


Abstract
Bovine leukemia virus (BLV)-induced tumoral development is a multifactorial phenomenon that remains incompletely understood. Here, we highlight the critical role of the cellular CCCTC-binding factor (CTCF) both in the regulation of BLV transcriptional activities and in the deregulation of the three-dimensional (3D) chromatin architecture surrounding the BLV integration site. We demonstrated the in vivo recruitment of CTCF to three conserved CTCF binding motifs along the provirus. Next, we showed that CTCF localized to regions of transitions in the histone modifications profile along the BLV genome and that it is implicated in the repression of the 5′Long Terminal Repeat (LTR) promoter activity, thereby contributing to viral latency, while favoring the 3′LTR promoter activity. Finally, we demonstrated that BLV integration deregulated the host cellular 3D chromatin organization through the formation of viral/host chromatin loops. Altogether, our results highlight CTCF as a new critical effector of BLV transcriptional regulation and BLV-induced physiopathology.
INTRODUCTION
Bovine leukemia virus (BLV) is a B-lymphotropic oncogenic deltaretrovirus infecting cattle. Infections are under control in Western Europe with the help of regulated sanitary rules whereas countries lacking these measures are still facing massive economical losses in the food and milk industries (https://wahis.oie.int/#/dashboards/country-or-disease-dashboard, 1). In addition to these economic issues, BLV shares common features with Human T-lymphotropic Virus 1 and 2 (HTLV-1 and -2), thereby constituting a convenient animal model to further study HTLV-1-dependent tumorigenesis in humans (2,3). Regarding BLV physiopathology, while the majority of BLV-infected animals remain asymptomatic lifelong, 30% of them will develop a persistent lymphocytosis and <5% will suffer from B-cell leukemia or lymphoma, termed enzootic bovine leukosis, leading to rapid death of BLV-infected cattle (3). Remarkably, to further study BLV-induced oncogenic mechanisms, BLV can be inoculated experimentally to sheep, who have been demonstrated to be more sensitive to BLV-associated oncogenic properties with 95% of the infected animals developing B-cell leukemia or lymphoma after a shorter period of incubation compared to bovines (4–6). Overall, a common feature of BLV infection is the viral latency, characterized by an absence of viremia (3,7,8), which probably enables the escape from the host immune system and ultimately tumor development (9). Mechanistically, we and others have demonstrated that this viral latency occurs through the repression of the RNA polymerase II-dependent (RNAPII) promoter located in the 5′long terminal repeat (5′LTR) by several mechanisms (10), including genetic mutations in important cis-regulatory regions (11,12), epigenetic modifications (13–17), as well as the sequestration of host cellular transcription factors for which binding sites have been previously identified along the 5′LTR (18–25). However, despite the strong repression affecting the 5′LTR, we and others have discovered and characterized two additional promoter activities (26–29). Indeed, BLV encodes a highly expressed miRNA cluster, responsible for the transcription of 10 viral miRNAs through a non-canonical process involving the RNA polymerase III (RNAPIII). In addition, the 3′LTR exhibits an important RNAPII-dependent promoter activity, responsible for the expression of three viral antisense transcripts (26,29). Functionally, the BLV miRNAs and antisense transcripts could be responsible for tumor progression by deregulating the host transcriptome (30,31) and producing viral/host chimeric transcripts (32), respectively, in infected animals. Altogether, these findings have provided new insights into alternative ways used by BLV to express parts of its genome, despite viral latency affecting the 5′LTR promoter activity, thereby bringing additional approaches to study BLV-induced leukemogenesis.
In the present report, we investigated the putative role of the cellular CCCTC-binding factor (CTCF) in the regulation of BLV RNAPII-dependent promoter activities through transcriptional and epigenetic mechanisms. We also investigated the implication of CTCF in cellular three-dimensional (3D) chromatin organization disruption through the formation of viral/host chromatin loops. Indeed, CTCF is a transcription factor ubiquitously expressed in all cell types and highly conserved across bilaterians. Functionally, CTCF has been demonstrated to play major roles in transcriptional and epigenetic regulation, mainly by organizing the cellular genome at the 3D level. CTCF-mediated regulations occur through the formation of chromatin loops, in cooperation with the cohesin multiprotein complex. Besides ‘high-order’ regulation, local and direct effects on gene expression have also been described, depicting CTCF as an important multifunctional protein (reviewed in (33–36)). Of note, CTCF has been demonstrated to be involved in the regulation of the infectious cycle of several DNA viruses such as Kaposi's sarcoma-associated herpesvirus (KSHV), human papillomavirus (HPV), and Epstein-Barr virus (EBV) (37). Regarding retroviruses, CTCF has been shown to be recruited to a unique proviral binding site located in the regulatory region of the HTLV-1 provirus (38). However, the functional role of CTCF recruitment in HTLV-1 gene expression regulation as well as in HTLV-1-induced tumorigenesis remains incompletely understood (38–43). In the present study, we demonstrate a critical role of CTCF as a regulator of BLV gene expression and possibly as a new determinant of BLV-induced leukemogenesis.
MATERIALS AND METHODS
Cell lines and primary cell samples
The ovine cell lines L267 (15) and YR2 (8,44) are clonal cell lines established from the T267 B-cell lymphoma and M395 B-cell leukemia, respectively, developed by a BLV-infected sheep injected with naked proviral DNA of an infectious BLV variant (5) displaying a wild-type sequence. These cell lines were maintained in Opti-MEM GlutaMAX medium (Life Technologies) supplemented with 10% FBS, 1 mM sodium pyruvate, 2 mM glutamine, non-essential amino acids 1× and 100 μg/ml kanamycin. The human 293T cell line (CRL-3216), obtained from the American Type Culture Collection (ATCC), was maintained in DMEM medium (Life Technologies) supplemented with 10% FBS, 1 mM sodium pyruvate, and 1% of penicillin-streptomycin. The Raji cell line, a human B-lymphoid Epstein-Barr virus-positive cell line derived from a Burkitt's lymphoma obtained from the AIDS Research and Reference Reagent Program (National Institute of Allergy and Infectious Disease [NIAID], National Institute of Health [NIH]), was maintained in RPMI 1640-Glutamax I medium (Life Technologies) supplemented with 10% FBS and 1% of penicillin–streptomycin. All cells were grown at 37°C in a humidified 95% air/5% CO2 atmosphere. Frozen sheep PBMCs were kindly provided by Anne Van den Broeke (Unit of Animal Genomics, GIGA, Université de Liège (ULiège), Belgium) and derived from BLV-induced B-cell leukemia (M2241L (32)).
Plasmid constructs
The episomal plasmids named X-luc, LTR_WT-S-luc and LTR_WT-AS-luc were previously described by our laboratory (26) under the name pREP-luc, pREP-LTRWT-S-luc and pREP-LTRWT-AS-luc. Mutation of the CTCF binding site was obtained by QuickChange Site-directed Mutagenesis (Stratagene) using a pair of mutagenic oligonucleotides (Supplementary Table S2) on a non-episomal plasmid pLTR_WT-luc (19). The resulting mutated LTR was isolated by digestion with SmaI and cloned in both orientations in the X-luc construct digested with BglII and blunt-ended to obtain the LTR_mFull-S-luc and LTR_mFull-AS-luc. These constructs were then linearized at the CTCF mutated site with AflII to generate the constructs LTR_m10-S-luc, LTR_m10-AS-luc, LTR_m5-S-luc LTR_m5-AS-luc by NEBuilder HiFi DNA Assembly (New England Biolabs) using specific oligonucleotides (Supplementary Table S2). Lentiviral vectors were constructed by replacement of the XbaI–AgeI fragment of a lentiviral pTRIPz plasmid (Horizon Discoveries) by the full-length LTR_WT, LTR_mFull, LTR_m10 or LTR_m5 amplified by PCR from the LTR_WT-S-luc, LTR_mFull-S-luc, LTR_m10-S-luc or LTR_m5-S-luc constructs, respectively (Supplementary Table S2). The envelope pVSG-G and packaging psPAX2 vectors were obtained from the Reference Reagent Program. For all constructs, the fragments cloned were fully sequenced by Sanger sequencing.
In silico motif search
Identification of CTCF binding motifs along the BLV provirus was performed using PWMScan (45). The CTCF motif MA0139.1 (MEME-derived JASPAR CORE 2018 vertebrates motif library) was searched on both strands of the BLV reference genome (Genbank: KT122858.1) using a p-value cut-off of 0.1, allowing overlapping matches. A high P-value cut-off of 0.1 was used to enable the representation of the most statistically significant CTCF binding sites by discrete peaks.
