

Abstract
Background:
Systemic inflammation amplifies neonatal hypoxic-ischemic (HI) brain injury. Azithromycin (AZ), an antibiotic with anti-inflammatory properties, improves sensorimotor function and reduces tissue damage after neonatal rat HI brain injury.
Objective:
To determine if AZ is neuroprotective in two neonatal rat models of inflammation-amplified HI brain injury.
Design/Methods:
Seven-day-old (P7) rats received injections of toll-like receptor agonists lipopolysaccharide (LPS) or Pam3Cys-Ser-(Lys)4 (PAM) prior to right carotid ligation followed by 50 min (LPS+HI) or 60 min (PAM+HI) in 8% oxygen. Outcomes included contralateral forelimb function (forepaw placing; grip strength), survival, %intact right hemisphere (brain damage), and a Composite Score incorporating these measures. We compared P35 outcomes in controls and groups treated with 3 or 5 AZ doses. Then, we compared P21 outcomes when the first (of 5) AZ doses was administered 1, 2, or 4 hours after HI.
Results:
In both LPS+HI and PAM+HI models, AZ improved sensorimotor function, survival, brain tissue preservation, and Composite Scores. Benefits increased with 5 vs. 3-dose AZ and declined with longer initiation delay.
Conclusion:
Perinatal systemic infection is a common comorbidity of neonatal asphyxia brain injury and contributes to adverse outcomes. These data support further evaluation of AZ as a candidate treatment for neonatal neuroprotection.
Introduction
Neonatal encephalopathy (NE) affects 1–4/1000 live term or near-term births in developed countries 1. Moreover, in low-middle income countries NE is up to 10-fold more frequent 2 and it is a leading cause of disability and early death 3. Although NE has often presumptively been attributed to antecedent fetal ischemia and/or hypoxia, there is increasing recognition of multiple potential contributing mechanisms and etiologies 4, 5. Maternal or fetoplacental infections are common predisposing factors or co-morbidities of neonatal encephalopathy 6, and are associated with heightened risks for adverse long-term neurodevelopmental outcomes 7. Yet, when infants present acutely with neonatal encephalopathy in the setting of maternal chorioamnionitis, the responsible pathogen is usually unknown and most often the neonate does not have positive blood or CSF cultures 8. Among the many intraamniotic organisms associated with chorioamnionitis in term infants 9, 10, the Gram-positive organism Group B Streptococcus is the most common invasive pathogen identified, and the Gram-negative E. coli is the second most common 11, 12. Thus, evaluation of interactions between both Gram-positive and Gram-negative organisms and acute hypoxic-ischemic injury in relevant neonatal models are important.
The complex pathophysiologic interactions between perinatal infection and acute hypoxic-ischemic brain injury can be modeled experimentally 13. The best-defined strategies incorporate systemic administration of a pro-inflammatory stimulus, most commonly a toll-like receptor (TLR) agonist, followed by a standard lesioning method to elicit unilateral hypoxic-ischemic brain injury. Hagberg and colleagues first showed that systemic injection of a very low dose of E. coli-derived lipopolysaccharide (LPS, a TLR4 agonist) heightened vulnerability to HI brain injury, elicited by unilateral carotid artery ligation and timed exposure to moderate hypoxia in immature (7 day old) rats 14. In the neonatal HI lesion model upon which this study was based, the severity of brain injury is dependent on duration of hypoxia exposure 15, 16; with antecedent LPS administration, the temporal thresholds for brain injury were substantially reduced 14, i.e. shorter durations of hypoxia resulted in brain injury. These findings suggested that systemic infections with Gram-negative bacteria could heighten susceptibility to neonatal HI brain injury. LPS-induced post-HI hypoglycemia was suggested as the underlying mechanism, but treatment with supplemental glucose only partially attenuated LPS-amplified brain injury 17. Subsequent work implicated LPS-activated microglia as mediators of amplified brain injury in the rodent model 18–20.
Neonates are susceptible to both Gram-negative and Gram-positive bacterial infections. Bacteria-derived lipoproteins that activate TLR2 represent a pivotal pathogenic mechanism in group B streptococcal sepsis 21. Interest in understanding whether Gram-positive bacteria-mediated TLR2 activation could exert similar deleterious effects to the E. coli-derived TLR4 agonist prompted complementary studies with a synthetic lipopeptide TLR2 agonist Pam3Cys-Ser-(Lys)4 (Pam3CSK4, “PAM”) 22–24. In fact, PAM pre-treatment also amplified HI brain injury, but distinct differences between the two pro-inflammatory stimuli emerged, with respect to both physiological adverse effects (more pronounced with LPS) and magnitude and temporal profile of brain cytokine expression 22.
Azithromycin (AZ), a generally safe, widely used antibiotic, has intrinsic anti-inflammatory properties 25, and accumulates in brain tissue 26, 27. Evidence of AZ’s safety in human neonates was provided by the results of a series of studies that were undertaken to evaluate the impact of AZ treatment on Ureaplasma eradication in premature infants <29 weeks gestation at birth 28–31. In view of the pivotal role of inflammation in the pathogenesis of diverse forms of neonatal brain injury 13, we initially evaluated AZ’s neuroprotective efficacy in the neonatal rat HI brain injury model, elicited by unilateral carotid artery ligation and subsequent hypoxia exposure; AZ treatment, even when initiated several hours after the end of hypoxia, improved functional outcomes and reduced brain injury 32. Congruent findings are reported in adult rodent stroke 33.
In this study, building on our findings and knowledge of AZ’s immune-modulatory properties, we hypothesized that AZ could attenuate inflammation-amplified HI injury and incorporated experiments to evaluate AZ neuroprotection in both LPS- and PAM-amplified HI brain injury models. We selected AZ treatment protocols that incorporated delayed onset of initial drug administration (from 1–4 hours after the end of hypoxia exposure) and multiple dose regimens (up to 5 doses) to model translationally feasible clinical scenarios.
Methods
Overview.
Our general approaches include randomizing concurrently lesioned animals from a single litter to drug-treated and saline-control groups, and distributing males and females evenly among groups. We only include non-lesioned littermate controls when we are evaluating a functional outcome that is non-lateralizing, such as a “cognitive” measure. Dose selection and timing of azithromycin administration were guided by results of prior studies in our laboratory 32, 34. In pilot experiments, based on our experiences and protocols from other laboratories 35–37, we identified LPS and PAM administration protocols (dose and timing), coupled with hypoxia-ischemia durations, that yielded brain injury and relatively low mortality.
All outcome assessments included comparisons of performance on two sensorimotor function measures (see below) and quantification of brain injury. We implemented a novel approach to integrate function and pathology measures, and mortality, by calculating composite outcome scores (see below). In initial neuroprotection experiments, sensorimotor testing was continued weekly until P35 and brain injury was evaluated in coronal histopathology sections. In subsequent experiments to evaluate the impact of timing of treatment onset on neuroprotective efficacy, outcomes were evaluated on P21. All protocols were approved by the University of Michigan Animal Care and Use Committee.
Animal lesioning.
Isofluorane-anesthetized P7 Wistar rats underwent right carotid artery ligation, recovered in incubators (90 min, at 36.5°C, Hovabator, GQF, Savannah, GA), were placed in covered acrylic containers partially submerged in a water-bath (36.5 °C), and exposed to 8% oxygen/balance nitrogen 32, for hypoxia durations as specified for each protocol below. They recovered in incubators (15 min, 36.5°C), and then returned to dams.
Pro-inflammatory Challenges:
To model the clinical scenario of hypoxic-ischemic (HI) brain injury in the setting of Gram-negative systemic infection, lipopolysaccharide (LPS, E. coli, O55:B5, Sigma-Aldrich, St. Louis, MO, 0.05 mg/kg), a Toll-like receptor 4 (TLR4) agonist, was injected intraperitoneally (i.p.), 2.5 h prior to carotid ligation, and 1.5 h post-ligation, exposure to 50 min 8% O2 began 20, 35; this protocol is designated as “LPS+HI”. To model concurrent Gram-positive systemic infection, the synthetic lipopeptide TLR2 agonist Pam3Cys-Ser-(Lys)4 (Pam3CSK4, PAM, Sigma-Aldrich, 0.5 mg/kg) was injected i.p. 4.5 h prior to carotid ligation, and 1.5 h post-ligation, exposure to 60 min 8% O2 started 37; this protocol is designated as “PAM+HI”.
Physiology measurements.
Animals were weighed on P7, on each subsequently scheduled injection day, and then weekly from P14. Rectal temperatures were uniformly measured (YSI thermometer 43T with probe 554; YSI, Yellow Springs, OH) before surgery, at the end of hypoxia, and 15 min, 60 min, 120 min, and 24 h later, and also at 48, 72 and 96 h in one LPS+HI experiment and in all PAM+HI experiments. Blood glucose was measured in representative litters at several time points after HI, including immediately before AZ, and up to 24 h after HI (Germaine Laboratories AimStrip Plus Blood Glucose Testing System and Test Strips, Germaine Laboratories, San Antonio, TX).
