


Abstract
The DNAs of bacterial viruses are known to contain diverse, chemically complex modifications to thymidine that protect them from the endonuclease-based defenses of their cellular hosts, but whose biosynthetic origins are enigmatic. Up to half of thymidines in the Pseudomonas phage M6, the Salmonella phage ViI, and others, contain exotic chemical moieties synthesized through the post-replicative modification of 5-hydroxymethyluridine (5-hmdU). We have determined that these thymidine hypermodifications are derived from free amino acids enzymatically installed on 5-hmdU. These appended amino acids are further sculpted by various enzyme classes such as radical SAM isomerases, PLP-dependent decarboxylases, flavin-dependent lyases and acetyltransferases. The combinatorial permutations of thymidine hypermodification genes found in viral metagenomes from geographically widespread sources suggests an untapped reservoir of chemical diversity in DNA hypermodifications.
Graphical Abstract
Graphical Abstract.
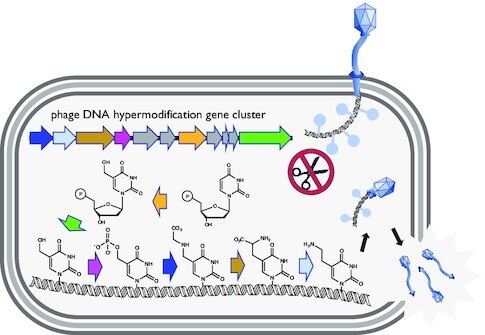
Pathways of thymidine hypermodification.
INTRODUCTION
Many bacteriophage isolates contain nucleobases in their virion DNA that are comparable in composition and complexity to the hypermodified bases of tRNA (1,2). These DNA hypermodifications likely function as viral countermeasures against host-encoded genome defense systems through steric interference of DNA binding by endonucleases (1,2). Examples of partial or complete substitutions of each of the canonical bases in DNA by hypermodified bases include glucosylmethylcytosine in the T-even phages (3), aminocarboxymethyladenine in phage Mu (4) and diverse queuosine-like 7-deazaguanine derivatives in multiple phage and bacterial species (5–7).
Chemically diverse hypermodified thymidines occur in the bacteriophages ΦW-14, SP10, ViI, M6 and others (8–11), the structures of which are shown in Figure 1A. The Delftia phage ΦW-14 contains α-putrescinylthymidine (hereafter referred to as Nα-putT) and the Bacillus phage SP10 contains α-glutamylthymidine (Nα-gluT). Nα-putT and Nα-gluT both extend from the 5-methyl group of thymine from an N-C bond. Salmonella phage ViI contains 5-aminoethoxy-2′-deoxymethyluridine (5-NeOmdU) and the Pseudomonas phage M6 contains 5-aminoethyl-2′-deoxyuridine (5-NedU), respectively substituting for a subset of their thymidines. For these latter two phages, the modifying substituents are connected to thymine through an ether (5-NeOmdU) or a C-C bond (5-NedU). The structures of these four hypermodified thymidines are shown at the pathway termini in Figure 1A. The chemical diversity of these modifications suggests an underlying diversity of enzymatic mechanisms involved in their formation.
Figure 1.

Thymidine hypermodification pathways, intermediates, cofactors, and products. Pathways of thymidine hypermodification (A). The thymidine hypermodifications discussed in this work utilize 5-hmdU, which is incorporated into DNA through steps occurring before and during DNA replication (as diagrammed in the grayed box) and proceed via a 5-PmdU common intermediate. Solid arrows and bolded enzyme names indicate experimentally verified activities, parentheses contain accession IDs for enzymes, and predicted molecular weights follow the abbreviations for the indicated nucleotide/nucleoside products. (B) HPLC/MS traces of nucleotide mixtures derived from mock treated 5-hmdU substrate DNA (lower trace) and M6 gp54 and ATP. Note, these samples were prepared by enzymatic hydrolysis of DNA in the absence of phosphatase activity. (C) HPLC/MS traces of nucleoside mixtures derived from reactions of 5-hmdU with M6 NedU biosynthetic enzymes and cosubstrates. Traces of no enzyme substrate DNA, synthetic Nα-GlyT and native M6 gDNA included for comparison. (D), HPLC/MS traces of nucleoside mixtures derived from reactions of 5-hmdU with ViI 5-NeOmdU biosynthetic enzymes. Traces from synthetic Nα-SerT (an isomer of O-SerT) and native ViI genomic DNA nucleosides included for comparison to enzymatically produced intermediates and final products, respectively. (E) HPLC/MS traces of nucleoside mixtures derived from reactions with PaMx11 5-AcNmdU biosynthetic enzymes. No enzyme DNA substrate control, and synthetic 5-NmdU standard shown for comparison.
Biosynthesis of thymidine hypermodifications occurs through mechanisms operating before and after DNA polymerization. During intracellular development, phages ΦW-14, SP10, ViI and M6 initially synthesize DNA containing 5-hmdU fully replacing thymidine using mechanisms similar to those of the Bacillus ‘hmU phages’ such as SPO1, 2C, SP8 and ϕe, which encode a suite of metabolic functions that eliminate dTTP from the deoxynucleotide triphosphate (dNTP) pool of their host and replace it with 5-hydroxymethyl-2′-deoxyuridine triphosphate (5-hmdUTP) (12). This pre-replicative pathway is depicted in the grayed boxed region of Figure 1A. Central to this pathway is 2′-deoxyuridine monophosphate (dUMP) hydroxymethylase, a virally encoded thymidylate synthase homolog producing 5-hydroxymethyl-2′-deoxyuridine monophosphate (5-hmdUMP) from dUMP (13–15). Phage encoded dTTPase (16) and dTMPase (17) deplete deoxythymidine mononucleotide pools and a phage encoded dCMP deaminase (18) supplies dUMP substrate for dUMP hydroxymethylase. A phage encoded deoxynucleotide monophosphate kinase phosphorylates 5-hmdUMP, and the resulting nucleotide diphosphate is converted to 5-hmdUTP by host deoxynucleotide diphosphate kinases (14). Phage encoded DNA polymerases then incorporate 5-hydroxymethyl-2′-deoxyuridine (5-hmdU) into DNA during replication.
However, DNA recovered from purified virions of SP10, ΦW-14, M6 and ViI contain no 5-hmdU. Instead, these thymidine hypermodifying phages convert 5-hmdU in sequence-specific contexts to either a hypermodified base (Figure 1A) or canonical thymine prior to packaging of the viral DNA into the phage capsids (8,19–21). Conversion of 5-hmdU to the hypermodified base in ΦW-14 and SP10 was reported to proceed by pyrophosphorylation of 5-hmdU in the DNA polymer (9,22). The pyrophosphoryl moiety was postulated to act as a leaving group during a nucleophilic substitution reaction with putrescine (putT) or glutamate (gluT). Using a combination of sensitive homology detection, genome context and comparative genomics, Aravind et al. identified a subfamily of P-loop kinases encoded by ΦW-14, SP10, ViI and M6 predicted to be a 5-hmU DNA kinase (5-HMUDK) synthesizing 5-pyrophosphoryloxymethyldeoxyuridine (5-PPmdU) (23). They also noted the co-occurrence with 5-HMUDK of genes predicted to encode a DNA glycosylase-like protein having a helix-hairpin-helix fold. These genes, often present as a paralogous pair, were named alpha-putrescinyl/glutamylthymidine pyrophosphorylases (aGPT-Pplase) after their predicted activity. Additionally, a predicted pyridoxal phosphate (PLP) dependent enzyme frequently co-occurs in viral genomes together with 5-HMUDK and aGPT-Pplase genes.
Previously, we demonstrated ex vivo biosynthesis of 5-NeOmdU by incubation of a 5-hmdU DNA substrate in a mixture of five crude lysates derived from recombinant Escherichia coli cultures expressing two paralogous predicted 5-HMUDKs, two paralogous aGPT-Pplases, and a predicted PLP-dependent enzyme (8). These results established the involvement of these genes in the biosynthesis of 5-NeOmdU; however, the source of the ethanolamine moiety and the order in which the enzymes participated in the conversion of 5-hmdU to 5-NeOmdU were unknown. Furthermore, the essentiality of either aGPT-Pplase in the pathway remained to be established. Building upon our ex vivo approach to reconstitution of thymidine hypermodification, here we repeated reactions leaving out components in various combinations to determine what enzymes were necessary and sufficient to the hypermodification reaction. We also reconstituted each of the enzymatic steps for in vitro syntheses of both 5-NeOmdU and 5-NedU using pure recombinant enzymes, substrates, and cofactors.
In this study, we identify the source of 5-NeOmdU and 5-NedU modifications to be serine and glycine respectively. We reveal enzymatic activities not previously seen modifying nucleobases in a polymeric DNA substrate: kinase (5-HMUDK), PLP-dependent decarboxylase, radical-SAM dependent isomerase and a novel amino acid:DNA transferase (AADT; encoded by the aforementioned aGPT-Pplase gene). Additionally, after observing a 5-HMUDK and an AADT encoded together with a flavin-adenine dinucleotide (FAD) dependent oxidoreductase and an acetyltransferase in the genome of the Pseudomonas phage PaMx11, we purified these enzymes and reconstituted the biosynthesis of a newly identified thymidine hypermodification, 5-acetylaminomethyl-2′-deoxyuridine (5-AcNmdU). Interestingly, the Pseudomonas phages PaMx11 and M6 both utilize a glycinylated thymidine intermediate in the synthesis of their respective hypermodifications. Chemically, this glycinylated thymidine is the DNA equivalent of the tRNA wobble uridine (U34) hypermodification 5-carboxymethylaminomethyluridine (cmnmU) suggesting parallels and/or adaptation of RNA hypermodification pathways for DNA modification. Looking beyond the cultured phages with known thymidine hypermodifications, we uncover homologs of these hypermodification pathway enzymes widely encoded in the viral metagenomic contigs of the IMG/VR2 (24,25) and GOV2.0 datasets (26). Their co-association with other predicted enzymatic activities, such as methylpyrimidine oxidizing iron/alpha-ketoglutarate (Fe/aKG) dependent dioxygenases, glutamidotransferases and amidoligases, in biosynthetic gene clusters akin to natural product biosynthesis (27) suggests that bacterial viruses have access to a gene-pool encoding an under-explored chemical diversity of nucleobase hypermodifications in DNA.
MATERIALS AND METHODS
Preparation of biotinylated DNA substrate for ex vivo hypermodification activity assay
SP8 genomic DNA was isolated and purified from ultracentrifugation-purified SP8 phage using phenol-chloroform extraction and alcohol precipitation procedures. The purified SP8 genomic DNA was digested to fragments using restriction endonuclease HpyCH4IV (NEB) according to manufacturer’s guidelines. SP8 DNA has 308 predicted HpyCH4IV cut sites distributed across the whole genome and the digested product contains fragments with wide range of sizes where the largest of which being 3.12 kb, and each having 5′ CG overhangs. Following heat-inactivation at 80°C for 20 min, the digest was purified using the Monarch® PCR & DNA Cleanup Kit (NEB). Fragmented SP8 genomic DNA at a final concentration of 200 ng/μl was further labeled with biotin and phosphorothioate at the two ends of a fragment by incubating 1 U/μL of Klenow fragment (3′–> 5′ exo–) DNA polymerase (NEB) with 50 μM biotin-16-dCTP (ChemCyte, Inc., San Diego, CA, USA) and 200 μM α-thio-dCTP (Trilink Bio Technologies, San Diego, CA, USA) at 37°C for 3 h in NEBuffer 2 (NEB). Biotin labeling allows specific DNA fragments to be recovered after enzymatic reaction by biding streptavidin coated magnetic beads (NEB); phosphorothioate labeling enhances the DNA fragments’ robustness against endogenous exonucleases in reactions containing cell lysates. After the labeling reaction, the biotinylated DNA product was purified using a QIAquick Nucleotide Removal Kit (Qiagen) or Monarch PCR & DNA Cleanup Kit (NEB).
