

Abstract
Clear evidence has shown that metal ions strongly connect and delicately tune the dynamic homeostasis in living bodies. They have been proved to be associated with protein structure, stability, regulation, and function. Even small changes in the concentration of metal ions can shift their effects from natural beneficial functions to harmful. This leads to degenerative diseases, malignant tumors, and cancers. Accurate characterizations and predictions of metalloproteins at the residue level promise informative clues to the investigation of intrinsic mechanisms of protein-metal ion interactions. Compared to biophysical or biochemical wet-lab technologies, computational methods provide open web interfaces of high-resolution databases and high-throughput predictors for efficient investigation of metal-binding residues. This review surveys and details 18 public databases of metal-protein binding. We collect a comprehensive set of 44 computation-based methods and classify them into four categories, namely, learning-, docking-, template-, and meta-based methods. We analyze the benchmark datasets, assessment criteria, feature construction, and algorithms. We also compare several methods on two benchmark testing datasets and include a discussion about currently publicly available predictive tools. Finally, we summarize the challenges and underlying limitations of the current studies and propose several prospective directions concerning the future development of the related databases and methods.
1. Introduction
Metal ions are certain atom compounds that usually form cations that have (a) positive electric charge(s). Metal ions play pivotal roles in protein structure, function, regulation, and stability [1, 2]. Common metal ions include zinc (Zn2+), calcium (Ca2+), magnesium (Mg2+), manganese (Mn2+), iron (Fe3+ or Fe2+), copper (Cu2+), cobalt (Co2+), sodium (Na+), potassium (K+), and nickel (Ni2+) ions. Recent estimates have shown that approximately 30%-40% of proteins require one or several metal cofactors to together express biological function [3]. The proportion varies in different types of organisms or tissues. For instance, K+ is mostly found inside the cell, while Na+ is abundant outside of the cell [4]. Mn2+ is found accumulated in leafy green plants [5]. In the human body, Ca2+ accounts for approximately 1.5% of total body weight. The bulk of Ca2+ is aggregated in bones and teeth [6].
Metal ion binding proteins, i.e., metalloproteins, play critical roles in a biological and chemical process in cellular reactions [7]. Inside the cell, the dynamic homeostasis of the metal ions is strongly connected and delicately tuned [8]. Reinhard et al. claimed that K+ and Na+ are involved in processing cell signaling, intercellular communication, and maintaining tissue electrolyte balance [9]. A small change in the concentration of metal ions may shift the effects of metal ions from natural beneficial to harmful [10]. A recent study pointed out that metalloproteins are associated with degenerative diseases, including Parkinson's disease and Alzheimer's disease [11]. For instance, α-synuclein (Cu2+-protein complex) constitutes the main component in Lewy bodies in Parkinson's disease [11]. Mn2+ and Fe3+ are responsible for inducing tangle pathology in Alzheimer's disease [8]. The aging of the brain or the development of diseases is associated with the deregulation of the management of metal ions [10]. Particularly, recent evidence indicates that if different types of metalloproteins interact in a certain salt solution, the potential galvanic erosion may dissolute the compound surface and result in inducing tumor formation [12, 13].
Elucidated protein-metal ion interactions rely in part on the advancement of various accurate characterizations and predictions of metalloproteins at the residue level. The traditional methods that are used to identify metal-binding conformation or binding residues include biophysics- or biochemistry-related wet-lab experiments, such as mass spectrometry [14], X-ray crystallography [15], and surface Plasmon resonance [16]. Since these technologies need expensive instruments, complex procedures, and elaborate labors, they shall benefit from the recent development of computation-aided methods.
We found 12 reviews that focused on the topic of exploring metal-binding residues or proteins in the past decade [7, 10, 11, 17–24]. Mallick et al. shed light on in silico methods including nine predictive tools and discussed the intrinsic mechanisms of metal-protein binding [24]. Thirumoorthy et al. investigated metallothionein isoforms and their role in pathophysiology [17]. They also provided the analysis of how metallothionein impact complex disease scenarios. In [18], the authors focused on structural variability and corresponding mechanisms of polymorphic amyloid oligomers complexed with metal ions. Bal et al. discussed ability constants, dissociation rates, and coordination chemistry of metal-binding residues in albumin [19]. Roohani et al. reviewed the literature related to zinc biochemical and physiological functions, metabolism, and zinc bioavailability in the human body [20]. The authors in [21] summarized the web tools that were proposed to identify metal-binding residues. Liu et al. systematically analyzed the structural features of Zn2+-binding sites and proposed an online predictor [22]. Akcapinar and Sezerman collected and surveyed computational toolboxes designed for the recognition of metal-binding sites or metalloproteins [7]. Quintanar and Kim summarized the research in degenerative diseases related in metal ions [11]. Witkowska and Rowińska-Żyrek overviewed the analytical and biophysical methods utilized for studies on metal-protein interactions [23]. Krzywoszyńska detailed the involvement of metal ions in signaling processes within the cell and its influence in health and disease [10]. Rauer et al. scrutinized computational approaches that are associated with the prediction of protein functional sites and also discussed metal-binding related works [25].
Broadly speaking, these reviews discuss some aspects of the predictive methods. Some of them provide sufficient coverage of databases and predictive models and discuss the challenges and limitations of considered approaches. These reviews bring informative clues for the following researchers in this field. From the pertinence of the research, the prediction of metal-binding can be divided into general and specific approaches. The former recognizes metal-binding residues without considering their types, while, the latter is aimed at identifying one or several specific metal-protein interactions. According to the basic design and scheme, we classify these methods into four categories, namely, learning-, docking-, template-, and meta-based methods.
This review covers a comprehensive set of 44 computation-based methods, and 25 of them were published in the past three years. Specifically, we survey 32 learning-based, 4 docking-based, 6 template-based, and 2 meta-based methods. Depending on whether the structure of a target protein is known or available, we further divide learn-binding methods into the structure- and sequence-based ones. We discuss their benchmark datasets, features, algorithms, and measurements, respectively. We also detail the docking-, template-, and meta-based methods and point out their advantages and limitations.
2. Public Databases for Metal Binding
The development in biochemistry and biophysics leads to a fast increasing number of protein-metal ion binding complexes. Figure 1 draws the top 10 metal-binding annotations in PDB. Our survey reveals that Zn2+, Ca2+, and Mg2+ occupy the top three prevalent metal ions. The Zn2+ is currently the best-explored and described metal ion [26]. Zn2+ participates in many biological processes, such as metabolism, immune system, neurotransmission, hormone secretion, and signaling [27]. According to a rough statistic, approximately 10% of eukaryotic proteins bind Zn2+ [28]. Ca2+ is mainly aggregated in bones and teeth vertebrates [29]. It helps form solid support structures through biomineralization [6]. Mg2+ is usually associated with solvent water molecules, which endow it with a good capability of binding affinity with proteins and movement. The solvation state of Mg2+ usually serves as the enzyme in which Mg2+ acts as a coenzyme [6].
Figure 1.
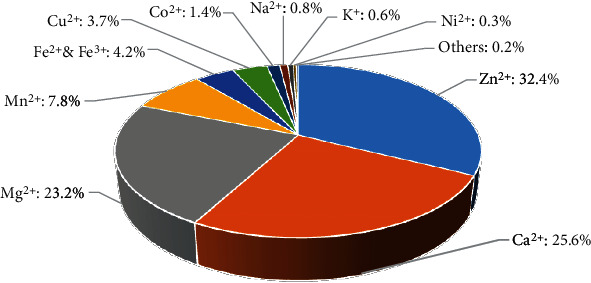
Fraction of top 10 metal-binding interactions that stored in PDB (date: December 20, 2021).
Besides RCSB PDB (https://www.rcsb.org/) [30], recent years have witnessed several specific databases that collect, categorize, and store these metal-protein interactions. Table 1 summarizes the publication year, considered metal ions, size of the database, web link, citations, and availability for the recently released database. We use citations as a one direct and good way to quantify the impact of these resources within the community [31]. The citation counts were collected from Google Scholar (https://scholar.google.com/) on December 20, 2021.
Table 1.
Summary of recently released database of metal ion binding interactions.
| Name | Year | Considered metal ions | Number of sites | Web link | Ref. | Citation | Availability |
|---|---|---|---|---|---|---|---|
| InterMetalDB | 2021 | All metal ion binding | 6,423 | https://intermetaldb.biotech.uni.wroc.pl/ | [26] | N/A | Yes |
| MeLAD | 2020 | All metal ion binding | N/A | https://melad.ddtmlab.org/ | [33] | 9 | Yes |
| ZincBindDB | 2019 | Zn | 24,992 | https://github.com/samirelanduk/ZincBindDB | [49] | 23 | Yes |
| MetalPDB (v2) | 2018 | All metal ion binding | N/A | http://metalweb.cerm.unifi.it | [34] | 90 | No |
| BioLiP | 2013 | All metal ion binding | 146,969 | https://zhanggroup.org/BioLiP/ | [36] | 446 | Yes |
| ZiFDB (v2) | 2013 | Zn | N/A | http://bindr.gdcb.iastate.edu/ZiFDB | [37] | 25 | No |
| MetalPDB (v1) | 2013 | All metal ion binding | N/A | http://metalweb.cerm.unifi.it | [35] | 108 | No |
| BioMe | 2012 | All metal ion binding | 20,307 | http://metals.zesoi.fer.hr | [39] | 30 | No |
| MetLigDB | 2011 | Zn, Mn, Fe, Ni, mg, cu, co, Mo | 732 | http://silver.sejong.ac.kr/MetLigDB | [40] | 13 | Yes |
| MIPS | 2010 | All metal ion binding | N/A | http://dicsoft2.physics.iisc.ernet.in/mips/ | [41] | 28 | Yes |
| MEDB | 2010 | All metal ion binding | N/A | http://www.uohyd.ernet.in/anambs/ | [42] | 14 | No |
| ZiFDB (v1) | 2009 | Zn | N/A | http://bindr.gdcb.iastate.edu/ZiFDB | [38] | 87 | No |
| MetalMine | 2009 | All metal ion binding | 412 | http://metalmine.naist.jp | [43] | 3 | No |
| Metal-MACiE | 2009 | All metal ion binding | N/A | https://www.ebi.ac.uk/thornton-srv/databases/Metal_MACiE/home.html | [44] | 60 | Yes |
| ZifBASE | 2009 | Zn | N/A | https://web.iitd.ac.in/~sundar/zifbase/ | [45] | 35 | Yes |
| MESPEUS | 2008 | Na, mg, K, ca, Mn, Fe, co, Ni, cu, Zn | 34,896 | http://eduliss.bch.ed.ac.uk/MESPEUS/ | [46] | 102 | No |
| MSDsite | 2005 | All metal ion binding | N/A | http://www.ebi.ac.uk/msd-srv/msdsite | [47] | 122 | Yes |
| MDB | 2002 | All metal ion binding | N/A | http://metallo.scripps.edu/ | [48] | 276 | No |
1We estimate the availability on December 1st, 10th, and 20th of 2021, respectively.
