

Abstract
Natural killer (NK) cells are innate lymphocytes that possess traits of adaptive immunity, such as memory formation. However, the molecular mechanisms by which NK cells persist to form memory cells are not well understood. Using single cell RNA sequencing, we identified two distinct effector NK cell (NKeff) populations following mouse cytomegalovirus (MCMV) infection. Ly6C– memory precursor (MP) NK cells displayed enhanced survival during the contraction phase in a Bcl2-dependent manner, and differentiated into Ly6C+ memory NK cells. MP NK cells exhibited distinct transcriptional and epigenetic signatures compared to Ly6C+ NKeff cells, with a core epigenetic signature shared with MP CD8+ T cells enriched in ETS1 and Fli1 DNA-binding motifs. Fli1 was induced by STAT5 signaling ex vivo, and increased levels of the pro-apoptotic factor Bim in early effector NK cells following viral infection. These results suggest that a NK cell-intrinsic checkpoint controlled by the transcription factor Fli1 limits MP NK formation by regulating early effector NK cell fitness during viral infection.
Introduction
The ability of the immune system to remember previous pathogen encounters by executing a specific and robust secondary response upon re-exposure to pathogen-associated antigens is termed immunological memory. During infection, this memory response is largely performed by the selective clonal proliferation and long-term persistence of adaptive lymphocytes that express somatically recombined antigen receptors (e.g. T and B cells). Adaptive lymphocytes form antigen-specific memory cells that are able to epigenetically maintain activation-induced transcriptional changes following clearance of pathogens1, 2. Coordination of stable epigenetic, transcriptional, and metabolic changes during T cell activation results in the cell-intrinsic capacity to form memory cells1, 3, 4. The kinetics of the adaptive immune response to infection consist of three distinct phases: clonal expansion, contraction and memory formation. During the contraction phase 90–95% of all expanded effector cells are eliminated through cell-intrinsic apoptosis, leaving memory T and B cells that persist long-term in the host5. Previous studies have demonstrated that expanded CD8+ T cell populations consist of shorter-lived terminal effectors (TEs) that display decreased persistence during the contraction phase, and a smaller proportion of memory precursors (MPs) that contribute to the self-renewing memory CD8+ T cell pool5–8. However, whether MP-like cell states exist within the innate immune system is unknown.
Natural killer (NK) cells are circulating group 1 innate lymphocytes (ILCs) that play a critical role during herpesvirus infection in mice and humans9–11. Although historically categorized as innate immune cells, circulating and tissue-resident group 1 ILCs can exhibit memory responses to mouse cytomegalovirus (MCMV)-associated glycoproteins through expression of germline encoded activating receptors12–14. Furthermore, NK cells exhibit clonal proliferation and persistence of a long-lived population of memory cells with enhanced protective capacity after secondary MCMV infection15, 16. In C57BL/6 mice, a subset of naive Ly49H+ NK cells initiate this adaptive response after recognition of the MCMV-encoded glycoprotein m15714, 17, 18. In both mice and humans, stochastically expressed germline-encoded activating and inhibitory receptors as well as developmental subsets generate naive NK cell diversity and heterogeneity during homeostasis12, 19, 20. Furthermore, heterogeneity within the naive Ly49H+ NK cell pool can influence NK cell responses to MCMV. This involves preferential expansion of NK cells with a history of recombination-activating gene (RAG) expression, NK cells that lack expression of killer cell lectin-like receptor G1 (KLRG1) or the inhibitory receptor NKR-P1B21–23, and NK cells with a longer history of NKp46 expression24. Although subsets of naive NK cells have been found to preferentially form Ly49H+ effector NK (NKeff) cells following MCMV infection, whether similar heterogeneity exists within expanded NKeff cells remains unresolved. Furthermore, whether a subset of NKeff cells preferentially survives to form memory cells, and the molecular mechanisms that regulate memory cell fate in NK cells remain poorly understood.
Using single cell RNA sequencing (scRNAseq), we identified an MP-like transcriptional state of NKeff cells following MCMV-driven expansion in vivo. We provide further evidence that Ly6C– NKeff cells preferentially survive the contraction phase to generate memory NK cells in a Bcl2-dependent manner. MP NK cells displayed distinct transcriptional and epigenetic signatures compared to Ly6C+ NKeff cells, with an overlapping epigenetic signature shared with MP CD8+ T cells enriched in ETS1 and Fli1 transcription factor binding motifs. Fli1 regulated MP NK cell formation by promoting Bim levels in early effector NK cells to decrease the fitness of expanding NK cells during viral infection. These results suggest that memory NK cells, similar to memory T cells, are generated by a subset of epigenetically distinct MP cells that preferentially survive during the contraction phase of the response to viral infection and identify Fli1 as a critical regulator of MP NK cell formation.
Results
Time-resolved scRNA-seq analysis reveals an MP-like NK cell state
To determine whether transcriptional heterogeneity exists within NKeff cells following viral infection, we adoptively transferred splenic Ly49H+ NK cells into recipient Klra8–/–(hereafter referred to as Ly49H–/–) mice and infected with MCMV. On day (D)7 post-infection (PI), adoptively transferred NKeff cells from the spleen were sorted and then profiled using 10x Genomics Chromium droplet scRNA-seq (Extended Data Fig. 1a). The resulting NKeff single cell dataset included 2,430 cells that were clustered based on differential expression of marker genes and visualized using a uniform manifold approximation and projection (UMAP) plot (Fig. 1a, Supplementary Table 1). Clustering analysis revealed 2 distinct clusters of NKeff cells defined by differential expression of transcription factors (Zeb2, Id2, Irf8, Runx3), regulators of cell survival (Gpx8, Shisa5, Bcl2), and cell surface proteins (Cx3cr1, Cd7, Ly6e) (Fig. 1c, Extended Data Fig. 1b). While previous studies have identified two subsets of naive mouse NK cells utilizing scRNA-seq19, these subsets did not account for the distinct clusters of NKeff cells identified within our dataset, because all Ly49H+ NK cells analyzed displayed a mature phenotype (CD27–CD11b+) on D7 PI (Extended Data Fig. 1c). Gene ontology enrichment analysis indicated that specific pathways were differentially regulated between the two NKeff cell clusters. Notably, significant terms related to regulation of apoptosis were enriched in cluster 1 in comparison to cluster 0, with cluster 1 expressing more transcripts of the pro-survival gene Bcl2 than cluster 0 (Fig. 1b,c). These results suggested that transcriptional heterogeneity existed within NKeff cells following viral infection, and that a specific effector cell state may preferentially persist during the contraction phase. Following MCMV-induced clonal proliferation, Ly49H+ NK cells undergo a contraction phase from D7 to D28 PI during which most NKeff undergo Bim-mediated apoptosis25. In order to address whether certain NKeff cell states could persist during the contraction phase, adoptively transferred Ly49H+ NK cells were sorted on D14 PI and profiled using scRNA-seq. Combining this dataset with Ly49H+ NK cells analyzed from D7 PI resolved 4,399 cells that formed six distinct clusters which were visualized by UMAP dimensional reduction (Fig. 1d, Supplementary Table 2). While the majority of cells comprising clusters 1,2 (Cx3cr1, Gpx8, Cma1) and 4 (Plac8, Gzmk, Thy1) were derived from D7 PI NKeff cells, Cluster 0 (Cx3cr1, Sell, Cma1) and 5 (CD69, Ifng, Nfkbia) predominantly contained cells from D14 PI NK cells and resembled terminally-differentiated and activated NK cells respectively (Fig. 1d, Extended Data Fig. 1d). Notably, cluster 3 (Shisa5, Bcl2a1b, Il2rb) contained a mix of cells from both D7 and D14 PI that were enriched in genes associated with response to stress and programmed cell death, suggesting that a distinct NKeff cell state may persist during the contraction phase (Fig. 1d, Extended Data Fig. 1d,e).
Figure 1. Single cell RNA sequencing reveals two clusters of NKeff cells following MCMV infection.
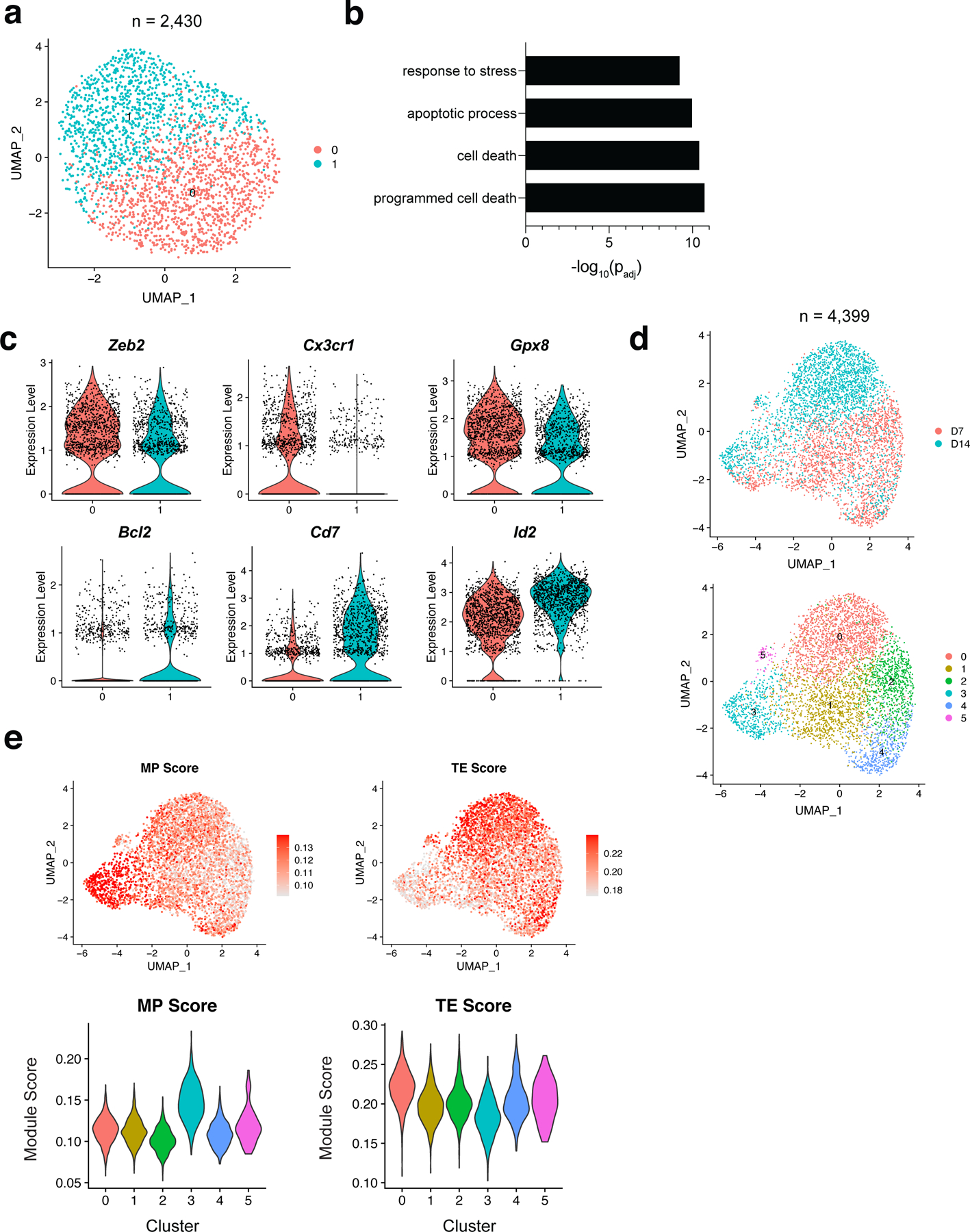
Ly49H+ NK cells were transferred into naive Ly49H–/– mice and infected with MCMV i.p. 16 hours later. TCRβ–CD3ε–NK1.1+Ly49H+KLRG1+ NK cells were sorted on D7 and D14 PI. Cells were immediately processed using 10x Genomics Chromium droplet single cell RNA sequencing. (a) UMAP plot of 2,430 D7 PI NKeff cells colored by cluster identities. Each dot represents an individual cell. (b) Gene Ontology (GO) enrichment analysis of upregulated genes in cluster 0 compared to cluster 1 from D7 PI NKeff cells. Terms were considered statistically significantly enriched if -log10(padj)<0.05. (c) Violin plots showing the relative expression of six differentially expressed genes in the two clusters of D7 PI NKeff cells. Each dot represents a cell. (d) UMAP plot of 4,399 cells combined from D7 and D14 PI NKeff cells, colored by timepoint analyzed (top) and cluster identity based on differential gene expression (bottom). Each dot represents an individual cell. (e) UMAP plot showing memory precursor (MP) and terminal effector (TE) CD8+ T cell gene module scores (top) and violin plots showing the distribution of MP and TE gene module scores for each cluster (bottom).
To test this hypothesis in silico, we utilized RNA velocity analysis26 to determine the time-resolved transcriptional fates of D7 PI NKeff cells (Extended Fig. 2a). Projection of the velocity field arrows onto the UMAP plot extrapolated future states of NKeff cells showing 4 distinct manually averaged trajectories (Extended Fig. 2b). Both clusters 2 and 4 transitioned through cluster 1 to cluster 3 (trajectory 1), while a subset of cluster 3 cells did not show directionality to any other cell state (trajectory 2) suggesting that cluster 3 represented a distinct cell state of NKeff (Extended Fig. 2a,b). RNA velocity analysis also suggested that cluster 0 represented an end stage of NKeff differentiation as both cluster 2 (trajectory 3) and a subset of cells from cluster 3 (trajectory 4) showed strong directionality toward this cell state (Extended Fig. 2a,b). Monocle pseudotime analysis further corroborated in silico trajectories 1 and 4, showing that cluster 1 likely represented a transition state between cluster 4 and 3 on D7 PI, and that cluster 3 cells differentiated to cluster 0 on D14 PI (Extended Fig. 2c). Trajectory 1 was defined by increased expression of Bcl2 and Il2rb, while trajectory 4 was defined by increased expression of genes associated with NK cell terminal maturation such as Zeb2 and Irf819, 27(Extended Fig. 2d). Because cluster 3 represented a distinct NKeff cell state that persisted during the contraction phase and could putatively differentiate to the majority of NK cells present on D14 PI, we then tested whether cluster 3 represented a MP-like NK cell state using MP and TE CD8+ T cell RNA-seq datasets described previously (GSE111902). Indeed, cluster 3 showed the highest enrichment for the MP CD8+ T cell gene expression module score, while cluster 0 scored the highest for the TE CD8+ T cell gene module score (Fig. 1e). Thus, these data suggest that D7 PI NKeff cells contain a mixture of MP-like and other NKeff cell states following viral infection.
Memory Ly6C+ NK cells are derived from Ly6C– MP NK cells
To determine whether a distinct MP NK cell subset could be identified in vivo, we analyzed cell surface expression of several genes predicted by our scRNA-seq dataset to be differentially expressed at D7 PI. While several cell surface proteins identified (CD44, Sca1, NKG2D, CD16) did not display bimodal expression in effector NK cells, there were clear positive and negative populations of Ly6C and CX3CR1 NKeff cells on D7 PI (Fig. 2a). To test whether either of these markers could identify a subset of NKeff cells with enhanced persistence during the contraction phase, we sorted congenically distinct Ly6C– and Ly6C+ or CX3CR1– and CX3CR1+ Ly49H+ NKeff cells at D7 PI and adoptively transferred equal mixtures of each into naive Ly49H–/– mice (Fig. 2b). While we did not observe a change in the frequencies of CX3CR1– and CX3CR1+ NKeff cells 12 days post-transfer (Extended Data Fig. 3a), there was a significant increase in the frequency of recovered Ly6C– compared to Ly6C+ NK cells on D12 following transfer. (Fig. 2c). These results were not due to differences in trafficking or proliferation of adoptively transferred Ly6C– NKeff cells, as the percentage of adoptively transferred Ly6C– and Ly6C+ Ly49H+ NK cells remained similar in peripheral organs at various timepoints PI, and we did not observe differences in the frequencies of Ki-67+ Ly6C– or Ly6C+ Ly49H+ NK cells on D7, D10, or D14 PI (Extended Data Fig. 3b–d). We also did not observe differences in single cell metabolic heterogeneity using SCENITH28, or differential induction of mitophagy through analysis of TMRE and MitoGreen staining between Ly6C+ and Ly6C– NKeff at various timepoints during the contraction phase29 (Extended Data Fig. 4a,b). Furthermore, D14 PI Ly6C+ and Ly6C– NKeff cells both displayed enhanced production of IFN-γ upon platebound antibody stimulation, and adoptively transferred D7 PI Ly6C+ and Ly6C– NKeff cells both rescued Ly49H–/– mice from lethal MCMV challenge (Extended Data Fig. 4c,d). The observed enhanced survival phenotype was found to be intrinsic to Ly6C– NKeff cells, as co-adoptive transfer of congenically distinct Ly6C– and Ly6C+ NKeff cells led to a higher ratio of Ly6C– to Ly6C+ NK cells 12 days post transfer into distinct Ly49H–/– hosts that previously received adoptively transferred naive NK cells and were infected with MCMV (Fig. 2d,e). These results suggest that Ly6C– NKeff cells persist to a greater extent than Ly6C+ NKeff cells during the contraction phase to preferentially form memory NK cells.
