

Abstract
Puberty marks the end of childhood and is a period when individuals undergo physiological and psychological changes to achieve sexual maturation and fertility. The onset of puberty is first detected as an increase in pulsatile secretion of gonadotropin-releasing hormone (GnRH). Pubertal onset is regulated by genetic, nutritional, environmental, and socio-economic factors. Disturbances affecting pubertal timing result in adverse health conditions later in life. Human genetic studies show that around 50–80% of the variation in pubertal onset is genetically determined. The genetic control of pubertal timing has been a field of active investigation in attempt to better understand the neuroendocrine control of this relevant period of life. Large populational studies and patient cohort-based studies have provided insights into the genetic regulation of pubertal onset. In this review, we discuss these discoveries and discuss potential mechanisms for how implicated genes may affect pubertal timing.
Keywords: puberty, genetics, GWAS, early puberty, delayed puberty
1. Introduction
Puberty is the developmental stage of physical and psychological maturation in which reproductive capacity, as well as adult stature, is attained. The hypothalamic-pituitary-gonadal (HPG) axis controls puberty and reproduction and is tightly regulated by a complex network of excitatory and inhibitory factors. Gonadotropin-releasing hormone (GnRH) neurons migrate from the nasal compartment to the hypothalamic preoptic area during embryogenesis. GnRH is released from axon terminals in the median eminence in a pulsatile manner to stimulate the secretion of luteinizing hormone (LH) and follicle-stimulating hormone (FSH) from the pituitary, which in turn act on the gonads to promote gametogenesis and the production of sex steroids (Figure 1 A–B). The HPG axis is active in the mid-gestational fetus but silenced towards the end of gestation. This restraint is removed at birth, leading to reactivation of the axis and an increase in gonadotropin concentrations. Subsequently, there is an active suppression of the axis during childhood. Studies in rodents and primates have shown that the initiation of puberty is orchestrated by an augmentation of GnRH through an enhancement of excitatory inputs and a reduction in inhibitory factors (1,2). Although genetic studies have increased our understanding of the regulation of the HPG axis, the exact components of this complex network regulating the reactivation of GnRH secretion, and consequently pubertal onset, is not entirely recognized.
Figure 1: Genes associated with pubertal timing across the lifespan.
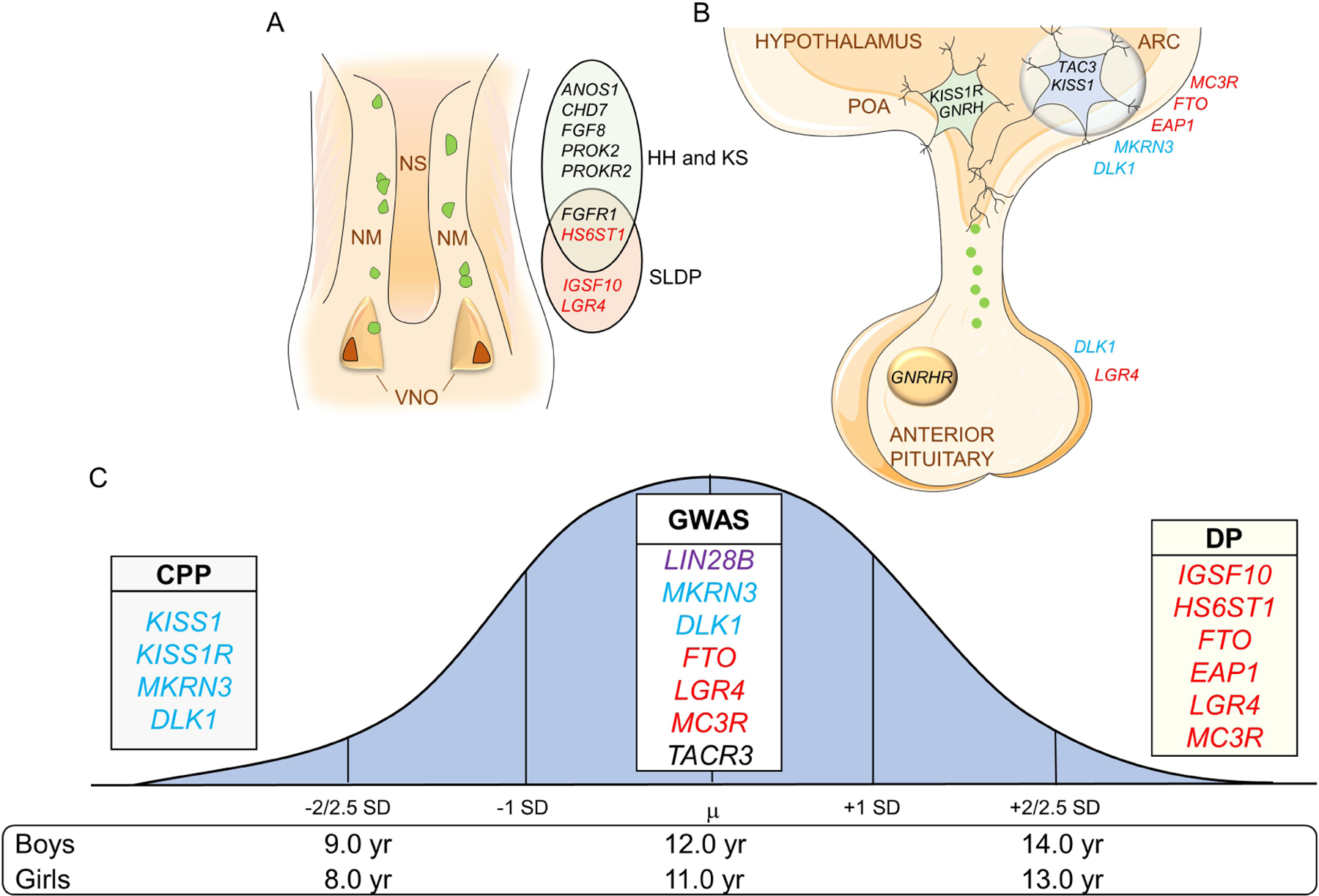
Genes in blue are associated with CPP, in red with DP, and genes in black are associated with HH and/or KS. Variants in LIN28B, in purple, have only been associated with pubertal onset in the normal population. A. Representation of the nasal placode at embryonic life and key genes important for GnRH neuronal migration. GnRH neurons (in green) will migrate alongside olfactory axons from the VNO to the forebrain at the level of the olfactory bulbs (not shown). Mutations in these genes can cause dysregulation in GnRH neuronal migration resulting in delayed puberty and/or HH-KS. B. Figure depicting the hypothalamic and pituitary regions involved in the central regulation of the reproductive axis and key genes controlling GnRH secretion. Kisspeptin-Neurokinin B-Dynorphin (KNDy) neuron is shown in blue in the ARC communicating with GnRH neuron (in green) in the POA. C. Normal distribution of the timing of puberty in the general population with the mean (µ) age of onset. ±2 to 2.5 standard deviation (SD) values indicate precocious or delayed onset of puberty. List of some of the important genes identified in genome-wide association studies (GWAS) and in patient-cohort based studies associated with timing of pubertal onset. VNO: vomeronasal organs; NM: nasal mesenchyme; NS: nasal septum; HH: hypogonadotropic hypogonadism; KS: Kallmann syndrome; SLDP: self-limited delayed puberty. ARC: Arcuate nucleus, POA: preoptic area, CPP: central precocious puberty, DP: delayed puberty.
