

SUMMARY
Monosodium urate crystals (MSUc) induce inflammation in vivo without prior priming, raising the possibility of an initial cell-autonomous phase. Here, using genome-wide transcriptomic analysis and biochemical assays, we demonstrate that MSUc alone induce a metabolic-inflammatory transcriptional program in non-primed human and murine macrophages that is markedly distinct to that induced by LPS. Genes uniquely upregulated in response to MSUc belong to lipid and amino acid metabolism, glycolysis, and SLC transporters. This upregulation leads to a metabolic rewiring in sera from individuals and mice with acute gouty arthritis. Mechanistically, the initiating inflammatory-metabolic changes in acute gout flares are regulated through a persistent expression and increased binding of JUN to the promoter of target genes through JNK signaling—but not P38—in a process that is different than after LPS stimulation and independent of inflammasome activation. Finally, pharmacological JNK inhibition limits MSUc-induced inflammation in animal models of acute gouty inflammation.
In brief
Cobo et al. show that uric acid crystals alone induce a unique inflammatory and metabolic phenotype in macrophages that is dependent on JUN and p-JUNSer63 upregulation via JNK in vitro and in vivo.
Graphical Abstract
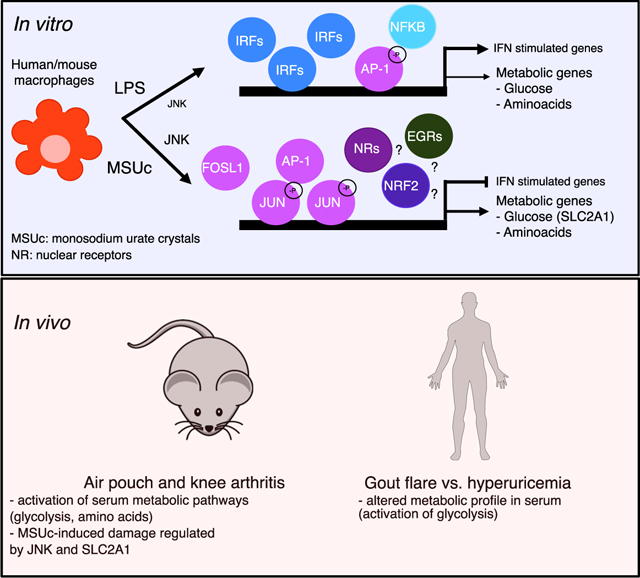
INTRODUCTION
Macrophages are the primary immune cells displaying an inflammatory response that is intended to restore tissue homeostasis after pathogen infection or tissue damage (Karin and Clevers, 2016; Mosser et al., 2021; Wynn and Vannella, 2016). A key feature of macrophages supporting their regulation of inflammatory responses, immunity, tissue homeostasis, and repair, is their high degree of plasticity in response to various microenvironmental stimuli (Atri et al., 2018; Chen et al., 2020). However, deregulated or persistent inflammation can promote irreversible tissue damage by shifting macrophage phenotypes. Therefore, understanding the mechanisms that govern macrophage biology is critical to effectively altering their functions for therapeutic purposes by blocking an unwanted pathway or reprogramming macrophages to phenotypes necessary for tissue homeostasis and repair responses (Kuznetsova et al., 2020).
Metabolic reprograming is a hallmark of macrophage activation (Biswas and Mantovani, 2012). Recent advances in transcriptomic and metabolomic studies have highlighted the link between metabolic rewiring of macrophages and their functional plasticity (Caputa et al., 2019; O’Neill and Pearce, 2016; Thapa and Lee, 2019). Many factors in the cell microenvironment activate key signaling pathways that modulate cellular metabolism to support macrophage differentiation and polarization. Indeed, pro-inflammatory macrophages utilize glycolysis and the pentose phosphate pathway to meet their ATP requirements, whereas oxidative phosphorylation (OXPHOS) as well as the fatty acid oxidation (FAO) pathways are downregulated. In contrast, in macrophages with anti-inflammatory properties, the Krebs cycle is intact, and their metabolic activity is characterized by enhanced amino acid uptake, enhanced FAO, and OXPHOS. These metabolic changes appear to be critical in the pathogenesis of inflammatory and autoimmune diseases (Bustamante et al., 2018; Sanchez-Lopez et al., 2019a; Weyand and Goronzy, 2017).
Gout is currently the most common cause of inflammatory arthritis. In gout, arthritis and bursitis are caused by tissue deposition of monosodium urate crystals (MSUc) (Dalbeth et al., 2016; Kuo et al., 2015). Macrophages, both specialized tissue-resident macrophages and derived from circulating monocytes, are thought to be important for initiating and driving the early pro-inflammatory phase of acute gout (Busso and So, 2010; Martin et al., 2009; So and Martinon, 2017). The study of the underlying mechanisms of gouty inflammation has yielded some insights into the control of pro-inflammatory cytokines, especially of IL-1b, after macrophage priming with TLR agonists, such as fatty acids, via TLR2 (Joosten et al., 2010) and inflammasome activation (Kingsbury et al., 2011; So and Martinon, 2017), secretion of other cytokines, including IL-8, and activation of signaling pathways, such as mitogen-activated protein kinase (MAPK) and nuclear factor κB (Liu et al., 2000; Lu et al., 2015). Of interest, Renaudin et al. (2020) recently described a role of glucose metabolism during the inflammatory IL-1β response induced by MSUc in primed THP1 cells, suggesting that metabolic changes in macrophages after MSUc activation are relevant in gout.
Since IL-1b plays an essential role in the pathogenesis of gout flares (Kingsbury et al., 2011; So and Martinon, 2017), nearly all mechanistic studies of MSUc-induced macrophage activation in vitro introduce a TLR-induced priming step. Yet, the inflammatory and metabolic effect that MSUc alone induce in non-primed primary macrophages and the mechanism by which macrophages respond to MSUc and activates this inflammatory and metabolic program is incompletely understood. While TLR-dependent signaling has been linked to the pathophysiology of gout, MSU crystals induce inflammation in vivo without prior priming, raising the possibility of an initial cell-autonomous phase. To investigate this possibility, we evaluated the transcriptional profile and its regulation in non-primed human monocyte-derived macrophages (mDMs) and murine bone marrow-derived macrophages (BMDMs) after MSU crystals alone. Our results indicate that MSUc alone induce a distinct transcriptional program, and that upregulation of JUN and p-JUNSer63 via JNK is necessary for upregulation of the inflammatory and metabolic transcriptional program induced by MSUc in both mDMs and BMDMs.
RESULTS
MSUc induce a distinct transcriptional program in both human and mouse non-primed macrophages
To address the genome-wide contribution of MSUc in non-primed macrophages, we performed RNA sequencing (RNA-seq) of macrophages differentiated from healthy human monocytes (mDMs) and of murine BMDMs stimulated with MSUc (250 μg/mL) or LPS (100 ng/mL) for 5 h. Principal-component analysis highlights the divergence of mDMs and BMDMs treated with MSUc from untreated cells or cells treated with LPS (Figure 1 A). Gene ontology analysis (Mootha et al., 2003; Subramanian et al., 2005) show those upregulated genes by MSUc versus PBS (1,480 in mDMs or 1,366 in BMDMs) belong to pathways related to inflammation. They also belong to metabolism (including lipid metabolism, transport of small molecules, metabolism of carbohydrates, and amino acids) and circadian clock at lesser degree (Figure 1B). Downregulated genes by MSUc. PBS (645 in mDMs or 1,076 in BMDMs) belong to organelle biogenesis and RNA transcription (Figure S1A).
Figure 1. MSUc activates a distinct transcriptional program.

(A) Principal-component analysis of human mDMs (left) or mouse BMDMs (right) treated with LPS (100 ng/mL) or MSUc (250 μg/mL) for 5 h showing the divergence of the transcriptomic program (n ≥ 5 donors in mDMs and n = 3 for BMDMs).
(B) Gene ontology analysis of genes upregulated in macrophages treated with MSUc versus PBS showing inflammatory gene sets as well as activation of nuclear receptor, NGF, circadian clock and metabolic signaling pathways.
(C) Venn diagram showing the overlap between genes upregulated in macrophages treated with LPS or MSUc.
(D) Examples of inflammatory genes upregulated in mDMs treated with LPS or MSUc showing upregulation of some genes in MSUc but greater upregulation in LPS.
(E) Gene ontology analysis of genes downregulated in MSUc versus LPS showing inflammatory gene sets including signaling by interferons.
(F) ELISA for CXCL2 and TNFA in mDMs and additionally CCL2 and CXCL1 for BMDMs in the supernatant of macrophages treated with LPS or MSUc showing increased concentration.
(G) Gene ontology analysis using REACTOME of genes upregulated in MSUc versus LPS showing metabolic gene sets as well as activation of NGF, receptors tyrosine kinase (RTKs), and circadian clock signaling pathways.
(H) Gene ontology analysis using REACTOME of genes uniquely upregulated by MSUc showing enrichment in genes sets of response to external stimuli activation of circadian clock, NGF, and metabolic signaling pathways (#p < 0.10, *p < 0.05, **p < 0.01).
We then compared the transcriptional program altered by MSUc with LPS. More than 4,000 genes were >2-fold differentially regulated (FDR < 0.05) in comparing MSUc-treated BMDMs with LPS-treated BMDMs. Similar differences were observed in mDMs, suggesting a distinct macrophage profile after MSUc stimulation (Figure S1B). Thirty-four percent (510/1480) in BMDMs and 44% (612/1366) in mDMs of genes upregulated by MSUc versus PBS were similarly upregulated by LPS versus PBS and 39.4% (392/645) or 65.4% (704/1076) were similarly downregulated in LPS versus PBS (Figures 1C and S1C). Common upregulated genes in LPS versus PBS and MSU versus PBS genes belong mainly to inflammatory signaling pathways (Figure S1D) including “CCLs,” “CXCLs,” and interleukin genes (Figures 1D and S1E). As expected, the degree of upregulation of inflammatory transcripts in LPS is greater than in MSUc (Figures 1D and 1E) and this was accompanied by higher secretion of inflammatory cytokines including CXCL2 and TNF in mDMs (Figure 1F) and BMDMs and CCL2, CXCL1 in BMDMs. Of note, activation of interferon-stimulated genes, including CXCL10, IFIT, and OAS genes, were only upregulated in LPS (Figures S1F and S1G). In line with this, signaling by Myd88 was required to a larger extent for the transcriptional program activated by LPS than MSUc (p = e–21 and p = e–69 or p = e–11 and p = e–36 for upregulated and downregulated in LPS or MSU, respectively) including Tnf, CXCL2, and CCL2 (Figures S1H and S1I).
Interestingly, a set of genes that were upregulated only in MSUc versus PBS belonged to metabolic pathways, including metabolism of lipids, carbohydrates, and amino acids, and transport of small molecules via SLC transporters (Figures 1G and 1H).
MSUc induce an altered metabolic profile both local and systemically characterized by activation of glycolysis and metabolism of amino acids
The preferential upregulation of metabolic signaling pathways led us to investigate the effect of MSUc in altering the metabolic profile of macrophages. RNA-seq showed de-regulation of several transcripts involved in metabolism of carbohydrates in macrophages treated with MSUc or LPS that were validated using qRT-PCR (Figure S2A), including upregulation of the glucose transporter SLC2A1 and PFKBP3 mRNA (Figure 2A) and SLC2A1 protein level using IF (Figure 2B) and WB (Figure S2A) in both BMDMs and mDMs treated with either MSUc or LPS. In addition, we performed one-dimensional nuclear magnetic resonance (1D 1H-NMR) in the culture medium and cell pellet of BMDMs and in the supernatant of mDMs at 4 and 8 h after MSUc or LPS treatment. We found increased concentration of several metabolites related to glucose metabolism, including acetate, citrate, formate, fumarate, lactate, and succinate, in the pellet and supernatant of BMDMs treated with MSUc but not LPS (except for increased lactate in BMDMs) (Figures 2C and 2D, respectively). Some of these metabolites also showed higher concentration in the supernatant of mDMs treated with MSUc including acetate, citrate, and lactate, and reduced concentration of succinate (Figure 2E). In addition to altered expression of genes involved in glucose metabolism, we found upregulation of genes related to amino acid transport in macrophages treated specially with MSUc, including SLCs transporters, such as SLC3A2, SLC1A4, SLC1A5, and SLC38A2, and genes associated with metabolism of amino acids, including genes involved in glycine, serine, and threonine metabolism (Figure 2F). These changes were accompanied by increased concentration in the supernatant and pellet of alanine, glutamate, glycine, phenylalanine, threonine, and tryptophan of BMDMs treated with MSUc but not LPS (Figures 2G and 2H, respectively). Some of these changes, including increased concentration of glutamate and glycine, were also observed in mDMs treated with MSUc; changes in concentration of other amino acids, such as aspartate, glutamine, leucine, and isoleucine, were only observed in mDMs treated with MSUc (Figure 2I).
