

Abstract
MET, the receptor for the hepatocyte growth factor (HGF), is strongly associated with resistance to tyrosine kinase inhibitors, key drugs that are used in the therapy of non–small cell lung cancer. MET contains 11 potential N‐glycosylation sites, but the site‐specific roles of these N‐glycans have not been elucidated. We report herein that these N‐glycans regulate the proteolytic processing of MET and HGF‐induced MET signaling, and that this regulation is site specific. Inhibitors of N‐glycosylation were found to suppress the processing and trafficking of endogenous MET in H1975 and EBC‐1 lung cancer cells and exogenous MET in CHO‐K1 cells. We purified the recombinant extracellular domain of human MET and determined the site‐specific N‐glycan structures and occupancy using mass spectrometry. The results indicated that most sites were fully glycosylated and that the dominant population was the complex type. To examine the effects of the deletion of N‐glycans of MET, we prepared endogenous MET knockout Flp‐In CHO cells and transfected them with a series of N‐glycan–deletion mutants of MET. The results showed that several N‐glycans are implicated in the processing of MET. The findings also suggested that the N‐glycans of the SEMA domain of MET positively regulate HGF signaling, and the N‐glycans of the region other than the SEMA domain negatively regulate HGF signaling. Processing, cell surface expression, and signaling were significantly suppressed in the case of the all‐N‐glycan–deletion mutant. The overall findings suggest that N‐glycans of MET affect the status and the function of the receptor in a site‐specific manner.
Keywords: glycosylation, hepatocyte growth factor (HGF), lung cancer, MET, tyrosine kinase inhibitor (TKI) resistance
N‐glycans of the SEMA domain of MET positively regulate hepatocyte growth factor (HGF) signaling, and the N‐glycans of the region other than the SEMA domain negatively regulate HGF signaling. Processing and signaling were significantly suppressed in the case of the all‐N‐glycan–deletion mutant. The overall findings suggest that N‐glycans of MET affect the status and the function of the receptor in a site‐specific manner.
![]()
Abbreviations
- EGFR
epidermal growth factor receptor
- FBS
fetal bovine serum
- HGF
hepatocyte growth factor
- nLC‐ESI‐MS/MS
nanoflow liquid chromatography‐electrospray ionization tandem mass spectrometry
- PNGase F
peptide N‐glycosidase F
- RTK
receptor tyrosine kinase
- SDS‐PAGE
sodium dodecyl sulfate polyacrylamide gel electrophoresis
- TKI
tyrosine kinase inhibitor
1. INTRODUCTION
MET is a receptor tyrosine kinase (RTK) for hepatocyte growth factor (HGF), and its functions have been implicated in biological events including embryogenesis and tissue regeneration. 1 , 2 , 3 , 4 It is initially synthesized as a single‐chain precursor, which is then glycosylated to form 195‐kDa pro‐MET. Pro‐MET is cleaved by furin, a calcium‐dependent serine protease in the Golgi compartment, to become mature‐MET, which is a disulfide‐linked heterodimer consisting of a 50‐kDa α subunit and a 145‐kDa β subunit. The whole α subunit and the N‐terminal region of β subunit form the SEMA domain, which is followed by a plexin‐semaphorin‐integrin (PSI) domain, four immunoglobulin‐plexin‐transcription factor (IPT) domains, a transmembrane domain, a juxtamembrane domain, and a tyrosine kinase domain. The binding of HGF to MET induces receptor homodimerization and autophosphorylation and is followed by the activation of downstream signal processes such as the PI3K/Akt pathway or the Ras/Erk pathway, which is responsible for causing various cellular events such as cell proliferation, migration, differentiation, and morphogenesis. The physiological activation of MET signaling is essential for embryologic and organ development. An aberrant activation of MET through gene amplification, tyrosine kinase domain mutations, and juxtamembrane splicing mutation has been observed in various types of cancer, including lung cancer. 5 , 6 , 7 , 8 It is particularly noteworthy that HGF overexpression and MET amplification are associated with resistance to epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors (TKIs), key drugs in lung cancer therapy. 9 , 10 Despite the fact that agents targeting HGF/MET signaling have been studied for many years, only a few drugs have been approved and are now being clinically applied. Peripheral edema is a main side effect, which is less common with other anticancer agents and leads to dose reduction or discontinuation of therapy. 11 , 12 To develop more effective anticancer agents that target MET, the detailed physical properties of MET need to be clarified.
Glycosylation is the most abundant post‐translational modification, and it is involved in the functional regulation of proteins. 13 , 14 , 15 , 16 Glycans control various types of physicochemical properties of proteins, such as structure, conformational flexibility, charge, and hydrophilicity. In the case of cell surface receptors, glycosylation has been shown to regulate each step in the signal transduction such as receptor trafficking to the cell surface, ligand binding, dimerization, phosphorylation, and endocytosis. 17 , 18 We have been examining the functions of N‐glycans on growth factor receptors 19 , 20 , 21 , 22 , 23 , 24 and have concluded that determining the site‐specific functions of glycans is crucial for elucidating the mechanisms responsible for the functional regulation of receptors. Human MET contains 11 potential N‐glycosylation sites, 1 and there are several reports suggesting that N‐glycans of MET are involved in the functional regulation of receptors. 25 , 26 , 27 , 28 However, the structures and the functions of these site‐specific N‐glycans of MET have not been examined. The aim of the present study was to further determine the mechanisms by which N‐glycans regulate MET. We determined the site‐specific glycosylation status by mass spectrometry, and found that most sites were fully glycosylated and the dominant population were complex‐type glycans. We also found that N‐glycans of the SEMA domain of MET positively regulate HGF signaling and the N‐glycans of the region other than the SEMA domain negatively regulate HGF signaling. The results indicate that N‐glycans regulate MET functions in a site‐specific manner.
2. MATERIALS AND METHODS
2.1. Reagents and antibodies
Tunicamycin, purchased from FUJIFILM Wako Pure Chemical Corporation, and NGI‐1, purchased from MedChemExpress, were dissolved in dimethyl sulfoxide. Recombinant human HGF (HEK293 derived, 100‐39H) was purchased from PEPRO TECH. The phospho‐specific monoclonal antibody to human MET (Tyr1234/1235, #3129), phospho‐specific polyclonal antibodies to Akt (Ser473, #9271) and Erk (Thr202/Tyr204, #9101), monoclonal antibodies to human MET (#3127, #8198), and polyclonal antibodies to Akt (#9272) and Erk (#9102) were purchased from Cell Signaling Technology. The monoclonal antibody to human β‐actin was purchased from Sigma Aldrich.
2.2. Cell cultures
Human lung adenocarcinoma cell lines H1975, human lung squamous carcinoma cell lines EBC‐1, and CHO‐K1 cell lines were obtained from the ATCC. The Flp‐In CHO cell line was obtained from Invitrogen. CHO‐K1, Flp‐In CHO, and EBC‐1 were maintained in Dulbecco's modified Eagle's medium (D‐MEM; Sigma Aldrich) supplemented with 10% (v/v) fetal bovine serum (FBS; EQUITECH‐BIO INC) and 1% (v/v) penicillin‐streptomycin. H1975 cells were maintained in RPMI 1640 (Sigma Aldrich) supplemented with 10% (v/v) FBS, 1% (v/v) MEM nonessential amino acid solution, and 1% (v/v) penicillin‐streptomycin.
