


Abstract
The mammalian cleavage factor I (CFIm) has been implicated in alternative polyadenylation (APA) in a broad range of contexts, from cancers to learning deficits and parasite infections. To determine how the CFIm expression levels are translated into these diverse phenotypes, we carried out a multi-omics analysis of cell lines in which the CFIm25 (NUDT21) or CFIm68 (CPSF6) subunits were either repressed by siRNA-mediated knockdown or over-expressed from stably integrated constructs. We established that >800 genes undergo coherent APA in response to changes in CFIm levels, and they cluster in distinct functional classes related to protein metabolism. The activity of the ERK pathway traces the CFIm concentration, and explains some of the fluctuations in cell growth and metabolism that are observed upon CFIm perturbations. Furthermore, multiple transcripts encoding proteins from the miRNA pathway are targets of CFIm-dependent APA. This leads to an increased biogenesis and repressive activity of miRNAs at the same time as some 3′ UTRs become shorter and presumably less sensitive to miRNA-mediated repression. Our study provides a first systematic assessment of a core set of APA targets that respond coherently to changes in CFIm protein subunit levels (CFIm25/CFIm68). We describe the elicited signaling pathways downstream of CFIm, which improve our understanding of the key role of CFIm in integrating RNA processing with other cellular activities.
INTRODUCTION
Most human genes have multiple sites where pre-mRNA 3′ end processing can occur to generate alternative transcript isoforms in different cell types and conditions (1). Alternative polyadenylation is a main contributor to the observed transcriptome diversity (2–5). Consistently, data from The Cancer Genome Atlas (TCGA) indicate that APA holds the highest prognostic value among all types of isoform variation in hepatocellular carcinoma (6).
A large class of APA isoforms are those that differ in the length of their 3′ untranslated regions (3′ UTRs). Mammalian cleavage factor I, a 3′ end processing complex conserved in multicellular organisms but absent from yeast (7), is one of the main regulators of 3′ UTR length (8–10). A CFIm tetramer composed of two 25 kDa subunits (CFIm25/CPSF5/NUDT21) and two larger subunits of 59 and/or 68 kDa (CFIm59/CPSF7 and CFIm68/CPSF6) associates with the RNA polymerase II (RNAPII) in the initial stages of transcription (11). Crosslinking and immunoprecipitation revealed that within individual genes, the most prominent peaks of CFIm binding are located in the vicinity of those poly(A) sites (PAS) that are ultimately used for the maturation of the messenger RNA (mRNA), indicating that the interaction of CFIm with high-affinity target sites promotes the 3′ end cleavage (9). These interaction sites are typically located distally in 3′ UTRs, in regions enriched in UGUA motifs (9,12). The Fip1 3′ end processing factor stabilizes the interactions of CFIm with the RNA (12), while the ubiquitination of the PCF11 component of the 3′ end processing complex by an ectopically activated MAGE-A11 ubiquitin ligase in cancer leads to the dissociation of CFIm25 from the complex (13).
The depletion of CFIm25 or CFIm68 subunits of CFIm leads to systematically shortened 3′ UTRs (9,10), whereas CFIm59 does not seem to impact the 3′ UTR length (9,14). The number of reported targets varies between tens to over a thousand among studies (9,10,15,16). The phenotypes observed upon perturbation of CFIm expression have been attributed to growth regulators in glioblastoma (15), chromatin-regulatory factors in somatic cell reprogramming (16), and metabolic enzymes in the activation of hematopoietic stem cells (17). While CFIm has emerged as an important regulator of cell fate decisions in normal and pathological contexts, its involvement in cancers is not fully understood (13,18). Reduced CFIm expression was reported to increase cell proliferation and promote glioblastoma and hepatocellular carcinoma formation (15,19,20), while opposite effects, reduced proliferation and increased apoptosis, have been reported in K562 leukemia cells (21).
To better understand how cells respond to the pervasive remodeling of the RNA pool that follows fluctuations in CFIm levels, we carried out a multi-omics characterization of HEK293 cells in which the CFIm25 and CFIm68 components of the CFIm complex were either stably overexpressed, or transiently repressed via siRNA-mediated knockdown. We demonstrate that hundreds of transcripts undergo reciprocal changes in 3′ UTR length in the knockdown (KD) and overexpression (OE) conditions, changes that are also very consistent between perturbations of CFIm25 and CFIm68. These targets cluster in specific cellular pathways, including stress signaling, cell cycle, RNA processing and miRNA-mediated repression. Many kinase-encoding transcripts undergo CFIm-dependent APA, which led us to globally estimate the changes in kinase activities upon CFIm perturbations from phosphoproteome measurements. Among the kinases whose activity changes we validate here, the stress-related ERK directly traces the CFIm expression, leading to expected changes in cell metabolism and growth. Specifically, real time growth estimates revealed that the ectopic expression of CFIm subunits promotes cellular proliferation, while the siRNA-mediated knockdown reduces growth in multiple cell lines. By regulating the processing of transcripts encoding miRNA pathway proteins, CFIm also modulates the miRNA activity. Interestingly, while the CFIm knockdown results in transcripts with shortened 3′ UTRs that can escape miRNA-dependent regulation, it also activates the biogenesis of miRNAs and their repressive activity on reporter constructs. Our study thus identifies key signaling pathways downstream of CFIm, explaining the impact of this 3′ end processing factor on fundamental cellular processes such as metabolism and growth.
MATERIALS AND METHODS
Cell culture, transfections, treatments and common reagents
For most experiments, wild type HEK293 cells were cultured as described before (22). For the overexpression of CFIm25 and CFIm68, the cDNAs were cloned into pcDNA5/FRT from Invitrogen. These were then stably integrated in the Flp-in recombination site of HEK293 cells (Flp-In™-293 Cell Line #R75007, Invitrogen). For RNAi, HEK293 cells were seeded at a density of 20% in six-well plates and all subsequent steps were done according to the ‘‘forward method’’ from the RNAiMAX protocol (Invitrogen). Following a 48 hr incubation, double-stranded siRNAs (starting from 30 pmol, from Dharmacon and Microsynth) were incubated with Lipofectamine RNAiMAX (Invitrogen) and added to the wells. Cells were harvested after 72 hours for further analysis. Western blotting was performed as described earlier (22). The HRP-labelled secondary antibodies were developed with SuperSignal™ West Pico PLUS Chemiluminescent Substrate (ThermoFisher Scientific #34580) or with SuperSignal™ West Femto Maximum Sensitivity Substrate (ThermoFisher Scientific #34095). LICOR IR680/800nm dye-labelled secondary antibodies were used for multiplexing several antibodies on the same membrane. All western blot images were documented with Azure c600 Gel documentation system equipped with a 8.3 MP CCD camera. Western blot quantifications were performed using the ImageJ software by quantifying the pixels of each band and normalizing against a housekeeping control. For comparison between conditions, all samples were normalised to the average levels of the corresponding control samples. Note that the loading control proteins (GAPDH/ACTIN) are shown multiple times in several figures, whenever a single membrane was re-utilised for staining of multiple candidates. Detailed information regarding antibodies and primers/oligos used for the study are listed in Supplementary Data.
Transcriptome profiling with poly(A)-enriched mRNA-seq
Total RNA was quality-checked on a Bioanalyser instrument (Agilent Technologies, Santa Clara, CA, USA) using the RNA 6000 Nano Chip (Agilent, Cat# 5067–1511) and quantified by spectrophotometry using a NanoDrop ND-1000 Instrument (NanoDrop Technologies, Wilmington, DE, USA). 1μg total RNA was used for library preparation with the TruSeq Stranded mRNA Library Prep Kit High Throughput (Cat# RS-122-2103, Illumina, San Diego, CA, USA). Libraries were quality-checked on the Fragment Analyser (Advanced Analytical, Ames, IA, USA) using the Standard Sensitivity NGS Fragment Analysis Kit (Cat# DNF-473, Advanced Analytical). The average concentration was 128 ± 12 nmol/l. Samples were pooled in equal molarity. Each pool was quantified by PicoGreen fluorometric measurement to be adjusted to 1.8 pM and used for clustering on a NextSeq 500 instrument (Illumina). Samples were sequenced using a NextSeq 500 High Output Kit 75-cycles (Illumina, Cat# FC-404-1005). Primary data analysis was performed with the Illumina RTA version 2.4.11 and base calling software version bcl2fastq-2.20.0.422.
Quantification of gene expression by poly(A)-enriched mRNA-seq
Human protein-coding and lincRNA genes from the Ensembl (23) release 90 annotation were stringently filtered for transcripts whose splice junctions ‘are supported by at least one non-suspect mRNA’ (Ensembl transcript support level 1). To minimize the chance of erroneous estimates of gene expression due to large changes in transcript length by 3′ UTR shortening the 3′-terminal exons of each transcript were discarded. Then, for every gene, we identified those regions that are annotated as belonging to an exon in all of the retained transcripts of that gene. Raw sequencing data in FASTQ format were processed with standard tools: cutadapt (version 1.16) (24) to remove adapters and poly(A)-tails from the reads, and STAR aligner (version 2.7.1a) (25) to map resulting fragments to the genome (assembly version GRCh38 with splice junction annotations derived from Ensembl release 90). The alignments were sorted and indexed with SAMtools (version 1.10) (26) and later used for plotting coverage profiles of distinct gene loci in RNA-seq samples with the Gviz R package (version 1.28.0; R software version 3.6.0) (27). For every gene, all reads with a single best reported alignment (‘unique mappers’) whose alignment start positions overlapped with any of the exclusively ‘exonic’ regions of that gene, prepared as described above, were summed up. The resulting gene count tables for each sequencing library were used as input for differential gene expression analyses with the edgeR (28) package (version 3.34.0; R version 4.1.0). First, genes with low counts were discarded by applying edgeR’s filterByExpr()function with default parameters across samples of all conditions to ensure that the same genes would be called for each comparison. Then, differentially expressed genes were identified in a pairwise manner between treated and control (‘wild type’) libraries, by applying the calcNormFactors(), estimateDisp(), exactTest() and topTags() functions with default parameters, yielding fold changes (log2) and corresponding P values and Benjamini-Hochberg-corrected (29) false discovery rates for every gene and for each comparison. The scripts used for this analysis are available from the github repository https://github.com/zavolanlab/CFI2021.
Quantification of relative PAS expression and average relative terminal exon lengths from poly(A)-enriched RNA-seq data
To quantify the relative usage of distinct poly(A) sites we applied the PAQR tool (30). The values were aggregated at the level of individual terminal exons to obtain the proportion of transcripts ending at individual positions in individual terminal exons. From these values we calculated a weighted average relative terminal exon length as the sum over all 3′ ends in the terminal exon, relative usage of the 3′ end multiplied by the length of the terminal exon ending at the respective site. We obtained quantification for 1′750 terminal exons with multiple poly(A) sites. The PAQR code is available from https://github.com/zavolanlab/PAQR2 and the source code for target identification and analysis from https://github.com/zavolanlab/CFI2021.
Quantification of relative PAS expression and average relative terminal exon lengths from 3′ end sequencing data
To identify targets of CFIm-mediated 3′ end processing based on 3′ end sequencing data, we used poly(A) site quantifications in relevant cellular systems from the PolyASite database (31). Briefly, this database contains 3′ end sequencing data from control and CFIm25/CFIm68-depleted HEK293 cells as well as control and CFIm25/CFIm68-depleted HeLa cells, both obtained with the A-seq method for 3′ end sequencing (32). PolyASite also contains data for HeLa control and CFIm68-depleted samples, generated with the PAPERCLIP method for 3′ end sequencing (33). Based on the ENSEMBL90 gene annotation, we extracted all annotated terminal exons, intersected the quantified poly(A) sites from the PolyASite database in the samples mentioned above, and then carried out the terminal exon length calculation as described in the previous section.
Selection of CFIm targets
We applied Principal Component Analysis (PCA) to per-sample average terminal exon lengths and calculated the projection on, as well as correlation of each terminal exon (treated as a vector in the space of samples) with principal component 1. We then selected those transcripts and genes whose exons exhibited higher than 0.9 correlation and higher than 10 projection scores (both in absolute values) as CFI targets. Almost all (855 of 858) of the transcripts underwent 3′ UTR shortening upon CFIm KD. These were the focus of our study.