Figure 2. MSUc alters the metabolic program of macrophages.

(A) Bar plot showing upregulation of genes involved in glucose metabolism in macrophages treated with MSUc (250 μg/mL) or LPS (100 ng/mL) at 5 h.
(B) Protein analysis by IF showing upregulation of SLC2A1 in macrophages treated with MSUc or LPS at 5 h. Signal for specific antibodies is pseudocolored in red. DAPI in blue is used to delineate DNA structures.
(C and D) Analysis of the concentration of glycolytic metabolites by 1D 1H-NMR in the pellet (C) or supernatant (D) or BMDMs treated with LPS or MSUc for 4 or 8 h showing activation of glycolysis in macrophages treated with MSUc to a higher degree than LPS (n ≥ 4/condition).
(E) 1D 1H-NMR showing increased levels of acetate, citrate, and lactate and reduced levels of succinate in the supernatant of mDMs treated with MSUc for 4 or 8 h (n = 3 donors/condition).
(F) Bar graphs showing upregulation of several amino acid transporters (top) or genes involved in glycine/serine/threonine metabolism (bottom) in macrophages treated with MSUc or LPS for 5 h assessed by RNA-seq.
(G and H) 1D 1H-NMR showing increased levels of glycine, threonine, and tryptophan in the pellet (G) and alanine, glutamate, phenylalanine, and tryptophan in supernatant (H) of BMDMs treated with MSUc or LPS for 4 or 8 h (n ≥ 4/condition).
(I) 1D 1H-NMR showing increased levels of glutamate and glycine, and reduced levels of aspartate, glutamine, isoleucine, and leucine in the supernatant of mDMs treated with MSUc for 4 or 8 h (n = 3 donors/condition) (#p < 0.10, *p < 0.05, **p < 0.01).
We next investigated whether MSUc activates these metabolic pathways in vivo. We first explored whether metabolism was altered in a murine air pouch model of acute gouty inflammation. Partial least squares discriminant analysis (PLS-DA) shows the difference in metabolic profile of the air pouch lavage of mice injected with MSUc as assessed by 1D 1H-NMR (Figure S3A). These changes include reduced concentration of glucose, trimethylamine, alanine, methionine, tyrosine, and phenylalanine, and an increase of dimethylsulfone (Figure S3B). Next, we determined whether local MSUc injection in the air pouch could induce an altered systemic metabolic profile in sera. PLS-DA separates the sera 1D 1H-NMR signal into two groups according to mice injected with PBS or with MSUc in the air pouch (Figure 3A). The two most important metabolites to discriminate between groups were acetate and trimethylamine and other changes in the metabolic profile include reduced concentration of citrate, formate fumarate, glucose, and lactate with MSUc (Figures 3B and 3C). Similar to the air pouch gout model in mice, we found a distinct metabolic profile of serum of patients suffering an acute gout flare compared with individuals with hyperuricemia (HU) but without current flare, including reduced levels of glucose, glutamine, tri-methylamine-N-oxide, and phosphocholine, and increased of dimethylamine (Figures 3D–3F). Of interest, glucose was one of metabolites that separated HU from gout patients.
Figure 3. MSUc leads to systemic metabolic changes.

(A) PLS-DA showing serum levels of metabolites of mice injected with PBS or MSUc (3 mg) in the air pouch as assessed by 1D 1H-NMR showing the divergence between groups (n ≥ 7 mice/group).
(B) The top 15 metabolites were ranked based on variable important projection (VIP) scores from the PLS-DA model (A). The blue and red squares indicate whether metabolite abundance was higher or lower between phenotypes.
(C) Bar graphs showing reduced concentration of several metabolites including acetate, citrate, formate, fumarate, glucose, lactate, and trimethylamine (TMA) in the serum of mice collected 8 h after injected with MSUc in the air pouch.
(D) PLS-DA showing serum levels of metabolites of individuals with gout flare versus HU as assessed by 1D 1H-NMR. Data show the divergence between groups (n = 11 individuals with gout and n = 13 individuals with HU).
(E) The top 15 metabolites were ranked based on VIP scores from the PLS-DA model (B). The blue and red squares indicate whether metabolite abundance was higher or lower between phenotypes.
(F) Bar graphs show reduced concentration of several metabolites including glucose, glutamine, phosphocholine, and TMA and increased dimethylamine in the serum of individuals with gout (#p < 0.10, *p < 0.05, **p < 0.01).
Since our data indicate systemic metabolic changes induced by MSUc, we next investigated in mice whether peripheral blood mononuclear cells (PBMCs) would have an altered transcriptional program when locally injected with MSUc in the air pouch. Indeed, MSUc injection in the air pouch altered the transcriptomic profile of PBMCs (Figure 3G), including upregulation of several genes coding for Slc transporters (Figure 3H) and inflammatory cytokines and adhesion molecules (Figure S3C). As expected, these changes were accompanied by increased concentration of cytokines CXCL1, CCL2, and IL-6 in blood and CXCL1, CXCL2, CCL2, and IL-6 in the air pouch lavage of mice injected with MSUc in the air pouch (Figures S3D and S3E, respectively). Our data indicate that MSUc trigger a metabolic program in macrophages that is also observed in animal models of gout and in serum from patients with acute gout flare. In addition, local acute gouty inflammation leads to increased cytokine levels and metabolic changes both locally and systemically, which are accompanied by a significantly altered metabolite transporter and inflammatory profile of PBMC.
MSUc de-regulate a transcription factor network featured by increased AP-1
To decipher transcriptional mechanisms by which MSUc induces an inflammatory and metabolic program of gene expression, we performed in silico promoter (−2,000 to +500 bp from the transcription start site) analysis of genes upregulated in MSUc or LPS using HOMER. The promoters of genes upregulated by MSUc were enriched in motifs for AP-1 members in both BMDMs and mDMs. Other transcription factors upregulated by MSUc were circadian clock proteins, MYC, NFILs, and the MITF/TFE family (Figure 4A upper panel), but not motifs for IRF, which were only displayed in the promoter of genes upregulated by LPS (Figure 4A, lower panel). Interestingly, macrophages treated with MSUc displayed increased expression of several AP-1 members including JUN, JUNB, JUND, ATF3, FOSB, FOSL1, and FOSL2 mRNA and JUN, p-JUNSer63, ATF3, FOSB, FOSL1, and FOSL2 protein levels but not IRFs, which were only upregulated in macrophages treated with LPS (Figures 4B–4D and S4). Some of them, including JUN, pJUNSer63 and FOSL1, displayed higher upregulation in MSUc than LPS.
Figure 4. MSUc leads to activation of AP-1 but not IRFs.

(A) Motif analysis using HOMER of the promoter (−2,000; +500 bp, from the transcription start site) of genes upregulated by LPS (100 ng/mL) or MSUc (250 μg/mL) for 5 h in mDMs or BMDMs showing enrichment in motifs for AP-1, MYC, MITF, NRF2, and nuclear receptors—but not IRFs—in the promoters of genes upregulated by MSUc.
(B) Bar plots showing upregulation of several AP-1 members—but not IRFs—in macrophages treated with MSUc for 5 h assessed by RNA-seq. Note the higher upregulation of AP-1 in MSUc compared with LPS except for ATF3 in mDMs.
(C) Quantification of nuclear expression of JUN, pJUNSer63, ATF3, FOSL1, FOSL2, FOSB, IRF1, and IRF7 in macrophages treated with MSUc or LPS for 5 h showing upregulation of AP-1 family members—but not IRF1 or IRF7—in macrophages treated with MSUc.
(D) Representative images of protein analysis by IF showing upregulation of JUN, pJUNSer63, FOSL1, and FOSB in macrophages treated with MSUc or LPS for 5 h used to generate plots in (C). Signal for specific antibodies is pseudocolored in red. DAPI in blue is used to delineate DNA structures. Note the higher upregulation of AP-1 in MSUc compared with LPS (#p < 0.10, *p < 0.05, **p < 0.01).
Activation of JNK is required for the inflammatory and metabolic changes induced by MSUc
The over-representation of AP-1 motifs in the promoters of upregulated genes by MSUc and the increased expression of several AP-1 members, including JUN, pJUNSer63, and Fosl1 suggest a role of JUN, JNK, and FOSL1 in the transcriptomic program activated by MSUc. Moreover, MSUc induce the upregulation of p-JNK, which is accompanied by sustained increase and nuclear shuttling of p-JNK (Figures 5A and S5A–S5C), suggesting a distinct activation of JNK in MSUc versus LPS. In line with this, treatment of BMDMs with SP600125, a JNK inhibitor (JNKi) (Bennett et al., 2001), ameliorated the gene upregulation in BMDMs treated with LPS but to a larger extent in BMDM treated with MSUc (p = e−49 for MSUc and p = e−34 for LPS) (Figure 5B). Similarly, incubation with siRNA against Jun or Fosl1 ameliorates the effect of MSUc and LPS (p = e−97 versus p = e−45 for LPS, respectively; and p = e−79 versus p = e−54 for MSUc, respectively) (Figure 5C), indicating that AP-1 activation via JUN-JNK and FOSL1 is required for the transcriptional program induced by MSUc. Moreover, a distinct distribution of changes LPS + JNKi differed to that observed in MSUc + JNKi versus MSUc compared with LPS + JNKi versus LPS or in macrophages treated with siJun or siFosl1 versus siControl (Figure S5D).
Figure 5. The inflammatory and metabolic program induced by MSUc is regulated through JNK.

(A) Protein analysis by WB of JNK and pJNK expression in BMDMs treated with LPS (100 ng/mL) or MSUc (250 μg/mL) at various time points. Data show prolonged pJNK expression in BMDMs treated with MSUc.
(B and C) Boxplots showing amelioration of gene expression after treatment with JNKi (20 μM) versus vehicle of genes upregulated by LPS or MSUc (B), genes upregulated only in LPS o rMSUc or commonly upregulated in LPS and MSUc for 5 h (C) (n ≥ 5/group). Data represent the distribution of fold change of upregulated genes in MSUc versus PBS or LPS versus PBS in cells treated with MSU + JNKi or LPS + JNKi.
(D) Gene ontology analysis using REACTOME of genes between 33% and 50% reduction of expression in MSUc + JNKi versus MSUc or LPS + JNKi versus LPS. Data show enrichment in inflammatory gene sets in LPS and MSUc and metabolic gene sets in MSUc.
(E) Gene ontology analysis using REACTOME of genes between 33% and 50% reduction of expression in siJun-MSUc versus siControl-MSUc or siJun-LPS versus siControl-LPS. Data show enrichment in inflammatory gene sets in LPS and MSUc gene sets.
(F–H) Expression analysis by RNA-seq (F and H) or qRT-PCR (G) of inflammatory and metabolic genes induced by MSUc or LPS showing reduction after treatment with JNKi in BMDMs (F) (n ≥ 5/group) or mDMs (G) (n = 3 donors/group), or in BMDMs after treatment with siJun or siFosl1 (H).
(I) Expression analysis by RNS-seq of Jun or Fosl1 in cells incubated with siControl or siJun or siFosl1 showing downregulation of Jun or Fosl1 in siJun or siFosl1 (#p < 0.10, *p < 0.05, **p < 0.01). Broken lines in (D and E) represents the cutoff for significance −log10(0.05) = 1.30.
