

Abstract
Coronavirus disease 2019 (COVID-19) persists and shook the global population where the endgame to this pandemic is brought on by developing vaccines in record-breaking time. Nevertheless, these vaccines are far from perfect where their efficiency ranges from 65 to 90%; therefore, vaccines are not the one only solution to overcome this situation, and apart from administration of vaccines, the scientific community is at quest for finding alternative solutions to incumber SARS-CoV-2 infection. In this study, our research group is keen on identifying a bioactive molecule that is independent in its mode of action from existing vaccines which can potentially target the SARS-CoV-2 virus replicative efficacy. Papain-like protease (PLpro) and main protease (Mpro) are the most lucrative targets of COVIDs against which the drugs can be developed, as these proteases play a vital role in the replication and development of viral particles. Researchers have modelled a compound such as GRL0617 and X77 as an inhibitor of Mpro and PLpro, respectively, but use of these compounds has several limitations on hosts like toxicity and solubility. Under the current study by deploying rigorous computational assessments, pool of microbial secondary metabolites was screened and handpicked to search a structural or functional analogue of GRL0617 and X77, with an idea to identify a compound that can serve as dual inhibitor for both PLpro and Mpro. From the manually curated database of known antiviral compounds from fungal origin, we found cytonic acids A and B to potentially serve as dual inhibitor of PLpro and Mpro.
Keywords: SARS-CoV-2, Antiviral, Papain-like protease (PLpro), Main protease (Mpro), Endophytes, Molecular dynamics simulations
Introduction
Coronavirus disease 2019 (COVID-19) manifests a range of physiological conditions on host, ranging from being entirely asymptomatic to minor influenza to cough, cold and sore throat to manifestations like acute respiratory distress, pneumonia and lung damage to even death [1, 2]. During the first wave of the infection, all affected countries endorsed various precautionary measures, including a total or partial lockdown of residents, implementation of social distancing, wearing of face masks and frequent hand sanitization. More significantly, individuals did not follow the prevention measures precisely and the number of affected individuals changed relying upon how powerful the prevention measures were. Additionally, these recommended measures have been headed to the detriment of the economy and more crucially, these severe control measures alone are not viable enough to stop the COVID-19 pandemic. This is vividly observed in both developing and developed countries, where people were hit by a third wave of coronavirus infection due to the lack of antiviral medication or vaccination, and as a result, the scientists from around the world are prompted to find effective medications and vaccines for COVID-19 treatment and prevention [1, 3].
In the last 1 year, COVID-19 vaccines are being developed using various strategies, executed from a typical inactivated and live-attenuated vaccines to more innovative messenger RNA (mRNA) and DNA technologies, like subunit vaccines [4–6]. For instance, BBV152 is a classical inactivated whole virion vaccine, while mRNA-1273 and mRNA-BNT162b2 are mRNA-based vaccines that target SARS-CoV-2 spike protein [5]. Herd immunity may hypothetically be achieved because of the high incidence of protection from infection after vaccination; however, there is no reliable assurance to it. Besides, there are yet various vaccination-related inquiries that are presently unaddressed, which include the length of a vaccination preventive effects and cross-protection against variants of interests (VOI) and variants of concerns (VOC) (Alpha, Beta, Gamma, Delta, Delta Plus, Epsilon, Eta, Theta, Iota, Kappa and Lambda), particularly the new VOC, Omicron (B.1.1.529) [7]. Mortality related with COVID-19 is fairly reducing as vaccination rates rise, but the list of variants is increasing as new mutations continue to evolve, challenging the whole vaccine regime [8].
While the vaccination drive continues, another promising strategy would be to foster new drugs that are compelling against SARS-CoV-2. Virus replication necessitates a certain set of functional and structural proteins. Malfunctioning or inhibition of these proteins can slow or stop infection or can also halt viral multiplication, culminating its spread, which is desired by researchers. As a result, medicines that can regulate the accessibility of these proteins should be pursued. SARS-CoV-2 is a 29-kb RNA genome that encodes two large overlapping polyprotein precursors (pp1a and pp1ab); these two translated stretches of inactive proteins are then treated upon by PLpro and Mpro which cleave pp1a and pp1b into various small fragments where each fragment serves as active viral protein that functions in replication, transcription and assembly of the virus nuclear material into the capsid protein coat (Fig. 1). Consequently, inhibiting these proteases may have a considerable impact on the viral machinery and the overall rate of viral infection. Scientists around the globe have well considered these proteases (Mpro and PLpro) as important drug targets for developing new medications or repurposing already existing drugs [9, 10], as inhibiting these proteases directly halts the functioning and replication of SARS-CoV-2, making the virus ineffective [11, 12].
Fig. 1.

Representation for addressing role of viral proteases PLpro and Mpro to produce non-structural proteins participating in viral replication and transcription
Bioactive and natural products derived from plants, microorganisms and animals are most often recommended to treat infectious disorders [13, 14]. The term ‘endophytes’ signifies microorganisms living inside the tissue of plant with imparting mutualistic behaviour. Considering the published literature, endophytes could be an untapped reservoir full of diverse resources of bioactive compounds [15]. Identifying a compound from reservoir that can block PLpro and Mpro is the rationale of this research which in itself is a challenging as well as exciting avenue.
The likelihood of discovering novel inhibitors using microbial databases of natural compounds has increased as computer hardware and software technologies have progressed. Several investigations have used computer-aided drug designing (CADD) tools where in its pipeline first the compounds are screened by ‘molecular docking’ approach which identifies compounds that can interact with important amino acids in the active site of protein, followed by ‘molecular dynamics (MD) simulations’ that perform a real-time simulation of interaction between screened ligand and protein under virtual physiological environment, and a blend of advanced in silico approaches to discover natural bioactive compounds from large databases that could be used as possible lead compounds [16–19]. The novelty of this piece of research work lies in the fact that antiviral metabolites from endophytic origin are very less investigated against the SARS-CoV-2 proteases (Mpro and PLpro), the reason being the scarcity of published literature and unavailability of off-the-shelf databases. To date, there are several secondary bioactive active metabolites identified from endophytes possessing ability to inhibit viral proteases, and they are emodin, ω-hydroxyemodin, ( +)-sclerotiorin, phomopsone B and phomopsone C [20–23], but such study for proteases of SARS-CoV-2 is lacking which serves as a research loophole which we are targeting in this research piece.
For this study, we have nit-picked literary evidence of antiviral compounds obtained from endophytic microorganisms and prepared a library to virtually screen them against SARS-CoV-2 Mpro and PLpro, as a dual inhibitor. This is done to ensure the optimal potential of the ligand library to act simultaneously on the activity of both proteases altogether. After screening the entire library, cytonic acids A and B were identified as PLpro and Mpro dual protease inhibitor. Under this research piece, screening hierarchy followed the use of protein–ligand docking, followed by pharmacophore hypothesis generation, which was then followed by absorption–distribution–metabolism–excretion–toxicity (ADMET) profiling and protein–ligand binding Gibbs free energy change calculation through molecular mechanics generalised Born surface area (MM-GBSA) assessment. Additionally, the dynamic nature of protein–ligand contacts was analysed using MD simulations. The simulation was undertaken for cytonic acid derivatives and control compounds for a 100-ns-long timescale. We predict that cytonic acids A and B as a dual-target therapeutic approach for COVID-19 could be more effective and would substantially reduce the use of combinational drugs as well as prevent the multi-drug dose load on the already immune-compromised and weakened host system.
Materials and methods
Protein preparation
PLpro protein of SARS-CoV-2 (PDB ID: 7CMD) co-crystalized form with GRL0617 and Mpro of SARS-CoV-2 (PDB ID: 6W63) co-crystalized form with X77 were retrieved from the protein databank (PDB) [24–26]. Here, GRL0617 and X77 are the known inhibitors for respective proteins and were used as reference. Proteins were imported to Schrödinger Maestro and were prepared in protein preparation wizard of Schrödinger Maestro [27]. In which, the addition of hydrogens was performed, and het state at pH 7.0 was created using Epik. Then, the protein was optimized with PROPKA keeping its parameter as pH = 7.0. On successful completion of these two steps, the minimization was performed using force field OPLS2005.
Metadata curation for ligand library development
From the available literature of research articles, the data of endophytic microorganisms and endophytic microorganisms and their bioactive metabolites was manually collected. Based on experimental antiviral effects of metabolites against various viruses such as influenza, H1N1, HIV, HCV, and EV-71, the 45 antiviral compounds were retrieved from published studies. Table 1 lists the 45 compounds present in the library, along with their isolation sources and bioactivity. All the ligands were retrieved in SDF format and were imported to the Schrödinger Maestro for ligand preparation for docking, where the LipPrep wizard was used, addition of hydrogens was performed, and ligand minimization was performed using force field OPLS2005. The files were exported after ligand minimization and subsequently used for molecular docking.
Table 1.
