

Abstract
RNA-guided nucleases from CRISPR-Cas systems expand opportunities for precise, targeted genome modification. Endogenous CRISPR-Cas systems in many prokaryotes are attractive to circumvent expression, functionality, and unintended activity hurdles posed by heterologous CRISPR-Cas effectors. However, each CRISPR-Cas system recognizes a unique set of protospacer adjacent motifs (PAMs), which requires identification by extensive screening of randomized DNA libraries. This challenge hinders development of endogenous CRISPR-Cas systems, especially those based on multi-protein effectors and in organisms that are slow-growing or have transformation idiosyncrasies. To address this challenge, we present Spacer2PAM, an easy-to-use, easy-to-interpret R package built to predict and guide experimental determination of functional PAM sequences for any CRISPR-Cas system given its corresponding CRISPR array as input. Spacer2PAM can be used in a ‘Quick’ method to generate a single PAM prediction or in a ‘Comprehensive’ method to inform targeted PAM libraries small enough to screen in difficult to transform organisms. We demonstrate Spacer2PAM by predicting PAM sequences for industrially relevant organisms and experimentally identifying seven PAM sequences that mediate interference from the Spacer2PAM-informed PAM library for the type I-B CRISPR-Cas system from Clostridium autoethanogenum. We anticipate that Spacer2PAM will facilitate the use of endogenous CRISPR-Cas systems for industrial biotechnology and synthetic biology.
Graphical Abstract
Graphical Abstract.

Spacer2PAM computationally backtracks the process of CRISPR spacer adaptation to identify potential PAM sequences for a given CRISPR-Cas system. These putative PAMs are then filtered based on alignment statistics to generate PAM predictions and inform PAM library design.
INTRODUCTION
Clustered regularly interspaced short palindromic repeats (CRISPR) CRISPR-associated (Cas) system-derived, RNA-guided nucleases have enabled an abundance of technologies (1–3), including gene editing. While CRISPR-Cas gene editing within eukaryotes using heterologous components, like Streptococcus pyogenes Cas9, proves effective across eukaryotic phylogenetic space (4), success of those same components remains unpredictable across prokaryotes (5–8). In fact, use of heterologous CRISPR-Cas effectors in prokaryotes poses three main hurdles. First, transformation and expression of functional effector proteins is difficult in many non-model prokaryotes. Many common CRISPR-Cas effectors are large in size requiring over 3 kb of DNA sequence to encode the expression construct which can further reduce already low transformation efficiencies (9). Thus, using these effectors decreases the chance of successful transformation before the editing event even takes place. Second, the functionality of heterologous effector complexes is not guaranteed in the target organism's cytosolic conditions. Enzymes are environmentally sensitive and demonstrate optimal activity within narrow physiological conditions. For example, the warm environment required by thermophiles can lead to inactivity of S. pyogenes Cas9 (10). Third, CRISPR-Cas effectors have the potential to demonstrate off target activities or unexplained toxicities. Heterologous CRISPR-Cas effectors can possess additional activities that can interfere with gene editing or viability in prokaryotes (5–8,11,12) because CRISPR-Cas effectors are often sourced from other prokaryotic systems. Taken together, these hurdles make difficult the adoption of CRISPR-Cas gene editing in the growing list of model and non-model prokaryotes relevant to industrial biotechnology and synthetic biology.
Endogenous CRISPR-Cas systems, which are prevalent throughout bacteria and archaea (13), inherently avoid many of the barriers to using heterologous CRISPR-Cas effectors. Native systems are encoded within the genome and are often constitutively expressed (14,15), adapted to function within their genome's cytosolic environment (16), and have evolved to interact with their genome's proteome without significant negative effects. In essence, using endogenous CRISPR-Cas systems presents unique opportunities for genome editing (14–18) and targeted antimicrobial applications (19–21) that otherwise would be inaccessible with current heterologous CRISPR-Cas effectors. However, identification of a functional protospacer adjacent motif (PAM) required for types I, II, and V CRISPR-Cas systems to target DNA (22) remains challenging when using endogenous CRISPR-Cas effectors. CRISPR-Cas effector complexes recognize a unique PAM or set of PAM sequences that is not easily gleaned from readily available information such as host organism or comparative genomics. Functional PAM identification thus requires empirical determination for each endogenous CRISPR-Cas system.
Current methods of PAM determination are often difficult to apply to CRISPR-Cas systems in prokaryotes, especially with multi-protein effector complexes, without robust genetic tools. The primary experimental method used to determine functional PAM sequences in these cases is the screening of a randomized, pooled PAM library in the organism encoding the CRISPR-Cas system (16). The library is sequenced before and after selection by the CRISPR-Cas system and the change in frequency of each PAM is calculated. Decreases in PAM frequencies are associated with successful targeting by the CRISPR-Cas system. Similarly, cell-free (23) and in vivo (24) heterologous expression of CRISPR-Cas effectors have been used to reconstitute CRISPR-Cas effectors and screen their PAM specificity, but these techniques are primarily applied to single protein effectors like those of type II and V CRISPR-Cas systems. Alternatively, researchers with limited resources or organisms that do not transform well enough to screen a randomized, pooled PAM library screen an unpooled PAM library (17). The unpooled nature of the library circumvents the need for large numbers of transformants but limits the throughput of PAM sequences that can be screened.
Computational methods can bypass the need for efficient DNA transformation to identify PAM sequences. Rather than observe the interference activity of a CRISPR-Cas system biochemically, computational methods can back trace the spacer adaptation process bioinformatically, guiding experimental design of a smaller subset of possible PAM sequences. Specifically, where a CRISPR-Cas system's adaptation machinery naturally samples invading nucleic acids for the presence of a PAM before integrating the adjacent protospacer into the CRISPR array (25), the process of nucleotide alignment can be used to identify the origin of CRISPR array spacers and query adjacent to the alignment for the identity of potential PAMs. By doing this process across all the spacers encoded by a CRISPR-Cas system's arrays and comparing all of the potential PAM sequences, frequent motifs can be observed and used to predict PAM preferences of that CRISPR-Cas system. Attempts at this process have been developed (17,26,27) but are often limited in their ability to identify functional PAMs, are difficult to interpret into actionable experiments, or are incomplete and require the use of multiple tools in a non-consolidated pipeline.
In this work, we develop, optimize, and apply Spacer2PAM, an R package built to identify and guide experimental determination of functional PAM sequences for any CRISPR-Cas system given its corresponding CRISPR array as input. This tool improves upon previous computational methods by implementing filter criteria to down select the number of sequence alignments, generating a more biologically relevant set of candidate PAM sequences and increasing the frequency of functional PAM predictions. We validate Spacer2PAM with 20 well-characterized CRISPR-Cas systems and optimize Spacer2PAM to output an experimentally actionable consensus PAM sequence, a score for the PAM prediction, and an optional sequence logo representing the sequences used to build the consensus. We then apply Spacer2PAM to predict PAM sequences for CRISPR-Cas systems from 10 organisms with uncommon carbon metabolism. Further, we use these predictions to determine and experimentally validate functional PAMs for the Clostridium autoethanogenum type I-B CRISPR-Cas system. Spacer2PAM offers an easy-to-use, accurate, and reproducible computational tool for PAM prediction that we anticipate will facilitate research into novel CRISPR-Cas systems and spur new synthetic biology applications.