Chromatin immunoprecipitation assays
ChIP assays were performed following the ChIP assay kit from EMD Millipore. Briefly, cells were cross-linked for 10 min at room temperature with 1% formaldehyde before lysis followed by chromatin sonication (Bioruptor Plus, Diagenode) to obtain DNA fragments of 200–400 bp. Chromatin immunoprecipitations were performed with chromatin from 6 × 106 cells or 1 × 106 cells for the epigenetic modifications and 5 μg of antibodies (Supplementary Table S3). Quantitative real-time PCR reactions were performed using 1/60 of the immunoprecipitated DNA and the TB Green Premix Ex Taq II (Takara). Relative quantification using the standard curve method was performed for each primer pair and 96-well Optical Reaction plates were read in a StepOnePlus PCR instrument (Applied Biosystem). Fold enrichments were calculated as percentages of input values. Primer sequences used for quantification (Supplementary Table S2) were designed using the software Primer 3.
ChIP-sequencing assays
ChIP assays were performed as described above. Recovered DNA was then used for library preparation using the Ovation Ultralow System v2 kit (NuGen) following the Manufacturer's instructions. Paired-end sequencing was then performed with the Illumina HiSeq 2000 instrument. More than 20 million of single reads were obtained for all libraries. Reads were mapped to a hybrid ovine genome (OAR v3.1) containing the BLV provirus sequence (GenBank: KT122858.1) at their respective insertion site and orientation (Supplementary Table S4) using Bowtie 2 v2.3.5.1 (46). The mapped reads with mapping quality score <15 and PCR duplicates were discarded using SAMtools v1.9 (47). CTCF peaks were called using MACS2 v2.2.5 (48) at a q-value cutoff of 0.01. CTCF motif and the orientation of each peak was identified using FIMO v5.1.1 (49) with motif MA0139.1 (50) with max-stored-scores 50 000 000. Bigwig files were generated with DeepTools v.3.3.2 (51) with a bin length of 10 bp, extending reads to 200 bp and CPM normalization. For analysis of Rad21 and CTCF localization on the viral genome, reads were mapped to a hybrid ovine genome (OAR v3.1) containing the BLV provirus sequence (GenBank: KT122858.1) at their respective insertion site and orientation with settings n1 k2 using Bowtie 2 v2.3.5.1. PCR duplicates were removed using SAMtools v1.0. Bigwig files were generated with DeepTools v.3.3.2 with a bin length of 10 bp, extending reads to 200 bp and CPM normalization.
Lentiviral production and transduction
VSV-G pseudotyped lentiviral particles were produced by transfection of 293T cells (5 × 106 /10 cm dish), with the different LTR-containing lentiviral constructs (9 μg), the pVSV-G (2.25 μg), and the psPAX2 (6.75 μg) vectors by calcium phosphate transfection method according to the manufacturer's protocol (Takara). Seventy-two hours post-transfection, viral stocks were harvested and concentrated 10× by ultracentrifugation. 293T cells (2 × 105) were then transduced in a 12 well-plate with 600 μl of concentrated lentiviral stock. Six hours post-transduction, the medium was replaced by 1ml of complete DMEM medium. Forty-eight hours post-transduction, puromycine selection (1 μg/ml) of stably infected clones was performed for 7 days before being harvested for ChIP assays as described above.
Transient transfection and luciferase assays
293T cells (2.5 × 105) were co-transfected with 900 ng of reporter constructs and 50 ng of pRL-TK (Promega) as an internal control by the calcium phosphate transfection method according to the manufacturer's protocol (Takara). Raji cells (3 × 106 cells) were co-transfected with 600 ng of reporter constructs and 50 ng of pRL-TK as an internal control using DEAE-dextran as described previously (23). Forty-eight hours post-transfection, cells were lysed and luciferase activities were measured using the DualGlo-luciferase reporter assay (Promega). Results were normalized for transfection efficiency using Renilla luciferase activities and total protein concentrations.
4C-seq
4C-seq experiments were performed as previously described (52). Briefly, 107 cells per sample were cross-linked in 2% formaldehyde for 10 min and the reaction was quenched by adding glycine at a final concentration of 0.13 M. Then, cells were lysed and the harvested cross-linked chromatin was digested using the restriction enzyme MboI (NEB). After a ligation step with the T4 DNA ligase (NEB), ligated samples were decross-linked and subjected to a second round of digestion using the restriction enzyme Csp6I (Thermo Scientific). Digested samples were then ligated using T4 DNA ligase (Roche) and purified before being used as a template for inverse PCR. Primers used for inverse PCR and library preparation are listed in Supplementary Table S2. Products were sequenced using Illumina sequencing (Illumina NextSeq 500) and reads were mapped to a hybrid ovine genome (OAR v3.1) containing the BLV provirus sequence (GenBank: KT122858.1) at the L267 or YR2 integration sites (Supplementary Table S4) and processed using pipe4C (52) (github.com/deLaatLab/pipe4C) with the following parameters: normalization to 1 million reads in cis, window size 21, top 2 read counts removed. Peaks were called using peakC (53) using default settings (alphaFDR = 0.1, qWr = 1). Coverage plots were generated using R (https://www.R-project.org/).
RESULTS
The BLV provirus contains three putative CTCF binding sites
In order to assess a putative role of CTCF in the regulation of BLV gene expression and physiopathology, we first performed in silico analysis of the BLV proviral genome for the presence of consensus CTCF binding sites. This in silico analysis performed on both strands of the viral genome revealed the presence of several putative CTCF binding sites (Figure 1). Among these putative CTCF binding sites, the two most relevant were located at the U5 region of both BLV LTRs (the 5′LTR and the 3′LTR), spanning from nucleotide (nt) +496 to nt +514 in the 5′LTR (hereafter called 5′LTRU5) and from nt +8685 to nt +8703 in the 3′LTR (hereafter called 3′LTRU5), respectively (nt +1 defined as the first nucleotide of the 5′LTR). In addition, a third CTCF site was observed in the regulatory region coding for the second exon of the viral Tax and Rex regulatory proteins (hereafter called Tax/Rex E2) spanning from nt +7515 to nt +7533. Of note, these putative CTCF binding sites were all located on the minus DNA strand of the BLV genome. Moreover, by performing computational analysis of several BLV strains referenced in the NCBI database, we showed an important conservation rate of these CTCF binding sites [81.6% (120/147) for the 5′LTR, 95.9% (141/147) for the Tax/Rex E2 and 84.3% (124/147) for the 3′LTR], thereby suggesting their potentially important role in BLV replication cycle and pathogenesis (Supplementary Figure S1 and Supplementary Table S1).
Figure 1.

In silico analysis of putative CTCF binding sites along the BLV provirus. CTCF motifs located either on the positive or on the negative strand are represented by black or red lines, respectively. The three BLV regions showing the highest similarity to the CTCF consensus motif (lowest p-value) are indicated by red arrows. The 5′ and 3′LTR are subdivided into three regions (U3, R, U5). Black arrows represent sense transcripts initiated from the 5′LTR. Grey arrows represent antisense transcripts initiated from the 3′LTR.
CTCF is recruited in vivo to three distinct regions along the BLV provirus
In order to determine whether the three putative CTCF binding sites identified in silico were able to recruit CTCF in vivo, we performed chromatin immunoprecipitation followed by high-throughput sequencing assays (ChIP-seq) using the BLV latently-infected B-lymphocytic ovine cell line L267. As shown in Figure 2A, we observed the in vivo recruitment of CTCF to both the 5′LTRU5 and the 3′LTRU5 and, to a lesser extent, to the Tax/Rex E2 region, thereby demonstrating that CTCF was recruited in vivo to the three regions containing the sites we identified by in silico analysis. To further validate these results, we next performed additional ChIP-seq experiments using another BLV latently-infected B-lymphocytic ovine cell line, referred to as the YR2 cell line. Of note, compared to the L267 cell line, which represents an epigenetic model for viral latency, BLV infection in the YR2 cell line is latent due to two E- to K- mutations in the viral Tax protein, which impair its transactivating activity (12). Our results, presented in Figure 2B, showed a recruitment profile of CTCF similar to the one observed in the L267 cell line. Indeed, CTCF was also recruited to the U5 region of both LTRs and, to a lesser extent, to the Tax/Rex E2 region. Finally, to validate CTCF recruitment to the BLV provirus in a more physiological model of BLV-infected cells, we performed additional ChIP-seq experiments using chromatin prepared from peripheral blood mononuclear cells (PBMCs) isolated from a BLV-infected sheep that developed leukemia. As shown in Figure 2C, our results were in good agreement with the ones obtained in the BLV-latently infected B-lymphocytic ovine cell lines L267 and YR2, thereby confirming, in the context of BLV-latently infected primary cells, the in vivo recruitment of CTCF to three distinct regions along the BLV provirus: to both the 5′LTRU5 and the 3′LTRU5 and, to a lesser extent, to the Tax/Rex E2 region. Additionally, we validated our ChIP-seq results by performing additional ChIP-qPCR experiments (Supplementary Figure S2). Taken together, our results demonstrate that the three CTCF sites identified in silico are at least in part responsible for the in vivo recruitment of CTCF to the BLV genome, thereby providing the first evidence of a putative function of CTCF in BLV transcriptional and epigenetic regulations.
Figure 2.