2.3. Protein sample preparation and Western blotting
Cells were harvested in lysis buffer (20 mmol/L HEPES, pH 7.4, 0.15 mol/L NaCl, 5 mmol/L ethylenediaminetetraacetic acid, 1% [w/v] Nonidet P‐40, 10% [w/v] glycerol, 5 mmol/L sodium pyrophosphate, 10 mmol/L NaF, 1 mmol/L sodium orthovanadate, 10 mmol/L β‐glycerophosphate, 1 mmol/L PMSF, 2 mg/mL aprotinin, 5 mg/mL leupeptin and 1 mmol/L dithiothreitol) and rotated for 30 minutes at 4°C and then centrifuged at 15 000 g for 10 minutes at 4°C; the resulting supernatant was used as a protein sample. The samples were separated by SDS‐PAGE and transferred to PVDF membranes (Millipore). The blots were probed with the indicated antibody, and the immunoreactive bands were visualized using a chemiluminescence reagent (Super Signal West Pico, Thermo Fisher Scientific). Densitometric analysis was performed by using a luminous analyzer.
2.4. Cell surface biotinylation assay
Cells were serum starved overnight, washed twice with ice‐cold PBS (+), and then incubated with 0.5 mg/mL of sulfo‐NHS‐LC‐biotin (Thermo Fisher Scientific) in PBS (+) for 30 minutes at 4°C. The reaction was quenched by washing the cells with Tris‐buffered saline and PBS (+), and cells were harvested in lysis buffer. For immunoprecipitation, Protein G Sepharose 4 fast flow (GE Healthcare Biosciences) was added to whole‐cell lysate with an anti‐MET monoclonal antibody (#8198, Cell Signaling Technology), rotated at 4°C overnight, and then washed with lysis buffer. For pulldown assay, streptavidin agarose (Invitrogen) was added to whole‐cell lysate, rotated at 4°C overnight, then washed with lysis buffer. Samples were subjected to Western blotting using indicated antibodies.
2.5. MET knockout cells
MET knockout cells were established following a previously described method with minor modifications. 29 Briefly, the Cas 9 protein (Thermo Fisher Scientific) and gRNA was transfected to CHO‐K1 cells or Flp‐In CHO cells by using Lipofectamine CRISPRMAX Cas9 Transfection Reagent (Invitrogen). The following gRNA was used: 5′‐AATGCCAGGUGACAGCACGGUGG‐3′ (Invitrogen). The knockout cells were obtained by single‐cell cloning by limiting dilution.
2.6. Establishment of MET‐transfected CHO‐K1 cells
Two methods were used to establish CHO‐K1 cells that stably express human MET. For a large amount of MET expression, human MET cDNA cloned in pCAGGS‐Flag vector was transfected into CHO‐METKO cells using Lipofectamine 2000 reagent (Thermo Fisher Scientific) with pcDNA 3.1 hygromycin, and stable‐expressing cells were selected using 600 μg/mL hygromycin B (Calbiochem). Single‐cell cloning was performed by a limiting dilution method. In the case of the N‐glycan–deleted mutant MET, the Flp‐In system (Invitrogen) was used for generating single‐copy isogenic cell lines. Human MET cDNA was subcloned into a pcDNA5/FRT expression vector, and asparagine residues in the N‐glycosylation sites were substituted with glutamine by site‐directed mutagenesis. The expression vectors were transfected into Flp‐In CHO‐METKO cells with pOG44 Flp‐recombinase expression vector (Invitrogen) using Lipofectamine 2000 (Invitrogen). The stable‐expressing cells were selected using 600 μg/mL hygromycin B.
2.7. Purification of recombinant human soluble MET (sMET)
For the sMET expression vector, myc‐His tag was introduced at the end of the extracellular domain (residue T932) of human MET and subcloned into a pcDNA5/FRT expression vector. To generate cells that stably express sMET, the Flp‐In system (Invitrogen) was used. Recombinant human sMET was purified following a previously described method. 22
2.8. Mass spectrometry (MS)
The purified sMET was reductively alkylated and digested with MS‐grade trypsin or chymotrypsin (Promega). The resultant peptides were desalted with a styrene divinylbenzene polymer tip column (GL Science) and a graphitized carbon tip column (GL Science) and were evaporated on a centrifugal evaporator. Obtained peptides were redissolved with 0.1% formic acid and subjected to Orbitrap Q Exactive Plus (Thermo Fisher Scientific) through an EASY‐nLC system equipped with a C18 column (0.075 mm × 125 mm, Nikkyo Technos). The sample was eluted with acetonitrile gradient from 0% to 35% in 135 minutes at the flow rate of 300 nL/minutes. Obtained MS data under the data‐dependent mode were processed using the MaxQuant 1.6.3.3 software for peptide searching. 30 , 31 Tandem mass spectrometry (MS/MS) spectra were assigned manually for determination of glycan structure, and the precursor intensities from the glycopeptide were integrated for the intensity‐based comparison with software MaxQuant and Xcalibur (Thermo Fisher Scientific). The MS data have been deposited to the ProteomeXchange Consortium via the PRIDE 32 partner repository with the dataset identifier PXD029295.
2.9. Cell proliferation assay
Cells were seeded in 96‐well plates at a density of 2000 or 4000 cells/well and incubated in D‐MEM supplemented with 10% (v/v) FBS at 37°C for 16 hours and then with100 ng/mL HGF in D‐MEM without FBS for 48 hours. Viable cell numbers were analyzed using WST‐1 (Takara Bio Inc).
2.10. Flow cytometry
Cells were stained with an FITC‐conjugated anti‐MET antibody (eBioclone97; Thermo Fisher Scientific) in PBS supplemented with 1% BSA and 2 mmol/L EGTA for 60 minutes at room temperature. After washing, an analysis was carried out using FACSCanto II (Becton Dickinson).
2.11. Immunofluorescence staining and confocal microscopy analysis
Cells cultured on chamber slide (Nunc) were fixed with ice‐cold methanol and blocked by treatment with 1% BSA in PBS. The resulting fixed cells were incubated with an antibody against MET (#8198, Cell Signaling Technology) and were labeled with Alexa Fluor 592–conjugated secondary antibodies (Thermo Fisher Scientific). The labeled cells were mounted with Prolong Gold with DAPI (Invitrogen), and fluorescent images were acquired with a ELYRA PS.1/LSM780 (Carl Zeiss).
2.12. Peptide N‐glycosidase F (PNGase F) treatment
For native conditions, purified sMET (2.5 μg) was incubated for 16 hours at 37°C with 100 mmol/L Tris‐HCl (pH 8.6) and 40 mU/mL PNGase F (Takara Bio Inc). For denaturing conditions, purified sMET (2.5 μg) was boiled in 500 mmol/L Tris‐HCl (pH 8.6), 0.5% SDS, and 0.1 mol/L 2‐mercaptoethanol for 3 minutes and incubated for 16 hours at 37°C with 100 mmol/L Tris‐HCl (pH 8.6), 1% NP‐40, and 20 mU/mL PNGase F. Samples were incubated with sample buffer with (reducing PAGE) or without (nonreducing PAGE) 2‐mercaptoethanol and subjected to SDS‐PAGE.
