

Abstract
Although cancer precision medicine has improved diagnosis and therapy, refractory cancers such as pancreatic cancer remain to be challenging targets. Clinical sequencing has identified the significant alterations in driver genes and traced their clonal evolutions. Recent studies indicated that the tumor microenvironment elicits alterations in cancer metabolism, although its involvement in the cause and development of genomic alterations has not been established. Genomic abnormalities can contribute to the survival of selected subpopulations, recently recognized as clonal evolution, and dysfunction can lead to DNA mutations. Here, we present the most recent studies on the mechanisms of cancer metabolism involved in the maintenance of genomic stability to update current understanding of such processes. Sirtuins, which are NAD+‐dependent protein deacetylases, appear to be involved in the control of genomic stability. Alterations of deleterious subpopulations would be exposed to selective pressure for cell survival. Recent studies indicated that a new type of cell death, ferroptosis, determines the survival of clones and exert cancer‐restricting or ‐promoting effects to surrounding cells in the tumor microenvironment. Suppressing genomic instability and eliminating deleterious clones by cell death will contribute to the improvement of cancer medicine. Furthermore, the elucidation of the mechanisms involved is seen as a bridgehead to the pharmacologic suppression of such refractory cancers as pancreatic cancer.
Keywords: ferroptosis, metabolism, oxidative stress, pancreatic cancer, sirtuins
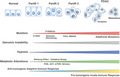
Genomic and metabolic alterations in pancreatic ductal adenocarcinoma (PDAC) development, representing the development of PDAC from pancreatic intraepithelial neoplasia (PanIN) 1, 2, and 3 in the precancer stages.
Abbreviations
- ACSL4
acyl‐CoA synthetase long‐chain family member 4
- APN
aminopeptidase N
- ATM
ataxia‐telangiectasia mutated
- BRG1
brahma‐related gene 1
- CD44v
CD44 variant
- CHD4
chromodomain helicase DNA‐binding protein 4
- DDB2
damage‐specific DNA‐binding protein 2
- DSB
DNA double‐strand break
- GLUD
glutamate dehydrogenase
- GOT1
aspartate transaminase
- GPX
glutathione peroxidase
- GSH
glutathione
- HIF1α
hypoxia‐inducible factor 1 subunit alpha
- HMGA2
high‐mobility group AT‐hook 2
- HOXA13
homeo box A13
- HR
homologous recombination
- IGF2BP1
insulin‐like growth factor 2 mRNA‐binding protein 1
- LIN28B
Lin‐28 homolog B
- LOXs
lipoxygenases
- LPCAT3
lysophosphatidylcholine acyltransferase 3
- PDAC
pancreatic ductal adenocarcinoma
- PD‐L1
programmed death‐ligand 1
- ROS
reactive oxygen species
- RPA1
replication protein A1
- SIRT1
sirtuin 1
- SLC3A2
solute carrier 3A2
- SLC7A11
solute carrier 7A11
- YTHDC2
YT521‐B homology domain containing 2
- α‐KG
α‐ketoglutarate
- γH2AX
H2A.X variant histone
1. INTRODUCTION
Pancreatic ductal adenocarcinoma (PDAC) is one of the most aggressive and lethal malignancies with a poor 5‐year survival rate of 5% due to a poor early diagnosis rate and limited response to treatments, despite the findings of recent extensive studies that stratified patients according to treatment to discover subtype‐specific therapies. 1 Clinical sequencing has elucidated three subtypes of PDAC, namely classical, quasi‐mesenchymal, and exocrine‐like ones, which correspond to their varying responses to therapy. 2
Clinical sequencing has shown that mutations of the oncogene KRAS occur in more than 90% of PDACs. Other major mutations observed include the SMAD family in the TGFb pathway and tumor suppressor genes, including TP53 and P16INK4A. 3 Recently, a study of metabolism in PDACs demonstrated that PDAC cells rely on the distinctive pathway in which glutamine supports PDAC growth through a KRAS‐regulated metabolic pathway. 4 Through this pathway, glutamine is converted to oxaloacetate by aspartate transaminase (GOT1), and oxaloacetate is converted further into malate and then pyruvate. The metabolic pathway is associated with an increased NADPH/NADP+ ratio that results in the maintenance of the cellular redox state. The KRAS pathway in PDAC is considered essential and indispensable, and the targeting oncogenes may provide novel therapeutic approaches to the treatment of PDACs. 4 However, few studies have elucidated the mechanisms of cancer metabolism alterations involved in genetic alterations, that is, whether they are a result of genomic instability, as is characteristic of malignant tumors. Dysfunction in the maintenance of genomic stability will elicit the clonal evolution of cancers. The control of cancer metabolism, genomic instability, and cell death is expected to be useful in creating a new era of cancer medicines against refractory cancer cell clones, such as those of PDACs (Figure 1).
FIGURE 1.
