor volume—baseline (cm3) | 1.75 | 2.42 | 14.4 | 0.79 | 0.025 |
| Contrast-enhancing tumor volume at first recheck (cm3) | 2.64 | 1.8 | 6.34 | 0.19 | — |
| Contrast-enhancing tumor volume at second recheck (cm3) | 3.31 | — | — | 0.0 | — |
| Contrast-enhancing tumor volume at third recheck (cm3) | — | — | — | 1.1 | — |
Imaging response
All subjects had a baseline MRI prior to treatment, and survivors underwent follow-up MRI every 4–6 weeks for response assessments. Within the dose-escalation cohort, the subject receiving the lowest dose (Dog 1) had PD at all timepoints. A small reduction in tumor volume (25%) was detected in the next subject (Dog 2) at the next highest dose 4–6 weeks after the first injection, which was classified as SD (Table 2). Dog 3 had a reduction in the contrast-enhancing region of the tumor, but little change in T2 FLAIR appearance of the mass, which was taken to indicate SD (14). Dog 4, treated at 20 μg, showed a volumetric change of 0.79 cm3 pretreatment to 0.19 cm3 (>75%; PR) after a single dose 5 weeks after the first injection (Table 2; Fig. 2). The dog then received a second dose of 20 μg; MRI 5 weeks later showed no contrast-enhancing tumor volume (CR). Fifteen weeks after the second injection, the glioblastoma recurred (Fig. 3A). At autopsy, recurrent glioblastoma was confirmed displaying typical features of neovascular proliferation (Fig. 3B) and abundant hemosiderin laden macrophages (Fig. 3C). The radiographic and outcome data suggest that there was evidence of efficacy for intratumoral injection of IACS-8779 at a dose of 20 μg.
Figure 2.

Dog 4 showing radiographic response to injection of 20 μg IACS-8779. Top row: baseline MR images; Bottom rows: congruent images at 6 weeks repeat MRI. From left to right: T1 precontrast axial, T1 postcontrast axial, and T2 FLAIR axial images.
Figure 3.

A, Dog 4 showing radiographic progression 15 weeks after second injection of IACS-8779. Top row and from left to right: T1 precontrast and postcontrast. Bottom row, left to right: T2 FLAIR and T2*. Note dark signal on T2 consistent with hemosiderin deposition in the tumor. B, Hematoxylin and eosin images of glioblastoma with microvascular proliferation at 100× magnification (scale bar = 100 μm). C, Hemosiderin laden macrophages at 200× magnification (scale bar = 50 μm).
Inflammatory responses
The last two subjects enrolled in the study fully recovered after anesthesia but began experiencing rapid neurologic decline within 8 hours after the procedure. The owner of the second dog treated at 20 μg of IACS-8779 (not included in Table 2) requested that the animal be compassionately euthanized following continued neurologic decline despite medical management for presumed intracranial hypertension. This dog was determined to have had an abscess, rather than a glioblastoma, postmortem. Because this underlying pathology was not yet known in the clinical study, the next and final dog (Dog 5) received a de-escalated dose of 15 μg IACS-8779. In Dog 5, sequential imaging showed evolution of an acute inflammatory response following a single intratumoral injection of 15 μg IACS-8779 as monitored through serial contrast CT (Fig. 4A). Midline shift and increased contrast enhancement are evident by 20 hours after injection and persisted despite medical management. Postmortem evaluation of the mass and surrounding tissues on Dog 5 showed perivascular inflammation (Fig. 4B), leptomeningeal inflammation (Fig. 4C), and a mixed inflammatory infiltrate of polymorphonuclear (PMN) leukocytes (Fig. 4D). The inflammatory response was myeloperoxidase positive (Fig. 4E).
Figure 4.