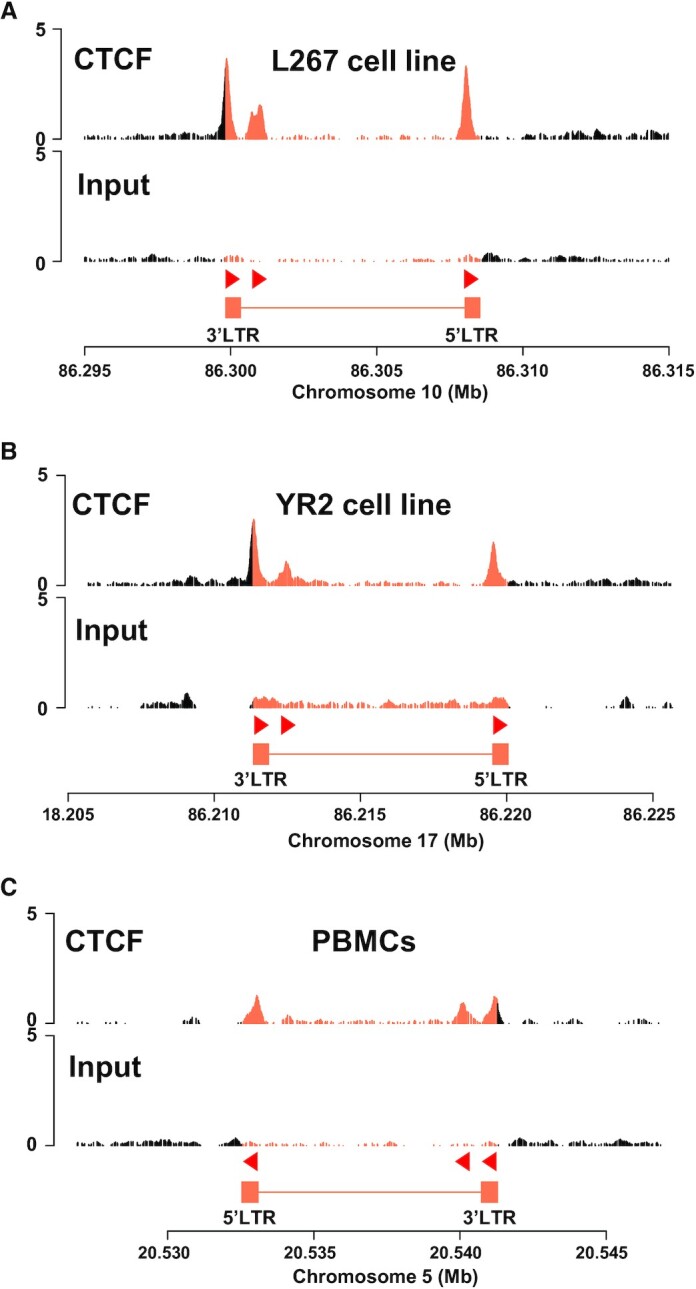
CTCF is recruited in vivo to three distinct regions along the BLV provirus. Chromatin prepared from BLV-infected (A) L267 cell line, (B) YR2 cell line, or (C) ovine PBMCs was immunoprecipitated with a specific antibody directed against CTCF. Recovered DNA or non-immunoprecipitated DNA (input) was then sequenced in paired-end and reads were mapped to a hybrid ovine genome containing the BLV provirus sequence at their respective insertion site and orientation (see Supplementary Table S4 for further details). The orientation of the CTCF motifs is indicated by red triangles.
CTCF binding differentially regulates RNAPII-dependent transcriptional activities arising from the BLV 5′ and 3′LTRs
Previous reports have demonstrated CTCF as an important transcription factor regulating gene expression through its ability to either activate or repress gene expression, as well as insulating an enhancer region or conversely bringing an enhancer close to its cognate promoter by the formation of long-range chromatin interactions (33–36,54). Based on the in vivo recruitment of CTCF to the 5′ and 3′ LTRs, which respectively exhibit sense and antisense RNAPII-dependent promoter activities, we next decided to investigate the putative functional role played by CTCF in these two transcriptional activities. To this end, we designed three different point mutations in the CTCF binding motif of the BLV LTR carefully avoiding the insertion of mutations in cis-regulatory elements previously described as critical for either sense or antisense LTR promoter activities (18–26) (Figure 3A). We then validated the effects of the designed mutations on CTCF recruitment in vivo, by performing ChIP-qPCR assays after transduction of HEK293T cells with lentiviral particles containing either the wild-type or the CTCF-mutated BLV LTRs. As positive and negative controls for CTCF immunoprecipitation, we also designed specific oligonucleotide primers hybridizing to two cellular genomic regions, MDM2 and GAPDH, previously reported to either recruit CTCF or not, respectively (55). As shown in Figure 3B, mutation of only the 10th nt in CTCF motif or a combined mutation of nt 3, 4, 5, 6, 9, 10 drastically reduced the in vivo recruitment of CTCF to the BLV LTRU5 binding site (3% or 0.03% of CTCF remaining compared to the WT, respectively), without affecting CTCF recruitment to other cellular regions. However, mutating the fifth nt alone only slightly reduced CTCF recruitment by 1.8-fold. Therefore, in order to evaluate the functional role of CTCF on both the 5′LTR sense and the 3′LTR antisense RNAPII-dependent transcriptional activities, we cloned either the wild-type or the CTCF-mutated BLV LTR upstream of the Firefly luciferase reporter gene in the sense or antisense orientation (representing the 5′ or 3′ LTR RNAPII-dependent promoter activity, respectively) into a modified pREP10 episomal vector (Figure 3C). Of note, using episomally replicating luciferase reporter constructs has the advantage to display the hallmarks of proper chromatin structure when transiently transfected into cells (56,57), an important feature allowing the study of transcriptional effects while taking into account the chromatin structure. To be in a relevant context of natural target cells corresponding to B cells, the reporter constructs were transiently transfected into a human B-lymphoïd cells, the Raji cell line. Forty-eight hours post-transfection, cells were lysed and assayed for luciferase activity. As shown in Figure 3D, when cloned in the sense orientation relative to the luciferase reporter gene, the RNAPII-dependent sense transcription was significantly increased when CTCF recruitment was abolished (constructs LTR_m10-S-luc and LTR_mFull-S-luc) while the sense transcription remained unchanged in the construct LTR_m5-S-luc, consistently with the level of recruitment of CTCF at the LTR. These results suggests that CTCF has a repressive role on the 5′LTR sense promoter activity. However, when cloned in the antisense orientation, the CTCF-mutated LTR promoter activity was significantly decreased when CTCF recruitment was abolished (constructs LTR_m10-AS-luc and LTR_mFull-AS-luc), while the antisense transcription remained unchanged in the construct LTR_m5-AS-luc, again consistently with the level of recruitment of CTCF at the LTR. These results suggest an activating effect of CTCF on the 3′LTR antisense promoter activity. We confirmed these data by additional transient transfection experiments into the HEK293T cell line, using the same experimental procedure (Supplementary Figure S3). Taken together, our results obtained in the context of transient transfections of episomal reporter constructs demonstrate a dual functional role of CTCF in the regulation of the BLV LTR promoter activities: a repressive role on the RNAPII-dependent sense transcription and an activating role on the RNAPII-dependent antisense transcription of the BLV retrovirus.
Figure 3.

CTCF binding differentially impacts BLV LTR promoter activities. (A) Schematic representation of the BLV LTR with the identified transcription start site for either the sense (nt +211) or the antisense (nt +460) transcription. The CTCF logo is indicated above the LTR_WT sequence. The three mutations in the CTCF binding site are highlighted in red and named as follows: LTR_m5; LTR_m10; LTR_mFull. The closest cis-regulatory element (BRE) is underlined. (B) Chromatin prepared from 293T cells stably transduced with either the wild-type BLV LTR or the mutated LTR was immunoprecipitated with a specific antibody directed against CTCF or with an IgG as background measurement. Purified DNA was then amplified with oligonucleotide primers hybridizing to either the BLV LTR or host cellular genes (GAPDH and MDM2). Results are presented as histograms indicating percentages of immunoprecipitated DNA compared to the input DNA (% IP/Input). Data are the mean ± SD from one representative of at least three independent experiments. (C) Illustration of the reporter constructs without LTR (X-luc) or with the WT or mutated LTR cloned in sense (S) or antisense (AS) orientation relative to the Firefly luciferase gene (luc). (D) The Raji B-cell line was transiently co-transfected with 600 ng of the reporter constructs and 50 ng of pRL-TK. Results are presented as histograms indicating relative luciferase activities compared to the value obtained with the LTR_WT-S-luc construct which was assigned to the value of 1. Data are the mean ± SD of at least three independent experiments. The Wilcoxon signed-rank (value of 1) or the Mann-Whitney statistical tests were used for sense and anti-sense orientations, respectively, with P > 0.05 = ns; P ≤ 0.05 = *; P ≤ 0.01 = **; P ≤ 0.001 = ***.