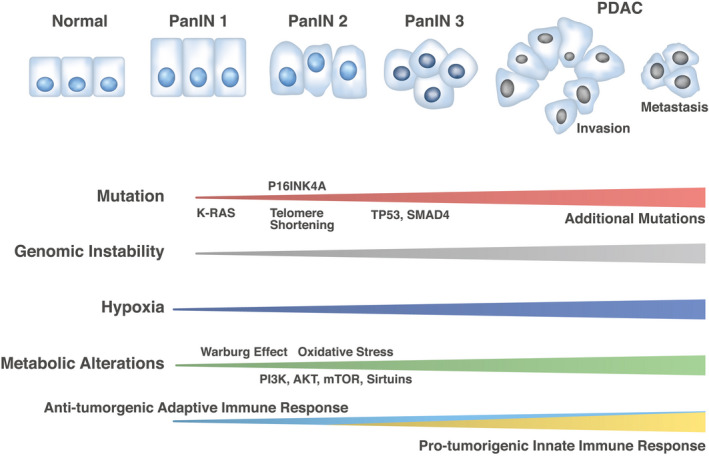
Genomic and metabolic alterations in pancreatic ductal adenocarcinoma (PDAC) development. The schema represents the development of PDAC from pancreatic intraepithelial neoplasia (PanIN) 1, 2, and 3 in the precancer stages. Clinical sequencing results indicate the mutation frequencies of KRAS (99%), TP53 (85%), and SMAD4 (55%) and telomere shortening (91%), although numerous additional mutations occur during the metastatic process. 59 Tissue abnormality has been associated with hypoxia and the Warburg effect at early and advanced stages of pancreatic carcinogenesis, 60 , 61 but recent studies have shown that cancer metabolism alterations such as phosphatidylinositol‐3 kinase (PI3K), AKT, mammalian target of rapamycin (mTOR), and sirtuins are supposed to be involved at least from PanIN 2 stages and can elicit oxidative stress and damage responses associated with genomic instability. 62 , 63 Such alterations may induce further mutations in advanced stages of PDAC. Antitumorigenic adaptive immune response may be involved in early stages of pancreatic carcinogenesis, whereas protumorigenic innate immune response may be involved in advanced stages of PDAC. 64 Oxidative stress controls the function of inflammatory cell types and mechanisms underlying genomic instability in PDAC. 65 As the effect of genomic instability, numerous neoantigens can be targets of immune cells 64 , 65
2. SIRTUINS CONTROL GENOMIC STABILITY IN PDACS
Although metabolism and DNA repair are considered different processes, and the point of contact of their mechanisms has not been clarified, recent studies have reported that several NAD+‐dependent signaling pathways regulate cell cycle progression and transcriptional regulation, as well as DNA repair, in response to genotoxic insult damages. NAD+‐dependent protein deacetylases, such as sirtuins, are involved in DNA damage responses, such as repair and recombination, during replication. 5
The sirtuin 1 (SIRT1) complex is involved in homologous recombination (HR) repair by reducing nucleosome density at DNA damage sites. 6 The ATPase domain of brahma‐related gene 1 (BRG1) and the zinc finger domain of SIRT1 interact with poly‐ADP ribose in response to DNA damage and are responsible for repairing broken DNA ends. At the damage sites, SIRT1 deacetylates BRG1 at lysine residues 1029 and 1033, which stimulates ATPase activity to promote HR. 6
SIRT2 and SIRT3 are reportedly involved in HR by utilizing I‐SceI endonuclease‐based green fluorescent protein reporter assays, which makes the precise introduction of DNA double‐strand breaks (DSBs) possible. This study indicates that SIRT2 or SIRT3 is involved in the recruitment of RAD51 to DSB sites, an essential step for RAD51‐dependent HR repair, as well as in the colocalization of the H2A.X variant histone (γH2AX) foci with replication protein A1 (RPA1). 7
The deacetylase SIRT6 is reportedly involved in chromatin remodeling and DNA damage response. SIRT6 was found recruited to sites of UV‐induced DNA damage and stimulated the repair of such damage by targeting damage‐specific DNA‐binding protein 2 (DDB2). 8 Moreover, SIRT6 is responsible for more efficient DNA DSB repair. A panel study on rodent species with diverse lifespans indicated that DSB repair and SIRT6 were optimized during the evolution of longevity, which provides new targets for antiaging interventions. 9 Furthermore, SIRT6 can coordinate with the chromatin remodeler chromodomain helicase DNA‐binding protein 4 (CHD4) to promote chromatin relaxation in response to DNA damage. 10
DNA damage can induce the activation of ataxia‐telangiectasia mutated (ATM) protein by TIP60 acetylation and autophosphorylation, which trigger the cascade of DNA damage response and repair, whereas SIRT7 is essential to the dephosphorylation and deactivation of ATM, suggesting that SIRT7 regulates ATM activity and DNA damage repair. 11 Given that DNA damage response defects are closely associated with genomic instability, an underlying hallmark of cancer, the appropriate functions of sirtuins are essential to the maintenance of DNA fidelity during replication and resultant genomic integrity.
In pancreatic cancer, SIRT1 regulates acinar‐to‐ductal metaplasia differentiation by deacetylating pancreatic transcription factor‐1a and β‐catenin, depending on its subcellular localization, and supports cancer cell viability, which can result in the sensitization of tumor cells to the SIRT1/2 inhibitor tenovin‐6. Thus, SIRT1 may be an important regulator and potential therapeutic target in pancreatic carcinogenesis. 12 Moreover, SIRT3 is involved in the hypoxia‐inducible factor 1 subunit alpha (HIF1α) pathway, which is regulated by the tumor suppressor profilin 1. The mechanism to the HIF1⍺ pathway is independent of its cytoskeleton‐related activity, although profilin1 was originally identified as an actin‐associated protein. 13 Furthermore, a study on SIRT3 and SIRT7 as biomarkers for determining patient outcome indicated tumor‐suppressing properties in the context of pancreatic cancer. 14 In addition, a previous study demonstrated that the loss of SIRT6 resulted in histone hyperacetylation at the Lin‐28 homolog B (LIN28B) promoter, MYC proto‐oncogene recruitment, and pronounced induction of LIN28B and downstream let‐7 target genes, high‐mobility group AT‐hook 2 (HMGA2), insulin‐like growth factor 2 mRNA‐binding protein 1 (IGF2BP1), and IGF2BP3. 15 The overall significance of SIRT1, SIRT3, SIRT6, and SIRT7 in cancers has indicated their use as therapeutic targets against pancreatic cancer 5 Nevertheless, the involvement of sirtuins in the control of genomic stability at exact molecular levels needs further elucidation, which would be important in the development of novel drugs (Figure 2).
FIGURE 2.
