


Abstract
Heterochromatin-associated gene silencing controls multiple physiological processes in malaria parasites, however, little is known concerning the regulatory network and cis-acting sequences involved in the organization of heterochromatin and how they modulate heterochromatic gene expression. Based on systematic profiling of genome-wide occupancy of eighteen Apicomplexan AP2 transcription factors by ChIP-seq analysis, we identify and characterize eight heterochromatin-associated factors (PfAP2-HFs), which exhibit preferential enrichment within heterochromatic regions but with differential coverage profiles. Although these ApiAP2s target euchromatic gene loci via specific DNA motifs, they are likely integral components of heterochromatin independent of DNA motif recognition. Systematic knockout screenings of ApiAP2 factors coupled with RNA-seq transcriptomic profiling revealed three activators and three repressors of heterochromatic gene expression including four PfAP2-HFs. Notably, expression of virulence genes is either completely silenced or significantly reduced upon the depletion of PfAP2-HC. Integrated multi-omics analyses reveal autoregulation and feed-forward loops to be common features of the ApiAP2 regulatory network, in addition to the occurrence of dynamic interplay between local chromatin structure and ApiAP2s in transcriptional control. Collectively, this study provides a valuable resource describing the genome-wide landscape of the ApiAP2 family and insights into functional divergence and cooperation within this family during the blood-stage development of malaria parasites.
INTRODUCTION
The complex life cycle of human malaria parasites involves a tightly regulated progression through well-defined stages including intraerythrocytic asexual replication, sexual differentiation and development in humans and mosquitoes (1). The stage-specific gene expression that occurs during each stage transition is achieved through complex and flexible transcriptional regulatory networks (2). Among the variety of alternative transcriptional control mechanisms previously described in malaria parasites, chromatin modification-mediated high-order chromatin structure and nuclear organization are thought to be the main determinants influencing the transcriptional activation or repression environment of genes in the nucleus (3–5). Such highly organized nuclear architecture provides a simplified and efficient platform to co-regulate various gene groups associated with different phenotypes.
In the genome of P. falciparum, which encodes approximately 5700 protein-coding or non-coding genes, over 500 genes are associated with heterochromatin. These genes are mainly composed of multi-gene families with clonally variant expression patterns, and are linked to various diverse phenotypes such as antigenic variation, pathogenesis, red blood cell invasion, and nutrient transport (6–9). In addition, there are a multiple single-copy heterochromatic genes, such as AP2-G, the master regulator of sexual commitment, which is thought to be co-regulated along with var genes by the heterochromatin environment (4,8,10,11). Nearly all heterochromatic genes are located within the specialized heterochromatic domains at subtelomeric or internal chromosomal regions which are marked by trimethylation of lysine 9 on histone H3 (H3K9me3) (12). H3K9me3 recruits heterochromatin protein 1 (HP1), a conserved regulator of heterochromatin formation for gene silencing (6,13). During the asexual blood stage, HP1 or H3K9me3 demarcate large heterochromatin domains from other euchromatic regions (9). Therefore, HP1 occupancy or H3K9me3 enrichment are markers of a transcriptionally repressive environment in heterochromatin regions in the parasite's genome (14).
In P. falciparum, the HP1-dependent heterochromatin environment is largely restricted to antigenic variant genes (var, rifin, stevor, and pfmc-2tm), phist, and other gene families associated with exported proteins (6). In addition, a group of non-coding ruf6 gene families undergo monoallelic expression, linked to the singular expression of var genes (15–17). As a primary example of HP1-dependent heterochromatin-mediated gene regulation, the strict mutually exclusive expression of ∼ 60 var genes encoding erythrocyte membrane protein 1 (PfEMP1) results in the clonal expression of antigens on infected red blood cells (RBCs) (18). Previous studies have revealed that high-order perinuclear heterochromatin structure determines the transcriptional silencing of most var genes by default, and local chromatin alteration at upstream regulatory regions is linked to switching of var expression (17,19). The single active member is located in a euchromatin microenvironment marked by H3K9ac and H3K4me2/3 in a specialized zone at the nuclear periphery. In addition to var genes, other variant gene families are also located in the heterochromatic region, arranged in tandem gene arrays in subtelomeric and a few internal chromosomal regions (20). Such gene arrangement facilitates not only simplified expression regulation of various multi-gene families, but also enables frequent recombination within each gene family, which in turn increases the sequence polymorphism of surface antigens (21,22).
Other epigenetic regulators such as Sir2a/b, Hda2, PfSETvs, and RUF6 ncRNAs have been found to be involved in heterochromatin formation and alternation via different mechanisms in P. falciparum, but the existence of heterochromatin-associated transcription factors remain to be investigated. Understanding of transcription factors in malaria parasites is limited as the majority of sequence-specific transcription factor families found in other eukaryotic organisms are appear to be absent in Plasmodium. However, a group of transcription factors containing the Apetala2 (AP2) DNA-binding domains, which was originally identified in plants, has expanded across the Apicomplexa phylum. To date, 27 members of the ApiAP2 family have been found in the genome of the human malaria parasite P. falciparum, and 26 of these have orthologs in rodent malaria parasite species (P. berghei and P. yoelii) (23–26). Growing evidence suggests that the ApiAP2 family of DNA binding proteins may be a major class of transcriptional regulators. For example, AP2-G controls sexual commitment whilst AP2-I regulates merozoite invasion of RBCs in P. falciparum (27,28). Two ApiAP2 members (PfSIP2 and PfAP2Tel) are associated with telomeric heterochromatin through binding telomere or subtelomeric sites (29,30).
Interestingly, a recent study using co-immunoprecipitation followed by LC-MS/MS analyses has found that one ApiAP2 member (PfAP2-HC) may be associated with the PfHP1 complex, but its biological functions remain elusive as no apparent transcriptome difference was observed in a PfAP2-HC knock-down line (8,31). Moreover, overexpression of the truncated version of another ApiAP2 protein (PfAP2-exp) with potential binding sites on the upstream regions of var genes upregulated a subset of heterochromatic genes including rifin and stevor (32). In addition, two recent works show that PfAP2-G2 and PfAP2-G5 may be also associated with heterochromatic genes in the regulation of gametocytogenesis (33,34). Together, these data suggest that ApiAP2 transcription factors may be involved in either the formation or maintenance of heterochromatin structure through interaction with the HP1 protein or the regulation of heterochromatic gene expression.
To gain an insight into the regulatory roles of ApiAP2 transcription factors in heterochromatic gene expression during the intraerythrocytic development, we systematically screened all ApiAP2s with unknown genomic occupancy. Twenty-three GFP tagged ApiAP2 transgenic lines and six ApiAP2 knockout lines were generated through CRISPR/Cas9 gene editing technique, which were subjected to chromatin immunoprecipitation sequencing (ChIP-seq) and RNA-seq analyses, respectively. By integrating ApiAP2 genome-wide binding profiles and knockout transcriptomes in addition to public ATAC-seq data (35), we comprehensively surveyed the regulatory roles and the network of heterochromatin-associated ApiAP2 transcription factors.
MATERIALS AND METHODS
Parasite culture
The Plasmodium falciparum 3D7 strain was cultured in fresh O-type human erythrocytes in complete RPMI 1640 medium (Gibco) with 0.5% Albumax I (Invitrogen) and a gas phase maintained under 5% CO2, 5% O2 and 90% N2 at 37°C (17). Ring stage cultures were regularly synchronized by 5% sorbitol treatment in most experiments. For transcriptomics analyses, parasites were more tightly synchronized to a 6-hour window by isolating schizonts over a 40% and 70% Percoll-sorbitol gradient followed by sorbitol treatment 6 hours post re-invasion.
Generation of transgenic lines
The pL6cs plasmid of the CRISPR/Cas9 system was modified to include sequences homologous to target regions as previously described (36,37). A guide RNA was first cloned into the vector between Xho I and Avr II restriction sites. To express endogenous C-terminal GFP tagged ApiAP2 transcription factor, the GFP coding region was placed between fragments homologous to the 3′ end and the 3′ untranslated region (UTR) of the AP2 gene, then cloned into pL6cs between the Afl II and Asc I sites. To disrupt AP2 functions fragments homologous to the upstream and the downstream region of the DNA binding domain were cloned into pL6cs between the Afl II and Asc I sites. Sequences of the guide RNA oligonucleotides and primers for the plasmid construction are listed in Supplementary Table S1.
Transfection of uninfected red blood cells was performed using 100 μg of pUF1-Cas9-BSD and 100 μg of pL6cs carrying homologous sequences, followed by the addition of purified schizonts (37). Subsequently, parasites were cultured in the presence of 2.5 nM WR99210 and 2 μg/mL BSD (or DSM1) until they were found in Giemsa's solution-stained thin blood smears. Knockout parasite lines were further cloned out by limiting dilution. Transgenic parasite lines were verified by diagnostic PCR and Sanger sequencing using the primers listed in Supplementary Table S1.
Western blotting
Sample preparation for western blotting was performed as previously described (17). Briefly, parasites were synchronized at the ring stage, and 200 μL of erythrocytes were collected at different stages of the next generation. Parasites were released from erythrocytes with 0.15% saponin, and boiled in Laemmli buffer at 100°C for 5 min. Extracts were separated on 6%, 8%, or 10% SDS-polyacrylamide gels, and then electroblotted onto Immobilon-P transfer membranes (Millipore). Probing the membranes with specific antibodies followed the standard protocol. Primary antibodies for western blotting included mouse anti-ty1 (used at 1:1000; Sigma, SAB4800032), rabbit anti-aldolase (1:2000; Abcam, ab207494), and rabbit anti-HP1 (1:2000; a gift from Artur Scherf, Institut Pasteur) (38). Horseradish peroxidase-conjugated secondary antibodies, namely goat anti-mouse IgG (Abcam, ab97040) and goat anti-rabbit IgG (Abcam, ab205718), were applied at 1:5000. Blot signals were detected using the ECL western blotting kit (GE healthcare).
Fluorescence imaging
Synchronized parasites were treated with 0.15% saponin and fixed with 4% paraformaldehyde (Sigma) for 10 minutes, then blocked with 1% BSA (Sigma) in PBS (Gibco) and subsequently incubated with antibodies on microscope slides. Primary antibody anti-ty1 was then applied for 1 h at room temperature, followed by the application of secondary antibodies Alexa Fluor 488 goat anti-mouse IgG (ThermoFisher Scientific, A11029) for 1 h at room temperature. The working dilution of primary and secondary antibodies was 1:1000 and 1:500, respectively. 5 μl sample was deposited on a microscope slide and covered with a coverslip. Images were taken using a Nikon A1R microscope at 100 × magnification, acquired via NIS Elements software and processed using Adobe Photoshop CS5.
Chromatin immunoprecipitation sequencing (ChIP-seq) and quantitative PCR (ChIP-qPCR)
ChIP-seq assays were carried out as previously described with minor modifications (28,39). In brief, synchronized parasites were harvested at the ring, trophozoite, and schizont stages respectively and crosslinked immediately with 1% paraformaldehyde (Sigma) by rotating for 10 min at 37°C, which was then quenched with 0.125 M glycine for 5 min on ice. Parasite nuclei released from infected red blood cells were sheared for 20–30 min using an M220 sonicator (Covaris) at 5% duty cycle, 200 cycles per burst, and 75 W of peak incident power to generate 100–500 bp fragments. 20 μL of the sheared chromatin was reserved as the input control. Chromatin was subsequently immunoprecipitated overnight at 4°C using 0.5 μg of antibodies against GFP, IgG (Abcam, ab171870), or HP1 in addition to protein A/G magnetic beads (ThermoFisher Scientific, 26162). After extensive washes, the immunoprecipitated materials were eluted with the elution buffer (1% SDS and 0.1M NaHCO3). Crosslinking was reversed by overnight incubation at 45°C, followed by treatment with RNase A at 37°C for 30 min and Proteinase K at 45°C for 2 h. The ChIPped DNA was finally purified according to the MinElute PCR purification kit (Qiagen, 28006) instructions.
