


Abstract
Restricting the localization of CENP-A (Cse4 in Saccharomyces cerevisiae) to centromeres prevents chromosomal instability (CIN). Mislocalization of overexpressed CENP-A to non-centromeric chromatin contributes to CIN in budding and fission yeasts, flies, and humans. Overexpression and mislocalization of CENP-A is observed in cancers and is associated with increased invasiveness. Mechanisms that remove mislocalized CENP-A and target it for degradation have not been defined. Here, we report that Cdc48 and its cofactors Ufd1 and Npl4 facilitate the removal of mislocalized Cse4 from non-centromeric chromatin. Defects in removal of mislocalized Cse4 contribute to lethality of overexpressed Cse4 in cdc48,ufd1 andnpl4 mutants. High levels of polyubiquitinated Cse4 and mislocalization of Cse4 are observed in cdc48-3, ufd1-2 and npl4-1mutants even under normal physiological conditions, thereby defining polyubiquitinated Cse4 as the substrate of the ubiquitin directed segregase Cdc48Ufd1/Npl4. Accordingly, Npl4, the ubiquitin binding receptor, associates with mislocalized Cse4, and this interaction is dependent on Psh1-mediated polyubiquitination of Cse4. In summary, we provide the first evidence for a mechanism that facilitates the removal of polyubiquitinated and mislocalized Cse4 from non-centromeric chromatin. Given the conservation of Cdc48Ufd1/Npl4 in humans, it is likely that defects in such pathways may contribute to CIN in human cancers.
Graphical Abstract
Graphical Abstract.
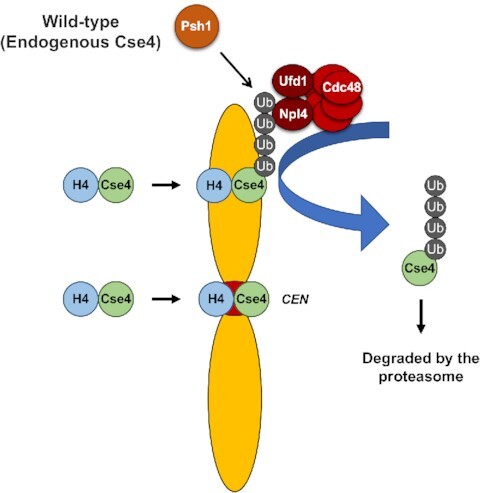
Psh1 ubiquitinates Cse4 (CENP-A in humans) which is mislocalized to non-centromeric regions under normal physiological conditions in budding yeast. Cdc48/Ufd1/Npl4 segregase associates with mislocalized Cse4 and targets it for degradation.
INTRODUCTION
Centromeres are specialized chromatin structures that are essential for faithful chromosome segregation. The kinetochore (centromeric DNA and associated proteins) provides an attachment site for microtubules for segregation of sister chromatids during mitosis. Centromeric localization of the evolutionarily conserved histone H3 variant CENP-A (Cse4 in Saccharomyces cerevisiae, Cnp1 in Schizosaccharomyces pombe, CID in Drosophila melanogaste and CENP-A in humans) serves as a platform for kinetochore assembly (1–3). Recruitment of Cse4/Cnp1/CID/CENP-A at the centromeres is regulated by the evolutionarily conserved CENP-A specific histone chaperone Scm3/CAL1/HJURP (Holliday Junction Recognition Protein) in budding and fission yeasts, flies, and humans, respectively (4–11).
Overexpression of CENP-A and its homologs lead to mislocalization to non-centromeric chromatin and contributes to chromosomal instability (CIN) in budding and fission yeasts, flies, and humans (12–18). Overexpression and mislocalization of CENP-A is observed in many cancers, which correlates with poor patient survival and increased risk of disease progression (19–26). Hence, characterization of pathways that prevent mislocalization of CENP-A will provide better diagnosis and treatment of CENP-A overexpressing cancers.
Studies with S. cerevisiae have identified pathways that prevent or promote mislocalization of Cse4. For example, post-translational modifications (PTMs) of Cse4 such as ubiquitination, sumoylation, and isomerization regulate steady-state levels of Cse4 and prevent its mislocalization to non-centromeric regions, thereby maintaining chromosomal stability (12,27–33). Ubiquitin-mediated proteolysis of Cse4 initiated by E3 ubiquitin ligases such as Psh1 (27,28), Slx5 (30,32,34), Ubr1 (32), SCF-Rcy1 (31) and SCF-Met30/Cdc4 (33) and by proline isomerase Fpr3 (29) regulate the cellular levels of Cse4. Psh1-mediated proteolysis of overexpressed Cse4 is regulated by several factors such as FACT (Facilitates Chromatin Transcription/Transactions) complex (35), CK2 (casein kinase 2) (36), HIR histone chaperone complex (37), and DDK (Dbf1-dependent kinase) complex (38). Mutants for genes that prevent mislocalization of Cse4 exhibit synthetic dosage lethality (SDL) when Cse4 is overexpressed from a GAL promoter (GAL-CSE4).
In addition to factors that prevent mislocalization of Cse4, defining pathways that promote mislocalization of Cse4 is also an area of active research. For example, evolutionarily conserved replication dependent Chromatin Assembly Factor 1 (CAF-1) promotes mislocalization of Cse4 to non-centromeric chromatin (39). We have recently shown that the interaction of overexpressed Cse4 with histone H4 facilitates the C-terminal sumoylation of Cse4, which promotes its interaction with CAF-1 and the deposition of Cse4 into non-centromeric chromatin (40,41). Mutants for genes that promote mislocalization of Cse4 suppress the SDL phenotype of psh1Δ GAL-CSE4 strain.
Despite advances in the characterization of factors that prevent or promote mislocalization of Cse4, the mechanism by which mislocalized Cse4 is removed from non-centromeric chromatin and targeted for degradation has not been clearly defined. We reasoned that mutants defective in removal of mislocalized Cse4 from non-centromeric chromatin may exhibit SDL with GAL-CSE4. Our previous genome-wide screen identified mutants that exhibit SDL with GAL-CSE4; among these, Psh1, HIR complex, DDK, SCF-Met30 and SCF-Cdc4 were top hits, which we have pursued in previous studies (33,37,38). In this study, we characterized the role of Cdc48, also identified from the screen. Cdc48 is referred to as p97/VCP (valosin-containing protein) in metazoans and is a highly conserved chaperone of the AAA (ATPase associated with diverse cellular activities)-ATPase family (42,43). Cdc48 plays an essential role in cellular processes such as endoplasmic reticulum-associated degradation (ERAD), spindle disassembly, membrane fusion, autophagy, and transcriptional control (42–46). Mechanistically, Cdc48/p97 acts as a ubiquitin-dependent segregase that extracts polyubiquitinated clients from macromolecular complexes and delivers them to the 26S proteasome for degradation (47,48). The cellular function and localization of the Cdc48/p97 ATPase is controlled by many cofactors (44,49,50); the best-characterized of which is the Ufd1–Npl4 heterodimer. Polyubiquitinated proteins can be directly targeted to the proteasome, whereas polyubiquitinated proteins that are embedded into cell membranes, chromatin-associated, or protein multi-subunit complexes often need to be segregated by the Cdc48Ufd1/Npl4 complex (43,45,46). Npl4 is responsible for the recognition of the substrate-attached ubiquitin chain in the initial step (51,52).
Here, we show that Cdc48Ufd1/Npl4 segregase is necessary for the removal of mislocalized Cse4 from non-centromeric regions. Loss-of-function mutants of CDC48, UFD1 and NPL4 show mislocalization of Cse4 to non-centromeric regions, accumulation of polyubiquitinated Cse4 in chromatin, and an enrichment of chromatin-bound Cse4, expressed from its endogenous promoter. Furthermore, Npl4, which is also enriched in chromatin, interacts with chromatin-bound Cse4 facilitated by the attached polyubiquitin chain. Taken together, we define a role for Cdc48Ufd1/Npl4 segregase in removing mislocalized Cse4 from non-centromeric regions and targeting Cse4 for degradation under normal physiological conditions.
MATERIALS AND METHODS
Yeast strains, plasmids and methods
Supplementary Table S1 and S2 describe the genotype of yeast strains and plasmids used for this study, respectively. Gene deletions and epitope-tagged alleles were constructed at the endogenous loci using standard PCR-based integration (53). All epitope tagging was confirmed by Western blot analysis.
Yeast cells were grown in rich media (YPD: 1% yeast extract, 2% bacto-peptone, 2% glucose) or synthetic complete (SC) media containing 2% glucose, 2% galactose or 2% sucrose + 2% galactose. For cell cycle assays, logarithmically growing cells in YPD at 25°C were treated with α-factor (3 μM) for G1 phase, hydroxyurea (0.2 M) for S phase, and nocodazole (20 μg/ml) for M phase arrests. Cell cycle arrest was confirmed by fluorescence-activated cell sorting (FACS) and microscopic analyses as described previously (54,55). To induce degradation of Cdc48 fused to the auxin-inducible degron, we added 3-indoleacetic acid (Sigma-Aldrich, I3750) to the medium at a final concentration of 5 mM. Chromosome spreads, protein stability assay, and plasmid retention assay were performed as described previously (33).