Expression of candidate thymidine hypermodification enzymes
Each predicted putative phage thymidine hypermodification candidate gene was cloned using PCR amplicons from native templates or synthesized with codon optimization for bacterial expression through Genscript (Genscript, Piscataway, NJ, USA). The synthesized genes were cloned into pET28b(+) vectors under the control of the T7 promoter (see Supplementary Table S1 for plasmid names and details). The sequence-confirmed constructs were used to transform E. coli strain NEB T7 Express for protein expression. LB medium was inoculated 1:50 of the culture volume from an overnight starting culture of the transformed strain and the culture was incubated at 37°C with agitation until it reached an OD600 ∼ 0.6. The culture was then cooled at RT before adding isopropyl β-D-1-thiolgalactopyranoside (IPTG) at a final concentration of 0.1 mM. After induction of expression, the culture was incubated at 16°C with shaking for overnight (∼16–20 h). Cells were pelleted by centrifugation. The supernatant was discarded, and the pellet was stored at -20°C until protein purification step.
Preparation of E. coli lysates containing candidate thymidine hypermodifying enzymes
To prepare cell lysate containing the recombinantly expressed putative thymidine modifying enzyme, the previously stored cell pellet was removed from the freezer and thawed on ice. The pellet was then added lysis buffer (10 mM Tris-HCl pH 8, 100 mM NaCl, 10 mM KCl and 1 mM phenylmethane sulfonyl fluoride [PMSF]) to resuspend. Cell disruption was carried out using a Q500 micro tip sonicator (Qsonica, Newtown, CT, USA) for 2 min with a 33% duty cycle and 30% power. The resulting lysate was clarified by centrifugation at 15 000 rpm, 4°C for 15 min, and the supernatant was collected. Expression of each gene product candidate was confirmed by SDS-PAGE analysis. Aliquots of lysates were frozen in liquid nitrogen and stored at −80°C until activity assay.
In vitro DNA modification assay
Biotinylated SP8 genomic DNA fragment (4 μg) was added to a mixture of total protein lysate or to a mixture of defined composition of purified protein containing approximately 20 μg total protein with other supplements such as ATP (1 mM), RNaseA (50 μg/μl) and reaction buffer (25 mM Tris-HCl pH7.5, 5 mM b-mercaptoethanol, 5 mM MgCl2, 25 mM KCl). The combined DNA–protein mixture was incubated at 37°C for 1 h and then quenched by addition of an equal volume of quenching/bead-binding buffer (20 mM Tris-HCl pH 8, 2 M NaCl, 5% polyvinyl alcohol [average MW 30 000–70 000] and 2 mM EDTA). Streptavidin-coated magnetic beads (NEB) were used to capture and separate the biotinylated SP8 DNA from endogenous nucleic acids. The beads (30 μl slurry) were first conditioned by washing with a bead-washing buffer (20 mM Tris-HCl pH 7.5, 0.5 M NaCl, 1 mM EDTA). Then the quenched DNA–protein reaction mixture was transferred to mix with the conditioned-beads. The mixture suspension was incubated at ambient temperature for 10 min after gentle mixing. The DNA-bound beads were captured by a magnet and the supernatant was discarded. Captured beads were further cleaned for another two wash cycles using bead-washing buffer then finally suspended in 43 μl of deionized water. To release the nucleosides from the beads for analysis, 5 μl of Nucleoside Digestion Mix Buffer (50 mM NaOAc pH 5.4, 1 mM ZnCl2) and 2 μl of Nucleoside Digestion Mix (NEB) were added to the bead suspension and incubated at 37°C for at least 2 h. The beads were removed using a magnet and the supernatant was collected and subjected to LC-MS analysis (see Chemical Methods). To estimate the ratio of phosphorylation of 5-hydroxymethyluridine, kinase-treated SP8 gDNA was digested to nucleotides by treatment with DNase I and P1 nuclease.
Purification of M6 gp53
Plasmid pYJL078 was co-transformed into E. coli NEB T7 Express with the iron sulfur cluster biogenesis chaperone expressing plasmid pDB1282 (28). Cells were cultured in TB medium with ampicillin (100 μg/ml) and kanamycin (50 μg/ml). The culture was incubated at 37°C and 250 rpm until the OD600 reached 0.3, when final concentrations of FeCl3 (25 μM), L-cysteine (300 μM) and L-arabinose (0.2% w/v) were added to the medium. When the OD600 reached 0.6, the culture was cooled to 18°C before the addition of IPTG to a final concentration of 0.1 mM and the mixing of flasks to remove headspace. The culture was allowed to incubate at 18°C with gentle agitation for an additional 18 h. The cells were subsequently harvested, sonicated and the clarified lysate was subjected to HisTrapHP column (GE Healthcare) according to the manufacturer’s protocol. N-terminal hexahistidine-tagged M6 gp53 was eluted with buffer containing 50 mM Tris pH 8, 200 mM NaCl, 500 mM imidazole and 1 mM dithiothreitol (DTT). M6gp53 was buffer exchanged into 50 mM Tris pH 8, 200 mM NaCl, 5 mM imidazole and 1 mM DTT before being subjected to a second round of the HisTrapHP column (GE Healthcare) purification, after which the eluent containing the desired protein was run through a HiLoad 16/600 Superdex 75 prep grade preparative gel filtration column (GE Healthcare). The sample was collected in buffer containing 25 mM Tris pH 8, 200 mM NaCl, and 5 mM DTT and then aliquoted and stored at -80°C.
Anaerobic FeS reconstitution of M6 gp53 for use in enzyme assays
It was noted that during purification, M6 gp53 retained rSAM enzymes’ characteristic brown color from the [4Fe-4S] cluster, suggesting retention of iron during aerobic purification. Analysis of the aerobically purified sample suggests 2.63 mol of Fe per protein molecule, determined according to published procedures (29,30); therefore, cluster reconstitution was employed based on a previously reported procedure to regenerate the rSAM enzyme’s [4Fe-4S] cluster (31). All reconstitution and further purification steps were performed within an anaerobic Coy chamber (approximately 95–97% N2, and 3–5% H2) using degassed or N2-purged samples. To initiate the reconstitution of the FeS clusters of M6gp53, the protein was reduced by the addition of 5 mM DTT at room temperature for 30 min. For cluster reconstitution, (NH4)2Fe(SO4)2 and Na2S were each added to the reduced protein at a 5 mol excess. The mixture was allowed to incubate at 12°C for 90 min. To remove excess iron sulfide particles and precipitated protein, the mixture was centrifuged at 5000 × g for 5 min. Any unbound iron sulfide was removed by passage through Micro Bio-Spin P-6 chromatography columns (Bio-Rad) that were buffer exchanged into 25 mM Tris (pH 8), 200 mM NaCl, and 5 mM DTT. Reconstituted M6gp53 had an iron content of 9.12 mole Fe per mol of M6gp53, which is agreeable with two [4Fe-4S] clusters per molecule of protein.
Modification in trans
Escherichia coli ATCC 700728 was transformed with a plasmid containing a modification gene of interest under the control of a Ptac promoter. The list of genes of interest is summarized on Supplementary Table S1. Overnight cultures of the resulting transformants were used to inoculate 50 ml LB medium in a conical shaking flask. The culture was incubated at 37°C until an OD600 of 0.1 was reached and subsequently induced by addition of IPTG to the medium at a final concentration of 0.1 mM. The cultures were incubated for another 20 min before infecting with phage CBA120 at an MOI ∼5. The infected E. coli cultures were further incubated with shaking at 37°C until lysis or for overnight. To collect the phages, the culture was centrifuged at 10 000 RCF at 4°C for 15 min to remove debris and unlysed cells. The clarified lysate was spot titer assayed and then further concentrated by precipitation addition of PEG8000 to 10% w/v and NaCl at 1 M final concentration. Following centrifugation at 12×000 RCF for 10 min, the pellet phages were resuspended in 5 ml of buffer containing 25 mM Tris-HCl pH 7.5, 75 mM NaCl and 10 mM MgCl2. Phage DNA was extracted, purified, digested to free nucleosides and subjected to LC/MS analysis as previously described (32).
Chemical methods
Unless otherwise specified, all chemicals were obtained from Sigma-Aldrich (St. Louis, MO) and used without further purification. 2′-deoxy-5-formyluridine was obtained from Carbosynth (Compton, UK). 5-Nα-glycinylthymidine (Nα-GlyT) and 5-Nα-serinylthymidine (Nα-SerT) were synthesized following a method adapted from Saavedra (33). Reaction yields were not optimized. Analytical reversed-phase HPLC was performed on an Agilent 1200 Series LC/MS System equipped with a G1315D Diode Array Detector and a 6120 Single Quadrupole Mass Detector in both positive (+ESI) and negative (-ESI) electrospray ionization modes. LC was performed on a Waters Atlantis T3 column (4.6 × 150 mm, 3 μm) with a gradient mobile phase consisting of aqueous ammonium acetate (10 mM, pH 4.5) and methanol. The relative abundance of each nucleoside was determined by UV absorbance. Preparative-scale HPLC purification was performed on a Waters 2535 System using a Waters Atlantis T3 OBD (10 μm, 50 × 250 mm) with a gradient mobile phase of aqueous ammonium acetate (10 mM, pH 4.5) and methanol. High-Resolution MS (HRMS) was performed on a Thermo Scientific Q Exactive Plus hybrid quadrupole-orbitrap mass spectrometer using direct injection. NMR experiments were performed by Novatia, LCC (Newtown, PA, USA).
((1-((2R,4S,5R)-4-hydroxy-5-(hydroxymethyl)tetrahydrofuran-2-yl)-2,4-dioxo-1,2,3,4-tetrahydropyrimidin-5-yl)methyl)glycine, Nα-GlyT: 2′-deoxy-5-formyluridine (92.5 mg, 1.0 eq) was suspended in 2.4 ml anhydrous ethanol, and glycine-tert-butyl ester (90.8 mg, 1.5 eq) was added to the solution. The reaction was stirred overnight at room temperature. NaBH4 (20.5 mg, 1.5 eq) was added, then stirred at room temperature for 4 h. Water (2.0 ml) was slowly added to quench the reaction. The crude reaction mixture was filtered and purified by preparative HPLC. A white solid (57.5 mg, 43% yield) was obtained after lyophilization. The resulting product was suspended in 1.0 ml anhydrous dichloromethane, then 1.0 ml TFA was added into the solution. The reaction was stirred at room temperature for 4 h. The solvent was evaporated, and the product dried under high vacuum overnight. The crude product was purified by preparative HPLC. The target product was isolated after lyophilization as a white powder (28.7 mg, 59% yield). ESI-HRMS calc. for C12H18N3O7, [M + H]+: m/z 316.1145, found m/z 316.1137. 1H NMR (500 MHz, DMSO-d6) δ 7.97 (s, 1H), 6.14 (t, J = 6.7 Hz, 1H), 4.25 (q, J = 4.3 Hz, 1H), 3.77 (q, J = 3.7 Hz, 1H), 3.60 (dd, J = 12.3, 3.9 Hz, 2H), 3.58 – 3.51 (m, 2H), 2.11 (t, J = 5.7 Hz, 2H). 13C NMR (126 MHz, DMSO-d6) δ 170.05, 163.20, 150.22, 139.99, 107.72, 87.54, 84.20, 70.18, 61.17, 49.78, 43.45.