Specifically, InterMetalDB collects and presents metal ion binding proteins from RCSB PDB. It uses MMseq2 [32] to cluster the structure chains with the 50% sequence identity. Then, it groups similar binding sites and selects the best-resolution structure as a representative. MeLAD is a metalloenzyme-ligand association database, which contains structural data, metal-binding pharmacophores, and ligand chemical similarity of metalloenzyme-ligand interactions [33]. MetalPDB details the local environment, three-dimensional (3D) structure, secondary structure, and solvent accessibility of the metal ion binding sites [34, 35]. BioLiP is a semimanually curated database, which includes protein-peptide, protein-nucleic acid, and protein-ligand annotations [36]. BioLiP stores and periodically updates all types of metal ion binding information from PDB. ZiFDB is a database that collects information about individual zinc fingers, engineered zinc-finger arrays, and related target sequences [37, 38]. BioMe provides a web interface for biologists to capture coordination numbers, distances, geometry, and percentage of monodentate and bidentate bound aspartic acid and glutamic acid carboxyl groups [39]. MetLigDB is specially designed to select chelating groups or chemical moieties that might be presented in the inhibitor of a metalloprotein [40]. MIPS stores the geometric information, macromolecular function, different chemical behavior of metals, and metalloproteins [41]. MEDB presents quantitative information on metal-binding sites in protein structures and can be used for the identification of trends or patterns in the metal-binding sites [42]. MetalMine automatically collects and categorizes different types of metal-binding sites that derived from the structures of protein-metal-ion complexes [43]. Metal-MACiE gathers all available metalloenzymes and includes structural and functional information of metal ions in the context of the catalytic mechanisms of these metalloenzymes [44]. ZifBASE deposits engineered and natural zinc finger proteins and provides sequences and structural features and associated potential target sites of these proteins [45]. MESPEUS [46] focuses on the geometry of metal sites in proteins at resolution ≤ 2.5 Å. It provides an open web interface for further identifying and displaying the metal sites. MSDsite deposits computation-based metal-binding geometries and residues [47]. MDB offers quantitative information about metalloproteins [48]. It provides functions to analyze the binding attributes such as metal-ligand bond distances and side-chain torsion angles in metal sites.
We show that twelve source databases are designed for all metal ion binding data. Two databases, namely, MetLigDB and MESPEUS, consider several types of metal ions. There are four specific zinc-binding-related databases. Our survey also reveals that BioLiP is the most favored database, given the fact that its citations are average about 56 (446/8≈56) per year. Moreover, we notice that only half of the databases are available. Thus, we recommend that future databases shall be chronically maintained, periodically updated, and easy expanded.
3. Method Development of Metal-Binding Prediction
Figure 2 illustrates the flowchart of computation-based methods for the prediction of metal-binding residues. Generally, based on the basic design and scheme, these methods can be categorized into four groups. The learning-based methods regard the identification of metal-binding residues as a typical classification problem and attempt to use machine learning or deep learning algorithms to construct prediction models. The docking-based approaches are aimed at finding proper binding conformation as well as the appropriate target binding residues by scanning protein surface. The scoring functions are introduced to assess the selected pockets and quantify the strength of binding affinity. The template-based methods are designed to select the optimal template structures for a given unknown protein. Then, they map and transfer the binding annotations from similar spatial conformation to the target protein. By contrast, the meta-based methods focus on combing the predictions from other methods in order to build more accurate predictors.
Figure 2.
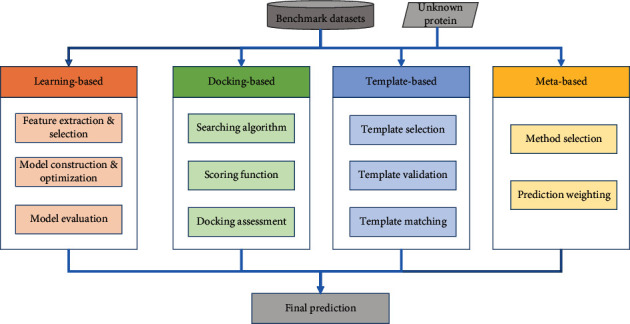
The flowchart of computation-based methods for prediction of metal-binding residues.
3.1. Benchmark Datasets
The sequences and structures of protein-metal ion complexes are available in public databases for the end-users to customize the benchmark datasets. As shown in Table 2, the considered methods use various numbers of sequences/chains, ranging from several dozens to thousands. Besides that, protein complexes with high resolution indicate relatively more comprehensive and accurate annotations of protein-metal ion interactions. According to our survey, 12 out of 23 sequence-based and 6 out of 9 structure-based methods filter the candidate complexes using high resolution with ≤3 Å. Some methods [50–57] remove the sequences/chains whose lengths are less than 50 residues (or 45 residues [58]) since they might be potential segments or peptides. To build an unbiased dataset, it is necessary to remove homologous or redundant proteins. The cutoff threshold which researchers choose varies from minimal 25% to maximal 90%. Generally, a higher identity means a higher chance in local alignments [59]. The literatures in [60, 61] point out that if a pair of proteins have a sequence identity lower than 30%, they have little chance to share the same biological processes. Three tools, namely, BLASTclust [62], PISCES [63], and CD-HIT [64], are mainly used to cluster homologous proteins.
Table 2.
Summary of learning-based methods.
| Type | Method1 | Ref. | Year | Metal ion binding2 | Dataset3 | Resolution | Sequence similarity (tool)4 | Prediction model5 | Cross-validation | Independent test | Measurements6 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Sequence-based | Liu et al. | [56] | 2020 | Zn, Cu, Fe, Co, Mn, Ca, Mg, Na, K | 5,340 | ≤3 Å | 30% (CD-HIT) | RF | 5-fold | √ | SN, SP, ACC, MCC |
| MIonSite | [90] | 2019 | Zn, Ca, Mg, Mn, Fe, Cu, Fe, Co, Na, K, Cd, Ni | 7,676 | N/A | 30% (CD-HIT) | SVM, AdaBoost | 5-fold | √ | SN, SP, ACC, MCC, AUC | |
| MPLs-Pred | [91] | 2019 | General metal ions | 1,492 | N/A | 30% (CD-HIT) | RF | 10-fold | √ | SN, SP, ACC, MCC | |
| SXGBsite | [92] | 2019 | Ca, Zn, Mg, Mn, Fe | 4,421 | N/A | 40% (PISCES) | GBM | 5-fold | √ | SN, SP, ACC, MCC, AUC | |
| Wang et al. | [50] | 2019 | Zn, Cu, Fe, Mn, Ca, Mg, Na, K | 5,146 | ≤3 Å | 30% (N/A) | SVM, SMO | 5-fold | √ | SN, SP, ACC, MCC | |
| znMachine | [51] | 2019 | Zn | 2,043 | ≤3 Å | 30% (BLASTclust) | SVM, NN | 5-fold | √ | SN, SP, ACC, MCC, PRE, AUC | |
| SSWPNN | [93] | 2019 | Zn | 213 | ≤2.5 Å | 70% (N/A) | SVM, NN | 5-fold | √ | SN, SP, PRE, F1, MCC, ACC | |
| ZinCaps | [94] | 2019 | Zn | 738 | ≤3 Å | N/A (N/A) | CN | 5-fold | √ | SN, SP, ACC, MCC, AUC | |
| Haberal and Oğul | [65] | 2018 | General metal ions | 2,727 | N/A | N/A (N/A) | CNN | 5-fold | √ | SN, ACC, PRE, F1 | |
| ZincBinder | [87] | 2018 | Zn | 738 | ≤2.5 Å | 30% (PISCES) | SVM | 5-fold | √ | SN, SP, ACC, MCC, AUC | |
| EC-RUS | [95] | 2017 | Ca, Mg, Mn, Fe, Zn | 4,421 | N/A | 40% (PISCES) | WSRC | 5-fold | √ | SN, SP, ACC, MCC, AUC | |
| Cao et al. | [52] | 2017 | Zn, Cu, Fe, Co, Mn, Ca, Mg, K, Na | 5,340 | ≤3 Å | 30% (CD-HIT) | SVM | 5-fold | √ | SN, SP, ACC, MCC | |
| Kumar | [96] | 2017 | Cu, Ca, Co, Fe, Mg, Mn, Ni, Zn | 3,922 | N/A | 50% (CD-HIT) | RF | 10-fold | √ | SN, SP, ACC, MCC | |
| DeepMBS | [97] | 2017 | General metal ions | 2,727 | ≤3 Å | N/A (N/A) | CNN | 5-fold | √ | SN, PRE, F1 | |
| Qiao et al. | [98] | 2017 | Ca | 2,239 | N/A | 30% (CD-HIT) | SVM | 5-fold | √ | SN, ACC, PRE, MCC, AUC | |
| IonCom | [99] | 2016 | Zn, Cu, Fe, Ca, Mg, Mn, Na, K | 1,374 | N/A | 30% (CD-HIT) | SVM, AdaBoost | 5-fold | √ | SN, SP, ACC, MCC | |
| Jiang et al. | [77] | 2016 | Ca | 1,885 | ≤3 Å | 25% (N/A) | SVM | 5-fold | √ | SN, SP, ACC, MCC | |
| TargetCom | [53] | 2016 | Cu, Fe, Zn | 1,373 | ≤3 Å | 40% (CD-HIT) | SVM, AdaBoost | 5-fold | √ | SN, SP, ACC, MCC | |
| OSML | [100] | 2015 | Ca, Zn, Mg, Mn, Fe | 4,421 | N/A | 40% (PISCES) | SVM | 5-fold | √ | SN, SP, ACC, MCC | |
| TargetS | [101] | 2013 | Ca, Zn, Mg, Mn, Fe | 4,421 | N/A | 40% (PISCES) | SVM, AdaBoost | 5-fold | √ | SN, SP, ACC, MCC, AUC | |
| ETMB-RBF | [102] | 2013 | General metal ions | 55 | N/A | 20% (BLASTclust) | RBFN | 10-fold | √ | SN, SP, ACC, MCC | |
| ZincExplorer | [103] | 2013 | Zn | 392 | ≤3 Å | N/A (N/A) | SVM | 5-fold | √ | SN, SP, PRE, MCC, AUPRC | |
| Horst et al. | [58] | 2010 | Ca | 635 | ≤2.1 Å | 35% (N/A) | LR | 10-fold | √ | MCC,AUC,AUPRC | |
| Structure-based | Nguyen et al. | [104] | 2021 | Mn, Fe, Co, Ni, Cu, Zn | 9,955 | ≤2.5 Å | 90% (N/A) | RF | 5-fold | × | ACC |
| TMP-MIBS | [54] | 2021 | General metal ions | 427 | N/A | 40% (CD-HIT) | RF | 10-fold | √ | SN, SP, ACC, MCC, AUC | |
| Zincbindpredict | [105] | 2021 | Zn | N/A | ≤ 2 Å | 40% (CD-HIT) | RF | 5-fold | √ | SN, PRE, F1, MCC | |
| Wang et al. | [81] | 2021 | Zn, Cu, Fe, Ca, Mg, Mn, Na, K, Co | 5,340 | ≤3 Å | 30% (N/A) | MLP,SVM | 5-fold | √ | SN, SP, ACC, MCC | |
| DELIA | [80] | 2020 | Ca, Mn, Mg | 3,966 | N/A | 30% (CD-HIT) | CNN | 5-fold | √ | SN, PRE, MCC, AUC | |
| Hu et al. | [57] | 2020 | Zn, Cu, Fe, Co, Mn, Ca, Mg, Na, K | 5,340 | ≤3 Å | 30% (CD-HIT) | GBM | 5-fold | √ | SN, SP, FPR, ACC, MCC | |
| MetalExplorer | [79] | 2017 | Ca, Co, Cu, Fe, Ni, Mg, Mn, Zn | 3,192 | ≤2.5 Å | 30% (CD-HIT) | RF | 5-fold | √ | SN, FPR, PRE, AUC, AURPC | |
| FINDSITE-metal | [55] | 2011 | Ca, Co, Cu, Fe, Mg, Mn, Ni, Zn | 860 | N/A | 35% (PISCES) | SVM | 2-fold | √ | ACC, SPC, PPV | |
| Zinc identifier | [78] | 2011 | Zn | 1,103 | ≤2.5 Å | N/A (N/A) | RF | 5-fold | √ | SN, PRE, SP, FPR, AUC, AUPRC |
1The name of each method is provided in either the publication or the last name of its first author. 2General metal ions mean that the related predictor does not differentiate the types of metal ion binding. Otherwise, we list the specific types of metal-binding in detail. 3The number represents the size of the benchmark dataset. 4The value reveals the protein similarity threshold in the benchmark dataset. The content in the blanket indicates the tool that is used for clustering proteins. 5SMO: sequential minimal optimization; SVM: support vector machine; WSRC: weighted sparse representation based classifier; NN: neural network; CN: capsule network; CNN: convolutional neural networks; RF: random forest; GBM: gradient boosting machine; RBFN: radial basis function networks; LR: logistic regression; MLP: multilayer perceptron. 6SN: sensitivity/recall; SP: specificity; ACC: accuracy; MCC: Matthew's correlation coefficient; PRE: precision; F1: F1-score; AUC: area under the ROC curve; AUPRC: area under the precision recall curve; FPR: false positive rate (FPR = 1-SP).