2.13. Binding assay with surface plasmon resonance analysis
Hepatocyte growth factor (5 μg/mL) in 10 mmol/L sodium acetate (pH 6.0) was immobilized on a sensor chip C1 of the Biacore 3000 system (Biacore) according to the manufacturer's instructions. For the running buffer, HBS‐EP buffer (GE Healthcare Bioscience) was used. sMET was injected at a flow rate of 30 μL/minutes. At the end of each cycle, the surface of the sensor chip was regenerated by injecting 1 mol/L NaCl. Sensorgrams of the interactions obtained by using various concentrations of sMET (12.5‐100 nmol/L) were analyzed. The association rate constant (ka) and the dissociation rate constant (kd) were calculated according to the BIAevaluation software (Version 3.1, Biacore AB).
3. RESULTS
3.1. Inhibitors of N‐glycosylation suppress the processing and trafficking of endogenous MET in human lung cancer cells and exogenous MET in CHO‐K1 cells
To examine the effects of inhibitors of N‐glycosylation on endogenous MET in lung cancer cells, we treated lung cancer cells H1975 (human lung adenocarcinoma cells harboring both exon 19 deletions and T790M EGFR mutant) and EBC‐1 (human lung squamous carcinoma cells with MET amplification) with tunicamycin and NGI‐1. As shown in Figure 1A, 195 kDa pro‐MET (upper band) and 145 kDa β subunit of mature‐MET (lower band) are visible in the Western blotting data for the nontreated samples. We observed that the lower bands in the tunicamycin‐treated samples were nearly invisible, which suggests that the tunicamycin treatment suppressed the processing of pro‐MET to mature‐MET, and the effect was much weaker in the case of the NGI‐1 treatment, especially in H1975 cells. Both tunicamycin and NGI‐1 caused a reduction in the molecular weight of pro‐MET due to their inhibitory effect on N‐glycosylation, and the effect was more obvious in the case of the tunicamycin treatment, which is consistent with findings reported in a previous study. 33 We next evaluated the effects of N‐glycosylation inhibitors on the trafficking of MET by cell surface biotinylation assays and found that the cell surface expression levels of MET were significantly reduced by the result of the tunicamycin treatment (Figure 1B). To examine the issue of whether N‐glycosylation inhibitors exhibit similar effects on exogenous MET, we established CHO‐K1 cells that stably express human MET. We first prepared endogenous MET knockout CHO‐K1 cells (CHO‐METKO) and then CHO‐METKO clone was further manipulated to stably express human MET (CHO‐MET; Figure 1C). As shown in Figure 1D,E, both the tunicamycin and NGI‐1 treatment exhibited effects on CHO‐MET that were similar to that for H1975 cells and EBC‐1 cells. These results indicated that reagents that inhibit N‐glycosylation reduced the processing and trafficking of MET, and these effects were correlated with the glycosylation inhibitory effects.
FIGURE 1.
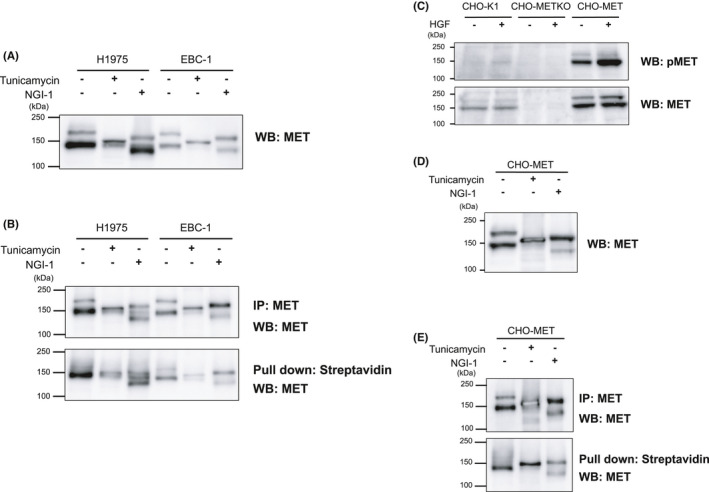
Inhibitors of N‐glycosylation suppress processing and trafficking of endogenous MET in human lung cancer cells and exogenous MET in CHO‐K1 cells. A, H1975 and EBC‐1 cells were serum starved with 2.5 μg/mL of tunicamycin or 10 nmol/L of NGI‐1 for 24 h. Whole‐cell lysates were prepared and subjected to Western blotting using an anti‐MET antibody. The data are representative of three independent experiments. B, E, Cell surface expression of MET. Cells were serum starved with 2.5 μg/mL of tunicamycin or 10 nmol/L of NGI‐1 for 24 h. Cells were incubated with 0.5 mg/mL of sulfo‐NHS‐LC‐biotin for 30 min at 4°C. Cell lysates were immunoprecipitated by an anti‐MET antibody or pulled down by streptavidin‐agarose. Samples were subjected to Western blotting using an anti‐MET antibody. The data are representative of three independent experiments. C, CHO‐K1 cells, endogenous MET knockout CHO‐K1 cells (CHO‐METKO), and human MET stably expressing CHO‐METKO cells (CHO‐MET) were serum starved for 16 h and stimulated with or without hepatocyte growth factor (HGF) (50 ng/mL) at 37°C for 5 min. Whole‐cell lysates were prepared and subjected to Western blotting using the indicated antibodies. The data are representative of three independent experiments. D, CHO‐MET was serum starved with 2.5 μg/mL of tunicamycin or 10 nmol/L of NGI‐1 for 24 h. The whole‐cell lysate was prepared and subjected to Western blotting using anti‐MET antibody
3.2. Identification of the site‐specific N‐glycosylation analysis of MET
As it was observed that N‐glycosylation is involved in the functional regulation of MET, a site‐specific N‐glycosylation analysis was carried out to determine glycan occupancy and glycan structure. We established CHO‐K1 cells stably expressing the extracellular domain of human MET (sMET) with a His‐tag using the Flp‐In system and purified sMET from the medium. An N‐glycomic analysis was then performed employing nanoflow liquid chromatography‐electrospray ionization tandem mass spectrometry (nLC‐ESI‐MS/MS). All of the expected glycopeptides containing potential sites for N‐glycosylation could be covered in trypsin or chymotrypsin digests of sMET. The MS intensity‐based profiles for the N‐glycans on sMET are shown in Figure 2A and Figure S1 and are summarized in Table 1. The degree of occupancy of N‐glycans at all 11 potential sites was relatively high (68.3%‐100%; Table 1). In particular, no unmodified (nonglycosylated) peptides containing N149, N405, N635, N785, and N879 were observed, indicating that the glycan occupancy of these sites was near 100%. The class of the N‐glycan structure observed at each site was predominantly of the complex type with the biantennary structure (Figure 2A). In most of the sites, more than 99% of the glycans were found to be fucosylated, except for N45 (0%), N202 (20.4%), and N785 (48.8%; Table 1). These site‐specific N‐glycosylation analyses indicated that sMET is highly modified by mature complex‐type N‐glycans. Figure 2B indicates a schematic representation of sMET with the predominant N‐glycans.