Selection of CFIm targets from 3′ end sequencing datasets
We applied a similar analysis to the terminal exon data from the 3′ end sequencing experiments mentioned above. The threshold on the correlation value was set such as to obtain a number of targets similar to that obtained from RNA-seq data. Specifically, the thresholds were 0.9 for HEK293 A-seq data, 0.8 for HeLa A-seq data and 0.95 for HeLa PAPERCLIP data. This yielded 867, 879, and 1071 target transcripts, respectively, for the three datasets.
UGUA frequency analysis
Terminal exons with exactly two poly(A) sites quantified by PAQR were used for the motif frequency analysis. First, we extracted sequences of 401 nucleotides (200 on each side of the PAS) from both proximal and distal PAS in each TE. We traversed each sequence recording the presence/absence of the UGUA motif at each position and then tabulated the counts at each position across all sites. These were plotted using a running average of 30 nucleotides, sliding by 1 nucleotide at a time.
Frequency analysis of UGUA motifs in genes from specific functional categories
From the genes whose terminal exon lengths we quantified with PAQR, we extracted those that were annotated with the Gene Ontology terms ‘Cellular Response to Stress’ and ‘Protein Transport’ (according to the STRING server (34)). We then separated these sets into CFIm targets and non-targets, and then carried out the UGUA motif analysis as described in the previous section.
Global proteome and phosphoproteome analysis by shotgun LC-MS
For each sample, 5 × 106 cells were washed twice with ice-cold 1x phosphate-buffered saline (PBS) and lysed in 100 μl urea lysis buffer (8 M urea (AppliChem), 0.1 M Ammonium Bicarbonate (Sigma), 1x PhosSTOP (Roche)). Samples were vortexed, sonicated at 4°C (Hielscher), shaken for 5 min on a thermomixer (Eppendorf) at room temperature and centrifuged for 20 min at 4°C full speed. Supernatants were collected and protein concentration was measured with BCA Protein Assay kit (Invitrogen). Per sample, a total of 300 μg of protein mass was reduced with tris(2-carboxyethyl)phosphine (TCEP) at a final concentration of 10 mM at 37°C for 1 hour, alkylated with 20 mM chloroacetamide (CAM, Sigma) at 37°C for 30 min and incubated for 4 h with Lys-C endopeptidase (1:200 w/w). After diluting samples with 0.1 M ammonium bicarbonate to a final urea concentration of 1.6 M, proteins were further digested overnight at 37°C with sequencing-grade modified trypsin (Promega) at a protein-to-enzyme ratio of 50:1. Subsequently, peptides were desalted on a C18 Sep-Pak cartridge (VAC 3cc, 500 mg, Waters) according to the manufacturer's instructions, split in peptide aliquots of 200 and 25 μg, dried under vacuum and stored at −80°C until further use.
For proteome profiling, sample aliquots containing 25 μg of dried peptides were subsequently labeled with an isobaric tag (TMT 10-plex, Thermo Fisher Scientific) following a recently established protocol (35). To control for ratio distortion during quantification, a peptide calibration mixture consisting of six digested standard proteins mixed in different amounts were added to each sample before TMT labeling. After pooling the TMT labeled peptide samples, peptides were again desalted on C18 reversed-phase spin columns according to the manufacturer's instructions (Macrospin, Harvard Apparatus) and dried under vacuum. TMT-labeled peptides were fractionated by high-pH reversed phase separation using a XBridge Peptide BEH C18 column (3,5 μm, 130 Å, 1 mm × 150 mm, Waters) on an Agilent 1260 Infinity HPLC system. Peptides were loaded on column in buffer A (ammonium formate (20 mM, pH 10) in water) and eluted using a two-step linear gradient starting from 2% to 10% in 5 min and then to 50% (v/v) buffer B (90% acetonitrile / 10% ammonium formate (20 mM, pH 10) over 55 min at a flow rate of 42 μl/min. Elution of peptides was monitored with a UV detector (215 nm, 254 nm). A total of 36 fractions were collected, pooled into 12 fractions using a post-concatenation strategy as previously described (36), dried under vacuum and subjected to LC–MS/MS analysis.
For phosphoproteome profiling, sample aliquots containing 200 μg of dried peptides were subjected to phosphopeptide enrichment using IMAC cartridges and a BRAVO AssayMAP liquid handling platform (Agilent) as recently described (37).
The setup of the μRPLC-MS system was described previously (35). Chromatographic separation of peptides was carried out using an EASY nano-LC 1000 system (Thermo Fisher Scientific), equipped with a heated RP-HPLC column (75 μm × 30 cm) packed in-house with 1.9 μm C18 resin (Reprosil-AQ Pur, Dr. Maisch). Aliquots of 1 μg total peptides were analysed per LC×MS/MS run using a linear gradient ranging from 95% solvent A (0.15% formic acid, 2% acetonitrile) and 5% solvent B (98% acetonitrile, 2% water, 0.15% formic acid) to 30% solvent B over 90 minutes at a flow rate of 200 nl/min. Mass spectrometry analysis was performed on a Q-Exactive HF mass spectrometer equipped with a nanoelectrospray ion source (both Thermo Fisher Scientific) and a custom made column heater set to 60°C. 3E6 ions were collected for MS1 scans for no >100 ms and analysed at a resolution of 120 000 FWHM (at 200 m/z). MS2 scans were acquired of the 10 most intense precursor ions at a target setting of 100 000 ions, accumulation time of 50 ms, isolation window of 1.1 Th and at resolution of 30 000 FWHM (at 200 m/z) using a normalized collision energy of 35%. For phosphopeptide enriched samples, the isolation window was set to 1.4 Th and a normalized collision energy of 28% was applied. Total cycle time was ∼1–2 s.
For proteome profiling, the raw data files were processed using the Mascot and Scaffold software and TMT reporter ion intensities were extracted. Phosphopeptide enriched samples were analysed by label-free quantification. Therefore, the acquired raw-files were imported into the Progenesis QI software (v2.0, Nonlinear Dynamics Limited), which was used to extract peptide precursor ion intensities across all samples applying the default parameters.
Quantitative analysis results from label-free and TMT quantification were further processed using the SafeQuant R package v.2.3.2. (https://github.com/eahrne/SafeQuant/) to obtain protein relative abundances. This analysis included global data normalization by equalizing the total peak/reporter areas across all LC–MS runs, summation of peak areas per protein and LC–MS/MS run, followed by calculation of protein abundance ratios. Only isoform specific peptide ion signals were considered for quantification. The summarized protein expression values were used for statistical testing of differences in expression of abundant proteins between conditions. Here, empirical Bayes moderated t-tests were applied, as implemented in the limma package (http://bioconductor.org/packages/release/bioc/html/limma.html) of R/Bioconductor. The resulting per protein and condition comparison P-values were adjusted for multiple testing using the Benjamini–Hochberg method.
Inference of kinase activity from phosphoproteome data
We used the Kinase Set Enrichment Analysis (KSEA) as described by Hernandez-Armenta et al. (38) and implemented in the R-package KSEA (https://github.com/evocellnet/ksea) to predict the kinase activity changes across conditions. The software takes as input the log2 fold-change in intensity of each phosphopeptide between two conditions, as well as kinase-substrate associations. It then determines whether the substrates of any of the kinases are enriched among the phosphopeptides with the largest change between conditions, and reports the −log10 of the P-value as a proxy of kinase regulatory activity (38). As only ∼6% of the quantified phosphopeptides in our data set have associated kinases in the PhosphoSitePlus database (39), we used weight matrix models of kinase substrate specificity to predict further associations as follows. Considering all of the peptide sequences 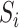 obtained in an experiment, the likelihood of a sequence
obtained in an experiment, the likelihood of a sequence 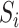 to have a binding site for a kinase
to have a binding site for a kinase 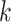 can be written as:
can be written as:
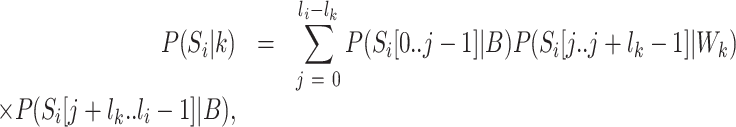 |
where 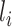 is the length of the peptide,
is the length of the peptide, 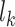 is the length of the weight matrix
is the length of the weight matrix 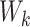 corresponding to kinase
corresponding to kinase 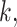 and
and 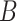 is the background model for the relative occurrence of amino acids (AA) in peptides (here we used the overall frequency of each AA in all peptides in the dataset). We constructed the weight matrices
is the background model for the relative occurrence of amino acids (AA) in peptides (here we used the overall frequency of each AA in all peptides in the dataset). We constructed the weight matrices 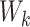 from all known kinase-substrate associations, taking a window of length
from all known kinase-substrate associations, taking a window of length 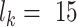 AA (±7 AA around the phospho site) for each kinase
AA (±7 AA around the phospho site) for each kinase 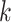 from the PhosphoSitePlus database (39). For completeness, we also included the possibility that the peptide does not correspond to any of the known WMs, i.e. explaining the peptide sequence entirely by the background model,
from the PhosphoSitePlus database (39). For completeness, we also included the possibility that the peptide does not correspond to any of the known WMs, i.e. explaining the peptide sequence entirely by the background model, 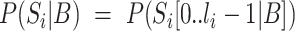 . From Bayes’ theorem, we have that the probability of a phosphorylated peptide
. From Bayes’ theorem, we have that the probability of a phosphorylated peptide 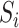 being explained by kinase
being explained by kinase 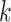 is given by
is given by 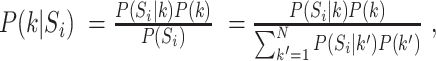 where
where 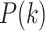 is the prior probability that a randomly selected phosphopeptide from the data is explained by kinase
is the prior probability that a randomly selected phosphopeptide from the data is explained by kinase 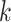 , and N is the number of kinases for which we have sequence specificity information (including the “background"). As we do not have prior information on
, and N is the number of kinases for which we have sequence specificity information (including the “background"). As we do not have prior information on 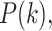 we assumed a uniform distribution, i.e.
we assumed a uniform distribution, i.e. 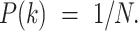 Finally, we have assigned to each phosphopeptide the kinase that had the highest posterior probability of explaining the peptide.
Finally, we have assigned to each phosphopeptide the kinase that had the highest posterior probability of explaining the peptide.
Real time proliferation assay
Cell growth was assayed using the xCELLigence system (RTCA, ACEA Biosciences, San Diego, CA). The background impedance of the xCELLigence system was measured for 12 s using 50 μl of cell culture media at room temperature in each well of an E-plate 16. After reaching 75% confluence, the cells were washed with PBS and detached from the flasks using a short treatment with trypsin/EDTA. Ten thousand cells were dispensed into each well of an E-plate 16. Growth and proliferation were monitored every 15 min up to 48 hrs via the incorporated sensor electrode arrays of the xCELLigence system, using the RTCA-integrated software according to the manufacturer's parameters. For the siRNA treatments, a lower number (3000) of cells were seeded and allowed to grow without interruption for a minimum of 42–48 h before the assay was briefly interrupted for the addition of the siRNA mixes or lipofectamine (for the mock treatment) to the corresponding wells. For the ERK inhibition assays, we used Ravoxertinib hydrochloride (GDC-0994 hydrochloride). This compound was validated as an orally bioavailable, selective inhibitor of ERK kinase activity, with a half-maximal inhibitory concentration (IC50) of 6.1 nM. We used a 10 mM solution in 1 ml of DMSO obtained from Medchem Express (Cat. No.: HY-15947A). The final concentration of the inhibitor used for seeding of cells was 6.1 nM in complete growth media. As control, an equivalent amount of DMSO was added to the cell culture medium. Ten thousand cells were counted from their culture flasks and mixed with Ravoxertinib hydrochloride or DMSO and seeded into the xCelligence plates as per standard protocol. All measurements were done with a minimum of five biological replicates.
qRT-PCR to estimate the abundance of RNAs and miRNAs
For mRNA quantifications, 50 ng of total RNA was used for reverse transcription following the manufacturer's protocol and cycling conditions (High-Capacity cDNA Reverse Transcription Kit, Thermo Fisher Scientific). Subsequently, the RT reaction was diluted 4-fold with water and subjected to q-PCR in a 96-well format, using primers specific to individual genes and GoTaq® qPCR Master Mix (Promega). The incubation and cycling conditions were set as described in the kit and the plates were analysed in a StepOnePlus Real-Time PCR System (Thermo Scientific). GAPDH was used as housekeeping control for relative estimation. Real-time analyses by two-step RT–PCR were carried out to quantify miRNA expression using the Thermo Scientific TaqMan chemistry-based miRNA assay system as performed earlier (40). Briefly, 25 ng of cellular RNA were used along with specific primers for human let7-a (assay ID 000377), miR-92a (assay ID 000431), miR-16 (assay ID 000391) and miR-19b (assay ID 000396). U6 snRNA (assay ID 001973) was used as an endogenous control. One third of the reverse transcription mix was subjected to PCR amplification with TaqMan® Universal PCR Master Mix No AmpErase (Thermo Scientific) and the respective TaqMan® reagents for target miRNA. Samples were analysed in PCR triplicates from at least two biological replicates of each condition, processed independently. The comparative Ct method which included normalization by the U6 snRNA, was used for each cell type for plotting of mean values with S.D.