CTCF localizes to histone marks transitions along BLV proviral genome
CTCF exhibits a broad range of functions and is enriched at boundaries of topologically associated domains (TADs). These TADs are regions where DNA contacts preferentially occur and which are often characterized by a similar epigenetic landscape (36,56). Even though the direct functional role of CTCF in delimitating histone modifications borders is still under debate (58), several studies have linked CTCF to the maintenance of distinct epigenetic profiles (59–61).
We have previously reported the epigenetic profile along the three BLV promoters in the context of the BLV latently-infected B-lymphocytic ovine cell line L267 (26). Here, to investigate whether CTCF could contribute to the establishment of a putative histone modifications border along the entire BLV provirus, we performed ChIP-qPCR experiments using chromatin from the BLV latently-infected B-lymphocytic ovine cell line L267 which was immunoprecipitated with specific antibodies directed against several histone post-translational modifications. As shown in Figure 4B, we observed an enrichment of histone post-translational marks associated with active transcription, i.e. acetylated histone 3 (AcH3), acetylated histone 3 lysine 9 (H3K9ac), acetylated histone 3 lysine 27 (H3K27ac), di-methylated histone 3 lysine 4 (H3K4me2), tri-methylated histone 3 lysine 4 (H3K4me3). These epigenetic marks encompassed the BLV proviral region delimitated by CTCF binding to the Tax/Rex E2 and the 3′LTRU5 regions. Of note, this activating epigenetic signature correlated with the constitutive antisense RNAPII-dependent transcription arising from the 3′LTR (26,29). Moreover, in agreement with the transcriptional silencing affecting the BLV 5′LTR, these results showed an absence of activating histone marks at the 5′LTR. Overall, our results demonstrated an epigenetic signature clustered by the regions to which CTCF was recruited.
Figure 4.

CTCF localizes to histone marks transitions along the BLV proviral genome. (A) Schematic representation of the BLV proviral genome with the localization of the ChIP-qPCR primers used. Chromatin prepared from BLV infected (B) L267 cells or (C) ovine PBMCs was immunoprecipitated with specific antibodies directed against histone H3 (PanH3), different histone post-translational modifications (H3Kme2, H3Kme3, AcH3, H3K9ac, H3K27ac) or with an IgG as background measurement. Results are presented as histograms indicating percentages of immunoprecipitated DNA compared to the input DNA (%IP/input) normalized to PanH3. Red dashed lines represent CTCF binding sites. Data are the means ± SD from one representative of at least three independent experiments.
Next, in order to strengthen our results showing a potential role of CTCF in the delimitation of a histone modifications border along the BLV provirus, we performed additional ChIP-qPCR experiments in the context of another BLV latently-infected B-lymphocytic cell line, the YR2 cell line (Supplementary Figure S4), and in a more physiological context of BLV infection corresponding to BLV-infected ovine PBMCs (Figure 4C). Our results confirmed an enrichment of activating histone post-translational modifications which were clustered by the two CTCF binding sites located in the Tax/Rex E2 region and in the 3′LTRU5, respectively. However, regarding the epigenetic profile along the 5′LTR and in opposition to what we observed in the L267 cell line, an accumulation of activating histone modifications was observed in the 5′LTR both in the YR2 cell line and in PBMCs. These observations were in agreement with previous results describing a weaker latency state in the YR2 cell line compared to the L267 cell line associated with a lower basal level of RNAPII-dependent sense transcription (13,26,32). However, since these activating histone modifications were not spreading downstream of CTCF binding to the 5′LTR, CTCF could also establish an epigenetic boundary contributing to BLV latency, a mechanism in agreement with the data we obtained by transient transfection assays (Figure 3D).
Taken together, our results showing the localization of CTCF at specific histone modifications transitions suggest that CTCF could be implicated in the maintenance of an active RNAPII-dependent antisense transcription. This would occur by the delimitation of a region characterized by the accumulation of activating histone marks, while maintaining the RNAPII-dependent 5′LTR promoter silent because preventing the spread of activating histone modifications.
The BLV provirus disrupts host three-dimensional chromatin organization by forming chromatin loops with host genomic regions
One of the most studied roles of CTCF is its involvement in the organization of 3D chromatin architecture, notably resulting in long-range interactions, forming structural domains and bringing enhancers/silencers close to their cognate promoters by forming chromatin loops between two CTCF binding sites (33,34,36,62,63). The formation of these chromatin loops requires the co-recruitment of the cohesin multiprotein complex to the CTCF binding sites mediated by a mechanism called loop extrusion (64). In this context, we decided to assess whether the BLV provirus was able to induce the formation of chromatin loops with the host genomic environment. To this end, we studied the co-recruitment of the cohesin complex to the previously identified regions of CTCF recruitment by performing ChIP-seq experiments in the two BLV latently-infected B-lymphocytic L267 and YR2 cell lines (Figure 5). In these two cellular models for BLV infection, we observed the in vivo recruitment of Rad21 (a structural component of the cohesin complex) to the end of both the 5′ and 3′LTRs (Figure 5A and B), as confirmed by ChIP-qPCR experiments (Supplementary Figure S5). Together, these results suggest a cooperative role of CTCF and cohesin in the formation of chromatin loops involving the BLV CTCF binding regions.
Figure 5.

The Rad21 subunit of the cohesin multiprotein complex is recruited to the BLV CTCF binding sites. Chromatin prepared from BLV-infected (A) L267 cell line or (B) YR2 cell line was immunoprecipitated with a specific antibody directed against Rad21. Recovered DNA or non-immunoprecipitated DNA (Input) were then sequenced in paired-end and reads were mapped to a hybrid ovine genome containing the BLV provirus sequence at their respective insertion site and orientation (see Supplementary Table S4 for further details). The orientation of the CTCF motifs is indicated by red triangles.
In order to assess whether the integrated BLV provirus was able to establish 3D chromatin structures with its host genome, we performed circular chromosome conformation capture (4C) experiments followed by high-throughput sequencing (4C-seq), a technique allowing the identification of physical contacts between a specific DNA region, called the viewpoint, and the entire cellular genome. Using the BLV-latently infected B-lymphotropic ovine cell lines L267 and YR2, we designed two specific viewpoints encompassing the CTCF binding site located in both LTRs. As shown in Figure 6, we clearly identified the presence of chromatin loops between the BLV provirus and its host genome in both the L267 and YR2 cell lines using a viewpoint encompassing the CTCF binding site located in the 5′LTR. In the context of the L267 cell line, we observed an interaction between the BLV provirus and a genomic region located approximatively 60 kb upstream of the BLV integration site (IS) (Figure 6A and Supplementary Figure S6A). Moreover, we observed that CTCF and cohesin co-localized at the genomic region physically interacting with the BLV provirus, further supporting their implication in the formation of the observed chromatin loop (Figure 6A). Similar 4C-seq profiles were obtained when we used the CTCF binding site located in the 3′LTR as the viewpoint (Supplementary Figure S7), thereby demonstrating that these proviral regions induced local topological changes in the host genome. In the context of the YR2 cell line, whose viral DNA is integrated at another location in the genome (Supplementary Table S4), 4C-seq data showed multiple physical contacts between the viral CTCF sites and the surrounding CTCF sites present in the host genome (Figure 6B and Supplementary Figure S6B). Altogether, our results demonstrated that the integrated BLV provirus modifies the host cellular 3D chromatin organization through the formation of viral/host chromatin loops. We speculate that these structures constitute a new mechanism used by the BLV leukemogenic retrovirus to perturbate the host 3D cellular genome organization and, as a consequence, the host cellular transcriptome.
Figure 6.

Chromatin contacts are established between BLV and cellular genomic regions. 4C-seq contact profiles averaged over three biological replicates in the context of (A) L267 cells or (B) YR2 cells using the BLV CTCF binding site of the 5′LTR as viewpoint. ChIP-seq or 4C-seq reads were mapped to a hybrid ovine genome containing the BLV provirus sequence at their respective insertion site and orientation (Supplementary Table S4). Reads mapping to the proviral genome are highlighted in orange. Statistically significant 4C-peaks are highlighted in green. Viewpoint is indicated by a red dashed line. Below the 4C plots, the Rad21 and CTCF ChIP-seq profiles of the extended studied region are shown. The orientation of the CTCF binding motifs is indicated by red (forward) or blue (reverse) triangles or by gray bars (not determined). The names of the surrounding host cellular genes close to the BLV integration site are presented.