MATERIALS AND METHODS
Prediction of PAM sequences
All CRISPR arrays were retrieved from CRISPRCasdb, part of CRISPR-Cas++, which can be found at https://crisprcas.i2bc.paris-saclay.fr/ (28). Alignment of CRISPR array spacers to genomes was done via BLAST (29) either programmatically using Spacer2PAM or manually through the web interface. The BLASTn algorithm was used and Eukaryotes (taxid:2759) were excluded from the search database. All other manipulations of sequence information and prediction of PAM sequences were completed using Spacer2PAM which is available at https://github.com/grybnicky/Spacer2PAM. Spacer2PAM requires the following dependencies: dplyr, ggplot2, ggseqlogo (30), taxonomizr, HelpersMG, httr, jsonlite, spatstat.utils, readr and seqinr. Prophage prediction uses the Phaster API (31).
Briefly, Spacer2PAM can be used by passing the CRISPR-Cas system's host organism name and a user-defined identifier to setCRISPRInfo, which sets the name of the CRISPR-Cas system and defines file output names. The user then chooses one of two options to input the CRISPR array spacer sequence data. If starting with a FASTA file containing each spacer as an individual sequence, the user may call FASTA2DF to arrange the spacer sequences and other user input information about the CRISPR spacers (array number, length of each array, direction of each array, and the array consensus repeat sequence) into a dataframe which is suitable for downstream analysis with Spacer2PAM. We recommend that the user then call DF2FASTA to generate another FASTA file containing all the spacer sequences. Although the user already supplied a FASTA file with the same sequence information, doing so ensures that the title of each sequence is compatible with downstream Spacer2PAM functions. Alternatively, a user may start with a formatted dataframe containing the headers ‘Strain’, ‘Spacers’, ‘Array.Orientation’, ‘Repeat’, ‘Array’, and ‘Spacer’ and pass it to DF2FASTA to generate a FASTA file containing the spacer sequences with the appropriate labels.
Next, the user then submits the sequences from the FASTA file for alignment to BLAST. This step can either be done programmatically or manually. To send a query to the BLAST server and retrieve the result, call FASTA2Alignment and pass the file location of the properly formatted FASTA file generated from DF2FASTA. While we recommend this method, some CRISPR-Cas systems may contain too many spacers and exceed the query length limit for the NCBI BLAST API. In this instance, FASTA2Alignment will return an error message and encourage the user to visit the BLAST web interface. If using the web interface, select the BLASTn algorithm and to exclude Eukaryotes (taxid:2759). This limits the alignment to relevant organisms and decreases both BLAST and Spacer2PAM computational time. Once the alignment is completed through the BLAST web server, the resulting hit table should be downloaded in .CSV format. The hit table file should then be passed to alignmentCSV2DF to convert it to a dataframe. Performing the alignment programmatically via FASTA2Alignment will generate this dataframe automatically without the need for alingmentCSV2DF.
The resulting dataframe should then be passed to joinSpacerDFandAlignmentDF. This function joins the spacer dataframe with the alignment dataframe, assigning spacer information to each alignment in the hit table. This function also converts the accession number of the alignment to the genus and species name of the organism that encodes the alignment sequence using the taxonomizr package. As the taxonomizr package requires the local download and set up of an SQL database, the user should be prepared to store the 65 GB (at time of writing this) database in a location stably accessible while running joinSpacerDFandAlignmentDF. The resulting joined dataframe is sufficient for PAM prediction by join2PAM, but we recommend calling Submit2Phaster if the user plans to select the prophage prediction option in join2PAM. Submit2Phaster interacts with the PHASTER prophage prediction web server to submit a nonredundant list of accession numbers from the joined dataframe for prophage detection. Depending on the volume of traffic on the PHASTER server, prediction can take minutes to weeks to complete.
Lastly, the joined dataframe is passed to join2PAM. This function is the core of Spacer2PAM and predicts a PAM sequence from the alignments generated by BLAST. Multiple combinations of filter sets can be run sequentially with a single call of join2PAM to enrich alignments to likely protospacers. These filtered alignments are then used to identify the genomes encoding the putative protospacer and the locations of potential PAMs. The algorithm then harvests these potential PAM sequences by taking the sequence upstream and downstream of the alignment based on the position of the alignment to the spacer and the user input flank length. This harvesting procedure accounts for alignments that do not include the ends of the spacer and appropriately adjusts the harvested sequence to ensure PAMs are not shifted. These sequences are then used to calculate significant nucleotide positions and determine frequent nucleotide identities at those positions, generating a PAM prediction. The output of join2PAM is the dataframe ‘collectionFrame’ that summarizes the filtering process and records the upstream and downstream predicted PAMs as well as their associated PAM score.
A template R script is provided in Supplementary File 1 to guide users on how to assemble a PAM prediction workflow using Spacer2PAM.
Plasmid construction
All individual plasmids and libraries in this work were generated by two-piece Gibson assembly using the GeneArt Seamless Plus kit. Linear backbone was generated by PCR of pMTL82254 using Kapa DNA polymerase Master Mix, purification by gel electrophoresis and extraction with Zymoclean Gel DNA recovery Kit. Linear dsDNA gBlocks ordered from IDT containing the PAM sequence upstream of C. autoethanogenum CRISPR array 1 spacer 19 were used as inserts. Gibson assembly products were transformed into chemically competent One Shot™ MAX Efficiency™ DH10B T1 Phage-Resistant Cells using standard procedures. DNA sequence was confirmed by Illumina MiSeq Sequencing V2 and V3 chemistry. All oligonucleotides and plasmids used in this study can be found in Supplementary Table S1.
Spacer2PAM-informed PAM prediction screening
Spacer2PAM was applied to the type I-B CRISPR-Cas system of C. autoethanogenum using the ‘Comprehensive’ method. The top 25% of high scoring PAM predictions were used to determine a set of 16 four-nucleotide PAM sequences that are likely to be functional (Supplementary Table S3). The Spacer2PAM-informed, unpooled PAM library constructs were transformed into E. coli HB101 carrying R702 (32) (CA434 (33)) in parallel. Conjugation of library members into C. autoethanogenum DSM 19630, a derivate of type strain DSM 10061, was performed as described earlier (33,34) using erythromycin (250 μg/mL) and clarithromycin (5 μg/mL) for plasmid selection in E. coli and C. autoethanogenum, respectively, and trimethoprim (10 μg/mL) as counter selection against E. coli CA434. Optical density of donor E. coli cultures were measured prior to addition to C. autoethanogenum cells. Transconjugant colonies were counted following 4 days of incubation at 37°C under 1.7 × 105 Pa gas (55% CO, 10% N2, 30% CO2, and 5% H2) in gas-tight jars. This was performed in biological triplicate, with 3 separate cultures of donor E. coli conjugated to aliquots of a single C. autoethanogenum culture.