To prepare sequencing libraries, 1.5 ng of ChIP-DNA was subjected to end-repair (Epicentre, ER81050), 3′ adenylation (NEB, M0212L), and adapter ligation (NEB, M2200L). Then after purification with Agencourt AMPure XP beads (Beckman Coulter), libraries were amplified using the KAPA HiFi PCR Kit (KAPA Biosystems, KB2500) under the following conditions: 1 minute of initial denaturation at 98°C, 12 cycles of 10 sec at 98°C and 1 min at 65°C, and 5 min of final extension at 65°C. Libraries were sequenced on an Illumina HiSeq X Ten platform to generate 150 bp pair-end reads. Two biological replicates were performed for each ChIP-seq data set.
ChIP-qPCR assays were conducted in biological triplicate and presented as fold changes of the examined samples relative to the negative controls (mock IP with non-immune IgG). The PCR thermal cycling started with 30 sec denaturation at 95°C, followed by 40 cycles of 5 sec at 95°C, 20 sec at 54°C, 7 sec at 56°C, 7 sec at 59°C, and 27 sec at 62°C. Primers were designed as previously described and listed in Supplementary Table S1 (40,41).
RNA extraction, RNA-seq, and quantitative reverse PCR (qRT-PCR)
Highly synchronous parasites were collected in TRIzol at the ring (10–16 hpi), trophozoite (24–30 hpi), and schizont (40–46 hpi) stages from the next cycle, respectively. Total RNA purification was achieve using the Direct-zol RNA Kit (Zymo Research). Libraries were prepared for strand-specific RNA sequencing by poly(A) selection with the KAPA mRNA Capture Beads (KAPA Biosystems) first and then RNA fragmentation to a size of 300–400 nucleotides. Subsequent library preparation steps followed the protocol of the KAPA Stranded mRNA-Seq Kit Illumina platform (KAPA Biosystems, KK8421). Libraries were sequenced on an Illumina HiSeq X Ten system to generate 150 bp pair-end reads.
For quantitative reverse PCR, 500–800 ng of total RNAs were reversely transcribed (Takara). Quantitative PCR assays were performed in biological triplicate using the primers shown in Supplementary Table S1 and Seryl-tRNA synthetase (PF3D7_0717700) as the endogenous control. The PCR thermal cycling program was the same as for ChIP-qPCR. Gene expression changes were quantified using the ΔΔCt method.
Co-immunoprecipitation
Co-immunoprecipitation assays were performed in two ways. In the conventional approach (42), parasite pellets were collected with 0.15% saponin, washed with PBS, resuspended in three volumes of lysis buffer (2 mM EDTA, 0.5 mM PMSF, 25 mM Tris-Cl, pH 7.5, 100 mM KCl, 0.05% NP-40, and protease inhibitor cocktail), and then homogenized with a sonicator (QSonica) at a 40% power for 4 min. Precleared supernatants were incubated overnight at 4°C with either anti-GFP or anti-IgG (Abcam, ab171870) in addition to protein A/G magnetic beads. The beads were then washed twice with the IPP500 buffer (500 mM NaCl, 0.05% NP-40, and 10 mM Tris-Cl, pH 8.0) and once with PBS. In the other approach, parasite fixation with 1% paraformaldehyde, preparation of nuclear lysates, and immunoprecipitation with either anti-GFP or anti-IgG followed the ChIP protocol described above. In both approaches, the immunoprecipitates were eluted in Laemmli buffer by boiling the beads at 100°C for 5min, resolved by SDS-polyacrylamide gels, and blotted with anti-HP1.
Read processing and mapping
To remove residual adapters and low-quality bases, sliding window trimming was performed on reads with Trimmomatic version 0.39 (43) using a window size of 4 and average quality of the window above 15. A minimum read length of 50 bp and average read quality above 20 were required after read clipping. Trimmed ChIP-seq reads were mapped to the P. falciparum 3D7 reference genome (release 47) using Bowtie2 version 2.4.2 and default parameters (44). HISAT2 version 2.2.1 (45) was employed to align trimmed RNA-seq reads to the 3D7 genome with the guide by the gene annotation, using default parameters except –max-intronlen 5000 –dta –rna-strandness RF.
ChIP-seq peak calling, annotation, and signal track generation
Peaks were identified using the callpeak function of MACS2 version 2.2.7.1 (46) with a q-value cutoff of 0.05 and fold enrichment above 1.5. The options –call-summits and –broad were applied to the detection of transcription factor and HP1 binding peaks respectively. Furthermore, to remove GFP background noises, transcription factor binding peaks with over 1.5 times the fold enrichment of the GFP background were kept for downstream analyses.
GenomicRanges version 1.44.0 (47) assigned target genes to peaks which overlapped the gene loci. The gene loci herein included 5′ UTRs within 3 kb upstream of the translation start sites, gene body, and 3′ UTRs within 0.5 kb downstream of the translation stop sites. To evaluate enrichment of peaks in specific genomic features, namely 5′ UTR, gene body, and 3′ UTR, we employed the permTest function of an R package regioneR version 1.24.0 (48) with parameters randomize.function = circularRandomizeRegions, ntimes = 1000, evaluate.function = numOverlaps, and alternative = ‘greater’. Over-representation analyses of malaria parasite metabolic pathways were performed on the target genes using clusterProfiler version 4.0.2 (49,50) (Benjamini-Hochberg (BH) adjusted P-value < 0.01). Moreover, we generated a matrix of target genes in which transcription factor binding to a target was scored as a ‘1’, and no binding as a ‘0’. Hierarchical clustering was then carried out using a R function hclust.
Peak summits were extended +/− 250 bp for identifying transcription factor binding motifs. If ChIP-seq replicates were available, only peaks detected in both replicates were considered. DREME version 5.1.1 (51) discovered enriched motifs between 6 bp and 10 bp in peaks as compared with random genomic regions of 500 bp. TOMTOM version 5.1.1 (52) was then employed to compare the de novo discovered DNA binding motifs to previously reported motifs (53) (E-value < 0.05). Normalized signal tracks of log2-transformed ChIP/input fold enrichment were generated with MACS2 bdgcmp function and visualized with Gviz version 1.36.2 (54). In addition, ChIP-seq enrichment at a given gene was calculated as the mean log2-transformed ChIP/input fold enrichment by default from 3 kb upstream of the translation start codon to 0.5 kb downstream of the translation stop codon, if not specified explicitly.
Gene expression quantification
StringTie version 2.1.2 (55) was used to count reads mapped to each gene. We retained genes expressed at a Counts Per Million (CPM) above 2 in no less than two samples. The filtered read count matrix was subsequently input to edgeR version 3.34.0 (56) to quantify gene expression levels in Fragments Per Kilobase per Million mapped fragments (FPKM) and analyze differential gene expression (fold change of > 2).
Mapping of highly repetitive regions
JDotter (57) was applied to genomic sequences of the most telomere-proximal 50,000 bp with default settings to create DNA dotplots and thereby map telomeric repeats and telomere-associated repetitive elements (TAREs) 1 to 6.
Statistical analyses
Differences between two groups were analyzed by two tailed t-tests, and multiple testing was corrected by the BH method.
Data visualization
In addition to Gviz, data visualization tools included Cytoscape version 3.8.2 and R (version 4.1.0) packages such as ggplot2 version 3.3.5, ggsignif version 0.6.2, and ComplexHeatmap version 2.8.0 (58,59).
RESULTS
Systematic screen identifies eight P. falciparum heterochromatin-associated ApiAP2 transcription factors
A total of eight P. falciparum ApiAP2 members have been investigated with respect to genome-wide occupancy thus far, namely PfSIP2, PfAP2-G, PfAP2-I, PfAP2-Tel, PfAP2-G2, PfAP2-HC, PfAP2-HS, and PfAP2-G5 (10,11,27,29–34,60). Among these, PfAP2-HC, PfAP2-G2, and PfAP2-G5 are present in the majority of heterochromatic regions with unknown functions for heterochromatic gene expression. In addition, PfSIP2 recognizes SPE2 motifs present in subtelomeric regions, and PfAP2Tel is exclusively distributed at telomeres via direct binding to repetitive sequences. To systematically explore the involvement of the ApiAP2 transcription factors in heterochromatin configuration, multi-omics analyses were performed at ring, trophozoite, and schizont stages, respectively, including genome-wide occupancy profiling by ChIP-seqs and transcriptome profiling by RNA-seqs, integrated with ATAC-seq characterization of accessible chromatin landscapes (35) (Figure 1A).
Figure 1.

Study overview. (A) Workflows of identification and characterization of heterochromatin-associated ApiAP2 factors (PfAP2-HFs) by ChIP-seq (left panel), RNA-seq (middle panel), and integrated multi-omics analyses (right panel). The GFP-tagging transgenic lines were generated by CRISPR-Cas9 gene editing technique for 23 pfap2 genes, and subjected to ChIP-seq analysis with anti-GFP antibody throughout the intraerythrocytic cycle (Ring, Trophozoite, Schizont). For functional study of ApiAP2 proteins, we adopted strategies of DNA-binding domain (DBD) depletion or open reading frame (ORF) disruption. Finally, we integrated our ChIP-seq and RNA-seq datasets with the public ATAC-seq data to identify those heterochromatin-associated transcription factors (PfAP2-HFs) with different regulatory functions. (B) Summary of identification of PfAP2-HFs in the ApiAP2 family of P. falciparum. Reported functions and binding profiles (27–34,60,83–85), ChIP-seq and knockout attempts, and expression levels during blood-stage development are listed for individual PfAP2 members. The underlined ApiAP2 members with unknown functions are named after their chromosomal locations. Expression levels at continuous intraerythrocytic stages including ring (R), trophozoite (T), and schizont (S) in the wild-type 3D7 strain were measured by RNA-seq and MudPIT liquid chromatography tandem mass spectrometry (86), respectively. The data were normalized as Z-scores.
Firstly, we aimed to profile the genome-wide occupancy of all ApiAP2 proteins except previously described telomere/subtelomere binding PfAP2-Tel and PfSIP2 (29,30), in which PfAP2-G and PfAP2-I were included as technical ChIP-seq controls. We first attempted to fuse a GFP tag to the C-termini of ApiAP2 proteins individually through CRISPR/Cas9 gene editing in the P. falciparum 3D7 strain (36) (Figure 1A). After at least two independent rounds of transfection, we successfully obtained a total of twenty-three transgenic lines carrying ApiAP2-GFP expression cassettes, all of which were verified by PCR and DNA sequencing of the fusion events (Supplementary Figure S1A). Though there were a trace of wild type parasites in the cultures of some transfectants, they in principle would not interfere with subsequent ChIP-seq assays. Furthermore, based on immunofluorescence assays and western blotting, eighteen PfAP2-GFP transgenic lines were selected for ChIP-seq analyses, as the rest showed either no or extremely low signals probably due to extremely low levels of expression of these apiap2 genes themselves (Figure 1B and Supplementary Figures S1B and S1C). Importantly, GFP tagging did not lead to appreciable changes in the transcript abundance (Supplementary Figure S1D). ChIP-seq assays were then performed on the eighteen authenticated GFP-tagged parasite lines using ChIP-grade anti-GFP antibody in rings, trophozoites, and schizonts. Technical controls PfAP2-G and PfAP2-I were analysed at the trophozoite stage when expression peaks. A transfectant with episomal GFP expression was used as a negative control to eliminate the ChIP background signal of the anti-GFP antibody.