Azithromycin (AZ) treatment.
Sterile lyophilized AZ (Fresenius Kabi USA, Lake Zurich, IL) was dissolved in sterile 0.9% saline, as required to achieve uniform i.p. injection volumes of 0.1ml per 10 gm body weight; routine intravenous injections are not feasible at this age. Controls were injected with 0.1 ml per 10 gm body weight of sterile saline. In both LPS+HI and PAM+HI models we compared P35 outcomes of 3-dose (initial 45 mg/kg at 2 h after HI, followed by 22.5 mg/kg at 24 and 48 h after HI, “AZ*3”), and 5-dose AZ regimens (adding 22.5 mg/kg doses at 72 and 96 h after HI, “AZ*5”) with outcomes in saline-injected controls (see Table 1). In subsequent experiments in each model, we evaluated the impact of the timing of treatment onset on the efficacy of the 5-dose AZ regimen, comparing initial dose administration at 1, 2, or 4 h after the end of HI with saline-injected controls; outcomes were assessed at P21 (see Table 1).
Table 1:
Azithromycin (AZ) Treatment Allocation.
| Group | Number of injections | Post-HI treatment (h after end of HI) | N | Deaths |
|---|---|---|---|---|
| LPS+HI a: 3 (AZ*3) vs. 5 (AZ*5) dose regimens b | ||||
| Saline | 5 | 2, 24, 48, 72, 96 | 15 | 1 |
| AZ*3 | 3 | 2, 24, 48 | 16 | 2 |
| AZ*5 | 5 | 2, 24, 48, 72, 96 | 16 | 1 |
| LPS+HI a: Initial-dose time-dependence of AZ*5 neuroprotection | ||||
| Saline | 5 | 2, 24, 48, 72, 96 | 12 | 4 |
| AZ*5 1h | 5 | 1, 24, 48, 72, 96 | 12 | 0 |
| AZ*5 2h | 5 | 2, 24, 48, 72, 96 | 12 | 0 |
| AZ*5 4h | 5 | 4, 24, 48, 72, 96 | 12 | 0 |
| PAM+HI c: 3 (AZ*3) vs. 5 (AZ*5) dose regimens b | ||||
| Saline | 5 | 2, 24, 48, 72, 96 | 8 | 2 |
| AZ*3 | 3 | 2, 24, 48 | 8 | 0 |
| AZ*5 | 5 | 2, 24, 48, 72, 96 | 8 | 1 |
| PAM+HI c: Initial-dose time-dependence of AZ*5 neuroprotection | ||||
| Saline | 5 | 2, 24, 48, 72, 96 | 5 | 1 |
| AZ*5 1h | 5 | 1, 24, 48, 72, 96 | 6 | 0 |
| AZ*5 2h | 5 | 2, 24, 48, 72, 96 | 5 | 0 |
| AZ*5 4h | 5 | 4, 24, 48, 72, 96 | 5 | 0 |
LPS+HI: Lipopolysaccharide (LPS, 0.05 mg/kg) was injected i.p., 2.5 h prior to right carotid artery ligation; 1.5 h later, exposure to 50 min 8% O2 began.
AZ*3: 45, 22.5, 22.5 mg/kg/dose; AZ*5: 45, 22.5, 22.5, 22.5, 22.5 mg/kg/dose. Saline and AZ injections were i.p.
PAM+HI: The synthetic lipopeptide TLR2 agonist Pam3Cys-Ser-(Lys)4 (PAM, 0.5 mg/kg) was injected i.p. 4.5 h prior to right carotid artery ligation, and 1.5 h later, exposure to 60 min 8% O2 began.
Sensorimotor function testing.
Performance was evaluated with two independent tests that consistently demonstrate asymmetric sensorimotor deficits in this model; videos of these tests are available on-line 32. Vibrissae-stimulated forepaw placing (10 trials/side) was tested weekly from P14 up to P21 or P35; one point was assigned for full and 0.5 points for partial extension (normal=10/10 bilaterally) 38, 39. Right forepaw placement was consistently normal, and only left forepaw scores are reported. Forepaw grip strength was measured (3 trials/side, Bioseb Grip Traction Meter model BIO-GS3, Forceleader DBA Bioseb, Pinellas Park, FL) on P21 (youngest age at which reliable measures can be obtained) or weekly from P21 to P35 39. In normal rats, grip strength increases from P21-P35 and remains bilaterally symmetric. To account for expected age-related increases in grip strength, results are expressed as left/right (L/R) forepaw grip strength ratios. After P7 HI lesioning, performance deficits on both measures are similar at P21 and P35 32 (also see Supplementary Data, Table S4).
Brain injury measures:
Animals were euthanized on P35 or P21 by i.p. pentobarbital injection (Fatal Plus ®, Vortech Pharmaceuticals, Dearborn, MI). In experiments that evaluated P35 outcomes, brains were rapidly frozen, and coronal brain sections (20 microns) were cresyl-violet stained. Bilateral cross-sectional areas of striatum, neocortex, hippocampus, and entire cerebral hemisphere were measured (using NIH Image J) on regularly spaced sections (mean 19 sections/brain; distance between sections 256 μm in LPS+HI experiments and 320 μm in PAM+HI experiments) from the level of the anterior genu to the posterior genu of the corpus callosum. Bilateral regional and hemisphere intact tissue volumes were calculated by summing areas and multiplying the sum by the distance between sections 39. As measures of injury severity, right-sided % Intact regional or whole hemisphere tissue volumes were expressed as [100*(right volume)/(left volume)]. In experiments that compared outcomes at P21, right and left cerebral hemispheres were separated and weighed; difference in hemisphere weight is a validated measure of hemisphere volume difference in this model 40.
For composite outcome score calculations (see below), either % Intact right hemisphere volume (P35) or % right hemisphere weight relative to left [100*(right/left hemisphere weights)] values (P21) were included.
Composite Outcome Score:
To generate a global outcome measure that would also take into account survival differences, we developed a 30-point composite outcome incorporating the two function measures, % Intact right hemisphere, and survival. The best possible Composite Score is 30. Scores were calculated as: {[left forepaw vibrissae score (maximum=10)] + [(L/R forepaw grip ratio) x 10] + [(% Intact right hemisphere)/10]}. Animals that died before completion of testing were assigned scores of 0 for each measure, and these values were included in all outcome assessments.
Data analysis:
Sequential rectal temperatures and body weights were compared by repeated measures ANOVA (using Prism 8.4.3, GraphPad Software, San Diego, CA, for all analyses). Impact of AZ treatment on blood glucose values after HI was evaluated by t-test or repeated-measures ANOVA. Non-parametric Kruskal Wallis testing was applied to compare sensorimotor measures, % intact right hemisphere and Composite score among AZ treatment groups and controls, since, by visual inspection, data were not normally distributed in many groups. To evaluate the decrement in AZ benefit as treatment delay increased, a post-hoc ANOVA linear test for trend was performed across ordered AZ 1h, 2h, 4h and NS control groups. 2-way ANOVA, factoring group and age, was applied to compare sensorimotor test scores at P21 and P35 in both models, and also, factoring treatment and sex, to evaluate for sex differences in the impact of AZ treatment on sensorimotor testing measures, % intact right hemisphere, and Composite Score. Repeated measures ANOVA, factoring region (as repeated measure) and treatment, was applied to evaluate regional specificity of AZ treatment effects.
Results
Physiology
In studies of P7 rat HI lesioning that incorporate hypoxia exposure durations of at least 90 min, typical mortality rates are <5%. Based on anticipated deleterious effects of LPS and PAM, in these “double-hit” models, hypoxia durations were shortened. Yet, in both models post-HI mortality was relatively high in saline-treated controls: LPS+HI (5/27; 19%); PAM+HI (3/13; 23%). In the LPS+HI model mortality was significantly reduced in AZ-treated groups (saline 5/27 vs. AZ 3/67; p<0.05 Fisher’s exact test). In the PAM+HI model, there was a non-significant trend to lower mortality in the AZ groups (saline 3/13 vs. AZ 1/32; p=0.07, Fisher’s exact test).
AZ treatment had no impact on body temperature for up to 4 days after HI (Supplementary Data Table S1, available online). Body weights from P8 through P21 or P35 did not differ between AZ-treated and control groups (not shown). Post-HI blood glucose concentrations differed between models; LPS-exposed animals were hypoglycemic whereas values in PAM-exposed animals were in the same range as controls. AZ treatment did not alter blood glucose concentrations in either group, and values in all groups were in the normal range at 24 h post-HI (see Supplementary Data Table S2).
Azithromycin improves outcomes of LPS-amplified hypoxic-ischemic injury.