Figure 2. Ly6C– NKeff cells preferentially persist following MCMV infection.
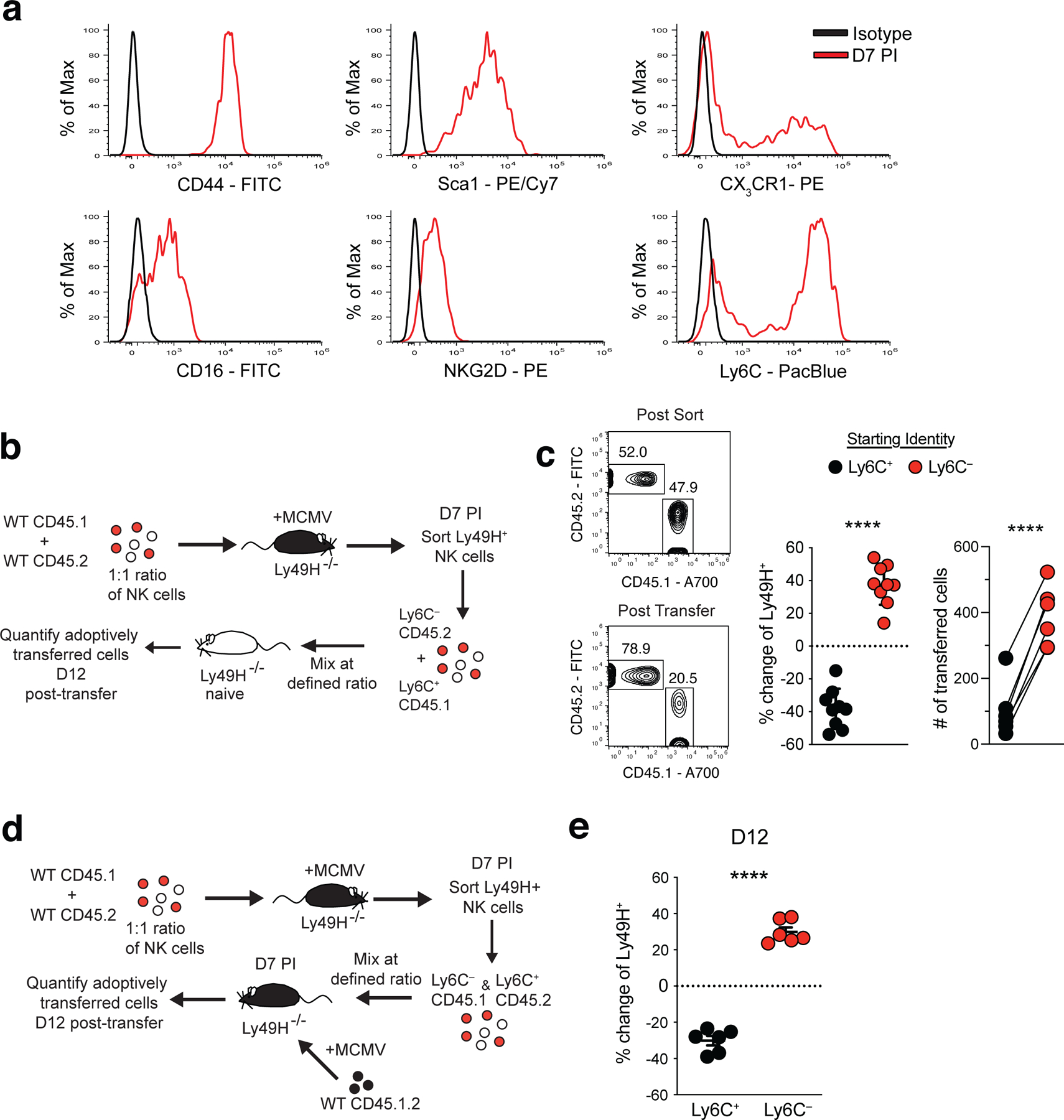
Splenic Ly49H+ NK cells were transferred into Ly49H–/– mice i.v. and infected i.p with MCMV 16 hours after adoptive transfer. (a) Histograms of indicated cell surface marker expression from D7 PI Ly49H+ NKeff cells (red) compared to isotype controls (black). (b) Experimental schematic. A mixture of purified WT splenic CD45.1 and CD45.2 Ly49H+ NK cells were transferred into Ly49H–/– mice and infected i.p with MCMV 16 hours after adoptive transfer. On D7 PI, TCRβ–CD3ε–NK1.1+Ly49H+KLRG1+CD45.1+Ly6C+ and TCRβ–CD3ε–NK1.1+Ly49H+KLRG1+CD45.2+Ly6C– NKeff were sorted and then transferred into naive Ly49H–/– hosts at a 1:1 ratio. Recipient spleens were harvested 12 days post transfer and analyzed by flow cytometry. (c) Representative flow cytometry plots and statistical analysis of co-adoptively transferred Ly6C+ (CD45.1) and Ly6C– (CD45.2) Ly49H+ NK cell subsets before transfer (post sort) and harvested from recipient Ly49H–/– mice on D12 (post transfer). Change from post sort frequency (middle) and numbers (right) of recovered cells 12 days post transfer. (d) Experimental schematic. Indicated CD45.1 and CD45.2 NKeff cell subsets were transferred into Ly49H–/– hosts that were previously adoptively transferred with CD45.1 x CD45.2 naive NK cells and infected with MCMV 7 days prior. (e) Quantification of the change in frequency of co-adoptively transferred Ly6C+ (CD45.1) and Ly6C– (CD45.2) Ly49H+ NK cells subsets harvested from recipient mice on D12 (post transfer). Data are representative of 2–3 independent experiments with n = 3 mice per group. Samples were compared using two-tailed Student’s t test with Welch’s correction, assuming unequal SD, and data points are presented as individual mice with the mean ± SEM (***p < 0.001, ****p < 0.0001).
Previous studies have suggested >95% of MCMV-induced memory NK cells are Ly6C+ on D28 PI30, 31. However, our adoptive transfer data suggested that Ly6C– NKeff give rise to the majority of memory NK cells present 12 days following transfer, suggesting that Ly6C+ NKeff likely do not represent MP NK cells (Fig. 2c,e). Notably, RNA velocity analysis indicated that a subset of MP-like NK cells transitioned to a terminal cell state that represented the majority of memory NK cells on D14 PI (Extended Data Fig. 2a–d). These results suggested that the Ly6C– NKeff population was likely enriched in the MP-like NK cell state identified by our scRNA-seq analysis. In support of this hypothesis, the percentage and numbers of adoptively transferred Ly6C– Ly49H+ NK cells declined over time in the blood (Fig. 3a–c) and peripheral organs by D28 PI (Extended Data Fig. 4e). Further examination of adoptively transferred Ly6C+ and Ly6C– NKeff cells demonstrated that while a majority of the surviving Ly6C+ memory NK cells were derived from D7 PI Ly6C– NKeff cells 12 days post transfer, ~80% of D7 Ly6C– NKeff cells upregulated Ly6C while Ly6C+ NKeff did not lose expression of Ly6C (Fig. 3d,e). These results suggest that D7 PI Ly6C– NKeff cells (referred to as MP NK cells hereafter) preferentially persist and differentiate into Ly6C+ memory NK cells during the contraction phase of the response to MCMV.
Figure 3. Ly6C– NKeff cells differentiate into Ly6C+ memory NK cells.
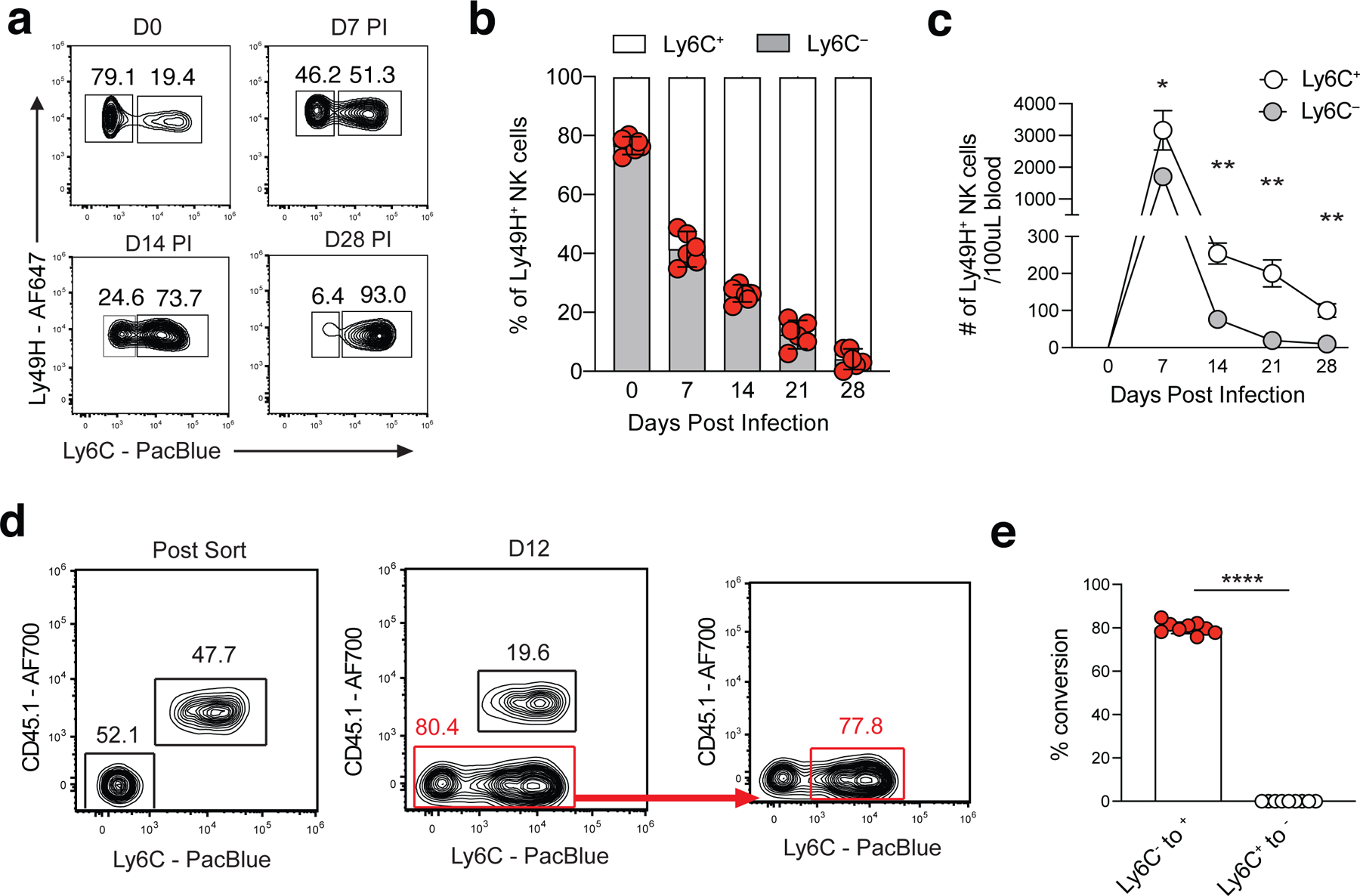
(a-c) Ly49H+ NK cells were adoptively transferred into Ly49H–/– mice and infected with MCMV i.p. 16 hours later. (a) Representative flow plots and (b) frequency of adoptively transferred Ly49H+ Ly6C– NK cells at indicated timepoints following MCMV infection. (c) Absolute numbers of Ly6C+ and Ly6C– Ly49H+ NK cells in the blood at indicated timepoints post MCMV infection. From left to right, p = 0.021392, p = 0.001225, p = 0.003876, p = 0.003876. (d) Representative flow plots showing Ly6C expression in indicated adoptively transferred NKeff cell subsets from Fig. 2C on D12 post transfer. (e) Frequency of Ly6C– to Ly6C+ conversion or vice versa at D12 post transfer. Data are representative of 2–3 independent experiments with a-c,e: n = 3 mice per group. Samples were compared using two-tailed Student’s t test with Welch’s correction, assuming unequal SD, and data points are presented as individual mice with the mean ± SEM (*p < 0.05, **p < 0.01, ****p < 0.0001).
Bcl2 is required for the survival of NKeff cells during contraction
To determine the mechanisms of MP NK cell persistence following MCMV infection, we adoptively transferred CD45.1+Ly49H+ NK cells into Ly49H–/– mice, infected with MCMV, sorted Ly6C+ and Ly6C– Ly49H+ NK cells from the spleens of the Ly49H–/– mice on D7 PI and performed RNA sequencing (RNA-seq). RNA-seq analysis identified 43 differentially expressed genes (DEGs) between Ly6C+ NKeff and MP NK cells, with MP NK cells displaying increased expression of Bcl2 and the transcription factor Zbtb33 (Fig. 4a,b and Supplementary Table 3). Conversely, Ly6C+ NKeff cells displayed increased expression of the cell cycle inhibitor Cdkn1b (Fig. 4b). Analysis of common DEGs between MP vs TE CD8+ T cells and MP NK cells vs Ly6C+ NKeff cells revealed higher expression levels of Bcl2 between MP CD8+ T cells and MP NK cells (Extended Fig. 5a). However, MP CD8+ T cells did not share an overlapping core transcriptional signature with MP NK cells. Instead, MP CD8+ T cells expressed high levels of Tcf7, Id3, P2rx7, and Espn, which have been associated with early memory T cell progenitor stemness programs 32 (Extended Fig. 5b). In confirmation of our RNA-seq dataset, MP NK cells expressed higher levels of the pro-survival molecule Bcl233, whereas pro-apoptotic Bim levels were similar when compared to Ly6C+ NKeff (Fig. 4c). Notably, bulk D7 PI NKeff cells displayed lower levels of Bcl2 compared to naive NK cells (Fig. 4d,e), and were highly dependent on Bcl2 activity for survival as evidenced by increased release of intracellular cytochrome c following Bcl2 inhibition ex vivo (Fig. 4f).To determine whether Bcl2 was required for the persistence of Ly49H+ NK cells during the contraction phase, we generated high efficiency CRISPR ribonucleoprotein (cRNP)-mediated deletion of Bcl2 in primary mature NK cells34 (Fig. 4g). Bcl2 cRNP-edited naive (CD45.2) Ly49H+ NK cells were co-adoptively transferred with control Rosa26-edited (CD45.1) Ly49H+ NK cells into Ly49H–/– mice and subsequently infected with MCMV. We observed a significant survival defect in Bcl2-deficient NK cells during the contraction phase (D7-D28 PI) (Fig. 4h). Together, these data suggest that higher levels of Bcl2 in MP NK cells may contribute to their enhanced persistence during the contraction phase of the response to MCMV infection.
Figure 4. MP NK cells are transcriptionally distinct and require Bcl2 for optimal survival during the contraction phase of the response to MCMV.