Microscopy analysis
Stellaris® FISH Probes, Custom Assay with CAL Fluor® Red 590 Dye targeting the Dicer Long Isoform and Stellaris® FISH Probes, Custom Assay with Quasar®670 Dye, targeting the common region of the transcript were obtained by utilizing the Stellaris RNA FISH Probe Designer (Biosearch Technologies, IncPetaluma, CA) available online at www.biosearchtech.com/stellarisdesigner). Cells were grown on coverslips coated with gelatin and subsequently fixed as done previously (22). FISH was performed as described on the website of the manufacturer derived from protocols established previously (https://biosearchassets.blob.core.windows.net/assets/bti_stellaris_protocol_adherent_cell.pdf) (41,42). Samples were imaged on a fast and stable inverted wide field microscope equipped with a MORE frame and enclosure, motorized XY-stage. Images were captured using a Hamamatsu ORCA flash 4.0 cooled sCMOS with the following parameters: Effective number of pixels: 2048 × 2048, Dynamic range: 16-bit, Quantum efficiency (peak): >70%, Read out noise: 1.9 electrons rms. The Objective used was a 60× TIRF APON with numerical aperture (NA) equal to 1.49. Illumination of the dyes was performed with 395/25, 550/15, 631/28 (nm) solid state light sources. The software used for the purpose of documentation was Live Acquisition 2.5. Images were exported to OMERO for documentation. Detection and analysis of spots were performed using automated pipelines developed in image analysis software IMARIS (BITPLANE). Prior to counting, the signal was deconvoluted using Huygens deconvolution software as per protocol recommended by the in-house imaging facility. Subsequently, images were transformed in IMARIS using SPOT and SURFACE detection modules according to the software-recommended steps. Following creation of spots and surfaces on a control image, the parameters were extrapolated for all other analysed images. Nuclear surface-overlapping spots were counted as nuclear signals, while all the others were counted towards cytosolic numbers. Each field of view was counted in aggregate and then normalised to the number of DAPI stained surfaces (after segmentation). For simplicity, we rounded the number of spots per nucleus to the closest integer before using these numbers for further calculations. A total of nine different fields of view from two independent biological replicates were utilised for statistics. Imaging of paraspeckles were performed using standard IF protocols as performed earlier (22) on an inverted Axio Observer Zeiss microscope (Zeiss) using a Plan Apochromat N 63×/1.40 oil DIC M27 objective with a Photometrics Prime 95B camera. Z-stack images were deconvoluted using ZEN software and further processed using the OMERO client server web tool for generating figures.
Luciferase assays
psiCHECK-2 Vector (Promega; C8021) was digested at XhoI–NotI restriction sites for insertion of the binding regions for the miRNA targets used in our analysis. Specifically, oligonucleotide constructs harboring a perfect match to the candidate miRNAs (hsa-miR-16–5p and hsa-miR-92a-3p) were inserted into the psiCHECK-2 vector MCS between XhoI and NotI for use as reporters. The sequence of the oligos used for the reporter constructs were TTGTAGTATTTTGCGCCAATATTTACGTGCTGCTAGTCGACCATTGTTAATC for the miR-16 Reporter and TGTAGTATTTTGACAGGCCGGGACAAGTGCAATAGTCGACCATTGTTAATC for the miR-92a Reporter. For the luciferase assay, 50 ng of the miRNA reporter construct or the undigested parent vector were transfected into HEK293 cells. siRNA treatment with oligos against CFIm25 or CFIm68 was performed 24 hours prior to transfection of the reporter plasmids. Cells were lysed at the 48 h mark post transfection using Passive Lysis Buffer (Promega) and 5 μl of each lysate was used for quantification of Renilla and Firefly luciferase expression. Firefly-normalised Renilla luciferase expression levels were used to compute fold-repression as described earlier (22).
Seahorse XF Real-Time ATP rate assay
For the seeding of cells, cell counting was performed and around 2650 cells were seeded in each well of a Agilent Seahorse XF96 Cell Culture Microplates. The plate was incubated for 72 h before the siRNA treatment was done. Measurement of ATP production rate in cells was performed using the Seahorse XF Real-Time ATP Rate Assay Kit according to the manufacturer's instructions. Briefly, Seahorse XF96 fluxpak cartridges were hydrated using Seahorse XF Calibrant Solution, 24 h pre-measurement. On the day of measurement, the culture medium was replaced with Seahorse XF DMEM medium (2 mM glutamine, 1 mM pyruvate, 10 mM glucose) and cells were incubated for 1 h at 37°C without additional CO2. Measurement was performed using the standard program for the ATP rate assay kit (Oligomycin injection after 18min, Rotenone/Antimycin A injection after 36 min). Acquired data were normalized to cell numbers via Hoechst33342 staining. Measurement of fluorescence intensity was performed using a Tecan Infinite® M1000 PRO.
Statistics
Samples were compared using the GRAPHPAD PRISM software t-test unless otherwise mentioned in the text. A P-value of less than or equal to 0.05 was considered significant and indicated on plots wherever applicable.
RESULTS
Wide-spread reciprocal changes in 3′ UTR length in CFIm KD and OE
As the overlap of CFIm targets reported in different studies is limited, we took advantage of prior observations that CFIm25 and CFIm68 have largely similar effects on 3′ UTR length (8–10) to establish a reference set of CFIm targets, specifically by identifying mRNAs whose 3′ UTRs undergo (1) similar changes in length upon perturbation of either CFIm25 or CFIm68, as well as (2) reciprocal length changes when the expression of these factors is reduced or increased. We therefore analysed HEK293 cell lines in which CFIm25 or CFIm68 were depleted by siRNA-mediated knockdown (Figure 1A and Supplementary Figure S1A) as well as HEK293 cell lines expressing FLAG-fusion constructs of either of the two CFIm subunits stably integrated into their genomes (9) (Figure 1B). After sequencing polyadenylated RNAs from these cell lines in 2–3 biological replicates per condition, we quantified the usage of tandem poly(A) sites in terminal exons (TE) with the PAQR tool (30) (Supplementary Figure S1B). The cumulative density functions (CDF) of average terminal exon length revealed the expected trend toward proximal PAS usage and short 3′ UTRs in CFIm KD cell lines (9,10,12,43), and a milder trend in the opposite direction in the OE conditions (Figure 1C). Principal component analysis of terminal exon length showed the expected condition-dependent clustering of the samples, and also that CFIm25 and CFIm68 affect the terminal exon (TE) length in similar ways. The first principal component (PC1), which explains over 90% of the variance in TE length data, reflects the level of CFIm expression (Figure 1D), as samples from CFIm25/68 KD and OE conditions are located at negative and positive coordinates on PC1, respectively. Therefore, we extracted our reference set of CFIm-dependent APA targets as those whose TE length aligned very well with PC1 (correlation > 0.9 in absolute value and projection > 10 in absolute value). We obtained 858 transcripts that satisfied these criteria, 855 of which had shorter 3′ UTRs upon CFIm25/68 KD (Supplementary Table S1, Figure 1E). The consistency of these results with previously reported effects of CFIm25/68 (9,10) supports the validity of our approach to CFIm target selection. Analysis of 3′ UTRs with exactly two PAS used across conditions showed that the CFIm-binding UGUA motif is more prevalent upstream of the distal PAS of transcripts that respond to CFIm perturbations (APA targets) compared to both the proximal PAS of these targets, as well as the proximal and distal PAS of transcripts that do not respond (non-targets, Figure 1F). Similar observations were made based on CFIm binding sites determined by crosslinking and immunoprecipitation (9). TIMP2, a previously documented target of CFIm (9,14,15), showed the preferential usage of a proximal poly(A) site in CFIm25/68 KD samples (Figure 1G). These results demonstrate that a large number of transcripts undergo coherent changes in PAS usage upon perturbation in CFIm25 and CFIm68 expression, forming a reference set of CFIm targets.
Figure 1.

Inference of a reference set of CFIm APA targets. (A, B) Western blots demonstrating the reduced expression of CFIm25 and CFIm68 in the KD (A) and increased expression in OE (B) HEK293 cell lines. Three biological replicates were generated for each condition. GAPDH was used as loading control. The expression of the 59 kDa component of CFIm, which does not influence the length of 3′ UTRs, was also measured. Shown are also quantifications of protein levels normalized to the mean expression in Control samples (±S.D.). Significant (<0.05) P-values computed from a two tailed t-test comparing each condition to Control are marked above individual columns. (C) Cumulative distributions of average terminal exon length, relative to the maximum given by the annotation (see Materials and Methods), in the different cell lines. P-values from two-sample KS-tests for the difference between the CDFs of the average TE length in CFIm25 OE and WT: 0.16, in CFIm68 OE and WT: 0.008, in CFIm25 KD and WT: 1.65e−28, and in CFIm68 KD and WT: 5.42e−64). (D) Principal component analysis of TE length. Each dot corresponds to a sample in the space defined by the first two principal components. (E) Selection of APA targets of CFIm: the vectors representing average length of individual TEs in all samples were projected onto the first principal component (from panel D) and the length of the projection (x-axis), as well as the correlation of these vectors (y-axis) were calculated. TEs for which both of these values were large in absolute value (marked by the red lines) were considered APA targets of CFIm. (F) Position-dependent frequency of occurrence of the CFIm-binding UGUA motif in the vicinity of proximal (dashed lines) and distal (full lines) sites of CFIm targets (blue) and non-targets (red). The curves represent running averages computed over 30 consecutive positions. (G) Genome browser tracks showing the coverage of the TE of TIMP2 (shown in the bottom track) by RNA-seq reads from two replicate experiments for each condition, with the two PAS that were quantified for this gene marked by black lines. The conditions are color-coded (color scheme as in panels C and D) and indicated on the y-axis.The y-axis shows the smoothened number of reads mapping along the TE, calculated by the GViz R package.
The CFIm knockdown increases the biogenesis and activity of miRNAs
As 3′ UTR shortening enables mRNAs to escape the repressive effect of miRNAs (44,45), we were intrigued by the conspicuous presence of key components of the miRNA pathway (DICER1, AGO2) among CFIm targets (Supplementary Table S1, Figure 2A, Supplementary Figure S2A and B). To verify changes in DICER1 isoform expression and further determine whether they occur in the nucleus as a result of APA, as opposed to the cytoplasm as a result of other mechanisms, we visualized the abundance and distribution of DICER1 3′ UTR isoforms within cells by single molecule RNA FISH (Figure 2B) in control and CFIm KD (25-KD and 68-KD) conditions. We used probes that selectively bind either to the distal end of the long 3′ UTR isoform (green), or to a region that is shared by the long and short 3′ UTR isoforms (red). The probes are expected to co-localize on the long isoform, which will appear yellow, whereas the short isoform, which lacks the sequence that can hybridize to the green probe, will only fluoresce in the red channel. The quantitative analysis of the relative number of RNA molecules hybridizing to the different probes in CFIm 25-KD/68-KD cells revealed a marked depletion of the long isoform already in the nuclear region, indicating the increased usage of the proximal poly(A) site of the DICER1 transcript upon CFIm knockdown. The longer isoform was also depleted in the cytosol in these conditions, where the overall number of DICER1 molecules was markedly higher than in the nucleus (Figure 2C and Supplementary Figure S2D). Western blotting showed that DICER1 protein expression also increases upon CFIm KD (Figure 2D), matching closely both the increased counts of DICER1 transcripts estimated from RNA-seq analysis and the imaging data (Figure 2C).