Figure 1, panels a-d, compare P35 function and brain injury measures, along with Composite Score in three LPS+HI groups: saline controls (NS), animals that received 3 doses of AZ (AZ*3), and animals that received 5 AZ doses (AZ*5). Left forepaw placement (scored in 10 trials, Fig. 1a) was markedly impaired in NS controls, significantly better in the AZ*3 group, and close to normal in the AZ*5 group (p<0.0001 Kruskal-Wallis; p<0.05 Dunn’s multiple comparisons test for all 3 between-group differences). Left(L)/Right (R) grip strength ratios (normal=1) were markedly reduced in NS controls, significantly higher in the AZ*5-treated group, and higher in the AZ*5 than AZ*3 group (p<0.0001 Kruskal-Wallis; p<0.05 Dunn’s multiple comparisons test for NS vs. AZ*5 and AZ*3 vs. AZ*5). The extent of right cerebral hemisphere volume loss (Fig 1.c) was more variable, and significantly different from controls only in the AZ*5 group (p<0.005 Kruskal-Wallis with Dunn’s multiple comparisons test, also see Table 2). Overall, Composite scores (best possible=30) were significantly higher than controls in both AZ*3 and AZ*5 groups (p<0.0001, Kruskal-Wallis), and better in AZ*5 than AZ*3 groups (p<0.05 Dunn’s).
Figure 1. Azithromycin (AZ) treatment Improves Outcomes after LPS-Amplified Neonatal Cerebral Hypoxia-Ischemia (HI).
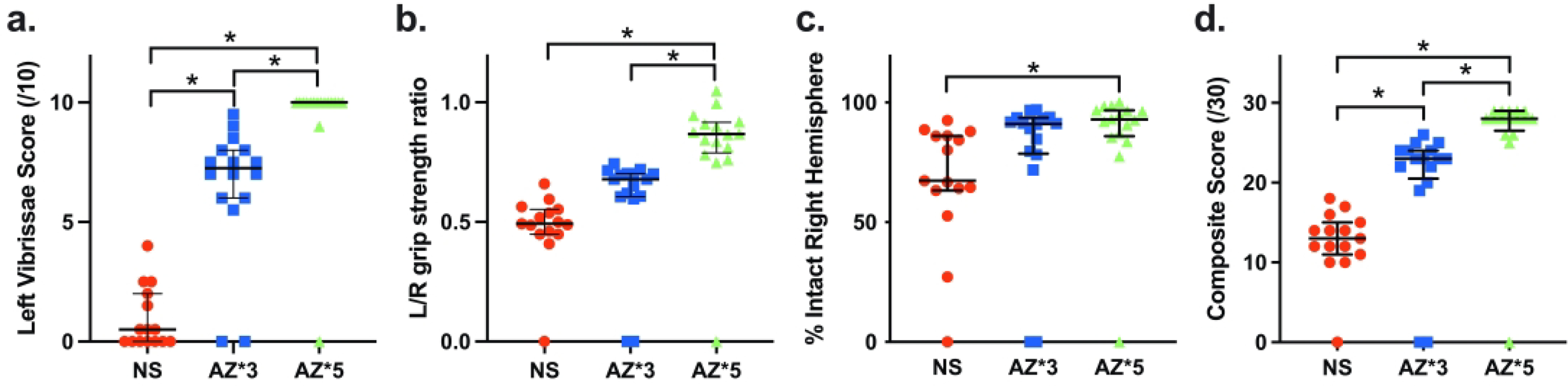
P7 rats received i.p. LPS injections 4 h prior to initiation of 50 min HI (LPS+HI), and were allocated to 3-dose AZ (AZ*3), 5-dose AZ (AZ*5), or saline control (NS) groups (see Methods). Panels a-c compare sensorimotor function and % residual intact right hemisphere volumes on P35; panel d compares outcomes with a 30-point Composite Score that incorporates these measures and also reflects survival (see Methods). Horizontal bars represent median and IQR. Both AZ treatments improved Composite Score vs. NS (d: p<0.0001 Kruskal Wallis test). Composite Score and functional performance were superior in AZ*5 vs. AZ*3 groups (a, b, d; *p<0.05 Dunn’s multiple comparisons test); all AZ*5 scores were superior to controls, as were AZ*3 sensorimotor test scores.
Table 2:
Reduction of region-specific neuropathology by a 5-dose Azithromycin (AZ) Regimen vs. a 3-dose Regimen, after LPS-amplified Hypoxia-Ischemia (LPS+HI) a or Pam3CSK4-amplified Hypoxia-Ischemia (PAM+HI) b.
| a. LPS-amplified HI a | |||||||||||
| Treatment | Side | P35 Volume c (mean ±SD, mm3) and %Intact Right c (mean ±SD) | |||||||||
| Cortex | Striatum | Hippocampus | Other d | Hemisphere | |||||||
| Volume | %Intact | Volume | %Intact | Volume | %Intact | Volume | %Intact | Volume | %Intact | ||
| Saline | Left | 118 ±6 | 36 ±3 | 13 ±3 | 141 ±10 | 287 ±81 | |||||
| Right | 79±30 | 62±29 | 20 ±8 | 53 ±28 | 10 ±3 | 72 ±27 | 115 ±24 | 76 ±25 | 209 ±83 | 67 ±25 | |
| AZ*3 | Left | 126 ±9 | 37 ±3 | 16±2 | 151 ±9 | 288±114 | |||||
| Right | 114±11 * | 80 ±32 | 30±7 * | 69±30 | 13 ±3 | 70 ±29 | 136±11 * | 79 ±32 | 256 ±103 | 78 ±31 | |
| AZ*5 | Left | 124±8 | 37 ±4 | 15±2 | 148 ±8 | 304 ±83 | |||||
| Right | 116 ±8 * | 87±24 † | 32 ±5 * | 81±24 † | 13±2 * | 80 ±23 | 139±14 * | 88 ±25 | 281 ±79 | 87 ±24 ‡ | |
| b. PAM-amplified HI b | |||||||||||
| Treatment | Side | P35 Volume c (mean ±SD, mm3) and %Intact Right c (mean ±SD) | |||||||||
| Cortex | Striatum | Hippocampus | Other d | Hemisphere | |||||||
| Volume | %Intact | Volume | %Intact | Volume | %Intact | Volume | %Intact | Volume | %Intact | ||
| Saline | Left | 110 ±9 | 40 ±3 | 17 ±2 | 129 ±11 | 222 ±138 | |||||
| Right | 66±14 | 45±30 | 18 ±4 | 34 ±22 | 11 ±3 | 48 ±31 | 97 ±17 | 56 ±35 | 144 ±93 | 48 ±31 | |
| AZ*3 | Left | 110 ±18 | 39 ±8 | 17 ±4 | 129 ±24 | 295 ±52 | |||||
| Right | 92 ±24 | 83 ±9 † | 29 ±9 * | 72±10 † | 13 ±3 | 81 ±10 † | 117 ±28 | 90 ±8 | 251 ±63 | 84 ±8 ‡ | |
| AZ*5 | Left | 112±18 | 39 ±8 | 16±2 | 128 ±19 | 258±112 | |||||
| Right | 97 ±22 * | 75±31 | 31 ±9 * | 68±29 † | 14±2 | 75 ±33 | 127±24 | 87 ±35 | 236 ±108 | 79 ±32 ‡ | |
LPS+HI: Lipopolysaccharide (LPS, 0.05 mg/kg) was injected intraperitoneally (i.p.), 2.5 h prior to right carotid artery ligation in seven-day-old (P7) rats, and 1.5 h later exposure to 50 min 8% O2 started (see Methods). Treatment began with i.p. injections 2 h after HI (n=15–16/group): AZ 3-dose (AZ*3: 45, 22.5, 22.5 mg/kg/dose at 2, 24 and 48 h) vs. 5-dose (AZ*5: 45, 22.5, 22.5, 22.5, 22.5 mg/kg/dose, 2, 24, 48, 72 and 96 h) vs. saline controls (5 injections, same times as AZ*5).
PAM+HI: The synthetic lipopeptide TLR2 agonist Pam3Cys-Ser-(Lys)4 (PAM, 0.5 mg/kg) was injected i.p. 4.5 h prior to right carotid artery ligation in seven-day-old (P7) rats, and 1.5 h later exposure to 60 min 8% O2 started (see Methods). Treatment regimens (n=8/group) were the same as in LPS+HI.
Bilateral regional and hemisphere volumes were calculated on postnatal day 35 (P35) from bilateral regional area measurements of intact-staining tissue in regularly spaced coronal sections (see Methods). Regional volumes, expressed as means ± SD, were compared by RM ANOVA, factoring region and treatment, with region as repeated measure. %Intact tissue on the right (R), compared to non-lesioned left (L), a measure of damage severity, was calculated for each region (and hemisphere) from bilateral volumes, as 100*(R/L); animals that die before P35 are assigned a volume of 0 and a %intact value of 0.
“Other” represents all other hemisphere tissue that was not designated as either cortex, striatum or hippocampus, and includes both grey matter regions (e.g. thalamus, globus pallidus, septum) and white matter tracts.