DISCUSSION
Despite the well described BLV latency affecting the viral 5′LTR promoter activity and preventing viral gene expression, it has been demonstrated that the transcriptional network regulating BLV gene expression is more complex than initially assumed since two additional promoter activities have been discovered: an RNAPIII-dependent promoter activity responsible for an active transcription of 10 viral micro-RNAs, and an RNAPII-dependent antisense transcription arising from the 3′LTR and allowing high expression levels of non-coding antisense transcripts (26–29). Altogether, these findings have highlighted the deep complexity of BLV transcriptional regulation and paved the way to discover new mechanisms allowing tumoral development. In the present report, we studied the implication of the cellular protein CTCF in this complex transcriptional network as well as its role in the delimitation of a specific histone modifications profile along the BLV provirus and the disruption of the 3D chromatin organization of infected cells. First, we identified in silico three highly conserved CTCF binding sites along the BLV provirus and demonstrated the in vivo recruitment of CTCF in BLV latently-infected B-lymphocytic ovine cell lines as well as in BLV-infected primary cells isolated from a leukemic sheep (Figure 1 and 2). Next, we investigated whether CTCF could act as a transcriptional regulator of either the 5′LTR sense or the 3′LTR antisense RNAPII-dependent transcriptional activities by introducing point mutations in the CTCF binding sites of the BLV LTR, thereby preventing its in vivo recruitment (Figure 3A and B). Importantly, we here demonstrated that CTCF exhibited opposite effects depending on the orientation of the RNAPII-dependent transcription. Indeed, our results showed that CTCF binding to the LTRU5 inhibited the RNAPII-dependent sense transcription, representative of the 5′LTR promoter activity, while it favors the RNAPII-dependent antisense transcription, representative of the 3′LTR promoter activity (Figure 3D). The opposite effects on BLV transcriptions observed in Figure 3D could be mainly explained by the localization of the CTCF binding site relative to the TSS. Indeed, Nora and colleagues have observed by genome-wide studies that CTCF acts as a transcriptional activator when bound to a CTCF binding site located ∼60 bp upstream of the TSS and in concordant orientation with the direction of transcription (58), a situation reminiscent of what is observed along the BLV genome where the BLV CTCF binding site is located 45 bp upstream of the antisense TSS and in concordant orientation with antisense transcription (Figure 3A). At the molecular level, through this promoter-proximal binding, CTCF could favor the recruitment of known interacting partners, such as the RNAPII subunit Rpb1 (65) or the chromatin remodeling enzyme BRG1 (Brahma-related gene 1) (66,67), with, as a result, the activation of the BLV antisense transcription arising from the 3′LTR. Moreover, when CTCF is located downstream of a TSS as observed in the BLV 5′LTR responsible for the sense transcription, CTCF has been shown to favor RNAPII pausing by stabilization of the negative elongation factor (NELF) and the DRB-sensitivity inducing factor (DSIF) (68), but also by reducing RNAPII processivity in vitro (69), thereby repressing the BLV sense transcription arising from the 5′LTR. Overall, our observations in the context of episomal reporter constructs suggest that CTCF is an activator of the 3′LTR antisense promoter activity, while it represses the 5′LTR sense promoter activity, thus favoring viral latency.
Among the multiple roles of CTCF described in the literature, its recruitment to boundaries delimitating distinct histone modifications profiles as well as its implication in the delimitation of TADs has been widely studied. In the retrovirology field, a previous study has reported a single CTCF binding site in the HLTV-1 pX regulatory region, acting as an epigenetic border by maintaining activating histone modifications and favoring antisense transcription (38). Even though these results were then refuted by the same group (39), a recent study has demonstrated by experimental infection of PBMCs with a CTCF-mutated HTLV-1 provirus and by the use of shRNAs, that CTCF binding to the HTLV-1 genome regulated the DNA and histone methylation profiles, in an integration site-depend fashion (43). In the present study, we studied the putative role of CTCF in the establishment of a specific histone modifications signature along the BLV provirus. Our results clearly showed a distinct epigenetic profile with activating histone modifications spreading from the 3′LTRU5 to the Tax/Rex E2 region, while these marks strongly decreased from this latter region to the 5′LTR. In addition, in the context of BLV-infected PBMCs, we also showed that these activating histone modifications were enriched along the 5′LTR but immediately dropped downstream. Interestingly, by comparing the distribution of histone marks along the BLV provirus with our data demonstrating the in vivo recruitment of CTCF, we hypothesized that CTCF recruitment to the 5′LTRU5 could prevent the spread of activating histone modifications, allowing the reinforcement of BLV latency while in the meantime favoring the antisense transcription through the establishment of an activating epigenetic landscape spreading from the CTCF binding site in the Tax/Rex E2 region to the 3′LTRU5. Altogether, our results suggest that CTCF could contribute to BLV 5′LTR silencing by acting not only at the transcriptional level but also at the epigenetic level. In contrast, CTCF could favor the RNAPII-dependent antisense transcription by acting as a transcriptional activator and by delimitating a region enriched in activating histone marks, thereby identifying CTCF as a potential new key regulator of BLV gene expression.
Despite the latest discoveries about the putative role of either BLV miRNAs (30,31) or chimeric viral/host transcripts arising from the 3′LTR (32), the precise mechanisms underlying BLV-induced leukemogenesis remain incompletely understood and could be a multifactorial phenomenon resulting from a combination of multiple cellular deregulations, thereby leading to the irreversible cellular transformation of infected B cells. Since CTCF has also been demonstrated to contribute to the chromatin organization at the 3D level, we here investigated whether such chromatin loops, induced by the inserted BLV CTCF binding sites, could contribute to BLV-associated pathogenesis. After performing 4C-seq experiments in the two BLV latently-infected B-lymphocytic ovine cell lines L267 and YR2, we showed that BLV was able to contact its host cellular environment, through the formation of viral/host chromatin loops established after the co-recruitment of CTCF and the cohesin multiprotein complex to the LTRs (as demonstrated by ChIP-seq data; Figures 2, 5 and 6). Together, our results indicate that the viral CTCF binding sites, randomly inserted into the host cellular genome following BLV integration, modify the natural host 3D chromatin architecture and, as consequence, could deregulate the expression of cellular genes that are critical for cell homeostasis and favor tumoral development. Indeed, deregulation of cellular gene expression through disruption of chromatin loops involving CTCF has frequently been associated with several diseases, including cancers (70–74). Moreover, the BLV LTR contains a well-described enhancer sequence (18–25), and therefore, bringing the viral LTR enhancer activity close to a cellular gene through such viral/host chromatin loops may directly deregulate gene expression, a situation already observed in the context of KSHV (75,76) and EBV (77) where viral CTCF-mediated chromatin loops determine viral promoter usage and latency type. In HTLV-1, the hypothesis by which the unique viral inserted CTCF binding site could deregulate the host chromatin architecture has been tested and has revealed clone-specific chromatin loops, which lead to the deregulation of genes as far as > 300 kb from the proviral integration site (40). However, the extent to which these abnormal chromatin loops and the subsequent gene expression deregulation contribute to the HTLV-1-induced adult T-cell leukemia (ATL) development is not yet clear (78). Our results presented here further support these observations. Indeed, our 4C-seq data in L267 ovine cells revealed a major contact between the BLV provirus and the host genome identified in the vicinity of the cellular CDC16 gene, which codes for a member of the anaphase-promoting complex (APC/C) known for its involvement in cell cycle progression through control of mitosis progression (79). By comparison, in the YR2 cell line, we observed a major contact established with the NOCT gene, encoding for a phosphatase involved in the regulation of cellular metabolism (80). Regarding the other minor contacts, we identified they encompassed other genes such as MGARP, NAA15 and RAB338. Altogether, we highlighted several putative deregulated cellular target genes which will be further investigated to evaluate their role in BLV-associated physiopathology. Of note, since BLV integration is a random process, the formation of such viral-host chromatin loops will also be a random process, a key feature that could explain that BLV-associated leukemogenesis occurs in only 5% of infected animals. Associated with the previously described roles of BLV miRNAs and chimeric viral/host transcripts in host transcriptome deregulation, we thus identified in the present report a potentially critical step leading to tumor development.
Finally, a major feature of BLV-induced B-cell tumors is the presence of an integrated provirus containing large deletions, often affecting the 5′LTR and therefore also explaining the viral latency phenomenon. Interestingly, a recent publication from the group of Anne Van den Broeke has demonstrated that the 3′LTR, in which we observed a high co-recruitment of CTCF and Rad21, is always conserved in tumor isolates, thereby demonstrating the critical importance of this proviral region in BLV physiopathology (32). Moreover, it has been observed that cancer driver genes are enriched upstream of BLV integration sites and are perturbated by the RNAPII-dependent antisense transcription arising from the 3′LTR (32). While these genes are relatively closer to the integrated provirus, the formation of viral-host chromatin loops, highlighted in the present report, could be an additional mechanism used by BLV to disrupt its host transcriptome, but at long-range distances. This hypothesis is strengthened by the fact that CTCF binding sites along the BLV provirus are in convergent orientation with the sites found upstream of the BLV integration site, thereby rendering the formation of chromatin loops highly probable (63).
Taken together, the present report identifies CTCF as a new regulator of BLV gene expression and associated physiopathology, by acting both at the transcriptional and epigenetic levels to specifically allow the RNAPII-dependent antisense transcription, while maintaining the 5′LTR promoter latent. In addition, our results pave the way for a critical role of CTCF binding along the BLV provirus to induce viral/host chimeric chromatin loops which could lead to the deregulation of the host cellular transcriptome and trigger tumoral development. Therefore, our results provide new insights into BLV transcriptional and epigenetic regulation as well as into BLV-associated leukemogenesis, thereby highlighting mechanisms used by retroviruses to regulate their own expression and to alter the host cellular transcriptome. A better understanding of these mechanisms should allow us to identify new strategies to cure BLV infection and, to a larger extent, decrease economical losses in endemic countries.