Figure 2.

CFIm KD increases the activity of miRNAs. (A) Genome browser tracks showing the coverage of DICER1 TE (bottom track) by RNA-seq reads from two replicate experiments for each condition, with the two PAS that were quantified for this gene marked by black lines. The conditions are color-coded (as in Figure 1) and also indicated on the y-axis. Y-axis shows the smoothened number of reads mapping along the TE, calculated by the GViz R package. (B) RNA fluorescence in situ imaging of DICER1 isoforms in Control and CFIm25 KD HEK293 cells with probes corresponding to the common region of the long and short 3′ UTRs (red) or to the region between the proximal and distal cleavage sites, thus present exclusively in the long 3′ UTR (green). Nuclei are marked with DAPI. Zoom-ins of the regions marked with dashed boxes are further shown both with the individual and merged channels. A snapshot of a digital representation of the actual image as processed in IMARIS is also depicted for reference. (C) Quantification of the copy number of the long and short 3′ UTR isoforms of DICER1 in the nucleus (left plot) and cytoplasm (right plot) of Control, CFIm25 and CFIm68 KD cells. Colocalization of the red and green signals reveals the presence of the long 3′ UTR isoform (yellow) whereas the signal from the red probe only reveals the presence of the shorter 3′ UTR isoform. mRNA copy numbers were estimated separately from the nucleus (overlapping with DAPI) and cytosol. Segregation of the signal was performed with IMARIS (see Methods). (D) Representative western blot showing the DICER1 expression in the Control, CFIm25 and CFIm68 KD cells. The quantification is relative to GAPDH. (E) qPCR measurements of let-7, miR-92a, miR-16 and miR-19b expression in CFIm25/68 KD cells relative to Control. ΔΔct values were calculated relative to U6 snRNA and then relative to the Control cells (where the ratio was set to 1). (F) Normalized Renilla luciferase expression of reporter mRNAs carrying binding sites for miR-16 and miR-92a in their 3′ UTRs, in Control, CFIm25 and CFIm68 KD cells, respectively. The Firefly luciferase expressed from the same construct was used as normalization control.
As DICER1 upregulation is predicted to increase the production of miRNAs, we measured the levels of three randomly-selected, ubiquitously-expressed miRNA by real time PCR, finding that they were indeed higher in CFIm KD cells relative to Control (Figure 2E). In contrast, despite the shortening of AGO2 3′ UTR as a result of CFIm KD, the AGO2 mRNA level only increased by 42/72% in CFIm25/68 KD relative to control (Supplementary Table S3), and the protein level changes measured by TMT proteomics were even smaller (27/24% in the same conditions). These differences were not detectable when AGO2 protein levels were compared by western blotting (Supplementary Figure S2A). To determine whether the increased miRNA biogenesis translates into increased miRNA-mediated repression, we measured the activity of dual luciferase reporters for two ubiquitously-expressed miRNAs, miR-16 and miR-92a. The reporter expression showed an increased miRNA activity in CFIm25/68 KD cells compared to mock-transfected Control samples, indicating that AGO2 levels were not limiting upon CFIm knockdown (Figure 2F). These results demonstrated the coherent effects of CFIm on the biogenesis and activity of miRNAs whereby the reduction in CFIm expression leads to increased miRNA-mediated repression of target reporters.
CFIm modulates signaling via CMGC kinases
To identify the molecular pathways whose components are APA targets of CFIm, we performed Gene Ontology enrichment analysis (Supplementary Table S1) with the clusterProfiler R package (46). Most enriched in CFIm targets were processes such as cellular response to stress and protein transport and modification (Figure 3A). Genes from these functional categories that we identified as targets exhibited the expected enrichment of the UGUA motif relative to those that are not CFIm targets according to our analysis (Supplementary Figure S3), indicating a sequence-specific effect of CFIm (Supplementary Figure S3). To further map the signaling events in which these targets participate, we measured the abundance of phosphopeptides by phosphoproteomics with IMAC enrichment (see Materials and Methods) in all of the HEK293 cell lines used in this study (Supplementary Table S2). Principal component analysis of the normalized phosphopeptide intensities showed that the OE samples separate well from Control as well as between CFIm components, while the KD samples separate well from Control, but less well between CFIm components (Figure 3B). Of the 22'707 phosphopeptides that were measured, 4′536 showed condition-dependent changes. We then sought to apply a recently developed method, kinase activity enrichment analysis (KSEA) (38) to identify kinases whose activity changes in a CFIm-dependent manner. KSEA is similar to the broadly-used Gene Set Enrichment Analysis (GSEA) (47), quantifying whether phophopeptides associated with a specific kinase are enriched among the phosphopeptides that undergo the largest change in abundance between two conditions. As described by Hernandez-Armenta et al. (38), we used the −log10 of the P-value, calculated from KSEA, as a proxy of the change in kinase activity with the sign indicating the direction of change of its associated phophopeptides between conditions. Along with changes in phosphopeptide levels, KSEA uses kinase-substrate interactions as input. Finding that only 6.7% of the phosphopeptides that we identified in our experiments are represented among known kinase-substrate interactions in the reference PhosphoSitePlus database (39), we first predicted additional kinase-substrate relationships using position-dependent weight matrix models of kinase substrate specificity (see Materials and Methods). KSEA then revealed pronounced changes in kinase activity in CFIm25/68 KD conditions and milder changes upon OE (Figure 3C). The more pronounced effects of the KD relative to OE on kinase activities mirror the response of 3′ UTR length to these perturbations (Figure 1C). Interestingly, 14 of 35 kinases with a significant activity change (KS-test P < 0.01) in at least one condition belonged to the CMGC family, which includes cyclin-dependent kinases (CDKs), mitogen-activated protein kinases (MAP kinases), glycogen synthase kinases (GSK) and CDK-like kinases (48). Only 21 of 173 kinases without a significant change were part of this family.
Figure 3.

CFIm has a large impact on the intra-cellular signaling landscape. (A) Gene Ontology analysis with the clusterProfiler R package (46) identifies biological processes that are significantly enriched in CFIm targets (FDR < 0.01). The y-axis shows the proportion of CFIm targets with a specific biological process annotation and the size of the oval is proportional to the absolute number. The color indicates the significance of the enrichment (FDR value). The 15 most significantly enriched GO categories are depicted. (B) Principal component analysis of phosphopeptide intensity data, showing the projection of Control, CFIm25/68 KD and OE samples on the first two principal components. (C) Kinase activity changes in KD and OE conditions, computed with the KSEA algorithm (see Methods). Shown are kinases estimated to have a significant activity change (KS-test P-value < 0.01) in at least one condition. The scale indicates both the statistical significance of the difference relative to Control samples (log10(P-value) of the KS-test) and the direction of the change (indicated by the sign). The sign is that of the mean of log2 fold-change in phosphopeptide intensity between conditions, taken over all phosphopeptides associated with a given kinase. (D, E) Western blots showing the response of the CK2A1 target motif (D) and of the phosphorylated SQSTM1 (E) to CFIm25/68 KD and OE, with associated quantifications (±S.D., P-values in the two-sided t-test). Values are calculated relative to the actin control, and then the ratios to the mean of Control samples are used to construct the bar graphs. The same blot was reprobed in the bottom panels of D and E.
KSEA predicted an increase in the activity of CK2A1/CK2A2, SYK, MAPKAPK2, CAMK2 and CK1D kinases upon CFIm25/68 KD (Figure 3C), but only CK2A2, MAPKAPK2, and CK1D exhibited a reciprocal change in activity upon CFIm25/68 OE. Focusing on the kinases whose activity changes reflected the change in CFIm expression, we evaluated the KSEA predictions by checking the patterns of phosphorylation of specific substrates. While we did not have any site that could be unambiguously attributed to CK1D in our data, we did find one of the best characterized substrates of MAPKAPK2, the Ser82 residue of the heat shock protein 27 (HSP27) (49). The phosphorylation level of this site was higher in CFIm25/68 KD (1.4-fold, Supplementary Table S2) and lower in CFIm25/68 OE (0.85/0.75, Supplementary Table S2) conditions compared to Control cells, consistent with the overall MAPKAPK2 activities predicted by KSEA. Taking advantage of an antibody that selectively labels all instances of the CK2 substrate consensus sequence, pS/pTD/EXD/E (the most crucial residues being those at positions +3 and +1 with respect to the phosphorylation site (50)), we also sought to independently validate the changes in this kinase's activity in our experimental conditions. Quantitative western blot analysis of the lysates obtained from KD and OE cells revealed an upregulation of total phosphorylation levels in the KD samples relative to the Control cell lysate, and no significant change upon OE, in agreement with the results obtained from the KSEA analysis (Figure 3D).
KSEA also predicted reciprocal changes in the activity of CMGC family kinases such as the mitogen-activated protein kinases JNK1/2 (MAPK8/9), P38D (MAPK13), ERK1/2 (MAPK3/1), and cyclin dependent-kinases CDK1/2/5 upon CFIm KD/OE, the activity decreasing in the KD and increasing in OE conditions (Figure 2C). Of these, transcripts corresponding to MAPK1, MAPK9 and MAPK13 are also in our reference set of CFIm targets (Supplementary Table S1). To independently validate the changes in MAPK13 activity we focused on its known target, Sequestosome-1, also known as ubiquitin-binding protein p62, which undergoes MAPK13-dependent phosphorylation at Thr269 and/or Ser272 in response to proteasomal stress (51). Both of these sites responded as expected in our phosphoproteome data, with decreased phosphorylation in the CFIm25/68 KD samples (fold-changes relative to Control 0.42/0.55 at Thr269, and 0.47/0.64 at Ser272, Supplementary Table S2) and increased phosphorylation in the CFIm25/68 OE conditions (fold-changes relative to Control 1.56/2.64 at Thr269, and 1.37/1.96 at Ser272, Supplementary Table S2). We observed similar changes in western blots, using an antibody that recognizes SQSTM1 only when phosphorylated at Thr269 and/or Ser272 (Figure 3E). Altogether, these results demonstrate that the level of CFIm is linked to the activity of CMGC kinases, some of which are encoded by transcripts that undergo CFIm-dependent APA.
CFIm-induced changes in cell proliferation reflect the activity of ERK1/2 kinases
The ERK/MAPK signaling pathway plays a key role in cell proliferation, differentiation and apoptosis (52). Activated by endoplasmic reticulum stress and unfolded protein response (53), this pathway can have both tumorigenic (54) and anti-tumorigenic (55) effects. These contrasting roles are reminiscent of the divergent changes in CFIm expression reported in various cancers (18). To validate the predicted change in ERK1/2 activity in our system, we estimated the levels of phosphorylated (Tyr202/Tyr204) ERK1/2 by western blotting. We found them to indeed be positively correlated with the CFIm25/68 expression level (Figure 4A). We then used a real time assay to determine the effect of CFIm on cell proliferation, which was also reported to differ between cell types (15,21). We found that the KD of CFIm25/68 reduced and the OE increased the growth of HEK293 cells (Figure 4B). To ascertain a compelling role of ERK signaling in the increased proliferative state of the CFIm25/68 OE cells, we used an inhibitor of ERK signaling, Ravoxertinib hydrochloride, at reported IC-50 concentrations (56). Cells seeded in the presence of the inhibitor had a conspicuous growth arrest and the growth patterns of the OE cells traced that of Control cells. In contrast, the DMSO treatment had no effect on the growth patterns (Figure 4C). We also verified the reduced growth phenotype of CFIm KD in other cell types, HeLa and LN-18 glioblastoma (Figure 4B), although for these cell lines the effects were milder than those observed in HEK293 cells.