A, Dog 5 demonstrating progressive midline shift and brain edema post injection of 15 μg IAC-8779. Post-injection, p.i. B, Hematoxylin and eosin images demonstrating an acute perivascular inflammatory response at 200× magnification (scale bar = 50 μm). C, Leptomeningeal inflammatory response at 100× magnification (scale bar = 100 μm). D, In the region of glioblastoma, there was diffuse infiltrating polynuclear mononuclear cells (PMN; example designated by red arrow) and macrophages (example designated by black arrow) at 200× magnification (scale bar = 50 μm). E, The inflammation within the tumor shows positive myeloperoxidase stain at 200× magnification (scale bar = 50 μm).
Discussion
Activation of the STING pathway has tremendous potential to improve antitumor immunity through multiple mechanisms. STING activation has been shown to produce efficacious antitumor immunity in a variety of rodent models of malignancy and as such there are several ongoing early-stage clinical trials (NCT04144140, NCT04109092, and NCT04609579) for solid tumors (15). Because of its key role in trafficking of T lymphocytes in the peritumoral microenvironment, the STING pathway is often envisioned as a modulator that can be leveraged to improve immunotherapeutic response to primary treatments such as immune checkpoint inhibitors. Notably, several of the initial clinical trials using STING agonists have not met expectations. As such, we have created our own STING agonists with enhanced potency (7). Our preclinical murine data indicate that these compounds, such as IACS-8803 and the closely related compound IACS-8779, may be highly effective as monotherapies, even for recalcitrant tumors such as glioma. Given the limitations of murine glioma model systems, including their clonotypic nature and small size, we initiated this clinical trial in dogs with spontaneously arising glioblastomas, based on the presence or absence of radiographic responses as a go/no-go pivotal study for determining whether we would advance these STING agonists to clinical trial testing in human glioma subjects. The cumulative data support our prioritization of human patients with glioma that have undergone a surgical debulking for phase I testing; investigational new drug–enabling studies are now underway. Most importantly, given the simple stereotactic delivery strategy and the marked volumetric reduction of the glioma in Dog 4, this strategy could suggest a viable treatment approach for deeply situated, unresectable gliomas that lack significant mass effect.
Clinical trials in dogs with spontaneous glioma can be combined with surgical resection (16), which may not be financially viable for most pet owners and comes with the risks of serious postoperative complications and mortality (17). Other canine clinical trials have focused on intratumoral delivery of conventional chemotherapeutic agents that have limited effectiveness in the treatment of glioma in humans (16, 18, 19). One recent study has shown safety and effectiveness of a novel combination of IL13 and Eph-A2 receptor–targeting cytotoxins in canines with gliomas (20). However, similar to human subjects, the challenge of intratumoral treatment of intracranial gliomas with nonimmunologic therapies is ensuring appropriate volume of distribution of the drug (21). A major potential advantage of local delivery of immunotherapy, such as the compounds used in this study, is that the immune response generated has been shown in model systems to act beyond the bounds of the initial injection (6), which could greatly simplify delivery and reduce the volume of drug needed for clinical effect. In our preclinical and clinical studies, very small volumes (100 and 50 μL, respectively) of injection were used, allowing for minimal local tissue disruption, minimal alteration of intratumoral and intracranial pressure, and little risk of infusate reflux.
The immune microenvironment of spontaneous canine gliomas has been evaluated by several groups. Immunohistochemically, canine gliomas have been found to be commonly infiltrated with CD3+ T cells and IBA1+ macrophages/microglia (22). At least one group has found a higher degree of CD3+ T cells, as well as FOXP3+ cells in high-grade gliomas compared with low-grade gliomas (23). Others have confirmed the presence of CD4+, CD8+, and FOXP3+ T cells in canine gliomas via flow cytometry (24). The presence of FoxP3+ T cells in canine gliomas suggests a pro-tumorigenic state of their immune microenvironment. This is supported by recent functional studies of glioma-associated macrophages/microglia in canine tumors, showing upregulation of TGFβ signaling and other pro-tumorigenic phenotypic markers (25). Only one of the subjects had analysis of the nature of the immune responses during the therapeutic window, whereas the others were during disease progression. In this former case, there was robust influx of both T cells and macrophages in the tumor microenvironment consistent with the mode of activity of STING agonists.