Randomized PAM library screening
The randomized, pooled PAM library was transformed into NEBExpress® E. coli and then purified by QIAprep Spin Miniprep Kit. An aliquot of this DNA was saved to determine PAM frequencies before exposure to the CRISPR-Cas system. Electroporation into C. autoethanogenum was performed as described previously (35,36). Following recovery, cells were pelleted by centrifugation at 4000 X g for 10 minutes, 9.5 mL of supernatant was discarded, and cells were resuspended in 500 μL YTF. Resuspensions were split by volume and spread on YTF 1.5% agar supplemented with 5 μG/mL clarithromycin, allowed to dry for ∼30 minutes, and incubated at 37°C for 4 days under 1.7 × 105 Pa gas (55% CO, 10% N2, 30% CO2, and 5% H2) in gas-tight jars. 2.5 mL of Luria broth was added to each plate and plates were scraped. Total DNA from the cell suspension was purified using the MasterPure™ Gram Positive DNA Purification Kit. PCR across the PAM and spacer was performed using Kapa DNA polymerase Master Mix followed by purification by gel electrophoresis (1.5% agarose) and extraction with Zymoclean Gel DNA recovery Kit. Extracts were quantified by Quant-iT (Thermo Fisher Scientific), diluted to 1 ng/μL, and prepared for sequencing following the Illumina 16S amplicon protocol starting at the Index PCR step. Ampure XP purified libraries were quantified by Quant-iT and sequenced using MiSeq Reagent Kit V3. Frequency of each PAM was determined by counting the occurrence of each PAM next to a correct protospacer sequence within the read. Briefly, all sequence reads are searched for the presence of the C. autoethanogenum Array 1 spacer 19 sequence and are binned as a forward read, reverse read, or does not contain the spacer. For all reads in the forward and reverse bins, the immediate 4 nucleotides upstream or downstream, respectively, are extracted. The sequences extracted from reverse reads are converted to their reverse complement to be compatible with the sequences extracted from forward reads and the two sets of sequences are combined. The frequency of each 4-nucleotide sequence in the combined list is then counted and recorded. The frequency of each PAM was converted to a relative frequency within the total library and the log2-fold change in relative frequency was calculated from exposure to the CRISPR-Cas system.
RESULTS
Spacer2PAM predicts functional PAMs from CRISPR array spacers
We set out to develop an easy-to-use computational framework for predicting and guiding experimental determination of functional PAMs from CRISPR array spacers. This framework, which we implement as a comprehensive R package, is called Spacer2PAM (Figure 1). With input of the CRISPR-Cas system's host organism, CRISPR array spacer sequences, the direction of each CRISPR array, and the consensus repeat sequence for each CRISPR array, Spacer2PAM performs a series of steps including sequence alignment, alignment filtering, potential PAM harvesting, identifying significant nucleotide positions, and identifying frequent nucleotide identities at those positions to output a PAM prediction. At the core of Spacer2PAM is an algorithm, join2PAM, which subjects the aligned sequences to six user set filtering steps to down select the number of alignments that are used in PAM prediction and improve the quality of PAM predictions. The first filter removes redundant alignments and any alignments to the organism that encodes the CRISPR-Cas system of interest. Removal of these alignments is important as their presence during prediction will return the CRISPR array repeat as the predicted PAM. The second through fifth filters remove alignments based on the number of gaps present in the alignment, E value of the alignment, the length of the alignment, and the start of the query sequence relative to the spacer sequence start, respectively. The sixth filter is optional, and filters based on whether the alignment occurs in a predicted prophage region in the query genome. While Spacer2PAM does not directly filter alignments based on mismatches, the number of mismatches is reflected in the e-value and mismatches at the end of a spacer are captured in the length and start position values. Spacer2PAM then outputs a consensus PAM sequences and associated PAM score.
Figure 1.

Overview of Spacer2PAM package functions. Functions are represented by boxes and data are represented by arrows. The user starts by inputting information about the CRISPR-Cas system via the setCRISPRInfo function. Next the user supplies either a FASTA or CSV file containing spacer information to the functions with the broken blue outlines. After programmatic or manual submission of spacer sequences to BLAST, the functions in Spacer2PAM are used to complete the rest of the data transformations and PAM analysis.
When predicting a PAM, there are two main factors to consider: positional significance and nucleotide identity. For a PAM to be functional, it must be in both the correct location relative to the protospacer as well as encode the right nucleotide sequence. To address these factors, join2PAM uses one method to determine significant nucleotide positions within the multiple sequence alignment and another method to determine what nucleotide is likely to be required at that position. To determine the significance of a position, the R score for each nucleotide is calculated. The formula for R score is shown below:
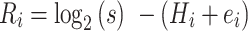 |
where s is the size of the nucleotide alphabet, Hi is the Shannon entropy at a nucleotide position i and ei is a small sample size correction factor based calculated by:
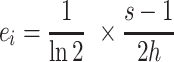 |
where h is the number of sequences in the multiple sequence alignment. Any nucleotide position that has an R score greater than one half standard deviation above the average R score across the flank length is deemed significant. Each significant position then passes to the second method, which determines the frequency of each of the four nucleotides at each significant position. If a nucleotide's frequency exceeds 25%, that nucleotide is added to the consensus PAM. Up to 3 nucleotides can be predicted at a position and are indicated by a ‘/’ in the predicted sequence. The PAM score is calculated by scaling the number of unique alignments hunique that were used to generate the consensus PAM prediction by the proportion of possible information content that the consensus PAM encodes as shown by:
 |
where nsig is the number of significant nucleotide positions, fb sig, i sig is the relative frequency of a predicted base b at significant position i, and Ri sig is the total information content encoded at significant position i. For example, if 25 alignments were used to generate a consensus PAM of CC and all 25 alignments encoded the CC motif, the resulting score would be close to 25. If there was disagreement between the sequences in the position of that predicted CC motif, the PAM score would decrease as those two positions would encode less total information content and the C in each position would occur at lower relative frequency. Spacer2PAM can also output a sequence logo of the upstream and downstream PAM predictions using the ggseqlogo package (30) and annotate it with the consensus PAM sequence and PAM score. If no spacer hits make it through the filter criteria, then no PAM is predicted. If there are few hits, a PAM is predicted, but it will have a low PAM score as the PAM score is the number of sequences used to generate the PAM prediction scaled by the information content of the PAM prediction. Ultimately, join2PAM outputs both an upstream and downstream PAM prediction conveyed by a nucleotide string, PAM score for each prediction, and a sequence logo of each multiple sequence alignment that was used to generate the PAM predictions.
Spacer2PAM was validated by predicting PAMs from the CRISPR array spacers of 20 CRISPR-Cas systems with known PAMs over a range of 256 filter criteria sets. Spacer2PAM is effective in predicting PAMs (Figure 2). These model CRISPR-Cas effectors have known PAM sequences and come from: Acinetobacter baumanii (37), Bacillus halodurans (38), Campylobacter jejuni (39), Clostridiodes difficile (40), Clostridium pasteurianum (17), Clostridium tyrobutyricum (41), Francisella tularensis (42), Gluconobacter oxydans (43), Hungateiclostridium thermocellum (16), Lactobacillus crispatus (15), Moraxella bovoculi (42), Neisseria meningitidis (24), Parvibaculum lavamentivorans (44), Pseudomonas aeruginosa (45), Staphylococcus aureus (44), Streptococcus canis (46), Streptococcus pasteurianus (44), Streptococcus pyogenes (47), Streptococcus thermophilus (48), and Treponema denticola (24). Out of the best PAM predictions for the 20 model systems used, Spacer2PAM predicted functional PAMs for 12. Functional PAMs are defined by sequences that would lead to interference in the presence of the CRISPR-Cas system, but the motif may be more restrictive than the true minimal PAM. The best predictions for the remaining 8 model systems yielded partial PAMs, meaning that the prediction is not functional but correctly identifies some positions and residues in the PAM without misidentifying any essential residues. Although these sequences are not functional, they still indicate part of the functional PAM and are valuable in limiting the nucleotide search space. From this analysis, there do not appear to be trends in how well Spacer2PAM performs based on CRISPR-Cas system type. Importantly, no incorrect PAM predictions were observed in this sample set.
Figure 2.

Spacer2PAM recapitulates PAMs from characterized CRISPR-Cas systems. Representative sequence logo of the most accurate 10-nucleotide PAM prediction for each of twenty CRISPR-Cas systems are shown. Predicted sequence, experimentally determined sequence, PAM score, and known CRISPR-Cas system type are indicated for each system. Functional (which are capable of mediating interference) and partial (which do not mediate interference, but do not misidentify any residue) predictions are outlined in blue and purple, respectively.