List of antiviral compounds of the endophytes with their metadata
| Sr. no | Secondary metabolite | Endophytic organism | Host | Location | Virus | Activity (IC50) | Reference |
|---|---|---|---|---|---|---|---|
| 1 | 6-O-Demethyl-4-dehydroxyaltersolanol A | Nigrospora sp. YE3033 | Aconitum carmichaelii | China | H1N1 influenza A | 2.59 ± 1.22 µg/mL | [61] |
| 2 | 4-Dehydroxyaltersolanol A | 8.35 ± 1.41 µg/mL | |||||
| 3 | Altersolanol B | 7.82 ± 1.86 µg/mL | |||||
| 4 | Chermesinone B | 0.80 ± 0.29 µg/mL | |||||
| 5 | Emodin | Aspergillus versicolor | Halimeda opuntia | Egypt | HCV protease | 22.5 ± 1.6 µg/mL | [20] |
| 6 | ω-Hydroxyemodin | Fusarium equiseti | Padina pavonica | Egypt | HCV protease | 10.71 ± 2.3 µM | [22] |
| 7 | ( +)-Sclerotiorin | Penicillium sclerotiorum PSU-A13 | Garcinia atroviridis | Thailand | HIV-1 integrase | 14.5 µg/mL | [23] |
| HIV-1 protease | 62.7 µg/mL | ||||||
| 8 | Cordycepin | Fusarium equiseti | Padina pavonica | Egypt | HCV protease | 22.3 µM | [22] |
| 9 | Ara-A | 24.5 µM | |||||
| 10 | Cyclic tetrapeptidecyclo-[phenyl alanyl-pro-leu-pro] | 29.4 µM | |||||
| 11 | 17-Demethyl-2,11-dideoxy-rhizoxin | 29.4 µM | |||||
| 12 | 5-Chloro-3,6-dihydroxy-2-methyl-1,4-benzoquinone | 34.4 µM | |||||
| 13 | Perlolyrine | 35.1 µM | |||||
| 14 | Cyclo (l-Pro-l-Val) | 23.2 µM | |||||
| 15 | Griseoxanthone C | 19.8 µM | |||||
| 16 | Stachybogrisephenone B | Stachybotry sp. HH1 ZDDS1F1-2 | Sponge | China | EV71 | 30.1 µM | [62] |
| 17 | Grisephenone A | 50.0 µM | |||||
| 18 | 3,6,8-Trihydroxy-1-methylxanthine | 40.3 µM | |||||
| 19 | Emerimidine A | Emericella sp. (HK-ZJ) | Aegiceras corniculatum | China | H1N1 influenza A | 42.07 µg/mL | [63] |
| 20 | Emerimidine B | 62.05 µg/mL | |||||
| 21 | Chloropupukeananin | Pestalotiopsis fici | Unknown | China | HIV-1 | 14.6 µM | [64] |
| 22 | Periconiasin G | Periconia sp. F-31 | Annona muricata | China | HIV-1 | 67.0 µM | [65] |
| 23 | Aspergilline A | Aspergillus versicolor | Paris polyphylla var. yunnanensis | China | Tobacco mosaic virus | 56.4 ± 3.8 μM | [60] |
| 24 | Aspergilline B | 47.3 ± 3.2 μM | |||||
| 25 | Aspergilline C | 35.6 ± 2.8 μM | |||||
| 26 | Aspergilline D | 38.9 ± 3.5 μM | |||||
| 27 | Aspergilline E | 33.6 ± 3.0 μM | |||||
| 28 | Cytonic acid A | Cytonaema sp. F32027 | Quercus sp. | UK | CMV protease | 43 μM | [58] |
| 29 | Cytonic acid B | 11 μM | |||||
| 30 | Phomopsone B | Phomopsis sp. CGMCC No.5416 | Achyranthes bidentata | China | HIV-1 | 7.6 μM | [21] |
| 31 | Phomopsone C | 0.5 μM | |||||
| 32 | Altertoxin V | Alternaria tenuissima QUE1Se | Quercus emoryi | USA | HIV-1 | 0.09 μM | [66] |
| 33 | Altertoxin I | 1.42 μM | |||||
| 34 | Altertoxin II | 0.21 μM | |||||
| 35 | Altertoxin III | 0.29 μM | |||||
| 36 | Xiamycin | Streptomyces sp. GT2002/1503 | Bruguiera gymnorrhiza | Germany | HIV-1 | NA | [67] |
| 37 | Xiamycin methyl ester | NA | |||||
| 38 | Neosartoryadin A | Neosartorya udagawae HDN13-313 | Avicennia marina | China | H1N1 influenza A | 66 μM | [68] |
| 39 | Neosartoryadin B | 58 μM | |||||
| 40 | Asperphenalenone A | Aspergillus sp. CPCC 400,735 | Kadsura longipedunculata | China | HIV-1 | 4.5 μM | [59] |
| 41 | Asperphenalenone D | 2.4 μM | |||||
| 42 | Cytochalasin Z8 | 9.2 μM | |||||
| 43 | Epicocconigrone A | 6.6 μM | |||||
| 44 | Alternariol 5-o-methyl ether | Colletotrichum sp. | NA | China | HIV-1 | NA | [69] |
| 45 | 8-Methyl emodin | Aspergillus versicolor | Halimeda opuntia | Egypt | HCV protease | 40.2 ± 2.3 µg/mL | [20] |
NA not available
Generation of E-pharmacophore model
E-pharmacophore hypothesis was generated separately using the crystal structure of (i) SARS-CoV-2 PLpro bound to an inhibitor GRL0617 with a resolution of 2.1 Å (PDB ID: 7CMD) and (ii) SARS-CoV-2 Mpro bound to an inhibitor X77 with a resolution of 2.1 Å (PDB ID: 6W63). During pre-processing of the complex, water molecules within 5 Å distance of the ligand were eliminated from the structure and the minimised complex was used to develop E-pharmacophore model [28, 29]. The E-pharmacophore model was developed using the Phase module (Phase, Schrödinger, LLC, New York, NY, 2021) [30]. For developing hypothesis, default pharmacophore properties like hydrogen bond acceptor (A), hydrogen bond donor (D), aromatic ring (R) and hydrophobicity (H) were mapped. On performing E-pharmacophore, two hypotheses were generated, one for each protein–ligand complex. These E-pharmacophore hypotheses were then used for screening of ligands.
E-pharmacophore-based virtual screening
A E-pharmacophore-based virtual screening was carried out on the prepared library of 45 compounds using the Phase module of the Schrodinger suite to develop a collection of compounds having requisite ligand characteristics aimed at best binding to PLpro and Mpro. This was achieved by mapping E-pharmacophore models on the library of 45 compounds.
Molecular docking
The prepared proteins as shown in earlier step were used for molecular docking. The docking grid at the exact coordinates as ligand GRL0617 in 7CMD for PLpro and X77 in 6W63 for Mpro was prepared using the Glide module of Schrödinger Maestro. The grid for PLpro was developed at the coordinates X − 34.22, Y − 11.61 and Z − 30.05, with the size 10 Å × 11 Å × 14 Å, while for Mpro, the grid was developed at the coordinates X − 20.58, Y 18.1 and Z − 26.98, with the size 12 Å × 14 Å × 15 Å and was prepared. For docking, the prepared ligands so obtained after E-pharmacophore-based screening (15 for PLpro and 18 for Mpro) were used. Moreover, native ligands GRL0617 for PLpro and X77 for Mpro were used as positive controls for docking with respective proteins, PLpro and Mpro. Docking level was set to ‘Extra Precision (XP)’, docking energies in negative values with unit kcal/mol were recorded, and the output file of docked poses was analysed for interactions in BIOVIA Discovery Studio (DS) visualizer. The ligands showing the highest negative binding docking scores and protein interactions were screened for further in silico assays including MM-GBSA and MD simulations.
MM-GBSA calculations
MM-GBSA determines the Gibbs free energy change in kcal/mol that predicts how spontaneous the interaction reaction can be between selected screened ligand and protein. The greater negativity in the values shows more spontaneous reaction. This ΔG value can be compared for with that of reference native ligand. If the values shown by screened selected ligand are more negative than by the reference ligand, then the binding spontaneity of the screened ligand is stronger than of the reference native ligand [31–34]. MM-GBSA assessment was performed using the Prime module of Schrodinger Maestro making use of OPLS2005 force field. The ΔGBind is the overall Gibbs free energy change value of the system, which is also bifurcated further into segments as ΔGCoulomb—Coulomb energy, ΔGHbond—hydrogen-bonding correction, ΔGLipo—lipophilic energy, ΔGPacking—pi-pi packing correction, ΔGvdW—Van der Waals energy, etc.
ADMET analysis
The ADMET properties of the GRL0617 and X77 (as controls) and top screened ligands (test endophytic molecules) were predicted using the pkCSM—pharmacokinetics server [35] which provides the theoretical values that are obtained by proven animal model turned in to finely tuned algorithms. Values for each of the parameters like water solubility, CaCO2 permeability, intestinal absorption (human), skin permeability, P-glycoprotein substrate, P-glycoprotein I and II inhibitor, blood–brain barrier (BBB) permeability, central nervous system (CNS) permeability, cytochrome p450 (CYP)2D6 and CYP3A4 substrate, CYP1A2-CYP2C19-CYP2C9-CYP2D6-CYP3A4 inhibitor, renal clearance, AMES toxicity, max. tolerated dose, hERG I and II inhibitor, oral rat acute and chronic toxicity, hepatotoxicity, skin sensitisation, T. pyriformis toxicity and minnow toxicity are obtained for each ligand under study by this assessment. This helps to understand and compare drugs, like properties of screened selected ligands in comparison with the reference molecules.
MD simulations
The Desmond module of Schrödinger Maestro was used to perform simulations of SARS-CoV-2 PLpro-ligand and Mpro-ligand complexes. Cytonic acid A and cytonic acid B were having efficient binding energy and proper ADMET profile and both were able to mimic interactions of native ligand with PLpro as well as Mpro; MD simulations were carried out for the following complexes: (I) PLpro-GRL0617, (II) PLpro-cytonic acid A, (III) PLpro-cytonic acid B, (IV) Mpro-X77, (V) Mpro-cytonic acid A and (VI) Mpro-cytonic acid B. For the simulation assessments, the complexes PLpro-GRL0617 and Mpro-X77 were taken as reference controls against which the simulation results of cytonic acid A and cytonic acid B with both the proteins were compared with. For MD simulations, the virtual simulation box was prepared into which complex was placed, and then, the simulation box was filled with water molecules with TIP3P model, after which the ion Cl− or Na+ was placed in the simulation box for neutralisation. MD simulations were performed with NPT ensemble at 300 K and 1.01 bar for 100 ns. Post MD simulation assessments were performed by assessing ligand receptor interaction profiles with respect to time, types and nature; the root mean square deviation (RMSD) and root mean square fluctuation (RMSF) assessments of trajectories were also analysed to evaluate the stability of protein–ligand complexes during the simulations run.
Results
Metadata assessment for ligand library development
The prepared library for this in silico analysis included specialized nit-picked literary evidenced endophytic compounds having in vitro anti-viral activity observed against influenza, H1N1, HIV, HCV and EV-71 viruses. Forty-five antiviral compounds obtained from endophytic microorganisms were used to virtually screen them against SARS-CoV-2 Mpro and PLpro, as a dual inhibitor. Table 1 contains information on the names of the compounds, their endophyte sources and their antiviral efficacy against particular viruses.
Structure‑based pharmacophore modelling
Receptor-pharmacophore based on the 3D structure of a target protein can provide detailed and accurate information on ligand interaction attributes. In totality, there are about 8 descriptors used for pharmacophore modelling; amongst which, hydrogen bond donors, acceptors, lipophilic regions and negative and positive ionizable groups along with aromatic rings are the most commonly used descriptors in pharmacophore modelling. For this study, 3D structure–based e-pharmacophores were prepared with the ‘Phase’ module which utilises the receptor–ligand pharmacophore generation procedure and discovered amino acids involved in ligand binding for a co-crystal GRL0617 inhibitor within the binding site of PLpro (Fig. 2a) and for a co-crystal X77 inhibitor within the binding site of Mpro (Fig. 2c). Hydrogen bond acceptor, hydrogen bond donor and pi-pi stacking of aromatic ring were amongst the five key 3D attributes of the generated e-pharmacophore for the evaluated protein. Figure 2b depicts the five 3D pharmacophore characteristics for the PLpro-GRL0617 complex that includes two donor hydrogen bonds, one acceptor hydrogen bond and one aromatic ring sphere, while Fig. 2d depicts the five 3D pharmacophore characteristics for the Mpro-X77 complex, including two acceptor hydrogen bonds and three aromatic ring spheres.
Fig. 2.

Illustration of E-pharmacophore features of receptor-ligand complex. a Representative features important in binding of GRL0617 ligand with receptor PLpro. b Type of pharmacophore features significant for interaction with PLpro. c Representative features important in binding of X77 ligand with receptor Mpro. d Type of pharmacophore features significant for interaction with Mpro
Pharmacophore‑based virtual screening analysis
The 45 potential anti-viral compounds of endophytes origin (Table 1) were screened using the E-pharmacophore hypothesis created from the GRL0617-PLpro complex and X77-Mpro. The ‘Phase’ screen score and corresponding binding site variables were used to filter these compounds. The Phase screen score is utilised to score and rank the matching compounds. This scoring function assesses both the quantity (if partial matching is permitted) and quality of ligand feature matching, where quality is defined by site, vector and volume scoring components. After the execution of the proposed pharmacophore hypothesis, a total of 15 molecules managed to pass this filter for the PLpro-GRL0167 complex (Table 2) and 18 molecules for the Mpro-X77 complex (Table 3).