Ubiquitin (Ub) pull-down assay using whole cell extracts
Levels of ubiquitinated endogenous Cse4 were determined with ubiquitin pull-down assay as described previously (56) with some modifications. Cell lysates were prepared from 50 ml culture of logarithmically growing cells in YPD at 25°C. Cells were pelleted, rinsed with sterile water, and suspended in 0.5 ml of Ub lysis buffer (20 mM Na2HPO4, 20 mM NaH2PO4, 50 mM NaF, 5 mM tetra-sodium pyrophosphate, 10 mM beta-glycerophosphate, 2 mM EDTA, 1 mM DTT, 1% NP-40, 5 mM N-ethylmaleimide, 1 mM PMSF, and protease inhibitor cocktail (Sigma-Aldrich, P8215)). Cells were homogenized with Matrix C (MP Biomedicals) using a bead beater (MP Biomedicals, FastPrep-24 5G). Cell lysates were clarified by centrifugation at 6000 rpm for 5 min and protein concentration was determined using a DC protein assay kit (Bio-Rad). Samples containing equal amounts of protein were brought to a total volume of 1 ml with lysis buffer (50 μl of aliquot was saved as input). The equal concentration of lysates was incubated with tandem ubiquitin binding entities (Agarose-TUBE1, Life Sensors, Inc., UM401) overnight at 4°C. Proteins bound to the beads were washed three times with TBS-T at room temperature and eluted in 2× Laemmli buffer at 100°C for 10 min. The eluted protein was resolved on a 4–12% Bis–Tris gel (Novex, NP0322BOX) and ubiquitinated Cse4 was detected by western blot using anti-HA (12CA5) antibody (Roche, 11583816001). Input samples were analyzed using anti-HA (12CA5) and anti-Tub2 (Basrai laboratory) antibodies.
Co-immunoprecipitation (Co-IP) and Ub pull-down assay using solubilized chromatin lysates
Cells were grown to logarithmic phase of growth in YPD at 25°C. Subcellular fractionation was performed using 50 OD600 cells as described previously (15) with minor modifications. Whole cell extracts (WCEs) were prepared from lysates before the sucrose gradient centrifugation. Chromatin pellets were resuspended in IP lysis buffer (50 mM Tris–HCl at pH 8.0, 5 mM EDTA, 1% Triton X-100, 150 mM NaCl, 50 mM NaF, 10 mM beta-glycerophosphate, 1 mM PMSF, 1× protease inhibitor cocktail) for Co-IP or in Ub lysis buffer for Ub pull-down assay, and bead beat with Matrix C (MP Biomedicals) using a bead beater (MP Biomedicals, FastPrep-24 5G) for 40 s. Chromatin lysates were clarified by centrifugation at 6000 rpm for 5 min. For Co-IP, equal amounts of chromatin lysates were incubated with anti-HA agarose (Sigma-Aldrich, A2095) overnight at 4°C. After washing with IP lysis buffer three times, beads were incubated with 2× Laemmli buffer at 100°C for 5 min. For Ub pull-down assay, equal amounts of chromatin lysates were incubated with tandem ubiquitin binding entities (Agarose-TUBE1, Life Sensors, Inc., UM401) overnight at 4°C. After washing with TBS-T three times, beads were incubated with 2× Laemmli buffer at 100°C for 10 min. Protein samples were analyzed by western blot. Primary antibodies were as follows: anti-HA (12CA5) mouse (Roche, 11583816001), anti-HA rabbit (Sigma-Aldrich, H6908), anti-Pgk1 mouse (Invitrogen, 459250), anti-H2B rabbit (Abcam, ab1790), anti-c-Myc (9E10) mouse (Sigma-Aldrich, M4439; Covance, MMS-150P), anti-Cdc48 rabbit (AS ONE International, Inc., 62-303) and anti-Cse4 rabbit (Strahl laboratory). Protein levels were quantified using Image Lab software (version 6.0.0) from Bio-Rad Laboratories, Inc (Hercules, CA).
ChIP-qPCR
Chromatin immunoprecipitations were performed as previously described (41) with modifications. Briefly, logarithmic phase cultures (120–150 OD600) were grown in sucrose/galactose (2% final concentration each) media for 3–3.5 h and were treated with formaldehyde (1%) for 20 min at 30°C followed by the addition of 2.5 M glycine for 5 min. Cell pellets were washed twice in 1× PBS then resuspended in 1.5 ml FA Lysis Buffer (1 mM EDTA pH8.0, 50 mM HEPES–KOH pH7.5, 140 mM NaCl, 0.1% sodium deoxycholate, 1% Triton X-100) with protease inhibitor cocktail (Sigma) and PMSF (1 mM). The cell suspension was split into three Lysing Matrix C tubes (MP Biomedicals) and lysed in a FastPrep-24 5G (MP Biomedicals) for 40 s ten times with rest on ice between every three consecutive beads-beating. The crude chromatin pellet was washed in FA Lysis Buffer twice. Each pellet was resuspended in 700 μl of FA Lysis Buffer and combined into one 5 ml tube. The chromatin suspension was sonicated on ice with a Branson digital sonifer 24 times at 20% amplitude with a repeated 15 s on/off cycle. After 10 min of centrifugation (12 000 rpm, 4°C), the supernatant was transferred to a new tube. 50 μl of the resulting solubilized chromatin was taken as input. The remaining solubilized chromatin (600 μl each) was incubated with anti-HA-agarose beads (Sigma, A2095), anti-Myc (Sigma M4439) bound or anti-GST (Invitrogen MA4-004) bound protein G magnetic beads (Invitrogen 10004D) overnight at 4–8°C. The beads were washed in 1 ml FA, FA-HS (500 mM NaCl), RIPA, and TE buffers for 5 min on a rotor two times each. The beads were suspended in ChIP Elution Buffer (25 mM Tris–HCl pH7.6, 100 mM NaCl, 0.5% SDS) and incubated at 65°C overnight. The beads were treated with proteinase K (0.5 mg/ml) and incubated at 50°C for 2 h followed by phenol/chloroform extraction and ethanol precipitation. The DNA pellet was resuspended in a total of 100 μl sterile water. Samples were analyzed by quantitative PCR (qPCR) performed with the 7500 Fast Real Time PCR System with Fast SYBR Green Master Mix (Applied Biosystems). The occupancy of the respective protein was calculated as % input. As indicated, two to three biological replicates were performed for each strain tested. Primers used for this study are listed in Supplementary Table S3.
RESULTS
Cdc48Ufd1/Npl4 segregase complex mutants exhibit synthetic dosage lethality (SDL) with GAL-CSE4 and show mislocalization of endogenously expressed Cse4
Mutations or deletions of genes that prevent mislocalization of Cse4 manifest synthetic dosage lethality (SDL) (27,28,30,33,35–38). We have previously used synthetic genetic arrays (SGA) in a genome-wide effort to identify pathways that prevent mislocalization of Cse4 (33,37). The SGA screen for SDL with GAL-CSE4 with essential gene mutants identified cdc48-3 mutant, which has two missense substitutions (P257L and R387K) in the D1 ATPase domain (Supplementary Figure S1A, (47)), as a strong genetic interactor (33). To confirm the SDL phenotype, we examined the growth of cdc48-3 strain with vector or GAL-CSE4 plasmid on plates containing glucose or galactose. The cdc48-3 strain exhibits SDL with GAL-CSE4 on galactose plates at the permissive temperature of 25°C (Supplementary Figure S1B). To test the allele specificity of the SDL phenotype, we examined growth of other cdc48 mutants (cdc48-1 and cdc48-4601). Sequence analysis of cdc48 alleles identified the mutations for each allele (Supplementary Figure S1A). Surprisingly, the two cdc48-1 strains from independent sources showed additional six mutations in YMB8734 when compared to YMB8735. Growth assays showed that all cdc48 mutants we examined exhibit SDL with GAL-CSE4 (Supplementary Figure S1B), suggesting that the GAL-CSE4 SDL phenotype is not specific to a particular allele of cdc48. Previous studies have shown that defects of Cse4 proteolysis contribute to GAL-CSE4-mediated SDL (27,28,30–33,38,56). To define a physiological role for Cdc48 in preventing mislocalization of Cse4, we examined the stability of endogenously expressed HA-Cse4 from its own promoter at the permissive temperature of 25°C. Protein stability assays showed that endogenous HA-Cse4 was rapidly degraded in the wild-type strain (t1/2 = 78 min) and was stabilized in the cdc48-3 strain (t1/2 = 127 min) (Supplementary Figures S1C and D). Thus, we conclude that defects in Cdc48 leads to GAL-CSE4 SDL and Cdc48 regulates proteolysis of Cse4 under normal physiological conditions in vivo.
Cdc48 cofactors Ufd1-Npl4 were previously shown to be required for degradation of Cdc48 substrates in the endoplasmic reticulum and for CMG helicase disassembly during DNA replication termination (42,57,58). We sought to examine whether UFD1 and NPL4 mutants exhibit SDL with Cse4 overexpression. Since npl4-1 mutant was not present on the array of temperature sensitive mutants used for the SGA screen, we decided to use an isogenic set of strains with wild-type, cdc48-3, ufd1-2 and npl4-1 for our analysis. Growth assays showed that ufd1-2 and npl4-1 strains also exhibit SDL with GAL-CSE4 on galactose plates at 25°C similar to that observed for the cdc48-3 strain (Figure 1A); albeit the growth defect was moderate in ufd1-2 strain, likely due to the weaker defect associated with the ufd1-2 allele at the permissive temperature. The SDL phenotypes are specific to GAL-CSE4, since we did not observe growth defects with overexpressed histone H3 or H4 (GAL-H3 or GAL-H4) in cdc48-3, ufd1-2, or npl4-1 strain (Supplementary Figure S2). The GAL-CSE4 dosage lethality is linked to mutations in the cdc48-3, ufd1-2 or npl4-1 strain as lethality and temperature sensitivity were suppressed by expressing the cognate wild-type gene in these mutants (Figure 1B).