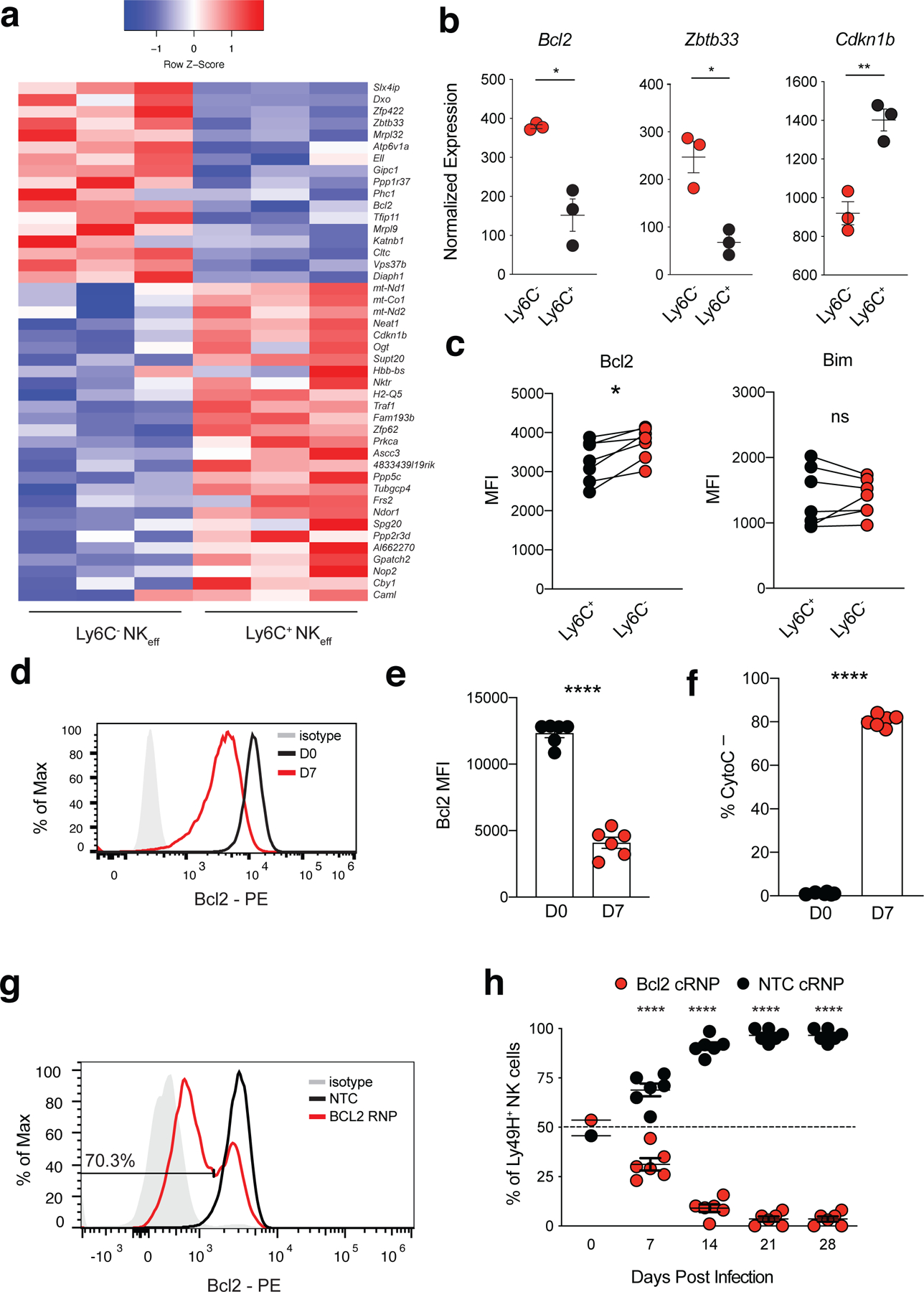
(a-b) Splenic Ly49H+ NK cells were transferred into Ly49H–/– mice i.v. and infected i.p with MCMV 16 hours after adoptive transfer. Splenic TCRβ–CD3ε–NK1.1+KLRG1+Ly49H+Ly6C+ and TCRβ–CD3ε–NK1.1+KLRG1+Ly49H+Ly6C– NKeff were sorted from recipient mice on D7 PI. Sorted NK cells were immediately processed for mRNA extraction, library preparation and sequencing. (a) Heatmap showing differentially expressed genes between Ly6C+ and Ly6C– D7 PI NKeff cells (padj < 0.05). (b) Normalized read counts of selected genes shown in (a). Left to right, p = 0.0304, p = 0.0183, p = 0.0043. (c) MFI of Bcl2 and Bim in D7 PI Ly6C+ and Ly6C– NKeff cells. Bcl2, p = 0.0180. (d) Representative histogram and (e) MFI of intracellular Bcl2 expression in naive (D0) and D7 PI Ly49H+ NKeff cells. (f) Quantification of the frequency of intracellular cytochrome c negative cells following Bcl2 inhibition with ABT-199 in naive and D7 PI Ly49H+ NKeff cells ex vivo. (g,h) IL-15 pre-activated congenically distinct NK cells were electroporated in the presence of either Rosa26 NTC cRNP (CD45.1) or Bcl2 cRNP (CD45.2) before being transferred i.v. into Ly49H-deficient recipients at a 1:1 ratio. (g) BCL2 protein expression shown by histogram of Bcl2 cRNP or Rosa26 NTC RNP CRISPR edited NK cells 3 days post gene edit ex vivo. (h) Recipient mice were infected i.p with MCMV 16 hours after adoptive transfer. Quantification of adoptively transferred Ly49H+ NTC cRNP or Bcl2 cRNP-edited NK cells in the blood of recipient mice at various timepoints PI. (b) Data are presented as individual biological replicates, n = 3 mice per group. (c,e,f,h) Data are representative of 2 independent experiments with n = 3 mice per group. Experiments were repeated 3 times total. Samples were compared using two-tailed Student’s t test with Welch’s correction, assuming unequal SD, and data points are presented as individual mice with the mean ± SEM ( *p < 0.05, **p < 0.01, ****p < 0.0001).
MP NK cells display a distinct epigenetic signature
To determine the mechanisms of MP NK cell formation following MCMV infection, we adoptively transferred CD45.1+Ly49H+ NK cells into Ly49H–/– mice, infected with MCMV, sorted Ly6C+ and Ly6C– Ly49H+ NK cells from the spleen of the Ly49H–/– mice on D7 PI and performed ATAC sequencing (ATAC-seq). ATAC-seq analysis identified 811 significantly differentially accessible (DA) peaks between Ly6C+ NKeff and MP NK cells (Fig. 5a, Supplementary Table 4). Using gene ontology analysis, we found that MP NK cells displayed an increase in accessible peaks associated with genes implicated in MAPK signaling while Ly6C+ NKeff cells showed increased accessibility in genes associated with the endosome and golgi (Fig. 5b). Next, we generated a core MP lymphocyte epigenetic signature consisting of 114 peaks that were significantly DA in MP CD8+ T and MP NK cells compared to TE CD8+ T cells and Ly6C+ NKeff (Fig. 5c, Supplementary Table 5). Peaks with greater accessibility in MP lymphocytes were associated with genes enriched in positive regulation of lymphocyte activation and differentiation such as the transcription factor Lef1, the chemokine receptor Cxcr3, and Bcl2 (Fig. 5d and Extended Data Fig. 6a–c). Together, these results suggest that MP NK cells display a common epigenetic signature shared with MP CD8+ T cells that is enriched in genes associated with T cell memory formation and effector function. To determine core transcriptional regulators of MP lymphocyte formation, we performed HOMER motif analysis of DA peaks shared between MP CD8+ T cells and MP NK cells. We found that MP DA peaks were enriched in ETS1 and Fli1 binding motifs compared to Ly6C+ NKeff and CD8+ TE cell DA peaks that were enriched in Tbet, Runx1 and Runx2 binding motifs (Fig. 5e). These data indicated that MP NK cells display a distinct epigenetic signature compared to Ly6C+ NKeff, and share a core epigenetic signature with MP CD8+ T cells.
Figure 5. MP NK cells share a core epigenetic signature with MP CD8+ T cells.
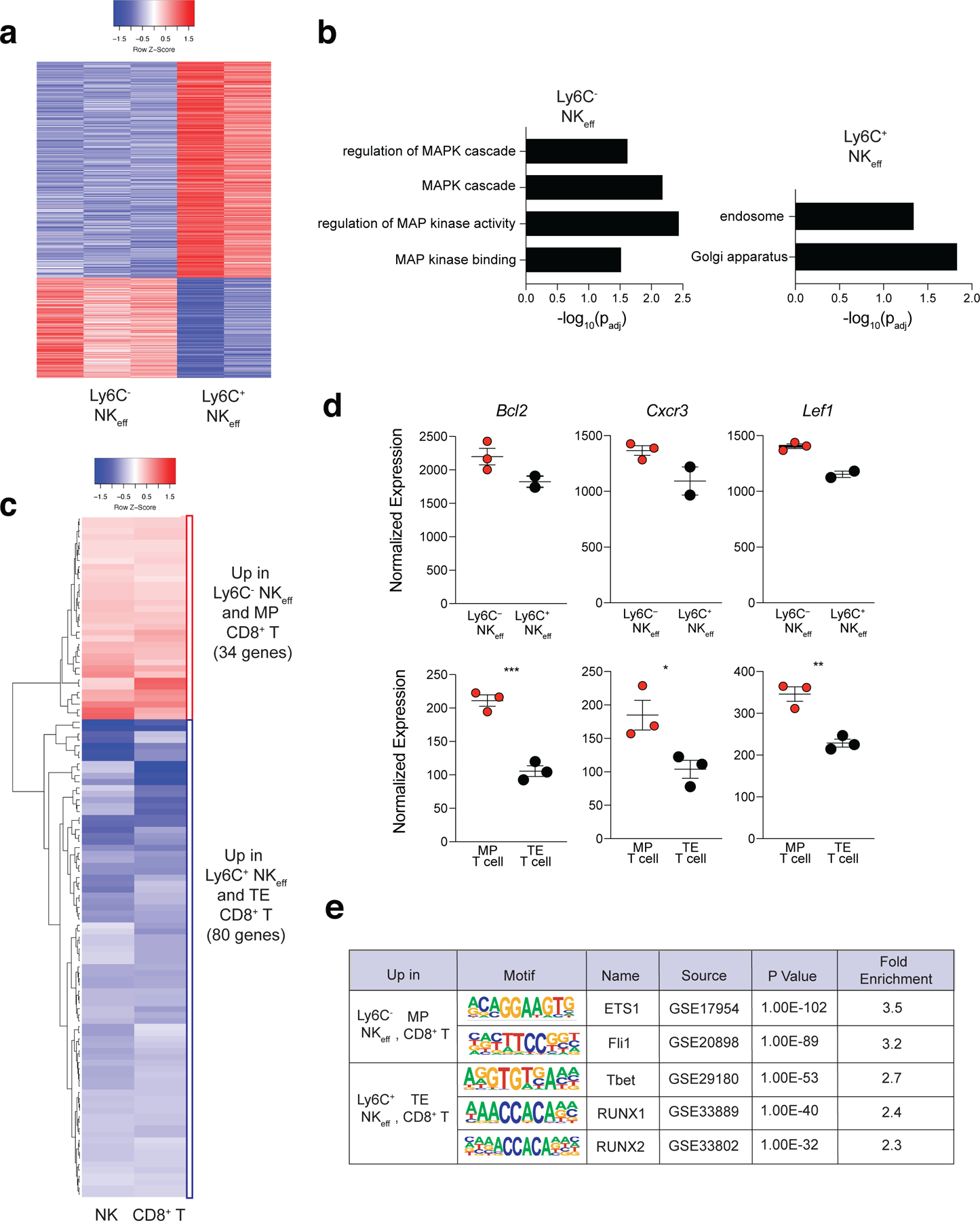
Splenic Ly49H+ NK cells were adoptively transferred into Ly49H–/– mice i.v. and infected i.p with MCMV 16 hours after adoptive transfer. Splenic TCRβ–CD3ε–NK1.1+KLRG1+Ly49H+Ly6C+ and TCRβ–CD3ε–NK1.1+KLRG1+Ly49H+Ly6C– NKeff were sorted from recipient mice on D7 PI. Sorted NK cells were immediately processed for ATAC library preparation and sequencing. (a) Heatmap showing differentially accessible peaks between Ly6C+ and Ly6C– NKeff cells. Peaks with padj < 0.15 were plotted. (b) GO enrichment analysis on genes related to differentially accessible peaks in Ly6C+ and Ly6C– NKeff cells. Terms were considered statistically significantly enriched if -log10(padj)<0.05. (c) Heatmap showing common differentially accessible peaks in the comparisons of Ly6C– to Ly6C+ NKeff cells and MP to TE CD8+ T cells (padj <0.05). (d) Normalized read counts of peaks related to selected genes which have differential accessibility between indicated NKeff cells (top) and T cells (bottom). Bottom row, left to right p = 0.0008, p = 0.0358, p = 0.0040. (e) HOMER motif analysis displaying common transcription factor binding site motifs in differentially accessible peaks between D7 PI Ly6C– NKeff and MP CD8+ T cells and D7 PI Ly6C+ NKeff and TE CD8+ T cells. Data are representative of n = 3 mice per group. Samples were compared using two-tailed Student’s t test with Welch’s correction, assuming unequal SD, and data points are presented as individual mice with the mean ± SEM ( *p < 0.05, **p < 0.01, ***p <0.001).
Our ATAC-seq analysis suggested that Fli1 may be a critical transcriptional regulator of MP NK cells. Therefore, we examined whether mature NK cells express Fli1 during MCMV infection. RNA-seq analysis demonstrated that Fli1 was repressed early during viral infection, but subsequently increased during the expansion phase of the response on D4 PI (Fig. 6a). A recent study found that IL-2 and IL-15 levels remain elevated in MCMV-infected tissues until D4 PI35, suggesting that these cytokines may induce Fli1 expression. Using the RNA-seq datasets from this study, we determined that splenic NK cells stimulated with IL-2 and IL-15 ex vivo induced Fli1 transcripts while IL-12 and IL-18 stimulated NK cells decreased Fli1 expression (Fig 6b). To determine whether IL-2 or IL-15 induce Fli1 levels in mature NK cells, we examined Fli1 protein by immunoblot of purified splenic NK cells after 48 h culture with increasing doses of IL-2 or IL-15. While we observed that Fli1 was induced by both IL-2 and IL-15 in a dose-dependent manner in splenic NK cells, IL-15 induced more Fli1 protein than IL-2 at each dose (Fig 6c). Further analysis of previously published ATAC-seq and STAT5 ChIP-seq data sets35 revealed that IL-2 and IL-15 stimulation of splenic NK cells increased accessibility of peaks within the Fli1 locus that overlapped with enriched STAT5 ChIP-seq peaks, suggesting that STAT5 may directly induce Fli1 expression in NK cells in response to IL-2 or IL-15 stimulation ex vivo (Fig 6d). To test this possibility, we stimulated splenic NK cells with IL-15 in the presence of a STAT5 inhibitor or vehicle control. Indeed, STAT5 inhibition resulted in a ~50% reduction of Fli1 protein (Fig 6e), suggesting that Fli1 protein levels are induced in mature NK cells by STAT5 signaling via IL-15 and/or IL-2 ex vivo.
Figure 6. Fli1 is induced by IL-15-mediated STAT5 signaling in mature NK cells.
