

Abstract
DSP‐7888 is an immunotherapeutic cancer vaccine derived from the Wilms’ tumor gene 1 (WT1) protein. This phase 1/2 open‐label study evaluated the safety and efficacy of DSP‐7888 dosing emulsion in patients with myelodysplastic syndromes (MDS). DSP‐7888 was administered intradermally (3.5 or 10.5 mg) every 2 weeks for 6 months and then every 2‐4 weeks until lack of benefit. Twelve patients were treated in phase 1 (3.5 mg, n = 6; 10.5 mg, n = 6), with no dose‐limiting toxicities reported. Thus, the 10.5 mg dose was selected as the recommended phase 2 dose, and 35 patients were treated in phase 2. Forty‐seven patients received ≥1 dose of the study drug and comprised the safety analysis set. The most common adverse drug reaction (ADR) was injection site reactions (ISR; 91.5%). Grade 3 ISR were common (58.8%) in phase 1 but occurred less frequently in 2 (22.9%) following implementation of risk minimization strategies. Other common ADR were pyrexia (10.6%) and febrile neutropenia (8.5%). In the efficacy analysis set, comprising patients with higher‐risk MDS after azacitidine failure in phases 1 and 2 (n = 42), the disease control rate was 19.0%, and the median overall survival (OS) was 8.6 (90% confidence interval [CI], 6.8‐10.3) months. Median OS was 10.0 (90% CI, 7.6‐11.4) months in patients with a WT1‐specific immune response (IR; n = 33) versus 4.1 (90% CI, 2.3‐8.1) months in those without a WT1‐specific IR (n = 9; P = .0034). The acceptable safety and clinical activity findings observed support the continued development of DSP‐7888 dosing emulsion.
Keywords: DSP‐7888, high‐risk myelodysplastic syndrome, myelodysplastic syndromes, Wilms’ tumor gene 1, WT1 peptide vaccine
Treatment options for patients with MDS after failure of hypomethylating agents, such as azacitidine (AZA), represent a significant therapeutic challenge. The prognosis for these patients is particularly poor. This phase 1/2 open‐label study demonstrated the safety and efficacy of DSP‐7888 dosing emulsion, an immunotherapeutic cancer vaccine derived from the WT1 protein, in patients with higher‐risk MDS after AZA treatment.

1. INTRODUCTION
Myelodysplastic syndromes (MDS) encompass a heterogeneous group of closely related diseases of pluripotent hematopoietic stem cells, characterized by peripheral blood cytopenia due to ineffective hematopoiesis. 1 The clinical course of MDS is extremely variable and may be indolent or rapidly progressive, with marked symptom burden and transformation into acute myeloid leukemia (AML). 2 , 3 In Japan, the estimated incidence of MDS is 3.8 cases per 100 000 for men and 2.4 cases per 100 000 for women. 4 However, incidence increases sharply with age, particularly in those aged over 70 years. 4
Therapeutic options for MDS are limited and are largely based on a patient’s age and prognosis, as determined by the International Prognostic Scoring System (IPSS) and the revised IPSS (IPSS‐R). 5 , 6 Allogeneic hematopoietic stem cell transplantation (allo‐HSCT) represents the only potentially curative option; however, most patients are not suitable candidates due to their advanced age. 7 For decades, the mainstay of treatment has been supportive care, including blood transfusions and hematopoietic factors, aimed at prolonging survival and improving quality of life. 8 The immunosuppressant agent lenalidomide and hypomethylating agents (HMA), including azacitidine (AZA) and decitabine, are approved for treatment of MDS globally. 9 , 10 Lenalidomide is approved for a small subset of MDS patients with chromosome 5q deletion. 9 In April 2020, luspatercept was approved by the US Food and Drug Administration for patients with very low to intermediate (Int) risk MDS. 11 AZA and decitabine have demonstrated improved response rates and prolonged time to AML transformation and survival compared to conventional care in randomized phase 3 trials, 12 , 13 , 14 and both are indicated in higher‐risk patients, although decitabine is not approved in Japan. 15 , 16 Approximately 40% of patients fail to respond to HMA, 17 and most responders experience disease relapse within 2 years. 13 Prognosis after relapse is particularly poor, with limited treatment options and median overall survival (OS) of less than 6 months. 18 , 19 Treatment of MDS after failure of HMA represents a significant therapeutic challenge.
Wilms’ tumor gene 1 (WT1) was originally isolated as a tumor suppressor gene based on its activity in Wilms’ tumors, 20 with later studies revealing its possible role as an oncogene. 21 , 22 , 23 , 24 Further, WT1 is overexpressed in hematological malignancies and solid tumors but minimally expressed in normal tissues and cells, making it a promising cancer vaccine target. 24 , 25 Our earlier generation WT1 peptide cancer vaccine, WT4869, consists of the modified version of WT1235‐243, 2M→Y peptide (comprising amino acids 235 to 243) derived from the WT1 gene product that is restricted to the human leukocyte antigen (HLA)‐A*24:02, present in approximately 60% of the Japanese population. In these patients, WT1235‐243, 2M→Y can induce WT1‐specific cytotoxic T lymphocytes (CTL). 26 , 27 WT1235‐243, 2M→Y is WT1235‐243 with the substitution of M with Y at the second amino acid position. Compared to natural WT1235‐243, WT1235‐243, 2M→Y has a higher binding affinity for HLA‐A*24:02 and induces WT1‐specific cytotoxic T lymphocytes (CTLs) more effectively. 28 , 29 , 30
DSP‐7888 is an investigational immunotherapeutic cancer vaccine comprising two WT1‐protein derived peptides, DSP‐7888‐K and DSP‐7888‐H. DSP‐7888‐K is a synthetic peptide containing WT1126‐134 and WT1235‐243, 2M→ Y , which has been modified by conversion of methionine to tyrosine at the second amino acid position. DSP‐7888‐H is a synthetic peptide with the same sequence as the naturally occurring peptide WT134‐51. DSP‐7888‐K elicits WT1‐specific CD8+ T cells targeting HLA‐A*02:01, HLA‐A*02:06, or HLA‐A*24:02. DSP‐7888‐H elicits WT1‐specific helper T cells in some subtypes of HLA‐DRB1, HLA‐DPB1, and HLA‐DQB1, 31 which enhance induction of WT1‐specific CD8+ T cells by DSP‐7888. Preliminary clinical activity has been shown with our earlier generation WT1 peptide cancer vaccine, WT4869, in a phase 1/2 study in patients with HLA‐A*24:02‐positive MDS, including those with higher‐risk (IPSS score ≥1.5) or lower‐risk (score <1.5) disease who were red blood cell transfusion‐dependent. 32 We hereby report the results of the phase 1/2 study evaluating the safety and efficacy of the peptide vaccine, DSP‐7888, in patients with MDS.
2. MATERIALS AND METHODS
2.1. Study design
This was an open‐label, single‐arm, multicenter, phase 1/2 study conducted in Japan (ClinicalTrials.gov: NCT02436252). The objective of phase 1 was to evaluate the safety and tolerability of DSP‐7888 dosing emulsion (hereafter referred to as “DSP‐7888”) in patients with MDS and determine the maximum tolerated dose (MTD) and recommended phase 2 dose (RP2D). Preliminary activity and potential biomarkers were also explored. The objective of phase 2 was to evaluate the safety and efficacy of DSP‐7888 in patients with higher‐risk MDS (IPSS score ≥1.5 + IPSS score <1.5 disease with myeloblasts ≥5%) after AZA failure using the RP2D. For the purposes of this study, AZA failure was defined as non–response to therapy (ie primary failure), loss of response (ie secondary failure), or intolerance due to adverse events (AE) as per Prébet et al (2011). 19 The IPSS score was evaluated at enrollment (ie following AZA failure) in this study. Potential biomarkers of safety and efficacy were also explored.