DATA AVAILABILITY
ChIP-seq and 4C-seq data have been deposited in the Gene Expression Omnibus with accession codes GSE178482 and GSE178483, respectively.
Supplementary Material
ACKNOWLEDGEMENTS
We acknowledge A. Van den Broeke (GIGA, University of Liège, Belgium) for generously providing BLV-infected ovine primary cells used in this work. We acknowledge the Lausanne Genomic Technology Facility of the University of Lausanne for performing high-throughput sequencing experiments and bioinformatical analyses. We thank all the members of the Van Lint laboratory for discussions and critical reading of the manuscript.
Contributor Information
Maxime Bellefroid, Service of Molecular Virology, Department of Molecular Biology (DBM), Université Libre de Bruxelles (ULB), Gosselies 6041, Belgium.
Anthony Rodari, Service of Molecular Virology, Department of Molecular Biology (DBM), Université Libre de Bruxelles (ULB), Gosselies 6041, Belgium.
Mathilde Galais, Service of Molecular Virology, Department of Molecular Biology (DBM), Université Libre de Bruxelles (ULB), Gosselies 6041, Belgium.
Peter H L Krijger, Oncode Institute, Hubrecht Institute-KNAW and University Medical Center Utrecht, Utrecht 3584, CT, The Netherlands.
Sjoerd J D Tjalsma, Oncode Institute, Hubrecht Institute-KNAW and University Medical Center Utrecht, Utrecht 3584, CT, The Netherlands.
Lorena Nestola, Service of Molecular Virology, Department of Molecular Biology (DBM), Université Libre de Bruxelles (ULB), Gosselies 6041, Belgium.
Estelle Plant, Service of Molecular Virology, Department of Molecular Biology (DBM), Université Libre de Bruxelles (ULB), Gosselies 6041, Belgium.
Erica S M Vos, Oncode Institute, Hubrecht Institute-KNAW and University Medical Center Utrecht, Utrecht 3584, CT, The Netherlands.
Sara Cristinelli, Institute of Microbiology, Lausanne University Hospital, University of Lausanne, Lausanne 1011, Switzerland.
Benoit Van Driessche, Service of Molecular Virology, Department of Molecular Biology (DBM), Université Libre de Bruxelles (ULB), Gosselies 6041, Belgium.
Caroline Vanhulle, Service of Molecular Virology, Department of Molecular Biology (DBM), Université Libre de Bruxelles (ULB), Gosselies 6041, Belgium.
Amina Ait-Ammar, Service of Molecular Virology, Department of Molecular Biology (DBM), Université Libre de Bruxelles (ULB), Gosselies 6041, Belgium.
Arsène Burny, Service of Molecular Virology, Department of Molecular Biology (DBM), Université Libre de Bruxelles (ULB), Gosselies 6041, Belgium.
Angela Ciuffi, Institute of Microbiology, Lausanne University Hospital, University of Lausanne, Lausanne 1011, Switzerland.
Wouter de Laat, Oncode Institute, Hubrecht Institute-KNAW and University Medical Center Utrecht, Utrecht 3584, CT, The Netherlands.
Carine Van Lint, Service of Molecular Virology, Department of Molecular Biology (DBM), Université Libre de Bruxelles (ULB), Gosselies 6041, Belgium.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
C.V.L. acknowledges funding from the Belgian National Fund for Scientific Research (F.R.S-FNRS, Belgium); Fondation Roi Baudouin; Internationale Brachet Stiftung (IBS); Walloon Region (Fonds de Maturation); Les Amis des Instituts Pasteur à Bruxelles, asbl.; University of Brussels (ULB - Action de Recherche Concertée (ARC) grant); ‘crédit de recherche’ grant of the F.R.S.-FNRS [J.0021.17]; Swiss National Science Foundation [314730_188877]; the laboratory of C.V.L. is part of the ULB-Cancer Research Centre (U-CRC); A.R. is a postdoctoral fellow (ULB ARC program); M.B. is a doctoral fellow from the Belgian ‘Fonds pour la formation à la Recherche dans l’Industrie et dans l’Agriculture (FRIA)’; M.G. is a postdoctoral fellow supported by ‘Les Amis des Instituts Pasteur à Bruxelles, asbl’; L.N. is supported by a ‘projet de recherche’ grant from the F.R.S.-FNRS; E.P. is a doctoral fellow from the Télévie program of the F.R.S.-FNRS; W.D.L. is founder and shareholder of Cergentis; C.V.L. is ‘Directeur de Recherches’ of the F.R.S.-FNRS. Published with the support of the “Fondation Universitaire de Belgique”
Conflict of interest statement. None declared.
REFERENCES
- 1. Bartlett P., Norby B., Byrem T.M., Parmelee A., Ledergerber J.T., Erskine R.J.. Bovine leukemia virus and cow longevity in michigan dairy herds. J. Dairy Sci. 2013; 96:1591–1597. [DOI] [PubMed] [Google Scholar]
- 2. Aida Y., Murakami H., Takahashi M., Takeshima S.N.. Mechanisms of pathogenesis induced by bovine leukemia virus as a model for human T-cell leukemia virus. Front. Microbiol. 2013; 4:328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gillet N., Florins A., Boxus M., Burteau C., Nigro A., Vandermeers F., Balon H., Bouzar A.B., Defoiche J., Burny A.et al.. Mechanisms of leukemogenesis induced by bovine leukemia virus: prospects for novel anti-retroviral therapies in human. Retrovirology. 2007; 4:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Merimi M., Ozkan Y., Cleuter Y., Griebel P., Burny A., Martiat P., Van den Broeke A.. Epigenetics and leukemia unraveling oncogenic processes in the BLV ovine model. Front. Biosci. 2009; 1:154–163. [DOI] [PubMed] [Google Scholar]
- 5. Willems L., Portetelle D., Kerkhofs P., Chen G., Burny A., Mammerickx M., Kettmann R.. In vivo transfection of bovine leukemia provirus into sheep. Virology. 1992; 189:775–777. [DOI] [PubMed] [Google Scholar]
- 6. Willems L., Burny A., Collete D., Dangoisse O., Dequiedt F., Gatot J.S., Kerkhofs P., Lefèbvre L., Merezak C., Peremans T.et al.. Genetic determinants of bovine leukemia virus pathogenesis. AIDS Res. Hum. Retrovir. 2000; 16:1787–1795. [DOI] [PubMed] [Google Scholar]
- 7. Lagarias D.M., Radke K.. Transcriptional activation of bovine leukemia virus in blood cells from experimentally infected, asymptomatic sheep with latent infections. J. Virol. 1989; 63:2099–2107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Van den Broeke A., Cleuter Y., Chen G., Portetelle D., Mammerickx M., Zagury D., Fouchard M., Coulombel L., Kettmann R., Burny A.. Even transcriptionally competent proviruses are silent in bovine leukemia virus-induced sheep tumor cells. Proc. Natl. Acad. Sci. U.S.A. 1988; 85:9263–9267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gillet N., Gutiérrez G., Rodriguez S.M., de Brogniez A., Renotte N., Alvarez I., Trono K., Willems L.. Massive depletion of bovine leukemia virus proviral clones located in genomic transcriptionally active sites during primary infection. PLoS Pathog. 2013; 9:1003687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Pluta A., Jaworski J.P., Douville R.N.. Regulation of expression and latency in BLV and HTLV. Viruses. 2020; 12:1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Willems L., Gegonne A., Chen G., Burny A., Kettmann R., Ghysdael J.. The bovine leukemia virus p34 is a transactivator protein. EMBO J. 1987; 6:3385–3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Van den Broeke A., Bagnis C., Ciesiolka M., Cleuter Y., Gelderblom H., Kerkhofs P., Griebel P., Mannoni P., Burny A.. In vivo rescue of a silent tax-deficient bovine leukemia virus from a tumor-derived ovine B-cell line by recombination with a retrovirally transduced wild-type tax gene. J. Virol. 1999; 73:1054–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Colin L., Dekoninck A., Reichert M., Calao M., Merimi M., Van Den Broeke A., Vierendeel V., Cleuter Y., Burny A., Rohr O.et al.. Chromatin disruption in the promoter of bovine leukemia virus during transcriptional activation. Nucleic Acids Res. 2011; 39:9559–9573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Merezak C., Reichert M., Van Lint C., Kerkhofs P., Portetelle D., Willems L., Kettmann R.. Inhibition of histone deacetylases induces bovine leukemia virus expression in vitro and in vivo. J. Virol. 2002; 76:5034–5042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Merimi a M., Klener P., Szynal M., Cleuter Y., Bagnis C., Kerkhofs P., Burny A., Martiat P., Van den Broeke A.. Complete suppression of viral gene expression is associated with the onset and progression of lymphoid malignancy: observations in bovine leukemia virus-infected sheep. Retrovirology. 2007; 4:51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Nguyen L.A., Calomme C., Wijmeersch G., Nizet S., Veithen E., Portetelle D., De Launoit Y., Burny A., Van Lint C.. Deacetylase inhibitors and the viral transactivator taxBLV synergistically activate bovine leukemia virus gene expression via a cAMP-responsive element- and cAMP-responsive element-binding protein-dependent mechanism. J. Biol. Chem. 2004; 279:35025–35036. [DOI] [PubMed] [Google Scholar]