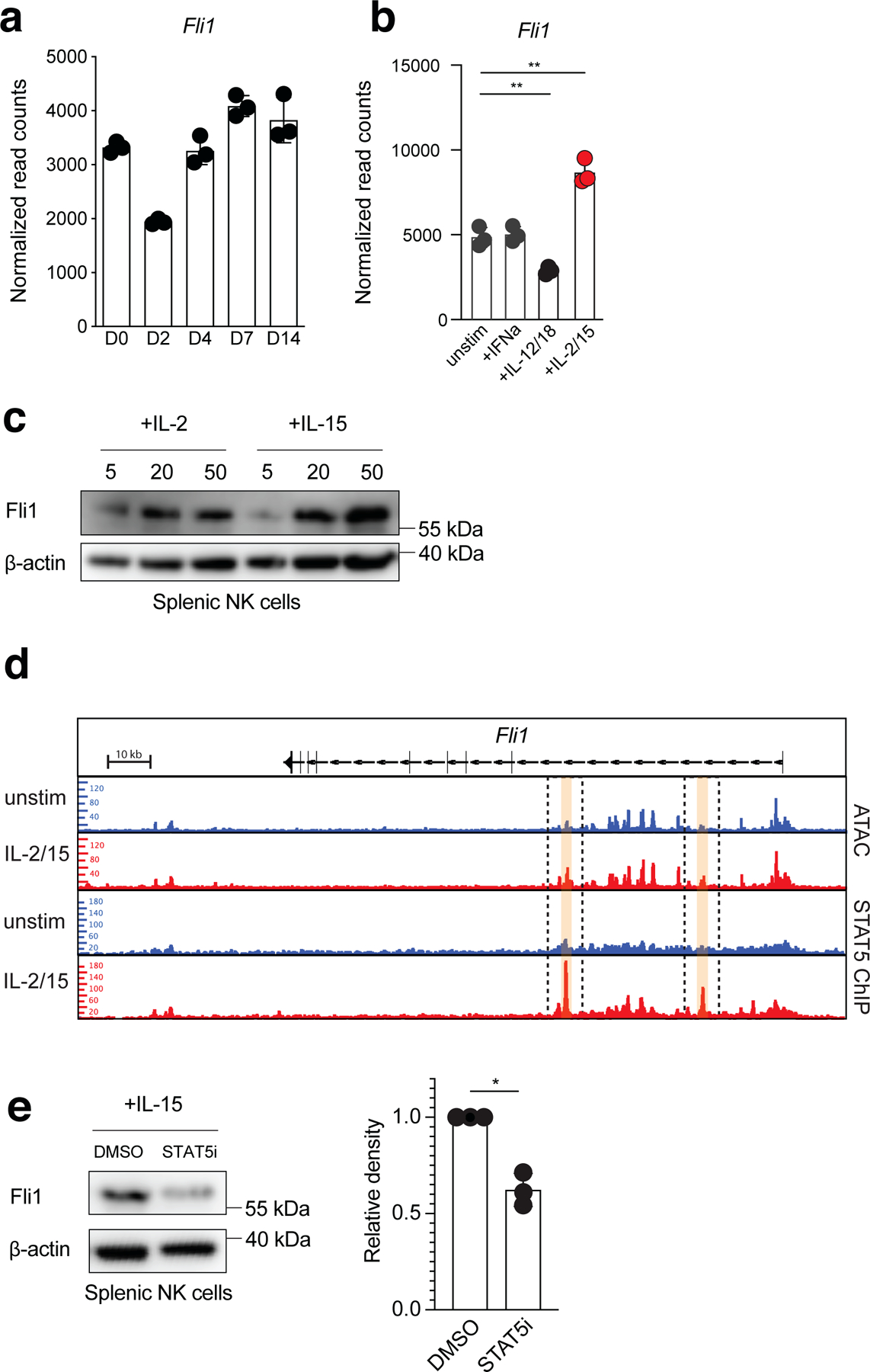
(a) Normalized read counts from bulk RNA-seq analysis of Ly49H+ NK cells at indicated time points PI. (b) Normalized read counts of Fli1 in splenic NK cells 3 hours after indicated cytokine stimulation ex vivo. Unstimulated vs +IL-2/IL-15, p = 0.0022. Unstimulated vs +IL-12/18, p = 0.0052. (c) Western blot showing Fli1 and β-actin loading control protein levels in unstimulated, IL-2-stimulated, or IL-15-stimulated splenic NK cells. (d) Upper two rows, ATAC-seq peaks in the Fli1 locus; Lower two rows, STAT5 ChIP-seq peaks in the Fli1 locus in unstimulated or IL-2/IL-15-stimulated splenic NK cells. (e) Western blot showing Fli1 and β-actin loading control protein levels in IL-15-stimulated + DMSO, or IL-15 stimulated + STAT5 inhibitor (CAS 285986–31-4) treated splenic NK cells. Samples were compared using two-tailed Student’s t test with Welch’s correction, assuming unequal SD. p = 0.0168. (a,b) Data points are presented as individual mice, n = 3 mice per group, with the mean ± SEM ( *p < 0.05, **p < 0.01). (c,e) Data represent n = 3 mice pooled per data point with the mean ± SEM ( *p < 0.05, **p < 0.01).
Fli1 increases BIM levels in early NKeff to limit MP NK cell formation
To determine the significance of Fli1 in regulating MP NK cells, we ablated Fli1 in mature NK cells using CRISPR cRNPs (Fig. 7a). We then adoptively transferred control or Fli1-edited Ly49H+ NK cells mixed at a 1:1 ratio into Ly49H–/– mice and infected with MCMV 16 hours later. After D3 PI, Fli1-deficient NK cells represented a greater frequency of adoptively transferred Ly49H+ NK cells at all time points analyzed and persisted in greater numbers in the blood and peripheral organs on D28 PI (Fig. 7b–d). While the frequency of Fli1-deficient NKeff cells increased compared to control edited cells on D7 PI, there was a greater impact of Fli1-deficiency on the frequency of MP NK cells compared to Ly6C+ NKeff. These results suggested that Fli1 restricts the formation of MP NK cells in addition to limiting NKeff formation (Fig. 7e). We hypothesized that the effects of Fli1 loss could be attributed to either increased proliferation or prolonged survival of adoptively transferred Ly49H+ cells. Analysis of adoptively transferred Fli1-deficient Ly49H+ NK cells on D3 PI did not reveal differences in CTV dilution, proliferation kinetics, or Ki-67 staining (Fig. 7f–h), suggesting that Fli1 does not control proliferation of Ly49H+ NK cells following viral infection. Instead, examination of Bim and Bcl2 protein by flow cytometry revealed that Fli1-deficient NK cells expressed significantly decreased levels of pro-apoptotic Bim versus control edited NK cells, while displaying similar levels of Bcl2 on D3 PI (Fig. 7i). This increased Bcl2/Bim ratio in Fli1-deficient NK cells coupled with their increased persistence in vivo suggested that Fli1 restricts the fitness of early effector NK cells through regulation of Bim levels during viral infection (Extended Data Fig. 7a,b).
Figure 7. Fli1 restricts the formation of MP NK cells during viral infection.
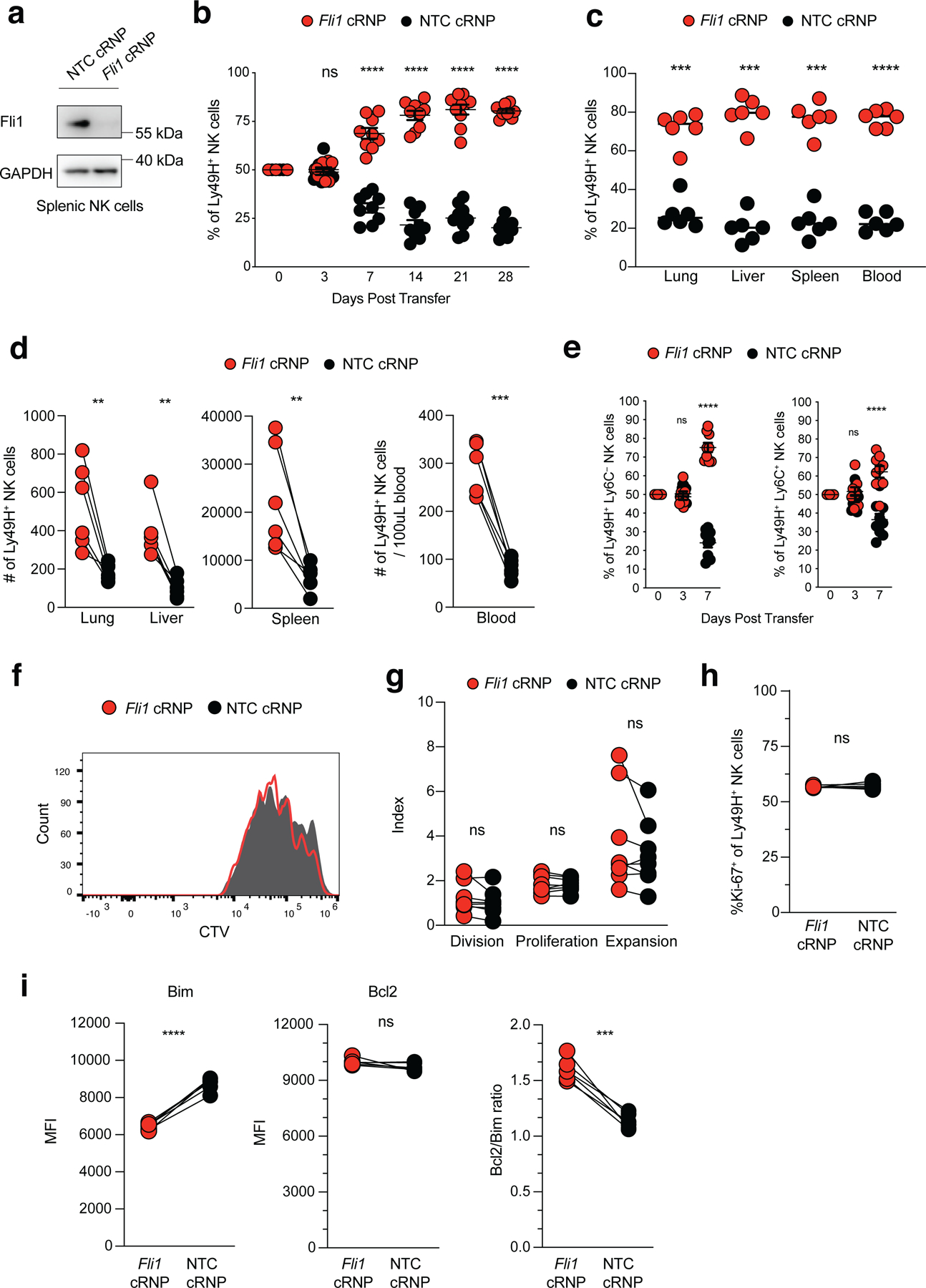
(a) Protein levels of Fli1 shown by western blot of Fli1 cRNP or Rosa26 NTC cRNP CRISPR edited NK cells 3 days after editing ex vivo. (b-i) IL-15 pre-activated congenically distinct NK cells were electroporated in the presence of either Rosa26 NTC cRNP (CD45.1) or Fli1 cRNP (CD45.2) before being transferred i.v. into Ly49H-deficient recipients at a 1:1 ratio. Recipient mice were infected i.p with MCMV 16 hours after adoptive transfer. (b) Quantification of adoptively transferred Ly49H+ NTC cRNP or Fli1 cRNP-edited NK cells in the blood of recipient mice at various timepoints PI. (c) The percentage and (d) number of Ly49H+ NTC cRNP or Fli1 cRNP-edited NK cells are quantified in the spleen, lung, liver, and blood of D28 recipient mice. For (c), lung p = 0.000999, liver p = 0.000192, spleen p = 0.000387, and blood p = 0.000028. For (d), lung p = 0.007766, liver p = 0.001366, spleen p = 0.0074, blood p = 0.0001. (e) The percentage of Ly6C– or Ly6C+ NK cells is shown in Ly49H+ NTC cRNP or Fli1 cRNP-edited NK cells at various timepoints PI. (f,g) Fli1 cRNP or Rosa26 NTC cRNP CRISPR edited NK cells were labeled with CTV dye before being transferred i.v. into Ly49H-deficient recipients and splenic NK cells were isolated on D3 PI. (f) Representative histograms displaying CTV dilution of Fli1 cRNP and NTC cRNP Ly49H+ NK cells. (g) Quantification of division, proliferation and expansion index calculated using FlowJo v10.7.2 software. (h,i) Splenic NK cells were isolated Day 3 PI. Dot plots displaying quantification of (h) %Ki-67+ and (i) MFI of Bim, Bcl2 and Bcl2/Bim ratio in Fli1 cRNP and NTC cRNP Ly49H+ NK cells. Bcl2/Bim p = 0.0002. (a) Data represent 2 independent experiments with: n = 3 mice per group. (b,e,i) Data represent 3 independent experiments with: n = 3 mice per group. (c,d,h) Data represent 2 independent experiments with: n = 3 mice per group. Samples were compared using two-tailed Student’s t test with Welch’s correction, assuming unequal SD, and data are presented as individual mice with the mean ± SEM (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001)
Discussion
We utilized single cell sequencing to identify a MP-like NK cell state following mouse cytomegalovirus (MCMV) infection. Ly6C– NKeff cells displayed enhanced survival during the contraction phase and were determined to be the main precursors of Ly6C+ memory NK cells. MP NK cells displayed distinct transcriptional and epigenetic signatures compared to Ly6C+ NKeff cells, with increased protein expression of Bcl2. While Bcl2 was required for the survival of Ly49H+ NK cells during the contraction phase, STAT5 signaling likely induced Fli1 in early effector NK cells to increase Bim levels and restrict the formation of MP NK cells.
Time-resolved trajectory analysis of our scRNA-seq data suggested that a subset of D7 PI NKeff cells transition to a cell state enriched in the MP CD8+ T cell transcriptional signature. Although bulk RNA-seq analysis demonstrated that MP CD8+ T cells and MP NK cells did not display overlapping gene signatures, they both display higher expression of Bcl2 transcript and protein, which is required for effector T cells to preferentially survive during the contraction phase of the anti-viral response36, 37. These data suggest that Mcl1 expression, which is required for NK cell survival during homeostasis33, 38, is not sufficient to inhibit apoptosis in NKeff cells due to the decreased levels of Bcl2 observed on D7 PI. The discrepancy between our bulk RNA-seq and scRNA-seq data may be explained by the possibility that D7 PI Ly6C– NKeff cells contain a mixture of transitional NKeff and MP NK cells rather than a homogenous MP cell state identified by scRNA-seq. Furthermore, because a small frequency of adoptively transferred Ly6C+ NKeff persist during the contraction phase, it is possible that D7 PI Ly6C+ NKeff contain a small fraction of transitional NKeff that survive to generate memory NK cells on D14 PI. Irrespective of these points, our adoptive transfer data suggest that the majority of memory NK cells present on D19 PI are derived from D7 PI Ly6C– NKeff cells, making this a functional MP population.
RNA velocity analysis suggested that MP-like NK cells undergo continuous differentiation to a terminally differentiated state on D14 PI, which could explain the constant decay of memory NK cell numbers observed in all studies to date following the expansion phase in response to MCMV infection39. Indeed, monocle trajectory analysis identified Zeb2 expression increasing towards the terminally differentiated memory NK cell state on D14 PI, and Zeb2 has been implicated in the terminal maturation of naive NK cells during homeostasis as well as the terminal differentiation of effector CD8+ T cells following viral infection27, 40, 41. However, our results indicate that Zeb2 is required for the expansion of NKeff cells, but not the terminal differentiation of MP-like NK cells (data not shown). Similarly, Id2 has been shown to maintain the terminal differentiation of effector CD8+ T cells through sustained repression of central memory-associated transcriptional programs in addition to regulating NK cell development and epigenetic regulation of effector functionality42–45. While we observed higher expression of Id2 in the D7 PI MP-like NK cell cluster from our scRNA-seq analysis, we did not observe an increase in MP NK cells following inducible deletion of Id2 during the contraction phase following MCMV infection (data not shown). Together, these findings suggest that there may be important epigenetic differences between effector NK and CD8+ T cells that dictate lineage specific terminal differentiation programs, although future studies will be necessary to support this hypothesis.
Our ATAC-seq dataset revealed that Ly6C+ NKeff and TE CD8+ T cells share common enrichment in Runx1, Tbet, and Runx2 binding motifs, with Ly6C– NKeff and MP CD8+ T cells show enrichment for Fli1 and ETS1 binding motifs. While Tbet and Runx1 are important for NK cell survival during homeostasis and MCMV infection46, 47, the precise roles of these transcription factors during NKeff terminal differentiation will need to be studied in further detail. In mouse NK cells, ETS-1 has been shown to be required for mature NK cell development, and can be induced by IL-2/IL-15 signaling in human NK cells48, 49. Similarly, we found that STAT5 signaling induced expression of Fli1 in mature mouse NK cells in response to IL-2 and IL-15 ex vivo. Interestingly, CRISPR-mediated deletion of Fli1 in mature naive NK cells leads to decreased Bim levels and a greater persistence of early effector NK cells following MCMV infection, with a larger proportion of MP NK cells persisting through the contraction phase as a result. This finding suggests that Fli1 acts as a repressor of MP NK cell formation, likely to limit bystander immunopathology of expanded Ly49H+ NK cells following clonal proliferation. Similarly, Fli1-deficient CD8+ T cells accumulate more effector cells following LCMV infection50, implicating Fli1 as a critical intrinsic checkpoint regulator of effector lymphocyte formation. While future studies will be needed to determine the epigenetic and/or transcriptional mechanisms by which Fli1 represses early effector lymphocyte fitness, these results identify an important regulator of effector lymphocyte formation. Thus, understanding the transcriptional and epigenetic pathways that induce MP states in NK cells could inform strategies aimed at enhancing adaptive NK cell adoptive immunotherapies.