3.2. The Validation and Evaluation Metrics
3.2.1. Cross-Validation and Independent Test
To construct a predictor with high accuracy and decent generalization ability, it is necessary to avoid potential overfitting. In practice, cross-validation and independent test are two popular ways (Table 2) to evaluate the proposed models [31]. Specifically, k-fold cross-validation is usually adopted on the training dataset when building the prediction model and optimizing the related parameters [61]. First, the training dataset is equally divided into k parts. The division can be done at residue level or protein level. Next, k-1 subsets are used to train the model, and the last one subset is used for testing. The procedure repeats k times until every subset is been predicted. The performance of the model is usually evaluated by averaging the results of the k repeats.
3.2.2. Performance Measures
According to Table 2, the measures that used to evaluate the performance of the predictors can be divided into binary value-based and propensity score-based ones. The former needs preset thresholds to compute the number of putative binding residues and nonbinding residues. These measures include sensitivity (SN)/recall/true positive rate (TPR), specificity (SP), false positive rate (FPR, FPR = 1-SP) precision (PRE), accuracy (ACC), F1-score (F1), and Matthew's correlation coefficient (MCC). They are defined as follows:
| (1) |
where TP (true positive) indicates the number of correctly recognized metal-binding residues, FP (false positive) means the number of non-metal-binding residues that are incorrectly predicted as metal-binding residues, TN (true negative) stands for the number of correctly predicted non-metal-binding residues, and FN (false negative) is the number of metal-binding residues that are incorrectly predicted as non-metal-binding residues.
The prediction of metal-binding residues is a typical imbalanced classification problem. That is, the number of metal-binding residues is much less than that of the non-metal-binding ones. Therefore, F1-score and MCC are regarded as key criteria since they are featured by assessing the prediction performance for both metal-binding and non-metal ion binding residues.
The propensity score-based measures include receiver operating characteristic curve (ROC curve) and precision-recall curve (PR curve). The ROC curve draws the TPR (true positive rate) against the FPR (false positive rate) at various thresholds. The AUC computes the area under the ROC curve and can be used to quantify the ROC curve. The PR curve plots PRE values on the y-axis and recalls values on the x-axis, and the AUPRC estimates the area under the PR curve.
3.3. Learning-Based Methods
Learning-based methods treat the recognition of metal-binding residues as a typical pattern recognition problem. Specifically, the metal-binding residues and nonbinding ones are encoded by using mathematical descriptors, i.e. features. According to the information that used to compute the features, the learning-based methods can be further categorized into sequence-based and structure-based methods. The former only needs simple protein sequences to extract features when encoding the binding residues. These features include sequence directly derived, evolutionary profile-based, and putative structure-based features, while the latter uses both sequence and native structure data to mathematically describe a binding residue. We make a comprehensive literature search and collect 23 sequence- and 9 structure-based methods that were published after the year 2010.
3.3.1. Feature Construction
(1) Sequence Directly Derived Features. We define sequence directly derived features as the ones that are computed from protein primary sequences without using any other information. In Figure 3, 14 out of 32 considered methods consider amino acid composition [50], which quantifies the relative difference in abundance of a given amino acid type [65, 66]. Amino acid pairs, or dipeptides, are based on the observation that amino acid pairs show different propensities in protein structure and function. For instance, pairs of lysine are found present in close spatial vicinity [67]. Moreover, the concept of k-spaced amino acid pairs is introduced in [68]. It calculates the amino acid pairs with k spaces between two residues. Our survey also shows that the majority of studies use physicochemical properties to describe the local environment of the metal-binding residues. The basic physicochemical environment of a metal-protein binding interface is reflected by the specific roles the metal plays in biostructural chemistry and protein function. These properties are crucial since they underpin many of the functional roles of metal ions. These properties include aliphatic [69], sulphur [70], aromatic [71], hydrophobic [72], charge [73], polar [74], positive [73], acidic [75], and hydroxylic [76]. The position-related features mainly consider the influence of the specifically located residues, such as autocross covariance [77] and sequence length [78, 79].
Figure 3.

Summary of the feature construction and selection for learning-based methods. The light green cells indicate sequence directly derived features. The light blue cells stand for profile-based features. The light red cells mean putative structure-based features. The light grey cells are native structure-based features. 1In the amino acid pair column, the cells without annotations indicate original amino acid pairs; the cells annotated using “k-spaced” means k-spaced amino acid pairs. 2PWM: position weight matrix; PSSM: position specific scoring matrix; PMS: position matrix scoring; PAM: point accepted mutation; EMS: evolutionary matrix scoring. 3TS: topology structure; DM: distance matrix; GTN: graph theoretic network; RRCG: residue-residue contact graphs. 4FFS: forward feature selection; EB: experience-based; BA: Boruta algorithm; mRMR: minimum-redundancy maximum-relevancy; MDGI: mean decrease Gini index [50–51, 53–58, 65, 77–81, 87, 90–105].
(2) Evolutionary Profile-Based Features. Recent studies [54, 56, 57, 80, 81] pointed out that functional or structural important residues tend to show higher evolutionary conservation. The conserved residues are usually involved in enzyme activity, ligand binding, or protein structural stability [82]. The conserved residues and regions can be identified by multiple sequence alignment [83]. These multiple sequence alignments, also named conservation profiles, include aligning families of homologous sequences and having knowledge of their evolutionary relationships [84]. For an unknown protein, although its accurate function is not available, it is expected that we can use its homologous proteins to speculate the function since they share the similar evolutionary profile [85]. Many studies use position-specific scoring matrix (PSSM), which is computed from PSI-BLAST [62], to quantify the evolutionary conservation. PSSM scores the substitution probability of each residue in the protein being substituted by other types of amino acids. Liu et al. [56] and Hu et al. [57] set different weights according to the positions of considered residues within the window and construct position weight matrix (PWM). Wang et al. proposed a customized position matrix scoring (PMS) algorithm, which uses known sequence patterns to describe the composition of amino acids at different positions [50]. Haberal and Oğul introduced a point accepted mutation (PAM) scoring matrix, which measures the rate at which point mutations that substitute one residue for another during evolution [65]. Jiang et al. adopted evolutionary matrix scoring (EMS) algorithm to extract the position conservation of amino acid residues from segments with low dimension feature parameters [77].
(3) Putative Structure-Based Features. For an unknown protein, although the accurate function is not available, it is expected that we can use its homologous proteins or template structures to speculate the structure. The secondary structure mainly involves α-helix, β-sheet, and coil, which are fundamental elements of protein tertiary structure [86]. Natively disordered or unstructured regions are proved to be associated with molecular assembly, protein translation, modification, and molecular recognition [78, 79, 87]. Previous studies [79, 87] indicate that disordered regions are strongly correlated with local solvent accessibility areas. Figure 3 reveals that 16 methods introduce secondary structure features and 3 approaches use disorder features, respectively. The secondary structure can be obtained from the primary sequence by using PSIPRED [88]. Putative intrinsic disorder data can be computed by using DISOPRED [89].
(4) Structure-Based Features. The structure-based features include descriptors that are computed from protein 3D structure. These features include solvent exposure, B-factor, spatial cluster properties, and native secondary structure. Compared with the abovementioned putative structure-based features, the native structure-based features are more accurate since they are directly computed by using residue coordinate data. Besides that, a residue contact network is also considered by some literature. In [79], two residues are defined as being in contact if the distance of their Cα atoms is less than a predefined cutoff distance of 6.5 Å. These features include clustering coefficient, degree, density, distance, topology structure, and graph theoretic network [55, 79, 80].
3.3.2. Sliding Window Optimization and Feature Selection
As shown in Figure 3, many methods adopt a sliding window scheme when they construct different types of features. It is because residues in proteins are influenced by adjacent residues. Besides that, binding residues tend to cluster together. If a central residue is a native-binding residue, its adjacent residues usually have a relatively higher chance to bind the same ligands. Usually, the residues with a long distance away have a lower impact on the central residues when compared with the residues with short distance. Figure 3 summarizes that 19 out of 23 methods use the sliding window scheme. The size of the shortest window is 3 [105], while the size for the longest one is 25 [94]. Some studies [50, 52, 53, 56, 57, 81, 90, 105] use more than one type of window because they consider different types of metal-binding residues. A long window means the introduction of more features.
However, a bigger number of features do not absolutely mean a better prediction performance [106, 107]. The existence of potential “bad” features may interfere with the classifiers and cause unpredictable consequences [108]. The so-called “bad” features include irrelevant and redundant ones. To avoid their terrible influences, it is necessary to perform feature selection before training the model [109]. Figure 3 reveals that 6 out of 32 methods adopt feature selection before training the model. These feature selection approaches include forward feature selection [79, 102], experience-based [104], Boruta algorithm [57, 81], minimum-redundancy maximum-relevancy [79], and mean decrease Gini index [78].
3.3.3. Prediction Algorithms
Learning-based methods use machine-learning or deep-learning-based algorithms to train the model and perform predictions [110]. As shown in Table 2, a variety of algorithms are introduced for solving the problem of correctly recognizing metal-binding residues. Support vector machine (SVM) is a popular machine learning algorithm in bioinformatical research. It is aimed at finding a hyperplane or decision boundary that can segregate a high-dimensional space [111]. Particularly, it uses kernel functions to reduce computation time to avoid strapping into dimension disaster [112]. Sequential minimal optimization (SMO) is an algorithm that is specially used for training support vector machines [113]. The procedure of training large data by SVM usually leads to a complex quadratic programming optimization problem [114]. SMO breaks large programming optimization problems into small ones, which endows SVM a good generalization on large data [113]. The idea of a neural network (NN) comes from the work system of neurons in the biological brain [115]. It learns the correlations between inputs and outputs, making generalizations and build models [116]. The NN algorithm assigns and adjusts different weights for neurons and edges as learning proceeds. The radial basis function network (RBFN) is a variant of the original NN [117]. It adopts radial basis functions as activation functions, which can be used for accelerating learning speed due to their universal approximation [118]. The multilayer perceptron (MLP) algorithm is an improved back propagation NN [119]. It mainly includes three procedures: forward propagation, error evaluation, and error backpropagation [120]. The MLP is featured by its strong generalization and fault tolerance [121]. Therefore, it is proved to be an efficient classification algorithm. The logistic regression (LR) adopts a logistic function to model the probability of an unknown sample being a certain class [122].
Our survey also reveals that the ensemble algorithms are favored by eight studies. The random forest (RF) aggregates the predictions of all the decision trees and performs decisions by most trees [123]. RF can be used for classification, regression, and optimization problems [124]. Adaptive boosting (AdaBoost) is aimed at combining weak learners with strong ones [125, 126]. The key point of AdaBoost is to ensure the diversities of individual learners, which makes it a good generalization ability [90, 127]. The gradient boosting machine (GBM) is another popular ensemble algorithm. During the iterative process, GBM dynamically increases the weight of wrong recognitions and reduces that of the correct ones [128–131]. It should be noted that GBM focuses on the sample residual of the previous iteration instead of the sample itself [132].