((1-((2R,4S,5R)-4-hydroxy-5-(hydroxymethyl)tetrahydrofuran-2-yl)-2,4-dioxo-1,2,3,4-tetrahydropyrimidin-5-yl)methyl)-D-serine, Nα-SerT: 2′-deoxy-5-formyluridine (42.6 mg, 1.0 eq) was suspended in 2.4 ml anhydrous ethanol, and O-tert-butyl-L-serine-tert-butyl ester (63.3 mg, 1.5 eq) was added to the solution. The reaction was stirred overnight at room temperature. NaBH4 (9.5 mg, 1.5 eq) was added, then stirred at room temperature for 4 h. Water (1.5 ml) was slowly added to quench the reaction. The crude reaction mixture was filtered and purified by preparative HPLC. A white solid (18.4 mg, 24% yield) was obtained after lyophilization. The resulting product was suspended in 1.0 ml of anhydrous dichloromethane, then 0.5 ml TFA was added into the solution. The reaction was stirred at room temperature for 4 h. The solvent was evaporated, and the product dried under high vacuum for overnight to yield brownish oil (13.7 mg, 92% purity by analytical HPLC). The crude product was used without further purification. ESI-MS calc. for C13H20N3O8 [M + H]+: m/z 346.1, found m/z 346.1; calc. for C13H18N3O8 [M − H]–: m/z 344.1, found m/z 344.2.
RESULTS
Phage-encoded 5-hydroxymethyluracil DNA kinases
In previous work by Aravind et al., comparative genomics was combined with highly sensitive homology detection to bioinformatically identify candidate DNA nucleobase kinases active on 5-hmdU in ΦW-14 and SP10 genomes (23) (see also Figure 1A). Homologs of these genes were also found to be encoded in other phages, including M6 and ViI. We cloned, expressed and purified gp54, the putative 5-hmdU DNA kinase of M6 (Supplementary Figure S1). Catalytic activity of M6 gp54 was assessed by incubating with genomic DNA containing 5-hmdU isolated from the Bacillus bacteriophage SP8 in the presence of 1 mM ATP. Phage SP8 genomic DNA fully substitutes thymidine with 5-hmdU and, thus, presents this non-canonical base in a variety of sequence contexts. Following purification to remove protein and low MW products, the kinase treated DNA was digested to free nucleotides using DNaseI and P1 nuclease and analyzed by liquid chromatography-mass spectrometry (LC-MS) to determine the masses of the dominant species in each peak.
As seen in Figure 1B, following treatment of the SP8 genomic DNA with purified M6 gp54, a new peak containing a species of 418 u (nominal mass) nucleotide eluting at ∼5 min was produced. A corresponding decrease in the amount 5-hmdUMP, which elutes at ∼10 min and exhibits a nominal mass of 388 u was observed, as can be determined from comparing to the integrated peak areas of the dA as an internal control. Under these conditions, nearly 23% of the 5-hmdU is converted to the new product. The 80 u mass difference between the new product and 5-hmdUMP corresponds to the gain of a phosphate group. Ratios of the integrated peak areas of the 5-hmdU to dA before and after treatment of the kinase indicated that approximately 23% of 5-hmdU bases were phosphorylated. The reaction produced only ADP as the other major product (Supplementary Figure S2).
A 418 u nucleotide with identical retention time as that produced by M6 gp54 is also observed in reactions using the predicted nucleobase kinases ViI gp67, ViI gp243, ΦW-14 gp37, SP10 gp186, as well as a C-terminal deletion of SP10 gp186 corresponding to the kinase domain alone (Supplementary Figure S5). In experiments using [γ-32P]-ATP, the radiolabeled phosphate is transferred to the DNA (Supplementary Figure S4A) as detected by phosphor imaging. Phosphorylation of the 5-hmdU DNA by M6 gp54 resulted in protection of the DNA from cleavage by the restriction endonuclease NcoI (Supplementary Figure S4B), providing further support that the phosphate is added internally to the DNA polymer.
In contrast with previous studies that suggested 5-PPmdU was the phosphorylation product (9,22) (which would have yielded 5-PPmdUMP in our method), only the singly phosphorylated nucleobase base form of 5-hmdU (i.e. 5-PmdUMP) was observed, even in the presence of excess (5 mM) ATP. Although triply phosphorylated nucleotides are highly polar, and therefore potentially lost in the void volume during HPLC separation, our chromatographic method was capable of resolving each of the canonical dNTPs (Supplementary Figure S3). Thus, the enzymatic transformations carried out by M6 gp54 and its aforementioned homologs allow one to conclude that they function as 5-hmdU DNA kinases (5-HMUDKs) that catalyze transfer of a single phosphate to the 5-hydoxyl moiety of 5-hmdU to produce 5-phosphomethyl-2′-deoxyuridine (5-PmdU). To the best of our knowledge, 5-HMDUK enzymes are the first kinases shown to work directly on the base of a pyrimidine nucleotide. The closest example of this chemistry is the hydroxymethylpyrimidine (HMP) kinase ThiD, which established precedent for the phosphorylation of free hydroxymethypyrimidines (34).
Enzymatic transfer of free amino acids to DNA
In our previous work, we have shown that a mixture of five cell extracts derived from individual E. coli cultures, each recombinantly expressing Salmonella phage ViI gp67, gp160, gp226, gp243 or gp247 (see also Table 1), when combined with a 5-hmdU DNA substrate (see Supplementary Figure S6 for a schematic outline of this experimental workflow) could produce a modified nucleoside indistinguishable in mass (301 u) and retention time from the native ViI thymidine hypermodification, 5-NeOmdU (8), as detected by LC/MS based nucleoside analysis (see Materials and Methods section for details). Using the same approach, we attempted to reconstitute the formation of the Pseudomonas phage M6 modification, 5-NedU, from a mixture of five lysates containing the M6 proteins gp51, gp52, gp53, gp54 and gp55. The Pfam assignments and Genbank accession numbers of these gene products are listed in Table 1 and their genomic context is illustrated in sequence 1 of Figure 2.
Table 1.
Proteins described in this study
| Phage | Protein (gp) | accession | Pfam/abbrev. | Function |
|---|---|---|---|---|
| M6 | gp51 | YP_001294559 | aGPT-Pplase2 | glycine:DNA transferase |
| M6 | gp52 | YP_001294560 | PLP | PLP-dependent decarboxylase |
| M6 | gp53 | YP_001294561 | rSAM | glycinyl-thymine isomerase |
| M6 | gp54 | YP_001294562 | P-loop kinase | 5-hmdU DNA kinase |
| PaMx11 | gp46 | YP_009196299 | aGPT-Pplase2 | glycine:DNA transferase |
| PaMx11 | gp47 | YP_009196300 | FAD | flavin-dependent lyase |
| PaMx11 | gp48 | YP_009196301 | AT | acetyltransferase |
| PaMx11 | gp49 | YP_009196302 | P-loop kinase | 5-hmdU DNA kinase |
| SP10 | gp186 | YP_007003443 | P-loop kinase | 5-hmdU DNA kinase |
| ViI | gp67 | YP_004327432 | P-loop kinase | 5-hmdU DNA kinase |
| ViI | gp226 | YP_004327553 | PLP | PLP-dependent decarboxylase |
| ViI | gp243 | YP_004327565 | P-loop kinase | 5-hmdU DNA kinase |
| ViI | gp247 | YP_004327568 | aGPT-Pplase2 | serine:DNA transferase |
| ΦW-14 | gp109 | YP_003358963 | aGPT-Pplase2 | putrescine:DNA transferase |
| ΦW-14 | gp37 | YP_003358891 | P-loop kinase | 5-hmdU DNA kinase |
Figure 2.

Thymidine hypermodification biosynthetic gene clusters of cultured phages and the global metavirome. Subgenomic regions spanning thymidine hypermodification genes encoded by cultured phages are depicted together with regions of meta-virome derived contigs having similar organization and therefore predicted to synthesize the same modification (sequences 1–13). Open reading frames are labeled with their functional name and/or Pfam annotation and homologous features share the same colors. Combinatorial permutations of hypermodification genes whose functions have been determined in this work are shown in sequences 14–25, including multi-domain fusions (sequences 14–16), combinations not previously observed in cultured phages (sequences 17–21), and association with predicted 5-methylpyrimidine dioxygenases of the Tet/JBP family (sequences 22–25) leading to the in situ formation of hydroxyl acceptor groups for subsequent nucleobase modifications. Sequences accession IDs labeled with the prefix ‘NC_’ were obtained from Genbank, those with the prefix ‘Station’ were obtained from GOV2.0, and all others were obtained from JGI IMG/VR2.
Instead of forming a nucleotide matching 5-NedU (271 u), our expression mixture unexpectedly yielded a new compound with a nominal mass of 315 u, exactly 44 u greater than the native 5-NedU nucleoside. Reactions excluding M6 gp52, gp53 and gp55, and consisting only of lysates expressing gp54 (the 5-HMUDK) and gp51 (Pfam annotation: aGPT-Pplase2) were sufficient to produce the 315 u product (Supplementary Figure S7). Reactions excluding gp54 or gp51 failed to form the 315 u nucleoside indicating each enzyme was necessary for its formation from a 5-hmdU DNA substrate and that the M6 clade 2 aGPT-Pplase, gp51, is likely responsible for group transfer to 5-PmdU and displacement of the phosphate (Supplementary Figure S7). The ViI clade 2 aGPT-Pplase, gp247, was similarly necessary for modification in lysate reconstitution experiments (Supplementary Figure S8). The 44 u mass difference between the M6 reaction product and the native hypermodified base, as well as the presence of predicted PLP-dependent enzymes encoded by both M6 and ViI genomes, suggests the nucleoside observed in the recombinant M6 lysate catalyzed reaction results from the accumulation of a carboxylated intermediate in the modification pathway. Indeed, a recombinant lysate mixture expressing four ViI proteins, excluding the predicted PLP-dependent ViI gp226 protein, produced a nucleoside of nominal mass 345 u, also 44 u greater than the native ViI 5-NeOmdU modification (Supplementary Figure S8). Conceptually, adding a carboxylate to the side groups present at C5 of 5-NedU and 5-NeOmdU implies thymidines potentially modified with glycine and serine, respectively, as intermediates in the formation of these hypermodifications.