One limitation of the current study was the presumptive diagnosis of high-grade glioma based on radiographic features. The clinical trial was initially designed to obtain a biopsy to confirm the pathologic diagnosis prior to administration of the STING agonist. However, this was ultimately judged to add too much risk to the trial, given that the canine subjects are companion animals, when presented to the owners for consent. As such, the criterion was removed from the study with the intent that the pathologic diagnosis would be confirmed at the time of tumor progression and authorized autopsy. Because STING agonists exert immune modulation within the local tumor microenvironment, systemic immune monitoring would have been unlikely to be informative and would have merely increased the cost and lack of compliance with follow-up visits, because these require the owner's commitment. However, another limitation of the current study is the absence of serial local and systemic immune response monitoring outside of that afforded by serial complete blood count monitoring every 4–6 weeks. Finally, the small sample size in this trial prevents us from being able to accurately estimate the expected response rate and AE rate in dogs with spontaneous gliomas. Although two dogs in this study experienced early poor outcomes, only one of them was ultimately judged to have been eligible for the trial. It was not possible to determine postmortem what, if any, role the clinical trial procedure or drug played in the patient's decline who had an abscess. Because histopathologic and genomic characterization of human gliomas prior to treatment selection is the standard of care, it is unlikely that human trial patients would be enrolled with uncertainty regarding their diagnosis.
Our study used the more potent IACS-8803 analog in healthy dogs for initial assessments of toxicity and the less potent IACS-8779 in dogs with spontaneous intracranial gliomas to reduce the expected intratumoral inflammatory response and risk of post-injection intracranial hypertension. In rodent models, two doses of 25 μg IACS-8803 show more robust local antitumor immunity and more effective clinical response than a single dose. We have demonstrated that small-molecule STING agonists are well tolerated for at least two cycles of injection, either subcutaneously in normal dogs at doses up to 100 μg separated by 2 weeks (IACS-8803), or intratumorally in dogs with spontaneously arising intracranial gliomas at doses up to 20 μg separated by 4–6 weeks (IACS-8779). The MTD of IACS-8779 in non-resected dogs was established at 15 μg IACS-8779. These data support further clinical studies optimizing dose and schedule in dogs with spontaneously arising gliomas, and translational clinical trials to human patients with glioma.
Authors' Disclosures
C. Horbinski reports grants from NIH during the conduct of the study. M.A. Curran reports grants and personal fees from ImmunoGenesis, Inc., ImmunoMet, Inc., as well as personal fees from Agenus, Inc., Alligator Bioscience, Inc., ImmunOs, Inc., Oncoresponse, Inc., Pieris, Inc., Nurix, Inc., Aptevo, Inc., Servier, Inc., Kineta, Inc., Salarius, Inc., Xencor, Inc., Amunix, Inc., Mereo, Inc., and Adagene, Inc. outside the submitted work. In addition, M.A. Curran has a patent for Methods and Composition for Localized Secretion of Anti-CTLA-4 Antibodies issued, licensed, and with royalties paid from multiple licensees; a patent for Dual Specificity Antibodies Which Bind Both PD-L1 and PD-L2 and Prevent Their Binding to PD-1 issued, licensed, and with royalties paid from ImmunoGenesis, Inc.; and a patent for Cyclic Dinucleotides as Agonists of Stimulator of Interferon Gene Dependent Signaling issued and licensed. A.B. Heimberger reports grants from NIH, Joan Traver Walsh Family Foundation, Dr. Marie Rose Foundation, Brockman Foundation, and Mr. Herb Simmons during the conduct of the study. A.B. Heimberger also reports personal fees from Caris Life Science and WCG Oncology; other support from Celldex Therapeutics, DNAtrix, Carthera, and Moleculin; and grants from Celularity and Codiak outside the submitted work. No disclosures were reported by the other authors.