FIGURE 2.
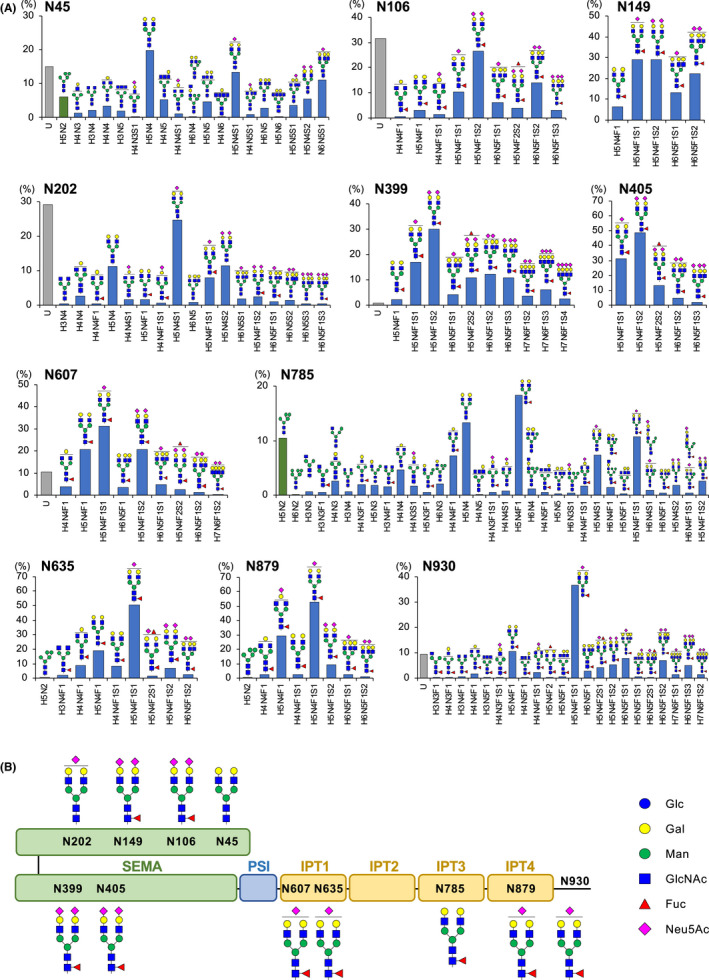
Intensity‐based N‐glycosylation profile of soluble MET (sMET) characterized by nanoflow liquid chromatography‐electrospray ionization tandem mass spectrometry (nLC‐ESI‐MS/MS). A, Site‐specific analysis of the N‐glycosylation of the extracellular domain of MET (sMET) expressed in Flp‐In CHO cells was performed as described in “Materials and Methods.” Relative abundance of unmodified peptides and estimated N‐glycoforms on the 11 potential N‐glycosylation sites are indicated. The labels on the horizontal axis of the bar charts represent glycan compositions. U, unmodified peptide; H, hexose; N, N‐acetylhexosamine (GlcNAc); F, deoxyhexose (fucose); S, sialic acid (Neu5Ac). Bar charts showing the relative abundance of unmodified peptide, high‐mannose–type glycoforms, and complex/hybrid‐type glycoforms are represented by gray, green, and blue, respectively. The mass spectrometry (MS/MS) spectra of the glycopeptides showing the highest intensities at each potential site are given in the “Supplementary Information.” B, Schematic representation of the predominant N‐glycans on each site of sMET. The symbols of the monosaccharides used in A and B are noted
TABLE 1.
Summary of the site‐specific profiles of N‐glycans on soluble MET (sMET)
| Site | Glycopeptide sequence | Digestive enzyme | Occupancy (%) | Fucosylation (%) | Sialylation (%) |
|---|---|---|---|---|---|
| N45 | YQLP N FTAETPIQNVILHEHHIFLGATNYIYVLNEEDLQK | Trypsin | 84.9 | 0 | 42.8 |
| N106 | A N LSGGVWK | Trypsin | 68.3 | 100 | 95.1 |
| N149 | HVFPH N HTADIQSEVHCIFS PQIEEPSQCPDCVVSALGAK | Trypsin | 100 | 100 | 93.6 |
| N202 | FINFFVGNTI N SSYFPDHPLHSISVR | Trypsin | 70.7 | 20.4 | 76.0 |
| N399 | CLQHFYGPNHEHCF N R | Trypsin | 99.1 | 100 | 97.8 |
| N405 | N SSGCEAR | Trypsin | 100 | 100 | 100 |
| N607 | VLLG N ESCTLTLSESTMNTLK | Trypsin | 89.4 | 100 | 68.5 |
| N635 | N MSIIISNGHGTTQY | Chymotrypsin | 100 | 99.8 | 69.7 |
| N785 | N FTVACQHR | Trypsin | 100 | 48.8 | 29.1 |
| N879 | KVG N KSCENIHL | Chymotrypsin | 100 | 99.8 | 68.0 |
| N930 | GKVIVQPDQ N F | Chymotrypsin | 90.4 | 100 | 80.5 |
3.3. N‐glycans on MET are implicated in processing of the receptor
Because inhibitors of N‐glycosylation affect various proteins, making it difficult to evaluate the precise effects of N‐glycosylation of MET, we examined N‐glycan–deletion mutants of MET. In this study, the Flp‐In system (Invitrogen) was used for generating single‐copy isogenic cell lines. In these experiments, we prepared endogenous MET knockout Flp‐In CHO cells (Flp‐In CHO‐METKO) and then prepared cells that stably express human wild‐type and mutated MET. As shown in Figure 3A, the Flp‐In CHO‐METKO cells did not respond to HGF stimulation, which was restored by the introduction of human wild‐type MET. Figure 3B indicates the schematic structure of the extracellular domain of human MET with 11 potential N‐glycosylation sites and a list of mutants in which the N‐glycans had been deleted: the deletion of N‐glycans in the α subunit (N45Q/N106Q/N149Q/N202Q, shown as 4N α 4Q), the deletion of N‐glycans in the SEMA domain (N45Q/N106Q/N149Q/N2020Q/N399Q/N405Q, shown as 6N SEMA 6Q), the deletion of N‐glycans in sites other than the SEMA domain (N607Q/N635Q/N785Q/N879Q/N930Q, shown as 5N not SEMA 5Q), the deletion of N‐glycans in the β subunit (N399/N405/N607Q/N635Q/N785Q/N879Q/N930Q, shown as 7N β 7Q), the deletion of N‐glycans in entire extracellular domain (N45Q/N106Q/N149Q/N2020Q/N399Q/N405Q/N607Q/N635Q/N785Q/N879Q/N930Q, shown as 11N all 11Q), and the deletion of N‐glycans on N399 and N405 (N399Q/N405Q). On examining the whole‐cell lysate of these mutants, it was found that the deletion of N‐glycans suppressed the processing of pro‐MET to mature‐MET (Figure 3C). This suppression was particularly significant in the 6N SEMA 6Q, 5N not SEMA 5Q, 7N β 7Q, and 11N all 11Q mutants. The suppression was less significant in mutants 4N α 4Q, and no suppression was observed in N399Q/N405Q. It was also observed that most of the total protein levels of MET in the N‐glycan–deletion mutants were significantly increased, especially in the 11N all 11Q mutant, suggesting that pro‐MET is resistant to protein degradation.