Figure 1.

Cdc48Ufd1/Npl4 segregase complex mutants exhibit SDL with GAL-CSE4 and show mislocalization of endogenous Cse4. (A) Overexpression of Cse4 results in SDL in cdc48-3, ufd1-2, and npl4-1 mutants. Wild-type, cdc48-3, ufd1-2 and npl4-1 cells containing either vector or GAL-CSE4 were spotted in five-fold serial dilutions on glucose (2%)- or galactose (2%)-containing synthetic medium selective for the plasmid. The plates were incubated at the permissive temperature of 25°C for 4 days. Three independent transformants for each strain are shown. (B) Plasmid (2μ)-borne CDC48, UFD1, and NPL4 can complement the GAL-CSE4 SDL phenotypes of cdc48-3, ufd1-2, and npl4-1 strains, respectively. Serial dilutions were conducted as described in A. The plates were incubated at 25°C and 37°C for 4–7 days. (C) Endogenous Cse4 is mislocalized in cdc48-3, ufd1-2 and npl4-1 strains. Localization of Cse4 was examined by chromosome spreads prepared from nocodazole (M) arrested wild-type, cdc48-3, ufd1-2 and npl4-1 cells. The cell cycle arrest was verified by FACS analysis (Supplementary Figure S3). DAPI (blue) and α-HA (red) staining were used to visualize DNA and Cse4 localization, respectively. Representative images of cells showing normal localization (counted as nuclei with one or two Cse4 foci) and mislocalization (counted as nuclei with more than two foci or a diffuse signal in the nucleus). (D) Quantification of Cse4 mislocalization as a percentage over total cell count. Error bars represent the standard deviation of multiple experiments. The increased mislocalization observed in the cdc48-3, ufd1-2 and npl4-1 strains was highly significant (one-way ANOVA, numbers of cells scored (n) = 627, 409, 418, 507, n = 503, 429, 340, 406, n = 349, 326, 555 and n = 330, 347, 448 for wild-type, cdc48-3, ufd1-2 and npl4-1, respectively in three or four independent experiments).
Previous studies have shown that the dosage lethality with GAL-CSE4 correlates with mislocalization of Cse4 to non-centromeric regions (39–41). In order to define the physiological role of Cdc48Ufd1/Npl4 segregase, we pursued all further studies with endogenous HA-Cse4 expressed from its own promoter grown at the permissive temperature of 25°C. Chromosome spreads were used to examine the localization of Cse4 as this technique removes soluble material and allows visualization of chromatin bound HA-Cse4. Wild-type, cdc48-3, ufd1-2 or npl4-1 cells were arrested in M phase with nocodazole to compare Cse4 localization in all strains in the same cell cycle stage (Figure 1C). Cell cycle arrest was confirmed by flow cytometry (Supplementary Figure S3). Consistent with clustering of kinetochores, immunofluorescence staining of HA-Cse4 showed one or two Cse4 foci coincident with DAPI (DNA) signal in the majority of wild-type cells (Figure 1C). Mislocalization of Cse4 was defined as cells showing more than two Cse4 foci or diffused Cse4 signals within DAPI stained region (Figure 1C). The percentage of cdc48-3, ufd1-2 or npl4-1 cells exhibiting Cse4 mislocalization was significantly higher than that observed in wild-type cells, confirming that Cdc48Ufd1/Npl4 activity is required to prevent Cse4 mislocalization (Figure 1D). Taken together, we conclude that Cdc48Ufd1/Npl4 complex mutants are hypersensitive to overexpression of Cse4 and show mislocalization of Cse4 under normal physiological conditions.
Chromatin-associated Cse4 contributes to SDL in the cdc48-3 strain
The ubiquitin ligase Psh1 constitutes one of the several Cse4 degradation pathways. Previous studies have shown that GAL-CSE4 dosage lethality in psh1Δ mutants is linked to enrichment of Cse4 in chromatin (27,28). If a similar mechanism applies to cdc48 mutants, we expected that preventing Cse4 mislocalization would suppress dosage lethality. To address this, we took advantage of our recent studies in which either reduced gene dosage of histone H4 or expression of cse4 K215/216R mutant (defective in C-terminal sumoylation) prevent mislocalization of overexpressed Cse4 and suppress dosage lethality of a psh1Δ GAL-CSE4 strain (40,41). We examined dosage lethality phenotypes of cdc48-3 mutants with and without deletion of one copy of H4 alleles (hhf1Δ or hhf2Δ) and observed suppression of synthetic dosage lethality (Figure 2A and Supplementary Figure S4). Moreover, overexpression of the sumoylation deficient Cse4 K215/216R also partially suppressed dosage lethality in the cdc48-3 mutants (Figure 2B and Supplementary Figure S5), similar to that observed in the psh1Δ GAL-cse4 K215/216R strain (40).
Figure 2.

Chromatin-bound Cse4 contributes to SDL. (A) Reduced dosage of histone H4 (hhf1Δ or hhf2Δ) suppresses the cdc48-3 GAL-CSE4 SDL. The indicated strains containing either vector or GAL-CSE4 were spotted in five-fold serial dilutions on glucose (2%)- or galactose (2%)-containing synthetic medium selective for the plasmid. The plates were incubated at 25°C for 4 days. (B) Overexpression of cse4 K215/216R mutant exhibits reduced SDL compared to GAL-CSE4 in cdc48-3 strain. Growth assay was conducted as described in A. The plates were incubated at 25°C for 5 days. (C) Chromatin-bound endogenous Cse4 is enriched in cdc48-3 and npl4-1 strains. Whole cell extracts (WCEs) prepared from equal numbers of logarithmically growing cells in YPD were fractionated into soluble and chromatin fractions. Cse4 levels in each fraction were monitored by Western blot analysis with anti-HA antibody. Pgk1 and histone H2B were used as markers for soluble and chromatin fractions, respectively.
Next, we examined if the severity of GAL-CSE4 SDL correlates with enrichment of Cse4 in chromatin. Subcellular fractionation was done using wild-type, cdc48-3, ufd1-2, and npl4-1 strains expressing endogenous HA-Cse4 grown at 25°C. Pgk1 and histone H2B served as controls for the soluble and the chromatin fractions, respectively. Levels of Cse4 in the soluble fraction were similar in all strains (Figure 2C and Supplementary Figure S6). In contrast, levels of chromatin-associated Cse4 in cdc48-3 and npl4-1 are significantly higher when compared to the wild-type strain. The level of chromatin-associated Cse4 is less pronounced in the ufd1-2 strain (Figure 2C and Supplementary Figure S6), consistent with the milder dosage lethality phenotype of the ufd1-2 GAL-CSE4 strain (Figure 1A). Taken together, these data suggest that increased chromatin-associated Cse4 causes dosage lethality in the cdc48-3 GAL-CSE4 strain.
Accumulation of polyubiquitinated Cse4 in cdc48-3, ufd1-2 and npl4-1 mutants
Previous studies have shown that defects in ubiquitin-mediated Cse4 proteolysis contribute to GAL-CSE4 dosage lethality in psh1Δ, doa1Δ and cdc7-7 strains and this phenotype can be suppressed by overexpression of UBI4 (encodes ubiquitin) presumably by driving Cse4 ubiquitination (38,56). To determine if defects in ubiquitination of Cse4 contribute to SDL in the cdc48-3 GAL-CSE4 strain, we examined the effect of UBI4 overexpression in the cdc48-3 GAL-CSE4 strain. In contrast to mutations that reduce Cse4 ubiquitination, the dosage lethality of cdc48-3 GAL-CSE4 was not suppressed by UBI4 overexpression (Supplementary Figure S7). This result suggests that Cdc48Ufd1/Npl4 is required for steps after the ubiquitination of Cse4.
The Cdc48Ufd1/Npl4 complex is an important factor in recognizing lysine 48-linked polyubiquitinated substrates for processing and delivery to the proteasomal degradation pathway (51). Tetraubiquitin is the minimum signal for efficient proteasomal targeting of the ubiquitinated substrate for degradation (59,60). Accordingly, a post-ubiquitination role for Cdc48Ufd1/Npl4 predicts an accumulation of polyubiquitinated Cse4 with more than four ubiquitin moieties in cdc48-3, ufd1-2 and npl4-1 mutants. To determine the status of Cse4 ubiquitination in vivo, we performed an affinity pull-down assay using agarose with tandem ubiquitin-binding entities (Ub+) from wild-type, cdc48-3, ufd1-2 and npl4-1 strains expressing endogenous HA-Cse4. Polyubiquitinated Cse4 corresponding to higher molecular weight species (>53 kDa) accumulated in cdc48-3 and npl4-1 mutants, and to a lesser extent in the ufd1-2 mutant, confirming that Cdc48Ufd1/Npl4 is dispensable for Cse4 polyubiquitination, but required for the processing of polyubiquitinated Cse4 (Figure 3A and Supplementary Figure S8A). The more modest effect observed in the ufd1-2 mutant is consistent with the lower phenotypic penetrance of this mutant at 25°C, as compared to cdc48-3 and npl4-1 (Figure 1A). A faster migrating species was also observed in all strains expressing HA-Cse4 (Figure 3A and Supplementary Figure S8A, asterisk), as reported previously (30,34,56). Since these faster migrating Cse4 species were similar in size to that in the input lanes, these represent unmodified HA-Cse4, which likely interacts with ubiquitinated proteins such as canonical histones. These results support a role of Cdc48Ufd1/Npl4 in the processing of polyubiquitinated Cse4; thus polyubiquitination of endogenous Cse4 is barely detectable in wild-type cells.