Following evidence from the lysate reconstitution experiments suggesting that phage M6 gp51 and ViI gp247, both annotated as clade 2 aGPT-Pplases, catalyze group transfer to a 5-PmdU DNA substrate, we set out to reconstitute these enzymes' activities from purified components in vitro. Purified M6 gp51 incubated with free glycine and 5-hmdU DNA previously treated with 5-hmU DNA kinase produced a new peak in the LC-MS traces with nominal mass of 315 u, demonstrating that the monophosphorylated thymine is chemically competent for further enzymatic modification (Figure 1C). To unambiguously show that glycine was being appended to the nucleobase, when the experiment was repeated with glycine-1,2–13C2, a product of 317 u was observed (Supplementary Figure S9) confirming the addition of isotopically labeled glycine. 5-NedU from phage M6 contains a C-C bond between the aminomethyl group and the C5 methyl of thymine. If glycine is the source of the aminomethyl group, one would expect that when glycine deuterated at C-2 is used, the final product would not retain both of the deuterons. However, M6 gp51 modification reactions using glycine doubly deuterated at the alpha carbon (i.e. glycine-2,2-d2) produced a species of 317 u showing retention of both deuterons (Supplementary Figure S10). These data suggest that an N-C bond is formed by appending the nucleophilic α-amine of glycine to the base to produce 5-Nα-glycinylthymidine (Nα-GlyT) on DNA. To test this possibility, Nα-GlyT was synthesized by reductive amination of 5-formyl-2′-deoxyuridine as described in the Supplementary methods and Supplementary Figure S11. As seen in Figure 1C and Supplementary Figure S11, this compound had identical mass and retention time to the nucleoside enzymatically produced by M6 gp51, suggesting that this intermediate is derived from 5-PmdU in the pathway leading to 5-NedU, as depicted in Figure 1A.
Amino acid transferase activity was also demonstrated in reactions with purified ViI gp247 (Figure 1D). A similar reaction, but with L-serine-(13C3,15N) produced a nucleoside with the expected 4 u additional mass (Supplementary Figure S12). The native ViI hypermodification contains an ether linkage between an ethanolamine moiety and the C5 methyl of thymidine, suggesting serine is initially appended to the base via its sidechain hydroxyl group to produce the nucleobase 5-O-serinylthymidine (O-SerT). However, based on mass alone, we could not initially rule out incorporation of serine by ViI gp247 into DNA through substitution reaction via the relatively more nucleophilic serine α-amine to produce 5-Nα-serinylthymidine (Nα-SerT). To rule out this latter possibility, a synthetically more accessible nucleoside standard consisting of serine modified thymidine containing an N-C linkage was synthesized by reductive amination of 5-formyluridine as described in the Supplementary Methods and in the scheme illustrated in Supplementary Figure S13. As seen in Figure 1D, this synthetic compound had a different retention time to the ViI enzymatic product, despite having identical mass, thus ruling out this isomer and indicating 5-O-serinylthymidine as the likely intermediate produced by ViI gp247 on the pathway to 5-NeOmdU as shown in Figure 1A.
Amino acid insertion via exocyclic C5 methylene intermediate
One can envision two possible mechanistic pathways to the formation of Nα-GlyT and O-SerT from 5-PmdU. The simplest is a displacement reaction, whereby a nucleophilic group from the incoming substrate displaces the phosphate at C5 to generate the product. The second would involve initial activation of the substrate by covalent attachment to an active site residue and concomitant elimination of the phosphate, which would then undergo Michael addition to form the product. Previously, a mechanism of polymer level biosynthesis of glutamyl-thymidine in the Bacillus phage SP10 proposed by Witmer in 1981 invoked the formation of a covalent intermediate by Michael addition of an active site cysteine thiol in the then unknown enzyme to position six of the pyrimidine ring in 5-PmdU (9), a mechanism reminiscent of classical thymidylate synthases (35). In this model, subsequent departure of the phosphate was proposed to result in the formation of an electrophilic exocyclic methylene at position five, which could be subject to attack by nucleophiles such as primary amines and hydroxyl groups.
Aravind et al. noted a highly conserved cysteine and neighboring glutamate in sequence alignments of clade 2 aGPT-Pplases, proposing these to be putative active site residues (23). An alignment of aGPT-Pplases highlighting this conserved cysteine and nearby glutamate is shown in Figure 3A. Site-directed mutation of either the cysteine or the proximal glutamate to alanine resulted in a loss of activity in both M6 gp51 and ViI gp247 in our in vitro assay (Supplementary Figure S14). If an aGPT-Pplase2 active site cysteine does form a covalent bond with C6 of 5-PmdU with concomitant formation of an exocyclic methylene, then one might expect that in the absence of the natural co-substrate the methylene might be accessible to exogenously added nucleophile or reductant. As shown in Figure 3B, incubation of M6 gp51with 5PhmdU and sodium borohydride in the absence of glycine resulted in the formation of thymidine, a result consistent with intermediacy of a methylene. A similar experiment using beta-mercaptoethanol (βME) resulted in the formation of a product with mass of 398 u identical to the mass of a putative βME adduct to the base (Figure 3B). Given that a thiol is a stronger nucleophile than a hydroxyl, we provisionally assigned this product as 5-(2-hydroxyethyl)thiomethyl-2′-deoxyuridine (5-heSmdU). Taken together, these data suggest the formation of a covalent intermediate and exocyclic methylene during the first half of the reaction between aGPT-Pplase2 and 5-PmdU as depicted in the pathway scheme shown in Figure 3D. In the case of gp247 of ViI, the exocyclic methylene is targeted by the hydroxyl of a serine side chain, whereas for M6 gp51, the attacking nucleophile is the α-amino group of glycine.
Figure 3.

Proposed mechanism of 5-hmdU glycinylation. (A) Multiple sequence alignment of the predicted active site of clade 2 aGPT-PPlase homologs colored according to degree of conservation and showing context of an essential cysteine predicted to form thioether and neighboring essential glutamate predicted to function as generalized acid. (B) Nucleotide analysis of 5-hmdU DNAs treated with M6 gp54 and gp51 in the presence of β-mercaptoethanol (βME) leads to the formation of adduct. (C) Nucleoside analysis of 5-hmdU DNA treated with gp54 and gp51 in the presence of sodium borohydride leading to the formation of deoxythymidine. Note, the HPLC method used did not separate the nucleoside form of the βME adduct from rA, though the monophosphate nucleotide form of this adducted species does resolve from rA, as seen in panel (B). (D) Proposed mechanism for glycinylation of 5-hmdU by M6 gp51 and PaMx11 gp46 and comparison of 5-AcNmdU synthesis to tRNA hypermodifications catalyzed by MnmEG and MnmC. Key to both pathways is the proposed formation of an exocyclic methylene at C5 that is the target of the modifying nucleophile, which is the alpha amine of glycine in this example.
Radical-mediated isomerization of Nα-GlyT
With the establishment of M6 gp51 as an amino acid transferase that generates 5-Nα-glycinylthymidine in DNA, we next sought to understand the enzymes that convert this intermediate into 5-NedU, the hypermodified thymine present in phage M6 virion DNA. As shown in sequence 1 of Figure 2, M6 and related phages encode two ORFs, gp52 (PLP) and gp53 (rSAM), in between the genes encoding gp51 (aGPT-Pplase2) and gp54 (P-loop kinase), which we have shown here to synthesize Nα-GlyT. The co-occurrence of these genes within the presumptive 5-NedU biosynthetic gene cluster across multiple related phage genomes (see also Supplementary Figure S15) strongly suggests that they are functionally linked. Sequence comparisons indicate that gp52 likely encodes a PLP-dependent enzyme whereas gp53 is a member of the radical SAM (rSAM) superfamily. Indeed, M6 gp53 gives high confidence match to the rSAM DNA repair enzyme spore product photolyase (SPL; pdb:4fhe) using the homology based, templated structure prediction method Phyre2 (36) (100% confidence, 13% identity) and aligns a putative iron-sulfur binding amino-acid sequence motif (37) of M6 gp53 (CTVGCAFC) beginning at residue 125 with the SPL Fe4S4 binding cluster (CMGHCHYC) beginning at residue 90. Radical SAM enzymes utilize a reduced [Fe4S4]+ cluster to homolytically cleave SAM and generate a reactive radical species, most commonly forming methionine and a putative 5′-deoxyadenosyl radical (5′-Ado•), the latter of which is used to initiate a wide variety of radical-based chemical transformations in processes such as vitamin, cofactor and secondary metabolite biosynthesis (38), activation of glycyl radical enzymes (39), and the respective post-transcriptional and post-translation modification of RNAs (40) and proteins (41–43).
Initial experiments where Nα-GlyT-containing DNA was treated with a lysate containing M6 gp53 under ambient conditions did not reveal any new species (Supplementary Figure S7). Given the notorious oxygen sensitivity of rSAM enzymes and the challenges in maintaining them in cofactor replete form, we opted to carry out FeS cluster and activity reconstitution experiments using pure recombinant gp53 inside an anaerobic chamber. M6 gp53 was expressed in an E. coli strain carrying the pDB1282 accessory plasmid (28) which supplies chaperones for FeS cluster biogenesis. Subsequently, pellets from these cultures were lysed and M6 gp53 purified by nickel affinity chromatography. Following purification, M6 gp53 was treated anaerobically as described in Materials and Methods to reconstitute its iron-sulfur clusters. The resulting protein solution was brown in color, a characteristic likely due to a [Fe4S4] cofactor associated with the enzyme. UV–Vis spectra of purified M6 gp53 before and after reconstitution showed an increase at ∼410 nm characteristic of iron-sulfur cluster containing proteins (Supplementary Figure S16). Iron quantitation of purified M6 gp53 yielded ∼9 Fe per protein, consistent with two 4Fe4S clusters, an observation supported by the occurrence of a second possible iron-sulfur binding motif in the M6 gp53 protein sequence. Purified M6 gp53 was incubated with Nα-GlyT-containing DNA oligos under reducing conditions and monitored for the formation of novel bases using LC-MS. As shown in Figure 1C, under these conditions, a new nucleoside was formed having a slightly shorter retention time but having the same mass (315 u) as the Nα-GlyT substrate. Control experiments where SAM or reductants were omitted prevented the reaction from occurring (Supplementary Figure S17).
The fact that the retention time of the nucleoside produced by gp53 changed while its nominal mass remained the same as Nα-GlyT indicated that the enzyme likely catalyzed an isomerization reaction. Given that 5-NedU has a primary amine whereas the nitrogen atom in Nα-GlyT is a secondary amine, and the fact that no other potential nitrogen donor besides SAM was present in the reactions, we suspected that gp53 likely catalyzes an N–C shift of the appended glycine residue. This would result in the formation of a Cα-GlyT, wherein the Cα atom becomes directly linked to the nucleobase through a C–C bond, thus exposing the amino group of the original amino acid as a primary amine. We therefore predict that the product nucleoside of the gp53 reaction is Cα-GlyT. Thus, we conclude that gp53 encodes an rSAM Nα-GlyT isomerase.
While the mechanism of the glycinylthymine isomerization reaction remains to be studied, we speculate that it may be akin to the fragmentation mechanism utilized by the B12-dependent carbon-skeleton mutases (e.g. methylmalonyl-CoA mutase and glutamate mutase). By analogy to those systems, 5′-Ado• would extract a hydrogen atom from the Cα position of glycine, leading to homolytic scission of N-5C bond to yield dehydroglycine and a 5-thymidyl-DNA radical. Reorientation of the dehydroglycine fragment before recombination would result in the formation of the C-Cα bond giving origin to Cα-GlyT. To the best of our knowledge, gp53 represents the first example of a radical SAM enzyme involved in the installation of a DNA modification. Prior to this work, spore photoproduct lyase was the only known member of the radical SAM superfamily member to be associated with DNA, as it functions in the resolution of UV-induced cyclobutane thymine dimers (44).