The phase 1 dose‐escalation part of the study involved a conventional 3 + 3 design, with sequential cohorts of three to six patients enrolled to receive DSP‐7888 at two dose levels (3.5; 10.5 mg). DSP‐7888 was administered as DSP‐7888 dosing emulsion (water in oil) containing DSP‐7888‐K, DSP‐7888‐H, and the adjuvant MONTANIDE ISA 51 VG, via intradermal injection (3.5 mg: two sites; 10.5 mg: six sites), with dosing repeated every 2 weeks for 6 months, followed by every 2‐4 weeks until lack of clinical benefit. For the 3.5 mg dose, the DSP‐7888 dosing emulsion included DSP‐7888‐K 2 mg and DSP‐7888‐H 1.5 mg in 200 μL of emulsion, administered across two sites. For the 10.5 mg dose, the 3.5 mg DSP‐7888 dosing emulsion was administered across six body sites, (ie a total DSP‐7888‐K dose of 6 mg and DSP‐7888‐H dose of 4.5 mg in 600 μL of emulsion).
During the dose‐limiting toxicity (DLT) evaluation period (ie within 29 days after the first DSP‐7888 dose), dose reductions were permitted in patients experiencing a DLT if the investigator deemed that continuation of DSP‐7888 would provide a potential benefit. After the DLT evaluation period in phase 1 or 2, dose reductions were permitted in patients experiencing an adverse drug reaction (ADR) that met the DLT criteria but did not meet discontinuation criteria (ie patient withdrawal, AE, and other reasons). Dose reductions were also permitted in patients experiencing an injection‐site reaction (ISR) if there was a concern that the reaction may worsen.
The study was conducted in accordance with the ethical principles of the Declaration of Helsinki, the International Council for Harmonisation Harmonised Tripartite Guideline for Good Clinical Practice, and the Institutional Review Board (IRB) regulations and following the applicable local regulatory requirements. The clinical study protocol, the investigator’s brochure, informed consent forms, and other study‐related documents were reviewed and approved by the IRB of all study sites. All patients provided written informed consent to participate in the study.
2.2. Patients
For phase 1 of the study, patients aged ≥20 years with a confirmed diagnosis of MDS (according to World Health Organization [WHO] 4th edition or French‐American‐British [FAB] classification, 33 , 34 excluding patients with chronic myelomonocytic leukemia [CMML] or refractory anemia with excess blasts in transformation [RAEB‐t]) and high‐risk (IPSS score ≥1.5) or low‐risk (IPSS score <1.5) disease requiring treatment other than supportive treatment were eligible for inclusion irrespective of previous AZA treatment.
For phase 2, patients aged ≥20 years with a confirmed diagnosis of MDS (according to WHO 4th edition or FAB classification [including CMML and RAEB‐t]) 33 , 34 and high‐risk (IPSS score ≥1.5) or low‐risk (IPSS score <1.5) disease with myeloblasts ≥5% were eligible if they had ≥1 previous cycles of AZA treatment with no subsequent treatment after AZA.
Other major inclusion criteria for both parts were a white blood cell count ≤12 000/mm3, an Eastern Cooperative Oncology Group Performance Status (ECOG PS) score of 0‐2, and HLA‐A*24:02, HLA‐A*02:01, or HLA‐A*02:06 positive.
Key exclusion criteria included: patients undergoing allo‐HSCT, those with concurrent autoimmune disease, those with a history of chronic or recurrent autoimmune disease, patients with grade ≥2 bleeding, and those undergoing long‐term systemic steroid therapy.
2.3. Assessments
The following data was collected at baseline, including demographics, MDS history (date of diagnosis, MDS classification, 33 , 34 and previous treatments), IPSS/IPSS‐R score before treatment, concurrent diseases, clinical symptoms, peripheral blood and bone marrow findings, and ECOG PS. Data on blood transfusions was collected from 8 weeks before the initial DSP‐7888 dose until completion of the final assessment.
2.3.1. Dose‐limiting toxicities and maximum tolerated dose
The MTD was defined as the highest dose at which no more than one out of six patients experienced a DLT, and the RP2D was determined by safety and biomarker activity. Dose‐limiting toxicities were assessed within 29 days after the first DSP‐7888 dose phase 1 and were defined as any ADR that met the following criteria: grade 4 febrile neutropenia (FN); grade 4 electrolyte abnormalities or grade 3 electrolyte abnormalities persisting for ≥7 days; grade 4 ISR or grade 3 ISR that the investigator deemed uncontrolled; grade 4 infections; or other grade ≥3 nonhematologic toxicities.
2.3.2. Safety
Adverse events and ADR were monitored throughout the study, according to the National Cancer Institute’s Common Terminology Criteria for Adverse Events version 4.03. Adverse drug reactions were defined as AE for which a causal relationship to DSP‐7888 could not be denied. Dose reductions or interruptions were permitted in the event of an AE considered possibly related to the study drug. For patients on a starting dose of 3.5 mg in 200 μL of emulsion across two injection sites outside of the DLT evaluation period, a reduction in the dose of the peptide to 1.75 mg in 100 μL of emulsion across 1‐2 injection sites or 0.875 mg in 50 μL of emulsion at one injection site was permitted. Patients requiring a dosage reduction from 3.5 mg in 200 μL of emulsion during the DLT evaluation period were discontinued from the study. For patients on a starting dose of 10.5 mg in 600 μL of emulsion across six injection sites, a reduction in the dose of the peptide to 3.5 mg in 200 μL of emulsion across two injection sites was permitted, or 1.75 or 0.875 mg were also permitted if 3.5 mg was not tolerated. Vital signs, laboratory variables, and 12‐lead electrocardiogram and chest X‐ray findings were also monitored.
2.3.3. Efficacy
Primary efficacy endpoint
The primary efficacy endpoint was OS, defined as the period from the date of DSP‐7888 initiation until the date of death from any cause. Patients alive at the time of analysis were censored at the last date known to be alive. As the median OS from historical data was 5.6 (95% CI, 5.0‐7.2) months, 19 the prespecified survival threshold was set at 7.2 months. Based on this, it was concluded that DSP‐7888 would be considered effective if the lower limit of the 90% CI for the median OS exceeded 7.2 months.
Secondary efficacy endpoints
Secondary efficacy endpoints were the hematological response rate and the disease control rate. Hematologic response was evaluated according to the International Working Group (IWG) 2006 response criteria. 35 The proportion of patients with hematologic improvement, cytogenetic response (both evaluated according to IWG 2006 response criteria), 35 transfusion independence, and AML transformation were also assessed. Transfusion independence was defined as ≥8 weeks without red blood cell and/or platelet transfusion. Time to AML transformation was calculated as the length of time from the date of DSP‐7888 initiation to the date of a definite AML diagnosis. For patients without AML transformation at the time of analysis, data was censored at the date of last sampling.