FIGURE 3.

N‐glycans of MET are implicated in receptor processing. A, Flp‐In CHO cells (Flp‐In CHO), endogenous MET knockout Flp‐In CHO cells (Flp‐In CHO‐METKO), and wild‐type human MET stably expressing Flp‐In CHO‐METKO cells (Flp‐In CHO‐MET WT) were serum starved for 16 h and stimulated with 50 ng/mL of hepatocyte growth factor (HGF) at 37°C for indicated time. Samples were subjected to Western blotting using indicated antibodies. B, Schematic structure of human MET with 11 potential N‐glycosylation sites and the list of N‐glycan–deletion mutants. C, Wild‐type and mutated MET–transfected Flp‐In CHO‐METKO were serum starved for 16 h, harvested in lysis buffer, and subjected to Western blotting using the indicated antibodies. The data are representative of three independent experiments
3.4. HGF‐induced signaling is downregulated in the transfectants of MET mutants in which the N‐glycans of the SEMA domain were deleted, and upregulated in the transfectants of MET mutants in which the N‐glycans of the region other than the SEMA domain were deleted
We next examined HGF‐induced signaling in N‐glycan–deleted mutant MET–transfected cells. As shown in Figure 4A,B, MET phosphorylation was suppressed in the transfectants of MET mutants 4N α 4Q, 6N SEMA 6Q, 5N not SEMA 5Q, 7N β 7Q, and 11N all 11Q. It was also observed that in the transfectants of MET mutants 4N α 4Q, 6N SEMA 6Q, and 11N all 11Q, phosphorylation levels of Akt and Erk were significantly reduced (Figure 4A). In contrast, phosphorylation levels of Akt and Erk were increased in the transfectants of MET mutants 5N not SEMA 5Q and 7N β 7Q (Figure 4B). The increase was particularly significant in the level of Erk phosphorylation in 5N not SEMA 5Q. As MET phosphorylation was reduced in these transfectants, it appears that the signaling pathway including other molecules should be involved. When N399Q/N405Q MET mutant–transfected cells were examined, no significant difference was observed between the wild‐type MET–transfected cells (Figure 4C).
FIGURE 4.
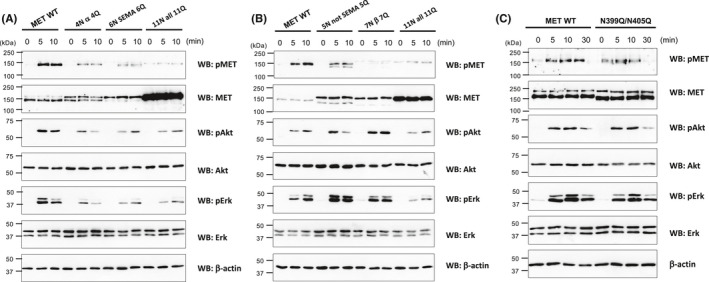
Downstream signaling is downregulated in the transfectants of MET mutants in which the N‐glycans of the SEMA domain were deleted and upregulated in the transfectants of MET mutants in which the N‐glycans of the region other than the SEMA domain were deleted. A‐C, Wild‐type and mutated MET–transfected Flp‐In CHO‐METKO were serum starved for 16 h and stimulated with 50 ng/mL of hepatocyte growth factor (HGF) at 37°C for indicated time. Samples were subjected to Western blotting using indicated antibodies. The data are representative of three independent experiments
3.5. Examination of 11N all 11Q mutant MET–transfected cells
We examined the biological effects of N‐glycans deletion and found that HGF‐induced cell proliferation was completely suppressed in 11N all 11Q–transfected cells (Figure 5A). We also examined the mechanisms by which downstream signaling is suppressed in 11N all 11Q–transfected cells. We repeated the flow cytometry, confocal microscopy analyses, and the cell surface biotinylation assay and found that the cell surface expression levels of the 11N all 11Q mutant MET were significantly reduced (Figure 5B) but not completely eliminated (Figure 5C,D). The phosphorylation levels of the cell surface 11N all 11Q mutant MET were also significantly reduced (Figure 5E).
FIGURE 5.
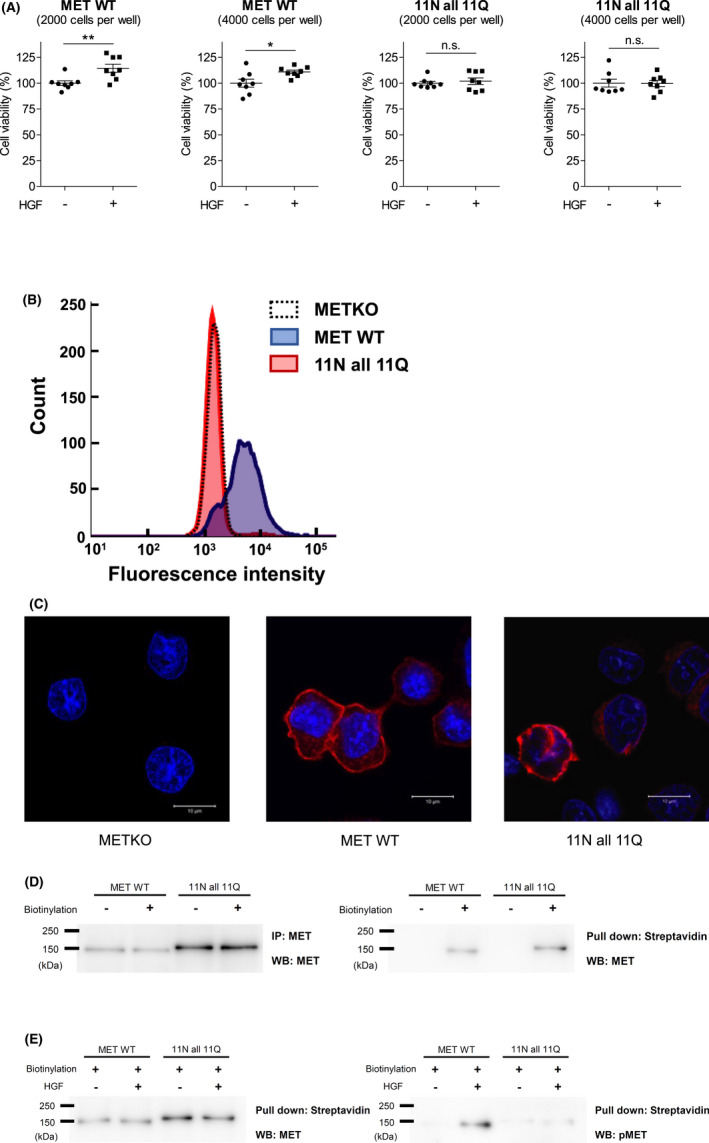
Examination of 11N all 11Q mutant MET–transfected cells. A, Cell proliferation assays were performed using wild‐type and 11N all 11Q mutant MET–transfected Flp‐In CHO‐METKO. The cells were seeded in 96‐well plates at a density of 2000 or 4000 cells/well and incubated in D‐MEM supplemented with 10% (v/v) FBS at 37°C for 16 h and then with 100 ng/mL hepatocyte growth factor (HGF) in D‐MEM without FBS for 48 h. Viable cell numbers were analyzed using WST‐1. Data are presented as mean ± SD (n = 8 wells per group). Student's t test was used for statistical comparisons. *P < .1; **P < .01; n.s., not significant. B, The cell surface expression of MET analyzed by flow cytometry. Flp‐In CHO‐METKO cells (METKO), wild‐type MET (MET WT)–, or 11N all 11Q mutant MET (11N all 11Q)–transfected Flp‐In CHO‐METKO cells were stained with FITC‐conjugated anti‐MET antibody and analyzed by flow cytometry. C, Confocal microscopy analysis of MET (red) and nuclear (cyan). Flp‐In CHO‐METKO cells (METKO), wild‐type MET (MET WT)–, or 11N all 11Q mutant MET (11N all 11Q)–transfected Flp‐In CHO‐METKO cells were subjected to immunofluorescent staining and confocal microscopy analysis. D, E, Cell surface MET and phosphorylated MET detected by biotinylation assay. Wild‐type MET (MET WT)– or 11N all 11Q mutated MET (11N all 11Q)–transfected Flp‐In CHO‐METKO cells were serum starved overnight and then incubated with or without 50 ng/mL HGF for 30 min at 4°C. After washing with PBS (+), the cells were incubated with 0.5 mg/mL of sulfo‐NHS‐LC‐biotin for 30 min at 4°C. The cell lysate was immunoprecipitated by an anti‐MET antibody or pulled down by streptavidin‐agarose. Samples were subjected to Western blotting using the indicated antibodies. The data are representative of three independent experiments