Supplementary Material
Imaging Technique
Acknowledgments
The study was designed by the investigators, who oversaw all data collection and interpretations. The funding agencies, including the NIH R01 NS120547, the Joan Traver Walsh Family Foundation, the Dr. Marnie Rose Foundation, the Brockman Foundation, and Mr. Herb Simmons, had no role in the data analysis, interpretation of the results, or writing of the article.
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
Note: Supplementary data for this article are available at Clinical Cancer Research Online (http://clincancerres.aacrjournals.org/).
Authors' Contributions
C.E. Boudreau: Conceptualization, data curation, formal analysis, writing–original draft. H. Najem: Formal analysis. M. Ott: Formal analysis. C. Horbinski: Formal analysis. D. Fang: Formal analysis. C.M. DeRay: Data curation, formal analysis. J.M. Levine: Conceptualization, data curation, formal analysis. M.A. Curran: Conceptualization, formal analysis. A.B. Heimberger: Conceptualization, formal analysis, writing–original draft.
References
- 1. Ager CR, Reilley MJ, Nicholas C, Bartkowiak T, Jaiswal AR, Curran MA. Intratumoral STING activation with T-cell checkpoint modulation generates systemic antitumor immunity. Cancer Immunol Res 2017;5:676–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Woo SR, Fuertes MB, Corrales L, Spranger S, Furdyna MJ, Leung MYK, et al. STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity 2014;41:830–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Downey CM, Aghaei M, Schwendener RA, Jirik FR. DMXAA causes tumor site-specific vascular disruption in murine non-small cell lung cancer, and like the endogenous non-canonical cyclic dinucleotide STING agonist, 2′3′-cGAMP, induces M2 macrophage repolarization. PLoS One 2014;9:e99988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sivick KE, Desbien AL, Glickman LH, Reiner GL, Corrales L, Surh NH, et al. Magnitude of therapeutic STING activation determines CD8(+) T cell-mediated anti-tumor immunity. Cell Rep 2018;25:3074–85. [DOI] [PubMed] [Google Scholar]
- 5. Demaria O, De Gassart A, Coso S, Gestermann N, Di Domizio J, Flatz L, et al. STING activation of tumor endothelial cells initiates spontaneous and therapeutic antitumor immunity. Proc Natl Acad Sci U S A 2015;112:15408–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Corrales L, Glickman LH, McWhirter SM, Kanne DB, Sivick KE, Katibah GE, et al. Direct activation of STING in the tumor microenvironment leads to potent and systemic tumor regression and immunity. Cell Rep 2015;11:1018–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ohkuri T, Ghosh A, Kosaka A, Zhu J, Ikeura M, David M, et al. STING contributes to antiglioma immunity via triggering type I IFN signals in the tumor microenvironment. Cancer Immunol Res 2014;2:1199–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Amin SB, Anderson KJ, Boudreau CE, Martinez-Ledesma E, Kocakavuk E, Johnson KC, et al. Comparative molecular life history of spontaneous canine and human gliomas. Cancer Cell 2020;37:243–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ager CR, Zhang H, Wei Z, Jones P, Curran MA, Di Francesco ME. Discovery of IACS-8803 and IACS-8779, potent agonists of stimulator of interferon genes (STING) with robust systemic antitumor efficacy. Bioorg Med Chem Lett 2019;29:126640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Veterinary cooperative oncology group - common terminology criteria for adverse events (VCOG-CTCAE) following chemotherapy or biological antineoplastic therapy in dogs and cats v1.1. Vet Comp Oncol 2016;14:417–46. [DOI] [PubMed] [Google Scholar]