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed and will be fulfilled by the Lead Contact, Timothy O’Sullivan (tosullivan@mednet.ucla.edu)
Method Details
Mice
Mice were bred at UCLA in accordance with the guidelines of the Institutional Animal Care and Use Committee (IACUC). The following mouse strains were used this study: C57BL/6 (CD45.2) (Jackson Labs, #000664), B6.SJL (CD45.1) (Jackson Labs, #002114), Klra8–/– (Ly49H-deficient). Experiments were conducted using 6–8 week old age- and gender-matched mice in accordance with approved institutional protocols.
MCMV infection
MCMV (Smith) was serially passaged through BALB/c hosts three times, and then salivary gland viral stocks were prepared with a dounce homogenizer for dissociating the salivary glands of infected mice 3 weeks after infection. Experimental mice in studies were infected with MCMV by i.p. injection of 7.5 x 103 plaque-forming units (PFU) in 0.5 mL of PBS. In other experiments, Ly49H–/– mice were intravenously injected with 25,000 sort purified D7 PI Ly49H+ NK cells or PBS as control. 7 days later, recipient mice were infected with MCMV by i.p. injection of 7.5 x 103 plaque-forming units (PFU) in 0.5 mL of PBS. Mice were monitored and weighed daily and sacrificed when body weight decreased 10% from initial weight.
Isolation and enrichment of mouse NK cells
Mouse spleens, livers, lungs, lymph nodes, and blood were harvested and prepared into single cell suspensions as described previously11. Splenic single cell suspensions were lysed in red blood cell lysis buffer and resuspended in EasySep™ buffer (Stemcell). To avoid depleting Ly6C+ NK cells we developed a custom antibody cocktail as follows: splenocytes were labeled with 10 μg per spleen of biotin conjugated antibodies against CD3ε (17A2), CD19 (6D5), CD8 (53–6.7), CD88 (20/70), Ly6G (1A8), SiglecF (S17007L), TCRβ (H57–597), CD20 (SA275A11), CD172a (P84) and magnetically depleted from total splenocyte suspensions with the use of anti-biotin coupled magnetic beads (Biolegend).
Cell sorting and Adoptive Transfer Experiments
Isolated splenic NK cells were sorted using Aria-H Cytometer. NK cells were sorted to > 95% purity. Approximately 2 × 105 enriched NK cells were injected intravenously into mice. In adoptive co-transfer experiments, equal numbers of Ly49H+ NK cells from each population (CD45.1+ and CD45.2+) were injected into recipients 16 hours prior to MCMV infection. In other experiments, TCRβ–CD3ε–NK1.1+Ly49H+KLRG1+CD45.1+CX3CR1+ and TCRβ–CD3ε–NK1.1+Ly49H+KLRG1+CD45.2+CX3CR1– or TCRβ–CD3ε–NK1.1+Ly49H+KLRG1+Ly6C+ and TCRβ–CD3ε–NK1.1+Ly49H+KLRG1+Ly6C– D7 PI NKeff were sorted and then transferred at a 1:1 ratio into naive or D7 PI Ly49H–/– hosts that had received CD45.1 x CD45.2 Ly49H+ NK cells 7 days prior to MCMV infection. Adoptively transferred cells were recovered by harvesting recipient mouse splenic NK cells, preforming magnetic enrichment, and analysis by flow cytometry at various time points post-transfer.
Guide RNA Design
Synthetic gRNAs were purchased from SYNTHEGO. gRNA sequences were derived from a mouse whole genome CRISPR library described previously52. 10 gRNA sequences from this dataset were ranked according to predicted indel percentage and low off-target score using the inDelphi machine-learning algorithm for each gene target53. The top 3–7 guides were validated for high indel percentages by Sanger sequencing and protein knockdown by western blot before utilization in experiments.
Electroporation and cRNP complex formation
gRNAs (Synthego) were diluted to 100 µM (100 pMol/μL) in 1x TE buffer (Sythego). 1.2 uL (120 pMol) of gRNA, 0.9 uL of 100 μM Alt-R® Cas9 Electroporation Enhancer (IDT) and 3.9 uL water were added to a 1.5 mL tube per sample for a total of 6 μL. 1 μL of recombinant Cas9 (20 pMol) (Synthego) was added to 5 μL water in a separate 1.5 mL tube. 6 μL of diluted Cas9 was added to 6 μL of gRNA-enhancer mixture for a total of 12 μL cRNP complex at a 1:3 molar ratio. The cRNP complex was allowed to incubate for at least 10 minutes at room temperature (RT) and electroporated using the Neon Transfection system (Thermo-Fisher) as described previously34, 54. Cells were then incubated at 37°C for either 10 minutes before adoptive transfer or 90 minutes before centrifugation and resuspension in complete media supplemented with 50 ng rmIL-15 for culturing in vitro. Cells were cultured in vitro for 3 days following electroporation prior to reading out gene editing efficiency by flow cytometry or sanger sequencing.
Ex vivo stimulation of lymphocytes
For plate-bound antibody stimulation experiments, ~5 x 105 isolated NK cells were stimulated with 4 mg/mL precoated antibody against NK1.1 (PK136) for 4 hours in complete media containing Brefeldin A (1:1000; BioLegend) and Monensin (2uM; BioLegend). Cells were cultured in media alone as a negative control. In cytokine stimulation experiments, isolated NK cells were incubated with various concentrations of mouse IL-15 or IL-2, in the presence or absence of 100 uM CAS 285986–31-4 STAT5 inhibitor (Millipore Sigma).
Adoptive transfer cRNP experiments
Adoptive NK cell co-transfer studies were performed by injecting a total of 1 x 106 NK cells; Rosa26 cRNP-edited WT, and gene x cRNP-edited WT NK cells purified from spleens of congenically distinct WT mice (CD45.1, CD45.1.2 or CD45.2) into Ly49H–/– mice 16 hours prior to MCMV infection.
Proliferation assays
CellTrace™ Violet (CTV) stock solution was prepared per the manufacturers’ instructions (Thermo) and diluted at 1:1000 in 37C PBS. Isolated NK cells were incubated in 0.5mL of diluted CTVsolution for 10 minutes at 37C. The solution was quenched with 10X the volume of CR-10 media. Cells were then washed and injected i.v. Division, proliferation and expansion indexes were quantified using FlowJo v10.7.2 software using the following calculations. Division Index: Total Number of Divisions / The number of cells at start of culture. Proliferation Index: Total Number of Divisions / Cells that went into division. Division Index: Total Number of Divisions / The number of cells at start of culture.
Flow Cytometry
Cells were analyzed for cell-surface markers using fluorophore-conjugated antibodies (BioLegend, eBioscience). Cell surface staining was performed in 1X PBS and intracellular staining was performed by fixing and permeabilizing using the eBioscience Foxp3/Transcription Factor kit for intranuclear proteins or BD Cytofix/Cytoperm kits for cytokines. Flow cytometry was performed using the Attune NxT Acoustic Focusing cytometer and data were analyzed with FlowJo v9.9.6 and v10.7.2 software (BD). Cell surface and intracellular staining was performed using the following fluorophore-conjugated antibodies: CD45.1 A700 1:300 (A20), CD45.1 PE/Cy7 1:400 (A20), CD45.2 FITC 1:400 (104), NK1.1 Percp/Cy5.5 1:100 (PK136), KLRG1 PE 1:400 (2F1), KLRG1 PE/Cy7 1:100 (2F1), TCRβ APC/Cy7 1:100 (H57–597), CD3ε APC/Cy7 1:100 (17A2), Ly49H A647 1:600 (3D10), Ly49H PE 1:300 (3D10), IFN-γ APC 1:100 (XMG1.2), Ly6C PacBlue 1:600 (HK1.4), CD44 FITC 1:100 (IM7), CD16 FITC 1:100 (93), Sca-1 PE/Cy7 1:100 (E13–161.7), CX3CR1 PE 1:100 (SA011F11), NKG2D PE 1:100 (CX5), BCL2 PE 1:50 (BCL/10C4), CD11b A700 1:100 (M1/70), CD27 APC 1:100 (LG.3A10), Bim PE 1:50 (C34C5), Ki-67 A700 1:100 (16A8), Cytochrome-C A647 1:100 (6H2.B4), CD122 PE 1:100 (5H4), CD49b PE/Cy7 1:100 (DX5). For mitochondrial dyes, NK cells were enriched from spleens as described above, stained with cell-surface antibodies, and then incubated with various dyes in Hank’s balanced salt solution plus Mg and Ca as follows: 100 nM Mitotracker Green (Life Technologies) for 30 min at 37°C to measure mitochondrial mass or 100 nM TMRE (Thermofisher) for 30 min at 37°C to measure mitochondrial membrane potential. BH3 profiling was performed as previously described55. Briefly, purified NK cells were resuspended in MEB buffer (150 mM Mannitol 10 mM HEPES-KOH, 50 mM KCl, 0.02 mM EGTA, 0.02 mM EDTA, 0.1 % BSA, 5 mM Succinate). 50μl of cell suspension (1 × 105 cells/well) were plated in wells holding 50 μL MEB buffer containing 0.002% digitonin and BCL2 inhibitor ABT-199. Plates were then incubated at 25°C for 50 min. Cells were then fixed with 4% paraformaldehyde for 10min, followed by neutralization with N2 buffer (1.7M Tris, 1.25M Glycine pH 9.1) for 5min. Samples were stained for 1 hour with 20 μL of staining solution (10% BSA, 2% Tween 20 in PBS) containing anti-cytochrome c (BioLegend). Immediately afterwards, cytochrome c release was quantified using Attune Flow Cytometer.
Single Cell Metabolism assay (SCENITH)
To profile single cell metabolic responses, 95 uL of purified NK cells in complete media were plated at 0.1–0.5x106 cells/mL in v-bottom 96-well plates. Experimental triplicates were performed in all conditions. Wells were then treated with Control (DMSO), 2-Deoxy-D-Glucose (DG) final concentration 100mM, Oligomycin (O) final concentration 1 mM, or a sequential combination of DG and O at the final concentrations mentioned. As negative control, the translation initiation inhibitor Harringtonine was added (Harringtonine, 2 mg/mL). Puromycin (final concentration 10 mg/mL) is added immediately after the metabolic inhibitor treatment. After puromycin treatment, cells were washed in cold PBS and stained for surface markers. Intracellular staining of puromycin was achieved using a custom anti-puromycin antibody27 was performed by incubating cells during 1h at 4°C diluted in permeabilization buffer.
PCR and Sanger Sequencing
DNA from NK cells was isolated using DNeasy Blood and Tissue kits (Qiagen). DNA concentration was measured using the NanoDrop OneC Microvolume UV-Vis Spectrophotometer (Thermo Scientific) and then diluted to 50 ng/μl in water before PCR amplification of cRNP-targeted genomic regions of approximately 500–1000 base pairs. PCR samples from WT and cRNP-edited cells were submitted for Sanger sequencing (GENEWIZ) and then indel percentage was calculated using ICE analysis (SYNTHEGO).
10x Library Preparation, Sequencing, and Alignment
Single cell RNA-sequencing libraries were generated with the Chromium Single Cell 3′ v2 (Day 7) and v3 (Day 14) assay (10X Genomics). Libraries were sequenced using the HighSeq 4000 platform (Illumina) to a depth of approximately 300 million reads per library with 2x50 read length. Raw reads were aligned to mouse genome (mm10) and cells were called using cellranger count (v3.0.2).
scRNA-seq Cell Clustering
The R package Seurat (v3.1.)56 was used to cluster the cells. Cells with less than 100 genes detected or more than 10% mitochondrial gene expression were first filtered out as low-quality cells. The gene counts for each cell were divided by the total gene counts for the cell and multiplied by a scale factor 10,000, then natural-log transformation was applied to the counts. The FindVariableFeatures function was used to select variable genes with default parameters. The ScaleData function was used to scale and center the counts in the dataset. Principal component analysis (PCA) was performed on the variable genes, and 20 principal components were used for cell clustering (resolution = 0.5) and uniform manifold approximation and projection (UMAP) dimensional reduction. The cluster markers were found using the FindAllMarkers function. Module scores were calculated using the AddModuleScore function with default parameters. In the comparison between D7 and D14 PI NK cells, the FindIntegrationAnchors and IntegrateData functions were used to find anchors and integrate the D7 and D14 PI datasets, and the other steps were the same as described above.
RNA velocity analysis
To estimate the RNA velocities of single cells, velocyto26 was used to distinguish unspliced and spliced mRNAs in each sample. The python package scVelo57 was then used to recover the directed dynamic information by leveraging RNA splicing information. Specifically, the data was first normalized using the filter_and_normalize function. The first- and second-order moments were computed for velocity estimation using the moments function. The velocity vectors were obtained using the velocity function. The velocities were projected into a lower-dimensional embedding using the velocity_graph function. Finally, the velocities were visualized in the UMAP embedding using the velocity_embedding_stream function. All scVelo functions were used with default parameters.
Pseudo-time trajectory construction
Pseudo-time trajectories were constructed using the R package Monocle58 (version 2.10.1). The raw counts for cells in the intended cell types were extracted and normalized by the estimateSizeFactors and estimateDispersions functions with the default parameters. Genes with average expression larger than 0.5 and detected in more than 10 cells were retained for further analysis. Variable genes were determined by the differentialGeneTest function with a model against the cell type identities. The top 2,000 variable genes with the lowest adjusted p value were used to order the cells. The orders were determined by the orderCells function, and the trajectory was constructed by the reduceDimension function with default parameters.
Bulk RNA Sequencing
RNA was isolated from the cells using RNeasy Mini kit (Qiagen) and used to generate RNA-seq libraries followed by sequencing using Illumina HighSeq 4000 platform (single end, 50bp). The reads were mapped with STAR59 (version 2.5.3.a) to the mouse genome (mm10). The counts for each gene were obtained by using –quantMode GeneCounts commands in STAR, and the other parameters during alignment were set to default. Differential expression analyses were carried out using DESeq260 (version 1.24.0) with default parameters. Genes with adjusted p value <0.05 were considered significantly differentially expressed. Sequencing depth normalized counts were used to plot the expression values for individual genes. The T cell bulk RNA-seq datasets were downloaded from GEO (GSE111902)61, and the same DESeq2 procedure was applied. Genes with the absolute log2 fold change >0.5 and adjusted p value <0.05 in both NK cell and T cell datasets were plotted in Figure 5C. Cytokine stimulated bulk NK RNA-seq datasets were downloaded from GEO (GSE140044)35.
ATAC-seq analysis
For ATAC-Seq, 50,000 cells per sample were lysed to collect nuclei and treated with Tn5 transposase (Illumina) for 30 minutes at 37°C with gentle agitation. The DNA was isolated with DNA Clean & Concentrator Kit (Zymo) and PCR amplified and barcoded with NEBNext High-Fidelity PCR Mix (New England Biolabs) and unique dual indexes (Illumina). The ATAC-Seq library amplification was confirmed by real-time PCR, and additional barcoding PCR cycles were added as necessary while avoiding overamplification. Amplified ATAC-Seq libraries were purified with DNA Clean & Concentrator Kit (Zymo). The purified libraries were quantified with Kapa Library Quant Kit (KAPA Biosystems) and quality assessed on 4200 TapeStation System (Agilent). The libraries were pooled based on molar concentrations and sequenced on an Illumina HighSeq 4000 platform (paired end, 100bp).