The promoter of genes downregulated in LPS + JNKi versus LPS were enriched in putative binding sites for IRFs, NFKB, and AP-1. Those downregulated in MSUc + JNKi versus MSUc were enriched in AP-1, MYC, NRF2, and circadian clock proteins (Figure S5E). Whereas gene sets recovered in LPS or both LPS and MSUc in cells treated with JNKi belonged to inflammatory signaling pathways, those uniquely ameliorated by MSUc were related to metabolism including metabolism of carbohydrates, glycosaminoglycan, transport of small molecules, and Slc-mediated transmembrane transport (Figure 5D). Similar recovery in inflammatory signaling pathways—but not metabolic signaling pathways—is observed in cells treated with siJun (Figure 5E), indicating that metabolic genes are more sensitive to changes in p-JNK, and that signaling by JNK is a major contributor to the regulation of the metabolic-inflammatory changes induced by MSUc.
Of note, treatment with JNKi only partially rescued the increased expression of metabolic genes, suggesting additional mechanisms involved (Figure 5F). Interestingly, several inflammatory genes, including Ccl3, Ccl9, Cxcl1, Cxcl2, and Tnfa, that were upregulated in LPS versus MSUc showed higher downregulation in MSUc + JNKi versus LPS + JNKi. Several metabolic genes, including Slc1a4, Slc2a1, and Slc7a11, that were upregulated to a similar extent in LPS and MSUc only showed downregulation in MSUc + JNKi. Some AP-1 members, such as Jun, Jund, Fosl1, Atf4, and Atf5, and the stress response gene Nqo1, that were upregulated in MSUc versus LPS also showed higher downregulation in MSUc + JNKi versus LPS + JNKi (Figure 5F). The ameliorated upregulation in MSUc + JNKi versus MSUc was also demonstrated for SLC2A1, SLC1A5, CXCL1, IL6, HK2, ENO2, and SLC38A2 in mDMs (Figure 5G). Similar to the treatment with JNKi, incubation with siJun ameliorates several inflammatory genes upregulated by LPS or MSUc, such as Ccl2, Ccl3, Ccl4, and Ccl7, and metabolic genes, such as Slc2a1 and Slc38a2, that were only ameliorated in MSU + JNKi are only ameliorated in siJun-MSUc but not in siFosl1-MSUc (Figure 5H). Interestingly, we find that JUN regulates Fosl1 expression and that FOSL1 regulates Jun expression (Figure 5I).
These mRNA changes were accompanied by reduced protein expression of JUN, JUNSer63, FOSL1, FOSB, and SLC2A1 in mDMs (Figures 6A and 6B), and additionally PFKBP3 in BMDMs (Figures S5F and S5G) treated with MSUc versus MSUc + JNKi as assessed by immunofluorescence and ELISA for CCL2, CXCL1, CXCL2, and TNF (Figure 6C). Of note, whereas JNKi reduced partially the increased secretion of cytokines by LPS, JNKi reduced to almost basal levels the secretion of cytokines by MSUc (Figure 6C). In addition, JNK inhibition also reduced the upregulation of some of the metabolic changes observed after MSUc stimulation, specifically JNK inhibition ameliorated the increase of creatinine phosphate, dimethylamine, glutamate, and lactate, but not acetate, formate, pyruvate (p = 0.26) or lysine (Figure 6D).
Figure 6. Increased JUN binding to the promoter of inflammatory and metabolic genes is regulated by JNK and is required for the activation of the inflammatory and metabolic program induced by MSUc.

(A and B) Protein analysis by IF of JUN, pJUNSer63, FOSL1, SLC2A1, and FOSB in mDMs treated with MSUc (250 μg/mL) or MSUc + JNKi (20 μM) for 5 h. Quantification of (B) corresponds to experiment shown in (A), and shows downregulation of protein expression in MSU + JNKi versus MSUc (n = 3/condition). Signal for specific antibodies is pseudocolored in red. DAPI in blue is used to delineate DNA structures.
(C) Protein analysis by ELISA of cytokines in the supernatant of BMDMs treated with LPS or MSUc overnight w/wo JNKi showing complete reduction in MSUc + JNKi versus MSUc and partial reduction in LPS + JNKi versus LPS (n = 3/group).
(D) Analysis of metabolites by 1D 1H-NMR in the culture medium of BMDMs treated with MSUc or MSUc + JNKi for 8 h showing varying degree of recovery (n = 3/condition).
(E and F) JUN ChIP in BMDMs (E) or mDMs (F) treated with LPS, MSUc, LPS + JNKi, or MSUc + JNKi and qPCR over regulatory regions of genes upregulated by LPS or MSUc. Data show higher levels of JUN binding in macrophages treated with MSUc that is reduced upon treatment with JNKi (n = 4/condition) (#p < 0.10, *p < 0.05, **p < 0.01).
Since phosphorylation by p38 is one of the major drivers of JUN phosphorylation (Shaulian and Karin, 2002), we next decided to investigate whether inhibition of p38 would have similar effect as inhibition of JNK. Interestingly, we did not observe amelioration on JUN or pJUNSer63 protein levels nor reduction on upregulated genes by MSUc in BMDMs treated with MSUc and p38 inhibitor (p38i)—except for Cxcl1—or recovery on the upregulation of FOSL1 or JUNB protein levels (Figures S6A–S6D). These data indicate that JNK—but not p38—regulates the inflammatory and metabolic program altered by MSUc.
We then investigated whether JNK-dependent gene expression is due to JUN binding to the regulatory regions of upregulated genes. We performed chromatin immunoprecipitation (ChIP) followed by qPCR of regions on open chromatin selected from publicly available ATAC-seq (Link et al., 2018). Interestingly, even though Ccl2, Ccl3, Cxcl2, and Pfkbp3 are more upregulated in LPS than in MSUc, we found similar or greater JUN enrichment in cells treated with MSUc than in LPS. In addition, genes that were only upregulated in MSUc displayed higher JUN occupancy, such as Slc38a2 and Nqo1. All the genes with reduced expression in MSUc + JNKi versus MSUc or LPS + JNKi versus LPS showed reduced JUN recruitment to their promoter regions after JNKi, including Slc1a4, Slc2a1, Slc7a11, Slc38a2, and Slc15a3, in a higher degree in MSUc + JNKi versus MSUc. Genes that were not affected by treatment with JNKi, such as Slc1a4, Slc2a1, or Slc7a11 in LPS + JNKi versus LPS did not display reduced JUN recruitment after treatment with JNKi (Figure 6E). Some of these findings were recapitulated in mDMs treated with JNKi, including reduced JUN recruitment to SLC2A1, ENO2, and SLC38A2 promoters (Figure 6F). Altogether, our data suggest that the inhibition of upregulated gene transcripts after MSUc by JNKi—but not p38i—is regulated through JUN recruitment to their promoter regions to a larger extent in genes upregulated by MSUc than by LPS.
Reduction of oxidative stress or blockage of inflammasome does not affect the activation of inflammatory or metabolic genes by MSUc
The interaction of MSUc with the plasma membrane of the cell promotes a cellular response that includes the production and release of reactive oxygen species and subsequent endoplasmic reticulum (ER) stress and oxidative stress (Choe et al., 2015. In line with this, we found that ER stress activated unfolded protein response (Sautin and Johnson, 2008; Zamudio-Cuevas et al., 2016), and NRF2 target genes were upregulated by MSUc (including Mafg, Atf4, Hsp70, and Soat2 mRNA and protein levels), and were also reduced by treatment with JNKi (Figures S6E–S6G). However, treatment of BMDMs with the antioxidant mixture butylated hydroxyanisole (BHA) reduced only partially the upregulation of JUN or pJUNSer63 protein levels (21% reduction in MSU + BHA versus MSU, p < 0.10) and did not alter the overexpression of inflammatory and metabolic genes by MSUc (Figures S6H–S6J), suggesting that JUN activation by oxidative stress or ER stress is not the principal etiology of the changes induced by MSUc. Moreover, despite the role of NLRP3 inflammasome in gout being well described (Lu et al., 2015; So and Martinon, 2017), we found that treatment of BMDMs with MCC950, a NLRP3 inflammasome inhibitor, did not reduce the expression of JUN, pJUNSer63, and FOSL1 protein levels nor affected the upregulation of gene expression by MSUc (Figure 6K). As expected, treatment with MCC950 ameliorated the increased secretion of BMDMs induced by LPS + MSUc (Figure S6K). Our data indicate that activation of the NLRP3 inflammasome is not required for upregulation of JUN and transcriptomic changes induced by MSU.
JNK and SLC2A1 transport inhibition ameliorated inflammation in animal models of acute gout
The previous results led us to investigate whether activation of JNK was required for the phenotype induced by MSUc in vivo In addition, since glucose metabolism was one of the top metabolic pathways deregulated by MSUc in both BMDMs and mDMs, we also tested whether the inhibition of the glucose transporter SLC2A1 improved animal models of gouty inflammation. We found increased expression of pJNK, SLC2A1, LDHA, ENO2, and iNOS in macrophages in the subcutaneous air pouch lining of mice injected with MSUc (Figures 7A, 7B, and S7A). Injection of MSUc in the air pouch lining led to a recruitment of leukocytes that was significantly lowered after treatment with JNKi (Figures 7C–7E) or BAY-876, a SLC2A1 inhibitor (Figures 7F–7H). Similarly, intra-articular injection of MSUc led to increased infiltrates of inflammatory cells that were significantly reduced after treatment with JNKi (Figures 7I and 7J) or BAY-876 (Figures 7K and 7L), as well as reduced serum cytokine levels of IL-6 and CCL2 (Figure 7M). Of note, we found that treatment of BMDMs with BAY-876 did not reduce the expression of JUN and pJUNSer63 protein levels or affect the upregulation of gene expression by MSUc (Figures S7B–S7D), suggesting that SLC2A1 is a critical downstream target gene of MSUc, but glycolysis activation is not responsible for upregulation of JUN and transcriptomic changes induced by MSU.
Figure 7. Signaling by JNK and SLC2A1 is required for the MSUc-induced damage in vivo.

(A and B) Protein analysis by IF showing expression of SLC2A1 (A) and pJNK (B) in macrophages in the subcutaneous cavity of the air pouch when injected with MSUc for 8 h(n = 2/group). Signal for specific antibodies is pseudocolored in red. DAPI in blue is used to delineate DNA structures.
(C) Histological analysis by H&E is showing the recruitment of inflammatory cells (arrows) induced by MSUc in the air pouch is reduced upon treatment with JNKi (15 mg/kg; n ≥ 4/group).
(D and E) Pathological assessment of inflammatory cell infiltrates of the air pouch (D) and neutrophil count of the air pouch lavage (E) showing reduction in MSUc + JNKi versus MSUc (n ≥ 4/group).
(F) Histological analysis by H&E is showing the recruitment of inflammatory cells (arrows) induced by MSUc in the air pouch is reduced upon treatment with BAY-876 (5 mg/kg; n ≥ 5/group).
(G and H) Pathological assessment of inflammatory cell infiltrates of the air pouch (G) and neutrophil count of the air pouch lavage (H) showing reduction in MSUc + BAY-876 versus MSUc (n ≥ 10/group).
(I and J) Histological analysis by H&E showing reduction in the recruitment of inflammatory cells (arrow) in the synovial cavity of mice injected with MSUc + JNKi (15 mg/kg) versus MSUc (n ≥ 10/group).
(K and L) Histological analysis by H&E showing reduction in the recruitment of inflammatory cells (arrow) in the synovium of mice injected with MSUc + BAY-876 (7.5 mg/kg) versus MSUc (n ≥ 7/group). (M) Protein analysis by ELISA of serum levels of IL-6 or CCL2 in mice injected with MSUc w/wo JNKi in the air pouch showing reduction in MSUc + JNKi versus MSUc (n = 4/group) (#p < 0.10, *p < 0.05, **p < 0.01).