DNA library preparation for next-generation sequencing succeeded except for PfAP2-06A, PfAP2-G4, and PfAP2-O2 at some stages (Figure 2A), due to low immunoprecipitation efficiency probably caused by extremely low protein levels of these transcription factors at these specific stages. ChIP-seq assays were conducted in two biological replicates, all of which showed good reproducibility with correlation coefficients above 0.85 (Supplementary Table S2). Intriguingly, out of the sixteen ApiAP2 transcription factors investigated in the present study, a total of eight showed significant interactions with heterochromatic regions at various levels (Pearson correlation coefficients between ApiAP2 and HP1 occupancy > 0.5, Figures 2B-C and Supplementary Figures S2 and S3). Furthermore, tight association with heterochromatin was detected in trophozoites and/or schizonts as previously observed for PfHP1 and PfSIP2 (9,29).
Figure 2.

Eight ApiAP2 transcription factors are associated with heterochromatin. (A) Overview of ChIP-seq assays for profiling ApiAP2 occupancies. Unqualified ChIP-seqs are marked in black. (B) Similarity between ApiAP2 transcription factors and PfHP1 in the genomic occupancy evaluated by Pearson correlation. (C) Log2-transformed ChIP/input fold enrichment and peaks of ApiAP2 transcription factors HFs and PfHP1 in chromosomes 7 and 12 as representatives at the schizont stage except for PfAP2-G5 at the trophozoite stage. In addition, log2-transformed ChIP/input fold enrichment of a GFP negative control is displayed at the bottom for each chromosome. Negative values of log2-transformed ChIP/input fold enrichment are not displayed to present succinct occupancy information. In the legend, PfAP2-HF-Is and PfAP2-HF-IIs are labeled with superscript I and II on the right side, respectively. The genomic regions framed in the red boxes (R1, R2 and R3) are further exemplified in Supplementary Figure S2. R, ring; T, trophozoite; S, schizont.
We denominated these eight ApiAP2 transcription factors hereafter as Plasmodium falciparum AP2 Heterochromatin-associated Factors (PfAP2-HFs), namely PfAP2-HC (PF3D7_1456000), PfAP2-G5 (PF3D7_1139300), PfAP2-O5 (PF3D7_1449500), PfAP2-12 (PF3D7_1239200), PfAP2-G2 (PF3D7_1408200), PfAP2-11A (PF3D7_1107800), PfAP2-O2 (PF3D7_0516800), and PfAP2-exp (PF3D7_1466400). To validate the ChIP-seq analyses, ChIP-qPCR assays were performed on the loci of the var genes, a representative gene family located in heterochromatic regions. Homologous sequences within the upstream and coding regions of the var genes allow the detection of interactions between ApiAP2 factors and var. Two independent degenerate primer sets were adopted to target these conserved upstream and exonic regions (40,41) (Supplementary Figure S4A). As expected, ChIP-qPCR confirmed occupancy of PfAP2-HFs at the var loci (Supplementary Figure S4B).
Unlike PfSIP2 and PfAP2-Tel which are recruited to more specific chromosomal regions (29,30), multiple PfAP2-HF binding sites were observed outside heterochromatic regions (Figure 2C and Supplementary Figures S2A, S3, and S5), indicating that the functions of these PfAP2-HF factors are not limited to the regulation of heterochromatin biology, i.e. heterochromatin assembly and maintenance or heterochromatic gene expression. We also note that although PfAP2-G2 and PfAP2-exp showed comparable or even higher expression levels at early stages, neither of them demonstrated as strong a preference for heterochromatic regions in rings as in trophozoites or schizonts (Figure 2B). This suggests that these PfAP2-HFs are involved in heterochromatin biology during schizogony.
PfAP2-HFs are differentially associated with heterochromatic regions in the P. falciparum genome
Given the significance of PfAP2-HF associations with PfHP1 and PfAP2-HF peak expression, our study focused on the schizont stage except for PfAP2-G5 which was scrutinized at the trophozoite stage. Intriguingly, five PfAP2-HFs were found throughout the heterochromatic regions, i.e. PfAP2-HC, PfAP2-G5, PfAP2-O5, PfAP2-12, and PfAP2-G2 (Figure 2C and Supplementary Figures S2A-S2D and S3). The rest of PfAP2-HFs, namely PfAP2-11A, PfAP2-O2, and PfAP2-exp are scattered with relatively lower signals (Figure 2C and Supplementary Figure S2A). We thereby further classified PfAP2-HFs into two subgroups: PfAP2-HF-I and PfAP2-HF-II.
It is known that PfHP1 mediates the epigenetic silencing of pfap2-g which encodes the master regulator of gametocytogenesis in malaria parasites (7,9). Here, we observed notable tethering of PfAP2-HC, PfAP2-G5, PfAP2-O5, and PfAP2-G2 together with PfHP1 to the upstream and coding regions of pfap2-g (fold enrichment > 2, Supplementary Figure S2E), which implies that these four PfAP2-HF factors may be involved in gametocytogenesis as upstream regulators of pfap2-g. In fact, a regulatory role of PfAP2-G5 in gametocyte production has recently been demonstrated (34). Interestingly, PfAP2-HC and PfAP2-O5 displayed similar occupancy profiles to that of PfHP1 which covered almost the entire pfap2-g locus, while PfAP2-G5 and PfAP2-G2 exhibited extremely low signals in the proximal upstream regulatory regions.
The P. falciparum genome encodes approximately 5700 genes, among which a total of 554 are distributed within chromosomal central and subtelomeric heterochromatin regions (6) (Supplementary Table S3). The majority of the rest of the genes are dispersed throughout euchromatic regions marked with H3K9ac or H3K4me3 modifications (61). All of the eight AP2-HF factors tended to target heterochromatic genes (BH adjusted hypergeometric test P-values < 5e-10, Supplementary Figure S5 and Table S4), but the PfAP2-HF-I members demonstrated a much stronger preference (enrichment ratios of heterochromatin targeting to euchromatin targeting > 10).
On the basis of function, heterochromatic genes can be categorized into antigenic variant genes (e.g. var, rifin, stevor, and pfmc-2tm), exported protein families (e.g. epf family, phist exported family, and hyp exported family), acs family, invasion genes, etc. Consistent with previous findings (32,62), interactions were observed between PfAP2-11A and var as well as between PfAP2-exp and antigenic variant genes (Figure 3A). A wide variety of heterochromatic genes were associated with PfAP2-HC, PfAP2-G5, PfAP2-O5, and PfAP2-G2, which targeted over half of the members in almost every family. PfAP2-12 occupancy was prevalent at antigenic variant genes, epf family, acs family, and rRNA genes. Considerable PfAP2-11A association was observed at epf, resa, and acs families. PfAP2-exp demonstrated association with most members of the var family, rifin family, var-associated ncRNA genes, resa family, and invasion genes. PfAP2-O2 exhibited less coverage, and was only significantly associated with the resa gene family.
Figure 3.

ApiAP2 transcription factor HFs are differentially tied up with heterochromatin. (A) Percentages of PfAP2-HF targets in each heterochromatic gene family. (B) Overall binding profiles of PfAP2-HFs and PfHP1 at the var genes with upstream A type promoters (top), upstream B type promoters (middle), and upstream C type promoters (bottom), respectively (P-values < 0.05). Significance of PfAP2-HF enrichment was compared to the GFP negative control. (C) Except for PfAP2-11A and PfAP2-O2, PfAP2-HFs bind to ruf6 loci (P-values < 0.05). Significance of PfAP2-HF enrichment at ruf6 loci from 100 bp upstream of translation start codon to 100 bp downstream of translation stop codon was compared to the GFP negative control. (D) Overall (left panel) and representative (right panel) binding profiles of PfAP2-HFs and PfHP1 at telomeres and subtelomeres. Tel, telomere; TR, telomere-associated repetitive element. (E) A schematic representation of PfAP2-HF occupancy at heterochromatin. Color codes of PfAP2-HFs and PfHP1 in (B-D) are displayed in front of (B).
Most var genes can be categorized into upsA, upsB, and upsC subtypes based on their positions on chromosomes and the sequence homology of their upstream regions (20). Several pieces of evidence suggest that the mutually exclusive expression of var genes involves multi-layer regulatory mechanisms (3,62–66). We assessed the distribution of each PfAP2-HF at var gene loci with respect to gene subtypes. Intriguingly, PfAP2-HFs associated with almost all var loci regardless of subtypes, except for the absence of PfAP2-11A and PfAP2-O2 in upsA-type var loci and PfAP2-O2 in upsC-type var loci (Figure 3B). Strong enrichment of PfAP2-HFs was also observed at the distal upstream regions of upsB-type var genes. Notably, PfHP1 exhibited a similar binding pattern. In addition, we investigated the non-coding ruf6 gene family, a critical regulator of virulence gene expression (15–17). The family members are located exclusively in 5′ UTRs of the chromosome-central var gene clusters, i.e. upsC-type var, which serves as the structural basis for their direct transcriptional regulation of the adjacent upsC-type var genes via local chromatin remodeling. RUF6 also interacts with distal heterochromatic regions, thereby derepressing subtelomeric var genes (17). Here, we observed that all PfAP2-HFs except PfAP2-11A and PfAP2-O2 bound to the ruf6 gene loci (Figure 3C), suggesting the involvement of RUF6 in PfAP2-HF mediated var gene expression. Given that PfAP2-11A and PfAP2-O2 were recruited to the heterochromatin mainly at the subtelomeres (Figure 2C and Supplementary Figure S2A), they may modulate the expression of virulence genes independent of the products of ruf6.
Signals of PfAP2-G2, PfAP2-G5, and PfAP2-12 were detected at the telomeres (average fold enrichment > 1.5, Figure 3D). Another noteworthy heterochromatic region lying between the telomere and subtelomeric variant genes consists of six different types of repetitive elements, namely TARE1-6, which produce telomere-associated long non-coding RNAs probably participating in telomeric heterochromatin formation and maintenance (67,68). In particular, TARE6 (also known as rep20) has been implicated in transcriptional silencing of adjacent var genes most likely via directing the perinuclear localization of gene loci (69). PfAP2-SIP2 regulates chromosome end biology by binding to SPE2 motifs present at TARE2/3 and a neighborhood between TARE6 and the adjacent upsB-type var gene (29). Surprisingly, all PfAP2-HFs were recruited to these two SPE2-rich zones, while HP1 covered the entire TARE1-6 (Figure 3D). This finding hints at the existence of an ApiAP2 complex involving PfAP2-HFs and PfAP2-SIP2/SPE2 interaction at two separate regions of the subtelomeres.
In summary, PfAP2-HFs are likely to play divergent roles in heterochromatin function (Figure 3E). Together with the previously characterized PfAP2-Tel, other ApiAP2 factors such as PfAP2-G2, PfAP2-G5, and PfAP2-12 may be also involved in telomere biology such as length maintenance. Colocalization of PfAP2-HFs with PfAP2-SIP2 in subtelomeric regions suggests they may co-ordinate in subtelomeric heterochromatin formation and genome integrity. In addition, regulation of virulence gene expression may involve different PfAP2-HFs.