ATAC-seq fastq files were trimmed to remove low-quality reads and adapters using Cutadapt62 (version 2.3). The reads were aligned to the reference mouse genome (mm10) with bowtie263 (version 2.2.9). Peak calling was performed with MACS264 (version 2.1.1). The peaks from all samples were merged into a single bed file, peaks from different samples that were closer than 10bp were merged into a single peak. HTseq65 (version 0.9.1) was used to count the number of reads that overlap each peak per sample. The peak counts were analyzed with DESeq260 (version 1.24.0) to identify differentially accessible genomic regions. Peaks with adjusted p value <0.15 were considered significantly differentially accessible. The peak counts were visualized with IGV, version 2.5.0. The differentially accessible peaks were analyzed using the findMotifsGenome.pl function from homer66 (version 4.9.1) to identify enriched cis-regulatory motifs of transcription factors. The T cell ATAC-seq datasets were downloaded from GEO (GSE111902), the same pipeline described above were used to analyze the datasets. Cytokine stimulated NK cell ATAC-seq datasets were downloaded from GEO (GSE140044) and visualized using the same pipeline described above. Peaks in the T cell ATAC-seq datasets with adjusted p value <0.05 were considered significantly differentially accessible. Significant peaks with the same change directions in the NK and T cell datasets were plotted in Figure 6C.
ChIP-seq analysis
STAT5 ChIP-seq datasets derived from IL-15/IL-2 stimulated splenic NK cells were downloaded from GEO (GSE140044)35 and visualized using IGV, version 2.5.0.
Western blot
Protein was extracted from enriched primary splenic NK cells using Pierce RIPA buffer (Thermo-Fisher) with Halt protease inhibitor cocktail (Thermo-Fisher) and protein concentration was quantified using the Pierce BCA Protein Assay kit (Thermo-Fisher). Samples were electrophoresed on NuPage Novex 4–12% Bis-Tris Protein Gels, transferred to PVDF membranes, and blocked for one hour at room temperature with 5% w/v nonfat milk in 1X TBS and 0.1% Tween-20. Immunoblots were performed using rabbit anti-Fli1 (Abcam ab133485), rabbit anti-β-actin (Cell Signaling CST4970), and rabbit anti-GAPDH (Millipore Sigma G9545). Proteins were detected using the SuperSignal West Pico PLUS ECL kit (Thermo-Fisher) and visualized using the Azure Biosystems c280 imager. Band density was quantified using ImageJ version 1.53.
Statistical Analyses
For graphs, data are shown as mean ± SEM, and unless otherwise indicated, statistical differences were evaluated using a Student’s t test with Welch’s correction to assume a non-normal variance in our data distribution. Samples were compared using the Log-rank (Mantel-Cox) test with correction for testing multiple hypotheses. p < 0.05 was considered significant. Graphs were produced and statistical analyses were performed using GraphPad Prism.
Data Availability
Sequencing datasets are accessible from GEO with accession number GSE176208. Original western blot scans can be found in source data.
Extended Data
Extended Data Fig. 1. Phenotypic and single cell sequencing analysis of naive, D7 PI and D14 PI Ly49H+ NK cells.
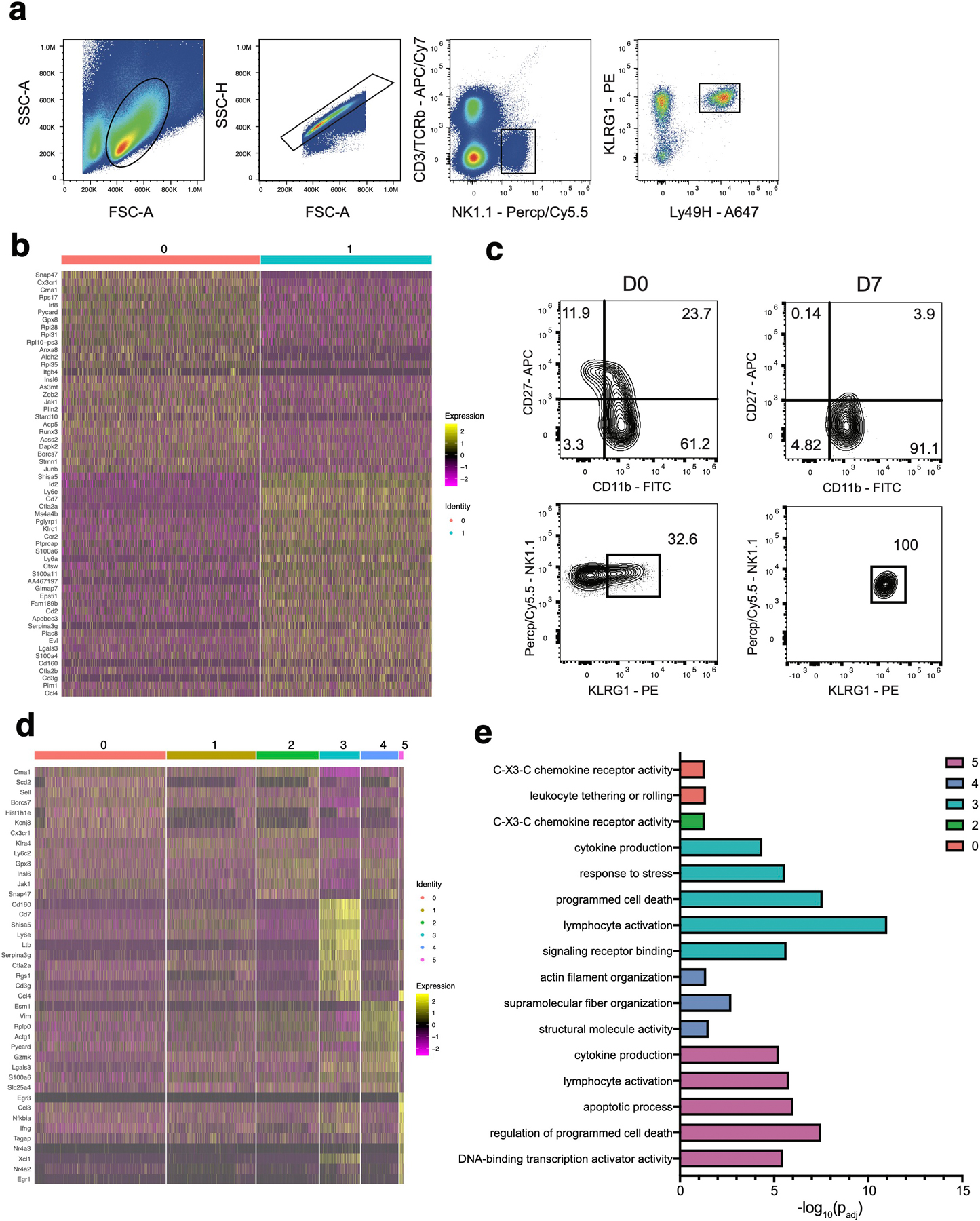
(a) Gating strategy to sort adoptively transferred TCRβ–CD3ε–NK1.1+KLRG1+Ly49H+ NK cells from the spleen of recipient Ly49H–/– mice on D7 and D14 following MCMV infection. (b-e) Ly49H+ NK cells were adoptively transferred into Ly49H–/– mice and infected with MCMV i.p. 16 hours later. TCRβ–CD3ε–NK1.1+Ly49H+KLRG1+ NK cells were sorted on D7 and D14 PI. Cells were immediately processed for single cell sequencing using 10x Genomics Chromium droplet single cell RNA sequencing. (b) Heatmap showing the top differentially expressed genes between the two clusters of D7 PI NK cells (padj < 0.05). (c) Representative flow plots of cell surface expression of CD27, CD11b, NK1.1 and KLRG1 on naive (left) and D7 PI (right) TCRβ–CD3ε–NK1.1+Ly49H+ NKeff cells. (d) Heatmap showing the top differentially expressed genes between the six clusters from NK cells at D7 and D14 PI (padj < 0.05). (e) GO enrichment analysis of marker genes for each cluster from (d). Terms were considered statistically significantly enriched if -log10(padj)<0.05. For (c-e), differentially expressed genes were identified using Wilcoxon Rank Sum test in Seurat v3.1.2.
Extended Data Fig. 2. Time-resolved putative differentiation pathways of NKeff and MP NK cells during MCMV infection.
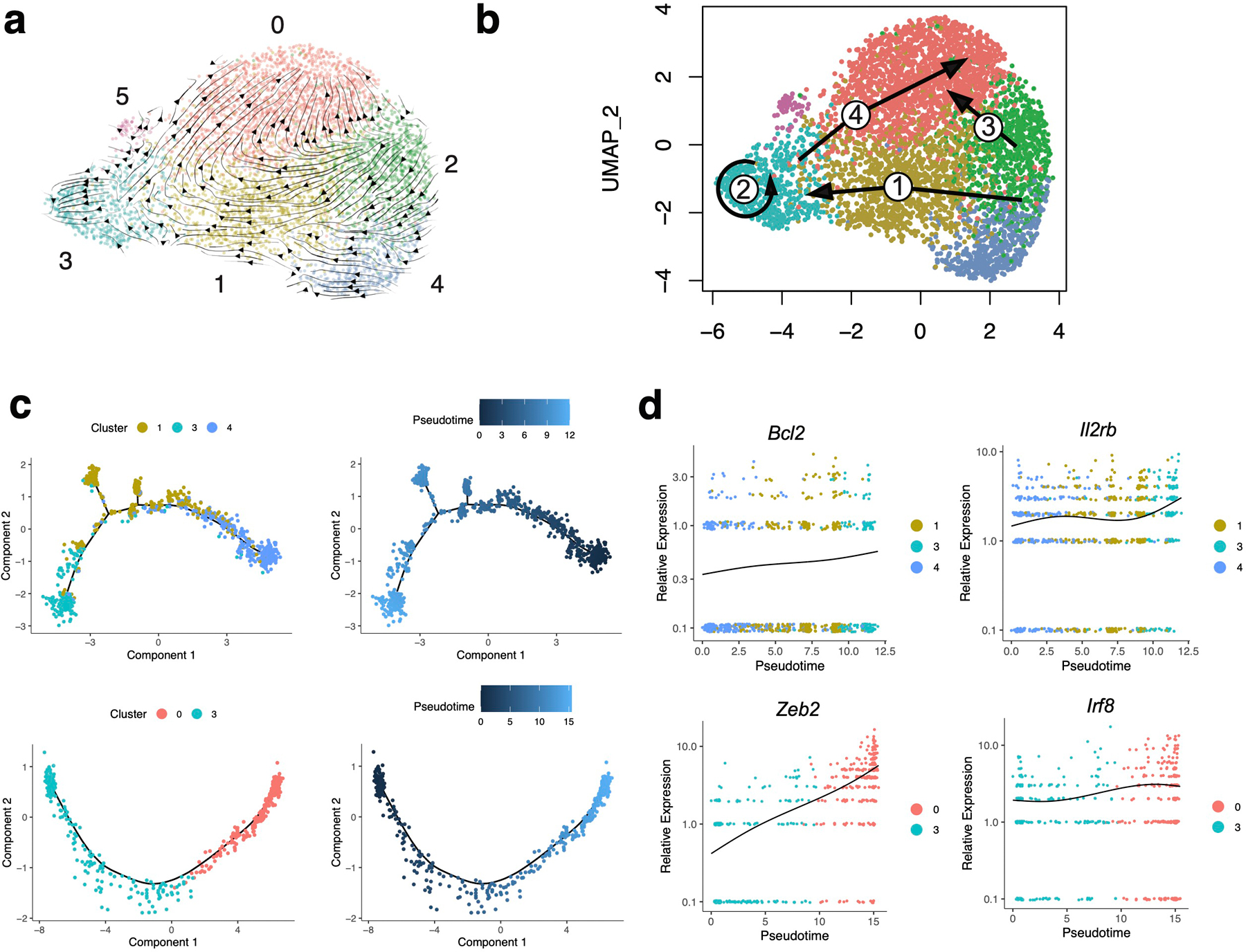
(a-d) Ly49H+ NK cells were adoptively transferred into Ly49H–/– mice and infected with MCMV i.p. 16 hours later. TCRβ–CD3ε–NK1.1+Ly49H+KLRG1+ NK cells were sorted on D7 and D14 PI. Cells were immediately processed for single cell sequencing using 10x Genomics Chromium droplet single cell RNA sequencing. (a) RNA-velocity analysis of D7 and D14 NKeff cell clusters with velocity field arrows projected onto the UMAP plot. (b) Arrows show the local average velocity evaluated on a regular grid and indicate the extrapolated future states of cells. (c) Monocle pseudotime analysis of NK cell clusters, indicating cluster identities (left) and pseudotime (right). (d) Scatter plot displaying relative expression (y-axis) of selected genes along pseudotime (x-axis).
Extended Data Fig. 3. NKeff subsets do not demonstrate differential trafficking or proliferation following MCMV infection.
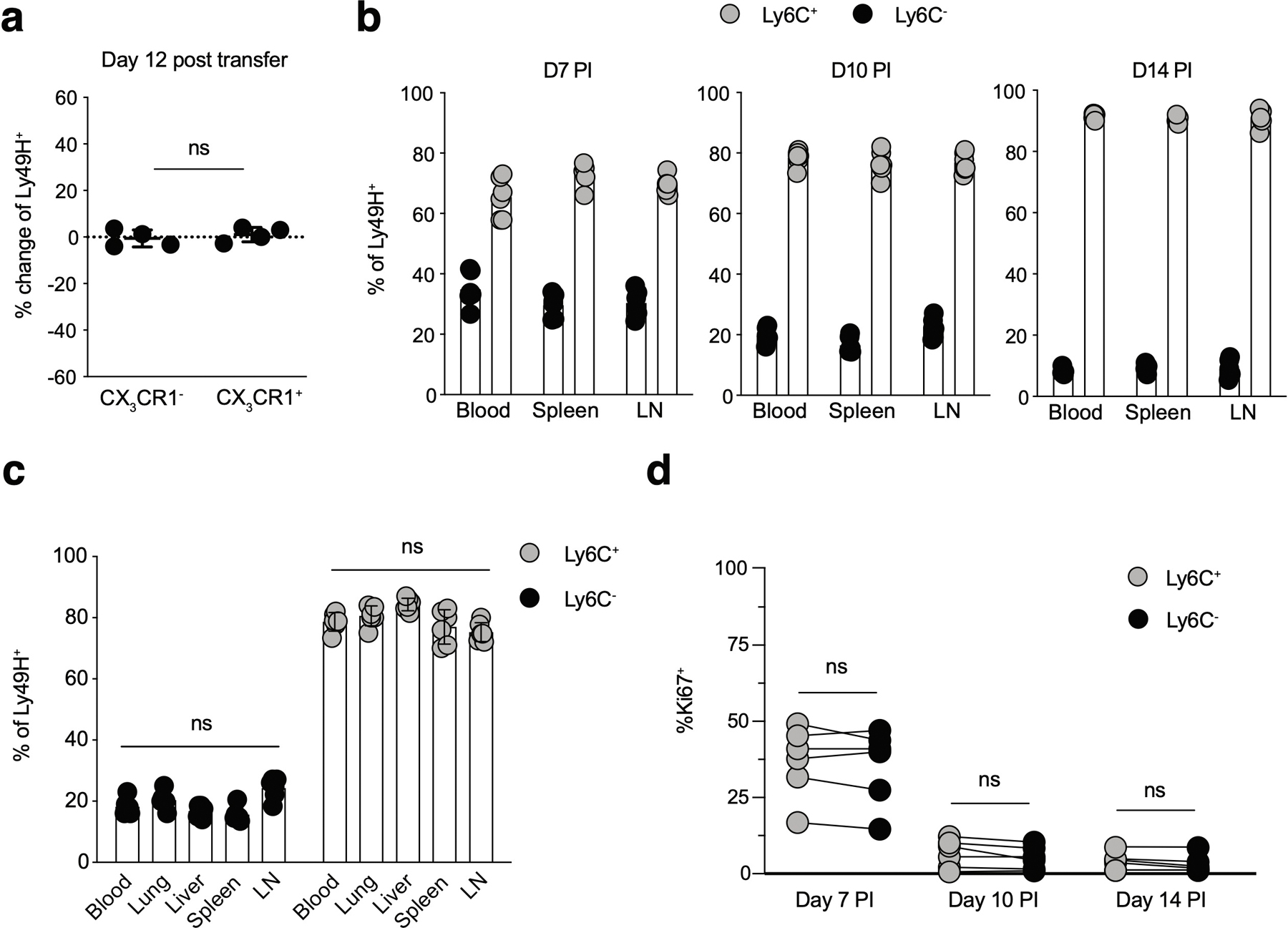
WT splenic CD45.1 and CD45.2 Ly49H+ NK cells were transferred into Ly49H–/– mice, and infected with MCMV i.p. 16 hours later. 7 days after MCMV infection, TCRβ–CD3ε–NK1.1+Ly49H+KLRG1+CD45.1+CX3CR1+ and TCRβ–CD3ε–NK1.1+Ly49H+KLRG1+CD45.2+CX3CR1– NKeff were sorted and then transferred into naive Ly49H–/– hosts at a 1:1 ratio. Recipient spleens were harvested 12 days post transfer and analyzed by flow cytometry. (a) Quantification of the change in frequency of CX3CR1+ (CD45.1) and CX3CR1– (CD45.2) Ly49H+ NK cells subsets from recipient mice on D12 (post transfer). (b-d) Splenic WT Ly49H+ NK cells were transferred into Ly49H–/– mice and infected with MCMV i.p. 16 hours later. (b) Quantification of Ly6C+ and Ly6C– Ly49H+ NK cells from the indicated peripheral organs at various timepoints PI and (c) D10 PI. (d) Quantification of Ki-67+ Ly49H+ splenic NK cells at D10 PI. (a) Data represent 2 independent experiments with n = 4 mice per group. (b-d) Data represent 2 independent experiments with n = 6 mice per group. Samples were compared using two-tailed Student’s t test with Welch’s correction, assuming unequal SD, and data points are presented as individual mice with the mean ± SEM (ns = not significant).