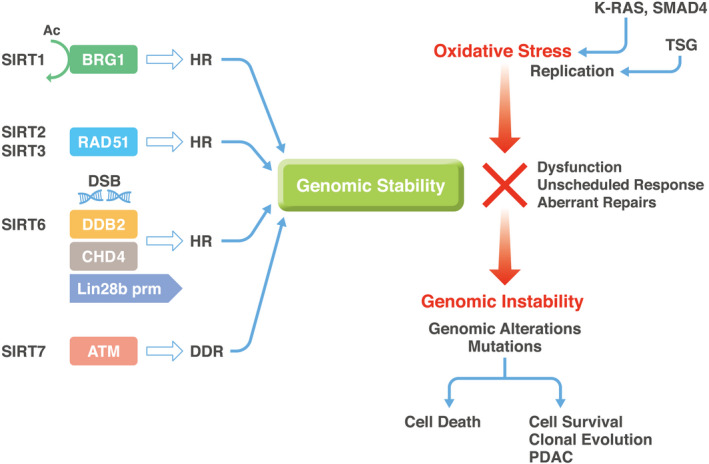
Sirtuins control genomic stability in pancreatic cells. The schema demonstrates the involvement of sirtuins in the maintenance of genomic stability in pancreatic cells. The information pertains not only to pancreatic ductal adenocarcinoma (PDAC), but also to other cancers, as shown in the main text. The schema indicates that oxidative stress gives rise to selection pressure for epithelial cells to determine their fate, whether repaired cells survive or not. Surviving cancer cells will lead to advanced PDAC. Importantly, this oxidative response may be involved in carcinogenesis in both early and advanced stages of PDAC. 64 Ac, acetyl; ATM, ataxia‐telangiectasia mutated; BRG1, brahma‐related gene 1; CHD4, chromodomain helicase DNA‐binding protein 4; DDB2, DNA‐binding protein 2; DDR, DNA damage response; DSB, DNA double‐strand break; HR, homologous recombination; Lin28b prm, Lin‐28 Homolog B (Lin28b) promoter; TSG, tumor suppressor genes, such as TP53 and P16INK4A
3. LIPID‐REACTIVE OXYGEN SPECIES CAN INDUCE FERROPTOSIS IN PDAC CELLS
Recent studies have indicated that the inevitable production of reactive oxygen species (ROS) during unlimited proliferation, a characteristic of tumors, is involved in the biological process of cells such as insults to DNA, repair of induced damage, and induction of cell death. 16 Given that a hallmark of cancer is unregulated proliferation, 17 , 18 a detailed understanding of the mechanism of tumor survival against physiological stimuli and therapeutic inductions of cancer cell death is needed. A new form of regulated cell death, ferroptosis, was discovered in 2012, 19 which involves the presence of intracellular iron and the accumulation of ROS. Recent studies have shown that such iron‐regulated form of cell death can be caused by the accumulation of lipid‐based ROS. 16 Previous studies demonstrated that unique features of ferroptosis were frequently attributed to other cell death pathways, such as necrosis, apoptosis, autophagy, and programmed cell death, but ferroptosis appears to play a central role in multiple pathologies. 16
These significant studies indicated that the following three factors can stimulate the ferroptotic pathway: (1) long‐chain polyunsaturated fatty acids contained in phospholipid membranes; (2) redox‐activated iron; and (3) defects in the lipid peroxide repair system. Certain molecular substances can also target important proteins and related metabolic pathways to trigger these processes, which are associated with cancer‐acquired drug resistance and immune evasion. 20
Among those factors, the redox activation system is well characterized. 20 The catalytic subunit of system XC − has a critical role in dependent antioxidant function. A recent study indicated that the import of oxidized cysteine (cystine) by the deletion of a solute carrier 7A11 (SLC7A11), the catalytic subunit of system XC −, induced PDAC‐selective ferroptosis, a form of cell death that results from the catastrophic accumulation of lipid‐based ROS, and resulted in the inhibition of PDAC growth; this molecule is involved in stemness of cancer cells. 21 Antioxidant function can also contribute to tumorigenesis by suppressing cell death during chemotherapy and radiation therapy. Antioxidant function is activated by the trans‐sulfuration pathway of one‐carbon metabolism, which is involved in various S‐adenosyl methionine‐induced methylation reactions. 22
Interestingly, a recent study indicated that the m6A reader YT521‐B homology domain containing 2 (YTHDC2), which can bind to methylated RNA and is frequently suppressed in lung adenocarcinoma, inhibits lung adenocarcinoma tumorigenesis by suppressing the catalytic subunit of system XC −‐dependent antioxidant function, indicating SLC7A11 is the direct target of YTHDC2. 23 Furthermore, another subunit of system XC −, solute carrier 3A2 (SLC3A2), was found equally important for YTHDC2‐induced ferroptosis. 24 The same study showed that YTHDC2 is involved in the m6A‐dependent destabilization of a transcription factor, homeobox A13 (HOXA13) mRNA, which plays a role in the promotion of SLC3A2 gene expression. YTHDC2 can be a powerful endogenous ferroptosis inducer that can suppress this solute carrier. As targeting system XC − is essential for the ferroptosis, YTHDC2 upregulation can be an alternative ferroptosis‐based cancer treatment. 24
In the downstream of system XC −, the glutathione peroxidase (GPX) family proteins reduce hydroperoxides at the expense of oxidizing two molecules of glutathione (reduced, GSH). Among the eight members of the GPX family proteins in mammals, four member proteins (GPX1, GPX2, GPX3, and GPX4) harbor a selenocysteine in their catalytic centers. 25 Specifically, GPX4 has a broader substrate preference and can reduce directly complex hydroperoxides. 20 Ferroptosis is mediated by GPX4 activity but also by acyl‐CoA synthetase long‐chain family member 4 (ACSL4), lysophosphatidylcholine acyltransferase 3 (LPCAT3), and lipoxygenases (LOXs). 20 The dysfunction of those pathways can elicit the induction and activation of innate immune cells, such as neutrophils, 26 and ferroptotic cells are efficiently engulfed by phagocytes, 27 suggesting that ferroptotic cancer cells can modulate tumor immunity. In PDACs, various forms of regulated cell death, such as apoptosis, necroptosis, ferroptosis, pyroptosis, and alkaliptosis, are involved in the process, from pathogenesis to therapeutic response. However, the exact mechanisms involving mutation burdens, such as KRAS mutations and tumor suppressors TP53 and P16INK4A, stress responses to hypoxia, poor nutrition, and chemotherapeutic reagents, and radiation exposure, remain to be understood (Figure 3). 28
FIGURE 3.