Figure 4.

CFIm expression level is linked to the activity of the ERK signaling pathway. (A) Western blot analysis of phospho-ERK1/2 (P-ERK1/2) levels in cell lysates from the KD and OE cell lines. The numbers below the blots indicate the levels of phospho(P)-ERK1/2 relative to the actin loading control. For the comparison across conditions, the ratio of P-ERK1/2 to actin in all samples was normalized to the average of the values from Control samples. (B) Proliferation index of cells with perturbed CFIm expression and Control cells. Graphs show the observed cell index values in a standard xCelligence real time growth analysis of Control and CFIm25/68 KD and OE HEK293 lines. LN18 and HeLa cells with siRNA-induced depletion of CFIm components were also measured. (C) The proliferation index of CFIm OE lines were compared to that of the Control line in the presence of ERK1/2 inhibitor ravoxertinib hydrochloride added at 6.1 nM final concentration in the growth medium. DMSO-containing medium was used as control, to ascertain the growth defects following the ERK1/2 inhibition. (D) Real time quantification of the rate of adenosine triphosphate (ATP) production via glycolysis and oxidative phosphorylation in live cells. Bar plots show the percentage of ATP produced via the glycolytic and oxidative phosphorylation routes in HEK293 cells in CFIm KD (top) and OE (bottom) relative to Control cells, estimated with the Seahorse ATP assay (see Methods). (E) Western blot estimation of the phosphorylated pyruvate dehydrogenase levels in CFIm25/68 KD/OE and Control HEK293 cells. (F) Estimation of cleaved PARP in lysates obtained from CFIm25/68 KD/OE and Control cells. The quantifications below the blots represent the fold changes of the respective targets in the various conditions in CFIm25/68 KD/OE relative to Control. The relative levels were calculated first by dividing the signal intensity to that of the actin loading control and then further normalized to the mean ratio in Control samples. The fold changes of the respective targets in CFIm25/68 KD/OE relative to Control were statistically significant (P-value in the two tailed t-test ≤ 0.05).
Both the upregulation (57) and downregulation (58) of ERK1/2 activity have been linked to the Warburg-like effect, the switch from oxidative phosphorylation to glycolysis in cellular energy production that is a hallmark of cancer (59). To determine whether the metabolic activity in our cell systems is consistent with changes in ERK1/2 activity, we compared the ATP production by glycolysis and oxidative phosphorylation in all conditions (WT, CFIm KD and OE) in a Seahorse ATP real-time rate assay (60). Indeed, we found the switch from oxidative phosphorylation to glycolysis in ATP production in both CFIm KD and OE cells compared to Control (Figure 4D), as reported for changes in ERK1/2 activity. The main enzyme that drives the carbon flux into mitochondria for the TCA cycle and oxidative phosphorylation is pyruvate dehydrogenase (PDH), whose inhibition leads to the Warburg effect (61). By western blotting, we found that the level of the inhibitory phosphorylation on Ser293 of PDH, known to be catalyzed by pyruvate dehydrogenase kinase 1 and 2 (PDHK1-2), was increased (Figure 4E) in both CFIm KD and OE conditions. Thus, perturbation in CFIm expression leads to metabolic shifts that are consistent with ERK1/2 activity changes converging on PDHK1-2 and PDH.
To better understand how CFIm KD and OE induce divergent changes in cell numbers but convergent changes in the metabolism of cells, we performed endpoint western blot analysis of cleaved PARP (Figure 4F). The cleavage of PARP-1 by caspases is a hallmark of apoptosis (62). The anti-correlation of cleaved PARP with CFIm expression levels indicates that the reduced cell culture growth in CFIm25/68 KD conditions is due to increased levels of apoptosis. In summary, these results demonstrate that CFIm promotes the growth of multiple cell types, primarily by suppressing apoptosis. Furthermore, both the cell proliferation and metabolic phenotype are consistent with changes in the ERK1/2 kinase activity.
3′ end sequencing reveals similar CFIm targets
We finally assessed the reproducibility of the CFIm target set with respect to the cellular system and the method for quantifying the 3′ end usage. Perturbations of CFIm25 and/or CFIm68 expression have been carried out not only in HEK293, but also in HeLa cells, where distinct methods for quantifying 3′ end usage were applied. We extracted 3′ end usage data upon CFIm KD in these systems from the PolyASite database (31), and, by applying the same target selection method (Supplementary Figure S4, also see Materials and Methods), we obtained ∼850–1000 genes whose TEs became shorter upon CFIm25/68 KD (Figure 5A–C) from each of these datasets. We then visualized their relationship in a Venn diagram (Figure 5D). The overlap of targets obtained by two distinct methods for PAS usage quantification or in two different cell systems was ∼20–40%. The majority (476 of 853) of genes in our core set are also identified as CFIm targets in another data set, while 51 are common to all data sets (significance of overlap from the SuperExactTest (63) P-value: 3.66e−101). Notably, this latter set includes kinases and kinase regulators such as MAPK1 (ERK2), Serine/threonine-protein kinase Chk1 (CHEK1), AMP-activated protein kinase 1 and 2 (AMPKA1-2), C-Jun-amino-terminal kinase-interacting protein 4 (SPAG9) and Receptor-interacting serine/threonine-protein kinase 2 (RIPK2) (Supplementary Table S4). These results indicate that the inferences we made based on the RNA-seq data in HEK293 data were robust, and in line with the growth phenotypes that we assessed above (Figure 4B).
Figure 5.

Overlap of CFIm targets identified with several methodologies from different cell types. (A, B) CDF of average relative TE length inferred from A-seq data in HEK293 cells (A) and HeLa cells (B). The P-value in the two-sample KS-test comparing the HEK293 CFIm25 KD and Control conditions is 3.04e−23, while for CFIm68 KD and Control conditions is 1.70e−43. The P-value for the two-sample KS-test comparing the HeLa CFIm25 KD and Control conditions is 8.75e−22, while for CFIm68 KD and Control conditions is 1.52e−28. (C) CDF of average relative TE length inferred from PAPERCLIP data in HeLa cells; the P-value of the two-sample KS-test comparing HeLa CFIm68 KD and Control conditions is 1.60e−58. (D) Venn diagram (94) showing the overlap of targets obtained from HEK293 RNA-seq, HEK293 A-seq, HeLa A-seq and HeLa PAPERCLIP data sets. The P-value of the overlap of 51 genes among all data sets is 3.66e-101 (SuperExactTest (63)). (E) 2D density plot of log2FC in average relative TE length measured with A-seq or RNA-seq in CFIm25 KD relative to Control cells. The Pearson and Spearman correlation coefficients are, respectively, 0.53 and 0.46 (P-values 6.27e−109 and 3.94e−81). (F) 2D density plot of log2FC in average relative TE length measured with A-seq or RNA-seq in the CFIm68 KD compared to Control cells. Pearson and Spearman correlation coefficients are, respectively, 0.59 and 0.54 (P-values 1.41e−142 and 3.09e−112), (G) Gene Ontology enrichment analysis with the clusterProfiler R package (46) identifies biological processes that are significantly enriched in CFIm targets that are common among 4 cell lines and 2 techniques for quantifying PAS usage (FDR < 0.05). The y-axis shows the proportion of CFIm targets with a specific biological process annotation and the size of the circle is proportional to the absolute number. The color indicates the significance of the enrichment (FDR value). (H) A graphical model of CFIm's impact on several cellular processes.
DISCUSSION
Our study makes two main contributions to the expanding field of alternative polyadenylation. First, we provide a reference set of APA targets of the CFIm 3′ end processing factor, the main regulator of 3′ UTR length known to date. These targets were stringently selected based on their consistent and coherent 3′ UTR length changes upon KD/OE of both CFIm25 and CFIm68 components of CFIm. They provide a basis for future analyses in other cell systems, and especially in cancers, where CFIm has been already implicated, with somewhat divergent roles (18). Second, our study uncovered a so-far uncharted layer of regulation downstream of the CFIm 3′ end processing complex, revealing that signaling pathways are extensively remodeled upon perturbations in CFIm expression. The activity of the ERK pathway essentially traces the CFIm expression level and can explain the proliferation, apoptosis and metabolic responses of cells to CFIm perturbations. Beyond these main findings, our results expand the knowledge of the interplay between RNA 3′ end processing and other cellular processes such as miRNA-mediated repression, as detailed below.
In spite of CFIm68 having similar 3′ UTR length regulatory functions, most prior studies focused on CFIm25, reporting a range of CFIm-dependent APA targets that varied ∼100-fold (15,64). To identify conserved functions of CFIm-dependent RNA processing in cell biology, here we constructed a reference set of CFIm targets by carrying out both the KD and the OE of not only CFIm25 but also the CFIm68 subunits of CFIm. We identified 858 transcripts with a highly consistent response across all of these conditions (Figure 1), 855 of which (from 853 genes) exhibited 3′ UTR shortening upon the KD of CFIm factors. The majority of these transcripts are also identified in other cellular systems or with other methods for poly(A) site usage quantification (Figure 5). The set includes well known CFIm targets like TIMP2 (9,14,65), DICER1 (15) and MECP2 (15,16) and, interestingly, paralogs of some reported targets, e.g. CCND2 and CHD6 in place of CCND1 and CHD9 (15,16). Along with the intersection of targets obtained by 3′ end sequencing data from HEK293 and HeLa cells being only partial (Figure 5), this latter finding may indicate that a subset of CFIm targets is cell type-specific. However, the targets identified in the same cellular system by distinct 3′ end sequencing methods are also not identical (Figure 5D), suggesting that differences in target sets could also be due to differences in the experimental design (e.g. PAPERCLIP-based 3′ end processing data was only available for the CFIm68 KD, and not for the CFIm25 KD). The changes in TE length estimated from the RNA-seq or the 3′ end sequencing data were well correlated, with Pearson correlation coefficients in the 0.5–0.6 range (Figure 5E-F), as observed before (30), underscoring the robustness of the target set.
The frequency of CFIm-binding UGUA motifs is ∼1.5–2-fold higher at the distal PAS of CFIm targets compared to the proximal PAS, in contrast to non-targets, where the two sites are not clearly distinguishable by the UGUA motif frequency (Figure 1). These results are in line with prior observations (9,12,64). The UGUA motif is strongly depleted downstream of the distal PAS, while at the proximal sites UGUA motifs occur with comparable frequency both upstream and downstream of the PAS. This supports a recently proposed model, whereby binding of CFIm to UGUA motifs flanking the proximal PAS leads to the looping of the RNA around the proximal PAS (66) and to an unproductive interaction with FIP1, which masks the site from cleavage. In contrast, the UGUA motif is only present upstream of the distal site of CFIm targets, leading to productive interaction with the other components of the 3′ end processing complex and 3′ end cleavage (64).
It was noted in a previous study that cell cycle-related genes such as cyclin D1 are targets of CFIm (15), linking APA to cellular signaling. Here, we found a strong enrichment of signaling-related proteins among the reference set of CFIm targets (Figure 3). We further predicted changes in the activity of many kinases upon perturbations in CFIm expression, particularly those from the CMGC family. We focused primarily on ERK1/2, because it can link the perturbation in CFIm level to multiple phenotypes reported in the context of cancer, including proliferation, apoptosis and glucose metabolism. ERK activity was positively correlated with the expression level of CFIm, consistent with the effects described in CFIm25-depleted K562 cells (21). Downregulation of ERK/MAPK has a negative effect on cellular growth and leads to a metabolic switch that favors glycolysis over oxidative phosphorylation (58), both of which we were able to demonstrate in CFIm KD conditions (Figure 4). Conversely, with real-time assays and cell-cycle analysis we showed that OE of CFIm promotes growth, as expected given the phosphorylation-dependent ERK/MAPK activation (67). Surprisingly, the ERK activity was anti-correlated with the activity of CK2A1, a kinase reported to contribute positively to ERK signaling, as part of the Kinase Suppressor of Ras 1 scaffolding complex (68) and as mediator of ERK nuclear translocation (69). Increased CK2A1 activity was observed in cancer cells (70), associated with drug resistance (71) and resistance to apoptosis (72). Although the mechanism behind seemingly discordant CK2A1 and ERK activities in our system remains to be determined, the upregulation of CK2A1 activity could contribute to the glycolytic switch that we paradoxically observed in CFIm KD conditions (73).