Decarboxylation of amino-acid:nucleobase conjugates by PLP-dependent enzymes
M6 gp52 encodes a predicted PLP-dependent enzyme immediately adjacent to the gene encoding the radical SAM enzyme that catalyzes the isomerization of Nα-GlyT to Cα-GlyT. While phage ViI does not encode a homolog of this radical SAM enzyme, it houses a homologous predicted PLP-dependent enzyme (gene 226). The product of this gene, ViI gp226, was previously shown to participate in the thymidine hypermodification reaction (8) (see also Supplementary Figure S8). PLP-dependent enzymes catalyze a variety of reactions involving amino acids and their secondary metabolite and neurotransmitter derivatives (45). Key to all these reactions is the availability of a primary amine that allows these molecules to reversibly form a Schiff base with a PLP-cofactor, causing the pKa of the adjacent Cα position to decrease to the point where it can be deprotonated under physiological conditions and initiate transamination, decarboxylation, racemization and other types of chemical transformations (46). Using both Phyre2 and I-TASSER structure/function prediction servers, the top matches to M6 gp52 were PLP-dependent enzymes. Both approaches align M6 gp52 sequences to active sites of known PLP-dependent enzymes and suggest that M6 gp52 residue K36 could be responsible for the internal aldimine to the PLP cofactor. Similar results were observed for ViI gp226 using Phyre2 through structurally templated alignment with tryptophan synthase beta subunit (pdb:1j0a; 98.9% confidence) and identified gp226 residue K92 as the potential source of ϵ-amino group for internal aldimine formation.
As the difference in both nominal mass and structure between Cα-GlyT and 5-NedU, as well as between O-SerT and 5-NeOmdU, corresponds to the loss of CO2, we expected M6 gp52 and ViI gp226 to be PLP-dependent decarboxylases. Both M6 gp52 and ViI gp226 were recombinantly expressed in E. coli and purified to homogeneity. Chromatographic fractions containing these proteins displayed a yellowish hue and their UV–Visible absorbance spectra showed peaks in the 410–425 nm range characteristic of PLP-dependent enzymes (Supplementary Figure S18). As shown in Figure 1C, incubation of Cα-GlyT-containing DNA with purified M6 gp52 produced a nucleoside with identical retention time and mass to those of the native M6 5-NedU. ViI gp226 was analogously able to decarboxylate a substrate containing O-SerT leading to a nucleoside with nominal mass 301 u (Figure 1D). Decarboxylation of stable isotope labeled O-SerT containing three 13C and one 15N resulted in the loss of a single carbon as evidenced by the formation of a 304 u nucleoside (Supplementary Figure S19). In contrast, gp52 did not react with the Nα-GlyT isomer produced by M6 gp51 (Supplementary Figure S7).
The observation that M6 gp52 did not convert Nα-GlyT until isomerized by M6 gp53 is logical given that this transformation unmasks a primary amine for formation of the Schiff base with the PLP cofactor. For ViI, this type of isomerization is not required prior to the decarboxylation step since the hypermodification intermediate contains serine ligated to the base via a C-O linkage leaving the alpha-amine available to form a Schiff base with the PLP cofactor. We thus established that M6 gp52 and gp53 are indeed associated with the biosynthesis of 5-NedU in M6 and related phages as well as the order in which these enzymes affect the conversion of 5-Nα-glycinylthymidine to finished hypermodified product. These results also establish ViI gp226 similarly performs a decarboxylation function leading to the mature modification.
Conversion of 5-Nα-glycinylthymidine to 5-acetylaminomethyl-2′-deoxyuridine
The PaMx11-like phages of Pseudomonads are closely related to M6 and encode a similar gene cluster (sequences 6–8 in Figure 2; see also Supplementary Figure S15) that likely installs a hypermodified thymine derivative in the genomes of these viruses. The presence of 5-HMUDK and clade 2 aGPT-Pplase genes in these genomes at locations syntenic to phage M6 indicates that these phages likely utilize Nα-glycinylthymidine as a precursor in the synthesis of a hypermodified base. Unlike M6, however, homologs of the PLP-dependent decarboxylase M6 gp52 and the radical SAM isomerase M6 gp53 are replaced in PaMx11 (and similar phages) by two different open reading frames, encoding gp47 and gp48, which exhibit sequence similarity to FAD-dependent oxidoreductases and acetyltransferases (AT), respectively. This observation suggests that the modified base found in PaMx11 and its relatives, although utilizing a similar biosynthetic pathway to generate Nα-GlyT, will be distinct from 5-NedU. To confirm that the clade 2 aGPT-Pplase of PaMx11 (gp46) can produce Nα-GlyT, E. coli lysates expressing PaMx11 gp46 and M6 gp54 (5-HMUDK) were incubated with a 5-hmdU containing DNA substrate. These reactions produced a nucleoside identical in mass and retention time to the nucleoside similarly produced by M6 gp51 (Supplementary Figure S20).
To identify the hypermodified thymine generated by the PaMx11 gene cluster, we purified the putative FAD-dependent oxidoreductases gp47 of PaMx11 and its associated acetyltransferase, gp48. Chromatographic fractions containing PaMx11 gp47 displayed a yellowish hue and UV-Vis absorption spectra displayed absorbance peaks at 365 nm and 450 nm characteristic of FAD-dependent enzymes (Supplementary Figure S21) (47). We next incubated substrate oligonucleotides containing Nα-GlyT residues with purified PaMx11 gp47 and/or gp48 either alone or in combination. Based on the predicted functions of the enzymes, we supplemented the reactions with FAD and acetyl-CoA to maximize the likelihood of observing unique nucleosides. As shown in Figure 1E, the combination of both enzymes produced a new nucleoside with a ∼28.5 min retention time and a 299 u nominal mass. Reactions containing gp48 alone exhibited no reaction with Nα-GlyT-containing oligos, whereas gp47 produced a new nucleoside with a ∼9.5 min retention time and a 257 u mass (Figure 1E). Treatment of this product oligo with gp48 recapitulated the 299 u nucleoside and, thus, established this enzyme as the likely ultimate step in the biosynthetic pathway of the unique hypermodified thymine in phage PaMx11.
The difference in mass between Nα-GlyT (315 u) and the product of the gp47 reaction (257 u) most strongly correlates with a loss of an acetoxy group from the former during the reaction. This observation suggests this intermediate product as 5-aminomethyl-2′-deoxyuridine (5-NmdU). That gp47 catalyzes the cleavage of the N-Cα bond of glycine in a reaction is reminiscent of the tRNA modification enzyme MnmC1 (Figure 3D) (48). Indeed, gp47 shares significant similarity with the FAD-containing domain of MnmC1 as revealed by analysis using Phyre2 (pdb:3pvc, 98.6% confidence, 18% sequence identity). The current mechanistic proposal for MnmC1 is that it oxidizes the N-Cα bond of the appended Gly residue to produce dehydroglycinylthymine, which non-enzymatically hydrolyzes to produce 5-aminomethyluracil and glyoxylate (48). On this basis, we propose that PaMx11 gp47 is a FAD-dependent glycinylthymine dehydrogenase.
The difference in mass between the final product of the gp47/gp48 reaction (299 u) and 5-NmdU (257 u) corroborates the addition of an acetyl group, indirectly supporting the classification of gp48 as an acetyltransferase based on the sequence comparisons (e.g. the top 20 Phyre2 matches are to GNAT-family acetyltransferases with >99% confidence). Within the context of the DNA polymer, the only functional group on 5-NmdU that is readily available for acetylation (without invoking tautomerization of the base) is the primary amine that has been unmasked by the action of gp47. The prospective acetylation of 5-NmdU is also consistent with our observation that gp48 only reacts with the product of gp47 and not with its Nα-GlyT precursor. There is precedent for N-acetyltransferases that act on nucleic acids. The human enzyme NAT10 acetylates cytidine 1842 in 18 S rRNA to generate N4-acetylcytidine (49). Finally, the mass of 5-acetylaminomethyl-2′-deoxyuridine (5-AcNmdU) is consistent with that observed for the product of the gp48 reaction. These data strongly suggest that gp48 is a bona fide 5-NmdU N-acetyltransferase. Although our attempts to obtain the Pseudomonas phage PaMx11 in order to purify its DNA for comparisons with the enzymatically produced 5-AcNmdU were unsuccessful, the data presented here strongly suggest that 5-AcNmdU is the native modification expressed in this subgroup of M6-like phages.
Global occurrence of diverse T-hypermodification pathways
Having established the functions of a suite of genes participating in the formation of at least three different thymidine hypermodifications, we next sought to recover viral sequences encoding homologs of these enzymes from metagenomes in order to document the occurrence of thymidine hypermodification genes as well as their co-association with other genes potentially involved in as-yet uncharacterized hypermodifications. We explored two metavirome sequence databases: the Joint Genome Institute’s Integrated Microbial Genomes Viral Resource version 2 (IMG/VR v2) (25) and the Global Oceanic Viromes 2.0 (GOV2.0) (26). IMG/VR2 contains 715 672 contigs encoding 16 215 899 proteins computationally identified as viral and retrieved from metagenome sequences archived at IMG. Global Oceanic Viromes 2.0 (GOV2.0) is an environmental meta-metagenome dataset encompassing 145 viromes sampled from the world’s oceans and containing 848 507 contigs encoding 12 486 732 proteins (sequence data available at https://datacommons.cyverse.org/browse/iplant/home/shared/iVirus/GOV2.0).
Given that 5-PmdU is a central intermediate to at least five different thymidine hypermodifications, we looked for genomes and genome fragments encoding 5-HMUDKs. Nucleotide sequences from these metavirome data sets were interrogated using tblastn and M6 gp54, ViI gp67 and ΦW-14 gp37 as query sequences with an e-value cutoff of 10–5. The contigs recovered from each of these searches were pooled and deduplicated yielding 352 sequences from IMG/VR2 ranging from 5.2 to 269 kb (and occurring at frequency of 4.92 × 10–4) and 713 sequences from GOV2.0 ranging from 5.0 to 94.4 kb in length (and occurring at frequency of 8.40 × 10–4). Open reading frames in the recovered contigs were predicted and annotated using Prokka (50) within JGI’s Kbase Knowledge Discovery Environment (KDE) (51). A fine-grained annotation was subsequently added by comparing each identified predicted coding sequence against the PfamA protein profile database (52) of hidden Markov models (HMMs) using an automated implementation of the hmmscan command from the HMMER biosequence analysis suite (53). Sub-genomic regions encoding clusters of thymidine hypermodification and accessory genes identified using this pipeline are shown in Figure 2.
Our inquiry into global metaviromes recovered contigs encoding gene clusters that we predict not only synthesize hypermodified thymidines using pathways uncovered in this work but also as-yet uncharacterized hypermodified structures that may use Nα-GlyT or O-SerT as an intermediate. For example, we found contigs whose gene content and organization (Figure 2, sequences 2 through 5) were identical or nearly identical to phage M6 (Figure 2, sequence 1) and therefore likely to make 5-NedU. Similarly, we found contigs (Figure 2, sequences 7 and 8) corresponding to phage PaMx11 (Figure 2, sequence 6) as well as SP10 (Figure 2, sequences 11 through 13). Among the contigs were interesting fusions between a 5-HMUDK and other hypermodification genes (Figure 2, sequences 14 through 16) such as a PLP-dependent enzyme and a triple fusion of 5-HMUDK, PLP-dependent decarboxylase, and a clade 2 aGPT-Pplase. Among contigs encoding 5-HMUDK and aGPT-Pplase2, we find a combinatorial diversity of contigs encoding combinations of rSAM enzymes, PLP-dependent decarboxylases, flavin dependent oxidoreductases, together with genes whose functions are not yet known but give Pfam matches to amidoligase and glutaminotransferase domains. Lastly, we observed that in many of these contigs, the modification genes were in the vicinity of dUMP hydroxymethylase, in accordance with this enzyme producing the 5-hmdU substrate for subsequent hypermodification. However, a group of sequences were found to lack a dUMP hydroxymethylase and instead encode a TET_JBP domain containing protein in the vicinity of the other putative T-hypermodifying genes. TET_JBP family proteins are iron/alpha-ketoglutarate (Fe/aKG)-dependent dioxygenases that hydroxylate 5-methylpyrimidines in DNA; TET utilizes 5-mC as a substrate, whereas JBP utilizes thymine (35). The absence of a nearby gene for producing 5-mC suggests the TET_JBP homologs encoded by these contigs convert thymine to hydroxymethyluracil in DNA, thereby producing the starting substrate for adjacently encoded thymidine hypermodification enzymes. Indeed, recent work has demonstrated that metavirome encoded TET_JBP oxidize methylpyrimidines in DNA in vivo and in vitro (54). The organization of these contigs implies a modular gene pool for generating combinatorial diversity of thymidine hypermodifications.