2.3.4. Biomarkers
Delayed‐type hypersensitivity (DTH) response to DSP‐7888‐K and DSP‐7888‐H was measured separately to confirm the induction of WT1‐specific immune response (IR). Suspensions of each peptide and a negative control solution were each injected intradermally into different sites of the same forearm, with the diameter of redness measured 2 days after injection. DTH assessments were performed within 28 days before the first dose of DSP‐7888 and on Day 3 of Cycles 3 and 13 and within 28 days after the last dose of DSP‐7888. Responses were classified according to the difference from the control as: (a) (−) <2 mm; (b) (±) ≥2 mm but <5 mm; (c) (+) ≥5 mm but <10 mm; (d) (2+) ≥10 mm but <15 mm; and (e) (3+) ≥15 mm. Positive DTH responses were considered DTH ±, +, 2+, and 3+ reactions.
Wilms’ tumor gene 1‐specific CD8+ T cell induction in peripheral blood was assessed to confirm the mechanism of action of DSP‐7888 and examine the relationship with safety and efficacy. The percentage of WT1‐specific CD8+ T cells was measured using an HLA tetramer assay, with assessments performed within 28 days before the first dose of DSP‐7888; on Day 15 of Cycles 2, 6, 12, and 18; every six cycles thereafter; and within 28 days after the last dose of DSP‐7888. Three tetramers provided by MEDICAL & BIOLOGICAL LABORATORIES were used in this study: (a) “T‐Select HLA‐A*24:02 modified WT1 Tetramer‐CYTWNQMNL‐PE”; (b) “T‐Select HLA‐A*02:01 WT1 Tetramer‐RMFPNAPYL‐APC”; and (c) HLA‐A*02:01 WT1 tetramer‐VLDFAPPGA‐PE, which was customized at the request of the sponsor. WT1‐specific CD8+ T cell induction was confirmed if the WT1 CD8+ tetramer+ T cell/Total CD8+ T cell ratio to baseline was ≥2 at any time and the WT1 CD8+ tetramer+ T cell was ≥10 at that time. If the WT1 tetramer+ CD8+ T cell at baseline was 0, WT1‐specific CD8+ T cell induction was confirmed when the WT1 tetramer+ CD8+ T cell was ≥10 at any time. If either a positive DTH‐K response or WT1‐specific CD8+ T cell induction (confirmed by HLA tetramer assay) was confirmed, the diagnosis was “WT1‐specific IR positive.” WT1 messenger RNA (mRNA) expression levels in the bone marrow and peripheral blood were also measured prior to treatment (≤28 days prior to first administration); on Day 15 before doses 2, 6, and 12; and following every six cycles thereafter.
With the exception of DTH response to WT1 peptides, biomarkers were measured at a central laboratory.
2.4. Statistical analysis
The full analysis set comprised all patients who received ≥1 dose of DSP‐7888 and the safety analysis set comprised all patients who received ≥1 dose of DSP‐7888, which contained a DTH solution. The efficacy analysis set included patients with higher‐risk MDS after AZA failure in phases 1 and 2 (ie some patients in the phase 1 part of the study who met the eligibility criteria for the phase 2 part and all patients in the phase 2 part of the study). For phase 1, a maximum of 12 patients was planned based on the 3 + 3 design. For patients with higher‐risk MDS who had failed prior AZA therapy, a maximum of 42 subjects was planned. This calculation was based on historical data reporting a median OS of 5.6 (95% CI, 5.0‐7.2) months in this patient population. 19 In the subgroup analysis of the phase 1/2 trial using our earlier vaccine, WT4869, a median OS of 13.0 (95% CI, 7.3‐NA) months was reported in 11 patients with higher‐risk MDS after AZA failure. 32 As DSP‐7888 was expected to be equivalent or more efficacious than WT4869, a median OS of 14.0 months was assumed, with the lower bound of the two‐sided 90% CI set at 80% above the upper threshold of 7.2 months based on historical data.
The Kaplan‐Meier method was used to calculate the median survival and time to AML transformation.
Subgroup analyses of efficacy were performed in patients with higher‐risk MDS by WT1‐specific IR.
All statistical analyses were performed using SAS software version 9.4 (SAS Institute).
3. RESULTS
3.1. Patient disposition
Between May 2015 and December 2019, 48 patients met the eligibility criteria and were enrolled, including 12 patients in phase 1 (six patients each in cohorts 1 and 2) and 36 patients in phase 2. Forty‐seven patients (phase 1, n = 12; phase 2, n = 35) received ≥1 dose of DSP‐7888 and comprised the full analysis set and safety analysis set, respectively (Figure 1). For the efficacy analysis set, 42 patients with higher‐risk MDS, defined as Int‐1 with myeloblasts ≥5%, Int‐2, or a high category based on IPSS, were included.
FIGURE 1.
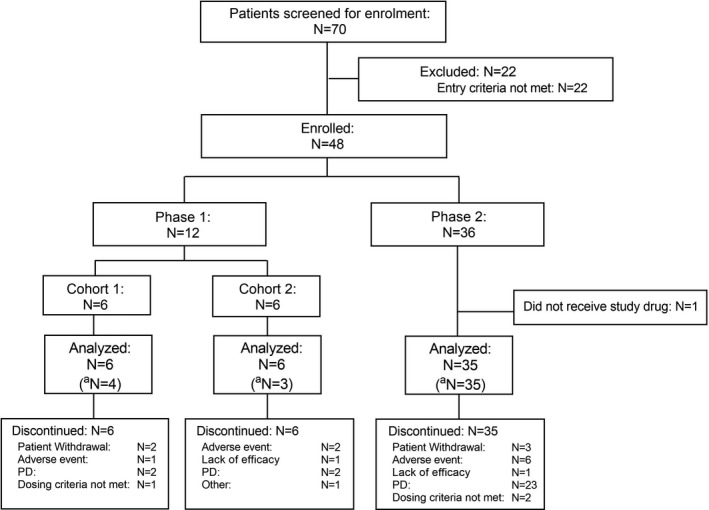
Patient disposition. aEfficacy analysis set comprises higher‐risk MDS patients who had failed azacitidine therapy (N = 42). FAS, full analysis set; MDS, myelodysplastic syndromes; PD, progressive disease; SAF, safety analysis set
Among the safety analysis set, all (100.0%) patients in phases 1 and 2 discontinued treatment with DSP‐7888. The primary reasons for treatment discontinuation were disease progression (PD; n = 27) and AE (n = 9), including two patients with ADR (myocarditis, n = 1; cellulitis and pyoderma gangrenosum, n = 1).
3.2. Patient characteristics
Baseline demographic and clinical characteristics of patients in the safety analysis set, and those with higher‐risk and other MDS, are presented in Table 1. The median (min‐max) age at study enrollment was 74.0 (52‐93) years, and most patients (n = 35, 74.5%) were male and had the HLA‐A*24:02 allele (n = 28, 59.6%). The majority of patients had IPSS Int‐2 (n = 28, 59.6%) and high‐risk (n = 9, 19.1%) disease, and 44.7% (n = 21) had a hemoglobin level <8 g/dL at baseline. A total of 53.2% (n = 25) of patients received transfusion of ≥4 units (ie 800 mL) of red blood cells within 56 days before treatment, and 27.7% (n = 13) of patients received transfusion of ≥10 units of platelets within 56 days before treatment. Among patients with higher‐risk MDS who had failed AZA (n = 42), the median (min‐max) duration of prior AZA therapy was 9.4 (0.3‐29.3) months, and the main reasons for discontinuation were primary (42.9%) and secondary (52.4%) treatment failure, followed by intolerance (4.8%).