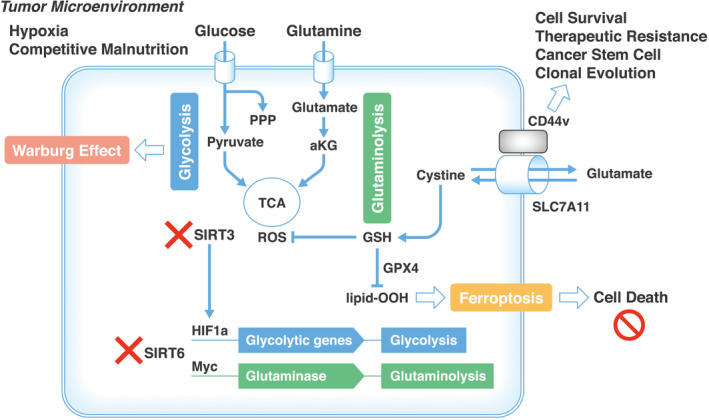
Oxidative stress response determines pancreatic ductal adenocarcinoma (PDAC) cell survival in the tumor microenvironment. The tumor microenvironment augments cancer metabolic flows of glycolysis and glutaminolysis; the former contributes to the Warburg effect and activation of PPP, which gives rise to the production of biomass such as nucleotides and lipids. The dysfunction of SIRT3 results in the stimulation of the HIF pathway, whereas the dysfunction of SIRT6 induces the transcription of glycolytic genes and glutaminase. The inevitable production of ROS from the mitochondria of living cells was reduced by GSH, which can lead to ferroptosis. The response to oxidative stress determines cell survival or death, which may contribute to clonal evolution. aKG, alpha‐ketoglutarate; CD44v, CD44 variant; GPX4, glutathione peroxidase 4; GSH, reduced glutathione, lipid‐OOH, lipid hydroperoxide; HIF1a, hypoxia‐inducible factor 1 subunit alpha; MYC, MYC proto‐oncogene, basic helix‐loop‐helix (BHLH) transcription factor; PPP, pentose phosphate pathway; ROS, reactive oxygen species; TCA, tricarboxylic acid cycle
4. SIRTUINS REGULATE FERROPTOSIS IN DAMAGED CELLS
A recent study has described the significant role of SIRT1 in the induction of ferroptosis in liver damage in mice. 29 A deficiency in intestinal SIRT1 mitigates ferroptosis, which protected mice from ethanol‐induced liver damage. This finding indicates that targeting SIRT1‐dependent ferroptosis signaling in the intestine and liver may be therapeutic against liver damage, 29 although the detailed mechanism remains to be elucidated. SIRT3 is reportedly involved in the induction of autophagy‐dependent ferroptosis via the activation of the activated protein kinase–mTOR pathway and GPX4 suppression, indicating that SIRT3 deficiency provides resistance against the inductions of high‐glucose–induced autophagy and its resulting ferroptosis. 30 Interestingly, a study on gallbladder cancer indicated that SIRT3 plays a role in the inhibition of AKT serine/threonine kinase 1–dependent mitochondrial metabolism, as well as in the blockade of epithelial‐mesenchymal transition, which thus can lead to tumor suppression by ferroptosis. 31 On the contrary, a study on traumatic brain injury indicated that SIRT2 can exert a suppressive function against TP53‐mediated ferroptosis. 32 A study using an experimental traumatic brain injury model suggests that the deacetylase function of SIRT2 is associated with the deacetylation of TP53, which inhibits TP53 expression and thus suppresses TP53‐mediated ferroptosis. Interestingly, TP53 deficiency alleviated the SIRT2 inhibition–induced exacerbation of ferroptosis 32 Considering that TP53 is inactivated in many cancers, whether SIRT2 may induce ferroptosis independently of TP53 expression remains to be further investigated. A recent study on pancreatic cancer in mice indicated that K147 of KRAS is a SIRT2‐specific deacetylation target site which was identified by mass spectrometry, and its acetylation status is associated with KRAS activity and tumor growth, suggesting that SIRT2 regulates the oncogene at the post‐translational modification level. 33 Moreover, a study on a PDAC model indicated that SIRT2 deficiency increases and prolongs a caerulein‐induced pancreatitis‐permissive phenotype, which is closely associated with the development of PDAC. 34 Notably, SIRT2 loss resulted in the induction of spontaneous KRAS mutations in mice. Furthermore, the pancreas in Sirt2‐/‐ mice exhibited a proinflammatory genomic signature. The loss of SIRT2 possibly enhanced the immune response to pancreatic injury by inflammation and induced an inflammatory phenotype permissive of the accumulation of cell clones carrying oncogenic KRAS mutations. 34 Taken together, these data suggest that sirtuins control the cell death process and are involved in the elimination of damaged cells and in the survival of abnormal cell clones with deleterious mutations in refractory cancers such as PDAC.