Hypermodification in trans produces viable phages having mixed modifications
The combinatorial permutation of thymidine hypermodification pathway genes seen in Figure 2 suggests that such modifications could be, to some extent, interchangeable among phages accessing a viral pangenome. Thus, we asked if a thymidine hypermodifying phage could acquire and tolerate heterologous hypermodifications in trans during infection of recombinant E. coli expressing a hypermodification gene from a different phage. As schematically illustrated in Figure 4A, E. coli cultures expressing recombinant aGPT-Pplase2 enzymes from either M6 (gp51), ΦW-14 (gp109) or green fluorescent protein (GFP) as a negative control were infected by coliphage CBA120 (which natively synthesizes 5-NeOmdU in its DNA). Following the infection, phage particles were purified from the cell culture supernatant and DNA was extracted from them to determine their nucleoside composition by LC/MS.
Figure 4.
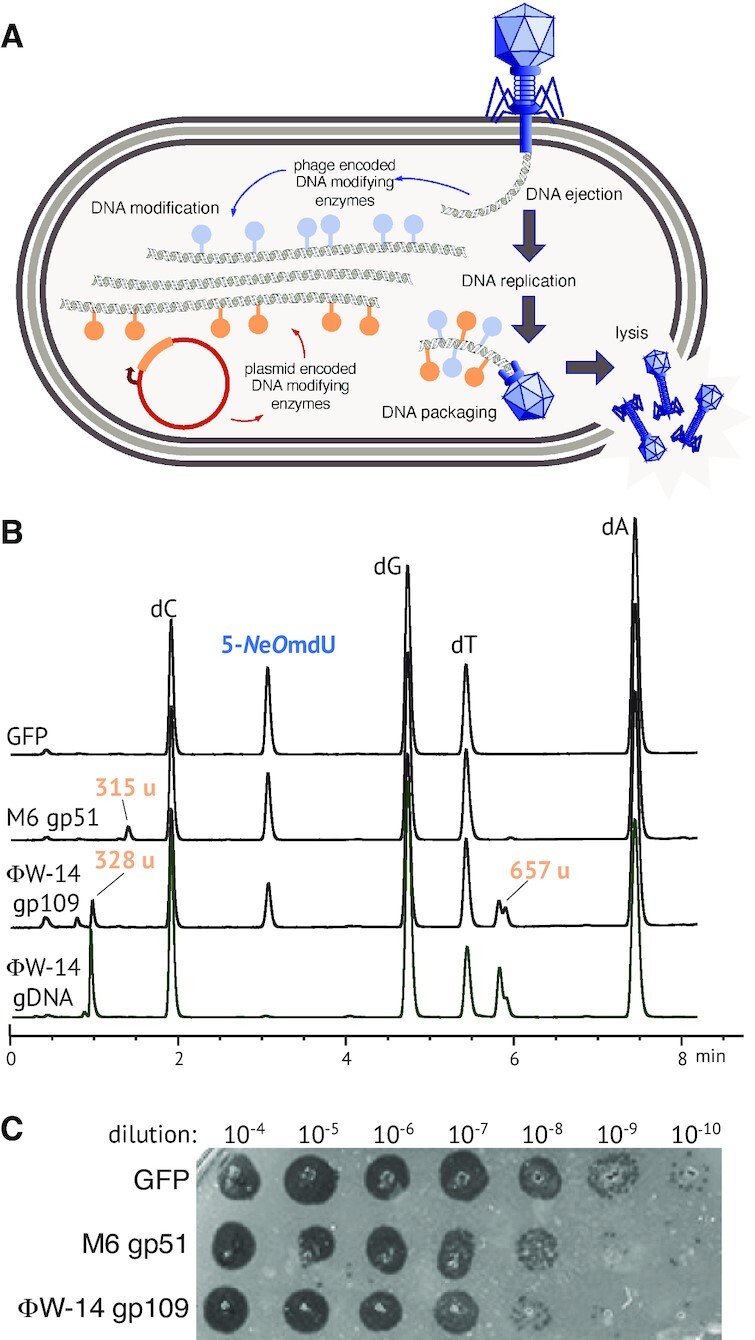
Heterologous thymidine hypermodification in trans. (A) Schematic of modification in trans experiment. Cells recombinantly expressing a thymidine hypermodifying enzyme infected by phage CBA120 undergo lytic development of the phage, during which time the phage will produce thymidine hypermodification intermediates potentially serving as a substrate for the heterologously expressed enzyme. Heterologously modified DNA is then packaged during virion morphogenesis and can be recovered from purified phage particles. (B) UPLC chromatograms and apparent masses of select peaks obtained from DNA isolated after infection of cells expressing either green fluorescent protein (GFP) as a negative control, M6 gp51 (producing Nα-GlyT) or ΦW-14 gp109 (predicted to produce Nα-putT). A trace obtained from a digest of native ΦW-14 genomic DNA is shown for comparison. (C) Serial dilutions of clarified lysates obtained after infection of cells expressing GFP, M6 gp51, or ΦW-14 gp109 were spotted on lawns of sensitive host.
As shown in Figure 4B, DNA extracted from phages produced during infection of M6 gp51 or ΦW-14 gp109 expressing cells yielded new nucleosides in addition to those produced by infection of GFP-expressing negative control cells. Phages recovered from a M6 gp51 expressing host strain produced a new nucleoside with an apparent mass of 315 u, identical to the Nα-glyT nucleoside observed during in vitro reactions with purified M6 gp51. Similarly, phages recovered from infections of host cells expressing ΦW-14 gp109 produced two new nucleoside species having nominal masses of 328 and 657 u. Comparison of this trace with one obtained from nucleoside digests of native phage ΦW-14 DNA show that these heterologous peaks have identical retention time and identical nominal mass as the native modifications. The 328 u nominal mass species matches the known mass of Nα-putT. However, the 657 u species has not been previously documented, and its exact composition is unknown, but could correspond to a dimer of thymidine linked by a polyamine. These data show that the CBA120 phage DNA ‘chassis’ is structurally capable of carrying additional modifications. Serial dilutions of phages shown in Figure 4C show the recovered phage are viable within two orders of magnitude relative to CBA120 recovered from infection of GFP expressing cells indicating that the heterologous modifications can be functionally tolerated as well. Of particular note, these results also indicate that the aGPT-Pplase2 gp109 of phage ΦW-14 is sufficient to produce Nα-putT in the context of CBA120 infection, consistent with its function as predicted by Aravind et al. (23).
DISCUSSION
The work presented here significantly expands the number of enzymes known to act directly on the nucleobases of DNA polymers. In addition to a nucleobase kinase, a PLP-dependent decarboxylase, a putative rSAM-dependent isomerase, a flavin-dependent oxidoreductase, and an acetyltransferase, we have demonstrated the activity of a family of enzymes that are amino-acid:DNA transferases (clade 2 aGPT-Pplases). The apparent mechanism of group transfer to C5 methyl by the clade 2 aGPT-Pplases of M6 and ViI adds these proteins to the list of enzymes using an active site thiolate to attack C6 of the pyrimidine nucleobase by Michael addition in order to effect chemistry at C5. This group includes SAM-dependent C5-cytidine DNA methyltransferases (12,35), SAM dependent C5-pyrimidine RNA methyltransferases (55), the folate/flavin dependent RNA methylases TrmFO and RlmFO (56) and the classical thymidylate synthases (including dUMP hydroxymethylases). aGPT-Pplases do not match any of these by homology and are predicted to have a DNA glycosylase-like helix-hairpin-helix fold (23). Further characterization of these enzymes might reveal the evolutionary path they took in acquiring this ability and may also indicate how other folds may similarly arrive at this mechanism. Unlike the SAM dependent methylases, which alkylate C5 by transfer of a saturated carbon, the classical thymidylate synthases, TrmFO, RlmFO and clade 2 aGPT-Pplases utilize an unstable exocyclic C5 methylene as reaction intermediates. However, in the thymidylate synthases, the methylene derives from transfer of carbon between a cationic N5-iminium isomer of methyltetrahydrofolate (mTHF) to C5 (57), whereas in clade 2 aGPT-Pplase, the methylene is formed by dehydration of a hydroxymethyl group already installed at C5, possibly assisted by the departure of the phosphate group, as shown in Figure 3D.
Our findings indicate genes encoding thymidine hypermodifying enzymes are widespread in natural populations of bacterial viruses and suggest that at least one in 2000 phages in any virome might contain a post-replicatively hypermodified thymidine synthesized from an initial phosphorylated thymidine analog (5-PmdU). We also observe other enzyme-encoding genes such as amidoligases and amidotransferases co-associated with the thymidine hypermodification genes characterized in this work, as well as other enzyme families not addressed in this study, suggesting many more thymidine modifying enzyme activities that await discovery. In addition to the variety of enzymes working in pathways to hypermodify thymidines, there also appears to be a collective combinatorial permutation of those genes when comparing individual phage genomes (as seen in Figure 2). For example, 5-HMUDK is a common intermediate to at least five thymidine hypermodifications, and Nα-Gly is shared between at least two hypermodification pathways.
Taken together, the evidence suggests the global viral pan-genome encodes an assortment of thymidine hypermodifying genes that can be modularly recombined into thymidine hypermodification pathways capable of synthesizing a diverse range of chemical structures appended to C5 of T-coding nucleobases in DNA. A similar portrait has emerged for the diverse modifications found at position 7 of the 7-deaza-2′-deoxyguanine substituted DNAs observed within phylogenetically diverse bacterial viruses (5–7,58). The clustering of genes involved in thymidine hypermodification and the diversity of their products is reminiscent of natural products produced by microbes as part of inter-species ‘chemical warfare’. DNA hypermodifications are resistant to a wide range of Type II restriction endonucleases in vitro and ostensibly protect the phage DNA from cellular defenses by their host (1,2,59). However, the similar degrees of resistance to cleavage between three different thymidine hypermodifications suggests that bacterial hosts may have other defenses, such as modification specific endonucleases (60–62), and can overcome steric hindrance to restriction endonucleases. These countermeasures may somehow be specific to each hypermodification and thus could drive diversification of a DNA ‘chemical phenotype’ by phages in response.