DISCUSSION
The transcriptional mechanisms underlying macrophage response to MSUc are largely unknown. Here, the investigation of genome-wide transcriptomic and bioinformatic analysis of the promoter of deregulated genes in macrophages revealed that MSUc activates a distinct inflammatory and metabolic program that is regulated through transcriptional activation, protein phosphorylation, and recruitment of JUN to the regulatory regions of inflammatory and metabolic genes, and this is regulated via JNK activity.
MSUc activate the inflammatory program of macrophages. However, priming cells either with LPS or other TLRs agonists hampers very much the understanding of the response to MSUc by macrophages. Our work demonstrates upregulation of inflammatory and metabolic genes in both mDMs and murine BMDMs by MSUc alone. Several metabolic pathways were upregulated after MSUc stimulation including carbohydrates metabolism, lipid metabolism and also nutrient, including amino acid, transport by the solute carrier (SLC) transporter family. The metabolic changes featured by SLC2A1-mediated glucose uptake yield crucial insights into how MSUc gouty inflammation exerts its pathogenic effect. Whereas some of those metabolic genes are similarly deregulated in macrophages treated with LPS mRNA, these macrophages do not display large deregulation on metabolite production (except for increased concentration of lactate). Although these differences are probably due to different timing, other options of control of metabolic pathways by LPS including protein stability and membrane localization are also possible.
Amino acid pathways, such as glycine, serine, and threonine, and the phenylamine pathway, and amino acid-related SLC transporters, were more obviously deregulated after MSUc than LPS stimulation, both in gene expression and metabolite levels. Prior studies have highlighted the important role of amino acids in macrophage phenotype and function. Amino acids, including glutamine, arginine, or glycine, were shown to control macrophage polarization and used differently in different macrophage subtypes. Some of these metabolic changes control macrophage polarization by epigenetically regulatingthe transcriptional activity of key genes (Phan et al., 2017). Whether MSUc activates a unique macrophage phenotype with a particular metabolic rewiring that helps with inflammation self-limitation and gout resolution, or whether targeting other SLC transporters play a key role in gout pathogenesis needs further studies. In addition, stable isotope-tracing metabolomics would be critical to understand the glucose and amino acid metabolic pathway activity, via the relative production and consumption of the labeled metabolites, and key differences between MSU- and LPS-induced metabolic shifts in macrophages.
Previous studies have shown the inflammatory program triggered by MSUc (Brovold et al., 2019; Chung et al., 2016; So and Martinon, 2017), and our current work, confirms previous work that showed that MyD88 was pivotal to MSUc-induced inflammation (Liu-Bryan et al., 2005). Recently a work by Renaudin et al. (2020) found that MSUc favored glycolytic activity of primed THP-1 cells. However, Renaudin’s work is limiting in identifying the mechanism involved in the MSUc effects in primary mouse and human macrophages. We provide evidence that AP-1 family signaling is required for activation of the transcriptional program by MSU. Enrichment of AP-1 motifs in the promoter of upregulated genes by MSUc was accompanied by upregulation of most AP-1 members at mRNA and protein level, including phosphorylation of JUN at the Ser63 residue. Activation of AP-1 activity is modulated through its dimer composition (Ameyar-Zazoua et al., 2005; Eferl and Wagner, 2003), which is determined by the regulation of the synthesis and stability of respective mRNA or through the regulation of protein stability (for example, via phosphorylation). Phosphorylation of AP-1 members modulates the transcriptional activity of AP-1 dimers (Karin, 1995) in a process that is mostly mediated by the JNK and p38 MAPK pathways that once translocated to the nucleus regulates the transcriptional activity of AP-1 dimers (Puigserver et al., 2001). Therefore, mechanisms that activate phosphorylation and/or mRNA synthesis would change the stoichiometry of AP-1 members and would dictate their dimer composition.
We found no evidence that MSUc induce a hyperphosphorylation of JNK compared with LPS. However, the prolonged nuclear pJNK cytosolic and nuclear shuttling in BMDMs exposed to MSUc suggests that increased pJUNSer63 could be due both to the unexpected increased expression of JUN mRNA, and/or increased shuttling of pJNK to the nucleus where JUN interacts with other AP-1 members. Of interest, steroidal and non-steroidal anti-inflammatory drugs that are used to treat acute flares of gout are known to abrogate JNK signaling (Adcock and Caramori, 2001; Amann and Peskar, 2002). Our finding that treatment with p38i has no effect in reducing JUN/pJUNSer63 or in ameliorating the transcriptomic effects induced by MSUc, suggest that p38 is not the main mechanism of JUN phosphorylation by MSUc. Regardless of the mechanism, phosphorylation of JUN on Ser 63/73, for example, potentiates its transcriptional activity by heterodimerization with FOS (Deng and Karin, 1994; Woodgett et al., 1993). Interestingly, we find that FOS is upregulated by MSUc but not by LPS (Log2-fold change −1.08 versus 0.5 in BMDMs or −2.31 versus 3.77 in mDMs after LPS or MSUc, respectively) and FOSB is the AP-1 member with largest upregulation in MSUc compared with LPS (Log2-fold change 0.44 versus 5.19 in BMDMs or 0.196 versus 6.85 in mDMs after LPS or MSUc, respectively). Transactivation of JUN is also potentiated by ATF2 through the binding of Rb or E1A to the ATF2-JUN dimer (Bhoumik et al., 2007; Shaulian and Karin, 2002; van Dam and Castellazzi, 2001). However, we find no upregulation of ATF2 in macrophages stimulated with MSUc, suggesting that this might not be the major cause of increased JUN activity.
Thus, our data point to a strong transcriptional activation of AP-1 genes by MSUc. The expression of several AP-1 components is under positive and negative AP-1 autoregulation and this feedback allows a finely tuned regulation of AP-1 member activity over time (Angel et al., 1988, 2001; Chiu et al., 1989; Hai and Curran, 1991). In pancreatic epithelial cells, for instance, Jun mRNA expression is regulated thorough the cooperative binding of the nuclear receptor NR5A2 and JUN itself (Cobo et al., 2018), suggesting that nuclear receptors might be playing a role in Jun upregulation by MSUc. Although siJun ameliorated the expression of some metabolic genes induced by MSUc, such as Slc2a1 or Slc38a2, most of the genes ameliorated by siJun or siFosl1 did not belong to metabolic signaling pathways. This discrepancy may be due to the remaining 40% of Jun or 50% of Fosl1 expression after incubation with siJun or siFosl1, respectively.
Our results using JUN ChIP demonstrate that the recruitment of JUN to the promoter of upregulated genes is greater in MSUc than LPS regardless of the degree of upregulation of the gene transcript. Interestingly, treatment with JNKi has greater effect over the upregulated genes induced by MSUc than by LPS; and, of those induced by LPS, JNKi has a greater effect in LPS-unique upregulated genes compared with those also upregulated by MSUc, suggesting that even when a gene is deregulated both by LPS and MSUc, JNK has greater importance for its regulation in MSUc than LPS. Our data provide evidence that treatment with JNKi could be therapeutically beneficial to overcome the damage associated with gouty inflammation. Dysregulation of the JNK pathway is associated with a wide range of immune and neurological disorders and cancer, and there is active research to develop better small molecules to inhibit JNK without generating major side effects (Bennett et al., 2001; Cicenas et al., 2017; Grassi et al., 2015; Hammouda et al., 2020; Hepp Rehfeldt et al., 2020; Meng and Huang, 2018; Montagut and Settleman, 2009). The motif analysis of the promoters of genes that are downregulated with JNKi versus vehicle suggest that, whereas the effect of JNKi on LPS-activated genes might be secondary to activation of IRFs, the effect of JNKi in MSUc-activated genes is mainly driven through modulation of AP-1 activity in cooperation with other transcription factors, such as NRF2, MITF, and circadian clock regulators. Experiments using macrophage-specific knockout mouse models would be helpful to address which transcription factor network is required for the inflammatory and metabolic alterations induced by MSUc in vitro and in vivo.
Despite the role of the NLRP3 inflammasome in gout disease being well described, we found no evidence that the inflammasome is required for the upregulation of inflammatory and metabolic genes by MSUc. We also found downregulation of Tlr3, Tlr8, and Tlr9 mRNA in BMDMs treated with MSUc and downregulation of TLR4, TLR5, and TLR6 mRNA in mDMs treated with MSUc, which, together with the significant differences with LPS program—including the lack of activation of IFN program by MSU—suggest that activation by TLRs is not the major driver of the phenotype observed in macrophages.
Finally, gout attacks are self-limiting, suggesting that certain regulatory mechanisms are present to modify the acute inflammatory response. Preliminary data showed that MSUc share a larger fraction of inflammatory genes with LPS at shorter time points, suggesting a transcriptional repression of the inflammatory program at later time points. Our data call for an alternative phenotype of macrophages exposed to MSUc. Genome-wide analysis of the transcriptomic effects of MSUc and LPS at different time points would help to address these points. Isolation of macrophages from the synovial cavity of patients with gout would be helpful to phenotypically characterize the effect of MSUc in resident macrophages.
Limitations of the study
There are few limitations in this study. (1) Additional experiments using genetic deletion of Jnk1 and Jnk2 will be helpful in further characterizing the role of JNK in the transcriptional program induced by MSUc. (2) Although the use of siRNA to knock down the expression of genes of interest is a common tool, the results should be interpreted with caution as the conditions for transfecting siRNA alter the transcriptional program of macrophages. (3) Cell death is observed in macrophages exposed to 250 μg of MSUc after 6 h which makes interpretation of some of the in vitro experiments difficult.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Monica Guma (mguma@health.ucsd.edu).
Materials availability
This study did not generate new unique reagents
Data and code availability
The accession number for the sequencing data reported in this paper is GEO: GSE191054.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human monocyte derived macrophages (mDM)
Human peripheral blood mononuclear cells (PBMC) were isolated from whole blood by Ficoll density gradient using Ficoll Plaque Premium (Sigma GE Healthcare, #17–544-02) or BD Vacutainer CPT Tubes (BD, #362753) as described somewhere else. PBMC were washed twice with HBSS (Gibco ThermoFisher, #14175095) containing 2% BSA, 1 mm EDTA. Monocytes were obtained by negative selection using a kit (StemCell, #19359). Monocytes were incubated with RPMI 1640 (Sigma Aldrich, #10–040) medium containing 10% de-complemented FBS (OmegaScientific, #FB-02), 1 mM Sodium Pyruvate (Gibco Thermofisher), Penicillin-Streptomycin (15140122, 1000 U/mL) and 25 ng/mL-50 ng/mL of recombinant human M-CSF (StemCell, #78057–2) for six days at 37°C and 5% CO2. Fresh media was added 48 h and four days after seeding. The mDM were incubated for five hours with 100 ng/mL of Kdo2 lipid KLA or 250 μg/mL of MSUc in RPMI 1640 medium containing 10% de-complemented FBS, 1 mM Sodium Pyruvate, Penicillin-Streptomycin and 25ng–50 ng/mL of recombinant human M-CSF. 20 μM of JNK inhibitor SP600125 (Tocris, #1496) was added to the media one hour prior treatment with MSUc.
Bone-marrow derived macrophages (BMDM)
Flushed bone marrow from both femurs and tibiasof C57BL/6or Myd88−/− mice were cultured in DMEM medium supplemented with 10% de-complemented FBS, 100 units/mL penicillin and 100 ug/mL streptomycin, 2 mM L-glutamine, and 20% of L929 conditioned media for for six days at 37°C and 5% CO2. On day six, BMDM were trypsinized and plated in DMEM medium supplemented with 10% de-complemented FBS, 2 mM L-glutamine, 100 units/mL penicillin and 100 ug/mL streptomycin, and 10 ng/mL recombinant murine M-CSF (Peprotech, #315–02) for one day prior being stimulated for five hours with 100 ng/mL of LPS or 250 μg/mL of MSUc. 20 mM of JNK inhibitor SP600125 (Tocris, #1496), 10 mM of BHA (Sigma, B1253), 20 nM of BAY-876 (Selleckchem, #S8452), 3 μM of p38 MAPK inhibitor SB203580 (Tocris, #1202) or NLRP3 inflammasome inhibitor MCC950 (1uM, Selleckchem, #S7809) were added to the media one hour prior to stimulation with LPS or MSUc.