Data showing an association between the ages at menarche in mothers and daughters and similar timing of puberty within ethnic groups are suggestive of genetic modulation of the reproductive endocrine axis. Human genetics studies suggest that around 50–80% of the variation in pubertal onset might be genetically determined (3,4). Nutritional status, adoption, geographical migration, and emotional well-being have also been shown to influence pubertal timing (5–7). Nutritional changes have an important role, as shown by the decrease in the average age of menarche between the mid-19th and the mid-20th centuries in the USA and in some European countries due to improvement in general health and nutrition (8). Later, human genetic studies revealed important factors linking the reproductive and metabolic systems (9,10).
Puberty onset varies between normal individuals and there is a 4–5-year window judged to be the normal age of pubertal development. The average timing of pubertal development is 8 to 13 years of age in Caucasian girls and 9 to 14 years of age in boys. (4). However, these ages are arbitrary and vary in different ethnicities. The mean age for onset of breast development in African-American girls is usually earlier than that in white girls (11). The onset of puberty before or after ± 2 to 2.5 standard deviations (SD) of the mean age is deemed pathological (Figure 1C) (4). It is unclear why boys enter puberty one to two years later than girls (12). Also, it is not clear why disorders of earlier puberty are exceedingly more common in girls than in boys, while disorders of delayed puberty are more common in boys than in girls. The first clinical sign of puberty onset in girls is breast development, and in boys, testicular enlargement. Testicular enlargement can be difficult to detect, which may contribute to undiagnosed cases of precocious puberty in boys. However, it seems like there are other unknown factors contributing to a higher frequency of precocious puberty in girls and delayed puberty in boys. In this review, we will discuss the genetic factors contributing to normal pubertal onset, Central Precocious Puberty (CPP), and Delayed Puberty (DP). Genes associated with hypogonadism will be discussed in another paper of this Series.
2. Genetic variants associated with age of onset of puberty in normal population
Genome-wide association and custom-genotyping array studies have identified several genetic variants enriched in the general population associated with puberty onset. Although menarche is regarded as the final marker of puberty in girls (11), it is nonetheless a milestone in female pubertal development that is easily recollected. For this reason, menarche is the pubertal marker used in most populational studies. In boys, age at voice breaking has been used as a similar marker (13).
In 2009, genome-wide association studies (GWAS) found polymorphisms near or in LIN28B to be significantly associated with age of menarche in girls (14–17). LIN28B is an RNA-binding protein whose major function is to repress the maturation of the large family of miRNAs, let-7, suggesting that miRNA regulatory pathways may participate in the control of puberty. Studies in mice demonstrated that genetic overexpression of the other member of the Lin28 family, Lin28A, resulted in a delay of pubertal onset, further supporting a role of the Lin28/let-7 pathway in pubertal control (18). However, the mechanism by which LIN28B controls GnRH secretion is unknown. It is suggested that LIN28B is involved in developmental mechanisms leading to pubertal onset (18). To date, one of the strongest signals associated with age at menarche and for voice breaking is located at LIN28B locus (13,19).
In 2014, GWAS and custom-genotyping arrays, taking into consideration the parent of origin of the variants, identified signals associated with age at menarche near imprinted genes, including DLK1 and MKRN3 which are both maternally imprinted and paternally expressed genes (20). Mutations in these genes were also identified in patients with CPP and will be discussed below (21,22). According to evolutionary theories, paternally expressed genes are predicted to increase the offspring demands. This is consistent with the finding that MKRN3 and DLK1 participate in the inhibition of puberty initiation, as loss-of-function mutations in these genes result in early pubertal development (21,23,24).
Most of the signals associated with puberal timing in GWAS studies were identified in genes expressed in the hypothalamus, highlighting the importance of the neuroendocrine regulation of pubertal onset (19,20). Some of these genes had been previously linked to the HPG axis such as GNRH1, FGFR1, TACR3 and FSHR (20,25,26). Furthermore, signals associated with pubertal timing and body mass such as FTO, LEPR, EC16B, TMEM18, NEGR1 and MC3R underscore the link between reproduction and metabolism (27). Genetic variants near MC3R have also been associated with adult height and reduced lean body mass (19,28). Further investigation of the MC3R variants revealed a strong signal at this gene locus with female carriers presenting with a 4.7-month delay in age at menarche (29). As we will discuss below, mutations in FTO and MC3R have also been associated with DP (27).
Improvement in methodologies and collaboration through larger populational studies yielded stronger correlations with pubertal markers. GWAS and custom-genotype studies identified additional signals indicating shared etiologies in both sexes between puberty timing and body mass index, fasting insulin levels, lipid levels, type 2 diabetes and cardiovascular disease (26). Furthermore, more recent studies linked several variants associated with puberty timing markers with risks for breast and endometrial cancers in women and prostate cancer in men (19,30).
In 2017, a large GWAS including not only girls with pubertal timing within the normal range, but also early and late pubertal timing of European ancestry revealed that the variants associated with puberty markers had disproportionate effects in a sex-discordant pattern. In females, the common genetic variants contribute more to early than late pubertal timing in girls, and the opposite was seen in males (19). This difference may also explain, at least partially, why precocious puberty appears to be more common in girls and delayed puberty in boys.
This fast approaching and successful progress derived by GWAS is constantly producing feasible candidates influencing pubertal timing. It also suggests, however, that many of these genetic variants have a low impact in the general population. These large-scale GWAS show that pubertal onset is highly polygenic, like other quantitative traits, highlighting the complexity of the genetic regulation of puberty timing (20,31).