Thymidine hypermodification resembles tRNA hypermodification in several ways. Superficially, like post-transcriptional modification of tRNA, biosynthesis of these complex DNA modifications proceeds via multiple enzymatic steps occurring in sequence on the polymer. Chemically, intermediates in the modification pathways of M6 and PaMx11 are DNA analogs of intermediates in the biosynthesis of xmn5 family of tRNA modifications found at U34 in the anti-codon wobble position, as diagrammed in Figure 3D. Mechanistically, M6 gp51 might provide insights into the function of MnmEG. Both enzymes attach glycine to methyluracil, albeit through different routes. M6 gp51 (and PaMx11 gp46) acts on a phosphorylated methyl group at C5 of uracil, with the phosphate as a leaving group and generating an electrophilic exocyclic methylene as illustrated in Figure 3D. This methylene is the target of nucleophilic attack by the amino group of glycine. In both cases, glycine is attached to a carbon bonded to C5 of the pyrimidine ring. In the hypermodified thymidines described here, this carbon is derived from mTHF and installed by a classical thymidylate synthase family enzyme, dUMP 5-hydroxymethyltransferase, at the nucleotide pool stage prior to DNA replication. For the tRNA modification cmnm5U, this carbon likely derives also from mTHF but possibly installed post-transcriptionally by MnmEG via a flavin-dependent methylation. MnmEG, TrmFO, RlmFO and ThyX are flavin-dependent uracil methylases (56,63). Flavin-dependent methyltransferases catalyze a transfer relay of a single carbon from the position 5 of methylenetetrahydrofolate to the C5 position of a nucleobase pyrimidine via an N5 iminium intermediate on the FADH2 cofactor (64,65). In the case of dUMP methylase ThyX, transfer of carbon from FADH2 N5 to uracil C5 results in an exocyclic methylene (66,67), which is subsequently reduced to methyl by hydride transfer from FADH2. Based on our observation that glycine likely attacks a transient exocyclic methylene during the formation of Nα-GlyT and O-SerT, if a similar exocyclic methylene occurs during tRNA modification by MnmEG, formation of cmnm5U could analogously be accomplished by the nucleophilic attack of glycine (or taurine in the case of tm5U synthesis), as proposed in Figure 3D. In PaMx11 DNA modification, the glycinylated thymidine is subsequently cleaved by gp47, a flavin-dependent lyase, producing 5-NmdU. PaMx11 gp47 is homologous to the MnmC1 domain of MnmC, which performs a similar reaction in tRNA. The evolutionary origins of this enzyme activity and whether they occurred first in RNA or DNA modification are not yet known, but this example of crosstalk between the DNA and RNA worlds follows precedents already set by SAM dependent methyltransferases such as DNMT2 and several enzymes involved in synthesis of 7-deazaguanosine derivatives in DNA and RNA. Further studies of DNA hypermodification will likely reveal other interesting chemistries and parallels between nucleobase modifications in DNA and RNA.
DATA AVAILABILITY
Protein and nucleic acid sequences described and analyzed in this work are publicly available through the databases and accession numbers indicated in the text. All other data are available in the main text or in the Supplementary Materials.
Supplementary Material
ACKNOWLEDGEMENTS
The authors gratefully acknowledge Dr Matthew Sullivan and Dr Ahmed Zayed of The Ohio State University for sharing with us a database containing the computational translations of all predicted coding sequences in the GOV 2.0 metavirome dataset. We appreciate helpful discussions and manuscript edits from Dr Christopher J. Noren, Dr Rebekah M.B. Silva, Dr William E. Jack, Dr Richard J. Roberts and Dr Elisabeth A. Raleigh. The authors are grateful for support from Donald G. Comb, James V. Ellard, and New England Biolabs, Inc., without which this work would not have been possible. This work is dedicated to the memory of Don Comb.
Contributor Information
Yan-Jiun Lee, Research Department, New England Biolabs, Inc., 240 County Road, Ipswich, MA, 01938, USA.
Nan Dai, Research Department, New England Biolabs, Inc., 240 County Road, Ipswich, MA, 01938, USA.
Stephanie I Müller, Research Department, New England Biolabs, Inc., 240 County Road, Ipswich, MA, 01938, USA.
Chudi Guan, Research Department, New England Biolabs, Inc., 240 County Road, Ipswich, MA, 01938, USA.
Mackenzie J Parker, Research Department, New England Biolabs, Inc., 240 County Road, Ipswich, MA, 01938, USA.
Morgan E Fraser, Research Department, New England Biolabs, Inc., 240 County Road, Ipswich, MA, 01938, USA.
Shannon E Walsh, Research Department, New England Biolabs, Inc., 240 County Road, Ipswich, MA, 01938, USA.
Janani Sridar, Research Department, New England Biolabs, Inc., 240 County Road, Ipswich, MA, 01938, USA.
Andrew Mulholland, Research Department, New England Biolabs, Inc., 240 County Road, Ipswich, MA, 01938, USA.
Krutika Nayak, Research Department, New England Biolabs, Inc., 240 County Road, Ipswich, MA, 01938, USA.
Zhiyi Sun, Research Department, New England Biolabs, Inc., 240 County Road, Ipswich, MA, 01938, USA.
Yu-Cheng Lin, Research Department, New England Biolabs, Inc., 240 County Road, Ipswich, MA, 01938, USA.
Donald G Comb, Research Department, New England Biolabs, Inc., 240 County Road, Ipswich, MA, 01938, USA.
Katherine Marks, Research Department, New England Biolabs, Inc., 240 County Road, Ipswich, MA, 01938, USA.
Reyaz Gonzalez, Chemistry Department, University of Massachusetts Boston, 100 William T. Morrissey Blvd. Boston, MA, 02125, USA.
Daniel P Dowling, Chemistry Department, University of Massachusetts Boston, 100 William T. Morrissey Blvd. Boston, MA, 02125, USA.
Vahe Bandarian, Department of Chemistry, University of Utah, 315 South 1400 East Salt Lake City, UT 84112, USA.
Lana Saleh, Research Department, New England Biolabs, Inc., 240 County Road, Ipswich, MA, 01938, USA.
Ivan R Corrêa, Jr, Research Department, New England Biolabs, Inc., 240 County Road, Ipswich, MA, 01938, USA.
Peter R Weigele, Research Department, New England Biolabs, Inc., 240 County Road, Ipswich, MA, 01938, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
New England Biolabs. Funding for open access charge: New England Biolabs.
Conflict of interest statement. Y.J.L., N.D., S.I.M., C.G., M.J.P., M.E.F., S.E.W., J.S,. A.M., K.N., Z.S., Y.C.L., D.G.C., K.M., L.S., I.C. and P.R.W. are/were employees of New England Biolabs, a manufacturer and vendor of molecular biology reagents. This affiliation does not affect the authors’ impartiality, adherence to journal standards and policies, or availability of data.
REFERENCES
- 1. Flodman K., Tsai R., Xu M.Y., Corrêa I.R., Copelas A., Lee Y.-J., Xu M.-Q., Weigele P., Xu S.. Type II restriction of bacteriophage DNA with 5hmdU-derived base modifications. Front Microbiol. 2019; 10:584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Huang L.H., Farnet C.M., Ehrlich K.C., Ehrlich M.. Digestion of highly modified bacteriophage DNA by restriction endonucleases. Nucleic Acids Res. 1982; 10:1579–1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lehman I.R., Pratt E.A.. On the Structure of the glucosylated hydroxymethylcytosine nucleotides of coliphages T2, T4, and T6. J. Biol. Chem. 1960; 235:3254–3259. [PubMed] [Google Scholar]
- 4. Swinton D., Hattman S., Crain P.F., Cheng C.S., Smith D.L., McCloskey J.A.. Purification and characterization of the unusual deoxynucleoside, alpha-N-(9-beta-D-2′-deoxyribofuranosylpurin-6-yl)glycinamide, specified by the phage Mu modification function. Proc. Natl. Acad. Sci. USA. 1983; 80:7400–7404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hutinet G., Kot W., Cui L., Hillebrand R., Balamkundu S., Gnanakalai S., Neelakandan R., Carstens A.B., Lui C.F., Tremblay D.et al.. 7-deazaguanine modifications protect phage DNA from host restriction systems. Nat. Commun. 2019; 10:5442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hutinet G., Swarjo M.A., Crécy-Lagard V.. Deazaguanine derivatives, examples of crosstalk between RNA and DNA modification pathways. RNA Biol. 2017; 14:1175–1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Crippen C.S., Lee Y.-J., Hutinet G., Shajahan A., Sacher J.C., Azadi P., Crécy-Lagard V., Weigele P.R., Szymanski C.M.. Deoxyinosine and 7-deaza-2'-deoxyguanosine as carriers of genetic information in the DNA of Campylobacter viruses. J. Virol. 2019; 93:307–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lee Y.-J., Dai N., Walsh S.E., Müller S., Fraser M.E., Kauffman K.M., Guan C., Corrêa I.R., Weigele P.R.. Identification and biosynthesis of thymidine hypermodifications in the genomic DNA of widespread bacterial viruses. Proc. Natl. Acad. Sci. USA. 2018; 115:201714812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Witmer H. Synthesis of deoxythymidylate and the unusual deoxynucleotide in mature DNA of Bacillus subtilis bacteriophage SP10 occurs by postreplicational modification of 5-hydroxymethyldeoxyuridylate. J. Virol. 1981; 39:536–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kropinski A.M., Bose R.J., Warren R.A.. 5-(4-Aminobutylaminomethyl)uracil, an unusual pyrimidine from the deoxyribonucleic acid of bacteriophage ΦW-14. Biochemistry. 1973; 12:151–157. [DOI] [PubMed] [Google Scholar]
- 11. Walker M.S., Mandel M.. Biosynthesis of 5-(4′, 5′-dihydroxypentyl) uracil as a nucleoside triphosphate in bacteriophage SP15-infected Bacillus subtilis. J. Virol. 1978; 25:500–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Weigele P., Raleigh E.A.. Biosynthesis and function of modified bases in bacteria and their viruses. Chem. Rev. 2016; 116:12655–12687. [DOI] [PubMed] [Google Scholar]
- 13. Wilhelm K., Rüger W.. Deoxyuridylate-hydroxymethylase of bacteriophage SPO1. Virology. 1992; 189:640–646. [DOI] [PubMed] [Google Scholar]
- 14. Witmer H., Dosmar M.. Synthesis of 5-hydroxymethyldeoxyuridine triphosphate in extracts of SP10c phage-infected Bacillus subtilis W23. Curr. Microbiol. 1978; 1:289–292. [Google Scholar]
- 15. Neuhard J., Maltman K.L., Warren R.A.. Bacteriophage ΦW-14-infected Pseudomonas acidovorans synthesizes hydroxymethyldeoxyuridine triphosphate. J. Virol. 1980; 34:347–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Roscoe D.H. Thymidine triphosphate nucleotidohydrolase: a phage-induced enzyme in Bacillus subtilis. Virology. 1969; 38:520–526. [DOI] [PubMed] [Google Scholar]
- 17. Aposhian H.V. A dTMPase found after infection of Bacillus subtilis with phage SP5C. Biochem. Biophys. Res. Commun. 1965; 18:230–235. [DOI] [PubMed] [Google Scholar]
- 18. Nishihara M., Chrambach A., Aposhian H.V.. The deoxycytidylate deaminase found in Bacillus subtilis infected with phage SP8*. Biochemistry. 1967; 6:1877–1886. [DOI] [PubMed] [Google Scholar]