Table 2.
Compounds managed to pass PLpro-GRL0167 complex pharmacophore hypothesis using Phase virtual screening
| Entry | Compound name | Number of sites matched | Matched ligand sites | Phase screen score |
|---|---|---|---|---|
| 1 | Stachybogrisephenone B | 4 | A(3) D(7) D(8) R(14) R(-) | 1.766 |
| 2 | 4-Dehydroxyaltersolanol A | 4 | A(5) D(10) D(9) R(13) R(-) | 1.723 |
| 3 | Aspergilline D | 4 | A(3) D(10) D(11) R(-) R(17) | 1.717 |
| 4 | Altersolanol B | 4 | A(4) D(9) D(8) R(11) R(-) | 1.669 |
| 5 | Aspergilline E | 4 | A(3) D(12) D(13) R(-) R(20) | 1.513 |
| 6 | Cytonic acid B | 4 | A(4) D(12) D(-) R(21) R(20) | 1.436 |
| 7 | Perlolyrine | 4 | A(1) D(-) D(4) R(6) R(9) | 1.435 |
| 8 | Neosartoryadin A | 4 | A(3) D(-) D(8) R(12) R(11) | 1.434 |
| 9 | Asperphenalenone D | 4 | A(7) D(13) D(12) R(23) R(-) | 1.419 |
| 10 | Cordycepin | 4 | A(4) D(8) D(7) R(-) R(12) | 1.400 |
| 11 | Asperphenalenone A | 4 | A(6) D(9) D(8) R(22) R(-) | 1.330 |
| 12 | Cytonic acid A | 4 | A(3) D(9) D(-) R(19) R(20) | 1.290 |
| 13 | Altertoxin I | 4 | A(1) D(9) D(8) R(-) R(12) | 1.244 |
| 14 | Aspergilline A | 4 | A(6) D(9) D(7) R(-) R(15) | 0.528 |
| 15 | Aspergilline C | 4 | A(6) D(9) D(7) R(-) R(17) | 0.505 |
Ligands having Phase screen score above 0.5 are screened
Table 3.
Compounds managed to pass Mpro-X77 complex pharmacophore hypothesis using Phase virtual screening
| Entry | Compound name | Number of sites matched | Matched ligand sites | Phase screen score |
|---|---|---|---|---|
| 1 | Altertoxin V | 4 | A(1) A(2) R(12) R(-) R(13) | 1.522 |
| 2 | Epicocconigrone A | 4 | A(3) A(4) R(17) R(-) R(18) | 1.428 |
| 3 | Stachybogrisephenone B | 4 | A(4) A(6) R(-) R(14) R(15) | 1.415 |
| 4 | Altertoxin II | 4 | A(2) A(3) R(12) R(-) R(11) | 1.39 |
| 5 | Alternariol 5-O-methyl ether | 4 | A(3) A(4) R(11) R(-) R(12) | 1.373 |
| 6 | Emodin-8-methyl ether | 4 | A(4) A(1) R(10) R(-) R(11) | 1.314 |
| 7 | Emodin | 4 | A(4) A(3) R(11) R(-) R(10) | 1.303 |
| 8 | Altertoxin III | 4 | A(2) A(6) R(12) R(-) R(13) | 1.285 |
| 9 | Altertoxin I | 4 | A(2) A(4) R(13) R(-) R(12) | 1.252 |
| 10 | Griseoxanthone C | 4 | A(3) A(4) R(11) R(12) R(-) | 1.235 |
| 11 | Perlolyrine | 4 | A(1) A(2) R(7) R(6) R(-) | 1.098 |
| 12 | Asperphenalenone A | 4 | A(6) A(1) R(23) R(-) R(22) | 1.094 |
| 13 | Asperphenalenone D | 4 | A(9) A(5) R(24) R(-) R(23) | 1.067 |
| 14 | Altertoxin VI | 4 | A(1) A(3) R(11) R(-) R(12) | 0.973 |
| 15 | Cytonic acids A | 4 | A(5) A(9) R(-) R(21) R(22) | 0.945 |
| 16 | Neosartoryadin B | 4 | A(3) A(4) R(-) R(16) R(17) | 0.939 |
| 17 | Neosartoryadin A | 4 | A(2) A(5) R(16) R(15) R(-) | 0.936 |
| 18 | Cytonic acid B | 4 | A(6) A(8) R(22) R(21) R(-) | 0.876 |
Ligands having Phase screen score above 0.5 are screened
Molecular docking analysis
After using Glide’s Receptor Grid Generation tool in Maestro to define the grid box, the minimised 3D molecular structure of the co-crystallised inhibitor GRL0617 was docked into the active site of viral PLpro. The molecular interaction of the co-crystallised inhibitor (GRL0617) with the binding site of the viral protease SARS-CoV-2-PLpro was discovered. In general, hydrophobic interactions induce GRL0617’s binding to SARS-PLpro, which results in protein inhibition. The aromatic rings’ 1-naphthyl moiety frames the primary contact as a pi–pi interaction with Tyr264 and Tyr268, is less exposed to solvent and fits into the cavity at the locus where the leucine at the P4 position is required. Furthermore, the 1-naphthyl moiety interacts with Pro247’s and Pro248’s side chains. The (R)-methyl group at GRL0167’s stereocenter is located inside the protein, between Tyr264 and Thr301 residues, in a small polar region. GRL0617 features a single aromatic ring with NH2 at R3 position, which is positioned at the cavity’s aperture, in addition to the 1-naphthyl moiety. The cavity is polar because of the presence of several polar groups, such as Gln269’s side chain oxygen and Tyr268’s hydroxyl group, which function as hydrogen bond acceptors in the interaction. After pharmacophore screening, a total of 15 microbial metabolites were investigated for molecular docking utilising Schrödinger’s Glide XP lead optimization methodology. Four fungal metabolites of endophytic origin, aspergilline E, cytonic acid A, cytonic acid B and 4-dehydroxyaltersolanol A, produced significant binding energies along with multiple conserved interactions with Tyr268 of SARS-CoV-2-PLpro. Each of these molecules produced the binding energies of − 7.7, − 7.655, − 7.292 and − 6.859 kcal/mol, respectively (Table 4). The interactions between these antiviral compounds and the protein (PLpro) are depicted in Fig. 3, and the structural features of GRL0617 and the top four antiviral metabolites are listed in Table 5.
Table 4.
Docking scores and the contributing binding residues of known PLpro inhibitor GRL-0617, Mpro inhibitor X77 and selected top antiviral metabolites generated using XP docking
| Compounds | Glide score (kcal/mol) | Contributing binding residues |
|---|---|---|
| SARS-CoV-2-PLpro | ||
| GRL-0617 (control) | − 6.441 | ASP164, GLN269, TYR268, TYR264, PRO247, PRO248, LEU162, GLY163, ARG166, MET208, TYR273, GLY271, CYS270, THR301 |
| Aspergilline E | − 7.77 | PRO247, TYR268, ASP164, GLY163, GLY271, TYR264, LEU162, TYR273, GLN269, GLY266, PRO248, ASN267, THR301, MET208 |
| Cytonic acid A | − 7.655 | GLU161, GLU167, LEU162, GLY163, ASP164, GLN269, CYS270, TYR264, TYR268, GLY266, PRO248, ASN267, MET208, ARG166, PRO247, GLY271, LYS157 |
| Cytonic acid B | − 7.292 | PRO247, PRO248, TYR273, ASN267, TYR264, GLU167, GLY266, GLY163, GLU161, LEU162, GLN269, ASP164, TYR268, MET208 |
| 4-Dehydroxyaltersolanol A | − 6.859 | PRO247, PRO248, TYR273, ASP164, GLY163, GLN269, GLU167, LYS157, LEU162, TYR268, TYR264, THR301 |
| SARS-CoV-2-Mpro | ||
| X77 (control) | − 8.52141 | MET49, ASP187, ARG188, PRO52, VAL42, CYS44, HIS41, THR25, LEU27, THR26, CYS145. ASN142, GLY143, SER144, HIS163, PHE140, LEU141, GLU166, HIS172, LEU167, TYR54, HIS164, GLN189, MET165, PRO168 |
| Cytonic acid B | − 9.998 | HIS41, ASP187, GLY170, GLU166, LEU167, ALA191, GLN192, GLN189, THR190, PRO168, ARG188, MET165, CYS145, ASN142, MET49, THR25, THR24, SER46, LEU27, GLY143, HIS164, THR26, TYR54 |
| Cytonic acid A | − 9.798 | CYS44, PRO52, MET49, TYR54, HIS41, ASN142, THR26, THR24, THR25, ASN119, GLY143, TYR118, LEU27, SER46, THR45, VAL42, GLU166, LEU167, PRO168, THR190, GLN192, ARG188, GLN189, HIS164, MET165, ASP187, ASP48 |
| Asperphenalenone D | − 8.969 | MET165, VAL186, HIS164, MET49, CYS44, THR45, SER46, THR25, THR24, GLY143, THR26, LEU27, CYS145, ASN142, SER144, GLN189, ASP187, PRO168, ALA191, THR190, LEU167, GLN192, ARG188 |
| Asperphenalenone A | − 6.2417 | MET165, CYS44, ARG188, GLN189, MET49, ASN28, LEU27, VAL42, THR26, ASN119, GLY143, LEU141, TYR118, ASN142, THR25, THR24, SER46, HIS41, HIS163, SER144, CYS145, GLU166 |
Fig. 3.

Interaction profile of GRL0617 and best docked four antiviral compounds with SARS-CoV-2-PLpro (amino acids without any bond interactions are interacting with Van der Waals forces)
Table 5.
Structural and chemical properties of screened antiviral compounds

Consequently, after successively establishing the grid box with Glide’s Receptor Grid Generation tool in Maestro, the minimised 3D molecular structure of the co-crystallised inhibitor X77 was docked into the active site of viral Mpro. The molecular interaction of the co-crystallised inhibitor (X77) with the binding site of the viral protease SARS-CoV-2-Mpro was observed. The co-crystallised binding site appears to be engaged in a number of hydrogen bonding, Pi stacking and hydrophobic interactions. The non-covalent inhibitor X77 forms a hydrogen bond with Asn142, Gly143, Glu166 and His41. Furthermore, the ligand is sandwiched between the benzene ring of Phe140 and the imidazole ring of His172, forming a pi-pi connection with Cys145 and His41. Also hydrophobic interactions with different amino acids were observed including Thr25, Leu27, Ser144, Phe140, Leu141, His172, Leu167, Tyr54, His164, Gln189, Met165 and Pro168 within the active site.