3.6. In vitro deletion of N‐glycans does not alter the binding affinity of HGF to the extracellular domain of MET
To further examine the mechanisms by which N‐glycan deletion affects downstream signaling, we examined ligand‐binding affinity using a surface plasmon resonance analysis. A PNGase F treatment decreased the molecular weight of the full length, β subunit, and α subunit of sMET, and although PNGase F cleaved N‐glycans more successfully under denaturing conditions, considerable amounts of N‐glycans were cleaved under native condition as well (Figure 6A). The binding kinetics of HGF were analyzed by surface plasmon resonance using the native‐condition PNGase F–treated sMET. The dissociation constant of sMET to HGF and the PNGase F–treated sMET to HGF were KD (kd/ka) = 4.75 and 5.35 nmol/L, respectively (Figure 6B). These results demonstrated that the once MET is processed to mature form, the deletion of N‐glycans does not alter the binding affinity of HGF.
FIGURE 6.
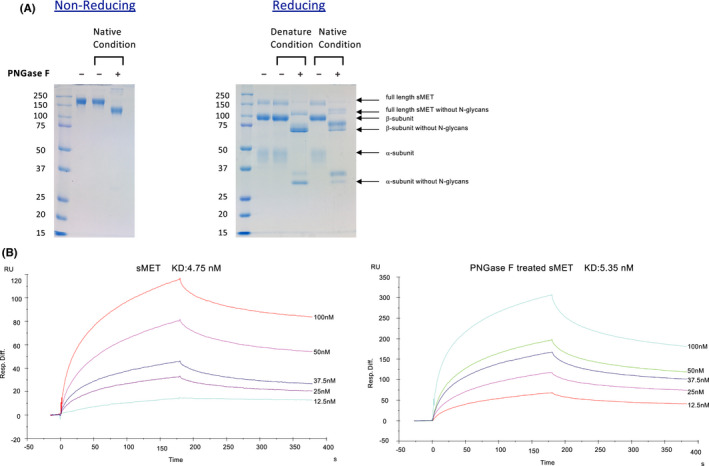
Ligand‐binding affinity of soluble MET (sMET) with and without N‐glycans. A, Purified recombinant human sMET produced in Flp‐In CHO cells was treated with PNGase F under denaturing conditions or native conditions as described in “Materials and Methods.” The samples were subjected to SDS‐PAGE followed by Coomassie Brilliant Blue R‐250 staining. Left panel, nonreducing PAGE. Right panel, reducing PAGE. B, Binding kinetics of hepatocyte growth factor (HGF) to sMET were evaluated by surface plasmon resonance analysis. HGF was immobilized on a sensor chip C1 and binding of sMET with (right panel) or without (left panel) PNGase F treatment under native condition to HGF was measured using HBS‐EP buffer as described in “Materials and Methods”
4. DISCUSSION
In this study, we show that N‐glycans have essential roles in MET processing and downstream signaling. By using N‐glycan–deletion mutants, we demonstrated that N‐glycans are involved in the processing of MET. The findings also suggest that the N‐glycans of the SEMA domain of MET positively regulate HGF signaling, and the N‐glycans of the region other than the SEMA domain negatively regulate HGF signaling. In the all‐N‐glycan–deletion mutant, processing and signaling were significantly suppressed. The cell surface expression levels of the all‐N‐glycan–deletion mutant were significantly reduced, and the phosphorylation levels of the receptors expressed on the cell surface were also suppressed. We also identified the structures of the N‐glycans of MET and demonstrated that the occupancy of most of the N‐glycosylation sites was considerably high, and the dominant population were complex type with sialic acids and core fucoses.
We first examined the effect of N‐glycosylation inhibitors on the status of MET and found that the extent of inhibition of MET processing and trafficking is correlated with the inhibitory effects of glycosylation (Figure 1). It was demonstrated that the glycosylation inhibitory effect of NGI‐1 is incomplete compared with tunicamycin. 33 It has been reported that tunicamycin and NGI‐1 suppress the processing and trafficking of MET, 28 , 34 and by comparing these two inhibitors, it was found that these effects were correlated with the glycosylation inhibitory effects.
As inhibitors of N‐glycosylation can affect various proteins, we examined N‐glycan–deletion mutants of MET. It was found that the processing of pro‐MET to mature‐MET was suppressed in most of the mutants (Figure 3C). The suppression was significant in the mutants 6N SEMA 6Q, 5N not SEMA 5Q, 7N β 7Q, and 11N all 11Q and was less significant in the mutant 4N α 4Q. No common mutations were found in the mutants 6N SEMA 6Q and 5N not SEMA 5Q. Therefore, it is difficult to determine the N‐glycan(s) responsibility in regulating MET processing, and we assumed that the deletion of multiple N‐glycans including those in the “not SEMA” region affect MET processing. The physiological significance of MET processing has not been elucidated yet; however, we conclude that N‐glycosylation regulates the status of MET in the ER or Golgi, and that it exerts an effect on MET processing.