Besides machine-learning algorithms, recent studies also use deep-learning methods in this research field. The convolutional neural network (CNN) is one of the most prevalent algorithms that is widely used in bioinformatics [133]. The CNN consists of three main layers, which are the convolutional layer, pooling layer, and fully connected layer [134–136]. Although the CNN is proved to be powerful in dealing with a variety of problems, it performs badly when facing samples with different sizes and orientations [137, 138]. To overcome this shortcoming, the capsule network (CN) is proposed to estimate features of objects by incorporating dynamic routing algorithms [139, 140]. Our review finds two studies use CNN [65, 97] and one uses CN [94].
3.4. Docking-Based Methods
The investigation on the protein-metal complex helps biologists to understand the mechanism of protein-metal interactions. Protein-ligand docking approaches are always based on molecular structure and are used to explore biomolecular interactions and mechanisms [141]. It can be adopted to predict binding conformation as well as the appropriate target binding residues [142, 143].
As shown in Figure 2, the docking-based methods mainly include three steps: searching algorithm, scoring function, and docking assessment [141]. The searching algorithm focuses on creating an optimum number of configurations that properly include the determined binding modes [144]. To reduce computation time, it is necessary to make a balance between the computational expense and the searching space. The scoring function includes a series of mathematical functions that quantify the strength of binding affinity [145]. The energy-based scoring functions are always introduced to score the potential interactions between the protein and the corresponding ligands [141]. The frequently used functions include empirical-based, knowledge-based, and consensus-based ones. Finally, the putative docking can be evaluated by using docking accuracy and the correlation between putative and native docking scores [145]. Figure 4 illustrates the structure of a calmodulin (PDB: 4HEX) that is secreted by Escherichia coli in Mus musculus [146]. Calmodulin is one of the most prevalent EF-hand calcium sensor proteins in eukaryotic cells [147]. It is a highly conserved and soluble protein, which activates enzymes and regulates many cellular functions. 4HEX has three Ca2+-binding and two Zn2+-binding sites. Ca2+-binding causes a change in calmodulin conformation opening both globular domains and exposing hydrophobic surfaces that form binding sites for the target enzymes. Figure 4 shows that these three Ca2+ are in the pockets. The binding pockets are half-closed and buried, which substantially limits the capability of Ca2+ to escape. Two Zn2+-binding sites are surrounded by a shell of hydrophilic groups that are embedded into a larger shell of hydrophobic groups. The amino acid side chains providing ligands to Zn2+ in these structures often form hydrogen bonds with other residues [147].
Figure 4.
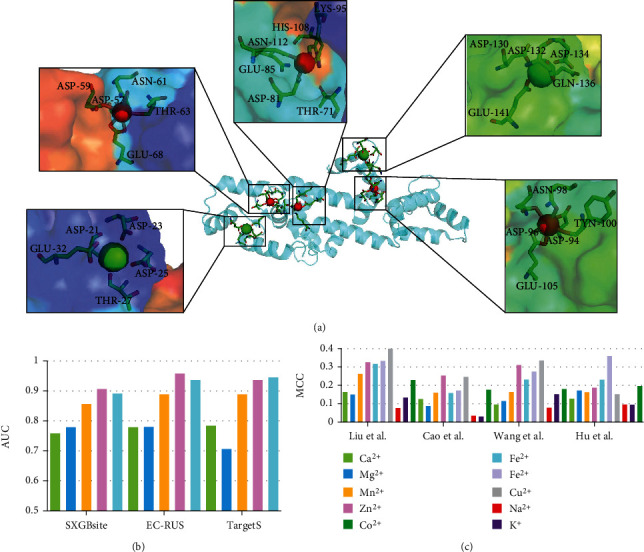
Ribbon and surface model of X-ray structure of Ca2+- and Zn2+-bound calmodulin (PDB: 4HEX) in Mus musculus. Red sphere represents bound zinc ion; green one indicates calcium ion; the spatial adjacent residues participating its coordination are shown by the stick model.
In [148], He et al. proposed a docking-based predictor named mFASD. It first explored the local biochemical environment of potential functional atoms and then measured the distances between the atoms and bound metal. mFASD also claimed that it can differentiate different types of metal-binding sites. Zhou et al. improved the FEATURE-based calcium model and used the grid scan algorithm to recognize binding sites [149]. GaudiMM [150] adopted a multiobjective genetic algorithm to search metal-binding sites in biological scaffolds. BioMetAll focused on the conformation of the potential metal-binding site, associated with the geometric organization of the protein backbone [151]. It was also proved to have good performance on the applications including the modulation and mutation of the metal-binding residues. Table 3 summarizes the key notes of the abovementioned 4 docking-based methods.
Table 3.
Summary of docking-based, template-based, and meta-based methods.
| Type | Method | Year | Notes |
|---|---|---|---|
| Docking-based | mFASD [148] | 2015 | Capture the characteristics of metal-binding sites and discriminate most types of these sites |
| Zhou et al. [149] | 2015 | Use a FEATURE-based calcium model and convert high scoring regions into specific site predictions | |
| GaudiMM [150] | 2019 | Find poses that satisfy metal-derived geometrical rules and use post optimizations | |
| BioMetAll [151] | 2020 | Predict metal-binding sites with particular motifs, determine transient sites in structures, and predict potential mutations to generate convenient sites | |
| Template-based | Deng et al. [152] | 2006 | Use a graph theory algorithm to find oxygen clusters of the protein (high potential for calcium binding) |
| Goyal et al. [153] | 2008 | Describe generation of 3D-structural motifs for metal-binding sites from the known metalloproteins | |
| Levy et al. [154] | 2009 | Analyze whether structural models based on remote homology are effective in predicting 3D metal binding sites | |
| FunFOLD [155] | 2011 | Use an automated method for ligand clustering and identification of binding residues | |
| FunFOLDQA [156] | 2012 | Use a fully automated agglomerative clustering approach for both ligand identification and residue selection | |
| FunFOLD2 [157] | 2013 | Propose a method that include protein-ligand binding prediction and quality assessment protocol | |
| Meta-based | Li et al. [158] | 2017 | Integrate the results of ZincExplorer [103], zincFinder [159], and zincPred [160] |
| IBayes_Zinc [161] | 2019 | Adopt Bayesian method and combine the predictions from ZincExplorer [103], zincFinder [159], and zincPred [160] |
3.5. Template-Based Methods
It is well known that protein structure determines function, and similar interface conformation indicates similar bound regions [162]. The template-based methods are based on the abovementioned hypothesis. Therefore, the most important thing for template-based methods is to find and validate proper structural templates. The fold recognition algorithms, which quantify the best matches from candidate templates, are commonly used to select the optimal template structures [163]. Next, the selected templates are used to map onto the target protein given the alignments with the template structures [164].
As shown in Table 3, Goyal and Mande analyzed the metal-binding sites by using structure templates and designing 3D motifs for several types of metal-binding interactions [153]. In [154], the authors analyzed whether structural models based on remote homology are effective in recognizing structural metal-binding residues based on simple protein primary sequences. Deng et al. applied a graph theory algorithm to identify, predict, and analyze calcium-binding residues [152]. However, it should be noted that this strategy produces good prediction performance when a decent complex is available as a template. If the template structure information is not available, this strategy might have poor predictions [164]. The FunFOLD was an automatic method that uses protein structure superposition of distantly related templates to a modelled protein for the clustering of ligands and prediction of metal binding residues [155]. The FunFOLDQA [156] approach determined the reliability of our FunFOLD [155] by assigning the quality assessment scores. FunFOLD2 was a web server that integrated cutting edge function and putative 3D structures to identify metal-binding residues [157].
3.6. Meta-Based Methods
The meta-based methods use a meta-learning strategy from fewer samples than traditional machine learning models. Since meta-based methods can only use limited data, they must ensure that the data is featured with high accuracy. As a result, a meta-based approach always directly combines the predictions of other methods. It uses weights or voting strategy on the available propensity scores or binary values. Thus, the meta-based method promises a robust accurate prediction on the metal-binding residues. In [158], Li et al. collected the predictions from ZincExplorer [103], ZincFinder [159], and ZincPred [160] (Table 3). Then, they built a linear regression model and optimized corresponding parameters on the training dataset. They claimed that the meta-model, which was named meta-zincPrediction, improves the AUPRC by about 2%~8%. IBayes_Zinc [161] was another meta-based predictor for the identification of zinc-binding residues (Table 3). It firstly computed the predictions of zinc-binding probabilities from ZincExplorer [103], ZincFinder [159], and ZincPred [160]. Next, IBayes_Zinc processed the missing attribute values and adopts Bayesian theory [165] to construct a meta-based model. The performance on the independent dataset proved that the MCC value of IBayes_Zinc was about 5~13% higher than the considered three predictors.
3.7. Prediction Results
This review surveys 44 computation-based methods. It is necessary to make a consensus comparison for these methods. However, since there is no standard benchmark dataset and some methods are not currently available, we use two datasets that are used by some methods to perform the evaluations. The first dataset is compiled by Yu et al. in [101], which includes five types of metal ions binding annotations. The second dataset is obtained from [52], consisting of ten types of metal ions binding annotations.
Figure 5 illustrates the predictive performance on two benchmark test datasets. Details are provided in Table S1 and Table S2 in Supplementary Materials, respectively; the corresponding results are sourced from [56, 57, 81, 92]. We notice that the predictors show relatively big differences in recognizing various types of metal-binding residues. On Yu et al.'s dataset [101], TargetS shows the best results in predicting Ca2+-, Zn2+-, and Mn2+-binding residues; EC-RUS [95] performs best in recognizing Mg2+-binding residues; OSML [100] achieves the highest MCC on Fe3+-binding predictions. Besides that, Figure 5(a) indicates that all five methods show a decent performance on recognizing Fe3+-binding residues (MCC values close or higher than 0.4), compared with MCC close or less than 0.2 on Ca2+ binding residues. Figure 5(b) draws the bars of AUC values for SXGBsite [92], EC-RUS [95], and TargetS [101], respectively. These three predictors all achieve high AUC scores (close or higher than 0.9) on Zn2+- and Fe3+-binding residues. Figure 5(c) summarizes the results of ten metal ions binding residues on Cao et al.'s dataset [52]. Among these predictors, Liu et al. [56] performs the best on Zn2+, Fe3+, and Cu2+, compared to [81], Wang et al. shows best on Zn2+ and Cu2+, and Hu et al. [57] achieves the highest on Fe2+. Interestingly, the binding residues associated with relatively inactive metal (Zn2+, Fe3+, and Cu2+) ions show relatively better results compared to that of the active metal ions (Na+ and K+). Particularly, four methods all give better results on Fe3+-binding residues than that on Fe2+-binding residues, which keep consistent with our observations as mentioned above.
Figure 5.
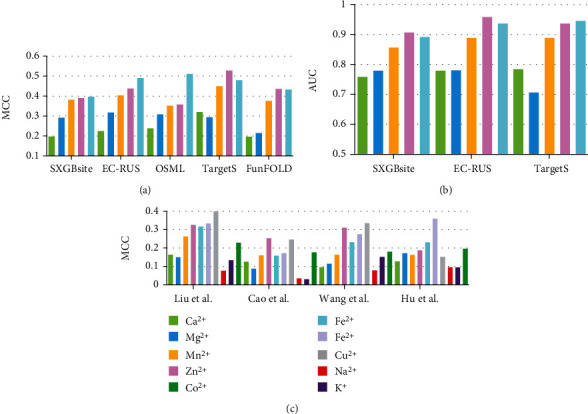
Comparative assessment of several predictors on two benchmark dataset. (a) and (c) indicate the MCC bar charts for considered methods on different metal ion binding residues on Yu et al.'s and Cao et al.'s testing datasets, respectively. (b) draws the AUC values of three predictors on corresponding metal ion binding residues.