Subjects
Patients meeting the 2015 ACR/EULAR gout classification criteria with hyperuricemia were recruited. Consecutive patients were recruited from the Rheumatology Outpatient Clinic. The study was approved by the VA Institutional Board Review and patients signed an informed consent. 11 patients with active gout flare (males, age of 58.7 ± 15.2, BMI of 31.8 ± 5.3, 7 white, 1 African American, 2 Asian and 1 Pacific Islander) and 13 patients with hyperuricemia (males, age of 57.9 ± 10.6, BMI of 31.2 ± 3.6, 4 White, 5 Pacific Islander, 2 African American, and 2 Asian), without a diagnosis of diabetes were included for NMR analysis. Patients were not on prednisone. Other co-morbidities including high blood pressure and dyslipidemia were similar in both groups. Non-fasting blood samples were collected in the clinic by research personnel into 10 mL BD Vacutainer Blood Collection Tubes containing spray-coated silica and a polymer gel for serum separation. After 30 min incubation at room temperature, tubes were centrifuged for 10 min at 2000xg and sera were transferred into 1.7 mL tubes and immediately frozen and stored at −80°C until analysis.
Mice
All animal experiments were performed in agreement with the Institutional Animal Care and Use Committee (IACUC). C57Bl/6 male mice (Jackson laboratories, Sacramento, CA, USA) were housed in a temperature-controlled room with a 06.00–18.00-h light cycle and consumed regular chow and tap water ad libitum. Mice were 8–12 weeks at start of the experiments, and randomized before starting treatment to reduce bias.
Air pouch model of inflammation
Dermal air pouches were established by injecting mice dorsally subcutaneously with 3 mL filtered (0.20 μm) air on day 0 and 3 (Sanchez-Lopez et al., 2019b; Stubelius et al., 2018). On day 7, BAY-876 (5 mg/kg, in 2.5%DMSO/40%PEG/5%Tween80) or JNK inhibitor (15 mg/kg, in 2.5%DMSO/30%PEG/10%Tween80) were injected intrapouch and one hour later, MSUc were injected (3 mg in 0.5 mL PBS). 2.5%DMSO/40%PEG/5%Tween80 or 2.5%DMSO/30%PEG/10%Tween80 was used as vehicle control in mice injected with BAY-876 or JNKi, respectively. After eight hours of MSUc intrpouch injection, pouch exudate was collected by rinsing with 1 mL endotoxin-free PBS, followed by 30 s of gentle massage. The collection was centrifuged (5 min, 450 × g) for analysis. Neutrophil quantification was performed after citospin and HE staining. Skin tissue covering the pouch was then excised and fixed in 10% formalin (Fisher #SF100–4) for subsequent haematoxylin and eosin (H&E) staining. Infiltrated cell area was scored using a qualitative score from 0 to 3 in a blinded manner for two independent readers. In some air pouch experiments, peripheral blood monocyte cells (PBMC) from whole blood by Ficoll density gradient using Ficoll Plaque Premium (Sigma GE Healthcare, #17–544-02) were isolated after eight hours of MSUc intrapouch injection, The number of mice used in each experiment is provided in the figure legends.
Intra-articular MSUc injection
BAY-876 (7.5 mg/kg) or JNK inhibitor (15 mg/kg) were injected intraperitoneally one hour prior the injection of PBS in one knee or 100 μg of MSUc in the other knee (Stubelius et al., 2018). Eight hours after the injection of PBS or MSU, the knees were collected, fixed and decalcified (10% EDTA, Fisher #BP2482–500). Slides were stained with H&E. Infiltrated cell area was scored using a qualitative score from 0 to 3 in a blinded manner for two independent readers. Two cutting depths were used per knee and the largest infiltrated area per treatment was used. The number of joints per group are provided in the figure legends. 2.5%DMSO/40%PEG/5% Tween80 or 2.5%DMSO/30%PEG/10%Tween80 was used as vehicle control in mice injected with BAY-876 or JNKi, respectively.
METHOD DETAILS
Reagents
PBS, DMEM (w/5% glutamine, 10% FBS, 5% penicillin), trypsin 0.25%, LPS (100 ng/mL), MSUc were prepared as described (Guma et al., 2009) suspended at 25 mg/mL in sterile, endotoxin-free phosphate buffered saline (PBS) and verified to be free of detectable lipopolysaccharide contamination by Limulus lysate assay (Lonza, Walkersville, Maryland, USA), MCSF (10 ng/mL, Peprotech), GLUT1 inhibitor BAY-876 (20 nM, Selleckchem, #S8452), p38 MAPK inhibitor SB203580 (3 uM, Tocris, #1202), JNK inhibitor SP600125 (20uM, Tocris, #1496), beta-hydroxybutyric acid (BHA; 10uM, Sigma, #B1253), NLRP3 inflammasome inhibitor MCC950 (1uM, Selleckchem, #S7809).
Knock down of gene expression in BMDM
Jun or Fosl1 expression was depleted from BMDMs using synthetic small interference RNAs (siRNAs, Dharmacon, SMARTPool) targeting mouse Jun (#L-043776–00-0005) or Fosl1(#L-040704–00-0005). siGENOME. Non-Targeting siRNA #2 (D-001210–02) was used as negative control. 2 million of differentiated BMDMs were transfected with 20 nM of specific siRNA using Lipofectamine™ RNAiMAX Transfection Reagent (Invitrogen, #13778150). Lipofectamine-siRNA complexes were formed by incubating 0.24 pmol of each siRNA with 24 mL of Lipofectamine reagent in Opti-MEM (Thermo Fisher Scientific) for 15 min at room temperature and then added into cells and incubated for 5 min at room temperature. The cells with the transfection complexes were seeded in 6 wells plates and cultured in RPMI 1640 containing 10% FBS, 1% penicillin/streptomycin supplemented with L-glutamine and 16.7 ng/ml M-CSF for two days and then treated with PBS, LPS or MSU.
RNA-seq
It was performed as described elsewhere (Gosselin et al., 2017; Seidman et al., 2020). Briefly, BMDM or mDM were lysed in TRIzol (ThermoFisher, #15596026). RNA isolation and DNase treatment were carried out using Direct-zol RNA MicroPrep kit (Zymoresearch, #11–33MB). 500ng-1 μg total RNA was enriched in poly-A tailed RNA transcripts by double incubation with Oligo d(T) Magnetic Beads (NEB, S1419S) and fragmented for 9 min at 94°C in 2X Superscript III first-strand buffer containing 10 mM DTT (Invitrogen, #P2325). The 10 μL of fragmented RNA was added to 0.5 μL of Random primers (Invitrogen, #48190011), 0.5 μL of Oligo d(T) primer (Invitrogen, #18418020), 0.5 μL of SUPERase inhibitor (Ambion, #AM2696), 1 μL of 10 mM dNTPs and incubated at 50°C for three minutes. Then, 5.8 μL of water, 1 μL of 10 mM DTT, 0.1 μL of 2 μg/μL Actinomycin D (Sigma, #A1410), 0.2 μL of 1% Tween 20 (Sigma) and 0.5 μL of SuperScript III (Invitrogen, #ThermoFisher 18080044) was added to the mix. Reverse-transcription (RT) reaction was performed at 25°C for 10 min followed by 50°C for 50 min. RT product was purified with RNAClean XP (Beckman Coulter, #A63987) and eluted in 10 μL in 0.01% Tween 20. The RNA-cDNA complex was then added to 1.5 μL of 10X Blue Buffer (Enzymatics, #B0110-L), 1.1 μL of dUTP mix (10 mM dATP, dCTP, dGTP and 20 mM dUTP), 0.2 μL of 5 μ/ml RNAseH (Enzymatics, #Y9220L), 1 μL of 10 U/μl DNA polymerase I (Enzymatics, #P7050L), 0.15 μL of 1% Tween 20 and 1.05 μL of nuclease free water; and incubated at 16 C for 2.5 h or overnight. The resulting dsDNA product was purified using 28 μL of SpeedbBead Magnetic Carboxylate (GE Healthcare, #651521050 50250) diluted in 20%PEG8000:2.5 M NaCl to a final 13% PEG concentration, washed twice with 80% etOH, air dry and eluted in 40 μL of 0.05% Tween 20. The purified 40 μL of dsDNA was end-repaired by blunting followed by A-tailing and adapter ligation as described elsewhere (Heinz et al., 2010) using BIOO Barcodes (BIOO Scientific, #514104), IDT TruSeq Unique Dual Indexes or Kapa Unique Dual-Indexed Adapters using 15 μL Rapid Ligation Buffer (Enzymatics, #L603-LC-L), 0.33 μL 1% Tween 20 and 0.5 μL T4 DNA ligase HC (Enzymatics, #L6030-HC-L). Libraries were amplified by PCR for 11–15 cycles using Solexa IGA and Solexa IGB primers (AATGATACGGCGACCACCGA and CAAGCAGAAGACGGCATACGA, respectively), purified using 1 μL of SpeedbBead Magnetic Carboxylate in 15.2 μL of 20% PEG8000:2.5 M NaCl, washed with 80% etOH and eluted in 0.05% Tween 20. Eluted libraries were quantified using a Qubit dsDNA HS Assay Kit and sequenced on a NextSeq 500 or Hi-Seq 4000 (Illumina, SanDiego, California). We have included the quality data (Table S1, related to Figure 1) and entire tpm spread sheet for each experiment as a supplementary table and deposit the primary sequencing data in GEO (GSE191054).
Analysis of RNA-Seq
FASTQ sequencing files were mapped to the mm10 or hg38 reference genomes for mouse samples or human samples, respectively using STAR with default parameters. Biological replicates were used in all experiments. Quantification of transcripts was performed with parameter −l 200 and analyzeRepeats.pl (HOMER) with parameters -condenseGenes -count exons -noadj. Principal Component Analysis (PCA) was obtained based on the Transcripts Per kilobase Million (TPM) on all genes of all samples. Differential expression analysis was performed using rnaDiff.R for genes with a minimum TPM of 0.5inat least two samples. Genes with log2 fold chance >1 or < (−1) and FDR <0.05 were considered as differentially expressed. Only libraries with >80% mapped reads were used for downstream analysis. A list of differentially expressed genes in human mDM and mouse BMDM is provided in Tables S2 and S3, respectively (related to Figure 1).
Gene Ontology Analysis
Analysis of signaling pathways regulated by differentially expressed genes was performed with the Molecular Signature Database (MSigDB) of Gene Set Enrichment Analysis (GSEA) (Liberzon et al., 2011, 2015; Subramanian et al., 2005) using the Investigate Gene Sets tool. Input gene identifiers were selected from the list of differentially expressed genes obtained by RNA-Seq. Significance of signaling pathways was generated by calculating the overlap between the gene list of interest and REACTOME, HALLMARKS or KEGG datasets as indicated in the Figure legends. A list of the gene sets of genes differentially expressed in human mDM or mouse BMDM is provided in Tables S4 and S5, respectively (related to Figure 1).
Promoter scanning analysis
In silico promoter analysis of differentially expressed genes was performed using the findMotifs.pl tool of HOMER searching for motifs of length between 8 and 10 nucleotides and from −2000 to +500 bp relative to the TSS, using 4 threads.
Gene expression analysis by retrotranscription and quantitative PCR (RT-qPCR)
Between 500 Kand 2 × 106 cells were lysed in Trizol (Invitrogen, #15596026). Total RNA was isolated using Direct-zol RNA MicroPrep kit (Zymoresearch, #11–33MB) and treated with DNasel on column. 500 ng-1 μg of RNA solution was enriched in poly-A tailed RNA by two consecutive incubations with Oligo d(T) Magnetic Beads (S1419S) or directly used for cDNA synthesis. cDNA was prepared according to the manufacturer’s specifications, using the SuperScript III Reverse Transcriptase (ThermoFisher, #18080093) or Superscript VILO cDNA master mix (Thermofisher, #11756050). RT-qPCR analysis was performed using the KAPA SYBR Fast PCR master mix (Sigma Aldrich, #KK4605) or SYBR Green PCR Master Mix (Applied Biosystems, #43–091-55) and an ABI-StepOne 96 well plate instrument (Applied Biosystems). Expression levels were normalized to HPRT mRNA levels using the ΔΔCt method. The sequence of the primers used can be found in Table S6.