Optimization of alignment filter criteria to improve Spacer2PAM performance
Though Spacer2PAM can predict functional PAMs for most of the CRISPR-Cas systems evaluated, the filter criteria that yielded the best result in each case varied between organisms. To determine generalized protocols in which Spacer2PAM should be used, we analyzed the outcome of all 256 sets of filter criteria (Figure 3A) for all 20 model CRISPR-Cas systems. In doing so, we define two ways in which Spacer2PAM can be used to inform PAM sequences for a given CRISPR-Cas system: ‘Quick’ or ‘Comprehensive.’
Figure 3.

Optimization of filter criteria enables generalized, ‘Quick’ prediction of functional PAMs. Data were generated by filtering alignments to 20 CRISPR-Cas systems with known PAMs through the 4 variable filters with 4 different cutoff values. A) Visual representations of each filter criterion. The blue line represents the spacer sequence and the red line represents the query sequence identified by BLAST. The Nucleotides Shorter than Spacer cutoff indicates the threshold value for the difference in alignment and spacer length. The Query Start cutoff indicates the threshold for the starting position of the alignment relative to the spacer. E Value (from BLAST) and Number of Gaps cutoffs are as their names imply. B) The fraction of PAM predictions that resulted in functional sequences out of total predictions is indicated by the fill of each tile with white and blue representing the least and most functional, respectively. C) The fraction of PAM predictions that resulted in incorrect sequences out of total predictions is indicated by the fill of each tile with blue and white representing the least and most incorrect, respectively.
If computational time or experimental resources are limited, Spacer2PAM can be used in a ‘Quick’ method with optimized filter criteria to suggest a single consensus sequence that is likely to be functional. The filter set chosen for down selecting alignments changes the accuracy of the PAM prediction. With the optimal filter set, Spacer2PAM predicted functional PAMs for 45% of CRISPR-Cas systems tested (Figure 3B) and the remaining 55% of predictions were partial matches for the known PAM (Figure 3C). If predicting a single PAM and not designing a targeted library, the user should use the following filter criteria: Number of Gaps cutoff of 0, E Value cutoff of 1.00, Nucleotides Shorter than Spacer cutoff of 3, and Query Start cutoff of 5. Using a Query Start cutoff of 5 or 7 performs equivalently in the sample set, but generally a stricter query start cutoff yields better predictions. It is worth noting that using this approach the PAM predicted is more likely to be functional, but also more restrictive than the true minimal PAM consensus.
Alternatively, Spacer2PAM can also be used in a ‘Comprehensive’ method to inform targeted PAM library design if computational time and experimental resources are available. By generating PAM predictions over a range of filter criteria, Spacer2PAM can explore the likely PAM space of a given CRISPR-Cas system more thoroughly than single filter set prediction can. Each prediction produces a consensus sequence and is assigned a PAM score which can be used to classify whether an individual PAM prediction should be considered for informing library design. Above a 75th percentile threshold, PAM predictions for the CRISPR-Cas systems evaluated were overwhelmingly at least partial matches to the known PAM (Figure 4A). When evaluating the PAM predictions in this scoring bracket, a targeted PAM library can be designed that holds positions supported by multiple predictions constant and varying other positions. This allows the user to change from a pooled, randomized library approach to experimentally simplified unpooled, defined, Spacer2PAM-informed library approach. Additionally, there is often diversity in the PAM prediction using a 75th percentile threshold, allowing for better identification of functional, but divergent PAMs. When this method was applied to the 20 model CRISPR-Cas systems, functional PAMs were identified in 100% of the proposed libraries and 85% of the libraries resulted in more than one functional sequence (Figure 4B, Supplementary Table S2).
Figure 4.

PAM score guides ‘Comprehensive’ PAM prediction. Data were generated by computing PAM predictions and scores over 256 sets of filter criteria for twenty CRISPR-Cas systems. A) Frequency is plotted against PAM Score for each system. The solid vertical line denotes the 75% percentile PAM score threshold for each CRISPR-Cas system. Blue, purple, and gray bars indicate functional, partial, and incorrect PAM predictions, respectively. The top 25% of PAM predictions seed the recommended PAM library for testing. B) Percentage functional PAM sequences within the recommended library are plotted for each CRISPR-Cas system determined by comparing known PAM motifs with members of the Spacer2PAM-informed library (Supplementary Table S2).
Despite this characterization of Spacer2PAM, we have not been able to identify a simple heuristic for prediction accuracy. While both the number of spacers and the number of alignments to those spacers make sense as factors affecting prediction strength, thresholding these values does not allow you to discern functional PAM predictions from partial or incorrect PAM predictions. While larger numbers of spacers and alignments are generally good for prediction, even low numbers of each can still perform well. For instance, the C. jejuni CRISPR array used only encodes 4 spacers that result in 81 alignments, but still predicts a functional prediction.
Application of Spacer2PAM for uncharacterized CRISPR-Cas systems
To evaluate the efficacy of the generalized protocols for Spacer2PAM, we applied both the ‘Quick’ and ‘Comprehensive’ methods to CRISPR-Cas systems with known and unknown PAM sequences. Out of the four characterized CRISPR-Cas systems from Thermobifida fusca YX, Clostridium butyricum JKY6D1, and Zymomonas mobilis ZM4 we tested, Spacer2PAM predicted functional PAM sequences for two of them using the ‘Quick’ method and all four were correctly predicted with the ‘Comprehensive’ method (Table 1). These results are consistent with the reported values from the ‘Quick’ and ‘Comprehensive’ methods reported above. Both methods were then applied to a variety of uncharacterized CRISPR-Cas systems occurring in organisms with unusual carbon metabolism (Table 1). These organisms could be used to convert carbon waste into valuable products. Identifying PAM sequences for their endogenous CRISPR-Cas systems could allow for genetic manipulation and genome modification to optimize these organisms for industrial biotechnology.
Table 1.
Prediction of PAM sequences for organisms with uncommon carbon metabolism. CRISPR array spacers were downloaded from CRISPRCasdb. The direction of each array was determined by literature evidence or annotation by CRISPRCasdb. The position of the first and last nucleotide in each prediction is indicated by subscripts. Slashes in a nucleotide sequence should be interpreted as ‘OR’, meaning that both of the nucleotides on either side of the slash are predicted at that position. Refer to Supplementary Table S3 for a complete version of this table
| Organism | CRISPR Type | Quick Prediction | Recommended Library | Know PAM |
|---|---|---|---|---|
| Thermobifida fusca YX | III-B | N-10NC/GNNCNNC/GN-1 | N-4GGN-1 | No PAM |
| I-E | N-10NNNNNC/GAAG-1 | S-4ANS-1 | W-3AK-1 | |
| Clostridium butyricum JKY6D1 | I-B | N-10NNNNNA/TA/TNA/T-1 | W-3WN-1 | T-3AA-1 & A-3CA-1* |
| Zymomonas mobilis ZM4 | I-F | N-10NNNNCNNNC-1 | N-4RSC-1 | C-2C-1 |
| Clostridium autoethanogenum DSM 10061 | I-B | A/T-10NTNNNNTNT-1 | T-4TNN-1 | N.D. |
| Clostridium beijerinckii a4a6934 | I-B | N-10A/TNA/TNNA/TNA/TN-1 | N-3WW-1 | N.D. |
| Clostridium saccharoperbutylacetonicum N1-504 | I-B | N-10NNA/TGNNNT/CA-1 | N-4CCN-1 | N.D. |
| Methylobacillus flagellatus KT | I-C | N-10A/GNNNNNTT/GN-1 | T-3NN-1 | N.D. |
| Methylocystis heyeri H2 | II-C | N+1C/GNNC/GNNNNN+10 | G+5SNN+8 | N.D. |
| Amycolatopsis sp. BJA-103 | I-E | N-10NNC/GNNC/GA/GNC/G-1 | R-3NS-1 | N.D. |
| Caldicellulosiruptor bescii DSM 6725 | Group I Repeat** | N-10NNANTNNNA-1 | N-3NA-1 | N.D. |
| Group II Repeat** | T-10NNNNNNNTN-1 | N-3TN-1 | N.D. |
*PAM is functional, but not all functional PAMs have been determined.