5. FERROPTOSIS DETERMINES CLONE SURVIVAL IN TUMOR MICROENVIRONMENT
As mentioned previously, the oxidative stress–dependent apoptotic pathway is critical in determining cell fate and cell death in the tumor microenvironment. Ferroptosis is possibly involved in the cellular response of surviving cell populations (ie, cancer stem cells) 35 after therapeutic exposure to chemotherapy and radiation, the pharmacological mechanism of action of which is mainly ROS induction in cancer cells. 36
Previous studies indicated that targeting SLC7A11 increased the ROS level and reduced cysteine and GSH levels, which subsequently attenuated the viability of cancer stem cells. This phenomenon was characterized by the induction of ferroptosis. Erastin, an inhibitor of SLC7A11, was found to hold a remarkably stronger cytotoxic effect on cancer stem cells. 37 SLC7A11 interacts with CD44 variant (CD44v), an adhesion molecule expressed in cancer stem‐like cells. 38 CD44 ablation resulted in the loss of SLC7A11 from the cell surface, thereby suppressing tumor growth in a gastric cancer mouse model with the activation of p38 (MAPK), a downstream target of ROS, and p21 (CIP1/WAF1) expression, indicating that CD44v functions as a regulator of ROS defense 38 and that ROS‐dependent ferroptosis may be involved in the selection of surviving clones after therapeutic interventions. Moreover, a study on head and neck squamous cell carcinoma indicated that the high expression of SLC7A11 and glutamine transporter SLC1A5 (ASCT2) is correlated with poor differentiation. 39 The administration of sulfasalazine, an SLC7A11 inhibitor, exhibited cytotoxicity to cancer cells via the induction of ASCT2‐dependent glutamine uptake and glutamate dehydrogenase (GLUD)‐mediated α‐ketoglutarate (α‐KG) production. This process consequently impaired GSH synthesis and enhanced oxidative phosphorylation in mitochondria, indicating oxidative damage. 39 In addition, another study on cell membrane–bound aminopeptidase N (APN) indicated that APN plays a role in the reduction of ROS levels in cancer stem cell clones. 40 The inhibition of APN resulted in the induction of cell death due to oxidative stress after exposure to chemotherapeutic agents. 41 Although multiple cell death pathways are suggested in therapeutic resistance in PDAC, 42 ROS‐dependent cellular responses possibly play a critical role in determining which cells survive. Monitoring by measurement or sequencing will help obtain an exact diagnosis and determine further therapeutic targets.
Given that intratumor heterogeneity is one of the causes of the intractability of cancers, the sustained survival of cancer stem cells and their derivative clones may be involved in the generation of tumor heterogeneity. 43 A super‐computational analysis of colorectal cancer revealed that driver gene aberrations were observed in patients with early‐stage cancer, whereas parental clones branched into numerous subclones in advanced‐stage cancer, with minor mutations such as neutral evolution. 43 A previous sequence analysis of PDAC demonstrated that clonal populations that give rise to distant metastasis are represented within the primary tumor tissues. The clonal populations are genetically evolved from nonmetastatic clones, suggesting that at least five more years are required for the acquisition of metastatic ability, 44 presumably via the accumulation of spatial and genetic divergences of mutations over time. 45 A deep sequence analysis indicated that epithelial remodeling via the expansion of clones with driver mutations is an inevitable consequence of normal aging and depends on lifestyle risks, such as tobacco or alcohol use. 46 A study on long‐term survivors of pancreatic cancer indicate the involvement of MUC16/CA125 neoantigens, which were lost during metastatic progression, suggesting neoantigen immunoediting. 47 Although many studies have suggested that oxidative stress response is involved in immunological monitoring mechanisms 48 and that ROS play an important role in T‐cell function, 49 further research is needed on the mechanism involving ferroptosis in the processes of these immunological surveillance mechanisms.
6. THERAPEUTIC TARGETING IN PDAC METABOLISM
Previous studies have indicated that targeting the metabolic differences between tumor and normal cells holds promise as a novel anticancer strategy. 50 Although evidence of the usefulness of therapeutic targeting in PDAC is accumulating, the previous studies actually identified several sirtuin modulators that functioned as activators or inhibitors that may be useful against several types of cancers. Resveratrol functions as an activator of SIRT1, SIRT3, and SIRT5. 51 , 52
Resveratrol functions as an activator of SIRT1, it induces protective autophagy in non–small cell lung cancer by inhibiting Akt/mTOR and activating p38‐MAPK. 53 Resveratrol and piceatannol (both activators of SIRT1, SIRT3, and SIRT5) upregulated programmed death‐ligand 1 (PD‐L1) expression in breast and colorectal cancer cells via histone acetylase 3/p300–mediated nuclear factor kappa‐light‐chain‐enhancer of activated B‐cells signaling, suggesting the usefulness of a combination of immune checkpoint therapy. 54 In contrast, nicotinamide inhibits SIRT2 and SIRT6 55 , 56 and is considered beneficial in the prevention of breast cancer recurrence. 57 In the sirtuin‐targeting strategy, family‐specific reagents will be necessary to be innovated, given that the standard therapeutic approaches are still not effective enough. For resectable PDAC cases, surgery followed by adjuvant chemotherapy with gemcitabine plus capecitabine is the standard of care. For borderline resectable and locally advanced unresectable PDACs, neoadjuvant protocols are utilized, whereas for metastatic PDACs, FOLFIRINOX (fluorouracil, irinotecan, and oxaliplatin) and nab‐paclitaxel–gemcitabine are standard treatment options in patients with good performance status. 58 We therefore emphasize that understanding the underlying mechanisms that allow sirtuins to show apparently two opposite anticancer roles should be one of our main challenges in developing effective PDAC treatment (Figure 4).
FIGURE 4.