The top four molecules from endophytes that showed to effectively bind with Mpro are cytonic acid B, cytonic acid A, asperphenalenone D and asperphenalenone A, respectively, with the binding energy of − 9.998 kcal/mol, − 9.798 kcal/mol, − 8.969 kcal/mol and − 6.2417 kcal/mol (Table 4). The compound with the best docking score, cytonic acid B, makes six hydrogen bonds with the amino acids, namely, Thr24, Thr26, Asn142, Glu166, Arg188 and Thr190; along with it forms Pi-anion interaction with Cys145. Moreover, hydrophobic interactions in the form of alkyl/pi-alkyl interaction with His41, Met49 and Met165 are being formed. Apart from these, Van der Waals interaction with this ligand is formed by 13 different amino acids as represented in Fig. 4. Cytonic acid A tends to form hydrogen bonds with Thr24, Asn142, Glu166 and Arg188, while alkyl/pi-alkyl interaction involves residues Cys44, Thr26, Met49 and Met165. Van der Waals interactions are also formed with several amino acid residues of the active site. The third best compound, based on the docking score, asperphenalenone D makes hydrogen bonds with five amino acids, namely, Thr25, Gly143, Cys145, Glu166 and Thr190. It forms hydrophobic interactions by making alkyl/pi-alkyl interaction with His41, Met49, Met165 and Pro168 along with making Van der Waals interactions with several amino acids in the active site of Mpro. Lastly, asperphenalenone D makes hydrogen bonds with His41, Asn119, Gly143 and Glu166, and makes hydrophobic interactions in the form of alkyl/pi-alkyl interactions with Met49, Tyr118, Leu141, Cys145 and Met165. Like other ligands, this also makes Van der Waals interactions with several other amino acids in the active site of Mpro. The interactions between these antiviral compounds and the protein (Mpro) are depicted in Fig. 4. Moreover, the ligand properties of the reference inhibitors and top screened ligands of endophytic origins for both PLpro and Mpro are represented in Table 5.
Fig. 4.

Interaction profile of X77 and best docked four antiviral compounds with SARS-CoV-2-Mpro (amino acids without any bond interactions are interacting with Van der Waals forces)
MM-GBSA analysis
The interaction of a receptor and a ligand modifies the energies of both the free receptor and the ligand (ΔGBind); these energies also have a substantial influence on the stability of the receptor-ligand complex. In general, negative energy reflects a system’s greater stability. Table 4 shows the various energies produced by MM-GBSA for 7CMD with native ligand GRL0617, as well as the top four docking hits. In this case, the binding energies of GRL0617 with both 7CMD (− 67.56 kcal/mol) are negative, indicating that the interactions are more spontaneous. Similarly, cytonic acid A is the most stable of the studied inhibitors, with a binding energy of − 58.96 kcal/mol, while cytonic acid B is the second most stable, with a binding energy of − 50.9 kcal/mol. The MM-GBSA energies of the remaining compounds examined were likewise negative, although the values were significantly higher, indicating a low occurrence rate for the complex. The compound aspergilline E had the greatest docking score with SARS-CoV-2-PLpro; however, its ΔGBind energy (− 50.81 kcal/mol) was determined to be greater than that of cytonic acid derivatives.
In a similar manner, for Mpro, considering the ΔGBind scores of reference compounds and ligands, the reference ligand X77 has the best ΔGBind score of − 79.89 kcal/mol, while of all the other test ligands of endophytic origins, cytonic acid A has the best ΔGBind score of − 75.30 kcal/mol, followed by cytonic acid B, asperphenalenone D and asperphenalenone A where they show the ΔGBind scores of − 67.68 kcal/mol, 65.91 kcal/mol and 50.46 kcal/mol respectively. Apart from ΔGBind energy, calculations for energy, hydrogen-bonding correction, lipophilic energy, pi-pi packing correction and Van der Waals energy is also provided in Table 6.
Table 6.
MM-GBSA binding free energy change profiles of ligands with SARS-CoV-2-PLpro and SARS-CoV-2-Mpro for docked complexes
| Ligand | ΔGBind (kcal/mol) | ΔGCoulomb (kcal/mol) | ΔGHbond (kcal/mol) | ΔGLipo (kcal/mol) | ΔGPacking (kcal/mol) | ΔGvdW (kcal/mol) |
|---|---|---|---|---|---|---|
| Ligands interacting with SARS-CoV-2-PLpro | ||||||
| GRL-0617 (control) | − 67.5637 | − 20.1695 | − 2.5442 | − 26.1400 | − 3.1440 | − 46.3768 |
| Aspergilline E | − 50.8186 | − 17.1949 | − 1.24148 | − 18.2587 | − 0.54685 | − 35.848 |
| Cytonic acid A | − 58.9626 | − 34.7886 | − 2.45573 | − 20.7273 | − 2.897 | − 42.5047 |
| Cytonic acid B | − 55.6721 | − 28.8001 | − 1.74414 | − 25.71 | − 3.10166 | − 52.1166 |
| 4-Dehydroxyaltersolanol A | − 38.3654 | − 22.0615 | − 3.34432 | − 14.1614 | − 1.84419 | − 31.2361 |
| Ligands interacting with SARS-CoV-2-Mpro | ||||||
| X77 (control) | − 79.8911 | − 35.3714 | − 1.8857 | − 17.6157 | − 3.0805 | − 59.5617 |
| Cytonic acid A | − 75.3087 | − 36.7368 | − 4.0966 | − 16.7277 | − 0.5834 | − 58.2717 |
| Cytonic acid B | − 67.6847 | − 41.9421 | − 3.7410 | − 13.1996 | − 1.6820 | − 54.3987 |
| Asperphenalenone D | − 65.9103 | − 24.9700 | − 2.4700 | − 16.5780 | − 1.7280 | − 50.2750 |
| Asperphenalenone A | − 50.4602 | − 10.6094 | − 1.8999 | − 11.8471 | − 2.2800 | − 52.6731 |
Coulomb, Coulomb energy; Hbond, hydrogen-bonding correction; Lipo, lipophilic energy; Packing, pi-pi packing correction; vdW, Van der Waals energy
ADMET analysis
Seven distinct models were used to measure the absorption property. CaCO2 permeability and intestinal absorption were assessed; except for cytonic acid A and cytonic acid B, all of the substances have greater CaCO2 permeability values. Furthermore, all of the tested compounds including cytonic acid A and cytonic acid B as well as control substances had the poorest skin permeability, implying low absorption. For the P-glycoprotein assay, the lead compounds were found to be the compatible substrates as they could potentially be able to pass across the membrane using ATP-binding cassette (ABC) transporter. All substances tested as inhibitors of P-glycoproteins I and II were negative, except aspergilline E. The VDss assay is used to optimise the measurement of the total quantity of medications required for uniform distribution of pharmaceuticals throughout the blood. The assay values for all the drugs were less than 0.45 log L/kg, indicating that less medication volume will be required, but the values for the reference inhibitors GRL0617 and X77 were positive integer values. All the chemicals studied had negative CNS permeability values, indicating that they are projected to have decreased CNS permeability. Metabolism of the test drugs within the body was evaluated and except for asperphenalenone D and asperphenalenone A, which are anticipated to interact with CYP3A4, all other test chemicals were determined to be negative for all seven distinct models of metabolism. The total clearance and renal OCT2 assays were used to determine the drugs’ excretion from the body. All the test chemicals, including GRL0167, tested negative for the renal OCT2 assay, indicating that none of them can be eliminated by organic cation transporter 2, apart from X77. Out of all ten distinct models that were used to measure toxicity, AMES test revealed that all four test molecules were negative, indicating that they are not carcinogenic or mutagenic, although GRL0617 was reported positive for this test. For cytonic acid A and cytonic acid B, maximum recommended tolerated dose values were found to be highest, that is 0.44 and 0.467 log(mg/kg/day), respectively. Table 7 contains the values of the ADMET analysis and properties of the test compounds along with reference compounds GRL0617 and X77.
Table 7.
ADMET properties of screened antiviral compounds
| Property | Model name | Predicted values | Unit | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| GRL0617 | X77 | Cytonic acid A | Cytonic acid B | Aspergilline E | 4-Dehydroxyaltersolanol A | Asperphenalenone D | Asperphenalenone A | |||
| Absorption | Water solubility | − 4.678 | − 2.878 | − 2.902 | − 2.914 | − 2.639 | − 2.009 | − 3.495 | − 3.746 | Numeric (log mol/L) |
| Absorption | CaCO2 permeability | 1.302 | 1.29 | − 1.108 | − 1.17 | 0.697 | 0.633 | − 0.093 | − 0.172 | Numeric (log Papp in 10–6 cm/s) |
| Absorption | Intestinal absorption (human) | 92.815 | 89.686 | 46.99 | 62.692 | 56.317 | 67.204 | 76.573 | 79.961 | Numeric (% absorbed) |
| Absorption | Skin permeability | − 2.785 | − 2.735 | − 2.735 | − 2.735 | − 2.735 | − 2.848 | − 2.735 | − 2.735 | Numeric (log Kp) |
| Absorption | P-glycoprotein substrate | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Categorical (yes/no) |
| Absorption | P-glycoprotein I inhibitor | No | Yes | No | No | Yes | No | Yes | Yes | Categorical (yes/no) |
| Absorption | P-glycoprotein II inhibitor | Yes | Yes | No | No | No | No | Yes | Yes | Categorical (yes/no) |
| Distribution | VDss (human) | 0.086 | 0.656 | − 1.267 | − 1.707 | 0.431 | − 0.006 | 0.454 | − 0.357 | Numeric (log L/kg) |
| Distribution | Fraction unbound (human) | 0 | 0.262 | 0.08 | 0.006 | 0.373 | 0.413 | 0.056 | 0.013 | Numeric (Fu) |
| Distribution | BBB permeability | 0.055 | − 0.899 | − 1.896 | − 1.782 | − 0.969 | − 0.844 | − 1.67 | − 1.404 | Numeric (log BB) |
| Distribution | CNS permeability | − 1.604 | − 2.31 | − 3.143 | − 3.183 | − 3.826 | − 3.494 | − 3.054 | − 2.722 | Numeric (log PS) |
| Metabolism | CYP2D6 substrate | No | No | No | No | No | No | No | No | Categorical (yes/no) |
| Metabolism | CYP3A4 substrate | Yes | No | No | No | No | No | Yes | Yes | Categorical (yes/no) |
| Metabolism | CYP1A2 inhibitor | Yes | No | No | No | No | No | No | No | Categorical (yes/no) |
| Metabolism | CYP2C19 inhibitor | Yes | No | No | No | No | No | No | No | Categorical (yes/no) |
| Metabolism | CYP2C9 inhibitor | Yes | No | No | No | No | No | No | No | Categorical (yes/no) |
| Metabolism | CYP2D6 inhibitor | No | No | No | No | No | No | No | No | Categorical (yes/no) |
| Metabolism | CYP3A4 inhibitor | Yes | Yes | No | No | No | No | Yes | Yes | Categorical (yes/no) |
| Excretion | Total clearance | 0.221 | 0.671 | 0.268 | 0.183 | 0.143 | 0.453 | − 0.035 | 0.071 | Numeric (log ml/min/kg) |
| Excretion | Renal OCT2 substrate | No | Yes | No | No | No | No | No | No | Categorical (yes/no) |
| Toxicity | AMES toxicity | Yes | Yes | No | No | No | No | No | No | Categorical (yes/no) |
| Toxicity | Max. tolerated dose (human) | − 0.043 | 0.601 | 0.44 | 0.467 | − 0.645 | − 0.078 | 0.229 | 0.191 | Numeric (log mg/kg/day) |
| Toxicity | hERG I inhibitor | No | No | No | No | No | No | No | No | Categorical (yes/no) |
| Toxicity | hERG II inhibitor | Yes | Yes | No | No | No | No | Yes | Yes | Categorical (yes/no) |
| Toxicity | Oral rat acute toxicity (LD50) | 2.472 | 2.396 | 2.499 | 2.444 | 2.302 | 1.923 | 2.253 | 2.042 | Numeric (mol/kg) |
| Toxicity | Oral rat chronic toxicity (LOAEL) | 0.462 | 1.528 | 2.897 | 2.891 | 3.695 | 2.734 | 1.602 | 2.462 | Numeric (log mg/kg_bw/day) |
| Toxicity | Hepatotoxicity | No | Yes | No | No | No | No | No | No | Categorical (yes/no) |
| Toxicity | Skin sensitisation | No | No | No | No | No | No | No | No | Categorical (yes/no) |
| Toxicity | T. pyriformis toxicity | 0.529 | 0.285 | 0.285 | 0.285 | 0.285 | 0.287 | 0.285 | 0.286 | Numeric (log ug/L) |
| Toxicity | Minnow toxicity | 1.936 | 2.563 | 1.089 | 0.011 | 4.392 | 4.044 | − 2.806 | − 4.272 | Numeric (log mM) |
Molecular dynamic simulations
Two isomeric chemicals, cytonic acids A and B, previously reported as inhibitors of human cytomegalovirus protease (hCMV), were observed to produce docking scores and MM-GBSA energies for both PLpro and Mpro. As a result, both compounds were subjected to an MD simulations experiment to determine the stability of the protein–ligand combination for 100 ns. In addition to the test substance (cytonic acids A and B), simulations of GRL0617 with SARS-CoV-2-PLpro and X77 with SARS-CoV-2-Mpro were run as a control set for each protein.