Figure 3.

Polyubiquitinated Cse4 is enriched in cdc48-3, ufd1-2, and npl4-1 mutants under normal physiological conditions. (A) Levels of Cse4 polyubiquitination, which are not specific to any cell cycle stage, are enhanced in cdc48-3, ufd1-2 and npl4-1 strains. Ub pull-down assay was performed using protein extracts from logarithmically growing cells, G1 phase cells synchronized with α-factor, S phase cells synchronized with hydroxyurea (HU), and M phase cells synchronized with nocodazole (Noc), in YPD at 25°C. The cell cycle arrest was verified by FACS analysis and cell morphology using propidium iodide-stained cells (Supplementary Figure S9). Input and ubiquitin pull down samples were analyzed using anti-HA (Cse4) and anti-Tub2 antibodies. Untagged Cse4 was used as a negative control. Asterisk shows nonmodified Cse4. (B) Levels of Cse4 polyubiquitination are enhanced by inactivation of Cdc48. Control (CDC48) and cdc48-aid strains were grown in YPD to logarithmic phase at 25°C, then treated with auxin at the indicated time point. For M phase arrested cells, nocodazole (Noc) was added to the media after 2 h auxin treatment. Ub pull-down assay was conducted as described in (A).
All our studies so far were done at the permissive temperature of 25°C to avoid cell cycle position effects, because complete inactivation of cdc48-3 at the non-permissive temperature of 37°C shows cell cycle arrest in metaphase due to defects in bipolar attachment of kinetochores to microtubules (61). Nevertheless, to rule out a possible indirect effect of cell cycle arrest on polyubiquitination of Cse4, we performed ubiquitin pull-down assays in wild-type and cdc48-3 strains that were arrested in G1 (α-factor treatment), S (hydroxyurea treatment), or M phase (nocodazole treatment). Flow cytometry and nuclear morphology confirmed the cell cycle arrest of the strain (Supplementary Figure S9). The levels of polyubiquitinated Cse4 in wild-type strain (>53 kDa) are barely detectable in any stage of cell cycle (Figure 3A and Supplementary Figure S8B). In contrast, levels of polyubiquitinated Cse4 (>53 kDa) were much higher in the cdc48-3 strain in G1, S and M phases. Hence, accumulation of polyubiquitinated Cse4 in cdc48-3 strain is not due to defects in cell cycle.
We next examined if the accumulation of polyubiquitinated Cse4 in cdc48-3 mutant is allele specific using an auxin-inducible degron for Cdc48, as described previously (57). Treatment with auxin for 2 h led to efficient depletion of Cdc48 in the cdc48-aid strain (Supplementary Figures S10A and B). Ubiquitin pull-down assays were done with logarithmically grown and M phase arrested cells with and without auxin treatment. Flow cytometry confirmed the cell cycle arrest of the cells (Supplementary Figure S11). The cdc48-aid cells show higher levels of Cse4 when compared to the control wild type CDC48 cells, suggesting a defect in Cse4 degradation (Supplementary Figures S10A, C and D). More importantly, we observed higher levels of polyubiquitinated Cse4 (>53 kDa) in logarithmically grown (–Noc) and M phase arrested (+Noc) cdc48-aid cells when compared to the control CDC48 cells and non-auxin treated cdc48-aid cells (Figure 3B and Supplementary Figure S12). Taken together, we conclude that accumulation of polyubiquitinated Cse4 in cdc48-3 strain is cell cycle independent and due to the defect in Cdc48 activity rather than an allele specific effect.
Chromatin-associated Cse4 accumulates in a polyubiquitinated form in cdc48-3 strain
Our results so far have shown that defects in the Cdc48Ufd1/Npl4 segregase cause dosage lethality with GAL-CSE4 and leads to enrichment of Cse4 in chromatin as well as accumulation of polyubiquitinated Cse4. Based on these results, we hypothesized that chromatin-associated Cse4 is polyubiquitinated in a cdc48-3 strain. We used Cse4 mutants where Cse4 Y193 was changed to alanine (Cse4 Y193A) or phenylalanine (Cse4 Y193F) to examine a correlation between dosage lethality, chromatin enrichment, and accumulation of polyubiquitinated Cse4 in the cdc48-3 strain. Cse4 Y193 is located at the center of alpha helix 2 and interacts with the alpha helix 2 of H4 (62). Phenylalanine (F) is identical to tyrosine (Y) except for the hydroxyl group present on Y. Interestingly, human CENP-A has a phenylalanine (F101), instead of tyrosine (Y193) (Figure 4A). We previously showed that the cse4 Y193A mutant exhibits a severe defect in Cse4 sumoylation and suppresses the Cse4 synthetic dosage lethality with psh1Δ mutants (41). Similarly, the structural mimic mutant cse4 Y193F, which shows low levels of Cse4 sumoylation, partially suppresses dosage lethality of psh1Δ (41). These results show that sumoylation of Cse4 promotes mislocalization to non-centromeric regions and mislocalized Cse4 contributes to the dosage lethality. Analogous to the effects of Cse4 Y193A/F in the psh1Δ strain (41), GAL-cse4 Y193A does not exhibit dosage lethality, whereas GAL-cse4 Y193F shows moderate lethality in the cdc48-3 strain (Figure 4B and Supplementary Figure S5).
Figure 4.

Chromatin-bound Cse4 is polyubiquitinated under normal physiological condition. (A) Sequence comparison of α2-helix domain of Cse4 and CENP-A. Human CENP-A has F101, instead of Y. (B) The Y193A mutation in Cse4 does not cause SDL in a cdc48-3 GAL-CSE4 strain. Growth assay was conducted using a cdc48-3 strain containing vector, GAL-CSE4, or GAL-cse4 Y193A/F. Five-fold serial dilutions of the indicated strain were plated on glucose (2%)- or galactose (2%)-containing synthetic medium selective for the plasmid. The plates were incubated at 25°C for 4 days. (C) Endogenous Cse4 Y193A exhibits reduced enrichment in chromatin. Whole cell extracts (WCEs) prepared from equal numbers of logarithmically growing cells in YPD were fractionated into soluble and chromatin fractions. Cse4 levels in chromatin fraction were monitored by Western blot analysis with anti-HA antibody. Histone H2B was used as marker for chromatin fraction. (D) Levels of chromatin-bound Cse4 from C were quantified in arbitrary density units after normalization to H2B. Error bars represent average deviation of two biological repeats. Statistical significance was assessed by one-way ANOVA (P = 0.0036) followed by Tukey's multiple comparisons test. (E) Endogenous Cse4 Y193A exhibits reduced Cse4 polyubiquitination. Ub pull-down assay was performed using protein extracts from logarithmically growing cells in YPD at 25°C. Input and ubiquitin pull down samples were analyzed using anti-HA (Cse4) and anti-Tub2 antibodies. Untagged Cse4 was used as a negative control. Asterisk shows nonmodified Cse4.
We next examined the levels of chromatin-associated endogenous Cse4 Y193A/F expressed from its own promoter in cdc48-3 mutants. Consistent with previous experiment (Figure 2C), wild-type Cse4 was enriched in chromatin in cdc48-3 mutant. Lower levels of Cse4 Y193A were present in chromatin (Figure 4C and D). On the other hand, no significant difference in chromatin enrichment was observed for the Cse4 Y193F mutant. Both cse4 Y193A/F alleles complement cse4Δ mutants and do not show temperature sensitivity (Supplementary Figure S13), suggesting that Cse4 Y193A/F is not defective for centromere function. These results show that dosage lethality correlates with enrichment in chromatin; lack of dosage lethality of GAL-Cse4 Y193A correlates with lower levels of this protein in the chromatin in cdc48-3 strain.
We next asked if the cdc48-3 GAL-cse4 Y193A/F dosage lethality phenotype and enrichment in chromatin correlate with levels of polyubiquitinated Cse4 in these strains. As described earlier (Figure 3A), we observed an accumulation of polyubiquitinated Cse4 in the cdc48-3 mutant; this was greatly reduced in cse4 Y193A but not affected in the cse4 Y193F strain (Figure 4E and Supplementary Figure S14). Taken together, our results suggest that Cse4 associated with non-centromeric chromatin is polyubiquitinated and Cdc48 activity is necessary to remove mislocalized, chromatin-associated, polyubiquitinated Cse4.
Npl4 interacts with chromatin-associated Cse4 in a cdc48-3 strain
Our model predicts that the Cdc48Ufd1/Npl4 segregase recognizes polyubiquitinated Cse4 to facilitate its removal from non-centromeric regions. Npl4 binds specifically to lysine 48-linked polyubiquitinated substrate and functions as a cofactor for the Cdc48 ATPase (51,52). We therefore used co-immunoprecipitation (Co-IP) experiments to test whether Npl4 binds chromatin-associated Cse4 in vivo. We reasoned that Npl4-Cse4 interaction may be transient in a wild-type strain due to removal of chromatin-associated Cse4. Hence, in addition to wild-type, we performed Co-IP experiments in cdc48-3 mutant, which we expected to enrich the recognition state because removal of mislocalized Cse4 is prevented by defects in segregase activity. Cse4 was immunopurified from chromatin fractions of cells expressing Npl4-Myc and HA-Cse4 and lysates were analyzed by anti-myc immunoblotting. An enrichment of not only Cse4, but also Npl4 and Cdc48 was observed in the chromatin when Cdc48 activity was reduced in cdc48-3 strains (Figure 5A and B). Importantly, Npl4 co-purified with Cse4 specifically in cdc48-3 mutant where the substrate recognition state is enriched (Figure 5C and Supplementary Figure S15).