**Arrays were grouped based on the nucleotide identity of the repeat sequence.
We further sought to validate Spacer2PAM by experimentally demonstrating the utility of the ‘Quick’ and ‘Comprehensive’ predictions for one of the uncharacterized CRISPR-Cas systems we predicted PAMs for in Table 1. Clostridium autoethanogenum was chosen because it is an industrially relevant microbe and obligate anaerobe with applications in sustainable chemical synthesis (49–51). Therefore, we applied Spacer2PAM to the three CRISPR arrays of the C. autoethanogenum type I-B CRISPR-Cas system to predict functional PAM sequences. Using the ‘Quick’ method, Spacer2PAM predicted a W-10NTNNNNTNT-1 PAM (Table 1). The results of the ‘Comprehensive’ method indicated a T-4TNN-1 library would likely yield a functional PAM (Supplementary Table S3). To test these predictions and determine the full range of PAMs recognized by the type I-B CRISPR-Cas system of C. autoethanogenum, we took two experimental approaches: (i) screening the 16-member T-4TNN-1 Spacer2PAM-informed library in an unpooled approach and (ii) screening a 256-member randomized 4-nucleotide PAM library using a pooled approach in C. autoethanogenum. Both methods involve exposing the plasmid-borne PAM library upstream of an actively targeted protospacer to the CRISPR-Cas system in vivo. If a PAM is recognized by the CRISPR-Cas interference machinery, plasmid cleavage will occur and the plasmid bearing antibiotic resistance will no longer be replicated. Each method differs in how the data are collected and evaluated (Figure 5A). Where the pooled library requires the use of NGS before and after screening to measure changes in PAM frequencies due to plasmid cleavage, the unpooled method only requires counting the number of C. autoethanogenum colonies that retain the antibiotic resistance due to unsuccessful CRISPR-Cas interference. Through the unpooled approach, we identified 7 sequences (T-4TGA-1, T-4TGT-1, T-4TTA-1, T-4TCG-1, T-4TCA-1, T-4TCT-1, and T-4TCC-1) that resulted in statistically lower (One-tailed Welch's T-test, p < 0.05) conjugation efficiencies than the non-targeting control PAM (A-4AAT-1) (Figure 5B). Using the pooled method, we determined that a consensus PAM sequence of N-4YCN-1 mediates interference and that there is little nucleotide dependence at the -4 position (Figure 5C, Supplementary Table S4). In testing Spacer2PAM predictions, we have validated that T-3CN-1 PAMs are recognized by the C. autoethanogenum type I-B CRISPR-Cas system with two lines of evidence.
Figure 5.

In vivo determination of functional PAMs in C. autoethanogenum. A) Plasmid-encoded PAM libraries were exposed to active CRISPR-Cas systems in vivo and then plated on selective media. Readout varied based on library approach. B) An unpooled T-4TNN-1 PAM library was screened by individually conjugating plasmid carrying a PAM variant and protospacer from E. coli to C. autoethanogenum. The non-targeting control PAM was A-4AAT-1. Blue indicates p-values less than 0.05 from a one-tailed Welch's t-test as compared to the non-targeting control. Data are shown in triplicate (n = 3) with three individual experiments, each plotted as a single point. C) A pooled N-4NNN-1 PAM library was screened in vivo by electroporation of plasmid into C. autoethanogenum. Nucleotide frequencies were calculated from NGS counts prior and after selection by the CRISPR-Cas system.
DISCUSSION
In this work, we present an easy-to-use, easy-to-interpret, and robust computational tool for predicting and guiding experimental determination of functional PAM sequences for CRISPR-Cas systems. We characterized the tool's performance to determine two methods of use. The ‘Quick’ method uses optimized filter criteria to generate a single consensus PAM using little computational time. The ‘Comprehensive’ method predicts 256 consensus PAMs over a range of filter criteria, which can then be down selected based on PAM score and used to inform a small PAM library, confining the sequence search space to experimentally feasible sizes for non-model organisms. The ‘Comprehensive’ method is 100% effective in predicting libraries containing a functional PAM in the test set, and both methods narrow the nucleotide search space and allow identification of functional PAMs experimentally more easily. This was exemplified by the ability of a 16-member, Spacer2PAM-informed library to identify 7 functional PAM sequences for the C. autoethanogenum type I-B CRISPR-Cas system.
While the PAM predicted for C. autoethanogenum type I-B CRISPR-Cas system by Spacer2PAM via the ‘Quick’ method was not universally functional, one in four PAM variants that fit the W-10NTNNNNTNT-1 consensus would have a C at the -2 position and be functional. This exemplifies that prediction using the ‘Quick’ method, though easy, should be used in combination with ‘Comprehensive’ method for the best predictive power. The ‘Comprehensive’ method was effective and seeded a library that led to the identification of several functional PAM sequences. While the T-4TNN-1 library suggested by Spacer2PAM excludes seven eighths of the consensus N-4YCN-1 variants by constraining the -3 and -4 positions to T, the limited library reduced the sequence space 16-fold and allowed functional PAMs to be determined experimentally in an unpooled manner without sequencing. This limitation of the search space to T at the -3 position instead of Y may be emblematic of some of the inherent constraints of spacer-based PAM prediction. First, the quality of PAM prediction by nucleotide alignment is dependent on representation of protospacers within the database. For organisms in which the mobile genetic element pool has not been well sequenced, there may be too few sequences present in the sequence database to predict the full range of functional PAMs. Alternatively, although both the adaptation and interference machinery of many CRISPR-Cas systems recognize a PAM, they may not experience selective pressures to maintain the exact same range of PAMs. The adaptation machinery, which probes invading DNA for a PAM and then integrates a protospacer adjacent to that PAM into the CRISPR array, is bounded to PAMs that the interference machinery recognizes. Otherwise, the connection between adaptation and interference required for CRISPR-Cas system function would break. On the other hand, the interference machinery's PAM is only bounded by not recognizing the CRISPR array repeat to prevent targeting of the CRISPR array and recognizing at least the adaptation-recognized PAM. As a result, the interference machinery may diversify to recognize other additional PAMs while maintaining the ability to recognize the same PAM that the adaptation machinery does. Doing so may even confer a benefit as broadened PAM recognition by the interference machinery could reduce the viability of potential escape mutations away from the adaptation-recognized PAM. As such, the interference-recognized PAM is likely broader than the adaptation-recognized PAM and predicting PAM sequences based on spacer sequences is likely to yield functional, but more restrictive PAMs than the full set that are recognized by the interference machinery.