The activity of another MAP kinase, MAPK13 (P38D), was also correlated with the CFIm25/68 levels (Figure 3). MAPK13 participates in key processes such as cell proliferation, differentiation, transcription regulation and development, and is overexpressed in a large set of human breast cancers (74,75). A main target of MAPK13 is SQSTM1 (p62), which MAPK13 phosphorylates on Ser269 and Thr272 (51), as we also found here (Figure 3E). Interestingly, our data includes 5 additional sites on SQSTM1, all 5 with increased phosphorylation in CFIm25/68 OE (Supplementary Table S2). As hyperphosphorylation of p62 is a marker for chemotherapy resistance in ovarian cancer cells (76), the increased phosphorylation of these sites is consistent with the increased growth of CFIm OE cells.
How do changes in 3′ UTR length lead to a remodeling of cellular signaling? Consistent with prior expectations (44), our data shows a small but significant increase in gene expression levels of CFIm targets compared to non-targets in the CFIm KD cells, indicating a small tendency toward increased stability of short 3′ UTR isoforms (77,78) (Supplementary Figure S5 and S6). We further calculated the Pearson correlation coefficient of gene expression changes with both changes in terminal exon length and changes in the proximal/distal PAS usage ratio. While small (<0.23), these correlation coefficients were significant (P-values < 0.01) and had the expected trend (negative correlation of terminal exon length and positive of proximal/distal ratio with gene expression, Supplementary Figure S6). The changes at the protein level were smaller (Supplementary Figure S5), for reasons that remain unclear. The OE of CFIm components did not lead to an opposite effect, namely reduction in target levels, which likely reflects the milder effect of OE on 3′ UTR length compared to the KD of CFIm. This is not surprising because the distal PAS are already preferentially used in HEK293 cells under control culture conditions (9), leading to limited lengthening of 3′ UTRs upon CFIm overexpression. Focusing on signaling-related targets, some of the key regulators that we analysed here, such as MAPK1 (ERK2), and MAPK13 did not show significant gene expression changes, while many others were upregulated upon CFIm25/68 KD. These include, for e.g. CK2A1, MAPK9 (JNK2), the TOR signaling pathway regulator (TIPRL) that negatively impacts JNK signaling by binding to MKK7 (79), the TAK1-interacting protein 27 (JAZF1), which inhibits cell proliferation and enhances apoptosis through its negative control of the TAK1/NF-KB signaling pathway (80), the MAP2K4/MKK4, an upstream activator of JNK signaling (81), and the NDFIP1 ubiquitin ligase activator involved in the ubiquitination of upstream activators of the JNK signaling pathway (82). In contrast to the RNAs, the abundance of the corresponding proteins was less affected by CFIm perturbations. This may be due to a lower sensitivity of the measurement technology, as the ∼2-fold change in abundance of DICER1 that we measured by WB was not apparent in the proteomics data. However, protein level changes were also not uniformly detectable for targets that we measured by WB (Figure 2D, Supplementary Figure S2C), suggesting additional post-transcriptional control of CFIm targets. The overall small protein-level changes could indicate that the phenotypic changes observed upon CFIm perturbations are due to a cumulative effect of small changes in many targets rather than to a small number of targets whose expression is strongly altered.
The similar 3′ UTR shortening in cancer and in CFIm KD conditions make CFIm a very appealing candidate for explaining the cancer-related remodeling of 3′ UTRs. Indeed, in glioblastoma and hepatocellular carcinoma (15,19,20), reduced CFIm expression has been implicated in 3′ UTR shortening and tumorigenesis. However, this relationship does not appear to be universal (18) and, in fact, the expression of 3′ end processing factors is typically higher in tumors compared to matched control tissues (Supplementary Figure S7A, (83)). Kaplan–Meier analysis (84) shows that the levels of CFIm25/68 are also not good predictors of cancer-free survival and that in the majority of cancer cohorts where a significant (P-value < 0.05) association between CFIm25/68 expression and survival can be detected, it is the high, not the low expression of CFIm that represents a risk factor (Supplementary Figure S7B, C). What could account for seemingly contrasting results regarding the CFIm expression and function in cancers? A hypothesis that can reconcile these observations is that the level of CFIm per se is not sufficient to predict the pattern of RNA processing in cancers, and that the RNA processing load of cells plays an equally important role; an increased processing load in proliferating cells may lead to transient CFIm deficiency in spite of its increased overall expression and this relationship may further be cell type-specific. This scenario has been reported for the U1 snRNA during neuronal activation, also leading to APA at proximal PAS (85). Of course, technical artifacts such as the variable degree of RNA degradation among samples may also lead to divergent results, due to erroneous estimates of CFIm expression levels (30). Nevertheless, the relationship between RNA processing demand and availability of 3′ end processing factors, especially in the context of cancer, warrants further studies.
Finally, our data show a complex effect of CFIm-dependent APA on the miRNA-mediated gene regulation. Much of the work on APA in the past decade has been motivated by the observation that 3′ UTRs become shorter in proliferating cells (44), presumably leading to the escape of the corresponding transcripts from miRNA regulation (44,45). That DICER1, the key enzyme in miRNA biogenesis, would be regulated in this manner to increase the production of miRNAs in these conditions is counter-intuitive, even though it has been observed before (15,45). Here, we found that CFIm KD leads to reduced expression of the long DICER1 3′ UTR isoform already in the nucleus, presumably via APA. The DICER1 protein expression increases in parallel to the transcript level. A further contribution to the increased miRNA biogenesis in CFIm KD condition may come from the reorganization of paraspeckles (PS), nuclear condensates that form around the long non-coding RNA (lncRNA) NEAT1 (86). Reduced CFIm levels lead to the production of a long isoform of NEAT1, called NEAT1_2, which nucleates the PS (87). NEAT1_2 also recruits the Drosha/DGCR8 Microprocessor complex to PS, where primary miRNAs interact closely with the NONO-PSF protein dimer (88), leading to increased miRNA biogenesis. Indeed, immunofluorescence analysis with an antibody targeting the PS-essential NONO protein (89) confirms the relationship between the CFIm level and the NEAT1_2-dependent size and organization of the PS (90) (Supplementary Figure S7D, E). The effector component of the miRNA pathway, AGO2, also undergoes CFIm-dependent APA, but without detectable protein-level changes (Supplementary Figure S2C, Supplementary Table S3). AGO2 does not seem to be limiting for miRNA repression in our systems, as the increased miRNA biogenesis upon CFIm KD is accompanied by a corresponding increase in their repressive activity, as demonstrated with reporter genes (Figure 2). These results demonstrate that CFIm organizes the miRNA-dependent regulatory layer by modulating both miRNA biogenesis and the subset of transcripts that are susceptible to miRNA-dependent regulation. It has been reported, for instance, that uncapped RNAs that are downstream products of cleavage at proximal PAS stably persist in the cell (91). These may alter the cellular milieu to trigger some of the signaling events that we observed, while the increased miRNA activity may serve to clear out some of these RNA species and counteract the cellular stress that they induce. How cells deal with globally increasing or decreasing RNA processing load is an important question to address in future studies.
In conclusion, our study has revealed a novel layer of CFIm-dependent gene regulation, mediated by numerous kinases, especially from the CMGC family. The ERK/JNK/MAPK pathways can explain many of the observed phenotypic changes caused by CFIm expression perturbations, including in cell proliferation, apoptosis and metabolism. We provide a reference set of transcripts that respond in a consistent manner to both KD and OE of CFIm25 and CFIm68 proteins and likely underlie the roles of CFIm in various cellular systems. Finally, we found that CFIm largely promotes cell growth, consistent with some, but not all of the previous studies of cancer systems. Given that the expression of 3′ end processing factors, and especially of CFIm is positively correlated with proliferation, our study integrates a variety of prior observations into a consistent framework. The exact mechanism that bridges the CFIm-mediated APA of several transcripts to the alteration in kinase activities is an interesting topic for future studies (Figure 5F), especially when considering the simultaneous changes in the miRNA biogenesis and, as suggested by our reporter assays, in the activity of the miRNA pathway.
DATA AVAILABILITY
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (92) partner repository with the dataset identifier PXD027034 and 10.6019/PXD027034. The source code to replicate all the analyses presented in this study is available at https://github.com/zavolanlab/CFI2021. Raw and processed RNA-seq data are available at Gene Expression Omnibus (GEO) (93) (https://www.ncbi.nlm.nih.gov/geo/) under accession number GSE179630.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Philipp Demougin from the Quantitative Genomics Facility for help with RNA sequencing and the sciCORE facility of the University of Basel for maintaining the high-performance computing network where all the computations have been carried out. M.B. is a recipient of the Fellowship for Excellence at the Biozentrum, University of Basel. We also like to acknowledge the support from Kai Schleicher and Laurent Guerard IMCF for their help in setting up a pipeline for IMARIS Quantifications and Konstantin Schneider-Heieck from the Christoph Handschin group for Seahorse assays.
Author contributions: Project design: S.G. and M.Z.; Experimental analyses: S.G. with help from B.D. (western blots) and G.M. (generation of OE lines); Computational analyses: M.A., M.B., A.B., A.K; with help from C.J.H. Proteomics measurements: A.S, K.B.; Manuscript writing: S.G., M.A., M.Z. with contributions from all authors; Funding acquisition: M.Z.
Contributor Information
Souvik Ghosh, Computational and Systems Biology, Biozentrum, University of Basel, Spitalstrasse 41, 4056 Basel, Switzerland.
Meric Ataman, Computational and Systems Biology, Biozentrum, University of Basel, Spitalstrasse 41, 4056 Basel, Switzerland; Swiss Institute of Bioinformatics, Biozentrum, University of Basel, Spitalstrasse 41, 4056 Basel, Switzerland.
Maciej Bak, Computational and Systems Biology, Biozentrum, University of Basel, Spitalstrasse 41, 4056 Basel, Switzerland; Swiss Institute of Bioinformatics, Biozentrum, University of Basel, Spitalstrasse 41, 4056 Basel, Switzerland.
Anastasiya Börsch, Computational and Systems Biology, Biozentrum, University of Basel, Spitalstrasse 41, 4056 Basel, Switzerland; Swiss Institute of Bioinformatics, Biozentrum, University of Basel, Spitalstrasse 41, 4056 Basel, Switzerland.
Alexander Schmidt, Proteomics Core Facility, Biozentrum, University of Basel, Spitalstrasse 41, 4056 Basel, Switzerland.
Katarzyna Buczak, Proteomics Core Facility, Biozentrum, University of Basel, Spitalstrasse 41, 4056 Basel, Switzerland.
Georges Martin, Computational and Systems Biology, Biozentrum, University of Basel, Spitalstrasse 41, 4056 Basel, Switzerland.
Beatrice Dimitriades, Computational and Systems Biology, Biozentrum, University of Basel, Spitalstrasse 41, 4056 Basel, Switzerland.
Christina J Herrmann, Computational and Systems Biology, Biozentrum, University of Basel, Spitalstrasse 41, 4056 Basel, Switzerland; Swiss Institute of Bioinformatics, Biozentrum, University of Basel, Spitalstrasse 41, 4056 Basel, Switzerland.
Alexander Kanitz, Computational and Systems Biology, Biozentrum, University of Basel, Spitalstrasse 41, 4056 Basel, Switzerland; Swiss Institute of Bioinformatics, Biozentrum, University of Basel, Spitalstrasse 41, 4056 Basel, Switzerland.