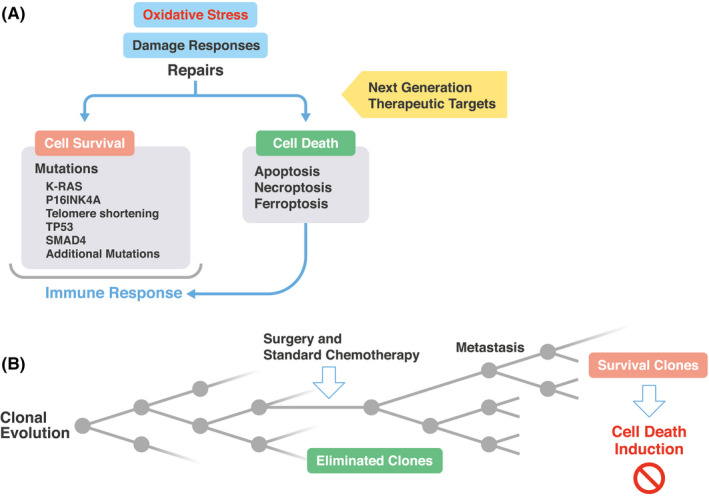
Oxidative stress induces damage response, and the selection of surviving clones presents new therapeutic targets against pancreatic ductal adenocarcinoma (PDAC). A, Oxidative stress induces damage responses in pancreatic cells, which can elicit the repair of deleterious alterations. The schema indicates that the resultant surviving cells harbor mutations or abnormalities, such as KRAS, P16INK4A, TP53, and SMAD4 and telomere shortening, whereas cell death was induced in heavily damaged cells, which induce immune response. 20 B, The surviving cells may expand, and dead cells will be eliminated from PDAC tissues, which will give rise to clonal evolution, as demonstrated by clinical sequencing analysis 44 , 45
Improving the treatment results of pancreatic cancer has been a longstanding goal in the oncological field. The genetic changes in pancreatic cancer are relatively simple, centered on KRAS drivers; however, their biological malignancy is extremely high. As analyzed in this paper, the metabolic characteristics of pancreatic cancer are closely related to genomic changes. Clarifying the mechanism for the selection and creation of dangerous clones is a major challenge in the medical treatment of pancreatic cancer. If the dangerous clones resulting from the oxidative stress response can be eradicated, current standard treatments can be combined to improve the outcome of pancreatic cancer.
DISCLOSURE
Partial institutional endowments were received by H.I. from Hirotsu Bio Science Inc. (Tokyo, Japan), Kinshu‐kai Medical Corporation (Osaka, Japan), IDEA Consultants Inc. (Tokyo, Japan), Kyowa‐kai Medical Corporation (Osaka, Japan), and Unitech Co. Ltd. (Chiba, Japan). K.O. is an employee of IDEA Consultants Inc. (Tokyo, Japan). The remaining authors do not have any conflict of interest. H.I. is a current associate editor in the editorial board of Cancer Science.
ACKNOWLEDGEMENT
We thank Professor Koshi Mimori, Kyushu University Beppu Hospital, Japan, for critically reviewing this manuscript. The authors are thankful to all lab members.
Takeda Y, Chijimatsu R, Ofusa K, et al. Cancer metabolism challenges genomic instability and clonal evolution as therapeutic targets. Cancer Sci. 2022;113:1097–1104. doi: 10.1111/cas.15279
Funding information
This work was supported in part by a Grant‐in‐Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science, and Technology (20H00541; 21K19526) to H.I. Partial support was received by H.I. from Princess Takamatsu Cancer Research Fund, Senshin Medical Research Foundation, and Mitsubishi Foundation.
DATA AVAILABILITY STATEMENT
Not applicable in this review article.
REFERENCES
- 1. Kleeff J, Korc M, Apte M, et al. Pancreatic cancer. Nat Rev Dis Primers. 2016;2:16022. [DOI] [PubMed] [Google Scholar]
- 2. Collisson EA, Sadanandam A, Olson P, et al. Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nat Med. 2011;17:500‐503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Makohon‐Moore A, Iacobuzio‐Donahue CA. Pancreatic cancer biology and genetics from an evolutionary perspective. Nat Rev Cancer. 2016;16:553‐565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Son J, Lyssiotis CA, Ying H, et al. Glutamine supports pancreatic cancer growth through a KRAS‐regulated metabolic pathway. Nature. 2013;496:101‐105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chiarugi A, Dölle C, Felici R, Ziegler M. The NAD metabolome — a key determinant of cancer cell biology. Nat Rev Cancer. 2012;12(11):741‐752. doi: 10.1038/nrc3340 [DOI] [PubMed] [Google Scholar]
- 6. Chen Y, Zhang H, Xu Z, et al. A PARP1‐BRG1‐SIRT1 axis promotes HR repair by reducing nucleosome density at DNA damage sites. Nucleic Acids Res. 2019;47:8563‐8580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yasuda T, Takizawa K, Ui A, et al. Human SIRT2 and SIRT3 deacetylases function in DNA homologous recombinational repair. Gene Cell. 2021;26:328‐335. [DOI] [PubMed] [Google Scholar]
- 8. Geng A, Tang H, Huang J, et al. The deacetylase SIRT6 promotes the repair of UV‐induced DNA damage by targeting DDB2. Nucleic Acids Res. 2020;48:9181‐9194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tian X, Firsanov D, Zhang Z, et al. SIRT6 is responsible for more efficient DNA double‐strand break repair in long‐lived species. Cell. 2019;177:622‐638.e622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hou T, Cao Z, Zhang J, et al. SIRT6 coordinates with CHD4 to promote chromatin relaxation and DNA repair. Nucleic Acids Res. 2020;48:2982‐3000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tang M, Li Z, Zhang C, et al. SIRT7‐mediated ATM deacetylation is essential for its deactivation and DNA damage repair. Sci Adv. 2019;5:eaav1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wauters E, Sanchez‐Arévalo Lobo VJ, Pinho AV, et al. Sirtuin‐1 regulates acinar‐to‐ductal metaplasia and supports cancer cell viability in pancreatic cancer. Cancer Res. 2013;73:2357‐2367. [DOI] [PubMed] [Google Scholar]