TABLE 1.
Demographic and baseline clinical characteristics
| Higher‐risk MDS after AZA failure (N = 42) | Other MDS (N = 5) | Total (N = 47) | |
|---|---|---|---|
| Sex, male, n (%) | 32 (76.2) | 3 (60.0) | 35 (74.5) |
| Age, years, median (range) | 74 (63‐93) | 76 (52‐89) | 74 (52‐93) |
| HLA Type (Class I), n (%) | |||
| A*02:01 | 4 (9.5) | 1 (20.0) | 5 (10.6) |
| A*02:01/A*24:02 | 5 (11.9) | 0 | 5 (10.6) |
| A*02:06 | 6 (14.3) | 1 (20.0) | 7 (14.9) |
| A*02:06/A*24:02 | 2 (4.8) | 0 | 2 (4.3) |
| A*24:02 | 25 (59.5) | 3 (60.0) | 28 (59.6) |
| ECOG PS, n (%) | |||
| 0 | 24 (57.1) | 4 (80.0) | 28 (59.6) |
| 1 | 16 (38.1) | 1 (20.0) | 17 (36.2) |
| 2 | 2 (4.8) | 0 | 2 (4.3) |
| WHO classification category, n (%) | |||
| RCUD (RA) | 0 | 1 (20.0) | 1 (2.1) |
| RCUD (RT) | 1 (2.4) | 0 | 1 (2.1) |
| RARS | 0 | 1 (20.0) | 1 (2.1) |
| RCMD | 3 (7.1) | 2 (40.0) | 5 (10.6) |
| RAEB‐1 | 16 (38.1) | 0 | 16 (34.0) |
| RAEB‐2 | 15 (35.7) | 1 (20.0) | 16 (34.0) |
| MDS‐U | 1 (2.4) | 0 | 1 (2.1) |
| N/A | 6 (14.3) | 0 | 6 (12.8) |
| FAB classification category, n (%) | |||
| RA | 5 (11.9) | 3 (60.0) | 8 (17.0) |
| RARS | 0 | 1 (20.0) | 1 (2.1) |
| RAEB | 30 (71.4) | 1 (20.0) | 31 (66.0) |
| RAEB‐t | 7 (16.7) | 0 | 7 (14.9) |
| IPSS risk category, n (%) | |||
| Low | 0 | 1 (20.0) | 1 (2.1) |
| Int‐1 | 5 (11.9) | 3 (60.0) | 8 (17.0) |
| Int‐2 | 27 (64.3) | 1 (20.0) | 28 (59.6) |
| High | 9 (21.4) | 0 | 9 (19.1) |
| N/A | 1 (2.4) | 0 | 1 (2.1) |
| IPSS Karyotype category, n (%) | |||
| Good | 12 (28.6) | 3 (60.0) | 15 (31.9) |
| Intermediate | 9 (21.4) | 2 (40.0) | 11 (23.4) |
| Poor | 20 (47.6) | 0 | 20 (42.6) |
| N/A | 1 (2.4) | 0 | 1 (2.1) |
| IPSS‐R risk category, n (%) | |||
| Low | 0 | 2 (40.0) | 2 (4.3) |
| Intermediate | 5 (11.9) | 2 (40.0) | 7 (14.9) |
| High | 13 (31.0) | 1 (20.0) | 14 (29.8) |
| Very high | 23 (54.8) | 0 | 23 (48.9) |
| N/A | 1 (2.4) | 0 | 1 (2.1) |
| IPSS‐R Karyotype risk category, n (%) | |||
| Good | 11 (26.2) | 3 (60.0) | 14 (29.8) |
| Intermediate | 13 (31.0) | 2 (40.0) | 15 (31.9) |
| Poor | 2 (4.8) | 0 | 2 (4.3) |
| Very poor | 15 (35.7) | 0 | 15 (31.9) |
| N/A | 1 (2.4) | 0 | 1 (2.1) |
| Bone marrow blasts, n (%) | |||
| ≤2% | 3 (7.1) | 2 (40.0) | 5 (10.6) |
| 3−<5% | 3 (7.1) | 2 (40.0) | 5 (10.6) |
| 5‐10% | 18 (42.9) | 0 | 18 (38.3) |
| >10% | 18 (42.9) | 1 (20.0) | 19 (40.4) |
| Hemoglobin, g/dL, n (%) | |||
| ≥10 | 8 (19.0) | 0 | 8 (17.0) |
| 8−<10 | 16 (38.1) | 2 (40.0) | 18 (38.3) |
| <8 | 18 (42.9) | 3 (60.0) | 21 (44.7) |
| Platelets, 1000/μL, n (%) | |||
| ≥100 | 5 (11.9) | 4 (80.0) | 9 (19.1) |
| 50−<100 | 16 (38.1) | 1 (20.0) | 17 (36.2) |
| <50 | 21 (50.0) | 0 | 21 (44.7) |
| Neutrophils, 1000/μL, n (%) | |||
| ≥0.8 | 15 (35.7) | 5 (100.0) | 20 (42.6) |
| <0.8 | 27 (64.3) | 0 | 27 (57.4) |
| Duration of prior AZA treatment, months, median (min‐max) | 9.4 (0.3‐29.3) | – | 9.4 (0.3‐29.3) |
| Duration from AZA treatment cessation to study drug administration, months, median (min‐max) | 1.6 (1.0‐49.4) | – | 1.6 (1.0‐49.4) |
| Type of AZA failure, n (%) | |||
| Primary failure a | 18 (42.9) | – | 18 (38.3) |
| Secondary failure b | 22 (52.4) | – | 22 (46.8) |
| AZA intolerance c | 2 (4.8) | – | 2 (4.3) |
AE, adverse event; AZA, azacitidine; CR, complete remission; ECOG, Eastern Cooperative Oncology Group; FAB, French‐American‐British, HI, hematologic improvement; HLA, human leukocyte antigen; IPSS, International Prognostic Scoring System; IPSS‐R, revised International Prognostic Scoring System; mCR, bone marrow complete remission; MDS‐U, MDS unclassifiable; PR, partial remission; RA, refractory anemia; RAEB, refractory anemia with excess blasts; RARS, refractory anemia with ringed sideroblasts; RCMD, refractory cytopenia with multilineage dysplasia; RCUD, refractory cytopenia with unilineage dysplasia; RN, refractory neutropenia; RT, refractory thrombocytopenia.
Defined as SD or PD.
Defined as failure after CR/mCR/PR/HI.
Defined as treatment discontinuation due to AE.
3.3. DSP‐7888 treatment
The median (min‐max) number of doses of DSP‐7888 was 7.0 (1.0‐43.0), the median (min‐max) treatment duration was 3.3 (0‐33.1) months, and the median (min‐max) total administered dose was 56.0 (10.5‐451.5) mg.
3.4. Safety
No DLT were observed in either the 3.5 or 10.5 mg dose cohorts in phase 1; therefore, the MTD was not determined. There was no significant difference in safety profile, including ISR, between 3.5 and 10.5 mg dose cohorts.