Figure 5.

Npl4 interacts with chromatin-bound Cse4 in a cdc48-3 mutant. (A) Endogenous Cse4, Npl4, and Cdc48 are enriched in chromatin of cdc48-3 strain. Chromatin was prepared from equal numbers of logarithmically growing cells. Levels of Cdc48, Npl4, and Cse4 in chromatin were monitored by Western blot analysis with anti-Cdc48, anti-Myc, and anti-HA antibodies, respectively. Histone H2B was used as marker for chromatin fraction. (B) Levels of chromatin-bound Cdc48, Npl4, and Cse4 from A were quantified in arbitrary density units after normalization to H2B. Error bars represent the standard deviation of three biological repeats. Statistical significance was assessed by unpaired t-test. (C) Npl4 interacts with chromatin-bound Cse4. In vivo interaction of Cse4 with Npl4 in chromatin was determined by Co-IP using chromatin lysates. α-Myc and α-HA antibodies were used to detect Npl4 and Cse4 on the Western blot, respectively.
Psh1 contributes to polyubiquitination of endogenous Cse4
E3 ubiquitin ligases such as Psh1 and Slx5 regulate ubiquitin-mediated proteolysis of overexpressed Cse4 (27,28,30–33). Our results for accumulation of polyubiquitinated endogenous Cse4 in cdc48-3 strain prompted us to examine the role of Psh1 and Slx5 in the polyubiquitination of endogenous Cse4, which is then targeted by Cdc48Ufd1/Npl4. Our results showed that deletion of PSH1 almost completely eliminated accumulation of polyubiquitinated endogenous Cse4 in cdc48-3 strain (Figure 6 and Supplementary Figure S16). In contrast, deletion of SLX5 did not show an obvious reduction of polyubiquitinated endogenous Cse4 in cdc48-3 strain (Supplementary Figure S17). We conclude that Psh1-mediated polyubiquitination of Cse4 is subsequently recognized by Cdc48Ufd1/Npl4 segregase for removal from chromatin and targeted for degradation.
Figure 6.

Psh1 contributes to polyubiquitination of endogenous Cse4 in cdc48-3 strain. Ub pull-down assay was performed using protein extracts from logarithmically growing cells in YPD at 25°C. Input and ubiquitin pull down samples were analyzed using anti-HA (Cse4) and anti-Tub2 antibodies. Untagged Cse4 was used as a negative control. Asterisk shows nonmodified Cse4.
Psh1 facilitates the association of Npl4 with mislocalized Cse4
Based on our results thus far, we hypothesize that Psh1 recognizes mislocalized Cse4, ubiquitinates these species at non-centromeric regions, and thereby generates a substrate for Cdc48Ufd1/Npl4 segregase for further processing. To test this hypothesis, we asked if Npl4 localizes to sites of mislocalized Cse4 in a Psh1-dependent manner. Previous studies have identified that promoters of SAP4 and RDS1 and pericentromeric R1 region of CEN3 are enriched for mislocalized Cse4 when it is overexpressed in a psh1Δ mutants (41,63). We examined the ubiquitination status of chromatin-associated Cse4 and the association of Cse4 and Npl4 at the non-centromeric regions in cdc48-3 and cdc48-3 psh1Δ strains overexpressing Cse4. We showed that polyubiquitinated chromatin-associated Cse4 is detected in a cdc48-3 strain and that the polyubiquitinated Cse4 is greatly reduced although not completely abolished in a cdc48-3 psh1Δ strain (Figure 7A and B).
Figure 7.

Psh1 facilitates the association of Npl4 with mislocalized Cse4. (A) Ubiquitinated chromatin-bound Cse4 is reduced in a cdc48-3 psh1Δ strain. Solubilized chromatin lysate was prepared from cdc48-3 GAL-CSE4 or cdc48-3 psh1Δ GAL-CSE4 strains. Cultures were grown in sucrose/galactose (2%) for 4hrs at 25°C to induce expression of Cse4 and Ub pull-down assay was performed using chromatin lysates. Ub pull down samples and chromatin-bound Cse4 were analyzed using anti-HA (Cse4) antibody. Untagged Cse4 was used as a negative control. Asterisk shows nonmodified Cse4. (B) Levels of chromatin-bound Cse4 ubiquitination from A were quantified in arbitrary density units after normalization to chromatin-bound Cse4. Error bars represent the standard deviation of three biological repeats. Statistical significance was assessed by unpaired t-test. (C) Cse4 is mislocalized upon overexpression of Cse4 in a cdc48-3 mutant. ChIP-qPCR was performed on chromatin lysate from cdc48-3 strain transformed with vector (endogenous Cse4) or GAL-CSE4 (overexpressed Cse4). Equal volume of solubilized, crosslinked chromatin from each strain was used for ChIP with anti-HA (Cse4), anti-Myc (Npl4) and anti-GST. qPCR was performed for association with CEN3, pericentromeric R1 region of CEN3, and SAP4 and RDS1 promoter regions. Enrichment of Cse4 is shown as a fold change over endogenous Cse4. Error bars represent standard deviation of the mean of two independent experiments. (D) Chromatin-bound Npl4 was significantly reduced at the promoters of SAP4 and RDS1 in a cdc48-3 psh1Δ strain. ChIP-qPCR was performed on chromatin lysate from cdc48-3 or cdc48-3 psh1Δ strain transformed with GAL-CSE4. Equal volume of solubilized, crosslinked chromatin from each strain was used for ChIP with anti-HA (Cse4) and anti-Myc (Npl4). Occupancy of Npl4 normalized to that of Cse4 was measured. Error bars represent standard deviation of the mean of three independent experiments.
We performed chromatin immunoprecipitation (ChIP)-qPCR experiments to examine if Cse4 is mislocalized to the non-centromeric regions in cdc48-3 strain. An enrichment of Cse4 was observed at the non-centromeric regions SAP4, RSD1 and R1 in a cdc48-3 mutant (Figure 7C). We next examined if association of Npl4 with Cse4 is dependent on Psh1 by comparing the non-centromeric occupancy of Npl4 in cdc48-3 and cdc48-3 psh1Δ strains. Steady state levels of Npl4 were not affected in a cdc48-3 psh1Δ strain (Supplementary Figure S18). Since chromatin-bound Cse4 was increased in a cdc48-3 psh1Δ strain, compared to a cdc48-3 strain (Supplementary Figure S19), we normalized the enrichment of Npl4 to Cse4 occupancy at each region. The ChIP-qPCR results showed that enrichment of Npl4 in the cdc48-3 psh1Δ strain was significantly reduced at SAP4 and RDS1, but not CEN3 and R1 region of CEN3 (Figure 7D). These results support our hypothesis that association of Npl4 to sites of Cse4 mislocalization is facilitated by Psh1-mediated polyubiquitination of Cse4.
We have previously shown that mislocalization of Cse4/CENP-A contributes to CIN in budding yeast and human cells (15,17,18). Hence, we examined if cdc48-3 strain exhibits CIN by measuring the ability of cells to retain a centromere-containing plasmid (pRS416 URA3) after growth in nonselective medium at 25°C (33,41). Plasmid retention was measured as the ratio of the number of colonies grown on SD-Ura versus SD+Ura medium. In wild-type strain, plasmid retention after 10 generations (10G) was about 80% (Supplementary Figure S20). In contrast, we observed reduced plasmid retention after 10 generations in nonselective medium (10G) in cdc48-3 strain. We conclude that Cdc48 contributes to the chromosomal stability. Taken together, our results show that Psh1 facilitates the association of Npl4 with mislocalized Cse4.
DISCUSSION
The incorporation of CENP-A into centromeric chromatin is essential for establishing centromere identity. Overexpressed CENP-A leads to its mislocalization to non-centromeric chromatin and contributes to aneuploidy in budding and fission yeasts, flies, and humans. Mechanisms that remove mislocalized CENP-A and target it for degradation have not been defined. Here, we report that Cdc48Ufd1/Npl4 segregase removes mislocalized Cse4 for degradation under normal physiological conditions in S. cerevisiae. We propose a model (Figure 8) in which Psh1-mediated polyubiquitination of mislocalized Cse4 facilitates the interaction with Cdc48Ufd1/Npl4 segregase via Npl4 thereby promoting the removal of mislocalized Cse4 and targeting it for proteasomal degradation.
Figure 8.

Molecular role of Cdc48Ufd1/Npl4 segregase in removal of mislocalized Cse4 from non-centromeric regions. Defects in several pathways contribute to mislocalization of Cse4 to non-centromeric regions and CIN. We propose that Psh1 contributes to polyubiquitination of mislocalized Cse4 and this facilitates the interaction between polyubiquitinated Cse4 and Cdc48Ufd1/Npl4 segregase complex via Npl4, resulting in the removal of Cse4 from non-centromeric regions for proteasomal degradation.