3.8. Publicly Available Tools
The publicly available standalone software or web server that implements the proposed approach provides convenience for biologists and researchers [79, 105, 122]. These tools help the community to repeat the results and build a platform for easy understanding and improvement. Table 4 summarizes the public availability of implementations for the considered methods. These 28 predictors are implemented as standalone software or web servers. Among these predictive tools, 16 (or 57%) of them are currently publicly available. Standalone software requires the biologists to build the same running environment. By contrast, the web server provides the most convenient since the users only need to submit their queries via the browser, and the server helps to do the computations. Three methods, namely, znMachine [51], ZincBinder [103], and FunFOLD [155], provide both web server and standalone software. TMP_MIBS [54] is designed to predict general metal-binding residues and deployed using the Python language. DELIA [80] requires PDB-formatted 3D coordinates input and produces both binary prediction and putative probability of a residue being potential specific metal-binding. BioMetAll uses a docking-based strategy to scan specific motifs, putative mutations, and binding residues. Another available docking-based method is mFASD, which distinguishes different types of metal-binding sites according to the interaction distances. MPLs-Pred [91], SXGBsite [92], MIonSite [90], OSML [100], EC-RUS [95], and TargetS [101] are all sequence-based predictive tools, which accepts FASTA-formatted input and produced the results of putative metal-binding residues. ZinCaps [94], SSWPNN [93], and ZincBinder [103] are specially designed for the identification of zinc-binding residues. FunFOLD [155], FunFOLDQA [156], and FunFOLD2 [157] are a series of template-based methods.
Table 4.
A breakdown of predictive tools of metal-binding residues.
1WS: web server; SS: standalone software. 2The availability was estimated on Dec 1st, 10th, and 20th of 2021, respectively.
4. Conclusions and Future Perspectives
This review summarizes the public database of metal ions binding interactions, discusses the architectures of computation-based methods for identifying binding residues, and comparatively evaluates four types of methods. Based on the observations made in this work, we propose a few recommendations for future research in this field:
First, the researchers should maintain and update the database regularly. This will significantly improve effectiveness and completeness for these databases and provide convenience for the computation-based methods, which depend on the accurate internal database. We expect a high-quality metal ion binding-related database with an advanced searching engine, high-speed download service, complete annotation information, etc. Particularly, a decent database should be designed to open for easy expanding and improvement. Second, standard benchmark datasets that related to general or ligand-specific metal-binding residues should be periodically compiled and made available. This will ensure consistent evaluation and comparative analysis of the performance of the existing and novel methods. Third, these predictors are expected to use delicate architectures and powerful algorithms. Since the differences between different types are quite small, the novel predictors shall not only correctly identify metal-binding residues but also distinguish different types of metal ions. Fourth, the authors of the metal-binding predictors are suggested to make their approaches publicly available, preferably as both webservers and standalone software. Particularly, high-throughput predictors promise a wide application among the research community since they can be used to perform large-scale computations, such as proteome-level predictions.
Acknowledgments
This work was supported in part by the National Natural Science Foundation of China (Nos. 62002307 and 61802329), by the Innovation Team Support Plan of University Science and Technology of Henan Province (No. 19IRTSTHN014), by the Key Scientific Research Project of Colleges and Universities in Henan Province (Grant No. 22A170019), by the Project of Science and Technology Department of Henan Province (No. 212102210392), and by Nanhu Scholars Program for Young Scholars of Xinyang Normal University.
Conflicts of Interest
The authors declare no conflict of interest.
Supplementary Materials
Table S1: comparative assessment of considered methods on Yu et al.'s independent testing dataset. Table S2: comparative assessment of considered methods on Cao et al.'s independent testing dataset.
References
- 1.Thompson K. H., Orvig C. Boon and bane of metal ions in medicine. Science . 2003;300(5621):936–939. doi: 10.1126/science.1083004. [DOI] [PubMed] [Google Scholar]
- 2.Aron A. T., Petras D., Schmid R., et al. Native mass spectrometry-based metabolomics identifies metal-binding compounds. Nature Chemistry . 2022;14(1):100–109. doi: 10.1038/s41557-021-00803-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Andreini C., Bertini I., Rosato A. Metalloproteomes: a bioinformatic approach. Accounts of Chemical Research . 2009;42(10):1471–1479. doi: 10.1021/ar900015x. [DOI] [PubMed] [Google Scholar]
- 4.Clausen M. J. V., Poulsen H. Metallomics and the Cell . Springer; 2013. Sodium/potassium homeostasis in the cell; pp. 41–67. [DOI] [PubMed] [Google Scholar]
- 5.Lambers H., Hayes P. E., Laliberte E., Oliveira R. S., Turner B. L. Leaf manganese accumulation and phosphorus-acquisition efficiency. Trends in Plant Science . 2015;20(2):83–90. doi: 10.1016/j.tplants.2014.10.007. [DOI] [PubMed] [Google Scholar]
- 6.Maret W., Wedd A. Binding, Transport and Storage of Metal Ions in Biological Cells . Royal Society of Chemistry; 2014. [DOI] [Google Scholar]
- 7.Akcapinar G. B., Sezerman O. U. Computational approaches forde novodesign and redesign of metal-binding sites on proteins. Bioscience Reports . 2017;37(2) doi: 10.1042/BSR20160179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kepp K. P. Alzheimer's disease: how metal ions define β-amyloid function. Coordination Chemistry Reviews . 2017;351:127–159. doi: 10.1016/j.ccr.2017.05.007. [DOI] [Google Scholar]
- 9.Reinhard L., Tidow H., Clausen M. J., Nissen P. Na+, K+-ATPase as a docking station: protein–protein complexes of the Na+, K+-ATPase. Cellular and Molecular Life Sciences . 2013;70(2):205–222. doi: 10.1007/s00018-012-1039-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Krzywoszyńska K., Witkowska D., Świątek-Kozłowska J., Szebesczyk A., Kozłowski H. General aspects of metal ions as signaling agents in health and disease. Biomolecules . 2020;10(10):p. 1417. doi: 10.3390/biom10101417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Quintanar L., Lim M. H. Metal ions and degenerative diseases . Springer; 2019. [DOI] [PubMed] [Google Scholar]
- 12.Tokar E. J., Benbrahim-Tallaa L., Waalkes M. P. 14 Metal ions in human cancer development. Metal Ions in Toxicology: Effects, Interactions, Interdependencies . 2015;8:p. 375. doi: 10.1515/9783110436624-019. [DOI] [PubMed] [Google Scholar]
- 13.Zhang J. Current advances in drug design and cancer research. Current Topics in Medicinal Chemistry . 2021;21(15):1307–1309. doi: 10.2174/156802662115210913102422. [DOI] [PubMed] [Google Scholar]
- 14.Potier N., Rogniaux H., Chevreux G., Van Dorsselaer A. Ligand-Metal Ion Binding to Proteins: Investigation by ESI Mass Spectrometry. Methods in Enzymology . 2005;402:361–389. doi: 10.1016/S0076-6879(05)02011-2. [DOI] [PubMed] [Google Scholar]
- 15.Handing K. B., Niedzialkowska E., Shabalin I. G., Kuhn M. L., Zheng H., Minor W. Characterizing metal-binding sites in proteins with X-ray crystallography. Nature Protocols . 2018;13(5):1062–1090. doi: 10.1038/nprot.2018.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shen D., Xu X., Wu H., et al. Metal ion binding to anticoagulation factor II from the venom of Agkistrodon acutus: stabilization of the structure and regulation of the binding affinity to activated coagulation factor X. Journal of Biological Inorganic Chemistry . 2011;16(4):523–537. doi: 10.1007/s00775-010-0752-9. [DOI] [PubMed] [Google Scholar]
- 17.Thirumoorthy N., Sunder A. S., Kumar K. M., Ganesh G., Chatterjee M. A review of metallothionein isoforms and their role in pathophysiology. World Journal of Surgical Oncology . 2011;9(1):1–7. doi: 10.1186/1477-7819-9-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Miller Y., Ma B., Nussinov R. Metal binding sites in amyloid oligomers: complexes and mechanisms. Coordination Chemistry Reviews . 2012;256(19-20):2245–2252. doi: 10.1016/j.ccr.2011.12.022. [DOI] [Google Scholar]
- 19.Bal W., Sokołowska M., Kurowska E., Faller P. Binding of transition metal ions to albumin: sites, affinities and rates. Biochimica et Biophysica Acta (BBA)-General Subjects . 2013;1830(12):5444–5455. doi: 10.1016/j.bbagen.2013.06.018. [DOI] [PubMed] [Google Scholar]
- 20.Roohani N., Hurrell R., Kelishadi R., Schulin R. Zinc and its importance for human health: an integrative review. Journal of research in medical sciences: the official journal of Isfahan University of Medical Sciences . 2013;18(2):144–157. [PMC free article] [PubMed] [Google Scholar]
- 21.Sobolev V., Edelman M. Web tools for predicting metal binding sites in proteins. Israel Journal of Chemistry . 2013;53(3-4):166–172. doi: 10.1002/ijch.201200084. [DOI] [Google Scholar]
- 22.Liu Z., Wang Y., Zhou C., Xue Y., Zhao W., Liu H. Computationally characterizing and comprehensive analysis of zinc-binding sites in proteins. Biochimica et Biophysica Acta (BBA)-Proteins and Proteomics . 2014;1844(1):171–180. doi: 10.1016/j.bbapap.2013.03.001. [DOI] [PubMed] [Google Scholar]
- 23.Witkowska D., Rowińska-Żyrek M. Biophysical approaches for the study of metal-protein interactions. Journal of Inorganic Biochemistry . 2019;199, article 110783 doi: 10.1016/j.jinorgbio.2019.110783. [DOI] [PubMed] [Google Scholar]
- 24.Mallick M., Sharan Vidyarthi A., Shankaracharya Tools for predicting metal binding sites in protein: a review. Current Bioinformatics . 2011;6(4):444–449. doi: 10.2174/157489311798072990. [DOI] [Google Scholar]
- 25.Rauer C., Sen N., Waman V. P., Abbasian M., Orengo C. A. Computational approaches to predict protein functional families and functional sites. Current Opinion in Structural Biology . 2021;70:108–122. doi: 10.1016/j.sbi.2021.05.012. [DOI] [PubMed] [Google Scholar]
- 26.Tran J. B., Krężel A. InterMetalDB: a database and browser of intermolecular metal binding sites in macromolecules with structural information. Journal of Proteome Research . 2021;20(4):1889–1901. doi: 10.1021/acs.jproteome.0c00906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cabrera A. J. R. Zinc, aging, and immunosenescence: an overview. Pathobiology of Aging & Age-related Diseases . 2015;5(1):p. 25592. doi: 10.3402/pba.v5.25592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Staats C. C., Kmetzsch L., Schrank A., Vainstein M. H. Fungal zinc metabolism and its connections to virulence. Frontiers in Cellular and Infection Microbiology . 2013;3:p. 65. doi: 10.3389/fcimb.2013.00065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Glimcher M. J. Bone: nature of the calcium phosphate crystals and cellular, structural, and physical chemical mechanisms in their formation. Reviews in Mineralogy and Geochemistry . 2006;64(1):223–282. doi: 10.2138/rmg.2006.64.8. [DOI] [Google Scholar]