- 13. Yao W, Ji S, Qin Y, et al. Profilin‐1 suppresses tumorigenicity in pancreatic cancer through regulation of the SIRT3‐HIF1alpha axis. Mol Cancer. 2014;13:187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. McGlynn LM, McCluney S, Jamieson NB, et al. SIRT3 & SIRT7: Potential novel biomarkers for determining outcome in pancreatic cancer patients. PLoS One. 2015;10:e0131344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kugel S, Sebastián C, Fitamant J, et al. SIRT6 suppresses pancreatic cancer through control of Lin28b. Cell. 2016;165:1401‐1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hirschhorn T, Stockwell BR. The development of the concept of ferroptosis. Free Radic Biol Med. 2019;133:130‐143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57‐70. [DOI] [PubMed] [Google Scholar]
- 18. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646‐674. [DOI] [PubMed] [Google Scholar]
- 19. Dixon S, Lemberg K, Lamprecht M, et al. Ferroptosis: an iron‐dependent form of nonapoptotic cell death. Cell. 2012;149:1060‐1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Friedmann Angeli JP, Krysko DV, Conrad M. Ferroptosis at the crossroads of cancer‐acquired drug resistance and immune evasion. Nat Rev Cancer. 2019;19:405‐414. [DOI] [PubMed] [Google Scholar]
- 21. Badgley MA, Kremer DM, Maurer HC, et al. Cysteine depletion induces pancreatic tumor ferroptosis in mice. Science. 2020;368:85‐89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Takeda Y, Chijimatsu R, Vecchione A, et al. Impact of one‐carbon metabolism‐driving epitranscriptome as a therapeutic target for gastrointestinal cancer. Int J Mol Sci. 2021;22(14):7278. doi: 10.3390/ijms22147278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ma L, Chen T, Zhang X, et al. The m6A reader YTHDC2 inhibits lung adenocarcinoma tumorigenesis by suppressing SLC7A11‐dependent antioxidant function. Redox Biol. 2021;38:101801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ma L, Zhang X, Yu K, et al. Targeting SLC3A2 subunit of system XC− is essential for m6A reader YTHDC2 to be an endogenous ferroptosis inducer in lung adenocarcinoma. Free Radic Biol Med. 2021;168:25‐43. [DOI] [PubMed] [Google Scholar]
- 25. Brigelius‐Flohé R, Maiorino M. Glutathione peroxidases. Biochim Biophys Acta. 2013;1830(5):3289‐3303. doi: 10.1016/j.bbagen.2012.11.020 [DOI] [PubMed] [Google Scholar]
- 26. Li W, Feng G, Gauthier JM, et al. Ferroptotic cell death and TLR4/Trif signaling initiate neutrophil recruitment after heart transplantation. J Clin Invest. 2019;129:2293‐2304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Klöditz K, Fadeel B. Three cell deaths and a funeral: macrophage clearance of cells undergoing distinct modes of cell death. Cell Death Discov. 2019;5:65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chen X, Zeh HJ, Kang R, et al. Cell death in pancreatic cancer: from pathogenesis to therapy. Nat Rev Gastroenterol Hepatol. 2021;18:804‐823. [DOI] [PubMed] [Google Scholar]
- 29. Zhou Z, Ye TJ, DeCaro E, et al. Intestinal SIRT1 deficiency protects mice from ethanol‐induced liver injury by mitigating ferroptosis. Am J Pathol. 2020;190:82‐92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Han D, Jiang L, Gu X, et al. SIRT3 deficiency is resistant to autophagy‐dependent ferroptosis by inhibiting the AMPK/mTOR pathway and promoting GPX4 levels. J Cellul Physiol. 2020;235:8839‐8851. [DOI] [PubMed] [Google Scholar]
- 31. Liu L, Li Y, Cao D, et al. SIRT3 inhibits gallbladder cancer by induction of AKT‐dependent ferroptosis and blockade of epithelial‐mesenchymal transition. Cancer Letter. 2021;510:93‐104. [DOI] [PubMed] [Google Scholar]
- 32. Gao J, Li Y, Song R. SIRT2 inhibition exacerbates p53‐mediated ferroptosis in mice following experimental traumatic brain injury. NeuroReport. 2021;32:1001‐1008. [DOI] [PubMed] [Google Scholar]
- 33. Song HY, Biancucci M, Kang H‐J, et al. SIRT2 deletion enhances KRAS‐induced tumorigenesis in vivo by regulating K147 acetylation status. Oncotarget. 2016;7:80336‐80349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Quan S, Principe DR, Dean AE, et al. Loss of Sirt2 increases and prolongs a caerulein‐induced pancreatitis permissive phenotype and induces spontaneous oncogenic Kras mutations in mice. Sci Rep. 2018;8:16501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Elgendy SM, Alyammahi SK, Alhamad DW, et al. Ferroptosis: An emerging approach for targeting cancer stem cells and drug resistance. Criti Rev Oncol Hematol. 2020;155:103095. [DOI] [PubMed] [Google Scholar]
- 36. Reya T, Morrison SJ, Clarke MF, et al. Stem cells, cancer, and cancer stem cells. Nature. 2001;414:105‐111. [DOI] [PubMed] [Google Scholar]
- 37. Xu X, Zhang X, Wei C, et al. Targeting SLC7A11 specifically suppresses the progression of colorectal cancer stem cells via inducing ferroptosis. Eur J Pharmaceut Sci. 2020;152:105450. [DOI] [PubMed] [Google Scholar]
- 38. Ishimoto T, Nagano O, Yae T, et al. CD44 variant regulates redox status in cancer cells by stabilizing the xCT subunit of system xc− and thereby promotes tumor growth. Cancer Cell. 2011;19:387‐400. [DOI] [PubMed] [Google Scholar]