Distinct patterns of PfAP2-HF recruitment to heterochromatin and euchromatin
Preferential binding of PfAP2-HFs to heterochromatic genes raises the question of how these transcription factors are recruited to the target gene loci. Hence PfAP2-HF binding patterns were investigated at euchromatic and heterochromatic genes, respectively. A larger percentage of PfAP2-HF binding sites were detected within upstream regions at euchromatic genes, compared with those at heterochromatic genes (Figures 4A-H panels i, ii, iv, and v and Supplementary Figure S6). Meanwhile, wider PfAP2-HF distribution and a preference for coding regions were generally observed at heterochromatic gene loci except for PfAP2-O2. The differential occupancy patterns imply that PfAP2-HF regulation of euchromatic and heterochromatic genes may involve distinct mechanisms.
Figure 4.

Distinct transcriptional regulatory patterns of heterochromatic and euchromatic genes by the heterochromatin associated AP2 transcription factors. (A-H) Top panel: (i) Log2-transformed ChIP/input fold enrichment at euchromatic gene loci; (ii) Peak distribution at euchromatic gene loci; (iii) Best DNA motif de novo discovered in the binding sites at euchromatin with E-values displayed at the top. Bottom panel: (i) Log2-transformed ChIP/input fold enrichment at heterochromatic gene loci; (ii) Peak distribution at heterochromatic gene loci; (iii) Most likely DNA motif de novo discovered in the binding sites at heterochromatin with E-values displayed above. PfAP2-HF-Is and PfAP2-HF-IIs are labeled with superscript I and II on the right side, respectively. TSS, translation start site; TES, translation end site.
To explore this issue, de novo motif discovery was performed to determine the DNA binding specificity of each individual PfAP2-HF for euchromatin and heterochromatin. Close inspection of the euchromatic binding sites identified one specific DNA binding motif for these PfAP2-HF factors (Figures 4A-H top panel iii and Supplementary Figure S7). Most of the discovered motifs are highly similar to those described by previous characterization using a protein binding microarray (PBM) in vitro (53) (Supplementary Figure S7). In particular, to our knowledge, this is the first report of the DNA binding consensus of PfAP2-O5. Notably, putative motifs found in the heterochromatic binding sites, which contain GCA repeats, are nearly identical for individual PfAP2-HF-I members (Figures 4A-H bottom panel vi). A lack of sequence specificity raises the possibility that these heterochromatic motifs may not constitute true binding consensuses. Recently, it has been reported that PfAP2-HC binds to heterochromatin loci independently of its AP2 domain (31). We therefore speculated that DNA binding domains might be required for the binding to euchromatin but not heterochromatin by some of the PfAP2-HF transcription factors such as PfAP2-HF-Is.
For PfAP2-11A and PfAP2-exp, putative target motifs present in euchromatin and heterochromatin shared highly similar sequences, suggesting these two ApiAP2 factors may be recruited into different chromatin environments via recognition of the same DNA motifs. Interestingly, two different motifs were detected in PfAP2-O2 bound euchromatic and heterochromatic loci, and the motif present in the latter loci showed a much higher matching score to the PBM identified motif (score = 2.66e-3). Considering PfAP2-O2 contains two DNA binding domains, it is likely that this factor binds to different chromatin environments via different domains.
Given the general role of HP1 in heterochromatin biology (6,70), we examined interactions between PfAP2-HFs and PfHP1 in the pfap2-hf-gfp transgenic lines. Weak physical interactions were detected for both PfAP2-HC and PfAP2-G5 (Supplementary Figures S8A and S8B). Detection sensitivity was dramatically improved with reasonable specificity by paraformaldehyde crosslinking prior to the preparation of nuclear extracts (Supplementary Figures S8A-S8C). Thus, PfAP2-HFs do not form stable complexes with PfHP1 as GDV1 (8) and probably loosely tether to PfHP1. Such weak multivalent interactions are common in the nucleus, and facilitate chromatin remodeling and transcriptional regulation (71). Alternatively, PfAP2-HFs may bind to the heterochromatin components that interact with PfHP1 rather than to PfHP1 itself.
PfPfAP2-HF factors are highly coordinated in modulating heterochromatic genes
We observed substantial colocalization of PfAP2-HFs, especially PfAP2-HF-Is, at heterochromatic regions (Figures 2B-C and Supplementary Figure S2). Indeed, collaboration among PfAP2-HF-Is was closer than other PfAP2-HFs (Figure 5A). PfAP2-HF-Is together targeted as many as 304 heterochromatic genes accounting for approximate 55% of all heterochromatic genes (Figure 5B and Supplementary Figure S9 and Table S5). An overwhelming majority of the heterochromatic genes were regulated by at least four PfAP2-HFs. Heterochromatin is indispensable for survival and transmission of parasites, especially host immune evasion (3), which requires tight transcriptional control such as through coordination of transcription factors like PfAP2-HFs. This is specially true for the virulence genes whose expression is likely associated with five or more PfAP2-HFs (Figures 5C and D and Supplementary Figures S9A-S9E). For instance, the major virulence gene family (var) was targeted by all eight PfAP2-HFs, and a significant number of members changed in expression level upon PfAP2-HF depletion (Figure 5D and Supplementary Figure S9A), as exemplified by PF3D7_0412700, the predominantly expressed var gene in the wild type 3D7 strain used in our study (18) (Supplementary Figures S10A and S10B). In addition, 494 heterochromatic genes are targeted by both PfAP2-G2 and PfAP2-G5 (Figure 5D), indicating a close collaboration between the two PfAP2-HFs, such as in co-regulation of early gametocyte development as recently described (33,34). Intriguingly, based on the 3D genome model (65), there seems to be spatial colocalization of PfAP2-HFs proximal to the telomere super-domain, hinting at the transcriptional co-regulation of these transcription factors (Supplementary Figure S11).
Figure 5.

Collaboration of PfAP2-HFs in modulating the heterochromatic genes. (A) Hierarchical clustering of PfAP2-HFs. (B) Classification of heterochromatic gene regulation by PfAP2-HFs. (C) Regulatory patterns of heterochromatic gene families by PfAP2-HFs. (A-C) are drawn on the basis of PfAP2-HF targeting heterochromatic genes. PfAP2-HF-Is and PfAP2-HF-IIs are labeled with superscript I and II on the right side, respectively. (D) Cross-talk between PfAP2-HFs in regulating heterochromatic genes. Sizes of transcription factor nodes are scaled proportionally to the numbers of their heterochromatic targets. Edges linking transcription factors are marked with double dark grey lines and scaled proportionally to their shared targets. PfAP2-HF regulation of the representative heterochromatic gene families are coloured individually and scaled according to BH adjusted enrichment P-values of either targets for direct targeting or differentially expressed genes for indirect regulation.
Cooperation patterns of PfAP2-HFs in regulating euchromatic genes were completely different from those in heterochromatic genes (Supplementary Figure S12A), which further supports the hypothesis that these transcription factors employ different strategies to control these two classes of genes. A considerable number of euchromatic genes were targeted by only one or two PfAP2-HFs (Supplementary Figure S12B and Table S6), indicating that PfAP2-HF regulation of euchromatic genes involves less cooperation between regulators. Thus, it is not surprising that cross-talk between PfAP2-HFs was much less common in the regulation of euchromatic genes (Supplementary Figure S12C) compared to that of heterochromatic genes (Figure 5D) and the euchromatic functions of PfAP2-HFs were divergent. We also note that PfAP2-HFs, especially PfAP2-G5, PfAP2-G2, PfAP2-exp, and PfAP2-O5, seem highly correlated with invasion-associated genes (Supplementary Figures S12D and S10C-S10F). The majority of invasion genes are targeted by different combinations of PfAP2-HFs. This may represent an example of the extra functions of PfAP2-HFs beyond regulation of heterochromatic gene expression.
PfAP2-HFs exert opposing effects on heterochromatic gene expression
To investigate the functions of PfAP2-HFs and other ApiAP2 factors in gene expression, we disrupted sixteen apiap2 genes individually through CRISPR/Cas9 editing (Figure 1A). Following transfection, drug selection, and cloning through limiting dilution, we obtained six knockout lines, including PfAP2-HCΔ, PfAP2-G5Δ, PfAP2-G2Δ, PfAP2-O2Δ, PfAP2-G4Δ, and PfAP2-HSΔ (Figure 1B and Supplementary Figure S13), which is generally consistent with previous transposon mutagenesis results in P. falciparum (72). However, we failed to disrupt PfAP2-exp as Martins et al. reported (32), although it was predicted to be mutable in its coding region. Among the disruptable ApiAP2 transcription factors, four belong to the PfAP2-HF family. To explore the roles of these ApiAP2 factors in gene transcription, their knockout lines were subjected to transcriptome analyses at ring, trophozoite, and schizont stages of the intraerythrocytic cycle with the 3D7 strain as the wild type control.
Subgroups of heterochromatic genes differ in their expression timing during the intraerythrocytic cycle (6,9). Given the majority of variant genes are highly transcribed from late ring to trophozoite stages, we first investigated the trophozoite stage. In agreement with the preference for heterochromatic genes, the disrupted PfAP2-HFs exhibited much stronger regulatory effects on heterochromatic gene expression compared to those on euchromatic genes in general (Figures 6A-H). Furthermore, consistent with recent reports (33,34), PfAP2-G5 and PfAP2-G2 mainly functioned as transcriptional repressors of heterochromatic genes (Figure 6M). Other PfAP2-HFs, namely PfAP2-HC and PfAP2-O2 both showed positive regulation of heterochromatic genes. Surprisingly, disruption of two non-PfAP2-HF members, namely PfAP2-G4 and PfAP2-HS, also resulted in dysregulation of heterochromatic gene expression (Figures 6I-M), though neither are associated with the heterochromatin environment in ChIP-seq analyses (Figure 2B and Supplementary Figure S5). This observation suggests that ApiAP2 transcription factors are capable of regulating heterochromatic gene expression in distinct manners either via direct binding or through an indirect cascade (Figure 5D). In addition, regulatory patterns of these disrupted ApiAP2 transcription factors were basically consistent throughout the asexual development (Supplementary Figure S14).
Figure 6.

Effects of disruptable ApiAP2 transcription factors, including PfAP2-HC, PfAP2-G5, PfAP2-G2, PfAP2-O2, PfAP2-G4, and PfAP2-HS, on transcription of heterochromatic genes. (A, C, E, G, I, and K) Differential gene expression in PfAP2-HCΔ, PfAP2-G5Δ, PfAP2-G2Δ, PfAP2-O2Δ, PfAP2-G4Δ, and PfAP2-HSΔ trophozoites, respectively (fold change of > 2). Dashed lines denote differential expression thresholds. Red open triangles and blue points are euchromatic and heterochromatic genes, respectively. (B, D, F, H, J, and L) PfAP2-HC, PfAP2-G5, PfAP2-G2, PfAP2-O2, PfAP2-G4, and PfAP2-HS exhibited stronger effects on heterochromatic genes than on euchromatic genes at the trophozoite stage. The boxes inside the violin plots represent the interquartile ranges with white diamonds indicating the medians. **** signifies P-value < 0.0001. (M) Expression changes of differentially expressed heterochromatic genes in PfAP2-HCΔ, PfAP2-G5Δ, PfAP2-G2Δ, PfAP2-O2Δ, PfAP2-G4Δ, and PfAP2-HSΔ trophozoite stage parasites, respectively. The boxes show the interquartile ranges with horizontal lines drawn in the middle indicating the medians. The ends of the upper and lower whiskers denote maximum and minimum values, respectively. (N) FPKM values of var transcripts at the ring stage.