Cdc48 is an abundant protein and is a member of the ATPase associated with diverse cellular activities (AAA) family that functions to maintain protein homeostasis. The cdc48-3 mutant shows synthetic dosage lethality with overexpressed Cse4. There are many substrates regulated by Cdc48 and hence we examined if the phenotypes of cdc48-3 strain are linked to mislocalization of Cse4. We recently reported that reduced mislocalization of Cse4 contributes to the suppression of lethality of psh1Δ GAL-CSE4 strain with either reduced gene dosage of histone H4 or cse4 K215/216R mutant (40,41). Consistent with these results, we observed that reduced gene dosage of histone H4 or expression of GAL-cse4 K215/216R suppresses the dosage lethality in the cdc48-3 strain. Hence, the dosage lethality of overexpressed Cse4 is due to mislocalization of Cse4 in the cdc48-3 strain. More importantly, our studies with endogenous Cse4 showed an enrichment in chromatin and mislocalization in cdc48-3, ufd1-2, and npl4-1 mutants at the permissive temperature of 25°C. Cdc48 associates with a large number of cofactors in diverse cellular processes, among which Ufd1 and Npl4 together with Cdc48 functions as a segregase. The enrichment in chromatin and mislocalization of endogenous Cse4 in all cdc48-3, ufd1-2 and npl4-1 mutants suggest that the Cse4-related phenotypes are specifically linked to defective segregase activity of Cdc48Ufd1/Npl4. We determined that cdc48-3 strain exhibits a CIN phenotype, however, this may not be solely due to mislocalization of Cse4 as Cdc48 has many other cell cycle regulated substrates (64,65).
Cdc48/p97 along with its cofactors acts as a ubiquitin-dependent segregase that extracts polyubiquitinated substrates from macromolecular complexes and targets them for degradation (44,49,50). Polyubiquitinated endogenous Cse4 is barely detectable in wild-type cells, probably due to its rapid removal by Cdc48Ufd1/Npl4 segregase and subsequent degradation. Studies with S. cerevisiae have identified multiple E3 ubiquitin ligases such as Psh1, Slx5, Ubr1, SCF-Rcy1 and SCF-Met30/Cdc4 (27,28,30–33). Each single mutant shows partial reduction of polyubiquitinated Cse4 when Cse4 is overexpressed. Our results are consistent with these reports as we observed reduced levels of polyubiquitinated chromatin-associated Cse4 in a cdc48-3 psh1Δ GAL-CSE4 strain. We propose that Psh1 is a major E3 ligase that contributes to polyubiquitination of chromatin-associated Cse4 and/or other E3 ligases may be functionally dependent on priming by Psh1 in a cdc48-3 strain. Alternatively, other E3 ligases may contribute to proteolysis of soluble Cse4 when Cse4 is overexpressed. Future studies will provide insights into how multiple E3 ligases and other factors target soluble versus chromatin bound Cse4 to prevent its mislocalization under normal physiological conditions.
Using cse4 Y193A/F mutants as a tool for Cse4 mislocalization, we uncovered a strong correlation between dosage lethality, chromatin enrichment, and accumulation of polyubiquitinated Cse4 in cdc48-3 strain. Both cse4 Y193A and cse4 Y193F alleles complement the cse4Δ strain, suggesting that sumoylation status of Cse4 does not affect haploid growth. These results also provide evidence that the role of Cdc48 segregase is specifically towards chromatin-bound mislocalized Cse4, but not centromeric Cse4. Consistent with the correlation between chromatin enrichment and accumulation of polyubiquitinated Cse4 in cdc48-3 cse4 Y193A/F mutants, we also showed that polyubiquitinated chromatin-associated Cse4 is detected in a cdc48-3 strain. We observed the enrichment of Cse4, Cdc48, and Npl4 in chromatin, the interaction of Npl4 with chromatin-associated Cse4, and the enrichment of Npl4 as well as overexpressed Cse4 at non-centromeric regions in the cdc48-3 mutant. Furthermore, the reduced enrichment of Npl4 at non-centromeric regions in cdc48-3 strain with deletion of PSH1, suggests that Cse4 that is polyubiquitinated by Psh1 is a substrate for Cdc48Ufd1/Npl4 segregase. These data support our model that mislocalized Cse4 is polyubiquitinated in a cdc48-3 strain and define a role for Cdc48Ufd1/Npl4 segregase in the removal of polyubiquitinated mislocalized Cse4 at non-centromeric regions even under normal physiological conditions (Figure 8).
The Cdc48 ATPase often acts upstream of the 26S proteasome. A recent study shows that cofactors Ufd1-Npl4 bind one unfolded ubiquitin and three to four folded ubiquitin molecules for stable binding (66). Cdc48 ultimately releases the unfolded, oligo-ubiquitinated polypeptides. In fact, a minimum of four ubiquitin moieties are required for recognition by the 26S proteasome for degradation (59,60). Our results are consistent with this conclusion as mono- or di- ubiquitination of Cse4 (around 53 kDa) were more abundant in wild-type cells due to rapid removal of polyubiquitinated Cse4 and higher molecular weight polyubiquitinated Cse4 signals (> 53 kDa) are enhanced in cdc48-3 and cdc48-aid strains.
The cdc48-3 mutant protein has two missense substitutions (P257L and R387K) in the D1 ATPase domain (47). It is of interest to examine if the ATPase activity of Cdc48 is important for the removal of mislocalized Cse4. The complementation of cse4Δ by cse4 Y193A/F suggests that polyubiquitination of Cse4 is not essential for centromeric roles of Cse4, and Cdc48 does not target centromeric Cse4 at the point centromeres of S. cerevisiae, which lacks heterochromatin. It is also possible that centromeric Cse4 is protected from Cdc48 activity due to its association with Scm3, similar to the protection of Scm3 associated Cse4 at centromeres from Psh1-mediated ubiquitination (55). It has been proposed that mislocalized Cse4 at non-centromeric regions is not associated with Scm3 and hence better targeted by E3 ligases for Ub proteasome-mediated degradation (67).
Non-proteolytic functions for the Cdc48 segregase have been demonstrated in various systems such as membrane extraction of transcription factors and dissociation of the SCF complex (68–72). Our data suggest that Cdc48Ufd1/Npl4 regulates the removal of Cse4 from non-centromeric chromatin with subsequent delivery to the proteasome for degradation. However, we do not have direct evidence for the involvement of Cdc48 in escorting Cse4 to the proteasome. It is possible that other proteasome shuttling factors such as Rad23, Ddi1, and Dsk2 (42) bind chromatin extracted ubiquitinated Cse4 for delivery.
In summary, we have defined a molecular role for Cdc48Ufd1/Npl4 segregase in the removal of mislocalized Cse4 from non-centromeric chromatin (Figure 8). Our studies expand the role of VCP/p97/CDC48 in the recognition of ubiquitinated substrates and extraction of misfolded proteins from multisubunit complexes or membranes such as ERAD to the regulation of spatial nucleosome composition. VCP/p97/CDC48 is a key regulator and provides the main quality control for soluble, as well as membrane- associated and chromatin-associated proteins. Mutations in p97 have been linked to a number of neurodegenerative diseases such as Alzheimer's and Parkinson's diseases. Overexpression of wild-type p97 is observed in numerous cancers including human melanomas and breast carcinoma, most likely due to the activation of protein degradation pathways in cancer cells (73,74). Inhibitors of p97 have been explored for treatment of various cancers (73,75), however the precise mechanism for their action has not been fully elucidated. We recently reported that mislocalization of overexpressed CENP-A leads to aneuploidy with karyotypic heterogeneity in human cells and xenograft mouse model (18). Our studies are important from a clinical standpoint given the poor prognosis of CENP-A overexpressing cancers. It is of interest to examine the molecular role of p97 as well as cofactors Ufd1 and Npl4 in preventing stable association of mislocalized CENP-A to non-centromeric regions and how defects in this pathway may contribute to aneuploidy in human cancers.
DATA AVAILABILITY
FACS data were deposited with Flow Repository under ID: FR-FCM-Z4RS.
Supplementary Material
ACKNOWLEDGEMENTS
We gratefully acknowledge Charles Boone, Michael Costanzo, Anastasia Baryshnikova, and Chad Myers for the SGA screen, Karim Labib for cdc48-aid strain, Brian D. Strahl for rabbit anti-Cse4 antibody, Kathy McKinnon of the National Cancer Institute Vaccine Branch FACS Core, Prashant K. Mishra of the Basrai laboratory for assistance with FACS analysis, and the members of the Basrai laboratory for helpful discussions and comments on the manuscript.
Contributor Information
Kentaro Ohkuni, Genetics Branch, Center for Cancer Research, National Cancer Institute, National Institutes of Health, Bethesda, MD 20892, USA.
Loran Gliford, Genetics Branch, Center for Cancer Research, National Cancer Institute, National Institutes of Health, Bethesda, MD 20892, USA.
Wei-Chun Au, Genetics Branch, Center for Cancer Research, National Cancer Institute, National Institutes of Health, Bethesda, MD 20892, USA.
Evelyn Suva, Genetics Branch, Center for Cancer Research, National Cancer Institute, National Institutes of Health, Bethesda, MD 20892, USA.
Peter Kaiser, Department of Biological Chemistry, School of Medicine, University of California, Irvine, CA 92697, USA.
Munira A Basrai, Genetics Branch, Center for Cancer Research, National Cancer Institute, National Institutes of Health, Bethesda, MD 20892, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health Intramural Research Program at the National Cancer Institute to M.A.B. and National Institues of Health [R01GM066164 to P.K.]. Funding for open access charge: NIH Intramural Research Program at the National Cancer Institute.
Conflict of interest statement. None declared.