3. Disturbances of pubertal timing
Approximately 5% of children have either precocious or delayed puberty, which is associated with adverse health and psychosocial outcomes (32,33). Early puberty has been associated with adverse health outcomes, including breast and endometrial cancer, obesity, type 2 diabetes, cardiovascular disease, short stature, and increased mortality (19,34–36). Data from the UK Biobank study from both men and women has demonstrated that delayed puberty also has great impacts on health in later life: late menarche (15–19 years) has been associated with significantly increased risk of early menopause, osteoporosis, malabsorption, low intelligence, asthma, and poor overall health (37,38). In addition, the same study observed an increased risk for cervical cancer, angina, myocardial infarction, high cholesterol, still birth, and depression. Later voice breaking in men was also significantly associated with anxiety disorders, chronic fatigue syndrome, depression, asthma, and poor overall health (37). Many previous studies have shown that patients with delayed puberty are at risk of short stature, and have a risk for decreased bone mineral density, and increased psychological distress (39–42).
4. Genetics of CPP
Precocious puberty encompasses benign variants of normal development, gonadotropin-independent precocious puberty, and gonadotropin-dependent precious puberty. Gonadotropin-dependent, or CPP, results from the early maturation of the hypothalamic-pituitary-gonadal axis. CPP is a rare disorder with a prevalence of 1 in 5,000 to 1 in 10,000 children. The diagnosis is based on physical exam findings indicating advancing puberty and on laboratory tests confirming central HPG axis activation. Several neurological disorders and chronic exposure to exogenous or endogenous sex steroids can cause CPP. Most cases of CPP in girls and up to 60% of cases in boys do not have a detectable CNS lesion and are described as idiopathic CPP. Some or most of these cases might have a genetic, metabolic, or environmental component, or a combination of these factors (4). Approximately 10% of CPP cases have a genetic cause identified.
Kisspeptin system
After several key regulators of GnRH secretion were identified in genetic studies of families with hypogonadism, a single mutation in KISS1R was identified in one girl with CPP (43). KISS1R was formerly known as GPR54, because it encodes a G-protein coupled receptor. KISS1R is expressed in GnRH secreting neurons. The only mutation in KISS1R associated with CPP, p.Arg386Pro, was identified in an adopted girl who had progressive thelarche from birth, suggesting early, persistent, and slightly increased estrogen secretion. Accelerated growth, skeletal maturation, and progression of breast development were noticed at the age of 7 years. The mechanism by which the p.Arg386Pro mutation, located in the C-terminal tail of the receptor, results in early puberty came from in vitro studies, which showed that the amino acid substitution led to prolonged activation of KISS1R intracellular signaling pathways in response to the ligand (43,44).
Kisspeptin, the ligand of KISS1R, is expressed in the arcuate and anteroventral periventricular hypothalamic nuclei. Physiological and pharmacological studies have shown that the kisspeptin ligand/receptor system is an essential regulator of GnRH secretion (45). Kisspeptin and neurokinin B are tightly regulated GnRH pulse frequency (Figure 1B)(46). One rare KISS1 variant, p.Pro74Ser, was identified in the heterozygous state in a boy who developed CPP at age 1 year, with high concentrations of basal LH and testosterone (47). Results of in vitro studies showed that the capacity to stimulate signal transduction was significantly greater for mutant p.Pro74Ser compared to wild type KISS1, suggesting that this variant might be more resistant to degradation, resulting in greater kisspeptin bioavailability (47). The mother and maternal grandmother of the boy carrying the mutation, also carried the p.Pro74Ser mutation in the heterozygous state, and they both had normal pubertal development. This finding suggests incomplete sex-dependent penetrance.
Physiological and pharmacological studies have shown that the kisspeptin ligand/receptor system is an essential part of the excitatory network that regulates GnRH secretion. It is important to mention that recent studies showed that the kisspeptin neurons are subjected to metabolic regulation, linking signals of caloric sufficiency to the reproductive axis (10).
MKRN3
Genetic causes for CPP remained limited to a few rare mutations for many years until whole exome sequencing analysis from 15 families with CPP identified loss-of-function mutations in MKRN3 in one-third of these families (21,43,47–49). MKRN3 mutations are the most common genetic defect associated with CPP to date with around 10% frequency, and an increased frequency in familial cases of CPP, ranging from 33 to 46% (21,50,51). MKRN3 mutations have been identified in several ethnic groups, and include severe mutations, resulting in premature truncation of the protein, and missense mutations, predicted to be loss of function by in vitro studies or software analysis (50,52). MKRN3 mutations reported in patients with CPP are described in Figure 2. MKRN3 encodes Makorin ring finger protein 3, an E3 ubiquitin ligase. MKRN3 is located on the long arm of chromosome 15, in a region with a cluster of imprinted genes. Some genes are paternally expressed only and are associated with Prader Willi syndrome (PWS) (53,54). MKRN3 is included in the cluster of paternal allele exclusive expression. Studies have shown that MKRN3 deletion is not required nor responsible for the PWS phenotype, with several cases of PWS presenting without deletion of MKRN3 (55–57). However, the loss of MKRN3 within the deletion causing PWS does not predict the pubertal phenotype, as not all patients with PWS with deletions including MKRN3 necessarily develop CPP (55,58,59). Patients with CPP and loss-of-function mutations in MKRN3 do not have any of the major features present in PWS. Following the imprinting pattern, all patients with CPP and mutations in MKRN3 were found to have inherited the mutations from their fathers when genetic analysis were possible. Paternal heterozygous deletions in the whole MKRN3 gene have been identified in two unrelated girls with non-syndromic CPP and obesity. Given the prevalence of obesity, it is difficult to draw any conclusions about the relevance of the deletion to this phenotype (60).
Figure 2. Schematic representation of MKRN3 protein structure and 59 mutations identified in patients with CPP.