Spacer2PAM differs from other spacer-based computational approaches to PAM prediction in that it employs alignment filtering and produces experimentally actionable outputs. To back track the process of spacer adaptation, Spacer2PAM uses nucleotide alignment through BLAST. While this process is central to Spacer2PAM and other spacer-based methods, nucleotide alignment is inherently sensitive to the length of the sequence submitted. When sequences are short, BLAST is more likely to identify alignments that are not biologically relevant by random chance despite the similarity in nucleotide sequence. As sequences lengthen, the chance of random alignment decreases. Since CRISPR array spacers are relatively short by nature, unfiltered alignments are prone to including biologically irrelevant sequences that then inhibit the ability of PAM prediction programs to identify PAM sequences. Spacer2PAM addresses this by using successive filter criteria to jettison alignments that are less likely to be biologically relevant based on alignments statistics. Though the absolute number of alignments used to generate the consensus PAM decreases, filtering enriches the alignments that are likely to lead to a functional PAM (Figure 3B, Supplementary Figure S1). When compared to CRISPRTarget (27) and CASPERpam (26), two unfiltered methods for predicting PAM sequences from CRISPR array spacers, the Spacer2PAM ‘Quick’ method outperforms these tools in predicting PAM sequences 12/20 and 11/16 times for known CRISPR-Cas systems and performs equally as well or better 17/20 and 13/16 times, respectively (Supplementary Tables S5 and S6). We measured the accuracy of each tool's predictions (Supplementary Tables S5 and S6) and found that Spacer2PAM produced the most functional predictions and the fewest incorrect predictions. Additionally, Spacer2PAM outputs predictions differently than some other programs. While many previous efforts use sequence logos to represent potential PAMs, users can interpret sequence logos in different ways. As a result, two researchers may attempt to use divergent PAMs experimentally despite applying the same prediction software. Spacer2PAM still provides the option to generate a sequence logo, but the standard output is a consensus PAM sequence and PAM score.
In addition to advances in PAM prediction, Spacer2PAM provides a rigorous and reproducible framework in which to choose PAMs for experimental determination. Multiple efforts to functionalize endogenous CRISPR-Cas systems for genome engineering have used manual interpretation of BLAST alignments to identify functional PAM sequences (16,17,41). Although this approach has yielded success in multiple organisms, it is difficult to reproduce as the researcher makes judgement calls to identify relevant BLAST results. Likewise, the effectiveness of the manual approach is difficult to gauge as it varies from researcher to researcher. This approach also suggested an N-3AA-1 PAM for the C. autoethanogenum type I-B CRISPR-Cas system (17) when our work indicates a Y-3CN-1 PAM mediates interference. Due to the intractable nature of manual interpretation, we are unable to determine the cause of this difference. Using Spacer2PAM, reporting the filter criteria used, and the hit table generated by BLAST provides a reproducible way in which to generate and report PAM predictions.
Even though the current version of Spacer2PAM outperforms other standard PAM prediction tools, there is room for future development. As join2PAM currently stands, the function removes all alignments to organisms with the same genus and species name as the origin of the CRISPR-Cas system. Although exclusion of alignments to CRISPR arrays is necessary to prevent returning the CRISPR array repeat as the predicted PAM, alignments to strains of the same organism that might not encode a CRISPR-Cas system or alignments that represent self-targeting spacers are excluded. Both of these are potential sources for additional sequences to be used in PAM prediction, especially given the high prevalence of self-targeting spacers across all CRISPR-Cas system types (52). Rather than filtering alignments by genus and species name, future versions of Spacer2PAM may filter by the location alignments within the original organism relative to CRISPR arrays. Likewise, the current iteration of join2PAM does not directly address the requirement of complementarity within a seed region of the R loop when filtering alignments. While the Query Start cutoff filter is meant to address this phenomenon, its current implementation does not consider mismatches in the seed sequence unless they occur continuously from the + 1 position of the alignment causing the start of the alignment to be shifted from the + 1 position. Future development of more specific filters within join2PAM may improve PAM prediction.
The determination of functional PAMs for the type I-B CRISPR-Cas system in C. autoethanogenum is important. This not only demonstrates the utility of Spacer2PAM, but also removes a large hurdle to the functionalization of the system for endogenous genome modification in the organism. Although a Y-3CN-1 PAM is likely functional, we recommend the use of T-3CN-1 PAMs for future use as they are supported by the results of both of our PAM library screens. While Cas9-based tools have been demonstrated previously in C. autoethanogenum (11,53) and used to vary the metabolic products it produces, the availability of endogenous tools increases the amount of nucleotide cargo that can be delivered while also modulating the genome. Likewise, it is also possible to design and introduce functional synthetic CRISPR arrays into the organism to endow it with resistance to mobile genetic elements such as bacteriophages which have traditionally plagued ABE fermentation processes (54).
We anticipate that the development of Spacer2PAM will encourage the functionalization of endogenous CRISPR-Cas systems for a variety of bacteria and archaea as well as help standardize the field. Likewise, Spacer2PAM also has the possibility of streamlining the process of characterizing novel heterologous CRISPR-Cas effectors. In both cases, Spacer2PAM represents a step forward that will enable better development of CRISPR-Cas technologies for use in prokaryotes and potential acceleration of applied technologies such as CRISPR-Cas-based antimicrobials.
DATA AVAILABILITY
Source code for Spacer2PAM as well as instructions are available via GitHub at https://github.com/grybnicky/Spacer2PAM. Illumina sequencing reads for the 4-nucleotide randomized PAM depletion experiment are available through SRA under BioProject accession number PRJNA755691. Further data available on request from the authors.
Supplementary Material
ACKNOWLEDGEMENTS
We would like to sincerely thank Logan Readnour for her help in library preparation and sequencing. We also thank the following investors in LanzaTech's technology: BASF, CICC Growth Capital Fund I, CITIC Capital, Indian Oil Company, K1W1, Khosla Ventures, the Malaysian Life Sciences, Capital Fund, L. P., Mitsui, the New Zealand Superannuation Fund, Novo Holdings A/S, Petronas Technology Ventures, Primetals, Qiming Venture Partners, Softbank China, and Suncor.
Contributor Information
Grant A Rybnicky, Chemistry of Life Processes Institute, Northwestern University, Evanston, IL, 60208, USA; Center for Synthetic Biology, Northwestern University, Evanston, IL, 60208, USA; Interdisciplinary Biological Sciences Graduate Program, Northwestern University, Evanston, IL, 60208, USA.
Nicholas A Fackler, LanzaTech Inc, Skokie, IL 60077, USA.
Ashty S Karim, Chemistry of Life Processes Institute, Northwestern University, Evanston, IL, 60208, USA; Center for Synthetic Biology, Northwestern University, Evanston, IL, 60208, USA; Department of Chemical and Biological Engineering, Northwestern University, Evanston, IL 60208, USA.
Michael Köpke, LanzaTech Inc, Skokie, IL 60077, USA.
Michael C Jewett, Chemistry of Life Processes Institute, Northwestern University, Evanston, IL, 60208, USA; Center for Synthetic Biology, Northwestern University, Evanston, IL, 60208, USA; Department of Chemical and Biological Engineering, Northwestern University, Evanston, IL 60208, USA; Robert H. Lurie Comprehensive Cancer Center, Northwestern University, Chicago, IL 60611, USA; Simpson Querrey Institute, Northwestern University, Chicago, IL 60611, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
This work was supported by the Department of Energy [DE-SC0018249, DE-AC02-05CH11231 to JGI], the Joint Genome Institute (JGI) Community Science Program (CSP) [CSP-503280], the David and Lucile Packard Foundation [2011-37152], the Camille Dreyfus Teacher-Scholar Program, and the National Science Foundation Graduate Research Fellowship Program [DGE-1842165 to GAR].