- 11. Ash K, Hayes GM, Goggs R, Sumner JP. Performance evaluation and validation of the animal trauma triage score and modified Glasgow Coma Scale with suggested category adjustment in dogs: a VetCOT registry study. J Vet Emerg Crit Care 2018;28:192–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bentley RT, Ober CP, Anderson KL, Feeney DA, Naughton JF, Ohlfest JR, et al. Canine intracranial gliomas: relationship between magnetic resonance imaging criteria and tumor type and grade. Vet J 2013;198:463–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chen AV, Wininger FA, Frey S, Comeau RM, Bagley RS, Tucker RL, et al. Description and validation of a magnetic resonance imaging-guided stereotactic brain biopsy device in the dog. Vet Radiol Ultrasound 2012;53:150–6. [DOI] [PubMed] [Google Scholar]
- 14. Okada H, Weller M, Huang R, Finocchiaro G, Gilbert MR, Wick W, et al. Immunotherapy response assessment in neuro-oncology: a report of the RANO working group. Lancet Oncol 2015;16:e534–e42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gogoi H, Mansouri S, Jin L. The age of cyclic dinucleotide vaccine adjuvants. Vaccines 2020;8:453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bentley RT, Thomovsky SA, Miller MA, Knapp DW, Cohen-Gadol AA. Canine (Pet Dog) tumor microsurgery and intratumoral concentration and safety of metronomic chlorambucil for spontaneous glioma: a phase I clinical trial. World Neurosurg 2018;116:e534–e42. [DOI] [PubMed] [Google Scholar]
- 17. Kohler RJ, Arnold SA, Eck DJ, Thomson CB, Hunt MA, Pluhar GE. Incidence of and risk factors for major complications or death in dogs undergoing cytoreductive surgery for treatment of suspected primary intracranial masses. J Am Vet Med Assoc 2018;253:1594–603. [DOI] [PubMed] [Google Scholar]
- 18. Freeman AC, Platt SR, Holmes S, Kent M, Robinson K, Howerth E, et al. Convection-enhanced delivery of cetuximab conjugated iron-oxide nanoparticles for treatment of spontaneous canine intracranial gliomas. J Neurooncol 2018;137:653–63. [DOI] [PubMed] [Google Scholar]
- 19. Young JS, Bernal G, Polster SP, Nunez L, Larsen GF, Mansour N, et al. Convection-enhanced delivery of polymeric nanoparticles encapsulating chemotherapy in canines with spontaneous supratentorial tumors. World Neurosurg 2018;117:e698–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Rossmeisl JH, Herpai D, Quigley M, Cecere TE, Robertson JL, D'Agostino RB, et al. Phase I trial of convection-enhanced delivery of IL13RA2 and EPHA2 receptor targeted cytotoxins in dogs with spontaneous intracranial gliomas. Neuro Oncol 2021;23:422–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sampson JH, Archer G, Pedain C, Wembacher-Schroder E, Westphal M, Kunwar S, et al. Poor drug distribution as a possible explanation for the results of the PRECISE trial. J Neurosurg 2010;113:301–9. [DOI] [PubMed] [Google Scholar]
- 22. Sloma EA, Creneti CT, Erb HN, Miller AD. Characterization of inflammatory changes associated with canine oligodendroglioma. J Comp Pathol 2015;153:92–100. [DOI] [PubMed] [Google Scholar]
- 23. Pi Castro D, Jose-Lopez R, Fernandez Flores F, Rabanal Prados RM, Mandara MT, Arus C, et al. Expression of FOXP3 in canine gliomas: immunohistochemical study of tumor-infiltrating regulatory lymphocytes. J Neuropathol Exp Neurol 2020;72:184–93. [DOI] [PubMed] [Google Scholar]
- 24. Filley A, Henriquez M, Bhowmik T, Tewari BN, Rao X, Wan J, et al. Immunologic and gene expression profiles of spontaneous canine oligodendrogliomas. J Neurooncol 2018;137:469–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Todebusch R, Grodzki AC, Dickinson PJ, Woolard K, Vinson N, Sturges B, et al. Glioma-associated microglia/macrophages augment tumorigenicity in canine astrocytoma, a naturally occurring model of human glioma. Neurooncol Adv 2021;3:vdab062. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Imaging Technique