Mihaela Zavolan, Computational and Systems Biology, Biozentrum, University of Basel, Spitalstrasse 41, 4056 Basel, Switzerland; Swiss Institute of Bioinformatics, Biozentrum, University of Basel, Spitalstrasse 41, 4056 Basel, Switzerland.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
SNF Grant [310030_189063]. Funding for open access charge: SNF.
Conflict of interest statement. None declared.
REFERENCES
- 1. Tian B., Hu J., Zhang H., Lutz C.S.. A large-scale analysis of mRNA polyadenylation of human and mouse genes. Nucleic Acids Res. 2005; 33:201–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Zhang H., Lee J.Y., Tian B.. Biased alternative polyadenylation in human tissues. Genome Biol. 2005; 6:R100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Shepard P.J., Choi E.-A., Lu J., Flanagan L.A., Hertel K.J., Shi Y.. Complex and dynamic landscape of RNA polyadenylation revealed by PAS-Seq. RNA. 2011; 17:761–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Derti A., Garrett-Engele P., Macisaac K.D., Stevens R.C., Sriram S., Chen R., Rohl C.A., Johnson J.M., Babak T.. A quantitative atlas of polyadenylation in five mammals. Genome Res. 2012; 22:1173–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Reyes A., Huber W.. Alternative start and termination sites of transcription drive most transcript isoform differences across human tissues. Nucleic Acids Res. 2018; 46:582–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wu F., Chen Q., Liu C., Duan X., Hu J., Liu J., Cao H., Li W., Li H.. Profiles of prognostic alternative splicing signature in hepatocellular carcinoma. Cancer Med. 2020; 9:2171–2180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Darmon S.K., Lutz C.S.. mRNA 3′ end processing factors: a phylogenetic comparison. Comp. Funct. Genomics. 2012; 2012:876893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kubo T., Wada T., Yamaguchi Y., Shimizu A., Handa H.. Knock-down of 25 kDa subunit of cleavage factor im in hela cells alters alternative polyadenylation within 3′-UTRs. Nucleic Acids Res. 2006; 34:6264–6271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Martin G., Gruber A.R., Keller W., Zavolan M.. Genome-wide analysis of pre-mRNA 3′ end processing reveals a decisive role of human cleavage factor i in the regulation of 3′ UTR length. Cell Rep. 2012; 1:753–763. [DOI] [PubMed] [Google Scholar]
- 10. Li W., You B., Hoque M., Zheng D., Luo W., Ji Z., Park J.Y., Gunderson S.I., Kalsotra A., Manley J.L.et al.. Systematic profiling of poly(a)+ transcripts modulated by core 3′ end processing and splicing factors reveals regulatory rules of alternative cleavage and polyadenylation. PLoS Genet. 2015; 11:e1005166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Venkataraman K., Brown K.M., Gilmartin G.M.. Analysis of a noncanonical poly(A) site reveals a tripartite mechanism for vertebrate poly(A) site recognition. Genes Dev. 2005; 19:1315–1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhu Y., Wang X., Forouzmand E., Jeong J., Qiao F., Sowd G.A., Engelman A.N., Xie X., Hertel K.J., Shi Y.. Molecular mechanisms for CFIm-mediated regulation of mRNA alternative polyadenylation. Mol. Cell. 2018; 69:62–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yang S.W., Li L., Connelly J.P., Porter S.N., Kodali K., Gan H., Park J.M., Tacer K.F., Tillman H., Peng J.et al.. A cancer-specific ubiquitin ligase drives mRNA alternative polyadenylation by ubiquitinating the mRNA 3′ end processing complex. Mol. Cell. 2020; 77:1206–1221. [DOI] [PubMed] [Google Scholar]
- 14. Kim S., Yamamoto J., Chen Y., Aida M., Wada T., Handa H., Yamaguchi Y.. Evidence that cleavage factor im is a heterotetrameric protein complex controlling alternative polyadenylation. Genes Cells. 2010; 15:1003–1013. [DOI] [PubMed] [Google Scholar]
- 15. Masamha C.P., Xia Z., Yang J., Albrecht T.R., Li M., Shyu A.-B., Li W., Wagner E.J.. CFIm25 links alternative polyadenylation to glioblastoma tumour suppression. Nature. 2014; 510:412–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Brumbaugh J., Di Stefano B., Wang X., Borkent M., Forouzmand E., Clowers K.J., Ji F., Schwarz B.A., Kalocsay M., Elledge S.J.et al.. Nudt21 controls cell fate by connecting alternative polyadenylation to chromatin signaling. Cell. 2018; 172:629–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sommerkamp P., Altamura S., Renders S., Narr A., Ladel L., Zeisberger P., Eiben P.L., Fawaz M., Rieger M.A., Cabezas-Wallscheid N.et al.. Differential alternative polyadenylation landscapes mediate hematopoietic stem cell activation and regulate glutamine metabolism. Cell Stem Cell. 2020; 26:722–738. [DOI] [PubMed] [Google Scholar]
- 18. Jafari Najaf Abadi M.H., Shafabakhsh R., Asemi Z., Mirzaei H.R., Sahebnasagh R., Mirzaei H., Hamblin M.R.. CFIm25 and alternative polyadenylation: conflicting roles in cancer. Cancer Lett. 2019; 459:112–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Han T., Kim J.K.. Driving glioblastoma growth by alternative polyadenylation. Cell Res. 2014; 24:1023–1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tan S., Li H., Zhang W., Shao Y., Liu Y., Guan H., Wu J., Kang Y., Zhao J., Yu Q.et al.. NUDT21 negatively regulates PSMB2 and CXXC5 by alternative polyadenylation and contributes to hepatocellular carcinoma suppression. Oncogene. 2018; 37:4887–4900. [DOI] [PubMed] [Google Scholar]
- 21. Zhang L., Zhang W.. Knockdown of NUDT21 inhibits proliferation and promotes apoptosis of human K562 leukemia cells through ERK pathway. Cancer Manag. Res. 2018; 10:4311–4323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ghosh S., Bose M., Ray A., Bhattacharyya S.N.. Polysome arrest restricts miRNA turnover by preventing exosomal export of miRNA in growth-retarded mammalian cells. Mol. Biol. Cell. 2015; 26:1072–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cunningham F., Achuthan P., Akanni W., Allen J., Amode M.R., Armean I.M., Bennett R., Bhai J., Billis K., Boddu S.et al.. Ensembl 2019. Nucleic Acids Res. 2019; 47:D745–D751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal. 2011; 17:10–12. [Google Scholar]
- 25. Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R.. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013; 29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Quinlan A.R., Hall I.M.. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010; 26:841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hahne F., Ivanek R.. Visualizing genomic data using gviz and bioconductor. Methods Mol. Biol. 2016; 1418:335–351. [DOI] [PubMed] [Google Scholar]
- 28. Robinson M.D., McCarthy D.J., Smyth G.K.. edgeR: a bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010; 26:139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Benjamini Y., Hochberg Y.. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. 1995; 57:289–300. [Google Scholar]
- 30. Gruber A.J., Schmidt R., Ghosh S., Martin G., Gruber A.R., van Nimwegen E., Zavolan M.. Discovery of physiological and cancer-related regulators of 3′ UTR processing with KAPAC. Genome Biol. 2018; 19:44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Herrmann C.J., Schmidt R., Kanitz A., Artimo P., Gruber A.J., Zavolan M.. PolyASite 2.0: a consolidated atlas of polyadenylation sites from 3′ end sequencing. Nucleic Acids Res. 2020; 48:D174–D179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Martin G., Schmidt R., Gruber A.J., Ghosh S., Keller W., Zavolan M.. 3′ end sequencing library preparation with A-seq2. J. Vis. Exp. 2017; 128:56129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hwang H.-W., Park C.Y., Goodarzi H., Fak J.J., Mele A., Moore M.J., Saito Y., Darnell R.B.. PAPERCLIP identifies MicroRNA targets and a role of cstf64/64tau in promoting Non-canonical poly(A) site usage. Cell Rep. 2016; 15:423–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Szklarczyk D., Morris J.H., Cook H., Kuhn M., Wyder S., Simonovic M., Santos A., Doncheva N.T., Roth A., Bork P.et al.. The STRING database in 2017: quality-controlled protein–protein association networks, made broadly accessible. Nucleic Acids Res. 2016; 45:D362–D368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ahrné E., Glatter T., Viganò C., Schubert C., Nigg E.A., Schmidt A.. Evaluation and improvement of quantification accuracy in isobaric mass tag-based protein quantification experiments. J. Proteome Res. 2016; 15:2537–2547. [DOI] [PubMed] [Google Scholar]
- 36. Wang Y., Yang F., Gritsenko M.A., Wang Y., Clauss T., Liu T., Shen Y., Monroe M.E., Lopez-Ferrer D., Reno T.et al.. Reversed-phase chromatography with multiple fraction concatenation strategy for proteome profiling of human MCF10A cells. Proteomics. 2011; 11:2019–2026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Post H., Penning R., Fitzpatrick M.A., Garrigues L.B., Wu W., MacGillavry H.D., Hoogenraad C.C., Heck A.J.R., Altelaar A.F.M.. Robust, sensitive, and automated phosphopeptide enrichment optimized for low sample amounts applied to primary hippocampal neurons. J. Proteome Res. 2017; 16:728–737. [DOI] [PubMed] [Google Scholar]
- 38. Hernandez-Armenta C., Ochoa D., Gonçalves E., Saez-Rodriguez J., Beltrao P.. Benchmarking substrate-based kinase activity inference using phosphoproteomic data. Bioinformatics. 2017; 33:1845–1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hornbeck P.V., Zhang B., Murray B., Kornhauser J.M., Latham V., Skrzypek E.. PhosphoSitePlus, 2014: mutations, PTMs and recalibrations. Nucleic Acids Res. 2015; 43:D512–D20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ghosh S., Guimaraes J.C., Lanzafame M., Schmidt A., Syed A.P., Dimitriades B., Börsch A., Ghosh S., Mittal N., Montavon T.et al.. Prevention of dsRNA-induced interferon signaling by AGO1x is linked to breast cancer cell proliferation. EMBO J. 2020; 39:e103922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Raj A., van den Bogaard P., Rifkin S.A., van Oudenaarden A., Tyagi S.. Imaging individual mRNA molecules using multiple singly labeled probes. Nat. Methods. 2008; 5:877–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Femino A.M., Fay F.S., Fogarty K., Singer R.H.. Visualization of single RNA transcripts in situ. Science. 1998; 280:585–590. [DOI] [PubMed] [Google Scholar]
- 43. Gruber A.R., Martin G., Keller W., Zavolan M.. Cleavage factor imis a key regulator of 3′ UTR length. RNA Biol. 2012; 9:1405–1412. [DOI] [PubMed] [Google Scholar]
- 44. Sandberg R., Neilson J.R., Sarma A., Sharp P.A., Burge C.B.. Proliferating cells express mRNAs with shortened 3′ untranslated regions and fewer microRNA target sites. Science. 2008; 320:1643–1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Mayr C., Bartel D.P.. Widespread shortening of 3′UTRs by alternative cleavage and polyadenylation activates oncogenes in cancer cells. Cell. 2009; 138:673–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Yu G., Wang L.-G., Han Y., He Q.-Y.. clusterProfiler: an r package for comparing biological themes among gene clusters. OMICS. 2012; 16:284–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S.et al.. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A. 2005; 102:15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hanks S.K., Hunter T.. The eukaryotic protein kinase superfamily: kinase (catalytic) domain structure and classification 1. FASEB J. 1995; 9:576–596. [PubMed] [Google Scholar]