REFERENCES
- 1. Torras-Llort M., Moreno-Moreno O., Azorin F.. Focus on the centre: the role of chromatin on the regulation of centromere identity and function. EMBO J. 2009; 28:2337–2348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Biggins S. The Composition, Functions, and Regulation of the Budding Yeast Kinetochore. Genetics. 2013; 194:817–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. McKinley K.L., Cheeseman I.M.. The molecular basis for centromere identity and function. Nat. Rev. Mol. Cell. Biol. 2016; 17:16–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Camahort R., Li B., Florens L., Swanson S.K., Washburn M.P., Gerton J.L.. Scm3 is essential to recruit the histone H3 variant Cse4 to centromeres and to maintain a functional kinetochore. Mol. Cell. 2007; 26:853–865. [DOI] [PubMed] [Google Scholar]
- 5. Mizuguchi G., Xiao H., Wisniewski J., Smith M.M., Wu C.. Nonhistone Scm3 and histones CenH3-H4 assemble the core of centromere-specific nucleosomes. Cell. 2007; 129:1153–1164. [DOI] [PubMed] [Google Scholar]
- 6. Stoler S., Rogers K., Weitze S., Morey L., Fitzgerald-Hayes M., Baker R.E.. Scm3, an essential Saccharomyces cerevisiae centromere protein required for G2/M progression and Cse4 localization. Proc. Natl. Acad. Sci. U.S.A. 2007; 104:10571–10576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Williams J.S., Hayashi T., Yanagida M., Russell P.. Fission yeast Scm3 mediates stable assembly of Cnp1/CENP-A into centromeric chromatin. Mol. Cell. 2009; 33:287–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Pidoux A.L., Choi E.S., Abbott J.K., Liu X., Kagansky A., Castillo A.G., Hamilton G.L., Richardson W., Rappsilber J., He X.et al.. Fission yeast Scm3: a CENP-A receptor required for integrity of subkinetochore chromatin. Mol. Cell. 2009; 33:299–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Foltz D.R., Jansen L.E., Bailey A.O., Yates J.R. 3rd, Bassett E.A., Wood S., Black B.E., Cleveland D.W.. Centromere-specific assembly of CENP-A nucleosomes is mediated by HJURP. Cell. 2009; 137:472–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Shuaib M., Ouararhni K., Dimitrov S., Hamiche A.. HJURP binds CENP-A via a highly conserved N-terminal domain and mediates its deposition at centromeres. Proc. Natl. Acad. Sci. U.S.A. 2010; 107:1349–1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chen C.C., Dechassa M.L., Bettini E., Ledoux M.B., Belisario C., Huen P., Luger K., Mellone B.G.. CAL1 is the Drosophila CENP-A assembly factor. J. Cell. Biol. 2014; 204:313–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Collins K.A., Furuyama S., Biggins S.. Proteolysis contributes to the exclusive centromere localization of the yeast Cse4/CENP-A histone H3 variant. Curr. Biol. 2004; 14:1968–1972. [DOI] [PubMed] [Google Scholar]
- 13. Heun P., Erhardt S., Blower M.D., Weiss S., Skora A.D., Karpen G.H.. Mislocalization of the Drosophila centromere-specific histone CID promotes formation of functional ectopic kinetochores. Dev. Cell. 2006; 10:303–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Moreno-Moreno O., Torras-Llort M., Azorin F.. Proteolysis restricts localization of CID, the centromere-specific histone H3 variant of Drosophila, to centromeres. Nucleic Acids Res. 2006; 34:6247–6255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Au W.C., Crisp M.J., DeLuca S.Z., Rando O.J., Basrai M.A.. Altered dosage and mislocalization of histone H3 and Cse4p lead to chromosome loss in Saccharomyces cerevisiae. Genetics. 2008; 179:263–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gonzalez M., He H., Dong Q., Sun S., Li F.. Ectopic centromere nucleation by CENP-A in fission yeast. Genetics. 2014; 198:1433–1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Shrestha R.L., Ahn G.S., Staples M.I., Sathyan K.M., Karpova T.S., Foltz D.R., Basrai M.A.. Mislocalization of centromeric histone H3 variant CENP-A contributes to chromosomal instability (CIN) in human cells. Oncotarget. 2017; 8:46781–46800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Shrestha R.L., Rossi A., Wangsa D., Hogan A.K., Zaldana .S., Suva E., Chung Y.J., Sanders C.L., Difilippantonio S., Karpova T.S.et al.. CENP-A overexpression promotes aneuploidy with karyotypic heterogeneity. J. Cell. Biol. 2021; 220:e202007195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tomonaga T., Matsushita K., Yamaguchi S., Oohashi T., Shimada H., Ochiai T., Yoda K., Nomura F.. Overexpression and mistargeting of centromere protein-A in human primary colorectal cancer. Cancer Res. 2003; 63:3511–3516. [PubMed] [Google Scholar]
- 20. Amato A., Schillaci T., Lentini L., Di Leonardo A.. CENPA overexpression promotes genome instability in pRb-depleted human cells. Mol. Cancer. 2009; 8:119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hu Z., Huang G., Sadanandam A., Gu S., Lenburg M.E., Pai M., Bayani N., Blakely E.A., Gray J.W., Mao J.H.. The expression level of HJURP has an independent prognostic impact and predicts the sensitivity to radiotherapy in breast cancer. Breast Cancer Res. 2010; 12:R18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Li Y., Zhu Z., Zhang S., Yu D., Yu H., Liu L., Cao X., Wang L., Gao H., Zhu M.. ShRNA-targeted centromere protein A inhibits hepatocellular carcinoma growth. PLoS One. 2011; 6:e17794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wu Q., Qian Y.M., Zhao X.L., Wang S.M., Feng X.J., Chen X.F., Zhang S.H.. Expression and prognostic significance of centromere protein A in human lung adenocarcinoma. Lung Cancer. 2012; 77:407–414. [DOI] [PubMed] [Google Scholar]
- 24. Lacoste N., Woolfe A., Tachiwana H., Garea A.V., Barth T., Cantaloube S., Kurumizaka H., Imhof A., Almouzni G.. Mislocalization of the centromeric histone variant CenH3/CENP-A in human cells depends on the chaperone DAXX. Mol. Cell. 2014; 53:631–644. [DOI] [PubMed] [Google Scholar]
- 25. Athwal R.K., Walkiewicz M.P., Baek S., Fu S., Bui M., Camps J., Ried T., Sung M.H., Dalal Y.. CENP-A nucleosomes localize to transcription factor hotspots and subtelomeric sites in human cancer cells. Epigenetics Chromatin. 2015; 8:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sun X., Clermont P.L., Jiao W., Helgason C.D., Gout P.W., Wang Y., Qu S.. Elevated expression of the centromere protein-A(CENP-A)-encoding gene as a prognostic and predictive biomarker in human cancers. Int. J. Cancer. 2016; 139:899–907. [DOI] [PubMed] [Google Scholar]
- 27. Hewawasam G., Shivaraju M., Mattingly M., Venkatesh S., Martin-Brown S., Florens L., Workman J.L., Gerton J.L.. Psh1 is an E3 ubiquitin ligase that targets the centromeric histone variant Cse4. Mol. Cell. 2010; 40:444–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ranjitkar P., Press M.O., Yi X., Baker R., MacCoss M.J., Biggins S.. An E3 ubiquitin ligase prevents ectopic localization of the centromeric histone H3 variant via the centromere targeting domain. Mol. Cell. 2010; 40:455–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ohkuni K., Abdulle R., Kitagawa K.. Degradation of centromeric histone H3 variant Cse4 requires the Fpr3 peptidyl-prolyl cis-trans isomerase. Genetics. 2014; 196:1041–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ohkuni K., Takahashi Y., Fulp A., Lawrimore J., Au W.C., Pasupala N., Levy-Myers R., Warren J., Strunnikov A., Baker R.E.et al.. SUMO-Targeted Ubiquitin Ligase (STUbL) Slx5 regulates proteolysis of centromeric histone H3 variant Cse4 and prevents its mislocalization to euchromatin. Mol. Biol. Cell. 2016; 27:1500–1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Cheng H., Bao X., Rao H.. The F-box protein Rcy1 is involved in the degradation of histone H3 variant Cse4 and genome maintenance. J. Biol. Chem. 2016; 291:10372–10377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cheng H., Bao X., Gan X., Luo S., Rao H.. Multiple E3s promote the degradation of histone H3 variant Cse4. Sci. Rep. 2017; 7:8565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Au W.C., Zhang T., Mishra P.K., Eisenstatt J.R., Walker R.L., Ocampo J., Dawson A., Warren J., Costanzo M., Baryshnikova A.et al.. Skp, cullin, F-box (SCF)-Met30 and SCF-Cdc4-mediated proteolysis of CENP-A prevents mislocalization of CENP-A for chromosomal stability in budding yeast. PLoS Genet. 2020; 16:e1008597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ohkuni K., Levy-Myers R., Warren J., Au W.C., Takahashi Y., Baker R.E., Basrai M.A.. N-terminal sumoylation of centromeric histone H3 variant Cse4 regulates its proteolysis to prevent mislocalization to non-centromeric chromatin. G3 (Bethesda). 2018; 8:1215–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Deyter G.M., Biggins S.. The FACT complex interacts with the E3 ubiquitin ligase Psh1 to prevent ectopic localization of CENP-A. Genes Dev. 2014; 28:1815–1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hewawasam G.S., Mattingly M., Venkatesh S., Zhang Y., Florens L., Workman J.L., Gerton J.L.. Phosphorylation by casein kinase 2 facilitates Psh1 protein-assisted degradation of Cse4 protein. J. Biol. Chem. 2014; 289:29297–29309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ciftci-Yilmaz S., Au W.C., Mishra P.K., Eisenstatt J.R., Chang J., Dawson A.R., Zhu I., Rahman M., Bilke S., Costanzo M.et al.. A genome-wide screen reveals a role for the HIR histone chaperone complex in preventing mislocalization of budding yeast CENP-A. Genetics. 