- 39. Okazaki S, Umene K, Yamasaki J, et al. Glutaminolysis‐related genes determine sensitivity to xCT‐targeted therapy in head and neck squamous cell carcinoma. Cancer Sci. 2019;110:3453‐3463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Haraguchi N, Ishii H, Mimori K, et al. CD13 is a therapeutic target in human liver cancer stem cells. J Clin Invest. 2010;120:3326‐3339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Toshiyama R, Konno M, Eguchi H, et al. Poly(ethylene glycol)–poly(lysine) block copolymer–ubenimex conjugate targets aminopeptidase N and exerts an antitumor effect in hepatocellular carcinoma stem cells. Oncogene. 2019;38:244‐260. [DOI] [PubMed] [Google Scholar]
- 42. Santofimia‐Castaño P, Iovanna J. Combating pancreatic cancer chemoresistance by triggering multiple cell death pathways. Pancreatology. 2021;21:522‐529. [DOI] [PubMed] [Google Scholar]
- 43. Mimori K, Saito T, Niida A, et al. Cancer evolution and heterogeneity. Annal Gastroenterol Surg. 2018;2:332‐338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Yachida S, Jones S, Bozic I, et al. Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature. 2010;467:1114‐1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Makohon‐Moore AP, Matsukuma K, Zhang M, et al. Precancerous neoplastic cells can move through the pancreatic ductal system. Nature. 2018;561:201‐205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Yokoyama A, Kakiuchi N, Yoshizato T, et al. Age‐related remodelling of oesophageal epithelia by mutated cancer drivers. Nature. 2019;565:312‐317. [DOI] [PubMed] [Google Scholar]
- 47. Balachandran VP, Łuksza M, Zhao JN, et al. Identification of unique neoantigen qualities in long‐term survivors of pancreatic cancer. Nature. 2017;551:512‐516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Muri J, Kopf M. Redox regulation of immunometabolism. Nature Rev Immunol. 2021;21:363‐381. [DOI] [PubMed] [Google Scholar]
- 49. Franchina DG, Dostert C, Brenner D. Reactive oxygen species: Involvement in T cell signaling and metabolism. Trend Immunol. 2018;39:489‐502. [DOI] [PubMed] [Google Scholar]
- 50. Martinez‐Outschoorn UE, Peiris‐Pagés M, Pestell RG, et al. Cancer metabolism: a therapeutic perspective. Nat Rev Clin Oncol. 2017;14:11‐31. [DOI] [PubMed] [Google Scholar]
- 51. Gertz M, Nguyen GTT, Fischer F, et al. A molecular mechanism for direct Sirtuin activation by resveratrol. PLoS One. 2012;7:e49761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Howitz KT, Bitterman KJ, Cohen HY, et al. Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature. 2003;425:191‐196. [DOI] [PubMed] [Google Scholar]
- 53. Wang J, Li J, Cao N, et al. Resveratrol, an activator of SIRT1, induces protective autophagy in non‐small‐cell lung cancer via inhibiting Akt/mTOR and activating p38‐MAPK. OncoTarget Therapy. 2018;11:7777‐7786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Lucas J, Hsieh T‐C, Halicka HD, et al. Upregulation of PD‐L1 expression by resveratrol and piceatannol in breast and colorectal cancer cells occurs via HDAC3/p300‐mediated NF‐κB signaling. Intern J Oncol. 2018;53:1469‐1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Tervo AJ, Kyrylenko S, Niskanen P, et al. An in silico approach to discovering novel inhibitors of human Sirtuin type 2. J Med Chemist. 2004;47:6292‐6298. [DOI] [PubMed] [Google Scholar]
- 56. Hu J, He B, Bhargava S, et al. A fluorogenic assay for screening Sirt6 modulators. Organ Biomol Chemist. 2013;11:5213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Dell'Omo G, Ciana P. Nicotinamide in the prevention of breast cancer recurrences. Oncotarget. 2019;10:5495‐5496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Neoptolemos JP, Kleeff J, Michl P, et al. Therapeutic developments in pancreatic cancer: current and future perspectives. Nat Rev Gastroenterol Hepatol. 2018;15:333‐348. [DOI] [PubMed] [Google Scholar]
- 59. Hu H‐F, Ye Z, Qin YI, et al. Mutations in key driver genes of pancreatic cancer: molecularly targeted therapies and other clinical implications. Acta Pharmacol Sin. 2021;42:1725‐1741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Jiang S‐H, Li J, Dong F‐Y, et al. Increased serotonin signaling contributes to the Warburg effect in pancreatic tumor cells under metabolic stress and promotes growth of pancreatic tumors in mice. Gastroenterology. 2017;153:277‐291.e219. [DOI] [PubMed] [Google Scholar]
- 61. Yang J, Ren BO, Yang G, et al. The enhancement of glycolysis regulates pancreatic cancer metastasis. Cell Mol Life Sci. 2020;77:305‐321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Halbrook CJ, Lyssiotis CA. Employing metabolism to improve the diagnosis and treatment of pancreatic cancer. Cancer Cell. 2017;31:5‐19. [DOI] [PubMed] [Google Scholar]
- 63. Qin C, Yang G, Yang J, et al. Metabolism of pancreatic cancer: paving the way to better anticancer strategies. Mol Cancer. 2020;19:50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Wörmann SM, Diakopoulos KN, Lesina M, et al. The immune network in pancreatic cancer development and progression. Oncogene. 2014;33:2956‐2967. [DOI] [PubMed] [Google Scholar]
- 65. Padoan A, Plebani M, Basso D. Inflammation and pancreatic cancer: Focus on metabolism, cytokines, and immunity. Int J Mol Sci. 2019;20:676. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable in this review article.