For LPS+HI (a.)
Right regional volumes were greater in AZ*5 than in controls for all 4 regions, and were greater in AZ*3 than controls for cortex, striatum and “other” (p<0.0001 overall by repeated measures ANOVA factoring treatment and region, with p<0.05 Tukey multiple comparison test for within-region comparisons).
%intact R was greater in cortex and striatum in AZ*5 than in controls, by post-hoc testing (p<0.05, Tukey multiple comparison test).
%intact R hemisphere was greater in AZ*5 than in controls (p<0.05 Kruskal Wallis with Dunn’s multiple comparisons test).
For PAM+HI (b.)
Right regional volumes were greater in AZ*5 than in controls for cortex and striatum, and were greater in AZ*3 than controls for striatum (p<0.05 overall by repeated measures ANOVA factoring treatment and region, with p<0.05 Tukey post-hoc multiple comparison test for within-region comparisons). There was no significant difference in mean R volumes between AZ*3 and AZ*5.
%intact R was greater in cortex, striatum and hippocampus in AZ*3, and greater in striatum in AZ*5 than in controls, by post-hoc testing (p<0.05, Tukey multiple comparison test).
%intact R hemisphere was greater in AZ*3 and AZ*5 than in controls (p<0.05 Kruskal Wallis with Dunn’s multiple comparisons test).
Figure 2, panels a-f, illustrate the features of P35 histopathology at the levels of striatum (a, c, e) and dorsal hippocampus (b, d, f) in samples from each of the three LPS+HI groups: saline-controls (a, b), the 3-dose AZ regimen (c, d; AZ*3), or the 5-dose AZ regimen (e, f; AZ*5). In the LPS+HI control (a, b), overall right hemisphere, striatal, and hippocampal atrophy are evident, as well as cortical thinning and cystic encephalomalacia. The AZ*3 sample (c, d) demonstrates greater tissue preservation, with mild right hemisphere and striatal atrophy, and minimal hippocampal atrophy. In the AZ*5 sample (e, f), only very mild right striatal (*) and hippocampal tissue loss are evident. Table 2.a provides results of regional morphometry to evaluate the extent and distribution of tissue injury in each treatment group. % Intact values, calculated as 100*(Right/Left) volumes were included as indices of tissue damage. Although there was variability in the extent of tissue damage within each group, as commonly encountered in the P7 rat HI model, these data confirmed that AZ*5 conferred significant neuroprotection across all regions while AZ*3 benefits were more variable.
Figure 2. Azithromycin (AZ) Treatment Attenuates LPS-Amplified Hypoxic-Ischemic (HI) Neuropathology.
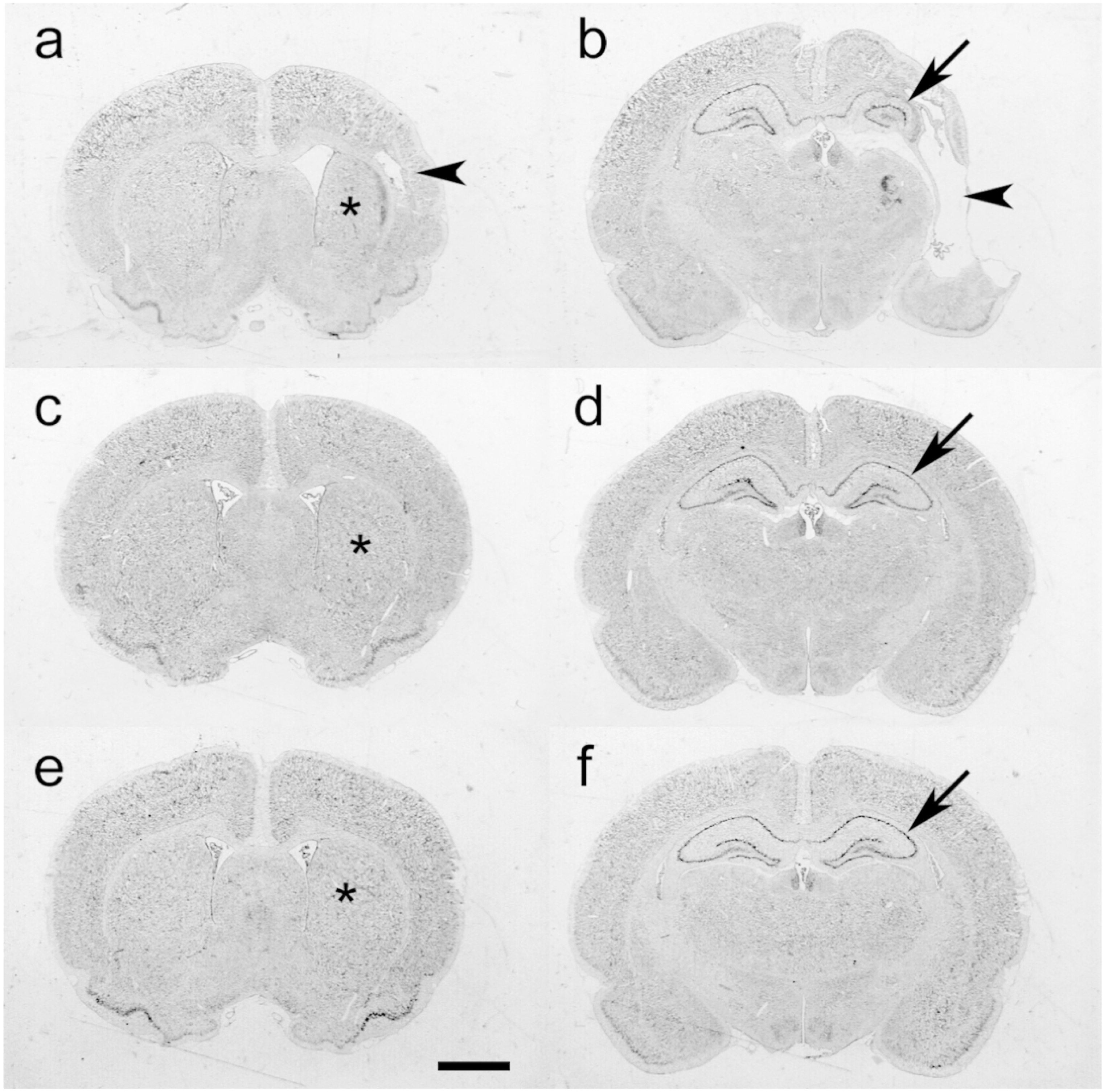
Panels a-f illustrate representative cresyl-violet stained coronal brain sections, obtained at P35, in samples from the 3 groups compared in Fig. 1. a–d. P7 rats received LPS injections followed by 50 min HI (LPS+HI; see Methods). Two h after the end of HI, animals started treatment with saline (a, b), the 3-dose AZ regimen (c, d; AZ*3), or the 5-dose AZ regimen (e, f; AZ*5). Sections a, c, and e are at the level of striatum (*); sections b, d and f are at the level of the dorsal hippocampus (arrows). In the LPS+HI control (a, b), note overall right hemisphere atrophy, striatal atrophy (*), marked hippocampal atrophy (arrow), and cortical thinning and cystic encephalomalacia (arrowheads). The AZ*3 sample (c, d) demonstrates mild right hemisphere and striatal atrophy (*), and minimal hippocampal atrophy (arrow). In a representative AZ*5 animal (e, f), only very mild right striatal (*) and hippocampal (arrow) tissue loss are evident. (Scale bar = 2 mm).
In the preceding experiments, a treatment onset time point 2 hours after the end of hypoxia was selected, based both on the goal of modeling a translationally relevant clinical scenario and also on knowledge that AZ treatment, initiated at this time point, was neuroprotective in P7 rats that underwent 90 min HI 32. To re-evaluate the impact of timing of treatment onset in the LPS+HI model, we focused on the AZ*5 regimen and compared outcomes on P21 when the first AZ dose had been administered 1, 2, or 4 h after the end of HI. Fig. 3, panels a-d, summarize the results. Median Composite scores at P21 (and each component measure) were superior to control values when AZ was initiated at 1 or 2 hours, but not at 4 hours after the end of HI (Fig. 3 a–d, p<0.0001, Kruskal-Wallis; * p≤0.0001 Dunn’s post-hoc). A linear decrement in AZ efficacy with increasing delay in treatment initiation was evident (p<0.0001 post-hoc linear test for trend and a trend toward some benefit vs. controls even in the 4 h group).
Figure 3. Impact of Time of Initiation on the Efficacy of Azithromycin Neuroprotection in LPS+HI.