The most common AE (≥20% incidence) during DSP‐7888 treatment are presented in Table 2. Adverse events occurred in all (n = 47, 100.0%) patients, including six patients (100.0%) each in cohorts 1 and 2 in phase 1 and 35 patients (100.0%) in phase 2.
TABLE 2.
Occurrence of common adverse events (with an incidence of ≥20%) a overall and by study cohort
| Phase 1 | Phase 2 (N = 35) | Total (N = 47) | ||
|---|---|---|---|---|
| Cohort 1 (N = 6) | Cohort 2 (N = 6) | |||
| Number of evaluable cases | 6 | 6 | 35 | 47 |
| Number of cases with AE | 6 (100.0) | 6 (100.0) | 35 (100.0) | 47 (100.0) |
| Occurrence of AE, SOC (PT) | ||||
| Blood and lymphatic system disorders | ||||
| Febrile neutropenia | 0 | 0 | 8 (22.9) | 8 (17.0) |
| Gastrointestinal disorders | ||||
| Nausea | 0 | ─ | ─ | ─ |
| Stomatitis | 2 (33.3) | 0 | 4 (11.4) | 6 (12.8) |
| General disorders and administration site conditions | ||||
| Injection‐site reaction | 6 (100.0) | 6 (100.0) | 31 (88.6) | 43 (91.5) |
| Pyrexia | 1 (16.7) | 1 (16.7) | 12 (34.3) | 14 (29.8) |
| Edema | 0 | 2 (33.3) | 1 (2.9) | 3 (6.4) |
| Infections and infestations | ||||
| Pharyngitis | 2 (33.3) | 0 | 1 (2.9) | 3 (6.4) |
| Sepsis | 0 | 2 (33.3) | 0 | 2 (4.3) |
| Injury, poisoning, and procedural complications | ||||
| Contusion | 0 | 2 (33.3) | 1 (2.9) | 3 (6.4) |
| Metabolism and nutrition disorders | ||||
| Iron overload | 1 (16.7) | 3 (50.0) | 6 (17.1) | 10 (21.3) |
| Nervous system disorders | ||||
| Dysgeusia | 2 (33.3) | 1 (16.7) | 1 (2.9) | 4 (8.5) |
| Skin and subcutaneous tissue disorders | ||||
| Rash | 2 (33.3) | 1 (16.7) | 1 (2.9) | 4 (8.5) |
| Asteatosis | 0 | 2 (33.3) | 2 (5.7) | 4 (8.5) |
Data are presented as n (%).
AE, adverse event; PT, preferred term; SAE, serious adverse event; SOC, system organ class.
Rows that contain any event with a frequency of ≥20% are presented.
Adverse drug reactions occurred in most (n = 43, 91.5%) patients, including six patients (100.0%) each in Cohorts 1 and 2 in phase 1 and 31 patients (88.6%) in phase 2 (Table 3). Adverse drug reactions leading to dose reduction or dose interruption occurred in 17 patients (36.2%) and seven patients (14.9%), respectively, and those leading to drug withdrawal occurred in two patients (4.3%). The most common ADR was ISR, occurring in 43 patients (91.5%), with a smaller proportion experiencing pyrexia (n = 5, 10.6%) and FN (n = 4, 8.5%). Adverse drug reactions were generally manageable, and no grade ≥4 ADR occurred in ≥5% of patients (Table 4). The most common grade 3 ADR was ISR (n = 15, 31.9%), followed by FN (n = 4, 8.5%).
TABLE 3.
Occurrence of common adverse drug reactions (with an incidence of ≥5%) a overall and by study cohort
| Phase 1 | Phase 2 (N = 35) | Total (N = 47) | ||
|---|---|---|---|---|
| Cohort 1 (N = 6) | Cohort 2 (N = 6) | |||
| Number of cases with ADR | 6 (100.0) | 6 (100.0) | 31 (88.6) | 43 (91.5) |
| Number of related SAE | 1 (16.7) | 2 (33.3) | 6 (17.1) | 9 (19.1) |
| Number of deaths related to study drug | 0 | 0 | 0 | 0 |
| Number of discontinuations due to ADR | 1 (16.7) | 0 | 1 (2.9) | 2 (4.3) |
| Number of dose reductions due to ADR | 2 (33.3) | 4 (66.7) | 11 (31.4) | 17 (36.2) |
| Number of dose interruptions due to ADR | 2 (33.3) | 1 (16.7) | 4 (11.4) | 7 (14.9) |
| Occurrence of ADR, SOC (PT) | ||||
| Blood and lymphatic system disorders | ||||
| Febrile neutropenia | 0 | 0 | 4 (11.4) | 4 (8.5) |
| Cardiac disorders | ||||
| Myocarditis | 1 (16.7) | 0 | 0 | 1 (2.1) |
| Supraventricular tachycardia | 0 | 1 (16.7) | 0 | 1 (2.1) |
| General disorders and administration site conditions | ||||
| Injection‐site reaction | 6 (100.0) | 6 (100.0) | 31 (88.6) | 43 (91.5) |
| Pyrexia | 1 (16.7) | 0 | 4 (11.4) | 5 (10.6) |
| Musculoskeletal and connective tissue conditions | ||||
| Myalgia | 1 (16.7) | 0 | 0 | 1 (2.1) |
| Nervous system disorders | ||||
| Dysgeusia | 2 (33.3) | 0 | 0 | 2 (4.3) |
Data are presented as n (%).
ADR, adverse drug reaction; PT, preferred term; SOC, system organ class.
Rows that contain any event with a frequency of ≥5% are presented.
TABLE 4.
Occurrence of common adverse drug reactions (with an incidence of ≥5%) a by severity
| Occurrence of ADR (SOC/PT) | Grade 1 | Grade 2 | Grade 3 | Grade 4 b | Grade 5 |
|---|---|---|---|---|---|
| Phase 1, Cohort 1 (N = 6) | |||||
| Cardiac disorders | |||||
| Myocarditis | 0 | 0 | 1 (16.7) | 0 | 0 |
| General disorders and administration site conditions | |||||
| Injection‐site reaction | 2 (33.3) | 1 (16.7) | 3 (50.0) | 0 | 0 |
| Pyrexia | 0 | 0 | 1 (16.7) | 0 | 0 |
| Musculoskeletal and connective tissue conditions | |||||
| Myalgia | 1 (16.7) | 0 | 0 | 0 | 0 |
| Nervous system disorders | |||||
| Dysgeusia | 0 | 2 (33.3) | 0 | 0 | 0 |
| Phase 1, Cohort 2 (N = 6) | |||||
| Cardiac disorders | |||||
| Supraventricular tachycardia | 0 | 0 | 1 (16.7) | 0 | 0 |
| General disorders and administration site conditions | |||||
| Injection‐site reaction | 2 (33.3) | 0 | 4 (66.7) | 0 | 0 |
| Phase 2 (N = 35) | |||||
| Blood and lymphatic system disorders | |||||
| Febrile neutropenia | 0 | 0 | 4 (11.4) | 0 | 0 |
| General disorders and administration site conditions | |||||
| Injection‐site reaction | 8 (22.9) | 15 (42.9) | 8 (22.9) | 0 | 0 |
| Pyrexia | 4 (11.4) | 0 | 0 | 0 | 0 |
| Total (N = 47) | |||||
| Blood and lymphatic system disorders | |||||
| Febrile neutropenia | 0 | 0 | 4 (8.5) | 0 | 0 |
| General disorders and administration site conditions | |||||
| Injection‐site reaction | 12 (25.5) | 16 (34.0) | 15 (31.9) | 0 | 0 |
| Pyrexia | 4 (8.5) | 0 | 1 (2.1) | 0 | 0 |
Data are presented as n (%).