- 17. Pierard V., Guiguen A., Colin L., Wijmeersch G., Vanhulle C., Van Driessche B., Dekoninck A., Blazkova J., Cardona C., Merimi M.et al.. DNA cytosine methylation in the bovine leukemia virus promoter is associated with latency in a lymphoma-derived B-cell line: potential involvement of direct inhibition of camp-responsive element (CRE)-binding protein/cre modulator/activation transcription. J. Biol. Chem. 2010; 285:19434–19449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Adam E., Kerkhofs P., Mammerickx M., Burny A., Kettmann R., Willems L.. The CREB, ATF-1, and ATF-2 transcription factors from bovine leukemia virus-infected b lymphocytes activate viral expression. J. Virol. 1996; 70:1990–1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Calomme C., Nguyên T.L.A., De Kiermer Y.L.V., Droogmans L., Burny A., Van Lint C.. Upstream stimulatory factors binding to an e box motif in the r region of the bovine leukemia virus long terminal repeat stimulates viral gene expression. J. Biol. Chem. 2002; 277:8775–8789. [DOI] [PubMed] [Google Scholar]
- 20. Dekoninck A., Calomme C., Nizet S., De Launoit Y., Burny A., Ghysdael J., Van Lint C.. Identification and characterization of a PU.1/Spi-B binding site in the bovine leukemia virus long terminal repeat. Oncogene. 2003; 22:2882–2896. [DOI] [PubMed] [Google Scholar]
- 21. Kiermer V., Van Lint C., Briclet D., Vanhulle C., Kettmann R., Verdin E., Burny A., Droogmans L.. An interferon regulatory factor binding site in the U5 region of the bovine leukemia virus long terminal repeat stimulates tax-independent gene expression. J. Virol. 1998; 72:5526–5534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Nguyên T.L.A., De Walque S., Veithen E., Dekoninck A., Martinelli V., De Launoit Y., Burny A., Harrod R., Van Lint C.. Transcriptional regulation of the bovine leukemia virus promoter by the cyclic AMP-response element modulator τ isoform. J. Biol. Chem. 2007; 282:20854–20867. [DOI] [PubMed] [Google Scholar]
- 23. Calomme C., Dekoninck A., Nizet S., Adam E., Nguyên T.L.-A., Van Den Broeke A., Willems L., Kettmann R., Burny A.. Overlapping CRE and e box motifs in the enhancer sequences of the bovine leukemia virus 5′ long terminal repeat are critical for basal and acetylation-dependent transcriptional activity of the viral promoter: implications for viral latency. J. Virol. 2004; 78:13848–13864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Xiao J., Buehring G.C.. In vivo protein binding and functional analysis of cis-acting elements in the U3 region of the bovine leukemia virus long terminal repeat. J. Virol. 1998; 72:5994–6003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Unk L., Kiss-toth E., Boros I.. Transcription factor AP-4 participates in activation of bovine leukemia virus long terminal repeat by p34 tax. Nucleic Acids Res. 1994; 22:4872–4875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Van Driessche B., Rodari A., Delacourt N., Fauquenoy S., Vanhulle C., Burny A., Rohr O., Van Lint C.. Characterization of new RNA polymerase III and RNA polymerase II transcriptional promoters in the bovine leukemia virus genome. Sci. Rep. 2016; 6:31125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Rosewick N., Momont M., Durkin K., Takeda H., Caiment F., Cleuter Y.. Deep sequencing reveals abundant noncanonical retroviral microRNAs in B-cell leukemia /lymphoma. Proc. Natl. Acad. Sci. U.S.A. 2013; 110:2306–2311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kincaid R.P., Burke J.M., Sullivan C.S.. RNA virus microRNA that mimics a B-cell oncomiR. Proc. Natl. Acad. Sci. U.S.A. 2012; 109:3077–3082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Durkin K., Rosewick N., Artesi M., Hahaut V., Griebel P., Arsic N., Burny A., Georges M., Van den Broeke A.. Characterization of novel bovine leukemia virus (BLV) antisense transcripts by deep sequencing reveals constitutive expression in tumors and transcriptional interaction with viral microRNAs. Retrovirology. 2016; 13:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gillet N., Hamaidia M., de Brogniez A., Gutiérrez G., Renotte N., Reichert M., Trono K., Willems L.. Bovine leukemia virus small noncoding RNAs are functional elements that regulate replication and contribute to oncogenesis in vivo. PLoS Pathog. 2016; 12:e1005588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Safari R., Jacques J.R., Brostaux Y., Willems L.. Ablation of non-coding RNAs affects bovine leukemia virus b lymphocyte proliferation and abrogates oncogenesis. PLoS Pathog. 2020; 16:e1008502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rosewick N., Durkin K., Artesi M., Marçais A., Hahaut V., Griebel P., Arsic N., Avettand-Fenoel V., Burny A., Charlier C.et al.. Cis-perturbation of cancer drivers by the HTLV-1/BLV proviruses is an early determinant of leukemogenesis. Nat. Commun. 2017; 8:15264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bastiaan Holwerda S.J., de Laat W.. CTCF: the protein, the binding partners, the binding sites and their chromatin loops. Philos. Trans. R. Soc. B Biol. Sci. 2013; 368:1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Phillips J.E., Corces V.G.. CTCF: master weaver of the genome. Cell. 2009; 137:1194–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Braccioli L., De Wit E.. CTCF: a Swiss-army knife for genome organization and transcription regulation. Essays Biochem. 2019; 63:157–165. [DOI] [PubMed] [Google Scholar]
- 36. Merkenschlager M., Nora E.P.. CTCF and cohesin in genome folding and transcriptional gene regulation. Annu. Rev. Genomics Hum. Genet. 2016; 17:17–43. [DOI] [PubMed] [Google Scholar]
- 37. Pentland I., Parish J.L.. Targeting CTCF to control virus gene expression: a common theme amongst diverse DNA viruses. Viruses. 2015; 7:3574–3585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Satou Y., Miyazato P., Ishihara K., Yaguchi H., Melamed A., Miura M.. The retrovirus HTLV-1 inserts an ectopic CTCF-binding site into the human genome. Proc. Natl Acad. Sci. U.S.A. 2016; 113:3054–3059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Miura M., Miyazato P., Satou Y., Tanaka Y., Bangham C.R.M.. Epigenetic changes around the pX region and spontaneous HTLV-1 transcription are CTCF-independent. Wellcome Open Res. 2018; 3:105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Melamed A., Yaguchi H., Miura M., Witkover A., Fitzgerald T.W., Birney E., Bangham C.R.M.. The human leukemia virus HTLV-1 alters the structure and transcription of host chromatin in cis. Elife. 2018; 7:e36245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Martinez M.P., Cheng X., Joseph A., Al-Saleem J., Panfil A.R., Palettas M., Dirksen W.P., Ratner L., Green P.L.. HTLV-1 CTCF-binding site is dispensable for in vitro immortalization and persistent infection in vivo. Retrovirology. 2019; 16:44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Cook L., Melamed A., Yaguchi H., Bangham C.R.. The impact of HTLV-1 on the cellular genome. Curr. Opin. Virol. 2017; 26:125–131. [DOI] [PubMed] [Google Scholar]
- 43. Cheng X., Joseph A., Castro V., Chen-Liaw A., Skidmore Z., Payton J.E., Ratner L., Edwards J.R., Ueno T., Fujisawa J.I.et al.. Epigenomic regulation of human T-cell leukemia virus by chromatin-insulator CTCF. PLoS Pathog. 2021; 17:e1009577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kettmann R., Cleuter Y., Gregoire D., Burny A.. Role of the 3′ long open reading frame region of bovine leukemia virus in the maintenance of cell transformation. J. Virol. 1985; 54:899–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ambrosini G., Groux R., Bucher P.. PWMScan: a fast tool for scanning entire genomes with a position-specific weight matrix. Bioinformatics. 2018; 34:2483–2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Langmead B., Salzberg S.L.. Fast gapped-read alignment with bowtie 2. Nat. Methods. 2012; 9:357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R.. The sequence alignment/map format and SAMtools. Bioinformatics. 2009; 25:2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Feng J., Liu T., Qin B., Zhang Y., Liu X.S.. Identifying chip-seq enrichment using MACS. Nat. Protoc. 2012; 7:1728–1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Grant C.E., Bailey T.L., Noble W.S.. FIMO: scanning for occurrences of a given motif. Bioinformatics. 2011; 27:1017–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Khan A., Fornes O., Stigliani A., Gheorghe M., Castro-Mondragon J.A., Van Der Lee R., Bessy A., Chèneby J., Kulkarni S.R., Tan G.et al.. JASPAR 2018: update of the open-access database of transcription factor binding profiles and its web framework. Nucleic. Acids. Res. 2018; 46:260–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ramírez F., Ryan D.P., Grüning B., Bhardwaj V., Kilpert F., Richter A.S., Heyne S., Dündar F., Manke T.. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 2016; 44:W160–W165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Krijger P.H.L., Geeven G., Bianchi V., Hilvering C.R.E., de Laat W.. 4C-seq from beginning to end: a detailed protocol for sample preparation and data analysis. Methods. 2020; 170:17–32. [DOI] [PubMed] [Google Scholar]