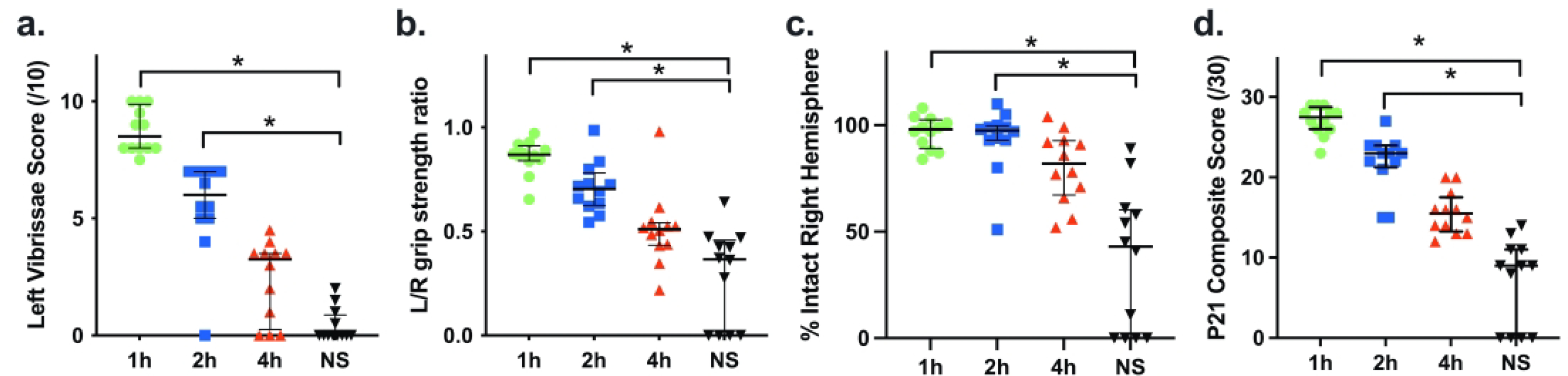
Outcomes of AZ*5 regimens with initial dose administration at 1, 2, or 4 h after the end of HI were compared with NS controls (a-d) were assessed at P21; %intact right hemisphere (c) was calculated using bilateral hemisphere weights. AZ*5 treatment initiated at 1 or 2 h post-HI, but not at 4 h, conferred improvements in each measure and Composite Score compared to controls (p<0.0001, Kruskal-Wallis, *p≤0.0001 Dunn’s). A linear decrement in AZ efficacy with increasing delay in treatment initiation was evident (p<0.0001 post-hoc linear test for trend).
We incorporated both P35 and P21 Composite scores to evaluate if there were sex-differences in AZ efficacy (Supplementary Data Table S3). Composite scores did not differ in saline controls (mean±SD: female: 10.1±6.0, male: 10.1±5.1) or in AZ*5-treated groups (female: 23.3±7.1; male: 22.4±7.2).
Azithromycin improves outcomes of PAM-amplified hypoxic-ischemic injury.
Note that based on prior studies and our pilot data (not shown) we implemented a PAM+HI model that differed from the LPS+HI model both with respect to time interval between PAM administration and subsequent HI lesioning (6 h, rather than 4h earlier), and HI duration (60 min rather than 50 min). Overall, AZ neuroprotective efficacy trends were similar in both models. Figure 4, panels a-d, compare P35 function and brain injury measures, along with Composite scores in three corresponding PAM+ HI groups: saline controls (NS), animals that received 3 doses of AZ (AZ*3), and animals that received 5 AZ doses (AZ*5). Controls all had significant sensorimotor deficits (Fig. 4 a, b), and right hemisphere brain damage (0 values represent animals that died prior to P35). Both AZ*5 and AZ*3 improved scores on all measures, compared with controls (p<0.005 Kruskal Wallis); however, there were no significant differences in outcomes between AZ*5 and AZ*3 groups in these analyses. Regional morphometry measures (Table 2. b) demonstrated similar trends, although significantly greater tissue preservation for both AZ treatments was only found in comparisons of striatal and hemisphere volumes (p<0.05, ANOVA and post-hoc Tukey’s tests).
Figure 4. Azithromycin (AZ) treatment Improves Outcomes after PAM-Amplified Neonatal Cerebral Hypoxia-Ischemia (HI).
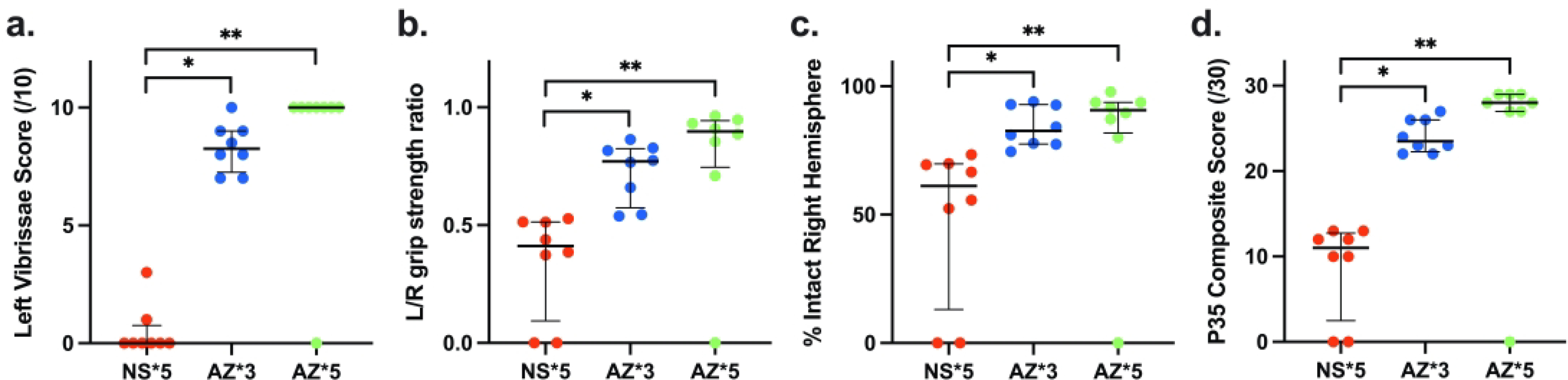
P7 rats received i.p. injections of Pam3CSK4 6 h prior to 60 min HI (PAM+HI), and were allocated to 3-dose AZ (AZ*3), 5-dose AZ (AZ*5), or saline control (NS) groups (see Methods). Panels a-c compare sensorimotor function and % intact right hemisphere volumes on P35 in the 3 groups; panel d compares outcomes with a 30-point Composite Score that incorporates these measures and also reflects survival (see Methods). Horizontal bars represent median and IQR. There was a significant AZ treatment benefit by all measures (p<0.005 Kruskal-Wallis). In both AZ treatment groups, for each measure results were superior to values in controls (* p<0.05, Dunn’s multiple comparisons test); scores did not differ significantly between the AZ*5 and AZ*3 groups.
Figure 5 illustrates histopathology in samples from each of the groups compared in Figure 4. a–d. at the levels of striatum (a, c, e) and dorsal hippocampus (b, d, f) in samples from each of the three PAM+HI groups: saline-controls (a, b), the 3-dose AZ regimen (c, d; AZ*3), or the 5-dose AZ regimen (e, f; AZ*5). In a control sample (a, b), right hemisphere, striatal (*), and hippocampal (arrow) atrophy along with cortical thinning (arrowhead, a) or cystic encephalomalacia (arrowhead, b) are again evident, similar to the LPS+HI pattern (compare with Figure 2. a–b). A representative AZ*3 sample (c, d) illustrates subtle right hemisphere tissue loss, striatal atrophy (*), and mild hippocampal atrophy together with pyramidal cell layer thinning (arrow). A representative AZ*5 sample (e, f) demonstrates mild right striatal (*) and hippocampal (arrow) tissue loss but no evidence of cortical injury.
Figure 5. Azithromycin (AZ) Treatment Attenuates PAM-amplified Hypoxic-Ischemic (HI) Neuropathology.