Chromatin immunoprecipitation (ChIP)
JUN ChIP was performed as described somewhere else (Eichenfield et al., 2016; Seidman et al., 2020). Briefly, BMDM or mDM were fixed with 3 mM Disuccinimidyl-glutarate, DSG (Proteochem, #C1104) in PBS for 30 min at room temperature followed by 10 min incubation with 1% formaldehyde (ThermoFisher, #28906) at room temperature. Then, fixation was quenched by adding 2.625 M of glycine to a final concentration of 125 mM. Cells were washed with 0.01%Triton X-100:PBS, scraped and centrifuged for 10 min at 3,000 rpm at 4°C and then washed once again with 0.01%Triton X-100:PBS, pelleted, snap frozen and stored at −80°C. For ChIP experiments, cells were thawed and permeabilized in 1 mL of ice-cold buffer containing 10 mM HEPES/KOH pH7.9, 85 mM KCl, 1 mM EDTA, 0.2% IGEPAL CA-630 (Sigma Aldrich, #I8896), 1X protease inhibitor cocktail (Sigma, #11836145001) and 1 mM PMSF for 10 min on ice. Cells were then spun down and lysed in 130 μL of lysis buffer containing 20 mM Tris/HCl pH7.5, 1 mM EDTA, 0.5 mM EGTA, 0.1% SDS, 0.4% Sodium Deoxycholate, 1% NP-40, 0.5 mM DTT, 1x protease inhibitor cocktail and 1 mM PMSF and chromatin was sheared by sonication. In all buffers, DTT, protease inhibitor and PMSF were added freshly. Cell lysates were sonicated in a 96 microTUBE Rack (Covaris, #500282) using a Covaris E220 for 22 cycles with the following settings: time, 60 s; duty 5.0; PIP, 140; cycles, 200; amplitude, 0.0; velocity, 0.0. Sonicated lysates were recovered and spun at 10,000 rpm for 10 min at 4°C to remove cell debris. One percent of sonicated lysate was kept as ChIP input for analysis. Immunoprecipitation mix consisting of Protein G Dynabeads (Invitrogen, #10003D) and 0.2 μg of JUN antibody (abcam, #32127) was added to sonicated chromatin solution and incubated overnight on a rotator at 4°C. Next day, immunocomplexes were placed on a magnet and bead complexes were washed for one minute with 150 μL of cold buffers as indicated: 3 times lysis buffer, 4 times with wash buffer containing 10 mM Tris/HCl pH7.5, 250 mM LiCl, 1 mM EDTA, 0.7% Na-Deoxycholate and 1% NP-40 alternative; 2 times with TET buffer containing 10 mM Tris/HCl pH 8.0,1 mM EDTA, 0.2% Tween 20; and 1 time with IDTE buffer containing 10 mM Tris/HCl pH 8.0 and 0.1 mM EDTA. Bead complexes were resuspended in 25 μL of TT buffer containing 10 mM Tris/HCl pH 8.0, 0.05% Tween 20. All wash buffers contained 1X Protease Inhibitor cocktail except for TT buffer. Crosslinks were reversed by adding 33.5 μL of mix containing 18.4 μL nuclease free water, 4 μL 10% SDS, 3 μL 0.5 M EDTA, 1.6 μL 0.2 M EGTA, 1 μL 10 mg/mL Proteinase K (Biolabs, #P8107S), 1 μL 10 mg/mL RNase A (ThermoFisher, #12091021), 4.5 μL5M NaCl by incubating at 55°Cfor 1 h followed by 75°C for 30 min. Dynabeads were removed and libraries were cleaned by adding 2 mLof SpeadBeads in 124 mLof 20% PEG 8000/1.5 M NaCl, washed by adding 150 mL80% EtOH, air dried and eluted in 15 μL of buffer containing 10 mM Tris/HCl pH 8.0 and 0.05% Tween 20. Enrichment was calculated as relative to input and to the negative region. A list of primers used for ChIP-qPCR is provided in Table S6.
Immunofluorescence (IF) in cells
Briefly, cells were plated in chambered slides after seven days of differentiation for murine BMDM or differentiated directly in chambered slides (Millipore, #C86024) in the case of human mDM. Cells were fixed with BD Cytofix/Cytoperm Buffer (BD, #BD554714) or 4% PFA containing 0.1% Saponin (Sigma, #47036) for 10 min at room temperature and then washed twice with HBSS containing 2% BSA and 1 mM EDTA. Cells were incubated in wash/permeabilization buffer (BD, #BD554714) for one hour at 4°C or kept at 4°C until the experiment was performed. Fixed BMDM were blocked using 3% BSA, 0.1% Triton-PBS for 30 min at room temperature and then with the primary antibody overnight at 4°C. For double IF, the corresponding antibodies were added simultaneously and incubated overnight at 4°C. Next day, mDM were washed with 0.1% Triton-PBS, incubated with the appropriate fluorochrome-conjugated secondary antibody and fluorescent probes. Nuclei were counter-stained with DAPI and Phalloidin (Abcam, #176759) was used to identify cell perimeter. After washing with 0.1% Triton-PBS, slides were mounted with Prolong Gold Antifade Reagent (Life Technology, #10144). Images were taken using a Leica SP8 with light deconvolution microscope or Leica TCS SPE microscope. Signal for specific antibodies was pseudocolored in red. DAPI was used to delineate DNA structures. In figures with IF, thicker outlines represent the cell perimeter and thinner outlines represent cell nucleus. A list of antibodies and fluorescent probes used for IF with their working concentration is shown below.
ATF3. CST, #D2Y5W. 1/100.
ATF4. CST. #D4B8. 1/100–1/200.
JUN. CST, #60A8. 1/100–1/200.
JUNB. Abcam, #245500. 1/100
pJUNSer63. CST, #2361. 1/100–1/200.
FOSL1. Santa Cruz, #376148. 1/150.
FOSL2. Santa Cruz, #166102. 1/150.
IRF1. Abcam, #191032. 1/100.
IRF7. Abcam, #115352. 1/100.
PFKFB3. Proteintech, #13763–1-AP
SLC2A1. Abcam, #115730. 1/100.
SOAT2. Santa Cruz, #59443. 1/50
Phalloidin. Abcam, #176759. 1/1000.
Donkey anti-mouse 488. ThermoFisher, #A21202. 1/200.
Donkey anti-rabbit 555. ThermoFisher, #A31572. 1/200.
Quantification of pixels intensity in ImageJ
ImageJ was used to quantify signal intensity of IF images. Briefly, three-colored images were split into single-colored images, nuclei were delineated using the freehand selection tool of ImageJ and intensity was calculated as the value of mean/area. Experiments were performed in duplicates. One representative experiment is shown.
Immunofluorescence (IF) in frozen sections
Air pouch skin tissues were fixed in a 1:1 acetone:methanol mixture for 10 min at room temperature. Sections were blocked for 1 h at room temperature in 3% BSA-PBS with 1% serum and 0.1% gelatin, and then incubated with primary antibody overnight at 4°C. After washing, Alexa Fluor secondary antibodies were added at 1:300 in 3% BSA-PBS for 1 h, followed by 1:1000 DAPI for 20 min. After washing with PBS, slides were mounted with FluroSave. The antibodies and working concentrations used are listed below.
F4/80. Invitrogen # MF48000 1/50
SLC2A1. Santa Cruz #sc-33781 1/50
p-JNK. Cell signaling #cs4668 1/50
Immunohistochemistry in paraffin sections (IHC)
IHC analysis were performed using 3 μM sections of formalin-fixed paraffin-embedded samples of air pouch skin. After deparaffinization and rehydration, antigen retrieval was performed by boiling in citrate buffer pH 6 for 30 min. After antigen retrieval, endogenous peroxidase was inactivated with 3% H202 in water for 10 min at room temperature. Sections were incubated with 4% BSA-PBS for 1 h at room temperature, and then with the specific primary antibody overnight at 4°C or 2 h at room temperature. After washing, the Envision secondary reagent (DAKO) was added for 30 min at room temperature and sections were washed three times with PBS. 3,30-Diaminobenzidinetetrahydrochloride (DAB) (Vector, #SK-4100) was used as a chromogen. Sections were lightly counterstained with hematoxylin, dehydrated and then mounted. A non-related IgG was used as a negative control. The antibodies and working concentrations used are listed below.
LDH. Santa Cruz, #sc-33781, 1/800
ENO2. Proteintech #104149–1-AP, 1/800
iNOS. Abcam ab53004, 1/2000
Enzyme-linked immunosorbent assay (ELISA)
Protein levels in the supernatant of 250,000 cells stimulated with LPS or MSUc in triplicates, or in the lavage or serum from mice after MSUc injection in the air pouch were measured by ELISA (R&D Systems) following manufacturer’s specifications.
Western blotting (WB)
For western blotting, proteins were extracted from 2 million cells unstimulated or stimulated with LPS or MSUc supplemented with protease inhibitors (Sigma Aldrich, #11697498001) Protein concentration was measured using the BCA reagent (Biorad, #500–0006). Proteins were resolved by standard SDS-PAGE gels and transfer to Immobilon PVDF membranes (Sigma, #IPVH00010) and incubated with specific antibodies overnight at 4°C. Membrane was then washed in PBS-Tween, incubated with Horseradish Peroxidase (HRP)-conjugated secondary antibodies and developed using Western Lighting Plus ECL reagent (Perkin) via autoradiography (Genesee, 30–507). Densitometry analysis of digitalized western blotting images was performed using Fiji software (NIH). The antibodies and working concentrations used are listed below.
SLC2A1. Santa Cruz, #sc-7903 1/500.
PFKBP3. Proteintech, #13763–1-AP 1/1000.
LDH. Santa Cruz, #sc-33781 1/500.
TUBULIN. Cell Signaling, #3873. 1/1000.
HPR-conjugated anti-mouse IgG. CST #7076. 1/2000.
HPR-conjugated anti-rabbit IgG. CST #7074. 1/2000.
Nuclear magnetic resonance (NMR) acquisition and processing
The metabolites from sera and supernatant were obtained by ultrafiltration (Tiziani et al., 2008) using a 3 KDa filter (3kDa Omega, Pall Corporation, #OD003C34) with a final standard concentration of 0.5 millimolar (mM) in a total volume of 50 μL. The standard used was 3-(trimethylsilyl)propionic-2,2,3,3-d4 acid, sodium salt (TSP-d4) in deuterium oxide (Aldrich Chemistry, #293040–25G). From cell pellets, the polar metabolites were extracted by the precipitation method (Wu et al., 2008) with a final TSP-d4 concentration of 0.06617 mM. The one-dimension nuclear magnetic resonance (1D 1H-NMR) spectra were recorded using a 600 MHz Bruker Avance III NMR spectrometer fitted with a 1.7 mm triple resonance cryoprobe. The standard Bruker pulse sequence “noesygppr1d” was used with a mixing time of 500 ms and 64 scans. A quality assurance procedure was performed before sample data acquisition, involving temperature checks and calibration as well as shim and water suppression quality. The data acquisition was obtained in the NMR facility of Skaggs School of Pharmacy and Pharmaceutical Science, University of California San Diego.
Metabolites identification and quantification
The identification of metabolites approach was performed using the software Chenomx NMR suite 8.5 professional (Chenomx Inc., Edmonton, Canada) version 11, which contents a library of metabolites that match the peaks of compound according to their chemical shift. Metabolites concentration were normalized according to the standard TSP-d4 and the concentrations were reported in micromolar (μM).