ADR, adverse drug reaction; PT, preferred term; SOC, system organ class.
Rows that contain any event with a frequency of ≥5% are presented.
An instance of Grade 4 pancytopenia occurred in one patient (2.1%) during the study.
Serious ADR occurred in 9 (19.1%) patients, including 3 (25.0%) patients in phase 1 and six patients (17.1%) in phase 2. In phase 1, one patient in Cohort 1 developed a serious ADR of ISR (not recovered [the patient died due to PD]), pyrexia (recovered) and myocarditis (recovering), and two patients in Cohort 2 developed serious ADR of ISR (not recovered [the patient discontinued due to lack of efficacy]/recovering, respectively), one of whom also developed supraventricular tachycardia (recovering). In phase 2, six patients developed serious ADR, including one patient who developed cellulitis (recovered) and pyoderma gangrenosum (recovered), four patients who developed FN (recovering [n = 1]; recovered [n = 3]), and one patient who developed pyrexia (recovered).
A total of three deaths (6.4%) occurred, including one patient (16.7%) who developed fatal sepsis (Cohort 2, phase 1) and two (5.7%) patients who developed splenic abscess/large intestinal perforation/peritonitis/ischemic colitis and non–small cell lung cancer, respectively (phase 2). In the patient who developed non–small cell lung cancer, the tumor was detected during phase 2 and was rapidly fatal within 1 month of diagnosis. However, no deaths were related to DSP‐7888.
The relationship between the ISR and WT1‐specific IR are presented in Figure 2. The proportion of patients who had a positive WT1‐specific IR increased with increasing ISR grade.
FIGURE 2.
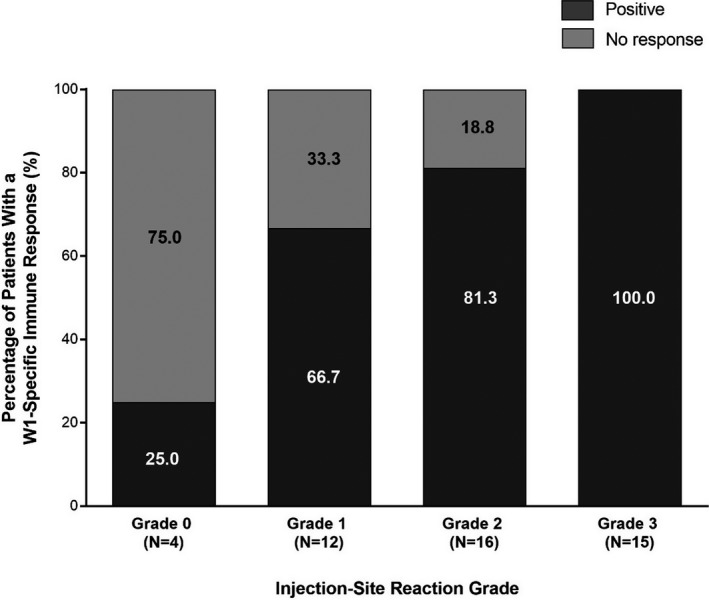
Relationship between injection‐site reactions and WT1‐specific immune responses. WT1, Wilms’ tumor gene 1
3.5. Efficacy
Clinical activity in the efficacy analysis set is summarized in Table 5. Median OS for patients in the efficacy analysis set was 8.6 months (90% CI, 6.8‐10.3 months; Figure 3). Treatment outcomes for each of the IPSS‐R karyotype categories are shown in Figure 4. When examined by WT1‐specific IR, the median OS was 10.0 (90% CI, 7.6‐11.4) months in patients with a positive WT1‐specific IR compared to 4.1 months (90% CI, 2.3‐8.1 months) in those without a response (P = .0034; Figure 5). A similar trend was observed in patients with a positive DTH response to DSP‐7888‐K (Figure 6) and those with a positive WT1‐specific CD8+ T cell response (Figure 7).
TABLE 5.
Summary of clinical activity in patients with higher‐risk MDS after AZA failure
| Clinical activity | Total (N = 42) |
|---|---|
| Response rate | 0 (0) |
| Disease control rate | 8 (19.0) |
| Clinical efficacy (CR, PR, mCR, HI) | 4 (9.5) |
| Median duration of disease control | 2.7 months |
| Hematological response | |
| Stable disease | 8 (19.0) |
| Progressive disease | 33 (78.6) |
| Not evaluable | 1 (2.4) |
| Median duration of hematological response a | 4.4 months |
| Hematologic improvement | |
| Erythroid | 2 (4.8) |
| Platelet | 1 (2.4) |
| Neutrophil | 1 (2.4) |
| Median (90% CI) overall survival | 8.6 (6.8‐10.3) months |
| Median (95% CI) time to AML transformation | 7.1 (3.8‐16.1) months |
Data are presented as n (%), unless otherwise specified. The best response was not evaluated in one patient. In those categories, the response rate, disease control rate, and hematologic improvement data were not available for one patient and the calculations only included 41 patients.
AML, acute myeloid leukemia; AZA, Azacitidine; CR, complete remission; HI, hematological improvement; mCR, bone marrow complete remission; PR, partial remission.
In patients with stable disease.
FIGURE 3.
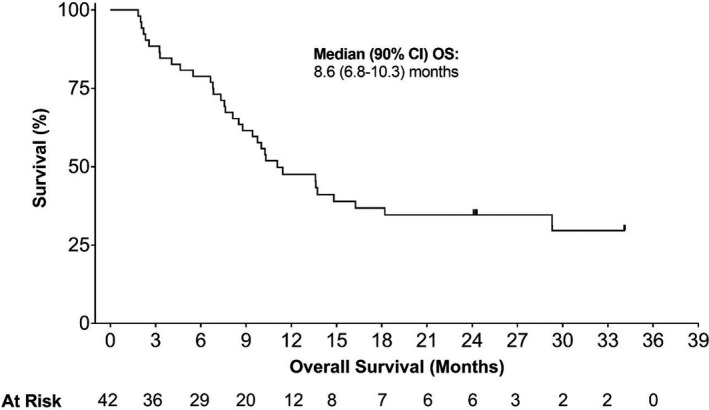
Overall survival in patients with higher‐risk MDS after AZA failure. AZA, Azacitidine; MDS, myelodysplastic syndromes; OS, overall survival
FIGURE 4.