- 49. Stokoe D., Campbell D.G., Nakielny S., Hidaka H., Leevers S.J., Marshall C., Cohen P.. MAPKAP kinase-2; a novel protein kinase activated by mitogen-activated protein kinase. EMBO J. 1992; 11:3985–3994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Meggio F., Pinna L.A.. One-thousand-and-one substrates of protein kinase CK2?. FASEB J. 2003; 17:349–368. [DOI] [PubMed] [Google Scholar]
- 51. Zhang C., Gao J., Li M., Deng Y., Jiang C.. p38δ MAPK regulates aggresome biogenesis by phosphorylating SQSTM1 in response to proteasomal stress. J. Cell Sci. 2018; 131:jcs216671. [DOI] [PubMed] [Google Scholar]
- 52. Guo Y.-J., Pan W.-W., Liu S.-B., Shen Z.-F., Xu Y., Hu L.-L.. ERK/MAPK signalling pathway and tumorigenesis. Exp. Ther. Med. 2020; 19:1997–2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Darling N.J., Cook S.J.. The role of MAPK signalling pathways in the response to endoplasmic reticulum stress. Biochim. Biophys. Acta. 2014; 1843:2150–2163. [DOI] [PubMed] [Google Scholar]
- 54. Smalley I., Smalley K.S.M.. ERK inhibition: a new front in the war against MAPK pathway–driven cancers?. Cancer Discov. 2018; 8:140–142. [DOI] [PubMed] [Google Scholar]
- 55. Gulmann C., Sheehan K.M., Conroy R.M., Wulfkuhle J.D., Espina V., Mullarkey M.J., Kay E.W., Liotta L.A., Petricoin E.F. 3rd. Quantitative cell signalling analysis reveals down-regulation of MAPK pathway activation in colorectal cancer. J. Pathol. 2009; 218:514–519. [DOI] [PubMed] [Google Scholar]
- 56. Blake J.F., Burkard M., Chan J., Chen H., Chou K.-J., Diaz D., Dudley D.A., Gaudino J.J., Gould S.E., Grina J.et al.. Discovery of (S)-1-(1-(4-Chloro-3-fluorophenyl)-2-hydroxyethyl)-4-(2-((1-methyl-1H-pyrazol-5-yl)amino)pyrimidin-4-yl)pyridin-2(1H)-one (GDC-0994), an extracellular signal-regulated kinase 1/2 (ERK1/2) inhibitor in early clinical development. J. Med. Chem. 2016; 59:5650–5660. [DOI] [PubMed] [Google Scholar]
- 57. Papa S., Choy P.M., Bubici C.. The ERK and JNK pathways in the regulation of metabolic reprogramming. Oncogene. 2019; 38:2223–2240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Grassian A.R., Metallo C.M., Coloff J.L., Stephanopoulos G., Brugge J.S.. Erk regulation of pyruvate dehydrogenase flux through PDK4 modulates cell proliferation. Genes Dev. 2011; 25:1716–1733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Vander Heiden M.G., Cantley L.C., Thompson C.B.. Understanding the warburg effect: the metabolic requirements of cell proliferation. Science. 2009; 324:1029–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Mookerjee S.A., Gerencser A.A., Nicholls D.G., Brand M.D.. Quantifying intracellular rates of glycolytic and oxidative ATP production and consumption using extracellular flux measurements. J. Biol. Chem. 2018; 293:12649–12652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. McFate T., Mohyeldin A., Lu H., Thakar J., Henriques J., Halim N.D., Wu H., Schell M.J., Tsang T.M., Teahan O.et al.. Pyruvate dehydrogenase complex activity controls metabolic and malignant phenotype in cancer cells. J. Biol. Chem. 2008; 283:22700–22708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Kaufmann S.H., Desnoyers S., Ottaviano Y., Davidson N.E., Poirier G.G.. Specific proteolytic cleavage of poly(ADP-ribose) polymerase: an early marker of chemotherapy-induced apoptosis. Cancer Res. 1993; 53:3976–3985. [PubMed] [Google Scholar]
- 63. Wang M., Zhao Y., Zhang B.. Efficient test and visualization of multi-set intersections. Sci. Rep. 2015; 5:16923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Schwich O.D., Blümel N., Keller M., Wegener M., Setty S.T., Brunstein M.E., Poser I., De Los Mozos I.R., Suess B., Münch C.et al.. SRSF3 and SRSF7 modulate 3′UTR length through suppression or activation of proximal polyadenylation sites and regulation of CFIm levels. Genome Biol. 2021; 22:82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Kubo T., Wada T., Yamaguchi Y., Shimizu A., Handa H.. Knock-down of 25 kDa subunit of cleavage factor im in hela cells alters alternative polyadenylation within 3′-UTRs. Nucleic Acids Res. 2006; 34:6264–6271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Yang Q., Coseno M., Gilmartin G.M., Doublié S.. Crystal structure of a human cleavage factor CFIm25/CFIm68/RNA complex provides an insight into poly(a) site recognition and RNA looping. Structure. 2011; 19:368–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Zhang W., Liu H.T.. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. 2002; 12:9–18. [DOI] [PubMed] [Google Scholar]
- 68. Ritt D.A., Zhou M., Conrads T.P., Veenstra T.D., Copeland T.D., Morrison D.K.. CK2 is a component of the KSR1 scaffold complex that contributes to raf kinase activation. Curr. Biol. 2007; 17:179–184. [DOI] [PubMed] [Google Scholar]
- 69. Plotnikov A., Chuderland D., Karamansha Y., Livnah O., Seger R.. Nuclear ERK translocation is mediated by protein kinase CK2 and accelerated by autophosphorylation. Cell. Physiol. Biochem. 2019; 53:366–387. [DOI] [PubMed] [Google Scholar]
- 70. Ruzzene M., Pinna L.A.. Addiction to protein kinase CK2: a common denominator of diverse cancer cells. Biochim. Biophys. Acta. 2010; 1804:499–504. [DOI] [PubMed] [Google Scholar]
- 71. Borgo C., Ruzzene M.. Role of protein kinase CK2 in antitumor drug resistance. J. Exp. Clin. Cancer Res. 2019; 38:287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Ahmad K.A., Wang G., Unger G., Slaton J., Ahmed K.. Protein kinase CK2–a key suppressor of apoptosis. Adv. Enzyme Regul. 2008; 48:179–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Silva-Pavez E., Tapia J.C.. Protein kinase CK2 in cancer energetics. Front. Oncol. 2020; 10:893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Wada M., Canals D., Adada M., Coant N., Salama M.F., Helke K.L., Arthur J.S., Shroyer K.R., Kitatani K., Obeid L.M.et al.. P38 delta MAPK promotes breast cancer progression and lung metastasis by enhancing cell proliferation and cell detachment. Oncogene. 2017; 36:6649–6657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Tan F.L.-S., Ooi A., Huang D., Wong J.C., Qian C.-N., Chao C., Ooi L., Tan Y.-M., Chung A., Cheow P.-C.et al.. p38delta/MAPK13 as a diagnostic marker for cholangiocarcinoma and its involvement in cell motility and invasion. Int. J. Cancer. 2010; 126:2353–2361. [DOI] [PubMed] [Google Scholar]
- 76. Nguyen E.V., Huhtinen K., Goo Y.A., Kaipio K., Andersson N., Rantanen V., Hynninen J., Lahesmaa R., Carpen O., Goodlett D.R.. Hyper-phosphorylation of sequestosome-1 distinguishes resistance to cisplatin in patient derived high grade serous ovarian cancer cells. Mol. Cell. Proteomics. 2017; 16:1377–1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Spies N., Burge C.B., Bartel D.P.. 3′ UTR-isoform choice has limited influence on the stability and translational efficiency of most mRNAs in mouse fibroblasts. Genome Res. 2013; 23:2078–2090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Gruber A.R., Martin G., Müller P., Schmidt A., Gruber A.J., Gumienny R., Mittal N., Jayachandran R., Pieters J., Keller W.et al.. Global 3′ UTR shortening has a limited effect on protein abundance in proliferating t cells. Nat. Commun. 2014; 5:5465. [DOI] [PubMed] [Google Scholar]
- 79. Song I.S., Jun S.Y., Na H.-J., Kim H.-T., Jung S.Y., Ha G.H., Park Y.-H., Long L.Z., Yu D.-Y., Kim J.-M.et al.. Inhibition of MKK7-JNK by the TOR signaling pathway regulator-like protein contributes to resistance of HCC cells to TRAIL-induced apoptosis. Gastroenterology. 2012; 143:1341–1351. [DOI] [PubMed] [Google Scholar]
- 80. Huang L., Cai Y., Luo Y., Xiong D., Hou Z., Lv J., Zeng F., Yang Y., Cheng X.. JAZF1 suppresses papillary thyroid carcinoma cell proliferation and facilitates apoptosis via regulating TAK1/NF-κB pathways. Onco. Targets. Ther. 2019; 12:10501–10514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Lee S., Rauch J., Kolch W.. Targeting MAPK signaling in cancer: mechanisms of drug resistance and sensitivity. Int. J. Mol. Sci. 2020; 21:1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Mund T., Pelham H.R.B.. Regulation of PTEN/Akt and MAP kinase signaling pathways by the ubiquitin ligase activators ndfip1 and ndfip2. Proc. Natl. Acad. Sci. U.S.A. 2010; 107:11429–11434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Schmidt R., Ghosh S., Zavolan M.. The 3′ UTR landscape in cancer. eLS. 2018; 10.1002/9780470015902.a0027958. [DOI] [Google Scholar]
- 84. Nagy Á., Munkácsy G., Győrffy B.. Pancancer survival analysis of cancer hallmark genes. Sci. Rep. 2021; 11:6047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Berg M.G., Singh L.N., Younis I., Liu Q., Pinto A.M., Kaida D., Zhang Z., Cho S., Sherrill-Mix S., Wan L.et al.. U1 snRNP determines mRNA length and regulates isoform expression. Cell. 2012; 150:53–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Visa N., Puvion-Dutilleul F., Bachellerie J.P., Puvion E.. Intranuclear distribution of U1 and U2 snRNAs visualized by high resolution in situ hybridization: revelation of a novel compartment containing U1 but not U2 snRNA in hela cells. Eur. J. Cell Biol. 1993; 60:308–321. [PubMed] [Google Scholar]
- 87. Naganuma T., Nakagawa S., Tanigawa A., Sasaki Y.F., Goshima N., Hirose T.. Alternative 3′-end processing of long noncoding RNA initiates construction of nuclear paraspeckles: LncRNA processing for nuclear body architecture. EMBO J. 2012; 31:4020–4034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Jiang L., Shao C., Wu Q.-J., Chen G., Zhou J., Yang B., Li H., Gou L.-T., Zhang Y., Wang Y.et al.. NEAT1 scaffolds RNA-binding proteins and the microprocessor to globally enhance pri-miRNA processing. Nat. Struct. Mol. Biol. 2017; 24:816–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Yamazaki T., Souquere S., Chujo T., Kobelke S., Chong Y.S., Fox A.H., Bond C.S., Nakagawa S., Pierron G., Hirose T.. Functional domains of NEAT1 architectural lncRNA induce paraspeckle assembly through phase separation. Mol. Cell. 2018; 70:1038–1053. [DOI] [PubMed] [Google Scholar]
- 90. Yamazaki T., Yamamoto T., Yoshino H., Souquere S., Nakagawa S., Pierron G., Hirose T.. Paraspeckles are constructed as block copolymer micelles. EMBO J. 2021; 40:e107270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Malka Y., Steiman-Shimony A., Rosenthal E., Argaman L., Cohen-Daniel L., Arbib E., Margalit H., Kaplan T., Berger M.. Post-transcriptional 3′-UTR cleavage of mRNA transcripts generates thousands of stable uncapped autonomous RNA fragments. Nat. Commun. 2017; 8:2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Perez-Riverol Y., Csordas A., Bai J., Bernal-Llinares M., Hewapathirana S., Kundu D.J., Inuganti A., Griss J., Mayer G., Eisenacher M.et al.. The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res. 2019; 47:D442–D450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Edgar R., Domrachev M., Lash A.E.. Gene expression omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002; 30:207–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Heberle H., Meirelles G.V., da Silva F.R., Telles G.P., Minghim R.. InteractiVenn: a web-based tool for the analysis of sets through venn diagrams. BMC Bioinf. 2015; 16:169. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (92) partner repository with the dataset identifier PXD027034 and 10.6019/PXD027034. The source code to replicate all the analyses presented in this study is available at https://github.com/zavolanlab/CFI2021. Raw and processed RNA-seq data are available at Gene Expression Omnibus (GEO) (93) (https://www.ncbi.nlm.nih.gov/geo/) under accession number GSE179630.