Subcloning may also contributes to expression changes, especially of clonally variant genes, although we didn’t observe apparent expression changes (fold change of > 2) between G7N and G7-10B wild-type subclones for most heterochromatic genes (data not shown). In addition, to address the possibility of differential expression attributable to low expression, we removed genes expressed at a FPKM value below 32 in both wild-type and knockout lines (Supplementary Figure S15A). As the transcript abundances of the silent var genes in the wild-type clone were generally lower than 32 in FPKM (Supplementary Figure S16A), here we adopted 32 as a cutoff to remove lowerly expressed genes. Basically, the regulatory patterns of the six disruptable ApiAP2 transcription factors observed in the highly expressed genes were consistent with those observed before low expression filtering, except for PfAP2-G4 (Supplementary Figures S15B and C). The median fold change of heterochromatic genes in PfAP2-G4Δ approximates to 1 (Supplementary Figure S15B). Low expression caused relatively large changes may explain the unexpected observation of heterochromatic gene expression changes in PfAP2-G4Δ. It is worthy of note that the knockout of PfAP2-G5 altered transcription of as many as 60 heterochromatic genes (Supplementary Figure S15B).
The primal virulence gene family var maintains strict mutually exclusive expression during the intraerythrocytic development (20). Here, we observed that PfAP2-HC depletion disrupted mutually exclusive expression of var genes completely (Figure 6N). The knockout silenced the entire repertoire of the var family, which was confirmed by quantitative reverse PCR in two parasite clones generated from two independent transfections (Supplementary Figures S16A and E). Regarding other ApiAP2s, only PfAP2-G2 knockout displayed a moderate effect on var expression, i.e. down-regulation of the active member. Meanwhile, the pfmc-2tm gene family was completely silenced as well, and transcript abundances of other variant gene families such as rifin and stevor were significantly reduced in the PfAP2-HC knockout line (Supplementary Figures S16B-D). More importantly, mutually exclusive expression of var was rescued when pfap2-hc was restored by a second-round CRISPR/Cas9 gene editing, though the predominantly expressed var gene switched to other members (Supplementary Figure S16F). The knockout and rescue results demonstrate an indispensable role of PfAP2-HC in the maintenance of the singular expression of var genes. Interestingly, this phenomenon was not observed in the PfAP2-HC knockdown line (31), indicating that PfAP2-HC regulation of variant gene expression probably does not rely heavily on its product dosage.
Dynamic regulation of transcriptional states by ApiAP2 transcription factors and chromatin accessibility
The expression levels of seven out of sixteen studied ApiAP2 transcription factors strongly correlated with that of their respective targets (|Pearson correlation| > 0.8, Figure 7A). Among them, the predicted overall effects of PfAP2-HC, PfAP2-G5, and PfAP2-O2 were consistent with knockout observations at at least one stage of the intraerythrocytic cycle (Supplementary Figure S17). We also note that PfAP2-HC, PfAP2-G5, PfAP2-O2, and PfAP2-HS exhibited opposite effects on target gene expression at different stages (Supplementary Figure S17), suggesting the dynamic and intricate regulation of transcription involving cooperation of transcription factors and possible additional regulatory layers such as local chromatin structures (6,7,12).
Figure 7.

Integrated analysis of ApiAP2 occupancy patterns throughout the intraerythrocytic cycle. (A) Pearson correlation between ApiAP2 transcription factors and their targets in expression levels. ApiAP2 expression (z-score standardized log2-transformed FPKM values) and target average expression across the intraerythrocytic cycle are displayed in solid and dashed lines, respectively. (B) Accessible fractions of 5′ UTR located ApiAP2 binding sites during the intraerythrocytic cycle. PfAP2-HF-Is and PfAP2-HF-IIs are labeled with superscript I and II on the right side, respectively. (C) High consistency in ATAC-seq detected accessible chromatin (35) at 5′ UTR of, PfAP2-exp occupancy at 5′ UTR of, and expression levels of invasion involved PfAP2-exp targets across the intraerythrocytic cycle. Profiles at PF3D7_1335100 are illustrated at the right side. R, ring; T, trophozoite; S, schizont.
Chromatin structures restrict the binding of transcription factors; on the other hand, transcription factor occupancy influences chromatin states as exemplified by pioneer factors (73). In addition, chromatin structures and transcription factor occupancy in 5′ UTRs largely determine transcriptional activity during P. falciparum development (2,3). Integration of ApiAP2 ChIP-seqs with ATAC-seqs (35) revealed that ApiAP2 occupancy at 5′ UTRs extensively accompanied accessible chromatin at trophozoite or schizont stages (Figure 7B). For instance, we observed a clear correlation among local chromatin accessibility at 5′ UTRs, PfAP2-exp occupancy, and transcript abundance throughout the intraerythrocytic cycle in invasion genes (Figure 7C). It is perhaps unsurprising that not all ApiAP2 targets were affected by ApiAP2 knockouts (Supplementary Figure S18), as transcriptional outputs integrate multiple regulatory layers (17,35,74,75). Functional redundancy and compensation may exsit in this family (31).
Intriguingly, only a small portion of PfAP2-HF-I binding sites fell within accessible chromatin (Figure 7B), suggesting that these PfAP2-HFs are more than a conventional transcription factor; they are also potentially integral components of chromatin. These chromatin-associated transcription factors may contribute to chromatin assembly and maintenance as reported in the recent studies of PfAP2-HC and PfAP2-G2 (31,33). This mechanism is not limited to Plasmodium spp. In fission yeast, for example, the ATF/CREB family of transcription factors are able to recruit histone deacetylases and the histone methyltransferase Clr4 to initiate heterochromatin assembly (76,77).
The appreciable concentration of ApiAP2 occupancy in the accessible chromatin reflects the capability of ApiAP2s to bind DNA. Therefore, we looked for DNA recognition motifs in the euchromatic binding sites (Supplementary Figure S7), given that the functions of some ApiAP2 transcription factors in heterochromatin do not rely on DNA binding. In general, each individual ApiAP2 transcription factor recognized highly similar DNA sequences throughout the intraerythrocytic cycle. Most of the discovered motifs are highly similar to those described by previous characterization using PBM in vitro (24). This is, to our knowledge, the first report on the DNA binding consensus of PfAP2-11B and PfAP2-O5. In addition, no motifs were discovered for PfAP2-06A either by PBM or by ChIP-seq.
Autoregulation and feedforward loops are prevalent in the coordinated ApiAP2 networks
The ap2-g gene may be regulated via a positive feedforward circuit (10,11). Such autoregulation and mutual regulation among ApiAP2 transcription factors may facilitate the parasites in regulating gene expression more precisely and in response to environmental stimuli more rapidly. To address this, we mapped the regulatory relationships among ApiAP2 transcripton factors at each stage of the intraerythrocytic cycle by integrating their binding profiles and knockout effects on transcription (Figures 8A-C). Autoregulatory loops were prevalent in ApiAP2 genes except pfap2-06a, pfap2-g4, and pfap2-o2. This type of regulatory motif, composed of one regulator that targets itself, decreases the biosynthetic cost of regulation and contributes to the tuning of response time to stimuli and gene expression stability (78). In addition, all ApiAP2 transcription factors under study were regulated by more than one ApiAP2 other than themselves. Transcriptional regulation of pfap2-06b and pfap2-hc appears to be under rigid control involving no less than eight ApiAP2s including themselves. We also note that PfAP2-HC, PfAP2-G2, and PfAP2-G5 participated in regulating many other ApiAP2s (number 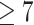 ). This finding may interpret the ‘side effect’ of the knockout of non-PfAP2-HF factors on the expression of heterochromatic genes (Figures 6I-M).
). This finding may interpret the ‘side effect’ of the knockout of non-PfAP2-HF factors on the expression of heterochromatic genes (Figures 6I-M).
Figure 8.

ApiAP2 regulatory networks in the intraerythrocytic cycle. (A-C) A relationship map of ApiAP2s based on ChIP-sequencing of these transcription factors at the ring, trophozoite, and schizont stages, respectively. ApiAP2 effects on transcription of their targets are indicated if RNA-seq assays of ApiAP2 knockouts are available. Types of relationship are listed in the right box beside (A). (D-F) Representative malaria parasite metabolic pathways (MPMP) targeted by ApiAP2 transcription factors at the ring, trophozoite, and schizont stages, respectively (BH adjusted enrichment P-values < 0.01). The color map is displayed beside (D). PfAP2-HF-Is and PfAP2-HF-IIs are labeled with superscript I and II on the right side, respectively. (G) ApiAP2 transcription factors collaborate via multi-output feedforward loops. Top panel: a simple positive feedforward loop consisting of two transcription factors A and B and a target C. Middle panel: PfAP2-G5 represses pfap2-g transcription, while PfAP2-G activates pfap2-g5 transcription. In addition, PfAP2-G regulates the expression of itself. PfAP2-G5 and PfAP2-G exert opposing effects on expression of early gametocyte (EG) genes. Bottom panel: PfAP2-G2 and PfAP2-HC target each other but do not seem to determine the expression level of the other. Autoregulation exists in both of these two transcription factors. PfAP2-G2 and PfAP2-HC co-activate var transcription.
PfAP2-HF-I factors showed the strongest preference for heterochromatin at the late stages (Figures 8D-F), which suggests a critical role for PfAP2-HFs in heterochromatin biology during schizogony. In addition, some PfAP2-HFs such as PfAP2-exp, PfAP2-G2, and PfAP2-G5 played more extensive roles. Notably, feedforward loops (for a simplified model, see the top panel of Figure 8G), which provide precise temporal control of gene activation and inactivation (78), were particularly prevalent in the regulatory networks (Figures 5D, 8A-F and Supplementary Figure S12C). This regulatory motif has been recently demonstrated in PfAP2-G5 and PfAP2-G co-regulation of P. falciparum gametocytogenesis (34) (Figure 8G middle panel). The present study revealed that PfAP2-G2 and PfAP2-HC also built a reiterative system of feedforward control to stimulate transcription of the var family (Figure 8G bottom panel).
Taken together, our data reveal the extensive cross-talk in the ApiAP2 network which may facilitate accurate tuning of stage-specific expression and homeostasis maintenance in the intraerythrocytic cycle of malaria parasites.
DISCUSSION
Members of the ApiAP2 protein family regulate gene expression during various physiological processes in malaria parasites (79,80). Functional investigation of these factors has shown that they may regulate gene expression in a positive (e.g. PfAP2-G, PfAP2-I, and PfAP2-HS) or negative manner (e.g. PfAP2-G2 and PfAP2-G5) (10,27,33,34,60,81). For PfAP2-G2 and PfAP2-G5, our study confirms their repressive function on gene transcription. Moreover, a number of ApiAP2 factors are also involved in local heterochromatin formation, genome integrity, and telomere biology, e.g. PfAP2-SIP2 and PfAP2-Tel. Considering the predominant role of ApiAP2 family in the dynamic gene expression in malaria parasites, here we addressed the functional diversity of these transcription factors during the asexal replicating stages.
One of the most intriguing finding of this study is that PfAP2-HFs function differently in euchromatin and heterochromain. While they may regulate euchromatic genes through recognition of specific DNA motifs as in PfAP2-G or PfAP2-I, the majority of these proteins are recruited to the heterochromain regions most probably via DNA-independent manner and are involved in the stage-specific expression of genes located in these regions. The exact reason why these PfAP2-HFs carrying various numbers of DBD abandon the classic DNA-protein interaction in heterochromain is still unclear, but it may be associated with the unusally extensive distribution of heterochromatin modification in the genome of malaria parasites. Therefore, PfAP2-HFs may be integral components of local heterochromatin rather than general transcription factors that bind to the UTR of target genes as described recently (31).