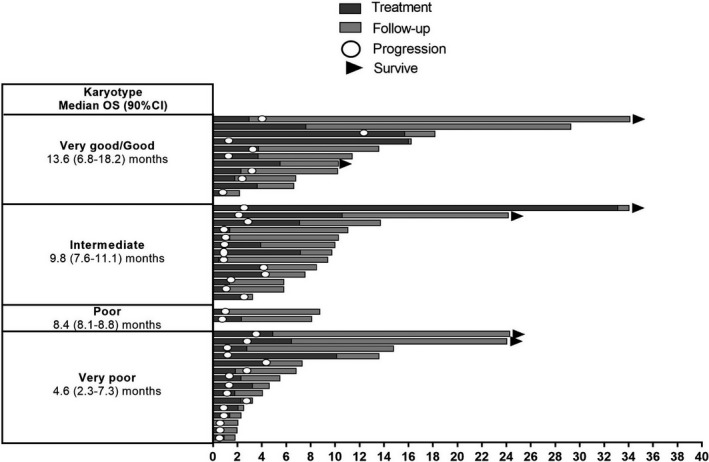
Summary of treatment and outcomes in patients with higher‐risk MDS after AZA failure according to the Revised International Prognostic Scoring System Karyotype. AZA, Azacitidine; MDS, myelodysplastic syndromes; OS, overall survival
FIGURE 5.
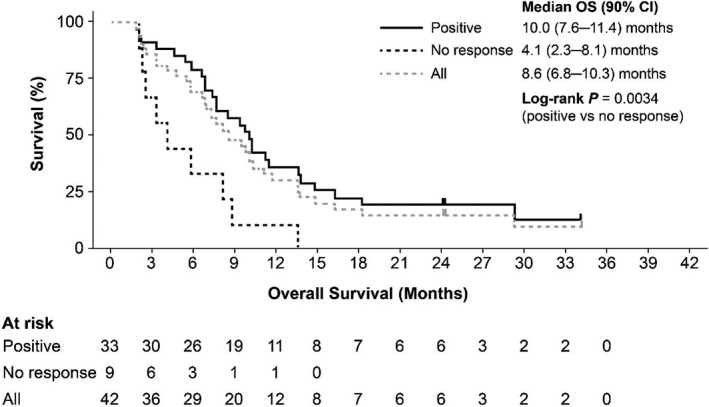
Overall survival in patients with higher‐risk MDS after AZA failure according to WT1‐specific immune response category. AZA, Azacitidine; MDS, myelodysplastic syndromes; OS, overall survival; WT1, Wilms’ tumor gene 1
FIGURE 6.
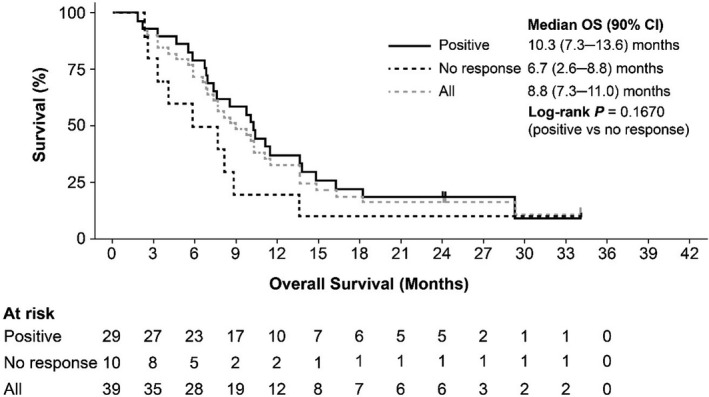
Overall survival in patients with higher‐risk MDS after AZA failure according to DTH response to DSP‐7888‐K. AZA, Azacitidine; DTH, delayed type hypersensitivity; MDS, myelodysplastic syndromes; OS, overall survival
FIGURE 7.
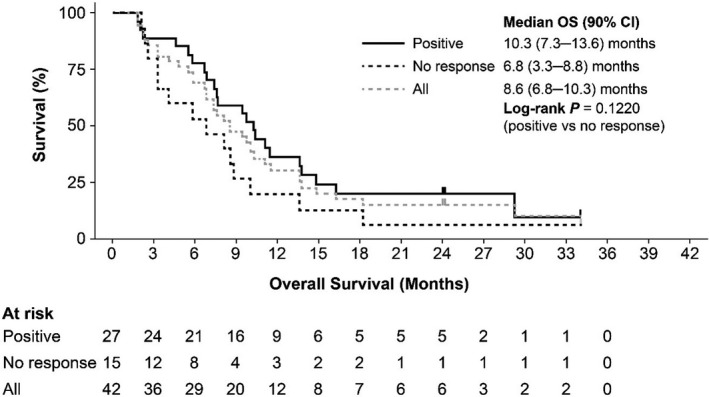
Overall survival in patients with higher‐risk MDS after AZA failure according to WT1‐specific CD8+ T cells. AZA, Azacitidine; MDS, myelodysplastic syndromes; OS, overall survival
Disease control (95% CI) was achieved in eight patients (19.0%) in the efficacy analysis set, and clinical efficacy (complete remission, partial remission, bone marrow complete remission, and hematologic improvement) was achieved in four patients (9.5%; Table 5). The best hematological response was SD in eight patients (19.0%) and PD in 33 patients (78.6%). The median (min‐max) duration of hematological response in patients with SD was 4.4 (3.8‐12.8) months. A small proportion of patients experienced erythroid hematologic improvement (n = 2, 4.8%), platelet hematologic improvement, and neutrophil hematologic improvement (n = 1, 2.4% each). No patients became transfusion independent and half (n = 21, 50.0%) of the patients had transformed to AML by the end of the study, with a median time to AML transformation of 7.1 months (95% CI, 3.8‐16.1 months).
3.6. Biomarkers
In phase 1, WT1‐specific CD8+ T cell induction was relatively higher in Cohort 2 (10.5 mg) than in Cohort 1 (3.5 mg; Figure 8). Figure S1 presents the results of the WT1 tetramer staining at baseline and on Day 15 of Cycle 2 in a patient who was WT1‐specific CD8+ T cell induction positive.
FIGURE 8.
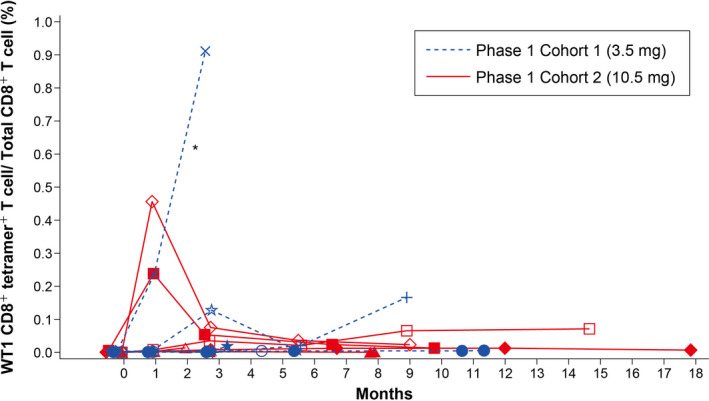
Individual plot of CTL induction with WT1 vaccine: Phase 1. *One patient in Cohort 1 (3.5 mg) was not evaluable because an accurate measurement of WT1 CD8+ tetramer+ by flow cytometry was hampered by background fluorescence. CTL, cytotoxic T lymphocyte; WT1, Wilms’ tumor gene 1
Delayed‐type hypersensitivity response was evaluated in 44/47 patients (93.6%) in the full analysis set and 39/42 patients (92.9%) in the efficacy analysis set. In the full analysis set, 33 patients (75.0%) and 28 patients (63.6%) experienced a response to the DSP‐7888‐K and DSP‐7888‐H peptides, respectively. In the efficacy analysis set, 29 patients (74.4%) and 24 patients (61.5%) patients experienced a response to the DSP‐7888‐K and DSP‐7888‐H peptides, respectively. Of 47 patients in the full analysis set who had WT1‐specific CD8+ T cell induction measured at baseline and after ≥1 study drug administration, 31 patients (66.0%) reported an increased percentage of WT1‐specific CD8+ T cell induction. A total of 37 patients (78.7%) experienced a positive WT1‐specific IR, defined as DTH‐K reaction‐positive or WT1‐specific CD8+ T cell positivity. In the efficacy analysis set, 27 patients (64.3%) reported an increased percentage of WT1‐specific CD8+ T cell induction, and 33 patients (78.6%) experienced a positive WT1‐specific IR. No significant differences in IR were observed between HLA‐A types (Table 6).