The RMSD values for all the frames in the trajectory were determined once the MD simulation was completed. The RMSD value of any simulation, in general, indicates the changes during the simulation of specific atoms with respect to their original state. The docked pose of the protein–ligand complex was used as a baseline state or reference pose to compute the RMSD values. The RMSD value of protein atoms is represented by the Y-axis (left) in Figs. 5 and 6. The equilibration of the protein backbone for PLpro-GRL0617 (Fig. 5) occurs at an RMSD value of 2.3 ± 0.5 Å, whereas the equilibration of the protein backbone for Mpro-X77 (Fig. 6) occurs at an RMSD value of 2.2 ± 0.6 Å. For proteins that are bigger and non-globular in shape, the value might exceed beyond 4.0 Å. For the smaller and most globular proteins, RMSD value is ideally supposed to be in the limit of 1.0–4.0 Å. Here, the RMSD value after equilibration is not exceeding 4 Å for all the complexes under study at the peak end which is an advantageous parameter to screen the lead against the known ligand. For the complex of PLpro-cytonic acid A (Fig. 5), RMSD value remains within the accepted limits of 2.0 ± 0.5 Å considering the size of protein, whereas for PLpro-cytonic acid B, RMSD value is equilibrated at 2.3 ± 0.5 Å. Similarly for complexes Mpro-cytonic acid A and Mpro-cytonic acid B (Fig. 6), the protein backbone RMSD never exceeds 3.5 Å. In the graphs of Fig. 5 right Y-axis represents the ligand RMSD value. ‘Lig fit Prot’ signifies the RMSD values of ligand with reference to protein backbone. Throughout the whole simulation, ‘Lig fit Prot’ value of the PLpro-GRL0617 (Fig. 5) complex is in the range of 1.5 ± 0.5 Å, whereas for the PLpro-cytonic acid A complex this value is found to be 5.5 ± 1 Å. For, PLpro-cytonic acid B this value is 4.5 ± 1 Å which is slightly lower than that of cytonic acid A which suggests the relatively higher stability of the complex. On the other hand, ‘Lig fit Lig’ value of GRL0617 is lower than that of both cytonic acid isomers which suggests changes in the binding pose of ligand, but the difference is miniscule, hence negligible. Such results show that GRL0617 is having relatively higher stability compared to the tested ligand in binding pocket at the given docking pose. The ‘Lig fit Prot’ values for Mpro-cytonic acid A and Mpro-cytonic acid B the RMSD values peak at 6.5 Å and 7.3 Å, respectively (Fig. 6). For all the ligands, the ‘Lig fit Prot’ implies that the interactions between protein and ligand are quite stable. Additionally, Figs. 7, 8, 9, 10, 11 and 12 provide pictorial insights onto the entire simulation results based on collected trajectories and 10,000 captured frameshifts.
Fig. 5.
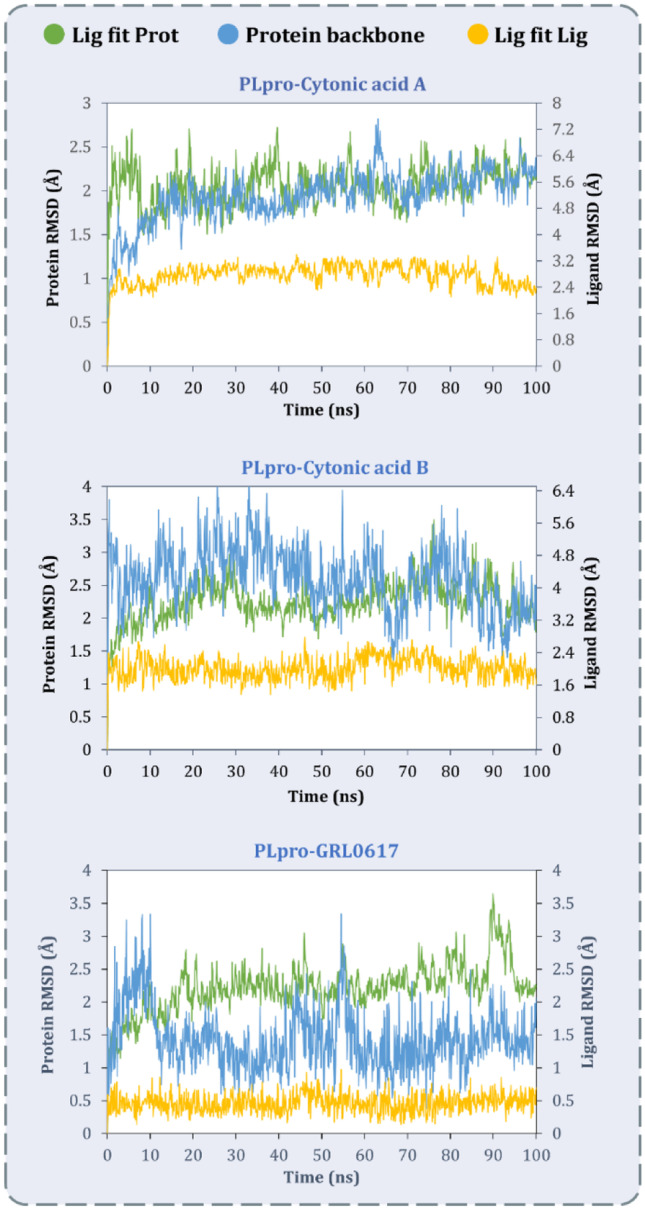
MD simulation protein–ligand interaction root-mean-square deviation (RMSD) profile of SARS-CoV-2-PLpro-cytonic acid A, SARS-CoV-2-PLpro-cytonic acid B and SARS-CoV-2-PLpro-GRL0617
Fig. 6.
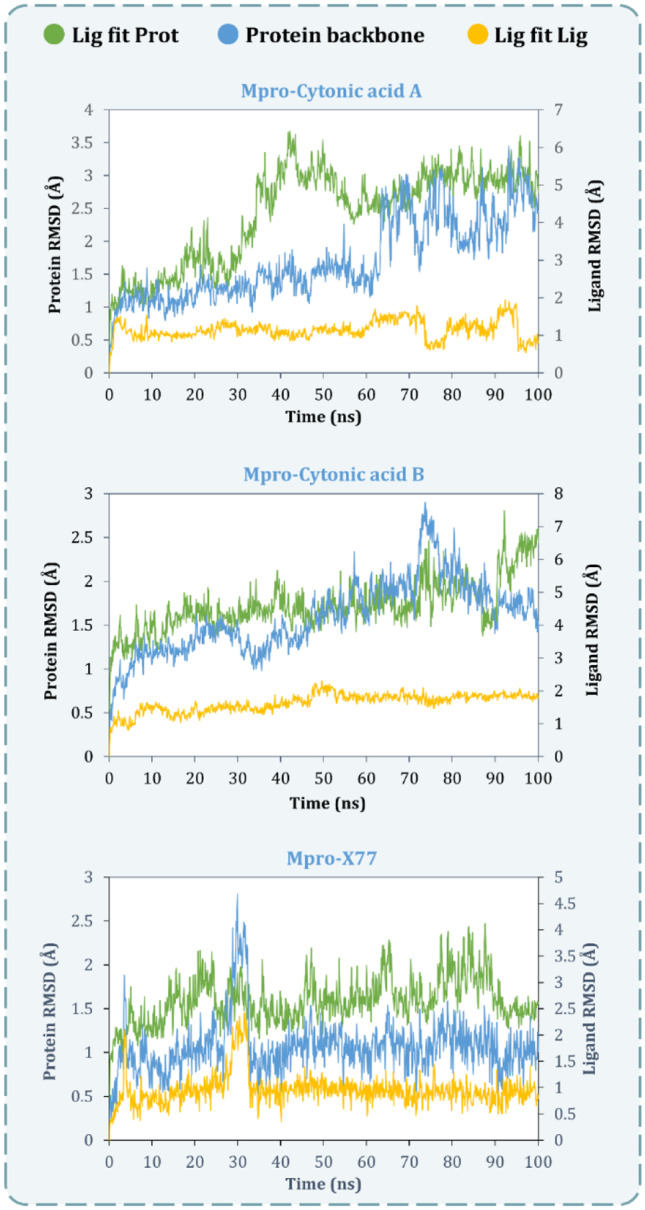
MD simulation protein–ligand interaction root-mean-square deviation (RMSD) profile of SARS-CoV-2-Mpro-cytonic acid A, SARS-CoV-2-Mpro-cytonic acid B and SARS-CoV-2-Mpro-X77
Fig. 7.

Protein–ligand interaction diagram for PLpro showing percent of total time involved for particular interaction during molecular dynamics
Fig. 8.

Protein–ligand interaction diagram for Mpro showing percent of total time involved for particular interaction during molecular dynamics
Fig. 9.

Protein–ligand interaction diagram showing interaction fraction of crucial interacting amino acids of PLpro
Fig. 10.

Protein–ligand interaction diagram showing interaction fraction of crucial interacting amino acids of Mpro
Fig. 11.

Timeline representation of the interactions of ligand with amino acids for the complexes SARS-CoV-2-PLpro-cytonic acid A, SARS-CoV-2-PLpro-cytonic acid B and SARS-CoV-2-PLpro-GRL0617
Fig. 12.