- 53. Geeven G., Teunissen H., de Laat W., de Wit E.. peakC: a flexible, non-parametric peak calling package for 4C and capture-C data. Nucleic Acids Res. 2018; 46:e91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Splinter E., Heath H., Kooren J., Palstra R.J., Klous P., Grosveld F., Galjart N., De Laat W.. CTCF mediates long-range chromatin looping and local histone modification in the β-globin locus. Genes Dev. 2006; 20:2349–2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Li J., Huang K., Hu G., Babarinde I.A., Li Y., Dong X., Chen Y.S., Shang L., Guo W., Wang J.et al.. An alternative CTCF isoform antagonizes canonical CTCF occupancy and changes chromatin architecture to promote apoptosis. Nat. Commun. 2019; 10:1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Van Der Vlag J., Den Blaauwen J.L., Sewalt R.G.A.B., Van Driel R., Otte A.P.. Transcriptional repression mediated by polycomb group proteins and other chromatin-associated repressors is selectively blocked by insulators. J. Biol. Chem. 2000; 275:697–704. [DOI] [PubMed] [Google Scholar]
- 57. Liu R., Liu H., Chen X., Kirby M., Brown P.O., Zhao K.. Regulation of CSF1 promoter by the SWI/SNF-like BAF complex. Cell. 2001; 106:309–318. [DOI] [PubMed] [Google Scholar]
- 58. Nora E.P., Goloborodko A., Valton A.L., Gibcus J.H., Uebersohn A., Abdennur N., Dekker J., Mirny L.A., Bruneau B.G.. Targeted degradation of CTCF decouples local insulation of chromosome domains from genomic compartmentalization. Cell. 2017; 169:930–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Lutz M., Burke L.J., Barreto G., Goeman F., Greb H., Arnold R., Schultheiss H., Brehm A., Kouzarides T., Lobanenkov V.et al.. Transcriptional repression by the insulator protein CTCF involves histone deacetylases. Nucleic Acids Res. 2000; 28:1707–1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Essafi A., Webb A., Berry R.L., Slight J., Burn S.F., Spraggon L., Velecela V., Martinez-Estrada O.M., Wiltshire J.H., Roberts S.G.E.et al.. A Wt1-controlled chromatin switching mechanism underpins tissue-specific wnt4 activation and repression. Dev. Cell. 2011; 21:559–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Cuddapah S., Jothi R., Schones D.E., Roh T.Y., Cui K., Zhao K.. Global analysis of the insulator binding protein CTCF in chromatin barrier regions reveals demarcation of active and repressive domains. Genome Res. 2009; 19:24–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Hsu S.C., Gilgenast T.G., Bartman C.R., Edwards C.R., Stonestrom A.J., Huang P., Emerson D.J., Evans P., Werner M.T., Keller C.A.et al.. The BET protein BRD2 cooperates with CTCF to enforce transcriptional and architectural boundaries. Mol. Cell. 2017; 66:102–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. De Wit E., Vos E.S.M., Wijchers P.J., Krijger P.H.L., Holwerda S.J.B., Valdes-quezada C., Verstegen M.J.A.M.. CTCF binding polarity determines chromatin looping. Mol. Cell. 2015; 60:676–684. [DOI] [PubMed] [Google Scholar]
- 64. Sanborn A.L., Rao S.S.P., Huang S.C., Durand N.C., Huntley M.H., Jewett A.I., Bochkov I.D., Chinnappan D., Cutkosky A., Li J.et al.. Chromatin extrusion explains key features of loop and domain formation in wild-type and engineered genomes. Proc. Natl. Acad. Sci. U.S.A. 2015; 112:6456–6465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Chernukhin I., Shamsuddin S., Kang S.Y., Bergström R., Kwon Y.-W., Yu W., Whitehead J., Mukhopadhyay R., Docquier F., Farrar D.et al.. CTCF interacts with and recruits the largest subunit of RNA polymerase II to CTCF target sites genome-wide. Mol. Cell. Biol. 2007; 27:1631–1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Marino M.M., Rega C., Russo R., Valletta M., Gentile M.T., Esposito S., Baglivo I., De Feis I., Angelini C., Xiao T.et al.. Interactome mapping defines BRG1, a component of the SWI/SNF chromatin remodeling complex, as a new partner of the transcriptional regulator CTCF. J. Biol. Chem. 2019; 294:861–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Valletta M., Russo R., Baglivo I., Russo V., Ragucci S., Sandomenico A., Iaccarino E., Ruvo M., De Feis I., Angelini C.et al.. Exploring the interaction between the swi/snf chromatin remodeling complex and the zinc finger factor ctcf. Int. J. Mol. Sci. 2020; 21:8950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Laitem C., Zaborowska J., Tellier M., Yamaguchi Y., Cao Q., Egloff S., Handa H., Murphy S.. CTCF regulates NELF, DSIF and P-TEFb recruitment during transcription. Transcription. 2015; 6:79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Shukla S., Kavak E., Gregory M., Imashimizu M., Shutinoski B., Kashlev M., Oberdoerffer P., Sandberg R., Oberdoerffer S.. CTCF-promoted RNA polymerase II pausing links DNA methylation to splicing. Nature. 2011; 479:74–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Krijger P.H.L., De Laat W.. Regulation of disease-associated gene expression in the 3D genome. Nat. Rev. Mol. Cell Biol. 2016; 17:771–782. [DOI] [PubMed] [Google Scholar]
- 71. Norton H.K., Phillips-Cremins J.E.. Crossed wires: 3D genome misfolding in human disease. J. Cell Biol. 2017; 216:3441–3452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Hnisz D., Weintrau A.S., Day D.S., Valton A.L., Bak R.O., Li C.H., Goldmann J., Lajoie B.R., Fan Z.P., Sigova A.A.et al.. Activation of proto-oncogenes by disruption of chromosome neighborhoods. Science. 2016; 351:1454–1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Flavahan W.A., Drier Y., Liau B.B., Gillespie S.M., Venteicher A.S., Stemmer-Rachamimov A.O., Suvà M.L., Bernstein B.E.. Insulator dysfunction and oncogene activation in IDH mutant gliomas. Nature. 2016; 529:110–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Liu E.M., Martinez-Fundichely A., Diaz B.J., Apostolou E., Sanjana N.E., Khurana Correspondence E.. Identification of cancer drivers at CTCF insulators in 1, 962 whole genomes. Cell Syst. 2019; 8:446–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Kang H., Wiedmer A., Yuan Y., Robertson E., Lieberman P.M.. Coordination of KSHV latent and lytic gene control by CTCF-cohesin mediated chromosome conformation. PLoS Pathog. 2011; 7:e1002140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Li D., Mosbruger T., Verma D., Swaminathan S.. Complex interactions between cohesin and CTCF in regulation of kaposi's sarcoma-associated herpesvirus lytic transcription. J. Virol. 2019; 94:1279–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Tempera I., Klichinsky M., Lieberman P.M.. EBV latency types adopt alternative chromatin conformations. PLoS Pathog. 2011; 7:e1002180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Bangham C.R.M., Miura M., Kulkarni A., Matsuoka M.. Regulation of latency in the human t cell leukemia virus, HTLV-1. Annu. Rev. Virol. 2019; 6:365–385. [DOI] [PubMed] [Google Scholar]
- 79. Wang Q., Moyret-Lalle C., Couzon F., Surbiguet-Clippe C., Saurin J.C., Lorca T., Navarro C., Puisieux A.. Alterations of anaphase-promoting complex genes in human colon cancer cells. Oncogene. 2003; 22:1486–1490. [DOI] [PubMed] [Google Scholar]
- 80. Estrella M.A., Du J., Chen L., Rath S., Prangley E., Chitrakar A., Aoki T., Schedl P., Rabinowitz J., Korennykh A.. The metabolites NADP+ and NADPH are the targets of the circadian protein nocturnin (Curled). Nat. Commun. 2019; 10:2367. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
ChIP-seq and 4C-seq data have been deposited in the Gene Expression Omnibus with accession codes GSE178482 and GSE178483, respectively.