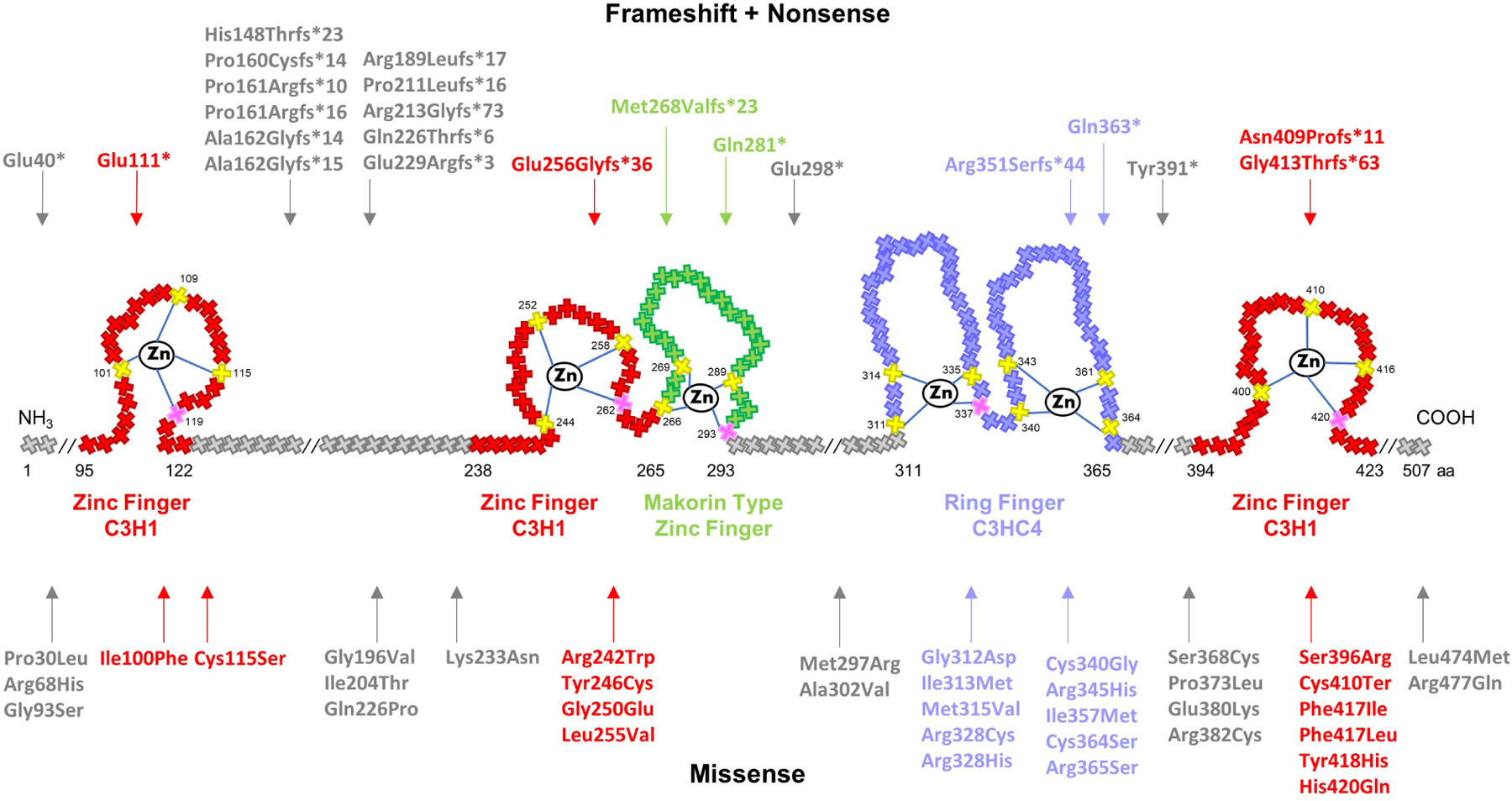
Crosses represent individual amino acids, and corresponding numbers indicate amino acid position. Yellow and pink crosses represent key cysteine and histidine amino acids, respectively, necessary for zinc ion interaction. RING finger C3HC4, in purple, is a protein binding domain responsible for ubiquitin ligase activity. Zinc Finger C3H1, in red, are RNA binding domains. Makorin type Zinc finger, in green, is a specific Cys–His domain identified in the proteins of the makorin family. Notably, 15 mutations (27%) were detected between the first two C3H1 domains, 11 of which are frameshift. Mutations tend to also cluster within the C3HC4 RING finger domain (20%)- the vast majority of which are missense.
Patients with CPP and loss-of-function MKRN3 mutations showed clinical and hormonal features consistent with premature activation of the HPG axis, including early pubertal signs such as breast development or testicular enlargement, advanced bone age, and elevated basal and/or GnRH-stimulated LH levels. Clinical and laboratory features between patients with CPP with or without MKRN3 mutations typically reported no significant differences (50). There were no differences in prevalence of obesity in patients with CPP and MKRN3 mutations compared to those with CPP without MKRN3 mutations (61,62). Additionally, a study that analyzed response to treatment did not show difference in expected family height in girls with CPP without MKRN3 mutations, demonstrating the efficacy of GnRH analog in preserving genetic adult height potential, regardless of the etiology of CPP (62).
MKRN3 expression is higher in the hypothalamus prior to sexual development with a decline during pubertal development in mice, rats and female non-human primates (52). Populational based studies showed that circulating MKRN3 protein levels decline in male and female humans prior to the onset of puberty (63,64). MKRN3 is expressed in kisspeptin neurons and inhibits transcription of the two important GnRH stimulators, KISS1 and TAC3 in vitro. MKRN3 is the first gene with mutations identified in humans with an inhibitory effect on GnRH secretion (52). It is also the first imprinted gene associated with non-syndromic CPP.
DLK1
Linkage analysis followed by whole-genome sequencing of a family with multiple members with CPP revealed a complex genomic defect in DLK1. A ~14 kb deletion, encompassing the entirety of exon 1, including the translational start site, and a 269-bp duplication of intron 3 was identified in four affected girls (two sisters, and two half-sisters) with biochemically confirmed CPP. (24). The paternal grandmother also had a history of premature menarche, suggesting that she may have also been affected. The father of the girls with CPP and DLK1 mutations did not developed CPP because DLK1 is also maternally imprinted and only expressed from the paternal allele. Familial segregation correlated perfectly with the maternal imprinting inheritance (24). Subsequently, three frameshift mutations in DLK1 were reported in five women from three families with CPP (79). Again, segregation analysis was consistent with the maternal imprinting of DLK1.
Similar to MKRN3, DLK1 is also located in a region of clustered imprinted genes and is expressed only from the paternal allele. DLK1 is located on chromosome 14q32 in a region associated with Temple syndrome (65). Temple syndrome is characterized by pre- and post-natal growth retardation, hypotonia, motor delay, small hands and CPP. Patients with CPP harboring DLK1 mutations don’t present features of Temple syndrome. DLK1 mutations resulted in absence of secreted protein in serum, as demonstrated by the undetectable levels in the serum of patients harboring these mutations (24,66).