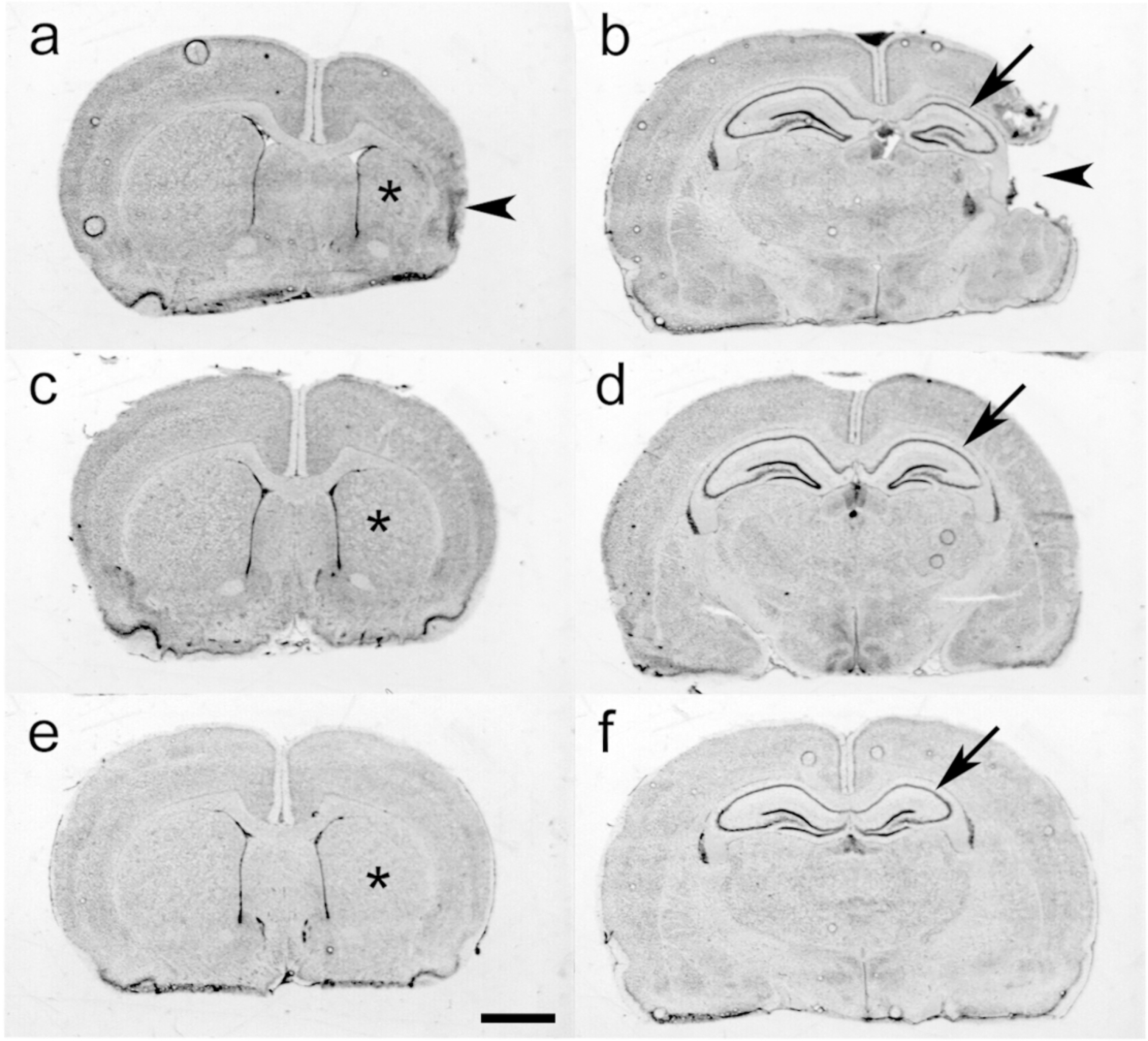
Panels a-f illustrate representative cresyl-violet stained coronal brain sections, obtained at P35, in samples from the 3 groups compared in Fig. 4. a–d. P7 rats received injections of Pam3CSK4, followed by initiation 60 min HI (PAM+HI; see Methods). Two h after the end of HI, animals started treatment with saline (a, b), the 3-dose AZ regimen (c, d; AZ*3), or the 5-dose AZ regimen (e, f; AZ*5). In a representative control sample (a, b), right hemisphere, striatal (*), and hippocampal (arrow) atrophy along with cortical thinning (arrowhead, a) or cystic encephalomalacia (arrowhead, b) are evident. A representative AZ*3 sample (c, d) illustrates subtle right hemisphere tissue loss, striatal atrophy (*), and mild hippocampal atrophy together with pyramidal cell layer thinning (arrow). A representative AZ*5 sample (e, f) demonstrates mild right striatal (*) and hippocampal (arrow) tissue loss but no evidence of cortical injury. (Scale bar = 2 mm).
Figure 6 a–d summarizes results of experiments that evaluated the impact of the timing of treatment onset on AZ*5 efficacy in the PAM+HI model; P21 outcomes were compared after AZ treatment onset 1, 2, or 4 h after the end of HI, and in NS controls. Treatment initiated at 1 or 2 hours after the end of HI improved outcomes with significantly higher median Composite scores than saline controls (Fig. 6 d, p<0.001, Kruskal-Wallis); scores were not significantly higher than controls when treatment was delayed for 4 h. For all measures there was a significant linear trend of decrement in AZ efficacy as delay to initiation increased from 1 h to 4 h vs. saline (p<0.05, ANOVA post-hoc linear test for trend).
Figure 6. Impact of Time of Initiation on the Efficacy of Azithromycin Neuroprotection in PAM+HI.
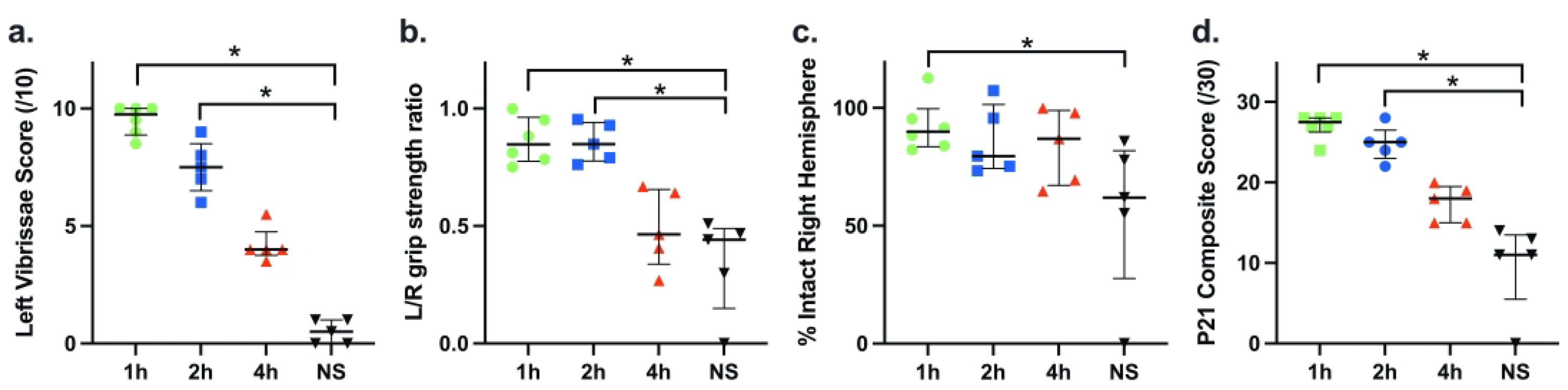
Outcomes of AZ*5 regimens with initial dose administration at 1, 2, or 4 h after the end of HI were compared with NS controls; outcomes were assessed at P21 (a-d) as in Fig. 3. AZ*5 treatment initiated at 1 or 2 h post-HI, but not at 4 h, conferred improvements in Composite Score (d) and sensorimotor test measures (a, b) (p<0.005, Kruskal-Wallis; * p<0.05 Dunn’s); %intact right hemisphere was only significantly greater than controls for the 1 h delay group (c; *p<0.05 Dunn’s). Overall, a linear decrement in AZ efficacy with increasing delay in treatment initiation from 1 h to 4 h vs. NS was evident (p<0.0001 post-hoc linear test for trend).
To evaluate if there were sex-differences in AZ efficacy we incorporated both P35 and P21 Composite scores (Supplementary Data Table S3). Composite scores differed between female and male controls (mean±SD: female: 5.7±6.3, male: 12.1±1.3); increased numbers of deaths in females accounted for this difference. Modified Composite scores (excluding deaths, which are assigned “0” in Composite scores) were also calculated to distinguish treatment effects on survival and brain damage severity. Modified Composite Scores did not differ between sexes (female: 11.3±1.5, male: 12.1±1.3) i.e. in surviving animals sensorimotor deficits and brain damage did not differ. AZ*5 treatment was associated with increased survival in females (saline: 3/6 vs AZ*5 9/10) and significantly improved both Composite and Modified Composite scores in both sexes.
To further evaluate the relationship between sensorimotor function measures at P21 and P35, we incorporated results from all experiments in which performance was assessed in the same animals at both time points and analyzed the data in two ways (see Supplementary Data Table S4 and Figure S1). In each model, for each test measure, results at P21 vs. P35 did not differ for any group, by 2-way ANOVA factoring treatment and age, with Tukey’s multiple comparison test. In addition, comparing scores for each animal at the two ages (n=64) revealed very strong relationships (simple linear regression: forepaw placing score: R2 =0.84, p<0.0001; L/R grip ratio: R2 =0.54, p<0.0001; see Supplementary Figure S1).
Discussion
In two complementary rodent models of inflammation-amplified neonatal hypoxic-ischemic brain injury, AZ treatment improved sensorimotor function and reduced brain damage. AZ treatment was more effective with longer durations (5 doses over 4 days vs 3 doses over 2 days) and with earlier onset of treatment (1 vs. 2 vs. 4 hours) after the end of hypoxia exposure. These results are congruent with both our previous findings in the neonatal rodent HI model (22) and reports in adult stroke and spinal cord injury models 33, 41. Evidence of AZ-mediated neuroprotection in both straight-forward HI 32 and inflammation-amplified HI models provide support for further evaluation of AZ as potential treatment for neonatal encephalopathy.