Timeline representation of the interactions of ligand with amino acids for the complexes SARS-CoV-2-Mpro-cytonic acid A, SARS-CoV-2-Mpro-cytonic acid B and SARS-CoV-2-Mpro-X77
Figure 7 represents the interactions made by reference ligand (GRL0617) and test compounds (cytonic acid A and cytonic acid B) with PLpro where the interaction types with percent interaction during 100-ns MD simulation run are represented. Similarly, for Mpro the interactions made by reference ligand (X77) and test compounds (cytonic acid A and cytonic acid B) are represented in Fig. 8. Figure 9 represents the interaction fraction of reference ligand (GRL0617) and test compounds (cytonic acid A and cytonic acid B) with PLpro while Fig. 10 represents the interaction fraction of reference ligand (X77) and test compounds (cytonic acid A and cytonic acid B) with Mpro. Hydrogen bonds, hydrophobic interactions such as Pi cation, pi-pi stacking, water bridges and ionic interactions made by ligand with amino acids of proteins during 100-ns MD simulation are represented in these figures. Amongst this variety of interactions, ionic interactions were found to be absent in both control and test ligand. For all the graphs in Figs. 9 and 10, the stacked bar charts are normalised over the course of the trajectory; for example, a value of 0.7 suggests that 70% of the simulation time of the specific interaction is maintained. Values over 1.0 are possible as some protein residue may make multiple contacts of same subtype with the ligand. All the bar charts in Figs. 9 and 10 suggest all the ligands (reference inhibitor and test compounds) form strong interactions with several amino acids of PLpro and Mpro respectively. At several instances, the interaction fraction shoots above the value of 0.8, which shows strong interaction of ligand with that amino acid.
Protein–ligand interaction timeline ligands interacting with PLpro and Mpro are represented in Figs. 11 and 12 respectively. These diagrams show the instance of ligand interacting with a particular amino acid with respect to time and intensity. For instance, in Fig. 11, GRL0617 interacts with Pro248, Tyr264, Try268 and Gln269 strongly and evenly through the 100 ns of simulation. Similarly, cytonic acid A and cytonic acid B also interact with various amino acids such as Glu161, Leu162, Asp164, Pro248, Tyr264, Tyr268, Gln269 and Tyr273 effectively. Similarly in Fig. 12, X77 effectively interacts with His41, Asn142, Gly143, His163 and Glu166 of Mpro. Here like PLpro, cytonic acid A and cytonic acid B interact with various amino acids of Mpro during the MD simulation. Thus, from the evidence of docking, MM-GBSA and MD simulations, it can be deduced that cytonic acid A and cytonic acid B might serve as dual inhibitor of PLpro and Mpro.
Discussion
Coronaviruses (CoVs) are diverse group of viruses that have potentials to infect various mammals including humans. These viruses have positive single strand of RNA as its genetic material. These CoVs have their structure architecture composed of proteins, namely, S-, E-, M- and N-protein. α, β, γ and δ are four genera into which these CoVs are distributed, whereby α and β are the ones that have nag of infecting humans as their S-protein has structural complementary with human’s ACE-2 receptor, enabling these viruses to attach human cell surface ACE-2 which is a vital interaction to cause infection [36–38]. Similarly, γ and δ CoVs have the tendency to infect Aves. As a reflection on the past, Middle East respiratory syndrome (MERS)-CoV and severe acute respiratory syndrome (SARS)-CoV deadly viruses belong to β genera of CoVs; also, the current pandemic causing SARS-CoV-2 belongs to β genera of CoVs. The recent variant Omicron that is ought to have very high infectivity is having high degree of mutations in S-protein which enables it to interact more efficiently with ACE-2; however, the target proteins used under present study, i.e. Mpro and PLpro, are identical with the previous variants of SARS-CoV-2; therefore, the research presented in this study is applicable to Omicron variant as well.
Aside from immunisation improvement with vaccine research, mainstream researchers have additionally given their energies and assets to distinguish intensifies that can counter the SARS-CoV-2 infection with alternative strategies [16, 39, 40]. Computationally driven molecular docking and MD recreations have been oftentimes used to identify compounds that possess potentials to interact with natural biochemistry of virus and halt its function [2, 41–43]. Assortment of metabolites as potential drugs from bacteria, moulds, yeast and plants has been investigated to restrain the urgent viral target proteins as their natural availability makes them unfathomably accessible. For example, pyranonigrin A, flaviolin and sterenin M are some secondary metabolites screened from moulds which by molecular docking and MD recreation studies with Mpro are recommended that they can restrain the viral replication by means of associating with this protein [44–46]. Besides, there are plethora of examinations successfully completed and published with in silico workflow utilizing sub-atomic powerful MD reenactment and docking assays that are very helpful in various different everyday issues of biological sciences [47–51]. Furthermore, these in silico methodologies have additionally been used for strength appraisal of hydroxychloroquine against different expected target proteins of SARS-CoV-2 [52]. In these computational examinations, the reasoning is extremely fundamental that in the event that any of these metabolites serving as potential drug leads can intrude with the viral proteins, they can thwart the typical life pattern of the viral particle [53–55].
Endophytic microbes are symbiotically associated with plants and have been proven to produce novel or analogues of host bioactive metabolites exhibiting a variety of biological activities including antiviral activity [56, 57]. However, compounds of specifically endophytic origin are not explored against SARS-CoV-2. In the current study, 45 compounds that have been reported active against viruses such as influenza, H1N1, HIV, HCV, hCMV and EV-71 were retrieved through literature and screened through rigorous computation workflow. Top compounds obtained after docking and MM-GBSA analysis for each protease (PLpro and Mpro) are known to have antiviral activity against different viruses. Top ligands for both the proteins in the present study, cytonic acids A and B, are known to possess in vitro inhibitory activities towards hCMV protease with the IC50 values of 43 µmol and 11 µmol, respectively. Both the cytonic acids were extracted from the fermented culture of endophytic fungus Cytonaema sp., which was isolated from the plant Quercus sp. (European oak) [58]. Other compounds interacting with Mpro are asperphenalenone A and asperphenalenone D, which were obtained from the ethyl acetate fraction of fermented culture of Aspergillus sp., an endophytic fungus isolated from the plant Kadsura longipedunculata. Asperphenalenone A and D displayed anti-HIV activity with IC50s of 4.5 and 2.4 µM, respectively [59]. Aspergilline E was extracted along with aspergillines A–D from Aspergillus versicolor, an endophyte isolated from the plant Paris polyphylla var. yunnanensis, collected from China. Aspergilline E showed weak antiviral activity towards tobacco mosaic virus with IC50 of 33.6 ± 3.0 μM [60]. In the present study, it interacts with the PLpro with multiple interaction types. Another compound having strong interaction with PLpro is 4-dehydroxyaltersolanol A, which was obtained from endophytic fungus Nigrospora sp. YE3033, harboured in Aconitum carmichaelii. 4-Dehydroxyaltersolanol A displayed in vitro antiviral activity towards H1N1 influenza A with IC50 of 8.35 ± 1.41 µg/mL [61]. With all these evidences, it is quiet insightful that cytonic acids A and B, asperphenalenone A, asperphenalenone D and aspergilline E have antiviral activities against different forms of viruses and could be of potential against SARS-CoV-2; of course, in vitro assays are needed to validate the findings, but such in silico study helps us to identify potential hits out of large pools of secondary metabolites and it is analogous of finding a purple wooden stick from the Amazon forest with ease.
The best docked compounds are cytonic acid A and cytonic acid B, which have significant interactions with both PLpro and Mpro proteases, according to the MD simulation studies. Considering the lack of treatment options for coronavirus infection, the current study proposes cytonic acids A and B as a dual inhibitor that interacts with critical amino acids in the binding site of PLpro and Mpro to inhibit their function, which could be useful in future in vitro and in vivo studies for COVID-19 therapeutics. Because we are dealing with infectious agents, in vitro validation of the suggested drugs mandates the use of Biosafety Level 4 (BSL4). Only an in silico investigation has been conducted. The information offered in this study will be beneficial to the scientific community. Even so, this work could potentially untap many more such exploratory work with endophytic compounds.
Acknowledgements
We acknowledge Education Department, Government of Gujarat, India, for providing research fellowship to Rohit Patel and Priyashi Rao under the ScHeme Of Developing High quality research (SHODH), and also, University Grants Commission (UGC), New Delhi, India, for providing the fellowship award of ‘CSIR-NET Junior Research Fellowship (JRF)’ to Jignesh Prajapati. The authors are thankful to the Department of Microbiology and Biotechnology, School of Sciences, Gujarat University, DST-FIST Sponsored Department, for providing necessary facilities to perform experiments. We acknowledge GSBTM, DST, Government of Gujarat, for providing Bioinformatics Node facility and Finishing School support. We acknowledge GUJCOST, DST, Government of Gujarat, for the provision of super-computing facility at the institute.
Author contribution
J.P. and D.G. conceived and designed the experiments; J.P., R.P. and P.R. performed the experiments; J.P., R.P. and D.G. analysed the data and wrote the manuscript; R.R. and M.S. supervised the research and revised the manuscript. All authors discussed the manuscript critically for important intellectual content and approved the final version of the manuscript.
Funding
This work was supported by Research Support Scheme (RSS) of Gujarat State Biotechnology Mission (GSBTM/JD(R&D)/610/20–21/365–371).
Availability of data and material
Library for the 3D structures of selected antiviral metabolites of endophyte origin is available from the corresponding author on reasonable request.
Prajapati, J., Patel, R., Rao, P., Saraf, M., Rawal, R. and Goswami, D. (2021). Perceiving SARS-CoV-2 Mpro and PLpro dual inhibitors from pool of recognized antiviral compounds of endophytic microbes: an in silico simulation study. The work has been submitted as a Preprint at: https://www.researchsquare.com/article/rs-829046/v1
Declarations
Conflict of interest
The authors declare no competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Rakesh Rawal, Email: rakeshrawal@gujaratuniversity.ac.in.
Dweipayan Goswami, Email: dweipayan.goswami@gujaratuniversity.ac.in.