QUANTIFICATION AND STATISTICAL ANALYSIS
Comparisons of quantitative data between 2 groups was carried out using two-tailed Student T test, and between more than 2 groups, using analysis of the variance with post-hoc Tukey test. Partial least-squares discriminant analysis (PLS-DA) was used to identify discriminant metabolites (Mendez et al., 2019). For RNA-Seq analysis, differentially expressed genes were obtained by DESeq2 (Love et al., 2014) using FDR <0.05 and log2 fold change of >1 or <(−1). Metabolites contributing to group discrimination between 2 phenotypes in the Partial Least Square Discriminant Analysis (PLS-DA) were depicted on the basis of having a variable important projection (VIP) score of >1. Box pots illustrate the median, Q1 and Q3 quartileof the data. Error bars in boxplots represent the lowest and highest data point within 1.5x Q1-Q3 range. Bar graphs represent the media. Error bars in bar graphs represent the Standard error of the mean. All plots were generated using Numbers (iWORK’09), R studio (version 2.15.2 [2012–10-26]), and MetaboAnalyst version 5.0 (Chong et al., 2019). (# = P <0.10; * = P <0.05; ** = P <0.01 compared to the control group).
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| ATF3 | CST | D2Y5W; RRID: AB_2799039 |
| ATF4 | CST | D4B8; RRID: AB_2616025 |
| JUN | CST | 60A8; RRID: AB_2130165 |
| JUNB | Abcam | 245500; RRID: AB_2904574 |
| pJUNSer63 | CST | 2361; RRID: AB_490908 |
| FOSL1 | Santa Cruz | 376148; RRID: AB_11012022 |
| FOSL2 | Santa Cruz | sc-166102, RRID:AB_2107079 |
| IRF1 | Abcam | ab191032; RRID: AB_2904575 |
| IRF7 | Abcam | ab115352, RRID:AB_10862356 |
| PFKFB3 | Proteintech | 13763-1-AP, RRID:AB_2162854 |
| SLC2A1 | Abcam | ab115730, RRID:AB_10903230 |
| SOAT2 | Santa Cruz | sc-59443, RRID:AB_10611790 |
| Donkey anti-mouse 488 | ThermoFisher | A-21202, RRID:AB_141607 |
| Donkey anti-rabbit 555 | ThermoFisher | A-31572, RRID:AB_162543 |
| LDH | Santa Cruz | sc-33781, RRID:AB_2134947 |
| ENO2 | Proteintech | 10149-1-AP, RRID:AB_2099180 |
| iNOS | Abcam | ab53004, RRID:AB_869050 |
| SLC2A1 | Santa Cruz | sc-7903, RRID:AB_2190936 |
| TUBULIN | CST | 3873, RRID:AB_1904178 |
| HPR-conjugated anti-mouse IgG | CST | 7076, RRID:AB_330924 |
| HPR-conjugated anti-rabbit IgG | CST | 7074, RRID:AB_2099233 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| KAPA SYBR FAST qPCR Master mix (2X) | Kapa Biosystems | Cat#07959427001 |
| Dynabeads Protein A | Thermo Fisher Scientific | Cat#10002D |
| Dynabeads Protein G | Thermo Fisher Scientific | Cat#10004D |
| SpeedBeads magnetic carboxylate modified particles | GE Healthcare | Cat#65152105050250 |
| TRIzol Reagent | Thermo Fisher Scientific | Cat#15596018 |
| Formaldehyde | Thermo Fisher Scientific | Cat#BP531-500 |
| Disuccinimidyl glutarate | ProteoChem | Cat#c1104-100mg |
| Oligo d(T)25 Magnetic Beads | NEB | Cat#S1419S |
| DTT | Thermo Fisher Scientific | Cat#P2325 |
| SUPERase-In | Ambion | Cat#AM2696 |
| Oligo dT primer | Thermo Fisher Scientific | Cat#18418020 |
| Agencourt RNA Clean XP Beads | Beckman Coulter | Cat#A63987 |
| 10 X Blue Buffer | Enzymatics | Cat#P7050L |
| DNA polymerase I | Enzymatics | Cat#P7050L |
| Random primers | Thermo Fisher Scientific | Cat#48190011 |
| SuperScript III Reverse Transcriptase | Thermo Fisher Scientific | Cat#18080044 |
| 5 X first-strand buffer | Thermo Fisher Scientific | Cat#18080044 |
| Actinomycin D | Sigma | Cat#A1410 |
| DNaseI | Worthington | Cat#LS002139 |
| SP600A125 | Tocris | #1496 |
| BAY-876 | Selleckchem | #S8452 |
| MAPK P38 inhibitor SB203580 | Tocris | #1202 |
| NLRP3 inflammasome inhibitor MCC950 | Selleckchem | #S7809 |
| BHA | Sigma | #B1253 |
| Lipofectamine™ RNAiMAX Transfection Reagent | Invitrogen | #13778150 |
| Ficoll Plaque Premium | Sigma GE Healthcare | #17-544-02 |
| Recombinant human M-CSF | Stem Cell | #78057-2 |
| BD Cytofix/Cytoperm Buffer | BD | #BD554714 |
| BD Wash and Permeabilization Buffer | BD | #BD554714 |
| Phalloidin | Sigma | #176759 |
| Prolong Gold Antifade Reagent | Life Technologies | #10144 |
|
| ||
| Critical commercial assays | ||
|
| ||
| Direct-zol RNA MicroPrep Kit | Zymo Research | Cat#R2062 |
| Qubit dsDNA HS Assay Kit | Invitrogen | Cat#Q32851 |
| Superscript III First-Strand Synthesis System | Thermo Fisher Scientific | Cat#18080051 |
| BD Vacutainer CPT Tubes | BD Biosciences | #362753 |
| BCA Assay | BioRad | #500–0006 |
| Western Lighting Plus ECL reagent | Perkin | #0RT2655/0RT2755 |
|
| ||
| Deposited data | ||
|
| ||
| Raw and Analyzed data | GEO | GSE191054 |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| C57BL/6 J mice | The Jackson Laboratory | RRID: IMSR_JAX:000664 |
| L929 | ATCC | CRL-6364 |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| MyD88−/− mice | The Jackson Laboratory | 009088 |
|
| ||
| Oligonucleotides | ||
|
| ||
| siControl | Dharmaphect | D-001210-02 |
| siJun | Dharmaphect | #L-043776-00–0005 |
| siFosl1 | Dharmaphect | #L-040704-00–0005 |
| Primers for qPCR and ChIP-qPCR | Table S6 | |
|
| ||
| Software and algorithms | ||
|
| ||
| Bowtie2 (Langmead and Salzberg, 2012) | http://bowtie-bio.sourceforge.net/bowtie2/ | |
| HOMER (Heinz et al., 2010) | http://homer.ucsd.edu/homer/ | |
| R package: DeSeq2 (Love et al., 2014) | https://bioconductor.org/packages/release/bioc/html/DESeq2.html | |
| STAR (Dobin et al., 2013) | https://github.com/alexdobin/STAR | |
| UCSC Genome Browser (Kent et al., 2002) | https://genome.ucsc.edu/ | |
| RStudio | RStudio | https://www.rstudio.com/ |
| Pathway Analysis (Liberzon et al., 2011, 2015; Subramanian et al., 2005) | GSEA | http://www.gsea-msigdb.org/gsea/login.jsp |
Highlights.
MSUc alone induces a unique strong macrophage activation and distinct phenotype
The transcriptional program induced by MSUc is regulated through AP-1 signaling
JNK or SLC2A1 inhibition ameliorates the in vivo damage associated with MSUc
These results force a reconsideration of initiating mechanisms in acute gout flares
ACKNOWLEDGMENTS
These studies were supported by NIH grant nos. AR073324 (to M.G.), 5R01 DK091183, P01 HL147835 (to C.K.G.), DK063491 (Sequencing Core), nos. AR060772 and AR075990 (to R.T.), no. T32AR064194 (to J.M.-S. and R.C.), the VA Research Service (to R.T.), VA Merit Review BX-002234-05 (to R.L.B.), and Foundation Leducq grant 16CVD01 (to C.K.G.). I.C. was supported by EMBO Long-Term Fellowship (ALTF 960-2018).
Footnotes
DECLARATION OF INTERESTS
Research grant Astra-Zeneca (R.T.) and consulting at SOBI, Selecta, Horizon, Allena, Astra-Zeneca (R.T.). Research grant Aspire-Pfizer and Novartis (M.G.).
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2022.110489.
REFERENCES
- Adcock IM, and Caramori G (2001). Cross-talk between pro-inflammatory transcription factors and glucocorticoids. Immunol. Cell Biol. 79, 376–384. [DOI] [PubMed] [Google Scholar]
- Amann R, and Peskar BA (2002). Anti-inflammatory effects of aspirin and sodium salicylate. Eur. J. Pharmacol. 447, 1–9. [DOI] [PubMed] [Google Scholar]
- Ameyar-Zazoua M, Wisniewska MB, Bakiri L, Wagner EF, Yaniv M, and Weitzman JB (2005). AP-1 dimers regulate transcription of the p14/p19ARF tumor suppressor gene. Oncogene 24, 2298–2306. [DOI] [PubMed] [Google Scholar]
- Angel P, Hattori K, Smeal T, and Karin M (1988). The jun proto-oncogene is positively autoregulated by its product, Jun/AP-1. Cell 55, 875–885. [DOI] [PubMed] [Google Scholar]
- Angel P, Szabowski A, and Schorpp-Kistner M (2001). Function and regulation of AP-1 subunits in skin physiology and pathology. Oncogene 20, 24132–2423. [DOI] [PubMed] [Google Scholar]
- Atri C, Guerfali FZ, and Laouini D (2018). Role of human macrophage polarization in inflammation during infectious diseases. Int. J. Mol. Sci. 19, 1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett BL, Sasaki DT, Murray BW, O’Leary EC, Sakata ST, Xu W, Leisten JC, Motiwala A, Pierce S, Satoh Y, et al. (2001). SP600125, an anthrapyrazolone inhibitor of Jun N-terminal kinase. Proc. Natl. Acad. Sci. U S A. 98, 13681–13686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhoumik A, Lopez-Bergami P, and Ronai Z (2007). ATF2 on the double - activating transcription factor and DNA damage response protein. Pigment Cell Res. 20, 498–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biswas SK, and Mantovani A (2012). Orchestration of metabolism by macrophages. Cell Metab. 15, 432–437. [DOI] [PubMed] [Google Scholar]
- Brovold H, Lund T, Svistounov D, Solbu MD, Jenssen TG, Ytrehus K, and Zykova SN (2019). Crystallized but not soluble uric acid elicits pro-inflammatory response in short-term whole blood cultures from healthy men. Sci. Rep. 9, 10513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busso N, and So A (2010). Mechanisms of inflammation in gout. Arthritis Res. Ther. 12, 206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bustamante MF, Oliveira PG, Garcia-Carbonell R, Croft AP, Smith JM, Serrano RL, Sanchez-Lopez E, Liu X, Kisseleva T, Hay N, et al. (2018). Hexokinase 2 as a novel selective metabolic target for rheumatoid arthritis. Ann. Rheum. Dis. 77, 1636–1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caputa G, Castoldi A, and Pearce EJ (2019). Metabolic adaptations of tissue-resident immune cells. Nat. Immunol. 20, 793–801. [DOI] [PubMed] [Google Scholar]
- Chen S, Yang J, Wei Y, and Wei X (2020). Epigenetic regulation of macrophages: from homeostasis maintenance to host defense. Cell Mol Immunol. 17, 36–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu R, Angel P, and Karin M (1989). Jun-B differs in its biological properties from, and is a negative regulator of, c-Jun. Cell 59, 979–986. [DOI] [PubMed] [Google Scholar]
- Choe JY, Park KY, and Kim SK (2015). Oxidative stress by monosodium urate crystals promotes renal cell apoptosis through mitochondrial caspase-dependent pathway in human embryonic kidney 293 cells: mechanism for urate-induced nephropathy. Apoptosis 20, 38–49. [DOI] [PubMed] [Google Scholar]
- Chong J, Wishart DS, and Xia J (2019). Using MetaboAnalyst 4.0 for comprehensive and integrative metabolomics data analysis. Curr. Protoc. Bioinformatics 68, e86. [DOI] [PubMed] [Google Scholar]
- Chung YH, Kim DH, and Lee WW (2016). Monosodium urate crystal-induced pro-interleukin-1beta production is post-transcriptionally regulated via the p38 signaling pathway in human monocytes. Sci. Rep. 6, 34533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cicenas J, Zalyte E, Rimkus A, Dapkus D, Noreika R, and Urbonavicius S (2017). JNK, p38, ERK, and SGK1 inhibitors in cancer. Cancers (Basel) 10, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobo I, Martinelli P, Flandez M, Bakiri L, Zhang M, Carrillo-de-Santa-Pau E, Jia J, Sanchez-Arevalo Lobo VJ, Megias D, Felipe I, et al. (2018). Transcriptional regulation by NR5A2 links differentiation and inflammation in the pancreas. Nature 554, 533–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalbeth N, Merriman TR, and Stamp LK (2016). Gout. Lancet 388, 2039–2052. [DOI] [PubMed] [Google Scholar]