Conflict of interest statement. N.A.F. and M.K. are employees of LanzaTech, a for-profit company with interest in commercial gas fermentation with C. autoethanogenum. M.C.J. is on the Scientific Advisory Board of LanzaTech, Inc. M.C.J.’s interests are reviewed and managed by Northwestern University in accordance with their competing interest policies. G.A.R and A.S.K. declare no competing interests.
REFERENCES
- 1. Barrangou R., Doudna J.A.. Applications of CRISPR technologies in research and beyond. Nat. Biotechnol. 2016; 34:17–20. [DOI] [PubMed] [Google Scholar]
- 2. Brandt K., Barrangou R.. Applications of CRISPR technologies across the food supply chain. Annu. Rev. Food Sci. Technol. 2019; 10:133–150. [DOI] [PubMed] [Google Scholar]
- 3. Dongen J.E.Van, Berendsen J.T.W., Steenbergen R.D.M., Rob M., Wolthuis F., Eijkel J.C.T., Segerink L.I. Biosensors and bioelectronics Point-of-care CRISPR /Cas nucleic acid detection: recent advances, challenges and opportunities. Biosens. Bioelectron. 2020; 166:112445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Shrock E., Güell M.. Torres-Ruiz R., Rodriguez-Perales S.B.T-.P.. Chapter six - CRISPR in animals and animal models. CRISPR in Animals and Animal Models. 2017; 152:Academic Press; 95–114. [DOI] [PubMed] [Google Scholar]
- 5. Sung J., Rok K., Pricilia C., Prabowo S., Ho J.. CRISPR /Cas9-coupled recombineering for metabolic engineering of corynebacterium glutamicum. Metab. Eng. 2017; 42:157–167. [DOI] [PubMed] [Google Scholar]
- 6. Rock J.M., Hopkins F.F., Chavez A., Diallo M., Chase M.R., Gerrick E.R., Pritchard J.R., Church G.M., Rubin E.J., Sassetti C.M.et al.. Programmable transcriptional repression in mycobacteria using an orthogonal CRISPR interference platform. Nat. Microbiol. 2017; 2:16274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lee Y.J., Hoynes-O’Connor A., Leong M.C., Moon T.S. Programmable control of bacterial gene expression with the combined CRISPR and antisense RNA system. Nucleic Acids Res. 2016; 44:2462–2473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhang S., Voigt C.A.. Engineered dCas9 with reduced toxicity in bacteria: implications for genetic circuit design. Nucleic Acids Res. 2018; 46:11115–11125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hanahan D. Studies on transformation of escherichia coli with plasmids. J. Mol. Biol. 1983; 166:557–580. [DOI] [PubMed] [Google Scholar]
- 10. Harrington L.B., Paez-espino D., Doudna J.A., Staahl B.T., Chen J.S., Ma E., Kyrpides N.C.. A thermostable cas9 with increased lifetime in human plasma. Nat. Commun. 2017; 8:1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nagaraju S., Davies N.K., Walker D.J.F., Köpke M., Simpson S.D.. Genome editing of clostridium autoethanogenum using CRISPR/Cas9. Biotechnol. Biofuels. 2016; 9:219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cho S., Choe D., Lee E., Kim S.C., Palsson B., Cho B.K.. High-Level dCas9 expression induces abnormal cell morphology in escherichia coli. ACS Synth. Biol. 2018; 7:1085–1094. [DOI] [PubMed] [Google Scholar]
- 13. Makarova K.S., Wolf Y.I., Alkhnbashi O.S., Costa F., Shah S.A., Saunders S.J., Barrangou R., Brouns S.J.J., Charpentier E., Haft D.H.et al.. An updated evolutionary classification of CRISPR-Cas systems. Nat. Rev. Microbiol. 2015; 13:722–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zheng Y., Han J., Wang B., Hu X., Li R., Shen W., Ma X., Ma L., Yi L., Yang S.et al.. Characterization and repurposing of the endogenous type I-F CRISPR – cas system of zymomonas mobilis for genome engineering. Nucleic Acids Res. 2019; 47:11461–11475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hidalgo-Cantabrana C., Goh Y.J., Pan M., Sanozky-Dawes R., Barrangou R.. Genome editing using the endogenous type i CRISPR-Cas system in lactobacillus crispatus. Proc. Natl. Acad. Sci. 2019; 116:15774–15783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Walker J.E., Lanahan A.A., Zheng T., Toruno C., Lynd L.R., Cameron J.C., Olson D.G., Eckert C.A.. Development of both type i – b and type II CRISPR /Cas genome editing systems in the cellulolytic bacterium clostridium thermocellum. Metab. Eng. Commun. 2020; 10:e00116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Pyne M.E., Bruder M.R., Moo-Young M., Chung D.A., Chou C.P.. Harnessing heterologous and endogenous CRISPR-Cas machineries for efficient markerless genome editing in clostridium. Sci. Rep. 2016; 6:25666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhou X., Wang X., Luo H., Wang Y., Wang Y., Zhang J.. Exploiting heterologous and endogenous CRISPR - Cas systems for genome editing in the probiotic clostridium butyricum. Biotechnol. Bioeng. 2021; 118:2448–2459. [DOI] [PubMed] [Google Scholar]
- 19. Gomaa A.A., Klumpe H.E., Luo M.L., Selle K., Barrangou R., Beisel L.. Programmable removal of bacterial strains by use of Genome- Targeting CRISPR-Cas systems. MBio. 2014; 5:e00928-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Selle K., Fletcher J.R., Tuson H., Schmitt D.S., Mcmillan L., Vridhambal G.S., Rivera A.J., Montgomery S.A., Fortier L., Barrangou R.et al.. In vivo targeting of clostridioides difficile using Phage- Delivered CRISPR-Cas3 antimicrobials. MBio. 2020; 11:e00019-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bikard D., Euler C.W., Jiang W., Nussenzweig P.M., Goldberg G.W., Duportet X., Fischetti V.A., Marraffini L.A.. Exploiting CRISPR-cas nucleases to produce sequence-specific antimicrobials. Nat. Biotechnol. 2014; 32:1146–1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Leenay R.T., Beisel C.L.. Deciphering, communicating, and engineering the CRISPR PAM ryan. J. Mol. Biol. 2017; 429:177–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Maxwell C.S., Jacobsen T., Marshall R., Noireaux V., Beisel C.L.. A detailed cell-free transcription-translation-based assay to decipher CRISPR protospacer-adjacent motifs. Methods. 2018; 143:48–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Esvelt K.M., Mali P., Braff J.L., Moosburner M., Yaung S.J., Church G.M.. Orthogonal cas9 proteins for RNA-Guided gene regulation and editing. Nat. Methods. 2013; 10:1116–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Barrangou R., Marraffini L.A.. CRISPR-Cas systems: prokaryotes upgrade to adaptive immunity. Mol. Cell. 2014; 54:234–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mendoza B.J., Trinh C.T.. In silico processing of the complete CRISPR-Cas spacer space for identification of PAM sequences. Biotechnol. J. 2018; 13:e1700595. [DOI] [PubMed] [Google Scholar]
- 27. Biswas A., Gagnon J.N., Brouns S.J.J., Fineran P.C., Brown C.M.. Bioinformatic prediction and analysis of crRNA targets CRISPRTarget. RNA Biol. 2013; 10:817–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Couvin D., Bernheim A., Toffano-nioche C., Touchon M., Rocha E.P.C., Vergnaud G., Michalik J., Bertrand N., Gautheret D., Pourcel C.et al.. CRISPRCasFinder, an update of CRISRFinder, includes a portable version, enhanced performance and integrates search for cas proteins. Nucleic Acids Res. 2018; 46:246–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Altschul S.F., Gish W., Miller W., Myers E.W., Lipman D.J.. Basic local alignment search tool. J. Mol. Biol. 1990; 215:403–410. [DOI] [PubMed] [Google Scholar]