Due to the critical function of the dynamic heterochromatin environment for gene expression-linked adaptation and development in malaria parasites, we conducted a systematic screen of heterochromatin-associated ApiAP2 factors via ChIP assays. Genome-wide occupancy analysis identified a total of eight PfAP2-HF factors involved in heterochromatin formation. Each PfAP2-HF was found to target genes located in both heterochromatic and euchromatic regions. DNA motif analysis further revealed that, at least for those targets in heterochromatin regions, these PfAP2-HFs are likely to be integral components of heterochromatin rather than direct DNA-binding proteins. Notably, they display weak physical interactions with PfHP1, which strongly suggests other heterochromatin compponents may recruite them into the heterochromatin directly. Intriguingly, we observed various knockout phenotypes of heterochromatin gene expression for three PfAP2-HFs. Among them, only PfAP2-HC and PfAP2-G2 are directly associated with the mutually exclusive expression of antigenic variant genes, supporting the hypothesis that the PfAP2-HF network involving differential or complementary fuctions for hetereochromatin gene expression.
Filarsky et al. found peptides of PfAP2-HC in all PfHP1 co-immunoprecipitation/mass spectrometry replicates and PfAP2-12 in one replicate, but their functions were not elucidated (8). The same authors also show that PfAP2-HC binds specifically to heterochromatin throughout the genome (31), and is recruited to heterochromatin regions in a DNA-binding independent mechanism. They concluded that PfAP2-HC regulates neither gene expression nor heterochromatin structure. Their data supports our hypothesis that PfAP2-HFs may be recruited to heterochromatin via an indirect pathway, i.e. through interaction with other heterochromatin compponents. We speculate that the reason they did not observe gene dysregulation may be that the extent of knockdown is not sufficient to produce measurable phenotypic changes. Indeed, our own data suggest that knoockdown parasite strains may be sub-optimal for the investigation of PfAP2-HFs. In contrast, gene knockouts, rather than knockdowns, clearly show that PfAP2-HC and PfAP2-G5 regulate expression of variant genes. Moreover, the dysregulation of target genes is associated with alteration of the local heterochromatin structure. Furthermore, binding of PfAP2-G2 within gene bodies correlates with H3K36me3 and HP1 (33). It is speculated that the occupancy of PfAP2-G2 in these heterochromatic gene bodies may serve as an alternative regulatory mechanism. The same study also showed that PfAP2-G2 may be a transcriptional repressor as it is known to be in rodent malaria parasites. In summary, previous reports support the main findings presented here that multiple ApiAP2 factors are involved in heterochromatin structure and regulate heterochromatic gene expression as transcription activators or repressors.
We also found that PfAP2-HFs preferentially recognized different heterochromatin regions including the non-coding subtelomeric region, subtelomeric gene-coding regions, and chromosomal gene-coding regions. Members of the PfAP2-HF-I subgroup are distributed in most heterochromatin regions whereas the other PfAP2-HFs mainly localize within non-coding subtelomeric regions, particularly in the locations surrounding the TARE2-4 region or between the first upsB-var gene and TARE6 (rep20) on the ends of chromosomes. These locations correspond to the SPE2 motif-enriched regions recognized by PfAP2-SIP2 (29). This suggests that the recruitment of PfAP2-HFs to the heterochromatin regions at chromosome ends may depend on the SEP2/SIP2 complex. Moreover, the long non-coding RNAs produced by TARE2-4 may also be associated with heterochromatin formation or maintenance as is the case with rep20 (69). Overexpression of PfAP2-SIP2 has no significant effect on global gene expression (29). Since most heterochromatic genes are suppressed by the heterochromatin environment, SIP2 knock-out or conditional disruption may release repression of these target genes. In addition to ApiAP2 factors, a conserved Pol III RNA polymerase-associated transcription factor TFIIIA, also called PfTRZ in P. falciparum, displays a similar binding pattern at the telomeres and the region adjacent to TARE6 (82). Therefore, ApiAP2 along with other transcription factors may be part of a complex network that regulates telomere functionality and/or heterochromatin formation and maintenance.
Taken together, our data reveal a complex heterochromatin structure involving multiple ApiAP2 factors with distinct functions for gene expression in human malaria parasites, contributing to a better understanding of the multi-layered mechanism of heterochromatic gene expression. It also provides a valuable resource for the genome-wide occupancy landscapes of these ApiAP2 factors over the intraerythrocytic development. Considering the lack of many classic transcriptional factors in the unicellular eukaryotic parasites, such datasets will be of great use in further mechanistic research on other phenotypes. The lack of the ApiAP2 family in mammals provides an opportunity for anti-malarial drug development. Our data will facilitate this process through target gene-based phenotypic identification for individual ApiAP2 proteins.
DATA AVAILABILITY
All raw sequencing data reported in this study have been deposited in GEO and are publicly accessible under accession GSE184659 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE184659). The genome-wide binding profiles of the sixteen studied ApiAP2 transcription factors are shown in UCSC genome browser sessions, including http://genome-asia.ucsc.edu/s/bzswmu/pf47_part1, and http://genome-asia.ucsc.edu/s/bzswmu/pf47_part2_new.
Supplementary Material
ACKNOWLEDGEMENTS
We are grateful to A. Scherf (Institut Pasteur, Paris) for providing anti-HP1 antibody, Zheng Bao (Southwest Medical University, China) for assisting in data deposition, and Ruping Sun (University of Minnesota, Minneapolis, MN) for his generous advice on data analysis.
Author Contributions: Q.Z., M.J. and J.C. conceived and designed experiments. X.S, C.W., Y.F., F.W., Y.Z., F.S., M.Z., X. H., G. Z. and J.T. performed the experiments of this study. M.J., S.Y. and G.G. performed all the bioinformatics analyses. Q.Z. and M.J. wrote the manuscript with contribution from X.S., X.Y., J.M., J.C. and R.C. All authors discussed and approved the manuscript.
Contributor Information
Xiaomin Shang, Laboratory of Molecular Parasitology, Key Laboratory of Spine and Spinal Cord Injury Repair and Regeneration of Ministry of Education, Tongji Hospital; Clinical Center for Brain and Spinal Cord Research, School of Medicine, Tongji University, Shanghai 200092, China; Department of Parasitology, Xiangya School of Medicine, Central South University, Changsha 410013, China.
Changhong Wang, Laboratory of Molecular Parasitology, Key Laboratory of Spine and Spinal Cord Injury Repair and Regeneration of Ministry of Education, Tongji Hospital; Clinical Center for Brain and Spinal Cord Research, School of Medicine, Tongji University, Shanghai 200092, China.
Yanting Fan, Laboratory of Molecular Parasitology, Key Laboratory of Spine and Spinal Cord Injury Repair and Regeneration of Ministry of Education, Tongji Hospital; Clinical Center for Brain and Spinal Cord Research, School of Medicine, Tongji University, Shanghai 200092, China; Department of Parasitology, School of Medicine, Northwest University, Xi’an, Shanxi 710069, China.
Gangqiang Guo, Laboratory of Molecular Parasitology, Key Laboratory of Spine and Spinal Cord Injury Repair and Regeneration of Ministry of Education, Tongji Hospital; Clinical Center for Brain and Spinal Cord Research, School of Medicine, Tongji University, Shanghai 200092, China.
Fei Wang, Laboratory of Molecular Parasitology, Key Laboratory of Spine and Spinal Cord Injury Repair and Regeneration of Ministry of Education, Tongji Hospital; Clinical Center for Brain and Spinal Cord Research, School of Medicine, Tongji University, Shanghai 200092, China.
Yuemeng Zhao, Laboratory of Molecular Parasitology, Key Laboratory of Spine and Spinal Cord Injury Repair and Regeneration of Ministry of Education, Tongji Hospital; Clinical Center for Brain and Spinal Cord Research, School of Medicine, Tongji University, Shanghai 200092, China.
Fei Sheng, Laboratory of Molecular Parasitology, Key Laboratory of Spine and Spinal Cord Injury Repair and Regeneration of Ministry of Education, Tongji Hospital; Clinical Center for Brain and Spinal Cord Research, School of Medicine, Tongji University, Shanghai 200092, China.
Jianxia Tang, National Health Commission Key Laboratory of Parasitic Disease Control and Prevention, Jiangsu Provincial Key Laboratory on Parasite and Vector Control Technology, Jiangsu Institute of Parasitic Diseases, Wuxi, 214064, China.
Xiaoqin He, National Health Commission Key Laboratory of Parasitic Disease Control and Prevention, Jiangsu Provincial Key Laboratory on Parasite and Vector Control Technology, Jiangsu Institute of Parasitic Diseases, Wuxi, 214064, China.
Xinyu Yu, National Health Commission Key Laboratory of Parasitic Disease Control and Prevention, Jiangsu Provincial Key Laboratory on Parasite and Vector Control Technology, Jiangsu Institute of Parasitic Diseases, Wuxi, 214064, China.
Meihua Zhang, National Health Commission Key Laboratory of Parasitic Disease Control and Prevention, Jiangsu Provincial Key Laboratory on Parasite and Vector Control Technology, Jiangsu Institute of Parasitic Diseases, Wuxi, 214064, China.
Guoding Zhu, National Health Commission Key Laboratory of Parasitic Disease Control and Prevention, Jiangsu Provincial Key Laboratory on Parasite and Vector Control Technology, Jiangsu Institute of Parasitic Diseases, Wuxi, 214064, China.
Shigang Yin, Laboratory of Neurological Diseases and Brain Function, The Affiliated Hospital of Southwest Medical University, Luzhou, China.
Jianbing Mu, Laboratory of Malaria and Vector Research, National Institute of Allergy and Infectious Diseases, National Institutes of Health, Rockville, MD 20892-8132, USA.
Richard Culleton, Division of Molecular Parasitology, Proteo-Science Centre, Ehime University, Matsuyama, Ehime 790-8577, Japan.
Jun Cao, National Health Commission Key Laboratory of Parasitic Disease Control and Prevention, Jiangsu Provincial Key Laboratory on Parasite and Vector Control Technology, Jiangsu Institute of Parasitic Diseases, Wuxi, 214064, China; Center for Global Health, School of Public Health, Nanjing Medical University, Nanjing 211166, China.
Mei Jiang, Laboratory of Molecular Parasitology, Key Laboratory of Spine and Spinal Cord Injury Repair and Regeneration of Ministry of Education, Tongji Hospital; Clinical Center for Brain and Spinal Cord Research, School of Medicine, Tongji University, Shanghai 200092, China; Department of Medical Genetics, Shanghai Tenth People's Hospital, School of Medicine, Tongji University, Shanghai 200072, China.
Qingfeng Zhang, Laboratory of Molecular Parasitology, Key Laboratory of Spine and Spinal Cord Injury Repair and Regeneration of Ministry of Education, Tongji Hospital; Clinical Center for Brain and Spinal Cord Research, School of Medicine, Tongji University, Shanghai 200092, China.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Key R&D Program of China [2018YFA0507300, 2020YFC1200105]; National Natural Science Foundation of China (NSFC) [81630063, 81971959, 31701282, 81971967, 82172302]; Jiangsu Provincial Project of Invigorating Health Care through Science, Technology and Education and the Jiangsu Provincial Commission of Health; the Shanghai Blue Cross Brain Hospital Co., Ltd. and Shanghai Tongji University Education Development Foundation.