DLK1 encodes the delta-like homolog 1 protein (DLK1) also known as Preadipocyte factor 1 (Pref-1), and is a transmembrane protein containing epidermal growth factor (EGF)‒like repeats homologous to the Notch/Delta/Serrate family. Animal and in vitro studies have demonstrated that DLK1 acts as an adipogenesis gatekeeper by preventing adipocyte differentiation (67). Indeed, Dlk1-null mice display accelerated weight gain and hyperlipidemia at adulthood, supporting antiadipogenic actions (66). Metabolic abnormalities, such as overweight, early-onset glucose intolerance, type 2 diabetes mellitus, and hyperlipidemia, appear to be more prevalent in women with CPP and DLK1 mutation than in those with CPP without DLK1 mutations. Two sisters with CPP and DLK1 mutation also exhibited polycystic ovary syndrome and infertility (66). The number of patients with CPP and DLK1 mutations is small, and it is difficult to make associations, but it is possible that DLK1 is a novel link between reproduction and metabolism.
5. Genetics of Delayed Puberty
DP is defined as the absence of testicular enlargement in boys or breast development in girls at an age that is 2 to 2.5 SD later than the normal population mean (68) (Figure 1 C). The pathogenesis of DP encompasses several conditions. An important differential diagnosis of DP is hypogonadism which occurs when there is a failure to go through puberty or incomplete pubertal development, and infertility in adult life. (68,69). Hypogonadism due to GnRH deficiency results in low levels of gonadotropin and is named hypogonadotropic hypogonadism (HH). When associated with abnormal sense of smell it is called Kallmann syndrome (KS). The most common cause of DP is constitutional delay of puberty which is called self-limited delayed puberty (SLDP) or constitutional delay of growth and puberty (CDGP) when associated with delayed growth. In CDGP and SLDP, pubertal onset is delayed but occurs spontaneously by the age of 18 years. SLDP can be sporadic or segregating within families, with up to 70% of families having an autosomal dominant pattern of inheritance suggesting a genetic component (70,71). Currently, there are no clinical available diagnostic tools to differentiate SLDP or CDGP from HH. A study showed that 16 children (13 boys and 3 girls) with delayed puberty that responded to kisspeptin-stimulation test with a rise in luteinizing hormone (LH) subsequently progressed through puberty, indicating that this test has the potential be used clinically in the future for differential diagnosis of SLDP and HH (72).
The genetic architecture of HH has been extensively explored and over 50% of patients with HH or KS has a genetic cause identified. It is not rare to identify relatives of patients with HH carrying the same gene mutations but presenting with only delayed puberty (73,74). One example is mutations in HS6ST1 that were found in 2% of patients with HH (74) and later, loss-of-function mutations were identified associated with familial SLDP. In this case, studies in C. elegans revealed that hs6t1 regulates neural branching in concert with other IHH-associated genes, including kal-1, the FGF receptor, and FGF suggesting an oligogenic interaction to determine the pubertal phenotype (74). Several other oligogenic defects have been associated with HH and SLDP (75). To add even more complexity, some patients with HH or KS can unexpectedly undergo reversal of the phenotype, even when carrying genetic mutations, indicating that the effects of a genetic defect can be overcome (76). In this section we will discuss genes associated with SLDP.
IGSF10
Loss-of-function mutations in IGSF10, encoding immunoglobulin superfamily member 10, were found to be causative of SLDP. Four variants in IGSF10 were found in 10 families and a total of 31 individuals with SLDP. Two amino-terminal variants were found in 20 individuals from six families with perfect segregation with the expected autosomal dominant pattern of inheritance. Two carboxyl-terminal variants were found in 11 individuals from four families, displaying incomplete penetrance and a possible de novo mutation. Mutated proteins resulted in failure of extracellular secretion in vitro and co-culture experiment showed that migration of immortalized GnRH neurons was reduced in Igsf10 knockdown studies in fibroblasts. In addition, Igsf10 knockdown in zebrafish dysregulated GnRH neuronal migration. Mutations in IGSF10 were also found in patients with hypothalamic amenorrhea, revealing a shared pathophysiology between SLDP and other forms of functional hypogonadism (77).
FTO
Rare heterozygous variants in FTO, encoding the fat mass and obesity protein, were found families with SLDP associated with extreme low BMI and maturational delay in growth in early childhood in the Finnish cohort (27). Two variants were identified in three families and inherited with the expected autosomal dominant pattern. Heterozygous Fto knockout mice displayed delayed vaginal opening (VO, i.e., pubertal marker for puberty initiation), but a similar body weight compared with wild-type animals. Variants in FTO locus were found to have the largest effect on BMI and obesity risk in GWAS studies (20). FTO is the first gene associated with obesity in GWAS with mutations associated with SLDP, remarking the strong relationship between energy balance and reproductive functions. It remains to be determined whether the effect of FTO on pubertal timing in self-limited delayed puberty is mediated via effects on body mass.
EAP1
EAP1, encoding the protein Enhanced at puberty 1, is highly expressed in the hypothalamus of male and female peri-pubertal rodents and non-human primate, and mRNA levels increase during puberty. EAP1 has been show to transactivate GnRH promoter (78). Two heterozygous rare variants, one missense and one in-frame deletion were found in EAP1 in two unrelated families with SLDP and segregated with the phenotype. A luciferase reporter assay showed that the EAP1 mutant proteins had reduced ability to trans-activate the GnRH promoter compared to wild-type EAP1 due to the inability to translocate to the nucleus (79).
LGR4
Rare variants in LGR4 were identified in 6 patients with familial SLDP in a Finnish cohort (80). LGR4 encodes Leucine-rich repeat-containing G protein-coupled receptor 4, a GPCR uniquely functioning through Wnt/b-catenin signaling. Lgr4 is expressed in key areas responsible for GnRH neuronal development. LGR4 mutant proteins showed impaired Wnt/β-catenin signaling, owing to defective protein expression, trafficking, and degradation. Mice deficient in Lgr4 had significantly delayed onset of puberty and fewer GnRH neurons compared with WT, whereas lgr4 knockdown in zebrafish embryos prevented formation and migration of GnRH neurons. Altogether these results demonstrate the pathogenicity of LGR4 variants resulting in SLPD.
MC3R
In a recent study, MC3R variants were associated with delayed pubertal onset, delayed growth during childhood, shorter adult stature, lower lean mass, and lower circulating levels of IGF1 in both females and males (29). MC3R encodes the G protein-coupled receptor melanocortin 3 receptor, which is activated by α-melanocyte-stimulating hormone (α-MSH). Functional annotation of missense variants identified in humans revealed that rarer variants caused a more severe signaling disruption and correlated with a greater delayed onset of puberty. Single-cell RNA sequencing of the arcuate nucleus in the hypothalamus showed that Mc3r is expressed in both kisspeptin-neurokinin B-dynorphin (KNDy) neurons and in growth hormone-releasing hormone (GHRH) neurons (29). Taken together, the evidence emerged from this study highlights the important role of MC3R in growth and reproduction.