2018; 210:203–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Eisenstatt J.R., Boeckmann L., Au W.C., Garcia V., Bursch L., Ocampo J., Costanzo M., Weinreich M., Sclafani R.A., Baryshnikova A.et al.. Dbf4-dependent kinase (DDK)-mediated proteolysis of CENP-A prevents mislocalization of CENP-A in Saccharomyces cerevisiae. G3 (Bethesda). 2020; 10:2057–2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hewawasam G.S., Dhatchinamoorthy K., Mattingly M., Seidel C., Gerton J.L.. Chromatin assembly factor-1 (CAF-1) chaperone regulates Cse4 deposition into chromatin in budding yeast. Nucleic Acids Res. 2018; 46:4440–4455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ohkuni K., Suva E., Au W.C., Walker R.L., Levy-Myers R., Meltzer P.S., Baker R.E., Basrai M.A.. Deposition of centromeric histone H3 variant CENP-A/Cse4 into chromatin is facilitated by its C-terminal sumoylation. Genetics. 2020; 214:839–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Eisenstatt J.R., Ohkuni K., Au W.C., Preston O., Gliford L., Suva E., Costanzo M., Boone C., Basrai M.A.. Reduced gene dosage of histone H4 prevents CENP-A mislocalization and chromosomal instability in Saccharomyces cerevisiae. Genetics. 2021; 218:iyab033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Finley D., Ulrich H.D., Sommer T., Kaiser P.. The ubiquitin-proteasome system of Saccharomyces cerevisiae. Genetics. 2012; 192:319–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Meyer H., Bug M., Bremer S.. Emerging functions of the VCP/p97 AAA-ATPase in the ubiquitin system. Nat. Cell. Biol. 2012; 14:117–123. [DOI] [PubMed] [Google Scholar]
- 44. Franz A., Ackermann L., Hoppe T.. Ring of change: CDC48/p97 drives protein dynamics at chromatin. Front. Genet. 2016; 7:73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bodnar N., Rapoport T.. Toward an understanding of the Cdc48/p97 ATPase. F1000Res. 2017; 6:1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. van den Boom J., Meyer H.. VCP/p97-mediated unfolding as a principle in protein homeostasis and signaling. Mol. Cell. 2018; 69:182–194. [DOI] [PubMed] [Google Scholar]
- 47. Verma R., Oania R., Fang R., Smith G.T., Deshaies R.J.. Cdc48/p97 mediates UV-dependent turnover of RNA Pol II. Mol. Cell. 2011; 41:82–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Raman M., Havens C.G., Walter J.C., Harper J.W.. A genome-wide screen identifies p97 as an essential regulator of DNA damage-dependent CDT1 destruction. Mol. Cell. 2011; 44:72–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Dargemont C., Ossareh-Nazari B.. Cdc48/p97, a key actor in the interplay between autophagy and ubiquitin/proteasome catabolic pathways. Biochim. Biophys. Acta. 2012; 1823:138–144. [DOI] [PubMed] [Google Scholar]
- 50. Hanzelmann P., Schindelin H.. The interplay of cofactor interactions and post-translational modifications in the regulation of the AAA+ ATPase p97. Front. Mol. Biosci. 2017; 4:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Tsuchiya H., Ohtake F., Arai N., Kaiho A., Yasuda S., Tanaka K., Saeki Y.. In vivo ubiquitin linkage-type analysis reveals that the Cdc48-Rad23/Dsk2 axis contributes to K48-linked chain specificity of the proteasome. Mol. Cell. 2017; 66:488–502. [DOI] [PubMed] [Google Scholar]
- 52. Sato Y., Tsuchiya H., Yamagata A., Okatsu K., Tanaka K., Saeki Y., Fukai S.. Structural insights into ubiquitin recognition and Ufd1 interaction of Npl4. Nat. Commun. 2019; 10:5708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Longtine M.S., McKenzie A. 3rd, Demarini D.J., Shah N.G., Wach A., Brachat A., Philippsen P., Pringle J.R.. Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast. 1998; 14:953–961. [DOI] [PubMed] [Google Scholar]
- 54. Mishra P.K., Au W.C., Choy J.S., Kuich P.H., Baker R.E., Foltz D.R., Basrai M.A.. Misregulation of Scm3p/HJURP causes chromosome instability in Saccharomyces cerevisiae and human cells. PLoS Genet. 2011; 7:e1002303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Mishra P.K., Guo J., Dittman L.E., Haase J., Yeh E., Bloom K., Basrai M.A.. Pat1 protects centromere-specific histone H3 variant Cse4 from Psh1-mediated ubiquitination. Mol. Biol. Cell. 2015; 26:2067–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Au W.C., Dawson A.R., Rawson D.W., Taylor S.B., Baker R.E., Basrai M.A.. A novel role of the N-terminus of budding yeast histone H3 variant Cse4 in ubiquitin-mediated proteolysis. Genetics. 2013; 194:513–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Maric M., Maculins T., De Piccoli G., Labib K.. Cdc48 and a ubiquitin ligase drive disassembly of the CMG helicase at the end of DNA replication. Science. 2014; 346:1253596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Maric M., Mukherjee P., Tatham M.H., Hay R., Labib K.. Ufd1-Npl4 recruit Cdc48 for disassembly of ubiquitylated CMG helicase at the end of chromosome replication. Cell. Rep. 2017; 18:3033–3042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Chau V., Tobias J.W., Bachmair A., Marriott D., Ecker D.J., Gonda D.K., Varshavsky A.. A multiubiquitin chain is confined to specific lysine in a targeted short-lived protein. Science. 1989; 243:1576–1583. [DOI] [PubMed] [Google Scholar]
- 60. Thrower J.S., Hoffman L., Rechsteiner M., Pickart C.M.. Recognition of the polyubiquitin proteolytic signal. EMBO J. 2000; 19:94–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Cheng Y.L., Chen R.H.. The AAA-ATPase Cdc48 and cofactor Shp1 promote chromosome bi-orientation by balancing Aurora B activity. J. Cell. Sci. 2010; 123:2025–2034. [DOI] [PubMed] [Google Scholar]
- 62. Zhou Z., Feng H., Zhuou B.-R., Ghirlando R., Hu K., Zwolak A., Miller Jenkins L.M., Xiao H., Tjandra N., Wu C.et al.. Structural basis for recognition of centromere histone variant CenH3 by the chaperone Scm3. Nature. 2011; 472:234–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Hildebrand E.M., Biggins S.. Regulation of budding yeast CENP-A levels prevents misincorporation at promoter nucleosomes and transcriptional defects. PLoS Genet. 2016; 12:e1005930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Meyer H., Popp O.. Role(s) of Cdc48/p97 in mitosis. Biochem. Soc. Trans. 2008; 36:126–130. [DOI] [PubMed] [Google Scholar]
- 65. Buchberger A. Roles of Cdc48 in regulated protein degradation in yeast. Subcell. Biochem. 2013; 66:195–222. [DOI] [PubMed] [Google Scholar]
- 66. Twomey E.C., Ji Z., Wales T.E., Bodnar N.O., Ficarro S.B., Marto J.A., Engen J.R., Rapoport T.A.. Substrate processing by the Cdc48 ATPase complex is initiated by ubiquitin unfolding. Science. 2019; 365:eaax1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Folco H.D., Desai A.. A PSHaver for centromeric histones. Mol. Cell. 2010; 40:351–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Hoppe T., Matuschewski K., Rape M., Schlenker S., Ulrich H.D., Jentsch S.. Activation of a membrane-bound transcription factor by regulated ubiquitin/proteasome-dependent processing. Cell. 2000; 102:577–586. [DOI] [PubMed] [Google Scholar]
- 69. Rape M., Hoppe T., Gorr I., Kalocay M., Richly H., Jentsch S.. Mobilization of processed, membrane-tethered SPT23 transcription factor by CDC48(UFD1/NPL4), a ubiquitin-selective chaperone. Cell. 2001; 107:667–677. [DOI] [PubMed] [Google Scholar]
- 70. Shcherbik N., Haines D.S.. Cdc48p. Npl4p/Ufd1p) binds and segregates membrane-anchored/tethered complexes via a polyubiquitin signal present on the anchors. Mol. Cell. 2007; 25:385–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Yen J.L., Flick K., Papagiannis C.V., Mathur R., Tyrrell A., Ouni I., Kaake R.M., Huang L., Kaiser P.. Signal-induced disassembly of the SCF ubiquitin ligase complex by Cdc48/p97. Mol. Cell. 2012; 48:288–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Lauinger L., Flick K., Yen J.L., Mathur R., Kaiser P.. Cdc48 cofactor Shp1 regulates signal-induced SCF(Met30) disassembly. Proc. Natl. Acad. Sci. U.S.A. 2020; 117:21319–21327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Huryn D.M., Kornfilt D.J.P., Wipf P.. p97: an emerging target for cancer, neurodegenerative diseases, and viral infections. J. Med. Chem. 2020; 63:1892–1907. [DOI] [PubMed] [Google Scholar]
- 74. Tang W.K., Xia D. Mutations in the human AAA(+) chaperone p97 and related diseases. Front. Mol. Biosci. 2016; 3:79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Vekaria P.H., Home T., Weir S., Schoenen F.J., Rao R.. Targeting p97 to disrupt protein homeostasis in cancer. Front. Oncol. 2016; 6:181. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
FACS data were deposited with Flow Repository under ID: FR-FCM-Z4RS.