By examining N‐glycan–deletion mutants of MET, it was demonstrated that the deletion of N‐glycans of the SEMA domain of MET reduced the HGF‐induced downstream signaling, and that the deletion of N‐glycans of the region other than the SEMA domain of MET increased this signaling (Figure 4A,B). In the all‐N‐glycan–deletion mutant, HGF signaling was significantly suppressed. The levels of phosphorylation of MET were significantly suppressed in 4N α 4Q, 6N SEMA 6Q, 5N not SEMA 5Q, 7N β 7Q, and 11N all 11Q. For 4N α 4Q, 6N SEMA 6Q, and 11N all 11Q, MET phosphorylation was correlated with downstream signaling. The cell surface expression levels of 11N all 11Q mutant MET were significantly reduced (Figure 5B) but not completely eliminated (Figure 5C,D). As the phosphorylation ratio of the cell surface 11N all 11Q mutant MET was also significantly reduced (Figure 5E), N‐glycan deletion appears to affect the function of the receptor, not only cell surface expression of the receptor. It was suggested that the in vitro deletion of N‐glycans of pro‐MET 35 and mature‐MET (Figure 6B) does not suppress the binding of HGF to MET. N45, N149, and N202 position blade 7, blade 2, and blade 3 of MET, respectively, which are located in the SEMA domain, which is important for MET dimerization. 36 MET mutation is more common in the SEMA domain in non–small cell lung cancer including V370D, which impaired MET dimerization. 29 , 37 From the data that downstream signaling is suppressed in the transfectants of MET mutants 4N α 4Q, 6N SEMA 6Q, and 11 all 11Q, it was assumed that N‐glycans in the SEMA domain are required for the proper conformation of MET to adopt the conformation needed for ligand binding or dimerization.
In the case of the mutants 5N not SEMA 5Q and 7N β 7Q, the HGF‐induced phosphorylation levels of MET were also suppressed (Figure 4B); however, as the downstream signaling is upregulated, ligand binding is not likely to be affected in these mutants, and a pathway including other molecules should be involved. It has been reported that MET catalyzes the phosphorylation of other RTKs, including the ErbB family in cancer cells. 38 , 39 Although we were not able to determine the counterpart, we assume that N‐glycans in the region other than the SEMA domain are required for inhibiting excessive interactions with other molecules on the cell surface.
As site‐specific glycosylation occupancy and the glycan structures of MET have not been examined so far, we prepared human sMET and analyzed the glycosylation status of MET. Among the 11 N‐glycosylation potential sites, the glycan occupancy of N149, N399, N405, N635, N785, and N879 were above 99%. There are three N‐glycosylation sites with >99% occupancy, N149, N399, and N405 in the SEMA domain. We have examined the N149Q/N399Q/N405Q N‐glycan–deleted mutant, but the effect on downstream signaling was much less significant compared with the 6N SEMA 6Q mutant (data not shown). Therefore, it is possible that the deletion of N‐glycans “to some extent,” “at once,” has an effect on signaling.
Regarding the structure of the N‐glycans of MET, it has been suggested that core fucose, bisecting GlcNAc, and sialic acid are involved in MET regulation. 25 , 40 , 41 , 42 Wang et al. reported that HGF signaling is attenuated in Fut8 knockout cells, 25 , 40 and Hyuga et al. 41 reported that the HGF‐induced phosphorylation of MET and Erk is upregulated in GnT‐III–transfected cells. The downregulation of ST6Gal‐I expression was reported to inhibit MET maturation and efficient trafficking to the cell surface in colon cancer cells. 42 As shown in Figure 2, most of the MET N‐glycans in CHO‐K1 cells were complex‐type biantennary N‐glycans that contained sialic acids and core fucoses, and it is possible that sialic acids and core fucose are involved in the functional regulation of MET.
MET is a key molecule in TKI resistance and is an important therapeutic target in lung cancer. We report herein that N‐glycosylation is involved in the functional regulation of MET. It is important to determine the role of the N‐glycosylation of MET in vivo, for example, by examining tumor formation of N‐glycan–deletion mutant MET–transfected cells in nude mice. Understanding the precise mechanisms by which N‐glycans regulate MET would provide clues for establishing new methods of controlling MET function.
DISCLOSURE
The authors declare that they have no conflict of interest with the contents of this article.
ETHICAL APPROVAL
This study does not include human or animal materials, and ethics approval was not required.
Supporting information
Fig S1
ACKNOWLEDGMENTS
We wish to thank Dr. Akiko Shiratsuchi and Hiromi Okamoto from Sapporo Medical University and Dr. Tsutomu Nakagawa from Health Sciences University of Hokkaido for valuable discussions. This work was supported by JSPS KAKENHI Grant Numbers 20K17218 and 21K06083.
Saitou A, Hasegawa Y, Fujitani N, et al. N‐glycosylation regulates MET processing and signaling. Cancer Sci. 2022;113:1292–1304. doi: 10.1111/cas.15278
Funding information
Grants‐in‐Aid for Scientific Research (KAKENHI), (Grant/Award Number: ‘20K17218, 21K06083’).
REFERENCES
- 1. Giordano S, Di Renzo MF, Narsimhan RP, Cooper CS, Rosa C, Comoglio PM. Biosynthesis of the protein encoded by the c‐met proto‐oncogene. Oncogene. 1989;4:1383‐1388. [PubMed] [Google Scholar]
- 2. Bottaro DP, Rubin JS, Faletto DL, et al. Identification of the hepatocyte growth factor receptor as the c‐met proto‐oncogene product. Science. 1991;251:802‐804. [DOI] [PubMed] [Google Scholar]
- 3. Birchmeier C, Birchmeier W, Gherardi E, Vande Woude GF. Met, metastasis, motility and more. Nat Rev Mol Cell Biol. 2003;4:915‐925. [DOI] [PubMed] [Google Scholar]
- 4. Zhang J, Babic A. Regulation of the MET oncogene: molecular mechanisms. Carcinogenesis. 2016;37:345‐355. [DOI] [PubMed] [Google Scholar]
- 5. Trusolino L, Bertotti A, Comoglio PM. MET signalling: principles and functions in development, organ regeneration and cancer. Nat Rev Mol Cell Biol. 2010;11:834‐848. [DOI] [PubMed] [Google Scholar]
- 6. Matsumoto K, Umitsu M, De Silva DM, Roy A, Bottaro DP. Hepatocyte growth factor/MET in cancer progression and biomarker discovery. Cancer Sci. 2017;108:296‐307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Comoglio PM, Trusolino L, Boccaccio C. Known and novel roles of the MET oncogene in cancer: a coherent approach to targeted therapy. Nat Rev Cancer. 2018;18:341‐358. [DOI] [PubMed] [Google Scholar]
- 8. Koch JP, Aebersold DM, Zimmer Y, Medova M. MET targeting: time for a rematch. Oncogene. 2020;39:2845‐2862. [DOI] [PubMed] [Google Scholar]
- 9. Yano S, Wang W, Li Q, et al. Hepatocyte growth factor induces gefitinib resistance of lung adenocarcinoma with epidermal growth factor receptor‐activating mutations. Cancer Res. 2008;68:9479‐9487. [DOI] [PubMed] [Google Scholar]