Summary
Human genetic studies resulted in the identification of common genetic variants associated with normal pubertal timing and rare variants or mutations causing CPP or SLDP. A significant number of genes such as MKRN3, DLK1, TACR3, FTO and MC3R have either variant in non-coding regions with a small contribution to the age at menarche likely due to small alteration in their transcription levels, or more severe variants in the coding regions resulting in loss-of-function of the protein causing a disorder of pubertal onset. The loss of maternally imprinted genes is associated with CPP which suggest an evolutionary mechanism regulating pubertal onset. The genetic control of DP is complicated by the facts that this trait is not rare and may be a spectrum of HH. Studies demonstrated an overlap of genetic defects associated with DP and HH and the importance of genes associated with GnRH neuronal development and function.
Practice points.
Early and late puberty can be associated with adverse health outcomes later in life.
MKRN3 is the most common cause of Central Precocious Puberty and genetic testing can assist in the diagnosis and familial genetic counseling.
It is important to consider MKRN3 imprinting pattern when analyzing pedigrees of families with CPP.
It is very important to diagnose the underlying cause of delayed puberty, especially to distinguish between self-limited DP and HH in adolescents, however, there are no ideal diagnostic tools currently.
Research agenda.
Expanding the studies in humans will indisputably identify additional genes affecting pubertal timing.
The genetic, epigenetic and environmental basis for the age of pubertal onset is an area of research where there is still much to be discovered and may bring future benefits for informed management of these patients.
Mechanistically targeted therapies should be pursued, especially with respect to the kisspeptin, tachykinin and MKRN3 system.
Financial Support:
AA was supported by National Institute of Health/Eunice Kennedy Shriver National Institute of Child Health and Human Development grant R00 HD 091381. AM was supported by the Lalor Foundation Fellowship Award. AA and AM are supported by NIH R01 HD082314.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kuiri-Hanninen T, Sankilampi U, Dunkel L. Activation of the hypothalamic-pituitary-gonadal axis in infancy: minipuberty. Hormone Research in Paediatrics 2014;82(2):73–80. [DOI] [PubMed] [Google Scholar]
- 2.Terasawa E, Fernandez DL. Neurobiological mechanisms of the onset of puberty in primates. Endocrine Reviews 2001;22(1):111–151. [DOI] [PubMed] [Google Scholar]
- 3.Palmert MR, Hirschhorn JN. Genetic approaches to stature, pubertal timing, and other complex traits. Molecular Genetics and Metabolism 2003;80(1–2):1–10. [DOI] [PubMed] [Google Scholar]
- 4.Abreu AP, Kaiser UB. Pubertal development and regulation. The Lancet Diabetes & Endocrinology 2016;4(3):254–264.* [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Juul A, Teilmann G, Scheike T, et al. Pubertal development in Danish children: comparison of recent European and US data. International journal of andrology Feb 2006;29(1):247–55; discussion 286–90. [DOI] [PubMed] [Google Scholar]
- 6.Teilmann G, Pedersen CB, Skakkebaek NE, et al. Increased risk of precocious puberty in internationally adopted children in Denmark. Pediatrics Aug 2006;118(2):e391–9. [DOI] [PubMed] [Google Scholar]
- 7.Mul D, Fredriks AM, van Buuren S, et al. Pubertal development in The Netherlands 1965–1997. Pediatric Research Oct 2001;50(4):479–86. [DOI] [PubMed] [Google Scholar]
- 8.Wyshak G, Frisch RE. Evidence for a secular trend in age of menarche. New England Journal of Medicine 1982;306(17):1033–1035. [DOI] [PubMed] [Google Scholar]
- 9.van der Klaauw AA, Farooqi IS. The hunger genes: pathways to obesity. Cell 2015;161(1):119–132. [DOI] [PubMed] [Google Scholar]
- 10.Navarro VM. Metabolic regulation of kisspeptin — the link between energy balance and reproduction. Nature Reviews Endocrinology 2020;16(8):407–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Biro FM, Huang B, Crawford PB, et al. Pubertal correlates in black and white girls. The Journal of Pediatrics 2006;148(2):234–240. [DOI] [PubMed] [Google Scholar]
- 12.Bianco S A potential mechanism for the sexual dimorphism in the onset of puberty and incidence of idiopathic central precocious puberty in children: sex-specific kisspeptin as an integrator of puberty signals. Frontiers in Endocrinology 2012;3:149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Day FR, Bulik-Sullivan B, Hinds DA, et al. Shared genetic aetiology of puberty timing between sexes and with health-related outcomes. Nature Communications 2015;6(1):1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Perry JR, Stolk L, Franceschini N, et al. Meta-analysis of genome-wide association data identifies two loci influencing age at menarche. Nature Genetics Jun 2009;41(6):648–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sulem P, Gudbjartsson DF, Rafnar T, et al. Genome-wide association study identifies sequence variants on 6q21 associated with age at menarche. Nature Genetics 2009;41(6):734–738. [DOI] [PubMed] [Google Scholar]
- 16.He C, Kraft P, Chen C, et al. Genome-wide association studies identify loci associated with age at menarche and age at natural menopause. Nature Genetics 2009;41(6):724–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ong KK, Elks CE, Li S, et al. Genetic variation in LIN28B is associated with the timing of puberty. Nature Genetics Jun 2009;41(6):729–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vazquez MJ, Daza-Dueñas S, Tena-Sempere M. Emerging roles of epigenetics in the control of reproductive function: Focus on central neuroendocrine mechanisms. Journal of the Endocrine Society 2021;5(11) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Day FR, Thompson DJ, Helgason H, et al. Genomic analyses identify hundreds of variants associated with age at menarche and support a role for puberty timing in cancer risk. Nature Genetics 2017;49(6):834–841.* [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Perry JRB, Day F, Elks CE, et al. Parent-of-origin-specific allelic associations among 106 genomic loci for age at menarche. Nature 2014;514(7520):92–97.* [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Abreu AP, Dauber A, Macedo DB, et al. Central precocious puberty caused by mutations in the imprinted gene MKRN3. New England Journal of Medicine 2013;368(26):2467–2475.