Extended Data Fig. 4. Ly6C+ and Ly6C– NKeff cells do not display differences in mitophagy, cell-intrinsic metabolism and memory functionality.
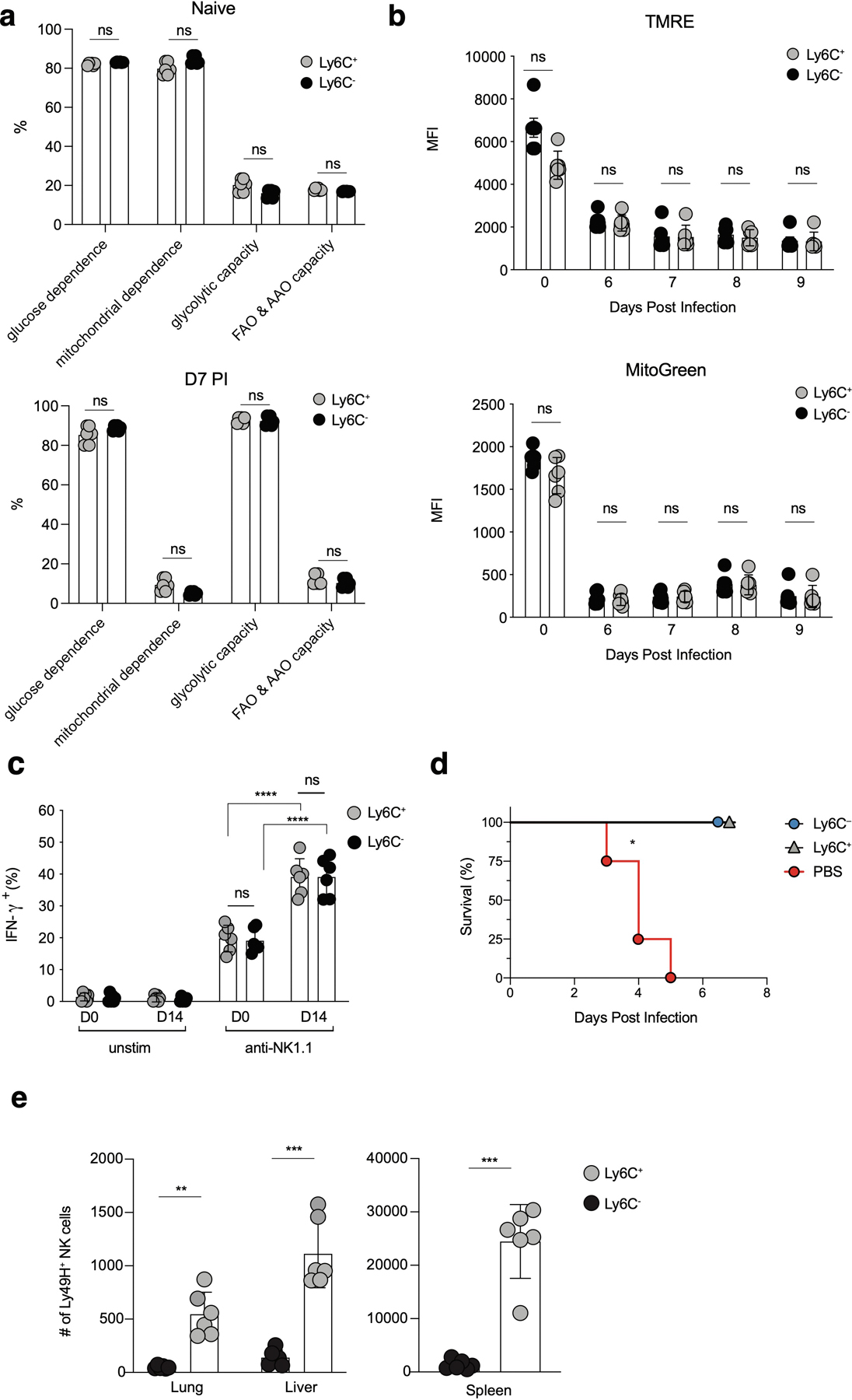
(a-e) Splenic Ly49H+ NK cells were transferred into Ly49H–/– mice i.v. and infected i.p with MCMV 16 hours after adoptive transfer. (a) Single cell metabolic analysis of naive (top) or D7 PI (bottom) NKeff using SCENITH. (b) MFI for tetramethylrhodamine ethyl ester (TMRE) staining (top) or MitoTracker Green staining (bottom) in adoptively transferred Ly49H+ NK cells from recipient spleens at the indicated time points PI. (c) Percentage of IFN-γ+ naive or adoptively transferred D14 PI Ly6C+ or Ly6C– Ly49H+ NK cells following no stimulation or 4hr stimulation ex vivo with anti-NK1.1 monoclonal platebound antibody. Ly6C+ p = 0.000083, Ly6C− p = 0.000101. (d) 2.5 x 103 D7 PI sorted TCRβ–CD3ε–NK1.1+KLRG1+CD45.1+Ly49H+Ly6C+ or TCRβ–CD3ε–NK1.1+KLRG1+CD45.1+Ly49H+Ly6C– NK cells were adoptively transferred i.v. into naive Ly49H–/– mice and infected with MCMV 7 days later. (d) Kaplan-Meier survival curves of Ly49H–/– mice that received PBS or indicated sorted NK cell populations i.v. p = 0.0174. (e) The number of Ly6C+ and Ly6C– Ly49H+ NK cells was quantified in various organs at D28 PI. Lung p = 0.001997, liver p = 0.000495, spleen p = 0.0003. (a-c,e) Data represent 2–3 independent experiments with n = 3 mice per group. Samples were compared using two-tailed Student’s t test with Welch’s correction, assuming unequal SD, and data are presented as individual points with the mean ± SEM. (d) Data are pooled from 2 independent experiments with n = 4–5 mice per group per experiment. Data points are presented as individual mice. Conditions were compared using the Log-rank (Mantel-Cox) test with correction for testing multiple hypotheses. (**p < 0.01, ***p < 0.001).
Extended Data Fig. 5. MP NK cells are transcriptionally distinct from MP CD8 T cells.
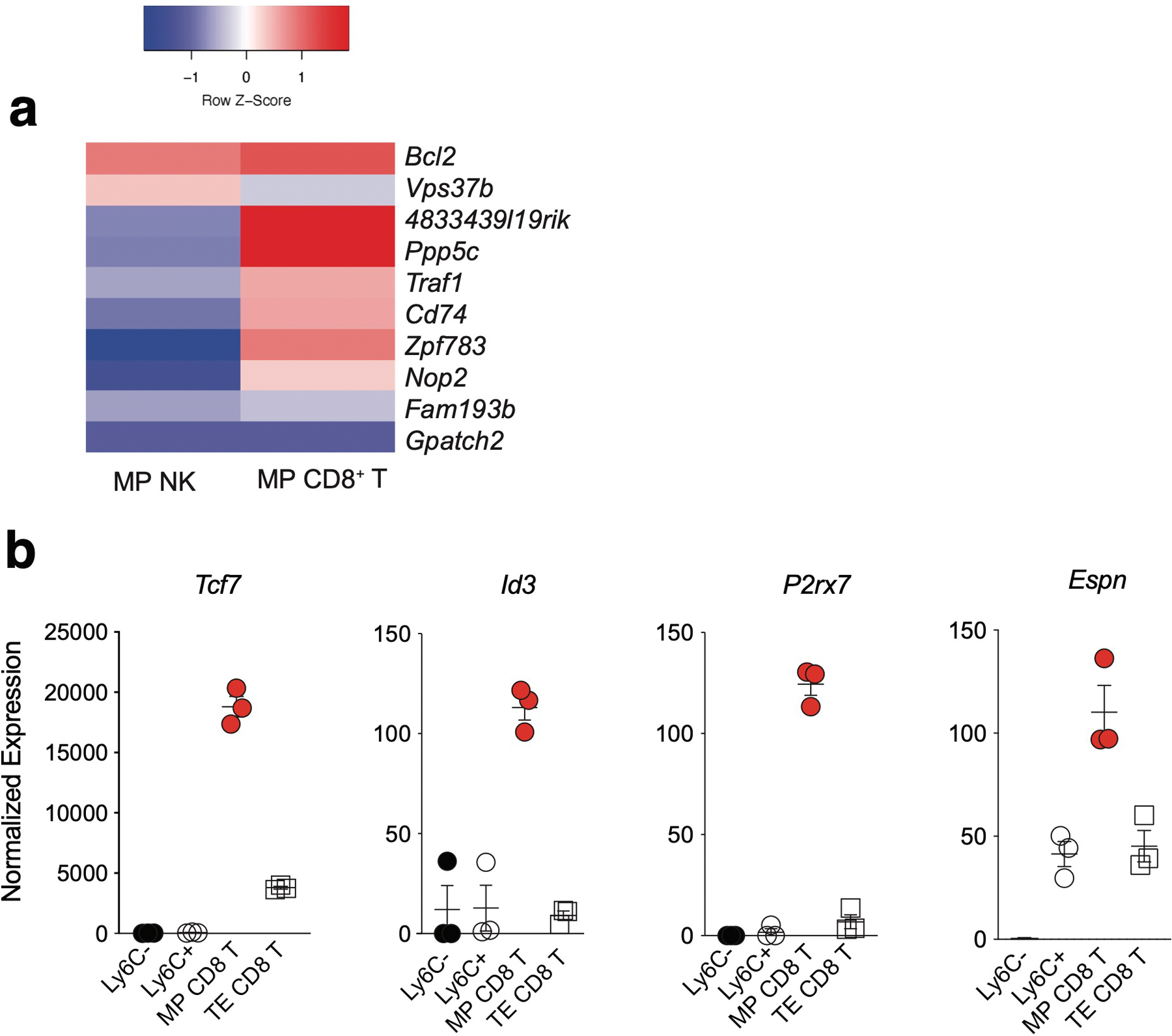
Splenic Ly49H+ NK cells were transferred into Ly49H–/– mice i.v. and infected i.p with MCMV 16 hours after adoptive transfer. Splenic TCRβ–CD3ε–NK1.1+KLRG1+Ly49H+Ly6C+ and TCRβ–CD3ε–NK1.1+KLRG1+Ly49H+Ly6C– NKeff were sorted from recipient mice on D7 PI. Sorted NK cells were immediately processed for mRNA extraction, library preparation and sequencing. (a) Differentially expressed genes common between MP Ly6C– NK and Ly6C+ NKeff cells compared to MP and TE CD8+ T cells (padj < 0.05). Differentially expressed genes were determined using the Wald test in DESeq2. (b) Normalized read counts of selected genes in indicated cell types. Data are representative of n = 3 mice per group, presented as mean ± SEM.
Extended Data Fig. 6. MP NK cells and MP CD8+ T cells display similar chromatin accessibility at specific gene loci.
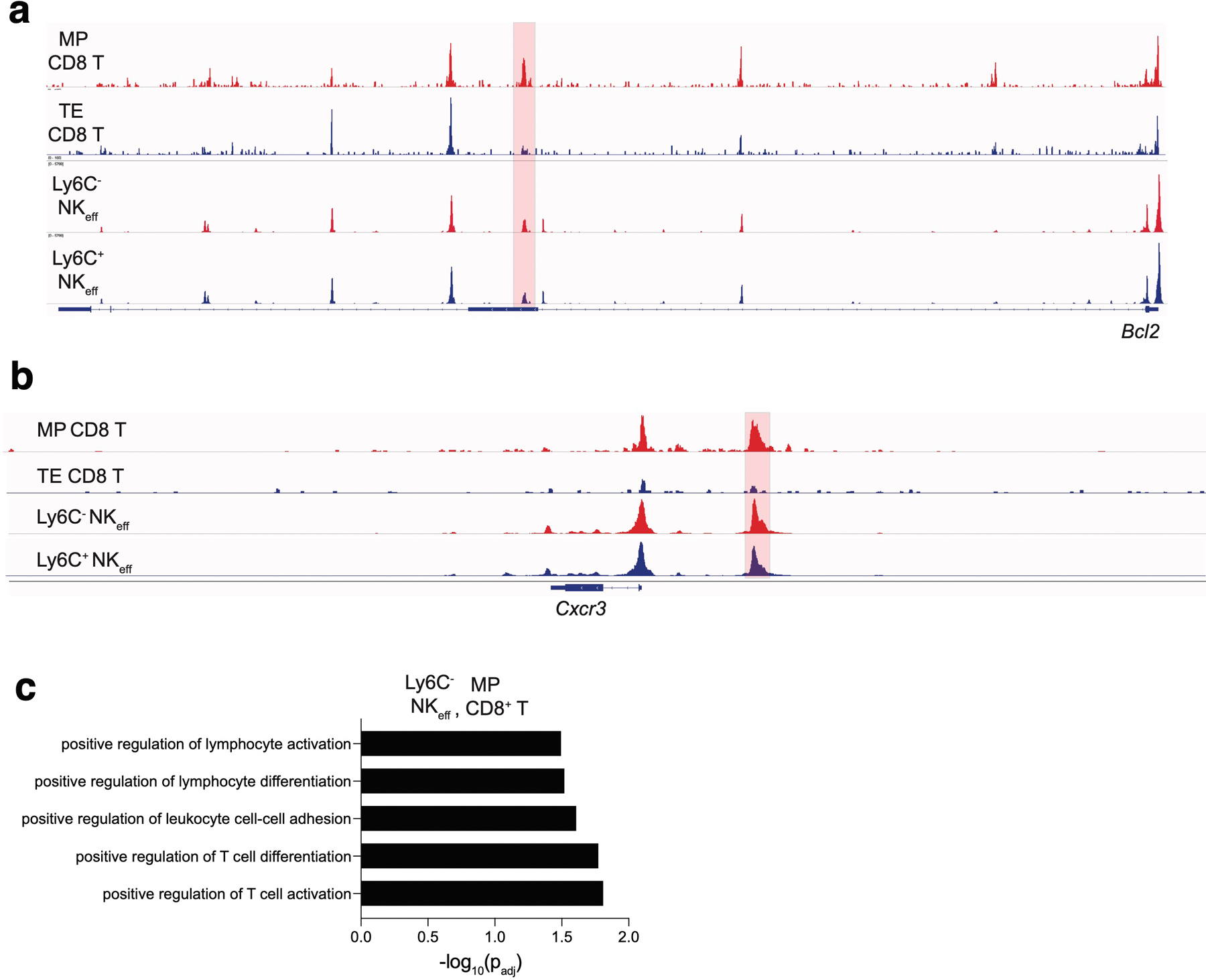
ATAC sequencing reads from Fig. 5 in the indicated cell subsets mapping to the (a) Bcl2 locus and (b) Cxcr3 locus. Highlighted peaks represent differentially accessible peaks (padj <0.15). For (a-b), differentially accessible peaks were identified using the Wald test in DESeq2. (c) GO term analysis from differentially accessible peaks shared between MP NK cells and MP CD8+ T cells. Terms were considered statistically significantly enriched if -log10(padj)<0.05. Data are representative of three independent replicates for MP and TE CD8+ T cells, D7 PI Ly6C– NKeff and two independent replicates for D7 PI Ly6C+ NKeff. GO term analysis was calculated using the cumulative hypergeometric distribution in HOMER.