Safety evaluation in both prior and current studies revealed no adverse effects of AZ treatment on core temperature, survival, or growth. More importantly, initial studies of AZ pharmacokinetics in critically ill premature infants enrolled in recent phase 1 and 2 clinical trials for treatment of pulmonary disease revealed no safety concerns 28, 30.
Although composite measures are often used to compare neurodevelopmental outcomes in clinical trials, they have not been widely applied in experimental neuroprotection studies. We implemented a Composite Score calculation to integrate two quantitative measures of sensorimotor function with measures of brain damage. Although there were no discordant trends among the individual outcome measures, this novel approach readily delineated informative trends. An important feature of the Composite Score was that mortality was robustly integrated (assigned lowest possible score). Thus, the deleterious effects of any intervention that improved function and/or tissue preservation at the expense of increasing death would be evident. Although the present studies did not include cognitive performance measures, it would be feasible to incorporate selected quantifiable test measures in future studies. Composite Scores yielded congruent outcomes when either % intact right hemisphere volumes (morphometry measures on P35) or right hemisphere weights (P21) were included.
Analysis of regional morphometry data did not reveal any selective vulnerability to inflammation-amplified injury in either model, and AZ reduced the extents of tissue damage in cortex, striatum, hippocampus, and in other regions that encompass white matter tracts.
Since sex differences in injury mechanisms or treatment effects have been reported in some neonatal rodent HI studies 42–45, sex was systematically evaluated in these experiments. In control LPS+HI groups, we found no differences in survival or injury severity. In PAM+HI controls, we noted an unexpected higher mortality in females (although sample sizes were small), and, in fact, AZ treatment was associated with higher survival rate. Moreover, in both models AZ neuroprotective efficacy did not differ between female and male animals.
Of interest, other clinically available drugs can also attenuate LPS-amplified HI brain injury in neonatal rodents, when treatment is initiated up to 30 minutes after HI. These include the anti-inflammatory and free radical scavenger N-acetylcysteine (efficacy lost with a 2-hour delay in treatment onset) 46; the immunosuppressive sphingosine-1-phosphate receptor agonist fingolimod (begun 30 min. after HI) 47; and the immunosuppressant rapamycin (administered immediately after HI) 48. Although no direct comparative efficacy studies among these drugs have been reported, the current data suggest that one advantage of AZ may be the longer duration of the therapeutic window for initiation of treatment than any of the other agents. As of March 2021, no reports have identified drugs that attenuate PAM-amplified HI brain injury, although immediate post-HI hypothermia is of benefit 37.
This well-established neonatal rodent model of hypoxic-ischemic brain injury has inherent limitations that have been discussed in reviews 49, 50. We relied on relatively simple, sensorimotor tests to assess functional outcomes; these tests are reliable, readily reproducible, and the supplementary data includes results to demonstrate that these tests identify deficits of similar magnitude over a two-week period (from P21 to P35).
We did not evaluate combined treatment with AZ and hypothermia, in view of the uncertainty of the translational relevance of the relatively brief (3–5 h) durations of hypothermia that are feasible experimentally in neonatal rats 51–53. Yet, the current results provide a strong impetus for future studies in larger animal models of neonatal inflammation-amplified asphyxial brain injury in which parenteral nutrition, physiologic monitoring, and much longer durations of hypothermia are feasible; data from neonatal piglets or fetal sheep would provide important complementary information about the safety and efficacy of AZ plus hypothermia combination treatment 54.
Our report does not address the mechanism(s) of AZ neuroprotection in the pathophysiologically complex models of inflammation-amplified HI injury. AZ has well-characterized anti-inflammatory properties that are independent of its antibiotic activity, and multiple studies suggest that its primary cellular targets are macrophage and microglia 25. In adult CNS injury models AZ neuroprotection is associated with promotion of a M2 or pro-resolution macrophage/microglia phenotype 33, 41. TLR2 and TLR4 are cell surface receptors with shared downstream intracellular signaling cascades that mediate both early pro-inflammatory and later regulatory and repair functions 55. We speculate that in both inflammation-amplified HI injury models AZ’s mode of action targets one or more shared downstream effector(s) of cell surface TLR signaling.
This report provides evidence of azithromycin’s potential efficacy as a treatment for neonatal encephalopathy. Equally important, our data raise important questions for future studies that will help clarify whether these findings support translation to early phase clinical trials. Priorities will include confirmation of safety and neuroprotective efficacy in complementary larger mammal models and in combination with clinically relevant hypothermia protocols.
Supplementary Material
Impact:
Azithromycin treatment decreases sensorimotor impairment and severity of brain injury, and improves survival, after inflammation-amplified hypoxic-ischemic brain injury, and this can be achieved even with a 2 hour delay in initiation.
This neuroprotective benefit is seen in models of inflammation-priming by both Gram-negative and Gram-positive infections.
This extends our previous findings, that azithromycin treatment is neuroprotective after hypoxic-ischemic brain injury in neonatal rats.
Statement of Financial Support:
Research reported in this publication was supported by the Michigan Institute for Clinical & Health Research (MICHR) that is supported by the National Institutes of Health under award number UL1TR002240. Research was also supported by the National Institutes of Health under award number R21 HD096251 (to J.D.E.B. and F.S.S.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. This research was also supported by The Reiter HIE Research Fund.
Footnotes
Disclosure: J.D.E.B., Y.L., I.A.D. and F.S.S. declare no conflict of interest.
Patient Consent: Not applicable and thus not required (preclinical animal model research).
References
- 1.Kurinczuk JJ, White-Koning M, Badawi N 2010. Epidemiology of neonatal encephalopathy and hypoxic-ischaemic encephalopathy. Early Hum Dev 86:329–338. [DOI] [PubMed] [Google Scholar]
- 2.Lee AC, et al. 2013. Intrapartum-related neonatal encephalopathy incidence and impairment at regional and global levels for 2010 with trends from 1990. Pediatr Res 74 Suppl 1:50–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Institute for Health Metrics and Evaluation Findings from the Global Burden of Disease Study 2017. (Institute for Health Metrics and Evaluation, Seattle, 2018). [Google Scholar]
- 4.Nelson KB, et al. 2012. Antecedents of neonatal encephalopathy in the Vermont Oxford Network encephalopathy registry. Pediatrics 130:878–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Aslam S, Strickland T, Molloy EJ 2019. Neonatal encephalopathy: Need for recognition of multiple etiologies for optimal management. Front Pediatr 7:142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McIntyre S, Blair E, Badawi N, Keogh J, Nelson KB 2013. Antecedents of cerebral palsy and perinatal death in term and late preterm singletons. Obstet Gynecol 122:869–877. [DOI] [PubMed] [Google Scholar]
- 7.Wu YW, et al. 2003. Chorioamnionitis and cerebral palsy in term and near-term infants. JAMA 290:2677–2684. [DOI] [PubMed] [Google Scholar]
- 8.Rao R, et al. 2020. Antimicrobial therapy utilization in neonates with hypoxic-ischemic encephalopathy (HIE): A report from the children’s hospital neonatal database (CHND). J Perinatol 40:70–78. [DOI] [PubMed] [Google Scholar]
- 9.Romero R, et al. 2015. Clinical chorioamnionitis at term I: Microbiology of the amniotic cavity using cultivation and molecular techniques. J Perinat Med 43:19–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Romero R, et al. 2021. Clinical chorioamnionitis at term X: Microbiology, clinical signs, placental pathology, and neonatal bacteremia - implications for clinical care. J Perinat Med 49:275–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sgro M, et al. 2020. Early-onset neonatal sepsis: Organism patterns between 2009 and 2014. Paediatr Child Health 25:425–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Polcwiartek LB, et al. 2021. Early-onset sepsis in term infants admitted to neonatal intensive care units (2011–2016). J Perinatol 41:157–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hagberg H, et al. 2015. The role of inflammation in perinatal brain injury. Nat Rev Neurol 11:192–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Eklind S, et al. 2001. Bacterial endotoxin sensitizes the immature brain to hypoxic-ischaemic injury. Eur J Neurosci 13:1101–1106. [DOI] [PubMed] [Google Scholar]