- Deng T, and Karin M (1994). c-Fos transcriptional activity stimulated by H-Ras-activated protein kinase distinct from JNK and ERK. Nature 371,171–175. [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eferl R, and Wagner EF (2003). AP-1: a double-edged sword in tumorigenesis. Nat. Rev. Cancer 3, 859–868. [DOI] [PubMed] [Google Scholar]
- Eichenfield DZ, Troutman TD, Link VM, Lam MT, Cho H, Gosselin D, Spann NJ, Lesch HP, Tao J, Muto J, et al. (2016). Tissue damage drives co-localization of NF-kappaB, Smad3, and Nrf2 to direct Rev-erb sensitive wound repair in mouse macrophages. Elife 5, e13024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gosselin D, Skola D, Coufal NG, Holtman IR, Schlachetzki JCM, Sajti E, Jaeger BN, O’Connor C, Fitzpatrick C, Pasillas MP, et al. (2017). An environment-dependent transcriptional network specifies human microglia identity. Science 356, eaal3222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grassi ES, Vezzoli V, Negri I, Labadi A, Fugazzola L, Vitale G, and Persani L (2015). SP600125 has a remarkable anticancer potential against undifferentiated thyroid cancer through selective action on ROCK and p53 pathways. Oncotarget 6, 36383–36399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guma M, Ronacher L, Liu-Bryan R, Takai S, Karin M, and Corr M (2009). Caspase 1-independent activation of interleukin-1beta in neutrophil-predominant inflammation. Arthritis Rheum. 60, 3642–3650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hai T, and Curran T (1991). Cross-family dimerization of transcription factors Fos/Jun and ATF/CREB alters DNA binding specificity. Proc. Natl. Acad. Sci. U S A. 88, 3720–3724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammouda MB, Ford AE, Liu Y, and Zhang JY (2020). The JNK signaling pathway in inflammatory skin disorders and cancer. Cells 9, 857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hepp Rehfeldt SC, Majolo F, Goettert MI, and Laufer S (2020). c-Jun N-terminal kinase inhibitors as potential leads for new therapeutics for alzheimer’s diseases. Int. J. Mol. Sci. 21, 9677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joosten LA, Netea MG, Mylona E, Koenders MI, Malireddi RK, Oosting M, Stienstra R, van de Veerdonk FL, Stalenhoef AF, Giamarellos-Bourboulis EJ, et al. (2010). Engagement of fatty acids with Toll-like receptor 2 drives interleukin-1beta production via the ASC/caspase 1 pathway in monosodium urate monohydrate crystal-induced gouty arthritis. Arthritis Rheum. 62, 3237–3248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karin M (1995). The regulation of AP-1 activity by mitogen-activated protein kinases. J. Biol. Chem. 270, 16483–16486. [DOI] [PubMed] [Google Scholar]
- Karin M, and Clevers H (2016). Reparative inflammation takes charge of tissue regeneration. Nature 529, 307–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, and Haussler D (2002). The human genome browser at UCSC. Genome Res. 12, 996–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kingsbury SR, Conaghan PG, and McDermott MF (2011). The role ofthe NLRP3 inflammasome in gout. J. Inflamm. Res. 4, 39–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo CF, Grainge MJ, Zhang W, and Doherty M (2015). Global epidemiology of gout: prevalence, incidence and risk factors. Nat. Rev. Rheumatol. 11, 649–662. [DOI] [PubMed] [Google Scholar]
- Kuznetsova T, Prange KHM, Glass CK, and de Winther MPJ (2020). Transcriptional and epigenetic regulation of macrophages in atherosclerosis. Nat. Rev. Cardiol. 17, 216–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberzon A, Birger C, Thorvaldsdottir H, Ghandi M, Mesirov JP, and Tamayo P (2015). The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 1, 417–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberzon A, Subramanian A, Pinchback R, Thorvaldsdottir H, Tamayo P, and Mesirov JP (2011). Molecular signatures database (MSigDB) 3.0. Bioinformatics 27, 1739–1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Link VM, Duttke SH, Chun HB, Holtman IR, Westin E, Hoeksema MA, Abe Y, Skola D, Romanoski CE, Tao J, et al. (2018). Analysis of genetically diverse macrophages reveals local and domain-wide mechanisms that control transcription factor binding and function. Cell 173, 1796–1809 e1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu R, O’Connell M, Johnson K, Pritzker K, Mackman N, and Terkeltaub R (2000). Extracellular signal-regulated kinase 1/extracellular signal-regulated kinase 2 mitogen-activated protein kinase signaling and activation of activator protein 1 and nuclear factor kappaB transcription factors play central roles in interleukin-8 expression stimulated by monosodium urate monohydrate and calcium pyrophosphate crystals in monocytic cells. Arthritis Rheum. 43, 1145–1155. [DOI] [PubMed] [Google Scholar]
- Liu-Bryan R, Scott P, Sydlaske A, Rose DM, and Terkeltaub R (2005). Innate immunity conferred by Toll-like receptors 2 and 4 and myeloid differentiation factor 88 expression is pivotal to monosodium urate monohydrate crystal-induced inflammation. Arthritis Rheum. 52, 2936–2946. [DOI] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu W, Xu Y, Shao X, Gao F, Li Y, Hu J, Zuo Z, Shao X, Zhou L, Zhao Y, et al. (2015). Uric acid produces an inflammatory response through activation of NF-kappaB in the hypothalamus: implications for the pathogenesis of metabolic disorders. Sci. Rep. 5, 12144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin WJ, Walton M, and Harper J (2009). Resident macrophages initiating and driving inflammation in a monosodium urate monohydrate crystal-induced murine peritoneal model of acute gout. Arthritis Rheum. 60, 281–289. [DOI] [PubMed] [Google Scholar]
- Mendez KM, Reinke SN, and Broadhurst DI (2019). A comparative evaluation of the generalised predictive ability of eight machine learning algorithms across ten clinical metabolomics data sets for binary classification. Metabolomics 15, 150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng L, and Huang Z (2018). In silico-in vitro discovery of untargeted kinase-inhibitor interactions from kinase-targeted therapies: a case study on the cancer MAPK signaling pathway. Comput. Biol. Chem. 75, 196–204. [DOI] [PubMed] [Google Scholar]
- Montagut C, and Settleman J (2009). Targeting the RAF-MEK-ERK pathway in cancer therapy. Cancer Lett. 283, 125–134. [DOI] [PubMed] [Google Scholar]
- Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E, Ridderstrale M, Laurila E, et al. (2003). PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinate downregulated in human diabetes. Nat. Genet. 34, 267–273. [DOI] [PubMed] [Google Scholar]
- Mosser DM, Hamidzadeh K, and Goncalves R (2021). Macrophages and the maintenance of homeostasis. Cell Mol Immunol. 18, 579–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Neill LA, and Pearce EJ (2016). Immunometabolism governs dendritic cell and macrophage function. J. Exp. Med. 213, 15–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phan AT, Goldrath AW, and Glass CK (2017). Metabolic and epigenetic coordination of T cell and macrophage immunity. Immunity 46, 714–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puigserver P, Rhee J, Lin J, Wu Z, Yoon JC, Zhang CY, Krauss S, Mootha VK, Lowell BB, and Spiegelman BM (2001). Cytokine stimulation of energy expenditure through p38 MAP kinase activation of PPARgamma coactivator-1. Mol. Cell 8, 971–982. [DOI] [PubMed] [Google Scholar]
- Renaudin F, Orliaguet L, Castelli F, Fenaille F, Prignon A, Alzaid F, Combes C, Delvaux A, Adimy Y, Cohen-Solal M, et al. (2020). Gout and pseudo-gout-related crystals promote GLUT1-mediated glycolysis that governs NLRP3 and interleukin-1beta activation on macrophages. Ann. Rheum. Dis. 79, 1506–1514. [DOI] [PubMed] [Google Scholar]
- Sanchez-Lopez E, Cheng A, and Guma M (2019a). Can metabolic pathways Be therapeutic targets in rheumatoid arthritis? J. Clin. Med. 8, 753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Lopez E, Zhong Z, Stubelius A, Sweeney SR, Booshehri LM, Antonucci L, Liu-Bryan R, Lodi A, Terkeltaub R, Lacal JC, et al. (2019b). Choline uptake and metabolism modulate macrophage IL-1beta and IL-18 production. Cell Metab. 29, 1350–1362 e1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sautin YY, and Johnson RJ (2008). Uric acid: the oxidant-antioxidant paradox. Nucleosides Nucleotides Nucleic Acids 27, 608–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seidman JS, Troutman TD, Sakai M, Gola A, Spann NJ, Bennett H, Bruni CM, Ouyang Z, Li RZ, Sun X, et al. (2020). Niche-specific reprogramming of epigenetic landscapes drives myeloid cell diversity in nonalcoholic steatohepatitis. Immunity 52, 1057–1074 e1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaulian E, and Karin M (2002). AP-1 as a regulator of cell life and death. Nat. Cell Biol. 4, E131–E136. [DOI] [PubMed] [Google Scholar]
- So AK, and Martinon F (2017). Inflammation in gout: mechanisms and therapeutic targets. Nat. Rev. Rheumatol. 13, 639–647. [DOI] [PubMed] [Google Scholar]
- Stubelius A, Sheng W, Lee S, Olejniczak J, Guma M, and Almutairi A (2018). Disease-triggered drug release effectively prevents acute inflammatory flare-ups, achieving reduced dosing. Small 14, e1800703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U S A. 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thapa B, and Lee K (2019). Metabolic influence on macrophage polarization and pathogenesis. BMB Rep. 52, 360–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiziani S, Emwas AH, Lodi A, Ludwig C, Bunce CM, Viant MR, and Gunther UL (2008). Optimized metabolite extraction from blood serum for 1H nuclear magnetic resonance spectroscopy. Anal Biochem. 377, 16–23. [DOI] [PubMed] [Google Scholar]
- van Dam H, and Castellazzi M (2001). Distinct roles of jun : fos and jun : ATF dimers in oncogenesis. Oncogene 20, 2453–2464. [DOI] [PubMed] [Google Scholar]
- Weyand CM, and Goronzy JJ (2017). Immunometabolism in early and late stages of rheumatoid arthritis. Nat. Rev. Rheumatol. 13, 291–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodgett JR, Pulverer BJ, Nikolakaki E, Plyte S, Hughes K, Franklin CC, and Kraft AS (1993). Regulation of jun/AP-1 oncoproteins by protein phosphorylation. Adv. Second Messenger Phosphoprotein Res. 28, 261–269. [PubMed] [Google Scholar]
- Wu H, Southam AD, Hines A, and Viant MR (2008). High-throughput tissue extraction protocol for NMR- and MS-based metabolomics. Anal Biochem. 372, 204–212. [DOI] [PubMed] [Google Scholar]
- Wynn TA, and Vannella KM (2016). Macrophages in tissue repair, regeneration, and fibrosis. Immunity 44, 450–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamudio-Cuevas Y, Martinez-Flores K, Fernandez-Torres J, Loissell-Baltazar YA, Medina-Luna D, Lopez-Macay A, Camacho-Galindo J, Hernandez-Diaz C, Santamaria-Olmedo MG, Lopez-Villegas EO, et al. (2016). Monosodium urate crystals induce oxidative stress in human synoviocytes. Arthritis Res. Ther. 18, 117. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession number for the sequencing data reported in this paper is GEO: GSE191054.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.