References
- 1.Shukla A, Parmar P, Kapoor G, Goswami D, Jha CK, Patel B, Saraf M (2021) Curse of La Corona: unravelling the scientific and psychological conundrums of the 21st century pandemic. Mol Divers (0123456789). 10.1007/s11030-020-10167-2 [DOI] [PMC free article] [PubMed]
- 2.Fakhar Z, Khan S, AlOmar SY, Alkhuriji A, Ahmad A. ABBV-744 as a potential inhibitor of SARS-CoV-2 main protease enzyme against COVID-19. Sci Rep. 2021;11(1):1–15. doi: 10.1038/s41598-020-79918-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Filsinger M, Freitag M, Erhardt J, Wamsler S (2021) Rally around your fellows: information and social trust in a real-world experiment during the corona crisis. 10.1080/03623319.2021.1954463, 1–15. 10.1080/03623319.2021.1954463
- 4.Forni G, Mantovani A, Forni G, Mantovani A, Moretta L, Rappuoli R, Vineis P. COVID-19 vaccines: where we stand and challenges ahead. Cell Death Differ. 2021;28(2):626–639. doi: 10.1038/s41418-020-00720-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kyriakidis NC, López-Cortés A, González EV, Grimaldos AB, Prado EO (2021) SARS-CoV-2 vaccines strategies: a comprehensive review of phase 3 candidates. npj Vaccines 6(1). 10.1038/s41541-021-00292-w [DOI] [PMC free article] [PubMed]
- 6.Zahid MN, Moosa MS, Perna S, Buti EB. A review on COVID-19 vaccines: stages of clinical trials, mode of actions and efficacy. Arab Journal of Basic and Applied Sciences. 2021;28(1):225–233. doi: 10.1080/25765299.2021.1903144. [DOI] [Google Scholar]
- 7.Kannan SR, Spratt AN, Cohen AR, Naqvi SH, Chand HS, Quinn TP, Singh K. Evolutionary analysis of the delta and Delta Plus variants of the SARS-CoV-2 viruses. J Autoimmun. 2021;124(July):102715. doi: 10.1016/j.jaut.2021.102715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang P, Nair MS, Liu L, Iketani S, Luo Y, Guo Y, Ho DD (2021) Antibody resistance of SARS-CoV-2 variants B.1.351 and B.1.1.7. Nature 593(7857):130–135. 10.1038/s41586-021-03398-2 [DOI] [PubMed]
- 9.Gajjar ND, Dhameliya TM, Shah GB (2021) In search of RdRp and Mpro inhibitors against SARS CoV-2: molecular docking, molecular dynamic simulations and ADMET analysis. J Mol Struct 1239. 10.1016/J.MOLSTRUC.2021.130488 [DOI] [PMC free article] [PubMed]
- 10.Silva LR, da Silva Santos-Júnior PF, de Andrade Brandão J, Anderson L, Bassi ÊJ, Xavier de Araújo-Júnior J, da Silva-Júnior EF (2020) Druggable targets from coronaviruses for designing new antiviral drugs. Bioorg Med Chem 28(22). 10.1016/J.BMC.2020.115745 [DOI] [PMC free article] [PubMed]
- 11.Nogara PA, Omage FB, Bolzan GR, Delgado CP, Aschner M, Orian L, Rocha JBT. In silico studies on the interaction between Mpro and PLpro from SARS-CoV-2 and ebselen, its metabolites and derivatives. Mol Inf. 2021;40(8):2100028. doi: 10.1002/MINF.202100028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Djokovic N, Ruzic D, Djikic T, Cvijic S, Ignjatovic J, Ibric S, Nikolic K. An integrative in silico drug repurposing approach for identification of potential inhibitors of SARS-CoV-2 main protease. Mol Inf. 2021;40(5):2000187. doi: 10.1002/MINF.202000187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thomford NE, Senthebane DA, Rowe A, Munro D, Seele P, Maroyi A, Dzobo K (2018) Natural products for drug discovery in the 21st century: innovations for novel drug discovery. Int J Mol Sci 19(6). 10.3390/ijms19061578 [DOI] [PMC free article] [PubMed]
- 14.Da A, Antonio S, Silveira L, Wiedemann M, Florêncio V, Florêncio Veiga-Junior F. Natural products’ role against COVID-19. RSC Adv. 2020;10(39):23379–23393. doi: 10.1039/D0RA03774E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Arora D, Sharma N, Singamaneni V, Sharma V, Kushwaha M, Abrol V, Gupta P. Isolation and characterization of bioactive metabolites from Xylaria psidii, an endophytic fungus of the medicinal plant Aegle marmelos and their role in mitochondrial dependent apoptosis against pancreatic cancer cells. Phytomedicine. 2016;23(12):1312–1320. doi: 10.1016/j.phymed.2016.07.004. [DOI] [PubMed] [Google Scholar]
- 16.Messaoudi O, Gouzi H, El-Hoshoudy AN, Benaceur F, Patel C, Goswami D, Bendahou M. Berries anthocyanins as potential SARS-CoV–2 inhibitors targeting the viral attachment and replication; molecular docking simulation. Egypt J Pet. 2021;30(1):33–43. doi: 10.1016/j.ejpe.2021.01.001. [DOI] [Google Scholar]
- 17.Rao P, Patel R, Shukla A, Parmar P, Rawal RM, Saraf M (2021) Identifying structural – functional analogue of GRL0617, the only well ‑ established inhibitor for papain ‑ like protease ( PLpro ) of SARS ‑ CoV2 from the pool of fungal metabolites using docking and molecular dynamics simulation. Mol Divers (0123456789). 10.1007/s11030-021-10220-8 [DOI] [PMC free article] [PubMed]
- 18.Dhameliya TM, Nagar PR, Gajjar ND (2022) Systematic virtual screening in search of SARS CoV-2 inhibitors against spike glycoprotein: pharmacophore screening, molecular docking, ADMET analysis and MD simulations. Mol Divers. 10.1007/S11030-022-10394-9 [DOI] [PMC free article] [PubMed]
- 19.Nagar PR, Gajjar ND, Dhameliya TM (2021) In search of SARS CoV-2 replication inhibitors: virtual screening, molecular dynamics simulations and ADMET analysis. J Mol Struct 1246. 10.1016/J.MOLSTRUC.2021.131190 [DOI] [PMC free article] [PubMed]
- 20.Hawas UW, El-Beih AA, El-Halawany AM. Bioactive anthraquinones from endophytic fungus Aspergillus versicolor isolated from red sea algae. Arch Pharmacal Res. 2012;35(10):1749–1756. doi: 10.1007/s12272-012-1006-x. [DOI] [PubMed] [Google Scholar]
- 21.Yang ZJ, Zhang YF, Wu K, Xu YX, Meng XG, Jiang ZT, Shao L (2020) New azaphilones, phomopsones A-C with biological activities from an endophytic fungus Phomopsis sp. CGMCC No.5416. Fitoterapia 145(March):104573. 10.1016/j.fitote.2020.104573 [DOI] [PubMed]
- 22.Hawas UW, Al-Farawati R, El-Kassem LTA, Turki AJ (2016) Different culture metabolites of the Red Sea fungus Fusarium equiseti optimize the inhibition of hepatitis C virus NS3/4A protease (HCV PR). Mar Drugs 14(10). 10.3390/md14100190 [DOI] [PMC free article] [PubMed]
- 23.Arunpanichlert J, Rukachaisirikul V, Sukpondma Y, Phongpaichit S, Tewtrakul S, Rungjindamai N, Sakayaroj J. Azaphilone and isocoumarin derivatives from the endophytic fungus Penicillium sclerotiorum PSU-A13. Chem Pharm Bull. 2010;58(8):1033–1036. doi: 10.1248/cpb.58.1033. [DOI] [PubMed] [Google Scholar]
- 24.Kumar BK, Faheem Sekhar KVGC, Ojha R, Prajapati VK, Pai A, Murugesan S (2020) Pharmacophore based virtual screening, molecular docking, molecular dynamics and MM-GBSA approach for identification of prospective SARS-CoV-2 inhibitor from natural product databases. J Biomol Struct Dyn 0(0):1–24. 10.1080/07391102.2020.1824814 [DOI] [PMC free article] [PubMed]
- 25.Rao P, Patel R, Shukla A, Parmar P, Rawal RM, Saraf M, Goswami D. Identifying structural–functional analogue of GRL0617, the only well-established inhibitor for papain-like protease (PLpro) of SARS-CoV2 from the pool of fungal metabolites using docking and molecular dynamics simulation. Mol Diversity. 2021 doi: 10.1007/s11030-021-10220-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fu Z, Huang B, Tang J, Liu S, Liu M, Ye Y, Huang H (2021) The complex structure of GRL0617 and SARS-CoV-2 PLpro reveals a hot spot for antiviral drug discovery. Nat Commun 12(1):1–12. 10.1038/s41467-020-20718-8 [DOI] [PMC free article] [PubMed]
- 27.Madhavi Sastry G, Adzhigirey M, Day T, Annabhimoju R, Sherman W. Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J Comput Aided Mol Des. 2013;27(3):221–234. doi: 10.1007/s10822-013-9644-8. [DOI] [PubMed] [Google Scholar]
- 28.Leach AR, Gillet VJ, Lewis RA, Taylor R. Three-dimensional pharmacophore methods in drug discovery. J Med Chem. 2010;53(2):539–558. doi: 10.1021/JM900817U. [DOI] [PubMed] [Google Scholar]
- 29.Sastry GM, Adzhigirey M, Sherman W. Protein and ligand preparation : parameters, protocols, and influence on virtual screening enrichments. J Comput Aided Mol Des. 2013;27:221–234. doi: 10.1007/s10822-013-9644-8. [DOI] [PubMed] [Google Scholar]
- 30.Dixon SL, Smondyrev AM, Rao SN. PHASE: a novel approach to pharmacophore modeling and 3D database searching. Chem Biol Drug Des. 2006;67(5):370–372. doi: 10.1111/J.1747-0285.2006.00384.X. [DOI] [PubMed] [Google Scholar]
- 31.Wang W, Donini O, Reyes CM, Kollman PA. Biomolecular simulations: recent developments in force fields, simulations of enzyme catalysis, protein-ligand, protein-protein, and protein-nucleic acid noncovalent interactions. Annu Rev Biophys Biomol Struct. 2001 doi: 10.1146/annurev.biophys.30.1.211. [DOI] [PubMed] [Google Scholar]
- 32.Wang J, Hou T, Xu X. Recent advances in free energy calculations with a combination of molecular mechanics and continuum models. Current Computer Aided-Drug Design. 2006;2(3):287–306. doi: 10.2174/157340906778226454. [DOI] [Google Scholar]
- 33.Kollman PA, Massova I, Reyes C, Kuhn B, Huo S, Chong L, Cheatham TE. Calculating structures and free energies of complex molecules: combining molecular mechanics and continuum models. Acc Chem Res. 2000;33(12):889–897. doi: 10.1021/ar000033j. [DOI] [PubMed] [Google Scholar]
- 34.Massova I, Kollman PA. Combined molecular mechanical and continuum solvent approach (MM- PBSA/GBSA) to predict ligand binding. Perspect Drug Discovery Des. 2000 doi: 10.1023/A:1008763014207. [DOI] [Google Scholar]
- 35.Pires DEV, Blundell TL, Ascher DB. pkCSM: Predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J Med Chem. 2015;58(9):4066–4072. doi: 10.1021/acs.jmedchem.5b00104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ren Z, Yan L, Zhang N, Guo Y, Yang C, Lou Z, Rao Z. The newly emerged SARS-Like coronavirus HCoV-EMC also has an “Achilles’ heel”: current effective inhibitor targeting a 3C-like protease. Protein Cell. 2013;4(4):248–250. doi: 10.1007/s13238-013-2841-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kumar A, Mehta V, Raj U, Varadwaj PK, Udayabanu M, Yennamalli RM, Singh TR. Computational and in-vitro validation of natural molecules as potential acetylcholinesterase inhibitors and neuroprotective agents. Curr Alzheimer Res. 2018;16(2):116–127. doi: 10.2174/1567205016666181212155147. [DOI] [PubMed] [Google Scholar]