- 10. Engelman JA, Zejnullahu K, Mitsudomi T, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007;316:1039‐1043. [DOI] [PubMed] [Google Scholar]
- 11. Paik PK, Felip E, Veillon R, et al. Tepotinib in non‐small‐cell lung cancer with MET Exon 14 skipping mutations. N Engl J Med. 2020;383:931‐943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wolf J, Seto T, Han JY, et al. Capmatinib in MET exon 14‐mutated or MET‐amplified non‐small‐cell lung cancer. N Engl J Med. 2020;383:944‐957. [DOI] [PubMed] [Google Scholar]
- 13. Helenius A, Aebi M. Intracellular functions of N‐linked glycans. Science. 2001;291:2364‐2369. [DOI] [PubMed] [Google Scholar]
- 14. Ohtsubo K, Marth JD. Glycosylation in cellular mechanisms of health and disease. Cell. 2006;126:855‐867. [DOI] [PubMed] [Google Scholar]
- 15. Varki A. Biological roles of glycans. Glycobiology. 2017;27:3‐49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Schjoldager KT, Narimatsu Y, Joshi HJ, Clausen H. Global view of human protein glycosylation pathways and functions. Nat Rev Mol Cell Biol. 2020;21:729‐749. [DOI] [PubMed] [Google Scholar]
- 17. Lau KS, Dennis JW. N‐Glycans in cancer progression. Glycobiology. 2008;18:750‐760. [DOI] [PubMed] [Google Scholar]
- 18. Takahashi M, Kizuka Y, Ohtsubo K, Gu J, Taniguchi N. Disease‐associated glycans on cell surface proteins. Mol Aspects Med. 2016;51:56‐70. [DOI] [PubMed] [Google Scholar]
- 19. Sato Y, Takahashi M, Shibukawa Y, et al. Overexpression of N‐acetylglucosaminyltransferase III enhances the epidermal growth factor‐induced phosphorylation of ERK in HeLaS3 cells by up‐regulation of the internalization rate of the receptors. J Biol Chem. 2001;276:11956‐11962. [DOI] [PubMed] [Google Scholar]
- 20. Shibukawa Y, Takahashi M, Laffont I, Honke K, Taniguchi N. Down‐regulation of hydrogen peroxide‐induced PKC delta activation in N‐acetylglucosaminyltransferase III‐transfected HeLaS3 cells. J Biol Chem. 2003;278:3197‐3203. [DOI] [PubMed] [Google Scholar]
- 21. Yokoe S, Takahashi M, Asahi M, et al. The Asn418‐linked N‐glycan of ErbB3 plays a crucial role in preventing spontaneous heterodimerization and tumor promotion. Cancer Res. 2007;67:1935‐1942. [DOI] [PubMed] [Google Scholar]
- 22. Takahashi M, Hasegawa Y, Ikeda Y, et al. Suppression of heregulin beta signaling by the single N‐glycan deletion mutant of soluble ErbB3 protein. J Biol Chem. 2013;288:32910‐32921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hasegawa Y, Takahashi M, Ariki S, et al. Surfactant protein D suppresses lung cancer progression by downregulation of epidermal growth factor signaling. Oncogene. 2015;34:838‐845. [DOI] [PubMed] [Google Scholar]
- 24. Umeda Y, Hasegawa Y, Otsuka M, et al. Surfactant protein D inhibits activation of non‐small cell lung cancer‐associated mutant EGFR and affects clinical outcomes of patients. Oncogene. 2017;36:6432‐6445. [DOI] [PubMed] [Google Scholar]
- 25. Wang Y, Fukuda T, Isaji T, et al. Loss of alpha1,6‐fucosyltransferase suppressed liver regeneration: implication of core fucose in the regulation of growth factor receptor‐mediated cellular signaling. Sci Rep. 2015;5:8264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gomes C, Osorio H, Pinto MT, Campos D, Oliveira MJ, Reis CA. Expression of ST3GAL4 leads to SLe(x) expression and induces c‐Met activation and an invasive phenotype in gastric carcinoma cells. PLoS One. 2013;8:e66737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wu YM, Liu CH, Huang MJ, et al. C1GALT1 enhances proliferation of hepatocellular carcinoma cells via modulating MET glycosylation and dimerization. Cancer Res. 2013;73:5580‐5590. [DOI] [PubMed] [Google Scholar]
- 28. Chen R, Li J, Feng CH, et al. c‐Met function requires N‐linked glycosylation modification of pro‐Met. J Cell Biochem. 2013;114:816‐822. [DOI] [PubMed] [Google Scholar]
- 29. Miao W, Sakai K, Sato H, et al. Impaired ligand‐dependent MET activation caused by an extracellular SEMA domain missense mutation in lung cancer. Cancer Sci. 2019;110:3340‐3349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tyanova S, Temu T, Cox J. The MaxQuant computational platform for mass spectrometry‐based shotgun proteomics. Nat Protoc. 2016;11:2301‐2319. [DOI] [PubMed] [Google Scholar]
- 31. Cox J, Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.‐range mass accuracies and proteome‐wide protein quantification. Nat Biotechnol. 2008;26:1367‐1372. [DOI] [PubMed] [Google Scholar]
- 32. Perez‐Riverol Y, Csordas A, Bai J, et al. The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res. 2019;47:D442‐D450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lopez‐Sambrooks C, Shrimal S, Khodier C, et al. Oligosaccharyltransferase inhibition induces senescence in RTK‐driven tumor cells. Nat Chem Biol. 2016;12:1023‐1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lopez Sambrooks C, Baro M, Quijano A, et al. Oligosaccharyltransferase inhibition overcomes therapeutic resistance to EGFR tyrosine kinase inhibitors. Cancer Res. 2018;78:5094‐5106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Stamos J, Lazarus RA, Yao X, Kirchhofer D, Wiesmann C. Crystal structure of the HGF beta‐chain in complex with the Sema domain of the Met receptor. EMBO J. 2004;23:2325‐2335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kong‐Beltran M, Stamos J, Wickramasinghe D. The Sema domain of Met is necessary for receptor dimerization and activation. Cancer Cell. 2004;6:75‐84. [DOI] [PubMed] [Google Scholar]
- 37. Sattler M, Reddy MM, Hasina R, Gangadhar T, Salgia R. The role of the c‐Met pathway in lung cancer and the potential for targeted therapy. Ther Adv Med Oncol. 2011;3:171‐184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Guo A, Villen J, Kornhauser J, et al. Signaling networks assembled by oncogenic EGFR and c‐Met. Proc Natl Acad Sci USA. 2008;105:692‐697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Liu X, Wang Q, Yang G, et al. A novel kinase inhibitor, INCB28060, blocks c‐MET‐dependent signaling, neoplastic activities, and cross‐talk with EGFR and HER‐3. Clin Cancer Res. 2011;17:7127‐7138. [DOI] [PubMed] [Google Scholar]
- 40. Wang Y, Fukuda T, Isaji T, et al. Loss of alpha1,6‐fucosyltransferase inhibits chemical‐induced hepatocellular carcinoma and tumorigenesis by down‐regulating several cell signaling pathways. FASEB J. 2015;29:3217‐3227. [DOI] [PubMed] [Google Scholar]
- 41. Hyuga M, Hyuga S, Kawasaki N, et al. Enhancement of hepatocyte growth factor‐induced cell scattering in N‐acetylglucosaminyltransferase III‐transfected HepG2 cells. Biol Pharm Bull. 2004;27:781‐785. [DOI] [PubMed] [Google Scholar]
- 42. Qian J, Zhu CH, Tang S, et al. alpha2,6‐hyposialylation of c‐Met abolishes cell motility of ST6Gal‐I‐knockdown HCT116 cells. Acta Pharmacol Sin. 2009;30:1039‐1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1