Extended Data Fig. 7. Proposed model of MP NK cell formation and mechanism of Fli1 induction.
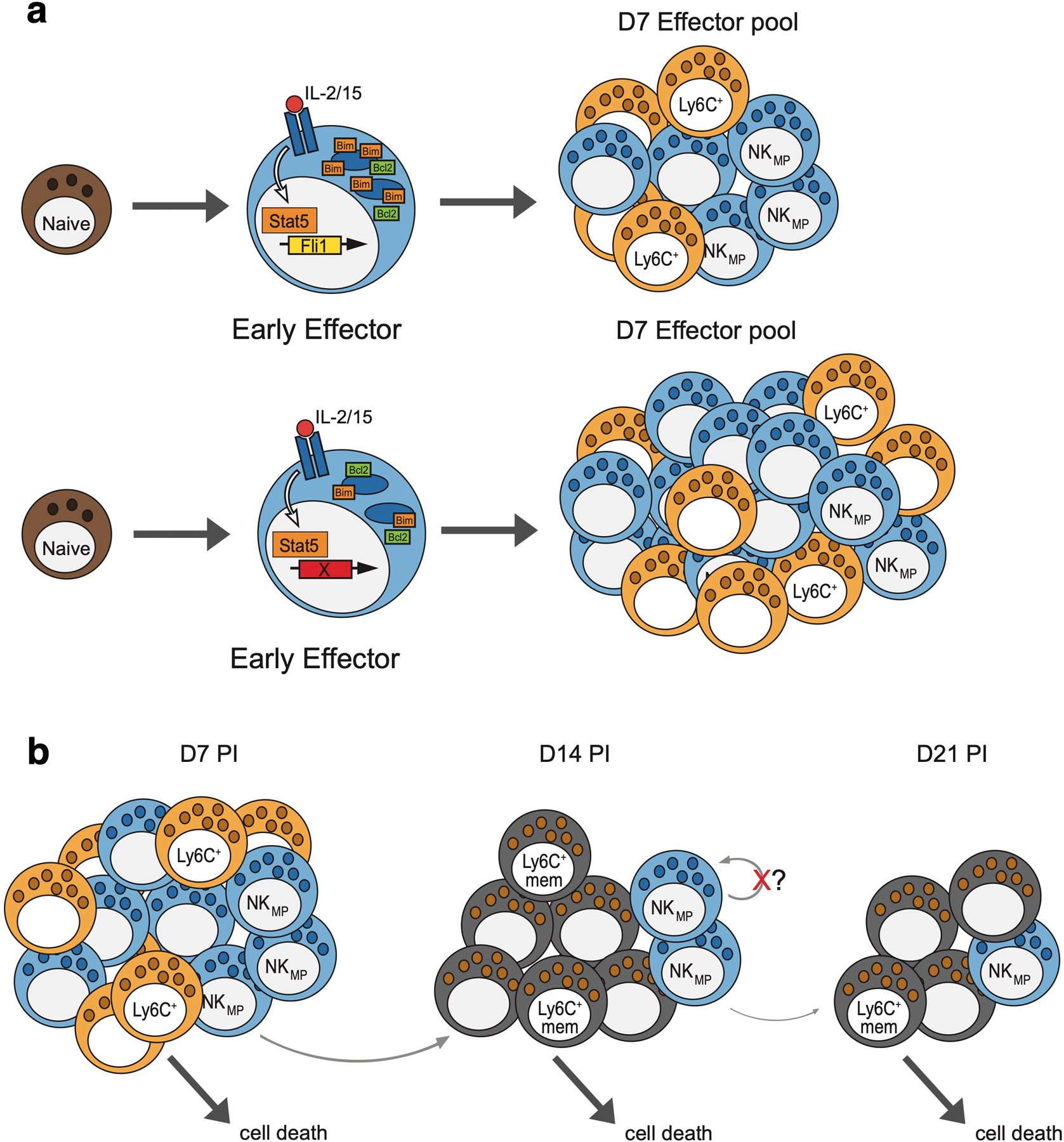
(a) IL-15 stimulated NK cells signal through STAT5 to induce Fli1 expression. During infection, Fli1 increases Bim levels to limit the number of early effector NK cells contributing to the D7 effector pool. In the absence of Fli1, Bim levels are reduced while BCL2 is unaffected, allowing more early effector NK cells to persist and form MP NK cells. (b) In the contraction phase post D7, MP NK cells continually give rise to Ly6C+ memory NK cells. The majority of D7 PI Ly6C+ NK cells undergo cell death, while a small number can become Ly6C+ memory cells. Lacking a stem-like transcriptional program, the pool of MP cells is depleted over time.
Supplementary Material
Acknowledgements
We thank members of the O’Sullivan and Sun labs for helpful discussion. T The Regents of the University of California have filed a provisional patent application with the U.S. Patent and Trademark Office towards methods for adoptive cell immunotherapy targeting Fli1 expression in NK cells. L.R., J.H.L., and T.E.O. are listed as inventors on this patent application. T.E.O. was supported by the NIH (AI145997) and UC CRCC (CRN-20-637105).
Footnotes
The authors declare no financial conflicts of interest.
Competing Interests Statement
The authors declare no competing interests.
References
- 1.Henning AN, Roychoudhuri R & Restifo NP Epigenetic control of CD8(+) T cell differentiation. Nat Rev Immunol 18, 340–356 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Youngblood B, Hale JS & Ahmed R T-cell memory differentiation: insights from transcriptional signatures and epigenetics. Immunology 139, 277–284 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bantug GR, Galluzzi L, Kroemer G & Hess C The spectrum of T cell metabolism in health and disease. Nat Rev Immunol 18, 19–34 (2018). [DOI] [PubMed] [Google Scholar]
- 4.Chang JT, Wherry EJ & Goldrath AW Molecular regulation of effector and memory T cell differentiation. Nat Immunol 15, 1104–1115 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kaech SM & Cui W Transcriptional control of effector and memory CD8+ T cell differentiation. Nat Rev Immunol 12, 749–761 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kaech SM et al. Selective expression of the interleukin 7 receptor identifies effector CD8 T cells that give rise to long-lived memory cells. Nat Immunol 4, 1191–1198 (2003). [DOI] [PubMed] [Google Scholar]
- 7.Joshi NS et al. Inflammation directs memory precursor and short-lived effector CD8(+) T cell fates via the graded expression of T-bet transcription factor. Immunity 27, 281–295 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sarkar S et al. Functional and genomic profiling of effector CD8 T cell subsets with distinct memory fates. J Exp Med 205, 625–640 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Biron CA, Byron KS & Sullivan JL Severe herpesvirus infections in an adolescent without natural killer cells. N Engl J Med 320, 1731–1735 (1989). [DOI] [PubMed] [Google Scholar]
- 10.Bukowski JF, Warner JF, Dennert G & Welsh RM Adoptive transfer studies demonstrating the antiviral effect of natural killer cells in vivo. J Exp Med 161, 40–52 (1985). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Weizman OE et al. ILC1 Confer Early Host Protection at Initial Sites of Viral Infection. Cell 171, 795–808 e712 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Riggan L, Freud AG & O’Sullivan TE True Detective: Unraveling Group 1 Innate Lymphocyte Heterogeneity. Trends Immunol 40, 909–921 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Weizman OE et al. Mouse cytomegalovirus-experienced ILC1s acquire a memory response dependent on the viral glycoprotein m12. Nat Immunol 20, 1004–1011 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sun JC, Beilke JN & Lanier LL Adaptive immune features of natural killer cells. Nature 457, 557–561 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.O’Sullivan TE, Sun JC & Lanier LL Natural Killer Cell Memory. Immunity 43, 634–645 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Grassmann S et al. Distinct Surface Expression of Activating Receptor Ly49H Drives Differential Expansion of NK Cell Clones upon Murine Cytomegalovirus Infection. Immunity 50, 1391–1400 e1394 (2019). [DOI] [PubMed] [Google Scholar]
- 17.Arase H, Mocarski ES, Campbell AE, Hill AB & Lanier LL Direct recognition of cytomegalovirus by activating and inhibitory NK cell receptors. Science 296, 1323–1326 (2002). [DOI] [PubMed] [Google Scholar]
- 18.Brown MG et al. Vital involvement of a natural killer cell activation receptor in resistance to viral infection. Science 292, 934–937 (2001). [DOI] [PubMed] [Google Scholar]
- 19.Crinier A et al. High-Dimensional Single-Cell Analysis Identifies Organ-Specific Signatures and Conserved NK Cell Subsets in Humans and Mice. Immunity 49, 971–986 e975 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Horowitz A et al. Genetic and environmental determinants of human NK cell diversity revealed by mass cytometry. Sci Transl Med 5, 208ra145 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Karo JM, Schatz DG & Sun JC The RAG recombinase dictates functional heterogeneity and cellular fitness in natural killer cells. Cell 159, 94–107 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rahim MM et al. Expansion and Protection by a Virus-Specific NK Cell Subset Lacking Expression of the Inhibitory NKR-P1B Receptor during Murine Cytomegalovirus Infection. J Immunol 197, 2325–2337 (2016). [DOI] [PubMed] [Google Scholar]
- 23.Kamimura Y & Lanier LL Homeostatic control of memory cell progenitors in the natural killer cell lineage. Cell Rep 10, 280–291 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Adams NM, Diaz-Salazar C, Dang C, Lanier LL & Sun JC Cutting Edge: Heterogeneity in Cell Age Contributes to Functional Diversity of NK Cells. J Immunol 206, 465–470 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Min-Oo G, Bezman NA, Madera S, Sun JC & Lanier LL Proapoptotic Bim regulates antigen-specific NK cell contraction and the generation of the memory NK cell pool after cytomegalovirus infection. J Exp Med 211, 1289–1296 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.La Manno G et al. RNA velocity of single cells. Nature 560, 494–498 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.van Helden MJ et al. Terminal NK cell maturation is controlled by concerted actions of T-bet and Zeb2 and is essential for melanoma rejection. J Exp Med 212, 2015–2025 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Arguello RJ et al. SCENITH: A Flow Cytometry-Based Method to Functionally Profile Energy Metabolism with Single-Cell Resolution. Cell Metab 32, 1063–1075 e1067 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.O’Sullivan TE, Johnson LR, Kang HH & Sun JC BNIP3- and BNIP3L-Mediated Mitophagy Promotes the Generation of Natural Killer Cell Memory. Immunity 43, 331–342 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bezman NA et al. Molecular definition of the identity and activation of natural killer cells. Nat Immunol 13, 1000–1009 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Min-Oo G & Lanier LL Cytomegalovirus generates long-lived antigen-specific NK cells with diminished bystander activation to heterologous infection. J Exp Med 211, 2669–2680 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Johnnidis JB et al. Inhibitory signaling sustains a distinct early memory CD8(+) T cell precursor that is resistant to DNA damage. Sci Immunol 6 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Viant C et al. Cell cycle progression dictates the requirement for BCL2 in natural killer cell survival. J Exp Med 214, 491–510 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Riggan L et al. CRISPR-Cas9 Ribonucleoprotein-Mediated Genomic Editing in Mature Primary Innate Immune Cells. Cell Rep 31, 107651 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wiedemann GM et al. Deconvoluting global cytokine signaling networks in natural killer cells. Nat Immunol 22, 627–638 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kurtulus S et al. Bcl-2 allows effector and memory CD8+ T cells to tolerate higher expression of Bim. J Immunol 186, 5729–5737 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dunkle A, Dzhagalov I, Gordy C & He YW Transfer of CD8+ T cell memory using Bcl-2 as a marker. J Immunol 190, 940–947 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sathe P et al. Innate immunodeficiency following genetic ablation of Mcl1 in natural killer cells. Nat Commun 5, 4539 (2014). [DOI] [PubMed] [Google Scholar]
- 39.Mujal AM, Delconte RB & Sun JC Natural Killer Cells: From Innate to Adaptive Features. Annu Rev Immunol 39, 417–447 (2021). [DOI] [PubMed] [Google Scholar]
- 40.Omilusik KD et al. Transcriptional repressor ZEB2 promotes terminal differentiation of CD8+ effector and memory T cell populations during infection. J Exp Med 212, 2027–2039 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dominguez CX et al. The transcription factors ZEB2 and T-bet cooperate to program cytotoxic T cell terminal differentiation in response to LCMV viral infection. J Exp Med 212, 2041–2056 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Omilusik KD et al. Sustained Id2 regulation of E proteins is required for terminal differentiation of effector CD8(+) T cells. J Exp Med 215, 773–783 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Delconte RB et al. The Helix-Loop-Helix Protein ID2 Governs NK Cell Fate by Tuning Their Sensitivity to Interleukin-15. Immunity 44, 103–115 (2016). [DOI] [PubMed] [Google Scholar]
- 44.Ikawa T, Fujimoto S, Kawamoto H, Katsura Y & Yokota Y Commitment to natural killer cells requires the helix-loop-helix inhibitor Id2. Proc Natl Acad Sci U S A 98, 5164–5169 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zook EC et al. Transcription factor ID2 prevents E proteins from enforcing a naive T lymphocyte gene program during NK cell development. Sci Immunol 3 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Madera S et al. Cutting Edge: Divergent Requirement of T-Box Transcription Factors in Effector and Memory NK Cells. J Immunol 200, 1977–1981 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rapp M et al. Core-binding factor beta and Runx transcription factors promote adaptive natural killer cell responses. Sci Immunol 2 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Grund EM, Spyropoulos DD, Watson DK & Muise-Helmericks RC Interleukins 2 and 15 regulate Ets1 expression via ERK1/2 and MNK1 in human natural killer cells. J Biol Chem 280, 4772–4778 (2005). [DOI] [PubMed] [Google Scholar]
- 49.Ramirez K et al. Gene deregulation and chronic activation in natural killer cells deficient in the transcription factor ETS1. Immunity 36, 921–932 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chen Z et al. In vivo CD8(+) T cell CRISPR screening reveals control by Fli1 in infection and cancer. Cell 184, 1262–1280 e1222 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
Methods Only References
- 51.Wang T et al. Gene Essentiality Profiling Reveals Gene Networks and Synthetic Lethal Interactions with Oncogenic Ras. Cell 168, 890–903 e815 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shen MW et al. Predictable and precise template-free CRISPR editing of pathogenic variants. Nature 563, 646–651 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hildreth AD, Riggan L & O’Sullivan TE CRISPR-Cas9 Ribonucleoprotein-Mediated Genomic Editing in Primary Innate Immune Cells. STAR Protoc 1, 100113 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Deng J et al. BH3 profiling identifies three distinct classes of apoptotic blocks to predict response to ABT-737 and conventional chemotherapeutic agents. Cancer Cell 12, 171–185 (2007). [DOI] [PubMed] [Google Scholar]
- 55.Stuart T et al. Comprehensive Integration of Single-Cell Data. Cell 177, 1888–1902 e1821 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bergen V, Lange M, Peidli S, Wolf FA & Theis FJ Generalizing RNA velocity to transient cell states through dynamical modeling. Nat Biotechnol 38, 1408–1414 (2020). [DOI] [PubMed] [Google Scholar]
- 57.Trapnell C et al. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat Biotechnol 32, 381–386 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dobin A et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Love MI, Huber W & Anders S Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yu B et al. Epigenetic landscapes reveal transcription factors that regulate CD8(+) T cell differentiation. Nat Immunol 18, 573–582 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kechin A, Boyarskikh U, Kel A & Filipenko M cutPrimers: A New Tool for Accurate Cutting of Primers from Reads of Targeted Next Generation Sequencing. J Comput Biol 24, 1138–1143 (2017). [DOI] [PubMed] [Google Scholar]
- 62.Langmead B & Salzberg SL Fast gapped-read alignment with Bowtie 2. Nat Methods 9, 357–359 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhang Y et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol 9, R137 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Anders S, Pyl PT & Huber W HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Heinz S et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 38, 576–589 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Sequencing datasets are accessible from GEO with accession number GSE176208. Original western blot scans can be found in source data.