- 15.Rice JE, Vannucci RC, Brierley JB 1981. The influence of immaturity on hypoxic-ischemic brain damage in the rat. Ann Neurol 9:131–141. [DOI] [PubMed] [Google Scholar]
- 16.Towfighi J, Yager JY, Housman C, Vannucci RC 1991. Neuropathology of remote hypoxic-ischemic damage in the immature rat. Acta Neuropathol 81:578–587. [DOI] [PubMed] [Google Scholar]
- 17.Eklind S, Arvidsson P, Hagberg H, Mallard C 2004. The role of glucose in brain injury following the combination of lipopolysaccharide or lipoteichoic acid and hypoxia-ischemia in neonatal rats. Dev Neurosci 26:61–67. [DOI] [PubMed] [Google Scholar]
- 18.Lehnardt S, et al. 2003. Activation of innate immunity in the CNS triggers neurodegeneration through a toll-like receptor 4-dependent pathway. Proc Natl Acad Sci U S A 100:8514–8519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang X, et al. 2009. Lipopolysaccharide sensitizes neonatal hypoxic-ischemic brain injury in a MyD88-dependent manner. J Immunol 183:7471–7477. [DOI] [PubMed] [Google Scholar]
- 20.Wang LW, Chang YC, Lin CY, Hong JS, Huang CC 2010. Low-dose lipopolysaccharide selectively sensitizes hypoxic ischemia-induced white matter injury in the immature brain. Pediatr Res 68:41–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Henneke P, et al. 2008. Lipoproteins are critical tlr2 activating toxins in group b streptococcal sepsis. J Immunol 180:6149–6158. [DOI] [PubMed] [Google Scholar]
- 22.Mottahedin A, et al. 2017. Systemic activation of toll-like receptor 2 suppresses mitochondrial respiration and exacerbates hypoxic-ischemic injury in the developing brain. J Cereb Blood Flow Metab 37:1192–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Falck M, et al. 2017. Hypothermic neuronal rescue from infection-sensitised hypoxic-ischaemic brain injury is pathogen dependent. Dev Neurosci 39:238–247. [DOI] [PubMed] [Google Scholar]
- 24.Falck M, et al. 2018. Neonatal systemic inflammation induces inflammatory reactions and brain apoptosis in a pathogen-specific manner. Neonatology 113:212–220. [DOI] [PubMed] [Google Scholar]
- 25.Parnham MJ, et al. 2014. Azithromycin: Mechanisms of action and their relevance for clinical applications. Pharmacol Ther 143:225–245. [DOI] [PubMed] [Google Scholar]
- 26.Shepard RM, Falkner FC 1990. Pharmacokinetics of azithromycin in rats and dogs. J Antimicrob Chemother 25 Suppl A:49–60. [DOI] [PubMed] [Google Scholar]
- 27.Jaruratanasirikul S, Hortiwakul R, Tantisarasart T, Phuenpathom N, Tussanasunthornwong S 1996. Distribution of azithromycin into brain tissue, cerebrospinal fluid, and aqueous humor of the eye. Antimicrob Agents Chemother 40:825–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Viscardi RM, et al. 2020. Randomised trial of azithromycin to eradicate ureaplasma in preterm infants. Arch Dis Child Fetal Neonatal Ed 105:615–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Viscardi RM, et al. 2013. Azithromycin to prevent bronchopulmonary dysplasia in ureaplasma-infected preterm infants: Pharmacokinetics, safety, microbial response, and clinical outcomes with a 20-milligram-per-kilogram single intravenous dose. Antimicrob Agents Chemother 57:2127–2133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Merchan LM, et al. 2015. Pharmacokinetics, microbial response, and pulmonary outcomes of multidose intravenous azithromycin in preterm infants at risk for ureaplasma respiratory colonization. Antimicrob Agents Chemother 59:570–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hassan HE, et al. 2011. Pharmacokinetics, safety, and biologic effects of azithromycin in extremely preterm infants at risk for ureaplasma colonization and bronchopulmonary dysplasia. J Clin Pharmacol 51:1264–1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Barks JDE, Liu Y, Wang L, Pai MP, Silverstein FS 2019. Repurposing azithromycin for neonatal neuroprotection. Pediatr Res 86:444–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Amantea D, et al. 2016. Azithromycin protects mice against ischemic stroke injury by promoting macrophage transition towards M2 phenotype. Exp Neurol 275 Pt 1:116–125. [DOI] [PubMed] [Google Scholar]
- 34.Barks JD, Liu Y, Silverstein FS 2019. Azithromycin reduces inflammation-amplified hypoxic-ischemic brain injury in neonatal rodents. Pediatric Academic Societies Meeting: Abstract 4155.4161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Berman DR, Liu YQ, Barks J, Mozurkewich E 2010. Docosahexaenoic acid confers neuroprotection in a rat model of perinatal hypoxia-ischemia potentiated by Escherichia coli lipopolysaccharide-induced systemic inflammation. Am J Obstet Gynecol 202:469 e461–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yang D, et al. 2013. Plasminogen activator inhibitor-1 mitigates brain injury in a rat model of infection-sensitized neonatal hypoxia-ischemia. Cereb Cortex 23:1218–1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Falck M, et al. 2018. Hypothermia is neuroprotective after severe hypoxic-ischaemic brain injury in neonatal rats pre-exposed to PAM3CSK4. Dev Neurosci 40:189–197. [DOI] [PubMed] [Google Scholar]
- 38.Felt BT, et al. 2002. Early appearance of functional deficits after neonatal excitotoxic and hypoxic-ischemic injury: Fragile recovery after development and role of the NMDA receptor. Dev Neurosci 24:418–425. [DOI] [PubMed] [Google Scholar]
- 39.Barks JD, et al. 2017. Maternal high-fat diet influences outcomes after neonatal hypoxic-ischemic brain injury in rodents. J Cereb Blood Flow Metab 37:307–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Andine P, et al. 1990. Evaluation of brain damage in a rat model of neonatal hypoxic-ischemia. J Neurosci Methods 35:253–260. [DOI] [PubMed] [Google Scholar]
- 41.Gensel JC, Kopper TJ, Zhang B, Orr MB, Bailey WM 2017. Predictive screening of M1 and M2 macrophages reveals the immunomodulatory effectiveness of post spinal cord injury azithromycin treatment. Sci Rep 7:40144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bona E, Hagberg H, Loberg EM, Bagenholm R, Thoresen M 1998. Protective effects of moderate hypothermia after neonatal hypoxia-ischemia: Short- and long-term outcome. Pediatr Res 43:738–745. [DOI] [PubMed] [Google Scholar]
- 43.Smith AL, Alexander M, Rosenkrantz TS, Sadek ML, Fitch RH 2014. Sex differences in behavioral outcome following neonatal hypoxia ischemia: Insights from a clinical meta-analysis and a rodent model of induced hypoxic ischemic brain injury. Exp Neurol 254:54–67. [DOI] [PubMed] [Google Scholar]
- 44.Chavez-Valdez R, Martin LJ, Razdan S, Gauda EB, Northington FJ 2014. Sexual dimorphism in BDNF signaling after neonatal hypoxia-ischemia and treatment with necrostatin-1. Neuroscience 260:106–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wood TR, et al. 2020. Variability and sex-dependence of hypothermic neuroprotection in a rat model of neonatal hypoxic-ischaemic brain injury: A single laboratory meta-analysis. Sci Rep 10:10833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang X, et al. 2007. N-acetylcysteine reduces lipopolysaccharide-sensitized hypoxic-ischemic brain injury. Ann Neurol 61:263–271. [DOI] [PubMed] [Google Scholar]
- 47.Yang D, et al. 2014. Blocking lymphocyte trafficking with FTY720 prevents inflammation-sensitized hypoxic-ischemic brain injury in newborns. J Neurosci 34:16467–16481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Srivastava IN, Shperdheja J, Baybis M, Ferguson T, Crino PB 2016. mTOR pathway inhibition prevents neuroinflammation and neuronal death in a mouse model of cerebral palsy. Neurobiol Dis 85:144–154. [DOI] [PubMed] [Google Scholar]
- 49.Northington FJ 2006. Brief update on animal models of hypoxic-ischemic encephalopathy and neonatal stroke. ILAR J 47:32–38. [DOI] [PubMed] [Google Scholar]
- 50.Hamdy N, Eide S, Sun HS, Feng ZP 2020. Animal models for neonatal brain injury induced by hypoxic ischemic conditions in rodents. Exp Neurol 334:113457. [DOI] [PubMed] [Google Scholar]
- 51.Barks JD, Liu YQ, Shangguan Y, Silverstein FS 2010. Phenobarbital augments hypothermic neuroprotection. Pediatr Res 67:532–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Osredkar D, et al. 2014. Hypothermia is not neuroprotective after infection-sensitized neonatal hypoxic-ischemic brain injury. Resuscitation 85:567–572. [DOI] [PubMed] [Google Scholar]
- 53.Patel SD, et al. 2015. Therapeutic hypothermia and hypoxia-ischemia in the term-equivalent neonatal rat: Characterization of a translational preclinical model. Pediatr Res 78:264–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhou KQ, Davidson JO, Bennet L, Gunn AJ 2020. Combination treatments with therapeutic hypothermia for hypoxic-ischemic neuroprotection. Dev Med Child Neurol 62:1131–1137. [DOI] [PubMed] [Google Scholar]
- 55.Luo L, Lucas RM, Liu L, Stow JL 2019. Signalling, sorting and scaffolding adaptors for toll-like receptors. J Cell Sci 133. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.