- 30. Wagih O. Ggseqlogo: a versatile r package for drawing sequence logos. Bioinformatics. 2017; 33:3645–3647. [DOI] [PubMed] [Google Scholar]
- 31. Arndt D., Grant J.R., Marcu A., Sajed T., Pon A., Liang Y., Wishart D.S.. PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Res. 2016; 44:W16–W21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Williams D.R., Young D.I., Young M.. Conjugative plasmid transfer from escherichia coli to clostridium acetobutylicum. Microbiology. 1990; 136:819–826. [DOI] [PubMed] [Google Scholar]
- 33. Woods C., Humphreys C.M., Rodrigues R.M., Ingle P., Rowe P., Henstra A.M., Köpke M., Simpson S.D., Winzer K., Minton N.P.. A novel conjugal donor strain for improved DNA transfer into clostridium spp. Anaerobe. 2019; 59:184–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Liew F., Henstra A.M., Köpke M., Winzer K., Simpson S.D., Minton N.P.. Metabolic engineering of clostridium autoethanogenum for selective alcohol production. Metab. Eng. 2017; 40:104–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Annan F.J., Al-Sinawi B., Humphreys C.M., Norman R., Winzer K., Köpke M., Simpson S.D., Minton N.P., Henstra A.M.. Engineering of vitamin prototrophy in clostridium ljungdahlii and clostridium autoethanogenum. Appl. Microbiol. Biotechnol. 2019; 103:4633–4648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Leang C., Ueki T., Nevin K.P., Lovley D.R.. A genetic system for clostridium ljungdahlii: a chassis for autotrophic production of biocommodities and a model homoacetogen. Appl. Environ. Microbiol. 2013; 79:1102–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Karah N., Samuelsen Ø., Zarrilli R., Sahl J.W., Wai S.N., Uhlin B.E.. CRISPR-cas subtype I-Fb in acinetobacter baumannii: evolution and utilization for strain subtyping. PLoS One. 2015; 10:e0118205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Leenay R.T., Maksimchuk K.R., Slotkowski R.A., Agrawal R.N., Gomaa A.A., Briner A.E., Barrangou R., Beisel C.L.. Identifying and visualizing functional PAM diversity across CRISPR-Cas systems. Mol. Cell. 2016; 62:137–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kim E., Koo T., Park S.W., Kim D., Kim K., Cho H.-Y., Song D.W., Lee K.J., Jung M.H., Kim S.et al.. In vivo genome editing with a small cas9 orthologue derived from campylobacter jejuni. Nat. Commun. 2017; 8:14500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Boudry P., Semenov E., Monot M., Datsenko K.A., Lopatina A., Sekulovic O., Ospina-Bedoya M., Fortier L.C., Severinov K., Dupuy B.et al.. Function of the CRISPR-cas system of the human pathogen: clostridium difficile. MBio. 2015; 6: 10.1128/mBio.01112-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zhang J., Zong W., Hong W., Zhang Z.T., Wang Y.. Exploiting endogenous CRISPR-Cas system for multiplex genome editing in clostridium tyrobutyricum and engineer the strain for high-level butanol production. Metab. Eng. 2018; 47:49–59. [DOI] [PubMed] [Google Scholar]
- 42. Zetsche B., Gootenberg J.S., Abudayyeh O.O., Slaymaker I.M., Makarova K.S., Essletzbichler P., Volz S.E., Joung J., van der Oost J., Regev A.et al.. Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell. 2015; 163:759–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Qin Z., Yang Y., Yu S., Liu L., Chen Y., Chen J., Zhou J.. Repurposing the endogenous type I-E CRISPR/Cas system for gene repression in gluconobacter oxydans WSH-003. ACS Synth. Biol. 2021; 10:84–93. [DOI] [PubMed] [Google Scholar]
- 44. Ran F.A., Cong L., Yan W.X., Scott D.A., Gootenberg J.S., Kriz A.J., Zetsche B., Shalem O., Wu X., Makarova K.S.et al.. In vivo genome editing using staphylococcus aureus cas9. Nature. 2015; 520:186–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Cady K.C., Bondy-Denomy J., Heussler G.E., Davidson A.R., O’Toole G.A. The CRISPR/Cas adaptive immune system of pseudomonas aeruginosa mediates resistance to naturally occurring and engineered phages. J. Bacteriol. 2012; 194:5728–5738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Pranam C., Noah J., M. J.J. Minimal PAM specificity of a highly similar spcas9 ortholog. Sci. Adv. 2021; 4:eaau0766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Jinek M., Chylinski K., Fonfara I., Hauer M., Doudna J.A., Charpentier E.. A programmable Dual-RNA guided DNA endonuclease in adaptive bacterial immunity. Science (80-.). 2012; 337:816–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Garneau J.E., Dupuis M.-È., Villion M., Romero D.A., Barrangou R., Boyaval P., Fremaux C., Horvath P., Magadán A.H., Moineau S.. The CRISPR/Cas bacterial immune system cleaves bacteriophage and plasmid DNA. Nature. 2010; 468:67–71. [DOI] [PubMed] [Google Scholar]
- 49. Karim A.S., Dudley Q.M., Juminaga A., Yuan Y., Crowe S.A., Heggestad J.T., Garg S., Abdalla T., Grubbe W.S., Rasor B.J.et al.. In vitro prototyping and rapid optimization of biosynthetic enzymes for cell design. Nat. Chem. Biol. 2020; 16:912–919. [DOI] [PubMed] [Google Scholar]
- 50. Fackler N., Heijstra B.D., Rasor B.J., Brown H., Martin J., Ni Z., Shebek K.M., Rosin R.R., Simpson S.D., Tyo K.E.et al.. Stepping on the gas to a circular economy: accelerating development of carbon-negative chemical production from gas fermentation. Annu. Rev. Chem. Biomol. Eng. 2021; 12:439–470. [DOI] [PubMed] [Google Scholar]
- 51. Liew F.(Eric), Nogle R., Abdalla T., Rasor B.J., Canter C., Jensen R.O., Wang L., Strutz J., Chirania P., De Tissera S.et al.. Carbon-negative production of acetone and isopropanol by gas fermentation at industrial pilot scale. Nat. Biotechnol. 2022; 10.1038/s41587-021-01195-w. [DOI] [PubMed] [Google Scholar]
- 52. Nobrega F.L., Walinga H., Dutilh B.E., Brouns S.J.J.. Prophages are associated with extensive CRISPR-Cas auto-immunity. Nucleic Acids Res. 2020; 48:12074–12084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Fackler N., Heffernan J., Juminaga A., Doser D., Nagaraju S., Gonzalez-Garcia R.A., Simpson S.D., Marcellin E., Köpke M.. Transcriptional control of clostridium autoethanogenum using CRISPRi. Synth. Biol. 2021; 6:ysab008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Jones D.T., Shirley M., Wu X., Keis S.. Bacteriophage infections in the industrial acetone butanol (AB) fermentation process further reading. J. Mol. Microbiol. Biotechnol. 2000; 2:21–26. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Source code for Spacer2PAM as well as instructions are available via GitHub at https://github.com/grybnicky/Spacer2PAM. Illumina sequencing reads for the 4-nucleotide randomized PAM depletion experiment are available through SRA under BioProject accession number PRJNA755691. Further data available on request from the authors.