- 30.Berman H. M., Westbrook J., Feng Z., et al. The protein data bank. Nucleic Acids Research . 2000;28(1):235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ye N. Survey of in-silico prediction of anticancer peptides. Current Topics in Medicinal Chemistry . 2021;21(15):1310–1318. doi: 10.2174/1568026621666210612030536. [DOI] [PubMed] [Google Scholar]
- 32.Hauser M., Steinegger M., Söding J. MMseqs software suite for fast and deep clustering and searching of large protein sequence sets. Bioinformatics . 2016;32(9):1323–1330. doi: 10.1093/bioinformatics/btw006. [DOI] [PubMed] [Google Scholar]
- 33.Li G., Su Y., Yan Y.-H., et al. MeLAD: an integrated resource for metalloenzyme-ligand associations. Bioinformatics . 2020;36(3):904–909. doi: 10.1093/bioinformatics/btz648. [DOI] [PubMed] [Google Scholar]
- 34.Putignano V., Rosato A., Banci L., Andreini C. MetalPDB in 2018: a database of metal sites in biological macromolecular structures. Nucleic Acids Research . 2018;46(D1):D459–D464. doi: 10.1093/nar/gkx989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Andreini C., Cavallaro G., Lorenzini S., Rosato A. MetalPDB: a database of metal sites in biological macromolecular structures. Nucleic Acids Research . 2012;41(D1):D312–D319. doi: 10.1093/nar/gks1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yang J., Roy A., Zhang Y. BioLiP: a semi-manually curated database for biologically relevant ligand–protein interactions. Nucleic Acids Research . 2012;41(D1):D1096–D1103. doi: 10.1093/nar/gks966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fu F., Voytas D. F. Zinc finger database (ZiFDB) v2. 0: a comprehensive database of C2H2 zinc fingers and engineered zinc finger arrays. Nucleic Acids Research . 2012;41(D1):D452–D455. doi: 10.1093/nar/gks1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fu F., Sander J. D., Maeder M., et al. Zinc finger database (ZiFDB): a repository for information on C2H2 zinc fingers and engineered zinc-finger arrays. Nucleic Acids Research . 2009;37(Database):D279–D283. doi: 10.1093/nar/gkn606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tus A., Rakipović A., Peretin G., Tomić S., Šikić M. BioMe: biologically relevant metals. Nucleic Acids Research . 2012;40(W1):W352–W357. doi: 10.1093/nar/gks514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Choi H., Kang H., Park H. MetLigDB: a web-based database for the identification of chemical groups to design metalloprotein inhibitors. Journal of Applied Crystallography . 2011;44(4):878–881. doi: 10.1107/S0021889811022503. [DOI] [Google Scholar]
- 41.Hemavathi K., Kalaivani M., Udayakumar A., Sowmiya G., Jeyakanthan J., Sekar K. MIPS: metal interactions in protein structures. Journal of Applied Crystallography . 2010;43(1):196–199. doi: 10.1107/S002188980903982X. [DOI] [Google Scholar]
- 42.Kumar Kuntal B., Aparoy P., Reddanna P. Development of tools and database for analysis of metal binding sites in protein. Protein and Peptide Letters . 2010;17(6):765–773. doi: 10.2174/092986610791190246. [DOI] [PubMed] [Google Scholar]
- 43.Nakamura K., Hirai A., Altaf-Ul-Amin M., Takahashi H. MetalMine: a database of functional metal-binding sites in proteins. Plant Biotechnology . 2009;26(5):517–521. doi: 10.5511/plantbiotechnology.26.517. [DOI] [Google Scholar]
- 44.Andreini C., Bertini I., Cavallaro G., Holliday G. L., Thornton J. M. Metal-MACiE: a database of metals involved in biological catalysis. Bioinformatics . 2009;25(16):2088–2089. doi: 10.1093/bioinformatics/btp256. [DOI] [PubMed] [Google Scholar]
- 45.Jayakanthan M., Muthukumaran J., Chandrasekar S., Chawla K., Punetha A., Sundar D. ZifBASE: a database of zinc finger proteins and associated resources. BMC Genomics . 2009;10(1):1–7. doi: 10.1186/1471-2164-10-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hsin K., Sheng Y., Harding M., Taylor P., Walkinshaw M. MESPEUS: a database of the geometry of metal sites in proteins. Journal of Applied Crystallography . 2008;41(5):963–968. doi: 10.1107/S002188980802476X. [DOI] [Google Scholar]
- 47.Golovin A., Dimitropoulos D., Oldfield T., Rachedi A., Henrick K. MSDsite: a database search and retrieval system for the analysis and viewing of bound ligands and active sites. Proteins: Structure, Function, and Bioinformatics . 2005;58(1):190–199. doi: 10.1002/prot.20288. [DOI] [PubMed] [Google Scholar]
- 48.Castagnetto J. M., Hennessy S. W., Roberts V. A., Getzoff E. D., Tainer J. A., Pique M. E. MDB: the metalloprotein database and browser at the Scripps Research Institute. Nucleic Acids Research . 2002;30(1):379–382. doi: 10.1093/nar/30.1.379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ireland S. M., Martin A. C. ZincBind—the database of zinc binding sites. Database . 2019;2019 doi: 10.1093/database/baz006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang S., Hu X., Feng Z., et al. Recognizing ion ligand binding sites by SMO algorithm. BMC Molecular and Cell Biology . 2019;20(S3):53–59. doi: 10.1186/s12860-019-0237-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yan R., Wang X., Tian Y., Xu J., Xu X., Lin J. Prediction of zinc-binding sites using multiple sequence profiles and machine learning methods. Molecular omics . 2019;15(3):205–215. doi: 10.1039/C9MO00043G. [DOI] [PubMed] [Google Scholar]
- 52.Cao X., Hu X., Zhang X., et al. Identification of metal ion binding sites based on amino acid sequences. PLoS One . 2017;12(8, article e0183756) doi: 10.1371/journal.pone.0183756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hu X., Wang K., Dong Q. Protein ligand-specific binding residue predictions by an ensemble classifier. BMC Bioinformatics . 2016;17(1):1–12. doi: 10.1186/s12859-016-1348-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Qu J., Yin S. S., Wang H. Prediction of metal ion binding sites of transmembrane proteins. Computational and Mathematical Methods in Medicine . 2021;2021:11. doi: 10.1155/2021/2327832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Brylinski M., Skolnick J. FINDSITE-metal: integrating evolutionary information and machine learning for structure-based metal-binding site prediction at the proteome level. Proteins: Structure, Function, and Bioinformatics . 2011;79(3):735–751. doi: 10.1002/prot.22913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liu L., Hu X., Feng Z., Wang S., Sun K., Xu S. Recognizing ion ligand–binding residues by random forest algorithm based on optimized dihedral angle. Frontiers in Bioengineering and Biotechnology . 2020;8:p. 493. doi: 10.3389/fbioe.2020.00493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hu X., Feng Z., Zhang X., Liu L., Wang S. The identification of metal ion ligand-binding residues by adding the reclassified relative solvent accessibility. Frontiers in Genetics . 2020;11:p. 214. doi: 10.3389/fgene.2020.00214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Horst J. A., Samudrala R. A protein sequence meta-functional signature for calcium binding residue prediction. Pattern Recognition Letters . 2010;31(14):2103–2112. doi: 10.1016/j.patrec.2010.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pearson W. R. An introduction to sequence similarity (“homology”) searching. Current Protocols in Bioinformatics . 2013;42(1) doi: 10.1002/0471250953.bi0301s42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Joshi T., Xu D. Quantitative assessment of relationship between sequence similarity and function similarity. BMC Genomics . 2007;8(1) doi: 10.1186/1471-2164-8-222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhang J., Chai H., Yang G., Ma Z. Prediction of bioluminescent proteins by using sequence-derived features and lineage-specific scheme. BMC Bioinformatics . 2017;18(1):p. 294. doi: 10.1186/s12859-017-1709-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Altschul S. F., Madden T. L., Schäffer A. A., et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Research . 1997;25(17):3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wang G., Dunbrack R. L., Jr. PISCES: a protein sequence culling server. Bioinformatics . 2003;19(12):1589–1591. doi: 10.1093/bioinformatics/btg224. [DOI] [PubMed] [Google Scholar]
- 64.Li W., Godzik A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics . 2006;22(13):1658–1659. doi: 10.1093/bioinformatics/btl158. [DOI] [PubMed] [Google Scholar]
- 65.Haberal I., Oğul H. Prediction of protein metal binding sites using deep neural networks. Molecular Informatics . 2019;38(7, article 1800169) doi: 10.1002/minf.201800169. [DOI] [PubMed] [Google Scholar]
- 66.Zhang J., Liang X., Zhou F., Li B., Li Y. TYLER, a fast method that accurately predicts cyclin-dependent proteins by using computation-based motifs and sequence-derived features. Mathematical Biosciences and Engineering . 2021;18(5):6410–6429. doi: 10.3934/mbe.2021318. [DOI] [PubMed] [Google Scholar]
- 67.Homchaudhuri L., Swaminathan R. Near ultraviolet absorption arising from lysine residues in close proximity: a probe to monitor protein unfolding and aggregation in lysine-rich proteins. Bulletin of the Chemical Society of Japan . 2004;77(4):765–769. doi: 10.1246/bcsj.77.765. [DOI] [Google Scholar]
- 68.Chen K., Kurgan L. A., Ruan J. Prediction of flexible/rigid regions from protein sequences using k-spaced amino acid pairs. BMC Structural Biology . 2007;7(1):1–13. doi: 10.1186/1472-6807-7-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Avrahami D., Oren Z., Shai Y. Effect of multiple aliphatic amino acids substitutions on the structure, function, and mode of action of diastereomeric membrane active peptides. Biochemistry . 2001;40(42):12591–12603. doi: 10.1021/bi0105330. [DOI] [PubMed] [Google Scholar]
- 70.Suliman M. E., Bárány P., Divino Filho J. C., et al. Influence of nutritional status on plasma and erythrocyte sulphur amino acids, sulph-hydryls, and inorganic sulphate in end-stage renal disease. Nephrology, Dialysis, Transplantation . 2002;17(6):1050–1056. doi: 10.1093/ndt/17.6.1050. [DOI] [PubMed] [Google Scholar]
- 71.Scheiner S., Tapas Kar A., Pattanayak J. Comparison of various types of hydrogen bonds involving aromatic amino acids. Journal of the American Chemical Society . 2002;124(44):13257–13264. doi: 10.1021/ja027200q. [DOI] [PubMed] [Google Scholar]
- 72.Strub C., Alies C., Lougarre A., Ladurantie C., Czaplicki J., Fournier D. Mutation of exposed hydrophobic amino acids to arginine to increase protein stability. BMC Biochemistry . 2004;5(1):9–9. doi: 10.1186/1471-2091-5-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Heard T. S., Weiner H. A regional net charge and structural compensation model to explain how negatively charged amino acids can be accepted within a mitochondrial leader sequence. Journal of Biological Chemistry . 1998;273(45):29389–29393. doi: 10.1074/jbc.273.45.29389. [DOI] [PubMed] [Google Scholar]
- 74.Kamtekar S., Schiffer J. M., Xiong H., Babik J. M., Hecht M. H. ChemInform Abstract: Protein design by binary patterning of polar and nonpolar amino acids. Science . 1994;25(10) doi: 10.1002/chin.199410253. [DOI] [PubMed] [Google Scholar]