* [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dauber A, Rosenfeld RG, Hirschhorn JN. Genetic evaluation of short stature. The Journal of Clinical Endocrinology & Metabolism 2014;99(9):3080–3092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Haig D Genomic imprinting and the evolutionary psychology of human kinship. Proceedings of the National Academy of Sciences 2011;108(Supplement_2):10878–10885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dauber A, Cunha-Silva M, Macedo DB, et al. Paternally inherited DLK1 deletion associated with familial central precocious puberty. The Journal of Clinical Endocrinology & Metabolism 2017;102(5):1557–1567.* [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tusset C, Noel SD, Trarbach EB, et al. Mutational analysis of TAC3 and TACR3 genes in patients with idiopathic central pubertal disorders. Arquivos Brasileiros de Endocrinologia & Metabologia 2012;56(9):646–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Day FR, Perry JR, Ong KK. Genetic regulation of puberty timing in humans. Neuroendocrinology 2015;102(4):247–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Howard SR, Guasti L, Poliandri A, et al. Contributions of function-altering variants in genes implicated in pubertal timing and body mass for self-limited delayed puberty. The Journal of Clinical Endocrinology & Metabolism Feb 1 2018;103(2):649–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wood AR, Esko T, Yang J, et al. Defining the role of common variation in the genomic and biological architecture of adult human height. Nature Genetics 2014;46(11):1173–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lam BYH, Williamson A, Finer S, et al. MC3R links nutritional state to childhood growth and the timing of puberty. Nature 2021;599(7885):436–441.* [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cancer CGoHFiB. Menarche, menopause, and breast cancer risk: individual participant meta-analysis, including 118 964 women with breast cancer from 117 epidemiological studies. The Lancet Oncology 2012;13(11):1141–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Elks CE, Perry JR, Sulem P, et al. Thirty new loci for age at menarche identified by a meta-analysis of genome-wide association studies. Nature Genetics Dec 2010;42(12):1077–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Widen E, Silventoinen K, Sovio U, et al. Pubertal timing and growth influences cardiometabolic risk factors in adult males and females. Diabetes Care Apr 2012;35(4):850–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.He C, Zhang C, Hunter DJ, et al. Age at menarche and risk of type 2 diabetes: results from 2 large prospective cohort studies. American Journal of Epidemiology Feb 1 2010;171(3):334–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Purdie DM, Green AC. Epidemiology of endometrial cancer. Review. Best practice & research: Clinical obstetrics & gynaecology Jun 2001;15(3):341–54. [DOI] [PubMed] [Google Scholar]
- 35.Elks CE, Loos RJ, Sharp SJ, et al. Genetic markers of adult obesity risk are associated with greater early infancy weight gain and growth. PLoS Medicine May 2010;7(5):e1000284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ong KK, Northstone K, Wells JC, et al. Earlier mother’s age at menarche predicts rapid infancy growth and childhood obesity. PLoS Medicine Apr 2007;4(4):e132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Day FR, Elks CE, Murray A, et al. Puberty timing associated with diabetes, cardiovascular disease and also diverse health outcomes in men and women: the UK Biobank study. Scientific Reports 2015;5:11208–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hollis B, Day FR, Busch AS, et al. Genomic analysis of male puberty timing highlights shared genetic basis with hair colour and lifespan. Nature Communications 2020;11(1) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhu J, Chan Y-M. Adult consequences of self-limited delayed puberty. Pediatrics 2017;139(6):e20163177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Albanese A, Stanhope R. Does constitutional delayed puberty cause segmental disproportion and short stature? European journal of pediatrics Apr 1993;152(4):293–6. [DOI] [PubMed] [Google Scholar]
- 41.Finkelstein JS, Neer RM, Biller BM, et al. Osteopenia in men with a history of delayed puberty. New England Journal of Medicine Feb 27 1992;326(9):600–4. [DOI] [PubMed] [Google Scholar]
- 42.Kaltiala-Heino R, Kosunen E, Rimpela M. Pubertal timing, sexual behaviour and self-reported depression in middle adolescence. Journal of Adolescence Oct 2003;26(5):531–45. [DOI] [PubMed] [Google Scholar]
- 43.Teles MG, Bianco SDC, Brito VN, et al. A GPR54-activating mutation in a patient with central precocious puberty. New England Journal of Medicine 2008;358(7):709–715.* [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bianco SDC, Vandepas L, Correa-Medina M, et al. KISS1R intracellular trafficking and degradation: Effect of the Arg386Pro disease-associated mutation. Endocrinology 2011;152(4):1616–1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Navarro VM, Tena-Sempere M. Kisspeptins and the neuroendocrine control of reproduction. Frontiers in Bioscience-Scholar 2011-01-01 2011;3(1):267–275. [DOI] [PubMed] [Google Scholar]
- 46.Dhillo WS. Kisspeptin, neurokinin B and new players in reproduction. Seminars in Reproductive Medicine 2019;37(04):153–154. [DOI] [PubMed] [Google Scholar]
- 47.Silveira LG, Noel SD, Silveira-Neto AP, et al. Mutations of the KISS1 gene in disorders of puberty. The Journal of Clinical Endocrinology & Metabolism 2010;95(5):2276–2280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tommiska J, Sørensen K, Aksglaede L, et al. LIN28B, LIN28A, KISS1, and KISS1R in idiopathic central precocious puberty. BMC Research Notes 2011;4(1):363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Krstevska-Konstantinova M, Jovanovska J, Tasic VB, et al. Mutational analysis of KISS1 and KISS1R in idiopathic central precocious puberty. Journal of Pediatric Endocrinology and Metabolism 2014;27(1–2):199–201. [DOI] [PubMed] [Google Scholar]
- 50.Valadares LP, Meireles CG, De Toledo IP, et al. MKRN3 mutations in central precocious puberty: A systematic review and meta-analysis. Journal of the Endocrine Society May 1 2019;3(5):979–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Simon D, Ba I, Mekhail N, et al. Mutations in the maternally imprinted gene MKRN3 are common in familial central precocious puberty. European Journal of Endocrinology 2016;174(1):1–8. [DOI] [PubMed] [Google Scholar]
- 52.Abreu AP, Toro CA, Song YB, et al. MKRN3 inhibits the reproductive axis through actions in kisspeptin-expressing neurons. Journal of Clinical Investigation Jul 20 2020; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jong MTC, Carey AH, Caldwell KA, et al. Imprinting of a RING zinc-finger encoding gene in the mouse chromosome region homologous to the Prader-Willi syndrome genetic region. Human Molecular Genetics 1999;8(5):795–803. [DOI] [PubMed] [Google Scholar]