Conflict of interest statement. None declared.
REFERENCES
- 1. Josling G.A., Williamson K.C., Llinás M.. Regulation of sexual commitment and gametocytogenesis in malaria parasites. Annu. Rev. Microbiol. 2018; 72:501–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hollin T., Le Roch K.G.. From genes to transcripts, a tightly regulated journey in plasmodium. Front Cell Infect Microbiol. 2020; 10:618454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hollin T., Gupta M., Lenz T., Le Roch K.G.. Dynamic chromatin structure and epigenetics control the fate of malaria parasites. Trends Genet. 2021; 37:73–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bunnik E.M., Cook K.B., Varoquaux N., Batugedara G., Prudhomme J., Cort A., Shi L., Andolina C., Ross L.S., Brady D.et al.. Changes in genome organization of parasite-specific gene families during the plasmodium transmission stages. Nat. Commun. 2018; 9:1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bunnik E.M., Venkat A., Shao J., McGovern K.E., Batugedara G., Worth D., Prudhomme J., Lapp S.A., Andolina C., Ross L.S.et al.. Comparative 3D genome organization in apicomplexan parasites. Proc. Natl. Acad. Sci. U. S. A. 2019; 116:3183–3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Brancucci N.M.B., Bertschi N.L., Zhu L., Niederwieser I., Chin W.H., Wampfler R., Freymond C., Rottmann M., Felger I., Bozdech Z.et al.. Heterochromatin protein 1 secures survival and transmission of malaria parasites. Cell Host & Microbe. 2014; 16:165–176. [DOI] [PubMed] [Google Scholar]
- 7. Coleman B.I., Skillman K.M., Jiang R.H.Y., Childs L.M., Altenhofen L.M., Ganter M., Leung Y., Goldowitz I., Kafsack B.F.C., Marti M.et al.. A plasmodium falciparum histone deacetylase regulates antigenic variation and gametocyte conversion. Cell Host Microbe. 2014; 16:177–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Filarsky M., Fraschka S.A., Niederwieser I., Brancucci N.M.B., Carrington E., Carrio E., Moes S., Jenoe P., Bartfai R., Voss T.S.. GDV1 induces sexual commitment of malaria parasites by antagonizing HP1-dependent gene silencing. Science. 2018; 359:1259–1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Fraschka S.A., Filarsky M., Hoo R., Niederwieser I., Yam X.Y., Brancucci N.M.B., Mohring F., Mushunje A.T., Huang X., Christensen P.R.et al.. Comparative heterochromatin profiling reveals conserved and unique epigenome signatures linked to adaptation and development of malaria parasites. Cell Host Microbe. 2018; 23:407–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kafsack B.F.C., Rovira-Graells N., Clark T.G., Bancells C., Crowley V.M., Campino S.G., Williams A.E., Drought L.G., Kwiatkowski D.P., Baker D.A.et al.. A transcriptional switch underlies commitment to sexual development in malaria parasites. Nature. 2014; 507:248–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sinha A., Hughes K.R., Modrzynska K.K., Otto T.D., Pfander C., Dickens N.J., Religa A.A., Bushell E., Graham A.L., Cameron R.et al.. A cascade of DNA-binding proteins for sexual commitment and development in plasmodium. Nature. 2014; 507:253–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lopez-Rubio J.J., Mancio-Silva L., Scherf A.. Genome-wide analysis of heterochromatin associates clonally variant gene regulation with perinuclear repressive centers in malaria parasites. Cell Host & Microbe. 2009; 5:179–190. [DOI] [PubMed] [Google Scholar]
- 13. Pérez-Toledo K., Rojas-Meza A.P., Mancio-Silva L., Hernández-Cuevas N.A., Delgadillo D.M., Vargas M., Martínez-Calvillo S., Scherf A., Hernandez-Rivas R.. Plasmodium falciparum heterochromatin protein 1 binds to tri-methylated histone 3 lysine 9 and is linked to mutually exclusive expression of var genes. Nucleic Acids Res. 2009; 37:2596–2606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cortés A., Deitsch K.W.. Malaria epigenetics. Cold Spring Harb. Perspect. Med. 2017; 7:a025528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Guizetti J., Barcons-Simon A., Scherf A.. Trans-acting GC-rich non-coding RNA at var expression site modulates gene counting in malaria parasite. Nucleic Acids Res. 2016; 44:9710–9718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Barcons-Simon A., Cordon-Obras C., Guizetti J., Bryant J.M., Scherf A.. CRISPR interference of a clonally variant GC-Rich noncoding RNA family leads to general repression of var genes in plasmodium falciparum. Mbio. 2020; 11:e03054-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Fan Y., Shen S., Wei G., Tang J., Zhao Y., Wang F., He X., Guo G., Shang X., Yu X.et al.. Rrp6 regulates heterochromatic gene silencing via ncRNA RUF6 decay in malaria parasites. Mbio. 2020; 11:e01110-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Duraisingh M.T., Skillman K.M.. Epigenetic variation and regulation in malaria parasites. Annu. Rev. Microbiol. 2018; 72:355–375. [DOI] [PubMed] [Google Scholar]
- 19. Batugedara G., Lu X.M., Bunnik E.M., Le Roch K.G. The role of chromatin structure in gene regulation of the human malaria parasite. Trends Parasitol. 2017; 33:364–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Scherf A., Lopez-Rubio J.J., Riviere L.. Antigenic variation in plasmodium falciparum. Annu. Rev. Microbiol. 2008; 62:445–470. [DOI] [PubMed] [Google Scholar]
- 21. Freitas-Junior L.H., Bottius E., Pirrit L.A., Deitsch K.W., Scheidig C., Guinet F., Nehrbass U., Wellems T.E., Scherf A.. Frequent ectopic recombination of virulence factor genes in telomeric chromosome clusters of p. falciparum. Nature. 2000; 407:1018–1022. [DOI] [PubMed] [Google Scholar]
- 22. Kirkman L.A., Deitsch K.W.. Recombination and diversification of the variant antigen encoding genes in the malaria parasite plasmodium falciparum. Microbiol Spectr. 2014; 2: 10.1128/microbiolspec.MDNA3-0022-2014. [DOI] [PubMed] [Google Scholar]
- 23. De Silva E.K., Gehrke A.R., Olszewski K., León I., Chahal J.S., Bulyk M.L., Llinás M.. Specific DNA-binding by apicomplexan AP2 transcription factors. Proc. Natl. Acad. Sci. USA. 2008; 105:8393–8398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Campbell T.L., De Silva E.K., Olszewski K.L., Elemento O., Llinas M.. Identification and genome-wide prediction of DNA binding specificities for the apiap2 family of regulators from the malaria parasite. PLoS Pathog. 2010; 6:e1001165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Modrzynska K., Pfander C., Chappell L., Yu L., Suarez C., Dundas K., Gomes A.R., Goulding D., Rayner J.C., Choudhary J.et al.. A knockout screen of apiap2 genes reveals networks of interacting transcriptional regulators controlling the plasmodium life cycle. Cell Host & Microbe. 2017; 21:11–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhang C., Li Z., Cui H., Jiang Y., Yang Z., Wang X., Gao H., Liu C., Zhang S., Su X.Z.et al.. Systematic CRISPR-Cas9-Mediated modifications of plasmodium yoelii apiap2 genes reveal functional insights into parasite development. Mbio. 2017; 8:e01986-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Santos J.M., Josling G., Ross P., Joshi P., Orchard L., Campbell T., Schieler A., Cristea I.M., Llinas M.. Red blood cell invasion by the malaria parasite is coordinated by the PfAP2-I transcription factor. Cell Host & Microbe. 2017; 21:731–741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Josling G.A., Russell T.J., Venezia J., Orchard L., van Biljon R., Painter H.J., Llinás M.. Dissecting the role of PfAP2-G in malaria gametocytogenesis. Nat. Commun. 2020; 11:1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Flueck C., Bartfai R., Niederwieser I., Witmer K., Alako B.T., Moes S., Bozdech Z., Jenoe P., Stunnenberg H.G., Voss T.S.. A major role for the plasmodium falciparum apiap2 protein pfsip2 in chromosome end biology. PLoS Pathog. 2010; 6:e1000784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sierra-Miranda M., Vembar S.S., Delgadillo D.M., Avila-Lopez P.A., Herrera-Solorio A.M., Amado D.L., Vargas M., Hernandez-Rivas R.. PfAP2Tel, harbouring a non-canonical DNA-binding AP2 domain, binds to plasmodium falciparum telomeres. Cell. Microbiol. 2017; 19:e12742. [DOI] [PubMed] [Google Scholar]
- 31. Carrington E., Cooijmans R.H.M., Keller D., Toenhake C.G., Bártfai R., Voss T.S.. The apiap2 factor PfAP2-HC is an integral component of heterochromatin in the malaria parasite plasmodium falciparum. Iscience. 2021; 24:102444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Martins R.M., Macpherson C.R., Claes A., Scheidig-Benatar C., Sakamoto H., Yam X.Y., Preiser P., Goel S., Wahlgren M., Sismeiro O.et al.. An apiap2 member regulates expression of clonally variant genes of the human malaria parasite plasmodium falciparum. Sci. Rep. 2017; 7:14042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Singh S., Santos J.M., Orchard L.M., Yamada N., van Biljon R., Painter H.J., Mahony S., Llinás M.. The pfap2-G2 transcription factor is a critical regulator of gametocyte maturation. Mol. Microbiol. 2021; 115:1005–1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Shang X., Shen S., Tang J., He X., Zhao Y., Wang C., He X., Guo G., Liu M., Wang L.et al.. A cascade of transcriptional repression determines sexual commitment and development in plasmodium falciparum. Nucleic Acids Res. 2021; 49:9264–9279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Toenhake C.G., Fraschka S.A., Vijayabaskar M.S., Westhead D.R., van Heeringen S.J., Bartfai R.. Chromatin accessibility-based characterization of the gene regulatory network underlying plasmodium falciparum blood-stage development. Cell Host Microbe. 2018; 23:557–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zhao Y., Wang F., Wang C., Zhang X., Jiang C., Ding F., Shen L., Zhang Q.. Optimization of CRISPR/Cas system for improving genome editing efficiency in plasmodium falciparum. Frontiers in Microbiology. 2020; 11:625862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ghorbal M., Gorman M., Macpherson C.R., Martins R.M., Scherf A., Lopez-Rubio J.J.. Genome editing in the human malaria parasite plasmodium falciparum using the CRISPR-Cas9 system. Nat. Biotechnol. 2014; 32:819–821. [DOI] [PubMed] [Google Scholar]