- 38.Yang H, Bartlam M, Rao Z. Drug design targeting the main protease, the Achilles heel of coronaviruses. Curr Pharm Des. 2006;12(35):4573–4590. doi: 10.2174/138161206779010369. [DOI] [PubMed] [Google Scholar]
- 39.Poustforoosh A, Hashemipour H, Tüzün B, Pardakhty A, Mehrabani M, Nematollahi MH. Evaluation of potential anti-RNA-dependent RNA polymerase (RdRP) drugs against the newly emerged model of COVID-19 RdRP using computational methods. Biophys Chem. 2021;272(January):106564. doi: 10.1016/j.bpc.2021.106564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Patel CN, Kumar SP, Pandya HA, Rawal RM. Identification of potential inhibitors of coronavirus hemagglutinin-esterase using molecular docking, molecular dynamics simulation and binding free energy calculation. Mol Diversity. 2021;25(1):421–433. doi: 10.1007/s11030-020-10135-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Guedes IA, Costa LSC, dos Santos KB, Karl ALM, Rocha GK, Teixeira IM, Dardenne LE. Drug design and repurposing with DockThor-VS web server focusing on SARS-CoV-2 therapeutic targets and their non-synonym variants. Sci Rep. 2021;11(1):1–20. doi: 10.1038/s41598-021-84700-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Basu A, Sarkar A, Maulik U. Molecular docking study of potential phytochemicals and their effects on the complex of SARS-CoV2 spike protein and human ACE2. Sci Rep. 2020;10(1):1–15. doi: 10.1038/s41598-020-74715-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Murugan NA, Kumar S, Jeyakanthan J, Srivastava V. Searching for target-specific and multi-targeting organics for Covid-19 in the Drugbank database with a double scoring approach. Sci Rep. 2020;10(1):1–16. doi: 10.1038/s41598-020-75762-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rao P, Shukla A, Parmar P, Rawal RM, Patel BV, Saraf M, Goswami D (2020) Proposing a fungal metabolite-flaviolin as a potential inhibitor of 3CLpro of novel coronavirus SARS-CoV-2 identified using docking and molecular dynamics. J Biomol Struct Dyn 0(0):1–13. 10.1080/07391102.2020.1813202 [DOI] [PubMed]
- 45.Prajapati J, Patel R, Goswami D, Saraf M, Rawal RM. Sterenin M as a potential inhibitor of SARS-CoV-2 main protease identified from MeFSAT database using molecular docking, molecular dynamics simulation and binding free energy calculation. Comput Biol Med. 2021;135:104568. doi: 10.1016/j.compbiomed.2021.104568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rao P, Shukla A, Parmar P, Rawal RM, Patel B, Saraf M, Goswami D. Reckoning a fungal metabolite, pyranonigrin A as a potential Main protease (Mpro) inhibitor of novel SARS-CoV-2 virus identified using docking and molecular dynamics simulation. Biophys Chem. 2020;264:106425. doi: 10.1016/j.bpc.2020.106425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ton A-T, Gentile F, Hsing M, Ban F, Cherkasov A (2020) Rapid identification of potential inhibitors of SARS-CoV-2 main protease by deep docking of 1.3 billion compounds. Mol Inf 39(8):2000028. 10.1002/MINF.202000028 [DOI] [PMC free article] [PubMed]
- 48.Alves VM, Bobrowski T, Melo-Filho CC, Korn D, Auerbach S, Schmitt C, Tropsha A. QSAR modeling of SARS-CoV Mpro inhibitors identifies sufugolix, cenicriviroc, proglumetacin, and other drugs as candidates for repurposing against SARS-CoV-2. Mol Inf. 2021;40(1):2000113. doi: 10.1002/MINF.202000113. [DOI] [PubMed] [Google Scholar]
- 49.Cavasotto CN, Di Filippo JI. In silico drug repurposing for COVID-19: targeting SARS-CoV-2 proteins through docking and consensus ranking. Mol Inf. 2021;40(1):1–8. doi: 10.1002/minf.202000115. [DOI] [PubMed] [Google Scholar]
- 50.Shukla A, Parmar P, Patel B, Goswami D, Saraf M. Breaking bad: better call gingerol for improving antibiotic susceptibility of Pseudomonas aeruginosa by inhibiting multiple quorum sensing pathways. Microbiol Res. 2021;252:126863. doi: 10.1016/J.MICRES.2021.126863. [DOI] [PubMed] [Google Scholar]
- 51.Rao P, Goswami D, Rawal RM. Revealing the molecular interplay of curcumin as Culex pipiens acetylcholine esterase 1 (AChE1) inhibitor. Sci Rep. 2021;11(1):1–18. doi: 10.1038/s41598-021-96963-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mukherjee S, Dasgupta S, Adhikary T, Adhikari U, Panja SS (2020) Structural insight to hydroxychloroquine-3C-like proteinase complexation from SARS-CoV-2: inhibitor modelling study through molecular docking and MD-simulation study. J Biomol Struct Dyn 1–13. 10.1080/07391102.2020.1804458 [DOI] [PMC free article] [PubMed]
- 53.Parmar P, Shukla A, Rao P, Saraf M, Patel B, Goswami D. The rise of gingerol as anti-QS molecule: darkest episode in the LuxR-mediated bioluminescence saga. Bioorg Chem. 2020;99(March):103823. doi: 10.1016/j.bioorg.2020.103823. [DOI] [PubMed] [Google Scholar]
- 54.Pandya PN, Kumar SP, Bhadresha K, Patel CN, Patel SK, Rawal RM, Mankad AU. Identification of promising compounds from curry tree with cyclooxygenase inhibitory potential using a combination of machine learning, molecular docking, dynamics simulations and binding free energy calculations. Mol Simul. 2020;46(11):812–822. doi: 10.1080/08927022.2020.1764552. [DOI] [Google Scholar]
- 55.Goswami D. Comparative assessment of RNA-dependent RNA polymerase (RdRp) inhibitors under clinical trials to control SARS-CoV2 using rigorous computational workflow. RSC Adv. 2021;11(46):29015–29028. doi: 10.1039/d1ra04460e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Prajapati J, Goswami D, Rawal RM (2021) Endophytic fungi: a treasure trove of novel anticancer compounds. Current Research in Pharmacology and Drug Discovery 100050. 10.1016/j.crphar.2021.100050 [DOI] [PMC free article] [PubMed]
- 57.Manganyi MC, Ateba CN. Untapped potentials of endophytic fungi: a review of novel bioactive compounds with biological applications. Microorganisms. 2020;8(12):1–25. doi: 10.3390/microorganisms8121934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Guo B, Dai JR, Ng S, Huang Y, Leong C, Ong W, Carté BK. Cytonic acids A and B: novel tridepside inhibitors of hCMV protease from the endophytic fungus Cytonaema species. J Nat Prod. 2000;63(5):602–604. doi: 10.1021/np990467r. [DOI] [PubMed] [Google Scholar]
- 59.Pang X, Zhao JY, Fang XM, Zhang T, Zhang DW, Liu HY, Yu LY (2017) Metabolites from the plant endophytic fungus Aspergillus sp. CPCC 400735 and their anti-HIV activities. J Nat Prod 80(10):2595–2601. 10.1021/acs.jnatprod.6b00878 [DOI] [PubMed]
- 60.Zhou M, Miao M-M, Du G, Li X-N, Shang S-Z, Zhao W, Gao X-M (2014) Aspergillines A−E, highly oxygenated hexacyclic indole−tetrahydrofuran−tetramic acid derivatives from Aspergillus versicolor. 10.1021/ol502307u [DOI] [PubMed]
- 61.Zhang SP, Huang R, Li FF, Wei HX, Fang XW, Xie XS, He J. Antiviral anthraquinones and azaphilones produced by an endophytic fungus Nigrospora sp. from Aconitum carmichaeli. Fitoterapia. 2016;112:85–89. doi: 10.1016/j.fitote.2016.05.013. [DOI] [PubMed] [Google Scholar]
- 62.Qin C, Lin X, Lu X, Wan J, Zhou X, Liao S, Liu Y (2014) Sesquiterpenoids and xanthones derivatives produced by sponge-derived fungus Stachybotry sp. HH1 ZSDS1F1–2. The Journal of Antibiotics advance online publication. 10.1038/ja.2014.97 [DOI] [PubMed]
- 63.Zhang G, Sun S, Zhu T, Lin Z, Gu J, Li D, Gu Q (2011) Antiviral isoindolone derivatives from an endophytic fungus Emericella sp. associated with Aegiceras corniculatum. Phytochemistry 72(11–12):1436–1442. 10.1016/j.phytochem.2011.04.014 [DOI] [PubMed]
- 64.Liu L, Liu S, Jiang L, Chen X, Guo L, Che Y. Chloropupukeananin, the first chlorinated pupukeanane derivative, and its precursors from Pestalotiopsis fici. Org Lett. 2008;10(7):1397–1400. doi: 10.1021/ol800136t. [DOI] [PubMed] [Google Scholar]
- 65.Zhang D, Tao X, Liu J, Chen R, Zhang M, Li L, Dai J (2016) Periconiasin G, a new cytochalasan with unprecedented 7/6/5 tricyclic ring system from the endophytic fungus Periconia sp. 10.1016/j.tetlet.2016.01.030
- 66.Bashyal BP, Wellensiek BP, Ramakrishnan R, Faeth SH, Ahmad N, Gunatilaka AAL. Altertoxins with potent anti-HIV activity from Alternaria tenuissima QUE1Se, a fungal endophyte of Quercus emoryi. Bioorg Med Chem. 2014;22(21):6112–6116. doi: 10.1016/j.bmc.2014.08.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ding L, Münch J, Goerls H, Maier A, Fiebig HH, Lin WH, Hertweck C. Xiamycin, a pentacyclic indolosesquiterpene with selective anti-HIV activity from a bacterial mangrove endophyte. Bioorg Med Chem Lett. 2010;20(22):6685–6687. doi: 10.1016/j.bmcl.2010.09.010. [DOI] [PubMed] [Google Scholar]
- 68.Yu G, Zhou G, Zhu M, Wang W, Zhu T, Gu Q, Li D. Neosartoryadins A and B, fumiquinazoline alkaloids from a mangrove-derived fungus Neosartorya udagawae HDN13-313. Org Lett. 2016;18(2):244–247. doi: 10.1021/acs.orglett.5b02964. [DOI] [PubMed] [Google Scholar]
- 69.Ding J, Zhao J, Yang Z, Ma L, Mi Z, Wu Y, Cen S. Microbial natural product alternariol 5-O-methyl ether inhibits HIV-1 integration by blocking nuclear import of the pre-integration complex. Viruses. 2017;9(5):1–14. doi: 10.3390/v9050105. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Library for the 3D structures of selected antiviral metabolites of endophyte origin is available from the corresponding author on reasonable request.
Prajapati, J., Patel, R., Rao, P., Saraf, M., Rawal, R. and Goswami, D. (2021). Perceiving SARS-CoV-2 Mpro and PLpro dual inhibitors from pool of recognized antiviral compounds of endophytic microbes: an in silico simulation study. The work has been submitted as a Preprint at: https://www.researchsquare.com/article/rs-829046/v1