- 75.Goodwin G. H., Sanders C., Johns E. W. A new group of chromatin-associated proteins with a high content of acidic and basic amino acids. FEBS Journal . 1973;38(1):14–19. doi: 10.1111/j.1432-1033.1973.tb03026.x. [DOI] [PubMed] [Google Scholar]
- 76.Zhang J., Ma Z., Kurgan L. Comprehensive review and empirical analysis of hallmarks of DNA-, RNA- and protein-binding residues in protein chains. Briefings in Bioinformatics . 2019;20(4):1250–1268. doi: 10.1093/bib/bbx168. [DOI] [PubMed] [Google Scholar]
- 77.Jiang Z., Hu X., Geriletu G., Xing H., Cao X. Identification of Ca2+-binding residues of a protein from its primary sequence. Genetics and Molecular Research . 2016;15(2) doi: 10.4238/gmr.15027618. [DOI] [PubMed] [Google Scholar]
- 78.Zheng C., Wang M., Takemoto K., Akutsu T., Zhang Z., Song J. An integrative computational framework based on a two-step random forest algorithm improves prediction of zinc-binding sites in proteins. PLoS One . 2012;7(11, article e49716) doi: 10.1371/journal.pone.0049716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Song J., Li C., Zheng C., Revote J., Zhang Z., Webb G. I. MetalExplorer, a bioinformatics tool for the improved prediction of eight types of metal-binding sites using a random forest algorithm with Two- Step feature selection. Current Bioinformatics . 2017;12(6):480–489. doi: 10.2174/2468422806666160618091522. [DOI] [Google Scholar]
- 80.Xia C.-Q., Pan X., Shen H.-B. Protein–ligand binding residue prediction enhancement through hybrid deep heterogeneous learning of sequence and structure data. Bioinformatics . 2020;36(10):3018–3027. doi: 10.1093/bioinformatics/btaa110. [DOI] [PubMed] [Google Scholar]
- 81.Wang S., Hu X., Feng Z., Liu L., Sun K., Xu S. Recognition of ion ligand binding sites based on amino acid features with the fusion of energy, physicochemical and structural features. Current Pharmaceutical Design . 2021;27(8):1093–1102. doi: 10.2174/1381612826666201029100636. [DOI] [PubMed] [Google Scholar]
- 82.Liu Y., Bahar I. Sequence evolution correlates with structural dynamics. Molecular Biology and Evolution . 2012;29(9):2253–2263. doi: 10.1093/molbev/mss097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Chai H., Zhang J. Identification of mammalian enzymatic proteins based on sequence-derived features and species-specific scheme. IEEE Access . 2018;6:8452–8458. doi: 10.1109/ACCESS.2018.2798284. [DOI] [Google Scholar]
- 84.Celniker G., Nimrod G., Ashkenazy H., et al. ConSurf: using evolutionary data to raise testable hypotheses about protein function. Israel Journal of Chemistry . 2013;53(3-4):199–206. doi: 10.1002/ijch.201200096. [DOI] [Google Scholar]
- 85.Chowdhury S., Zhang J., Kurgan L. In silico prediction and validation of novel RNA binding proteins and residues in the human proteome. Proteomics . 2018;18(21-22, article 1800064) doi: 10.1002/pmic.201800064. [DOI] [PubMed] [Google Scholar]
- 86.Zhang J., Chai H., Guo S., Guo H., Li Y. High-throughput identification of mammalian secreted proteins using species-specific scheme and application to human proteome. Molecules . 2018;23(6):p. 1448. doi: 10.3390/molecules23061448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Srivastava A., Kumar M. Prediction of zinc binding sites in proteins using sequence derived information. Journal of Biomolecular Structure and Dynamics . 2018;36(16):4413–4423. doi: 10.1080/07391102.2017.1417910. [DOI] [PubMed] [Google Scholar]
- 88.McGuffin L. J., Bryson K., Jones D. T. The PSIPRED protein structure prediction server. Bioinformatics . 2000;16(4):404–405. doi: 10.1093/bioinformatics/16.4.404. [DOI] [PubMed] [Google Scholar]
- 89.Ward J. J., Sodhi J. S., McGuffin L. J., Buxton B. F., Jones D. T. Prediction and functional analysis of native disorder in proteins from the three kingdoms of life. Journal of Molecular Biology . 2004;337(3):635–645. doi: 10.1016/j.jmb.2004.02.002. [DOI] [PubMed] [Google Scholar]
- 90.Qiao L., Xie D. MIonSite: ligand-specific prediction of metal ion-binding sites via enhanced AdaBoost algorithm with protein sequence information. Analytical Biochemistry . 2019;566:75–88. doi: 10.1016/j.ab.2018.11.009. [DOI] [PubMed] [Google Scholar]
- 91.Lu C., Liu Z., Zhang E., He F., Ma Z., Wang H. MPLs-Pred: predicting membrane protein-ligand binding sites using hybrid sequence-based features and ligand-specific models. International Journal of Molecular Sciences . 2019;20(13):p. 3120. doi: 10.3390/ijms20133120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Zhao Z., Xu Y., Zhao Y. SXGBsite: prediction of protein–ligand binding sites using sequence information and extreme gradient boosting. Genes . 2019;10(12):p. 965. doi: 10.3390/genes10120965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Li H., Pi D., Chen C., Li H. A novel prediction method for zinc-binding sites in proteins by an ensemble of SVM and sample-weighted probabilistic neural network. IEEE Access . 2019;7:186147–186157. doi: 10.1109/ACCESS.2019.2960374. [DOI] [Google Scholar]
- 94.Essien C., Wang D., Xu D. Capsule Network for Predicting Zinc Binding Sites in Metalloproteins. 2019 IEEE International Conference on Bioinformatics and Biomedicine (BIBM); 2019; San Diego, CA, USA. pp. 2337–2341. [DOI] [Google Scholar]
- 95.Ding Y., Tang J., Guo F. Identification of protein–ligand binding sites by sequence information and ensemble classifier. Journal of Chemical Information and Modeling . 2017;57(12):3149–3161. doi: 10.1021/acs.jcim.7b00307. [DOI] [PubMed] [Google Scholar]
- 96.Kumar S. Prediction of metal ion binding sites in proteins from amino acid sequences by using simplified amino acid alphabets and random forest model. Genomics & informatics . 2017;15(4):162–169. doi: 10.5808/GI.2017.15.4.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Haberal İ., Oğul H. Deepmbs: prediction of protein metal binding-site using deep learning networks. 2017 Fourth International Conference on Mathematics and Computers in Sciences and in Industry (MCSI); 2017; Corfu, Greece. pp. 1–25. [DOI] [Google Scholar]
- 98.Qiao L., Xie D. Sequence-based protein-Ca2+ binding site prediction using SVM classifier ensemble with random under-sampling. 2017 International Conference on Progress in Informatics and Computing (PIC); 2017; Nanjing, China. pp. 86–90. [DOI] [Google Scholar]
- 99.Hu X., Dong Q., Yang J., Zhang Y. Recognizing metal and acid radical ion-binding sites by integrating ab initio modeling with template-based transferals. Bioinformatics . 2016;32(21):3260–3269. doi: 10.1093/bioinformatics/btw396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Yu D.-J., Hu J., Li Q.-M., Tang Z.-M., Yang J.-Y., Shen H.-B. Constructing query-driven dynamic machine learning model with application to protein-ligand binding sites prediction. IEEE Transactions on Nanobioscience . 2015;14(1):45–58. doi: 10.1109/TNB.2015.2394328. [DOI] [PubMed] [Google Scholar]
- 101.Yu D.-J., Hu J., Yang J., Shen H.-B., Tang J., Yang J.-Y. Designing template-free predictor for targeting protein-ligand binding sites with classifier ensemble and spatial clustering. IEEE/ACM Transactions on Computational Biology and Bioinformatics . 2013;10(4):994–1008. doi: 10.1109/TCBB.2013.104. [DOI] [PubMed] [Google Scholar]
- 102.Ou Y.-Y., Chen S.-A., Wu S.-C. ETMB-RBF: discrimination of metal-binding sites in electron transporters based on RBF networks with PSSM profiles and significant amino acid pairs. PLoS One . 2013;8(2, article e46572) doi: 10.1371/journal.pone.0046572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Chen Z., Wang Y., Zhai Y.-F., Song J., Zhang Z. ZincExplorer: an accurate hybrid method to improve the prediction of zinc-binding sites from protein sequences. Molecular Bio Systems . 2013;9(9):2213–2222. doi: 10.1039/c3mb70100j. [DOI] [PubMed] [Google Scholar]
- 104.Nguyen H., Kleingardner J. Identifying metal binding amino acids based on backbone geometries as a tool for metalloprotein engineering. Protein Science . 2021;30(6):1247–1257. doi: 10.1002/pro.4074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Ireland S. M., Martin A. C. Zincbindpredict—prediction of zinc binding sites in proteins. Molecules . 2021;26(4):p. 966. doi: 10.3390/molecules26040966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Zhang J., Zhang Y., Li Y., Guo S., Yang G. Identification of cancer biomarkers in human body fluids by using enhanced physicochemical-incorporated evolutionary conservation scheme. Current Topics in Medicinal Chemistry . 2020;20(21):1888–1897. doi: 10.2174/1568026620666200710100743. [DOI] [PubMed] [Google Scholar]
- 107.Iqbal M. J., Faye I., Samir B. B., Md Said A. Efficient feature selection and classification of protein sequence data in bioinformatics. The Scientific World Journal . 2014;2014:12. doi: 10.1155/2014/173869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Wang L. Independent Component Analyses, Compressive Sampling, Wavelets, Neural Net, Biosystems, and Nanoengineering X . International Society for Optics and Photonics; 2012. Feature selection in bioinformatics; p. p. 8401. [Google Scholar]
- 109.Saeys Y., Inza I., Larranaga P. A review of feature selection techniques in bioinformatics. Bioinformatics . 2007;23(19):2507–2517. doi: 10.1093/bioinformatics/btm344. [DOI] [PubMed] [Google Scholar]
- 110.Shastry K. A., Sanjay H. A., Sanjay H. Statistical Modelling and Machine Learning Principles for Bioinformatics Techniques, Tools, and Applications . Springer; 2020. Machine learning for bioinformatics; pp. 25–39. [DOI] [Google Scholar]
- 111.Pisner D. A., Schnyer D. M. Machine Learning . Elsevier; 2020. Support vector machine; pp. 101–121. [DOI] [Google Scholar]
- 112.Liu L., Shen B., Wang X., Wang X. Advanced Technologies, Embedded and Multimedia for Human-Centric Computing . Springer; 2014. Research on kernel function of support vector machine; pp. 827–834. [DOI] [Google Scholar]
- 113.Platt J. Microsoft Research Technical Report; 1998. Sequential minimal optimization: a fast algorithm for training support vector machines. [Google Scholar]
- 114.Huang X., Shi L., Suykens J. A. Sequential minimal optimization for SVM with pinball loss. Neurocomputing . 2015;149:1596–1603. doi: 10.1016/j.neucom.2014.08.033. [DOI] [Google Scholar]
- 115.Lancashire L. J., Lemetre C., Ball G. R. An introduction to artificial neural networks in bioinformatics-application to complex microarray and mass spectrometry datasets in cancer studies. Briefings in bioinformatics . 2009;10(3):315–329. doi: 10.1093/bib/bbp012. [DOI] [PubMed] [Google Scholar]
- 116.Kim B.-H., Yu K., Lee P. C. Cancer classification of single-cell gene expression data by neural network. Bioinformatics . 2020;36(5):1360–1366. doi: 10.1093/bioinformatics/btz772. [DOI] [PubMed] [Google Scholar]
- 117.Lee T.-Y., Chen S.-A., Hung H.-Y., Ou Y.-Y. Incorporating distant sequence features and radial basis function networks to identify ubiquitin conjugation sites. PLoS One . 2011;6(3, article e17331) doi: 10.1371/journal.pone.0017331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Zainuddin Z., Kumar M. Radial basis function neural networks in protein sequence classification. Malaysian Journal of Mathematical Sciences . 2008;2(2):195–204. [Google Scholar]
- 119.Taud H., Mas J. Geomatic Approaches for Modeling Land Change Scenarios . Springer; 2018. Multilayer perceptron (MLP) pp. 451–455. [DOI] [Google Scholar]