- 19. Markewych O., Casella E., Dosmar M., Witmer H.. Deoxythymidine nucleotide metabolism in Bacillus subtilis W23 infected with bacteriophage SP1Oc: preliminary evidence that dTMP in SP10c DNA is synthesized by a novel, bacteriophage-specific mechanism. J. Virol. 1979; 29:61–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Witmer H., Wiatr C.. Polymer-level synthesis of oxopyrimidine deoxynucleotides by Bacillus subtilis phage SP10: characterization of modification-defective mutants. J. Virol. 1985; 53:522–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Maltman K.L., Neuhard J., Lewis H.A., Warren R.A.. Synthesis of thymine and alpha-putrescinylthymine in bacteriophage ΦW-14-infected Pseudomonas acidovorans. J. Virol. 1980; 34:354–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Maltman K.L., Neuhard J., Warren R.A.. 5-[(Hydroxymethyl)-O-pyrophosphoryl]uracil, an intermediate in the biosynthesis of alpha-putrescinylthymine in deoxyribonucleic acid of bacteriophage ΦW-14. Biochemistry. 1981; 20:3586–3591. [DOI] [PubMed] [Google Scholar]
- 23. Iyer L.M., Zhang D., Burroughs A.M., Aravind L.. Computational identification of novel biochemical systems involved in oxidation, glycosylation and other complex modifications of bases in DNA. Nucleic Acids Res. 2013; 41:7635–7655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Paez-Espino D., Eloe-Fadrosh E.A., Pavlopoulos G.A., Thomas A.D., Huntemann M., Mikhailova N., Rubin E., Ivanova N.N., Kyrpides N.C.. Uncovering Earth's virome. Nature. 2016; 536:425–430. [DOI] [PubMed] [Google Scholar]
- 25. Paez-Espino D., Roux S., Chen I.-M.A., Palaniappan K., Ratner A., Chu K., Huntemann M., Reddy T.B.K., Pons J.C., Llabrés M.et al.. IMG/VR v.2.0: an integrated data management and analysis system for cultivated and environmental viral genomes. Nucleic Acids Res. 2019; 47:D678–D686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gregory A.C., Zayed A.A., Conceição-Neto N., Temperton B., Bolduc B., Alberti A., Ardyna M., Arkhipova K., Carmichael M., Cruaud C.et al.. Marine DNA viral macro- and microdiversity from pole to pole. Cell. 2019; 177:1109–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zallot R., Oberg N.O., Gerlt J.A.. ‘Democratized’ genomic enzymology web tools for functional assignment. Curr. Opin. Chem. Biol. 2018; 47:77–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Johnson D.C., Dean D.R., Smith A.D., Johnson M.K.. structure, function, and formation of biological iron-sulfur clusters. Biochemistry. 2005; 74:247–281. [DOI] [PubMed] [Google Scholar]
- 29. Fish W.W. Rapid colorimetric micromethod for the quantitation of complexed iron in biological samples. Methods Enzymol. 1988; 158:357–364. [DOI] [PubMed] [Google Scholar]
- 30. Stookey L. Ferrozine: a new spectrophotometric reagent for iron. Anal. Chem. 1970; 42:779–781. [Google Scholar]
- 31. Bridwell-Rabb J., Zhong A., Sun H.G., Drennan C.L., Liu H.. A B12-dependent radical SAM enzyme involved in oxetanocin A biosynthesis. Nature. 2017; 544:322–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lee Y.-J., Weigele P.R.. Detection of modified bases in bacteriophage genomic DNA. Methods Mol. Biol. 2021; 2198:53–66. [DOI] [PubMed] [Google Scholar]
- 33. Saavedra J.E. Reductive alkylation of β-alkanolamines with carbonyl compounds and sodium borohydride. J. Org. Chem. 1985; 50:2271–2273. [Google Scholar]
- 34. Mizote T., Nakayama H.. Purification and properties of hydroxymethylpyrimidine kinase from Escherichia coli. Biochim. et Biophys. Acta. 1989; 991:109–113. [DOI] [PubMed] [Google Scholar]
- 35. Parker M.J., Lee Y.-J., Weigele P.R., Saleh L.. 5-Methylpyrimidines and their modifications in DNA. Comprehensive Natural Products III, Reference Module in Chemistry, Molecular Sciences and Chemical Engineering. 2020; Elsevier; 465–488. [Google Scholar]
- 36. Kelley L.A., Mezulis S., Yates C.M., Wass M.N., Sternberg M.J.E.. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015; 10:845–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sofia H.J., Chen G., Hetzler B.G., Reyes-Spindola J.F., Miller N.E.. Radical SAM, a novel protein superfamily linking unresolved steps in familiar biosynthetic pathways with radical mechanisms: functional characterization using new analysis and information visualization methods. Nucleic Acids Res. 2001; 29:1097–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Frey P.A., Hegeman A.D., Ruzicka F.J.. The radical SAM superfamily. Crit. Rev. Biochem. Mol. Biol. 2008; 43:63–88. [DOI] [PubMed] [Google Scholar]
- 39. Shisler K.A., Broderick J.B.. Glycyl radical activating enzymes: structure, mechanism, and substrate interactions. Arch. Biochem. Biophys. 2014; 546:64–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kimura S., Suzuki T.. Iron–sulfur proteins responsible for RNA modifications. BBA-Mol. Cell Res. 2015; 1853:1272–1283. [DOI] [PubMed] [Google Scholar]
- 41. Mahanta N., Hudson G.A., Mitchell D.A.. Radical S-adenosylmethionine enzymes involved in RiPP biosynthesis. Biochemistry. 2017; 56:5229–5244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Benjdia A., Balty C., Berteau O.. Radical SAM enzymes in the biosynthesis of ribosomally synthesized and post-translationally modified peptides (RiPPs). Front. Chem. 2017; 5:87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Dong M., Zhang Y., Lin H.. Noncanonical radical SAM enzyme chemistry learned from diphthamide biosynthesis. Biochemistry. 2018; 57:3454–3459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Yang L., Nelson R.S., Benjdia A., Lin G., Telser J., Stoll S., Schlichting I., Li L.. A radical transfer pathway in spore photoproduct lyase. Biochemistry. 2013; 52:3041–3050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Du Y.-L., Ryan K.S.. Pyridoxal phosphate-dependent reactions in the biosynthesis of natural products. Nat. Prod. Rep. 2018; 36:430–457. [DOI] [PubMed] [Google Scholar]
- 46. Eliot A.C., Kirsch J.F.. Pyridoxal phosphate enzymes: mechanistic, structural, and evolutionary considerations. Annu. Rev. Biochem. 2004; 73:383–415. [DOI] [PubMed] [Google Scholar]
- 47. Schwinn K., Ferré N., Huix-Rotllant M.. UV-visible absorption spectrum of FAD and its reduced forms embedded in a cryptochrome protein. Phys. Chem. Chem. Phys. 2020; 22:12447–12455. [DOI] [PubMed] [Google Scholar]
- 48. Moukadiri I., Prado S., Piera J., Velázquez-Campoy A., Bjork G.R., Armengod M.E.. Evolutionarily conserved proteins MnmE and GidA catalyze the formation of two methyluridine derivatives at tRNA wobble positions. Nucleic Acids Res. 2009; 37:7177–7193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ito S., Horikawa S., Suzuki T., Kawauchi H., Tanaka Y., Suzuki T., Suzuki T.. Human NAT10 is an ATP-dependent RNA acetyltransferase responsible for N4-acetylcytidine formation in 18 S ribosomal RNA (rRNA). J. Biol. Chem. 2014; 289:35724–35730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Seemann T. Prokka: rapid prokaryotic genome annotation. Bioinformatics. 2014; 30:2068–2069. [DOI] [PubMed] [Google Scholar]
- 51. Arkin A.P., Cottingham R.W., Henry C.S., Harris N.L., Stevens R.L., Maslov S., Dehal P., Ware D., Perez F., Canon S.et al.. KBase: The United States Department of Energy systems biology knowledgebase. Nat. Biotechnol. 2018; 36:566–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. El-Gebali S., Mistry J., Bateman A., Eddy S.R., Luciani A., Potter S.C., Qureshi M., Richardson L.J., Salazar G.A., Smart A.et al.. The Pfam protein families database in 2019. Nucleic Acids Res. 2019; 47:D427–D432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Eddy S.R. Accelerated profile HMM searches. PLoS Comput. Biol. 2011; 7:e1002195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Burke E.J., Rodda S.S., Lund S.R., Sun Z., Zeroka M.R., O’Toole K.H., Parker M.J., Doshi D.S., Guan C., Lee Y.-J.et al.. Phage-encoded ten-eleven translocation dioxygenase (TET) is active in C5-cytosine hypermodification in DNA. Proc. Natl. Acad. Sci. USA. 2021; 118:e2026742118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Motorin Y., Helm M.. RNA nucleotide methylation. Wiley Interdiscipl. Rev. 2011; 2:611–631. [DOI] [PubMed] [Google Scholar]
- 56. Lombard M., Hamdane D.. Flavin-dependent epitranscriptomic world. Arch. Biochem. Biophys. 2017; 632:28–40. [DOI] [PubMed] [Google Scholar]
- 57. Carreras C.W., Santi D.V.. The catalytic mechanism and structure of thymidylate synthase. Annu. Rev. Biochem. 1995; 64:721–762. [DOI] [PubMed] [Google Scholar]
- 58. Thiaville J.J., Kellner S.M., Yuan Y., Hutinet G., Thiaville P.C., Jumpathong W., Mohapatra S., Brochier-Armanet C., Letarov A.V., Hillebrand R.et al.. Novel genomic island modifies DNA with 7-deazaguanine derivatives. Proc. Natl. Acad. Sci. USA. 2016; 113:E1452–E1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Flodman K., Corrêa I.R., Dai N., Weigele P., Xu S.. In vitro type II restriction of bacteriophage DNA with modified pyrimidines. Front. Microbiol. 2020; 11:604618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Lutz T., Flodman K., Copelas A., Czapinska H., Mabuchi M., Fomenkov A., He X., Bochtler M., Xu S.. A protein architecture guided screen for modification dependent restriction endonucleases. Nucleic Acids Res. 2019; 47:9761–9776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Bair C.L.C., Black L.W.L.. A type IV modification dependent restriction nuclease that targets glucosylated hydroxymethyl cytosine modified DNAs. J. Mol. Biol. 2007; 366:768–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Machnicka M.A., Kaminska K.H., Dunin-Horkawicz S., Bujnicki J.M.. Phylogenomics and sequence-structure-function relationships in the GmrSD family of Type IV restriction enzymes. BMC Bioinform. 2015; 16:336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Mishanina T.V., Yu L., Karunaratne K., Mondal D., Corcoran J.M., Choi M.A., Kohen A.. An unprecedented mechanism of nucleotide methylation in organisms containing thyX. Science. 2016; 351:507–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Bou-Nader C., Cornu D., Guerineau V., Fogeron T., Fontecave M., Hamdane D.. Enzyme activation with a synthetic catalytic co-enzyme intermediate: nucleotide methylation by flavoenzymes. Angew. Chem. Int. Ed. 2017; 56:12523–12527. [DOI] [PubMed] [Google Scholar]
- 65. Piano V., Palfey B.A., Mattevi A.. Flavins as covalent catalysts: new mechanisms emerge. Trends Biochem. Sci. 2017; 42:457–469. [DOI] [PubMed] [Google Scholar]
- 66. Mishanina T.V., Corcoran J.M., Kohen A.. Substrate activation in flavin-dependent thymidylate synthase. J. Am. Chem. Soc. 2014; 136:10597–10600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Choi M., Karunaratne K., Kohen A.. Flavin-dependent thymidylate synthase as a new antibiotic target. Molecules. 2016; 21:654–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Protein and nucleic acid sequences described and analyzed in this work are publicly available through the databases and accession numbers indicated in the text. All other data are available in the main text or in the Supplementary Materials.