- 38. Chen P.B., Ding S., Zanghì G., Soulard V., DiMaggio P.A., Fuchter M.J., Mecheri S., Mazier D., Scherf A., Malmquist N.A.. Plasmodium falciparum pfset7: enzymatic characterization and cellular localization of a novel protein methyltransferase in sporozoite, liver and erythrocytic stage parasites. Sci. Rep. 2016; 6:21802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lopez-Rubio J.J., Siegel T.N., Scherf A.. Genome-wide chromatin immunoprecipitation-sequencing in plasmodium. Methods Mol. Biol. 2013; 923:321–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kaestli M., Cockburn I.A., Cortés A., Baea K., Rowe J.A., Beck H.P.. Virulence of malaria is associated with differential expression of plasmodium falciparum var gene subgroups in a case-control study. J. Infect. Dis. 2006; 193:1567–1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Rottmann M., Lavstsen T., Mugasa J.P., Kaestli M., Jensen A.T., Müller D., Theander T., Beck H.P.. Differential expression of var gene groups is associated with morbidity caused by plasmodium falciparum infection in tanzanian children. Infect. Immun. 2006; 74:3904–3911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Liu H., Cui X.-Y., Xu D.-D., Wang F., Meng L.-W., Zhao Y.-M., Liu M., Shen S.-J., He X.-H., Fang Q.et al.. Actin-related protein arp4 regulates euchromatic gene expression and development through H2A.Z deposition in blood-stage plasmodium falciparum. Parasit Vectors. 2020; 13:314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bolger A.M., Lohse M., Usadel B.. Trimmomatic: a flexible trimmer for illumina sequence data. Bioinformatics. 2014; 30:2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Langmead B., Salzberg S.L.. Fast gapped-read alignment with bowtie 2. Nat. Methods. 2012; 9:357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kim D., Langmead B., Salzberg S.L.. HISAT: a fast spliced aligner with low memory requirements. Nat. Methods. 2015; 12:357–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zhang Y., Liu T., Meyer C.A., Eeckhoute J., Johnson D.S., Bernstein B.E., Nusbaum C., Myers R.M., Brown M., Li W.et al.. Model-based analysis of chip-Seq (MACS). Genome Biol. 2008; 9:R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lawrence M., Huber W., Pages H., Aboyoun P., Carlson M., Gentleman R., Morgan M.T., Carey V.J.. Software for computing and annotating genomic ranges. PLoS Comput. Biol. 2013; 9:e1003118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Gel B., Diez-Villanueva A., Serra E., Buschbeck M., Peinado M.A., Malinverni R.. regioneR: an R/Bioconductor package for the association analysis of genomic regions based on permutation tests. Bioinformatics. 2016; 32:289–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ginsburg H. Progress in in silico functional genomics: the malaria metabolic pathways database. Trends Parasitol. 2006; 22:238–240. [DOI] [PubMed] [Google Scholar]
- 50. Yu G., Wang L.G., Han Y., He Q.Y.. clusterProfiler: an r package for comparing biological themes among gene clusters. OMICS. 2012; 16:284–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Bailey T.L. DREME: motif discovery in transcription factor chip-seq data. Bioinformatics. 2011; 27:1653–1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Gupta S., Stamatoyannopoulos J.A., Bailey T.L., Noble W.S.. Quantifying similarity between motifs. Genome Biol. 2007; 8:R24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Campbell T.L., De Silva E.K., Olszewski K.L., Elemento O., Llinas M.. Identification and genome-wide prediction of DNA binding specificities for the apiap2 family of regulators from the malaria parasite. PLoS Pathog. 2010; 6:e1001165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Hahne F., Ivanek R.. Visualizing genomic data using gviz and bioconductor. Methods Mol. Biol. 2016; 1418:335–351. [DOI] [PubMed] [Google Scholar]
- 55. Pertea M., Kim D., Pertea G.M., Leek J.T., Salzberg S.L.. Transcript-level expression analysis of RNA-seq experiments with HISAT, stringtie and ballgown. Nat. Protoc. 2016; 11:1650–1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Robinson M.D., McCarthy D.J., Smyth G.K.. edgeR: a bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010; 26:139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Brodie R., Roper R.L., Upton C.. JDotter: a java interface to multiple dotplots generated by dotter. Bioinformatics. 2004; 20:279–281. [DOI] [PubMed] [Google Scholar]
- 58. Shannon P., Markiel A., Ozier O., Baliga N.S., Wang J.T., Ramage D., Amin N., Schwikowski B., Ideker T.. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003; 13:2498–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Gu Z., Eils R., Schlesner M.. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics. 2016; 32:2847–2849. [DOI] [PubMed] [Google Scholar]
- 60. Tintó-Font E., Michel-Todó L., Russell T.J., Casas-Vila N., Conway D.J., Bozdech Z., Llinás M., Cortés A.. A heat-shock response regulated by the PfAP2-HS transcription factor protects human malaria parasites from febrile temperatures. Nat Microbiol. 2021; 6:1163–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Gupta A.P., Chin W.H., Zhu L., Mok S., Luah Y.H., Lim E.H., Bozdech Z.. Dynamic epigenetic regulation of gene expression during the life cycle of malaria parasite plasmodium falciparum. PLoS Pathog. 2013; 9:e1003170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Zhang Q., Huang Y., Zhang Y., Fang X., Claes A., Duchateau M., Namane A., Lopez-Rubio J.J., Pan W., Scherf A.. A critical role of perinuclear filamentous actin in spatial repositioning and mutually exclusive expression of virulence genes in malaria parasites. Cell Host Microbe. 2011; 10:451–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Amulic B., Salanti A., Lavstsen T., Nielsen M.A., Deitsch K.W.. An upstream open reading frame controls translation of var2csa, a gene implicated in placental malaria. PLoS Pathog. 2009; 5:e1000256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Zhang Q., Siegel T.N., Martins R.M., Wang F., Cao J., Gao Q., Cheng X., Jiang L., Hon C.-C., Scheidig-Benatar C.et al.. Exonuclease-mediated degradation of nascent RNA silences genes linked to severe malaria. Nature. 2014; 513:431–435. [DOI] [PubMed] [Google Scholar]
- 65. Lu B., Liu M., Gu L., Li Y., Shen S., Guo G., Wang F., He X., Zhao Y., Shang X.et al.. The architectural factor HMGB1 is involved in genome organization in the human malaria parasite plasmodium falciparum. Mbio. 2021; 12:e00148-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Deitsch K.W., Dzikowski R.. Variant gene expression and antigenic variation by malaria parasites. Annu. Rev. Microbiol. 2017; 71:625–641. [DOI] [PubMed] [Google Scholar]
- 67. Sierra-Miranda M., Delgadillo D.M., Mancio-Silva L., Vargas M., Villegas-Sepulveda N., Martínez-Calvillo S., Scherf A., Hernandez-Rivas R.. Two long non-coding RNAs generated from subtelomeric regions accumulate in a novel perinuclear compartment in plasmodium falciparum. Mol. Biochem. Parasitol. 2012; 185:36–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Broadbent K.M., Park D., Wolf A.R., Van Tyne D., Sims J.S., Ribacke U., Volkman S., Duraisingh M., Wirth D., Sabeti P.C.et al.. A global transcriptional analysis of plasmodium falciparum malaria reveals a novel family of telomere-associated lncRNAs. Genome Biol. 2011; 12:R56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Voss T.S., Healer J., Marty A.J., Duffy M.F., Thompson J.K., Beeson J.G., Reeder J.C., Crabb B.S., Cowman A.F.. A var gene promoter controls allelic exclusion of virulence genes in plasmodium falciparum malaria. Nature. 2006; 439:1004–1008. [DOI] [PubMed] [Google Scholar]
- 70. Fraschka S.A., Filarsky M., Hoo R., Niederwieser I., Yam X.Y., Brancucci N.M.B., Mohring F., Mushunje A.T., Huang X., Christensen P.R.et al.. Comparative heterochromatin profiling reveals conserved and unique epigenome signatures linked to adaptation and development of malaria parasites. Cell Host &Microbe. 2018; 23:407–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Sabari B.R., Dall’Agnese A., Young R.A.. Biomolecular condensates in the nucleus. Trends Biochem. Sci. 2020; 45:961–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Zhang M., Wang C., Otto T.D., Oberstaller J., Liao X., Adapa S.R., Udenze K., Bronner I.F., Casandra D., Mayho M.et al.. Uncovering the essential genes of the human malaria parasite plasmodium falciparum by saturation mutagenesis. Science. 2018; 360:eaap7847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Voss T.C., Hager G.L.. Dynamic regulation of transcriptional states by chromatin and transcription factors. Nat. Rev. Genet. 2014; 15:69–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Ruiz J.L., Tena J.J., Bancells C., Cortés A., Gómez-Skarmeta J.L., Gómez-Díaz E.. Characterization of the accessible genome in the human malaria parasite plasmodium falciparum. Nucleic Acids Res. 2018; 46:9414–9431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Komaki-Yasuda K., Okuwaki M., Kano S., Nagata K., Kawazu S.. 5' sequence- and chromatin modification-dependent gene expression in plasmodium falciparum erythrocytic stage. Mol. Biochem. Parasitol. 2008; 162:40–51. [DOI] [PubMed] [Google Scholar]
- 76. Jia S., Noma K., Grewal S.I.. RNAi-independent heterochromatin nucleation by the stress-activated ATF/CREB family proteins. Science. 2004; 304:1971–1976. [DOI] [PubMed] [Google Scholar]
- 77. Yamada T., Fischle W., Sugiyama T., Allis C.D., Grewal S.I.. The nucleation and maintenance of heterochromatin by a histone deacetylase in fission yeast. Mol. Cell. 2005; 20:173–185. [DOI] [PubMed] [Google Scholar]
- 78. Alon U. Network motifs: theory and experimental approaches. Nat. Rev. Genet. 2007; 8:450–461. [DOI] [PubMed] [Google Scholar]
- 79. Painter H.J., Campbell T.L., Llinás M.. The apicomplexan AP2 family: integral factors regulating plasmodium development. Mol. Biochem. Parasitol. 2011; 176:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Jeninga M.D., Quinn J.E., Petter M.. ApiAP2 transcription factors in apicomplexan parasites. Pathogens. 2019; 8:47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Yuda M., Iwanaga S., Kaneko I., Kato T.. Global transcriptional repression: an initial and essential step for plasmodium sexual development. Proc. Nat. Acad. Sci. USA. 2015; 112:12824–12829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Bertschi N.L., Toenhake C.G., Zou A., Niederwieser I., Henderson R., Moes S., Jenoe P., Parkinson J., Bartfai R., Voss T.S.. Malaria parasites possess a telomere repeat-binding protein that shares ancestry with transcription factor IIIA. Nat Microbiol. 2017; 2:17033. [DOI] [PubMed] [Google Scholar]
- 83. Miao J., Wang C., Lucky A.B., Liang X., Min H., Adapa S.R., Jiang R., Kim K., Cui L.. A unique GCN5 histone acetyltransferase complex controls erythrocyte invasion and virulence in the malaria parasite plasmodium falciparum. PLoS Pathog. 2021; 17:e1009351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Poran A., Notzel C., Aly O., Mencia-Trinchant N., Harris C.T., Guzman M.L., Hassane D.C., Lemento O.E., Kafsack B.F.C.. Single-cell RNA sequencing reveals a signature of sexual commitment in malaria parasites. Nature. 2017; 551:95–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Shang X., Wang C., Shen L., Sheng F., He X., Wang F., Fan Y., He X., Jiang M.. PfAP2-EXP2, an essential transcription factor for the intraerythrocytic development of plasmodium falciparum. Front. Cell Dev. Biol. 2021; 9:782293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Oehring S.C., Woodcroft B.J., Moes S., Wetzel J., Dietz O., Pulfer A., Dekiwadia C., Maeser P., Flueck C., Witmer K.et al.. Organellar proteomics reveals hundreds of novel nuclear proteins in the malaria parasite plasmodium falciparum. Genome Biol. 2012; 13:R108. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All raw sequencing data reported in this study have been deposited in GEO and are publicly accessible under accession GSE184659 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE184659). The genome-wide binding profiles of the sixteen studied ApiAP2 transcription factors are shown in UCSC genome browser sessions, including http://genome-asia.ucsc.edu/s/bzswmu/pf47_part1, and http://genome-asia.ucsc.edu/s/bzswmu/pf47_part2_new.