- 120.Ramchoun H., Idrissi M. A. J., Ghanou Y., Ettaouil M. Multilayer perceptron: architecture optimization and training. The International Journal of Interactive Multimedia and Artificial Intelligence . 2016;4(1):26–30. doi: 10.9781/ijimai.2016.415. [DOI] [Google Scholar]
- 121.Lorencin I., Anđelić N., Španjol J., Car Z. Using multi-layer perceptron with Laplacian edge detector for bladder cancer diagnosis. Artificial Intelligence in Medicine . 2020;102, article 101746 doi: 10.1016/j.artmed.2019.101746. [DOI] [PubMed] [Google Scholar]
- 122.Zhang J., Zhang Y., Ma Z. In silico prediction of human secretory proteins in plasma based on discrete firefly optimization and application to cancer biomarkers identification. Frontiers in Genetics . 2019;10:p. 542. doi: 10.3389/fgene.2019.00542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Schonlau M., Zou R. Y. The random forest algorithm for statistical learning. The Stata Journal . 2020;20(1):3–29. doi: 10.1177/1536867X20909688. [DOI] [Google Scholar]
- 124.Qi Y. Ensemble Machine Learning . Springer; 2012. Random forest for bioinformatics; pp. 307–323. [Google Scholar]
- 125.Zhang J., Chai H., Gao B., Yang G., Ma Z. HEMEsPred: structure-based ligand-specific heme binding residues prediction by using fast-adaptive ensemble learning scheme. IEEE/ACM Transactions on Computational Biology and Bioinformatics . 2018;15(1):147–156. doi: 10.1109/TCBB.2016.2615010. [DOI] [PubMed] [Google Scholar]
- 126.Lu H., Gao H., Ye M., Wang X. A hybrid ensemble algorithm combining Ada Boost and genetic algorithm for cancer classification with gene expression data. IEEE/ACM Transactions on Computational Biology and Bioinformatics . 2021;18 doi: 10.1109/TCBB.2019.2952102. [DOI] [PubMed] [Google Scholar]
- 127.Chen P., Pan C. Diabetes classification model based on boosting algorithms. BMC Bioinformatics . 2018;19(1):1–9. doi: 10.1186/s12859-018-2090-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Yan C., Wu F.-X., Wang J., Duan G. PESM: predicting the essentiality of miRNAs based on gradient boosting machines and sequences. BMC Bioinformatics . 2020;21(1):1–9. doi: 10.1186/s12859-020-3426-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Rawi R., Mall R., Kunji K., Shen C.-H., Kwong P. D., Chuang G.-Y. PaRSnIP: sequence-based protein solubility prediction using gradient boosting machine. Bioinformatics . 2018;34(7):1092–1098. doi: 10.1093/bioinformatics/btx662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Deng L., Pan J., Xu X., Yang W., Liu C., Liu H. PDRLGB: precise DNA-binding residue prediction using a light gradient boosting machine. BMC Bioinformatics . 2018;19(S19):135–145. doi: 10.1186/s12859-018-2527-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Zhao X., Zhang J., Ning Q., Sun P., Ma Z., Yin M. Identification of protein pupylation sites using bi-profile Bayes feature extraction and ensemble learning. Mathematical Problems in Engineering . 2013;2013:7. doi: 10.1155/2013/283129.283129 [DOI] [Google Scholar]
- 132.Ayyadevara V. K. Pro Machine Learning Algorithms . Springer; 2018. Gradient boosting machine; pp. 117–134. [Google Scholar]
- 133.Li X., Li S., Wang Y., Zhang S., Wong K.-C. Identification of pan-cancer Ras pathway activation with deep learning. Briefings in Bioinformatics . 2021;22(4):p. bbaa 258. doi: 10.1093/bib/bbaa258. [DOI] [PubMed] [Google Scholar]
- 134.Albawi S., Mohammed T. A., Al-Zawi S. Understanding of a convolutional neural network. 2017 International Conference on Engineering and Technology (ICET); 2017; Antalya, Turkey. pp. 1–6. [DOI] [Google Scholar]
- 135.Ghosh A., Sufian A., Sultana F., Chakrabarti A., De D. Recent Trends and Advances in Artificial Intelligence and Internet of Things . Springer; 2020. Fundamental concepts of convolutional neural network; pp. 519–567. [DOI] [Google Scholar]
- 136.Yang Y., Hou Z., Ma Z., Li X., Wong K.-C. iCircRBP-DHN: identification of circRNA-RBP interaction sites using deep hierarchical network. Briefings in Bioinformatics . 2021;22(4, article bbaa 274) doi: 10.1093/bib/bbaa274. [DOI] [PubMed] [Google Scholar]
- 137.Zhang Q., Zhang M., Chen T., Sun Z., Ma Y., Yu B. Recent advances in convolutional neural network acceleration. Neurocomputing . 2019;323:37–51. doi: 10.1016/j.neucom.2018.09.038. [DOI] [Google Scholar]
- 138.Wang Y., Yang Y., Ma Z., Wong K.-C., Li X. EDCNN: identification of genome-wide RNA-binding proteins using evolutionary deep convolutional neural network. Bioinformatics . 2022;38(3):678–686. doi: 10.1093/bioinformatics/btab739. [DOI] [PubMed] [Google Scholar]
- 139.Wang D., Liang Y., Xu D. Capsule network for protein post-translational modification site prediction. Bioinformatics . 2019;35(14):2386–2394. doi: 10.1093/bioinformatics/bty977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Li X., Li S., Huang L., Zhang S., Wong K.-c. High-throughput single-cell RNA-seq data imputation and characterization with surrogate-assisted automated deep learning. Briefings in Bioinformatics . 2022;23(1, article bbab 368) doi: 10.1093/bib/bbab368. [DOI] [PubMed] [Google Scholar]
- 141.Du X., Li Y., Xia Y.-L., et al. Insights into protein–ligand interactions: mechanisms, models, and methods. International Journal of Molecular Sciences . 2016;17(2):p. 144. doi: 10.3390/ijms17020144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Salha D., Andaç M., Denizli A. Molecular docking of metal ion immobilized ligands to proteins in affinity chromatography. Journal of Molecular Recognition . 2021;34(2):p. e 2875. doi: 10.1002/jmr.2875. [DOI] [PubMed] [Google Scholar]
- 143.Smith R. D., Engdahl A. L., Dunbar J. B., Jr., Carlson H. A. Biophysical limits of protein–ligand binding. Journal of Chemical Information and Modeling . 2012;52(8):2098–2106. doi: 10.1021/ci200612f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Huang S.-Y., Zou X. Advances and challenges in protein-ligand docking. International Journal of Molecular Sciences . 2010;11(8):3016–3034. doi: 10.3390/ijms11083016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Huang S.-Y., Grinter S. Z., Zou X. Scoring functions and their evaluation methods for protein–ligand docking: recent advances and future directions. Physical Chemistry Chemical Physics . 2010;12(40):12899–12908. doi: 10.1039/c0cp00151a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Kumar V., Chichili V. P. R., Tang X., Sivaraman J. A novel trans conformation of ligand-free calmodulin. PLoS One . 2013;8(1, article e54834) doi: 10.1371/journal.pone.0054834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Permyakov E. A. Metal Binding Proteins. Encyclopedia . 2021;1(1):261–292. doi: 10.3390/encyclopedia1010024. [DOI] [Google Scholar]
- 148.He W., Liang Z., Teng M., Niu L. mFASD: a structure-based algorithm for discriminating different types of metal-binding sites. Bioinformatics . 2015;31(12):1938–1944. doi: 10.1093/bioinformatics/btv044. [DOI] [PubMed] [Google Scholar]
- 149.Zhou W., Tang G. W., Altman R. B. High resolution prediction of calcium-binding sites in 3D protein structures using FEATURE. Journal of Chemical Information and Modeling . 2015;55(8):1663–1672. doi: 10.1021/acs.jcim.5b00367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Sciortino G., Garribba E., Rodriguez-Guerra Pedregal J., Maréchal J.-D. Simple coordination geometry descriptors allow to accurately predict metal-binding sites in proteins. ACS Omega . 2019;4(2):3726–3731. doi: 10.1021/acsomega.8b03457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Sánchez-Aparicio J.-E., Tiessler-Sala L., Velasco-Carneros L., Roldán-Martín L., Sciortino G., Maréchal J.-D. Bio met all: identifying metal-binding sites in proteins from backbone preorganization. Journal of Chemical Information and Modeling . 2021;61(1):311–323. doi: 10.1021/acs.jcim.0c00827. [DOI] [PubMed] [Google Scholar]
- 152.Deng H., Chen G., Yang W., Yang J. J. Predicting calcium-binding sites in proteins—a graph theory and geometry approach. Proteins: Structure, Function, and Bioinformatics . 2006;64(1):34–42. doi: 10.1002/prot.20973. [DOI] [PubMed] [Google Scholar]
- 153.Goyal K., Mande S. C. Exploiting 3D structural templates for detection of metal-binding sites in protein structures. Proteins: Structure, Function, and Bioinformatics . 2008;70(4):1206–1218. doi: 10.1002/prot.21601. [DOI] [PubMed] [Google Scholar]
- 154.Levy R., Edelman M., Sobolev V. Prediction of 3D metal binding sites from translated gene sequences based on remote-homology templates. Proteins: Structure, Function, and Bioinformatics . 2009;76(2):365–374. doi: 10.1002/prot.22352. [DOI] [PubMed] [Google Scholar]
- 155.Roche D. B., Tetchner S. J., McGuffin L. J. FunFOLD: an improved automated method for the prediction of ligand binding residues using 3D models of proteins. BMC Bioinformatics . 2011;12(1):1–20. doi: 10.1186/1471-2105-12-160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Roche D. B., Buenavista M. T., McGuffin L. J. FunFOLDQA: a quality assessment tool for protein-ligand binding site residue predictions. PLoS One . 2012;7(5, article e38219) doi: 10.1371/journal.pone.0038219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Roche D. B., Buenavista M. T., McGuffin L. J. The FunFOLD2 server for the prediction of protein–ligand interactions. Nucleic Acids Research . 2013;41(W1):W303–W307. doi: 10.1093/nar/gkt498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Li H., Pi D., Wu Y., Chen C. Integrative method based on linear regression for the prediction of zinc-binding sites in proteins. IEEE Access . 2017;5:14647–14656. doi: 10.1109/ACCESS.2017.2731872. [DOI] [Google Scholar]
- 159.Passerini A., Andreini C., Menchetti S., Rosato A., Frasconi P. Predicting zinc binding at the proteome level. BMC Bioinformatics . 2007;8(1):1–13. doi: 10.1186/1471-2105-8-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Shu N., Zhou T., Hovmöller S. Prediction of zinc-binding sites in proteins from sequence. Bioinformatics . 2008;24(6):775–782. doi: 10.1093/bioinformatics/btm618. [DOI] [PubMed] [Google Scholar]
- 161.Li H., Pi D., Chen C. An improved prediction model for zinc-binding sites in proteins based on Bayesian method. Tehnički vjesnik . 2019;26(5):1422–1426. doi: 10.17559/TV-20190730093945. [DOI] [Google Scholar]
- 162.Ohue M., Matsuzaki Y., Shimoda T., Ishida T., Akiyama Y. Highly precise protein-protein interaction prediction based on consensus between template-based and de novo docking methods. BMC Proceedings . 2013;7 doi: 10.1186/1753-6561-7-S7-S6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Deng H., Jia Y., Zhang Y. Protein structure prediction. International Journal of Modern Physics B . 2018;32(18, article 1840009) doi: 10.1142/S021797921840009X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Fiser A. Template-based protein structure modeling. Computational Biology . 2010;673:73–94. doi: 10.1007/978-1-60761-842-3_6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Karni E. Foundations of Bayesian theory. Journal of Economic Theory . 2007;132(1):167–188. doi: 10.1016/j.jet.2005.08.005. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1: comparative assessment of considered methods on Yu et al.'s independent testing dataset. Table S2: comparative assessment of considered methods on Cao et al.'s independent testing dataset.