- 54.Jong MT, Gray TA, Ji Y, et al. A novel imprinted gene, encoding a RING zinc-finger protein, and overlapping antisense transcript in the Prader-Willi syndrome critical region. Human Molecular Genetics May 1999;8(5):783–93. [DOI] [PubMed] [Google Scholar]
- 55.Angulo MA, Butler MG, Cataletto ME. Prader-Willi syndrome: a review of clinical, genetic, and endocrine findings. Journal of Endocrinological Investigation 2015;38(12):1249–1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kanber D, Giltay J, Wieczorek D, et al. A paternal deletion of MKRN3, MAGEL2 and NDN does not result in Prader–Willi syndrome. European Journal of Human Genetics 2009;17(5):582–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cassidy SB, Schwartz S, Miller JL, et al. Prader-Willi syndrome. Genetics in Medicine 2012;14(1):10–26. [DOI] [PubMed] [Google Scholar]
- 58.Costa RA, Ferreira IR, Cintra HA, et al. Genotype-phenotype relationships and endocrine findings in prader-willi syndrome. Frontiers in Endocrinology 2019;10:864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Maina EN, Webb T, Soni S, et al. Analysis of candidate imprinted genes in PWS subjects with atypical genetics: a possible inactivating mutation in the SNURF/SNRPN minimal promoter. Journal of Human Genetics 2007;52(4):297–307. [DOI] [PubMed] [Google Scholar]
- 60.Meader BN, Albano A, Sekizkardes H, et al. Heterozygous deletions in MKRN3 cause central precocious puberty without Prader-Willi syndrome. The Journal of Clinical Endocrinology & Metabolism 2020;105(8):2732–2739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Seraphim CE, Canton APM, Montenegro L, et al. Genotype-phenotype correlations in central precocious puberty caused by MKRN3 mutations. The Journal of Clinical Endocrinology & Metabolism Dec 31 2020; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ramos C, Macedo D, Canton A, et al. Outcomes of patients with central precocious puberty due to loss-of-function mutations in the MKRN3 gene after treatment with gonadotropin-releasing hormone analog. Neuroendocrinology 2020;110(7–8):705–713. [DOI] [PubMed] [Google Scholar]
- 63.Busch AS, Hagen CP, Almstrup K, et al. Circulating MKRN3 levels decline during puberty in healthy boys. The Journal of Clinical Endocrinology & Metabolism Jun 2016;101(6):2588–93. [DOI] [PubMed] [Google Scholar]
- 64.Hagen CP, Sorensen K, Mieritz MG, et al. Circulating MKRN3 levels decline prior to pubertal onset and through puberty: A longitudinal study of healthy girls. The Journal of Clinical Endocrinology & Metabolism May 2015;100(5):1920–6. [DOI] [PubMed] [Google Scholar]
- 65.Prasasya R, Grotheer KV, Siracusa LD, et al. Temple syndrome and Kagami-Ogata syndrome: clinical presentations, genotypes, models and mechanisms. Human Molecular Genetics 2020;29(R1):R107–R116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gomes LG, Cunha-Silva M, Crespo RP, et al. DLK1 is a novel link between reproduction and metabolism. The Journal of Clinical Endocrinology & Metabolism 2019;104(6):2112–2120. [DOI] [PubMed] [Google Scholar]
- 67.Smas CM, Sul HS. Pref-1, a protein containing EGF-like repeats, inhibits adipocyte differentiation. Cell 1993;73(4):725–734. [DOI] [PubMed] [Google Scholar]
- 68.Palmert MR, Dunkel L. Clinical practice. Delayed puberty. New England Journal of Medicine Feb 2 2012;366(5):443–53. [DOI] [PubMed] [Google Scholar]
- 69.Sedlmeyer IL, Palmert MR. Delayed puberty: analysis of a large case series from an academic center. The Journal of Clinical Endocrinology & Metabolism Apr 2002;87(4):1613–20. [DOI] [PubMed] [Google Scholar]
- 70.Wehkalampi K, Widen E, Laine T, et al. Patterns of inheritance of constitutional delay of growth and puberty in families of adolescent girls and boys referred to specialist pediatric care. The Journal of Clinical Endocrinology & Metabolism Mar 2008;93(3):723–8. [DOI] [PubMed] [Google Scholar]
- 71.Sedlmeyer IL, Hirschhorn JN, Palmert MR. Pedigree analysis of constitutional delay of growth and maturation: determination of familial aggregation and inheritance patterns. The Journal of Clinical Endocrinology & Metabolism Dec 2002;87(12):5581–6. [DOI] [PubMed] [Google Scholar]
- 72.Chan Y-M, Lippincott MF, Sales Barroso P, et al. Using kisspeptin to predict pubertal outcomes for youth with pubertal delay. The Journal of Clinical Endocrinology & Metabolism 2020;105(8):e2717–e2725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Pitteloud N, Quinton R, Pearce S, et al. Digenic mutations account for variable phenotypes in idiopathic hypogonadotropic hypogonadism. Journal of Clinical Investigation 2007;117(2):457–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tornberg J, Sykiotis GP, Keefe K, et al. Heparan sulfate 6-O-sulfotransferase 1, a gene involved in extracellular sugar modifications, is mutated in patients with idiopathic hypogonadotrophic hypogonadism. Proceedings of the National Academy of Sciences of the United States of America Jul 12 2011;108(28):11524–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sykiotis GP, Plummer L, Hughes VA, et al. Oligogenic basis of isolated gonadotropin-releasing hormone deficiency. Proceedings of the National Academy of Sciences 2010;107(34):15140–15144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sidhoum VF, Chan Y-M, Lippincott MF, et al. Reversal and relapse of hypogonadotropic hypogonadism: Resilience and fragility of the reproductive neuroendocrine system. The Journal of Clinical Endocrinology & Metabolism 2014;99(3):861–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Howard SR, Guasti L, Ruiz-Babot G, et al. IGSF10 mutations dysregulate gonadotropin-releasing hormone neuronal migration resulting in delayed puberty. EMBO Moluclar Medicine Jun 2016;8(6):626–42.* [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Heger S, Mastronardi C, Dissen GA, et al. Enhanced at puberty 1 (EAP1) is a new transcriptional regulator of the female neuroendocrine reproductive axis. Journal of Clinical Investigation Aug 2007;117(8):2145–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Mancini A, Howard SR, Cabrera CP, et al. EAP1 regulation of GnRH promoter activity is important for human pubertal timing. Human Molecular Genetics Apr 15 2019;28(8):1357–1368.* [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Mancini A, Howard SR, Marelli F, et al. LGR4 deficiency results in delayed puberty through impaired Wnt/beta-catenin signaling. JCI Insight Jun 4 2020;5(11)* [DOI] [PMC free article] [PubMed] [Google Scholar]