TABLE 6.
Summary of immune response in patients with higher‐risk MDS after AZA failure and by HLA type
| Clinical activity | Total (N = 47) | Higher‐Risk MDS after AZA failure (N = 42) | A*02:01 or A*02:06 (N = 10) | A*02:01/A*24:02 or A*02:06/A*24:02 (N = 7) | A*24:02 (N = 25) |
|---|---|---|---|---|---|
| Maximum DSP‐7888‐K response grade | n = 44 | n = 39 | n = 10 | n = 7 | n = 22 |
| − | 11 (25.0) | 10 (25.6) | 4 (40.0) | 1 (14.3) | 5 (22.7) |
| ± | 7 (15.9) | 5 (12.8) | 1 (10.0) | 0 | 4 (18.2) |
| + | 11 (25.0) | 11 (28.2) | 4 (40.0) | 2 (28.6) | 5 (22.7) |
| 2+ | 6 (13.6) | 6 (15.4) | 0 | 1 (14.3) | 5 (22.7) |
| 3+ | 9 (20.5) | 7 (17.9) | 1 (10.0) | 3 (42.9) | 3 (13.6) |
| Maximum DSP‐7888‐H response grade | n = 10 | n = 7 | n = 22 | ||
| − | 16 (36.4) | 15 (38.5) | 4 (40.0) | 4 (57.1) | 7 (31.8) |
| ± | 4 (9.1) | 3 (7.7) | 0 | 0 | 3 (13.6) |
| + | 6 (13.6) | 6 (15.4) | 3 (30.0) | 0 | 3 (13.6) |
| 2+ | 12 (27.3) | 10 (25.6) | 1 (10.0) | 2 (28.6) | 7 (31.8) |
| 3+ | 6 (13.6) | 5 (12.8) | 2 (20.0) | 1 (14.3) | 2 (9.1) |
| WT1‐specific CD8+ T cells | n = 10 | n = 7 | n = 25 | ||
| Positive | 31 (66.0) | 27 (64.3) | 7 (70.0) | 6 (85.7) | 14 (56.0) |
| No response | 16 (34.0) | 15 (35.7) | 3 (30.0) | 1 (14.3) | 11 (44.0) |
| WT1‐specific immune response | n = 10 | n = 7 | n = 25 | ||
| Positive | 37 (78.7) | 33 (78.6) | 8 (80.0) | 6 (85.7) | 19 (76.0) |
| No response | 10 (21.3) | 9 (21.4) | 2 (20.0) | 1 (14.3) | 6 (24.0) |
Number of patients (%) are shown.
AZA, azacitidine; DTH, delayed type hypersensitivity; HLA, human leukocyte antigen; MDS, myelodysplastic syndromes; WT1, Wilms’ tumor gene 1.
The expression level of WT1 mRNA in bone marrow was slightly decreased from the baseline (median [min‐max]: 12 500 [200‐210 000]) at Cycle 6 (median [min‐max]: 7450 [300‐120 000]) but had increased by the end of the study (median [min‐max]: 23 000 [300‐280 000]). Similarly, the expression level of WT1 mRNA in the peripheral blood did not decrease substantially from the baseline (median [min‐max]: 2900 [50‐140 000]) to Cycle 6 (median [min‐max]: 2400 [50‐75 000]) but was elevated by the end of the study (median [min‐max]: 11 000 [50‐250 000]).
4. DISCUSSION
This phase 1/2 study showed the safety and clinical activity of DSP‐7888 in patients with higher‐risk MDS who had failed prior AZA treatment. No DLT were observed at either the 3.5 or 10.5 mg dose, and there was no significant difference in safety profile, including ISR, between dose cohorts. In addition, WT1‐specific CD8+ T cell induction was relatively higher in Cohort 2 (10.5 mg) than in Cohort 1 (3.5 mg; Figure 8). Thus, 10.5 mg was selected as the RP2D based on the favorable safety profile and biomarker activity.
The most common ADR was ISR (in 91.5% of patients), which was manageable in all patients and did not lead to treatment discontinuation. Grade 3 ISR were reported in 7/12 patients (58.3%) in phase 1. Therefore, risk minimization measures for ISR were implemented for phase 2, including guidance on dose reduction, extension of dosing intervals, and/or treatment of ISR with corticosteroids, as required. Further, the guidance recommends selection of several injection site areas surrounding regional lymph nodes (ie upper arm, lower abdomen, and femoral). These measures likely contributed to the reduction in grade 3 ISR (8/35 patients, 22.9%) in phase 2. Febrile neutropenia occurred in a small proportion of patients (n = 4, 8.5%). However, two out of four patients had low neutrophil counts (<1000/μL) before DSP‐7888 treatment. Since cytopenias are inherent to MDS, it is difficult to assess the extent to which neutropenia was therapy‐related or part of MDS progression. It is also difficult to differentiate FN from post–vaccine pyrexia in patients with MDS. One (16.7%) patient developed myocarditis during the phase 1 part of our study. The patient had initially developed an injection‐site reaction, followed by pyrexia and viral pharyngitis and subsequently developed myocarditis. Although viral infections are recognized as the most frequent cause of myocarditis, 36 a causal relationship to DSP‐7888 could not be denied and, thus, it was considered an ADR.
In terms of clinical activity, disease control was achieved in eight patients (19.0%) in the efficacy analysis set. The median OS was 8.6 months (90% CI, 6.8‐10.3 months) in patients with higher‐risk MDS after AZA therapy. Although the lower bound of the 90% CI in our study (6.8 months) was marginally lower than the upper bound of the 95% CI reported in historical data (median OS: 5.6 [95% CI, 5.0‐7.2] months), 19 survival was prolonged when compared against historical data. 19 , 29 Further, the median OS reported in this study was shorter than the 13.0 months reported with WT4869. 32 However, this is not entirely unexpected considering that the previous result with WT4869 was from a subgroup analysis with a small sample size (n = 11), and only 42.3% (n = 11) of patients in that study had higher‐risk MDS and had previously failed AZA therapy. The median time to AML transformation was 7.1 months (95% CI, 3.8‐16.1 months). Patients with all IPSS‐R karyotypes experienced extended survival compared with the best supportive care arm in the phase 3 rigosertib study, 37 which was more pronounced in the “very good/good,” “intermediate” and “poor” karyotype groups compared with the “very poor” karyotype (Figure 4). Due to its mechanism of action, DSP‐7888 treatment is expected to be particularly suited to less aggressive rather than rapidly aggressive disease due to the time needed to induce immunity and produce a clinical response.
A total of 78.7% of patients experienced a positive WT1‐specific IR. Injection‐site reactions were the most common ADR and were positively associated with WT1‐specific IR. Compared with patients without a WT1‐specific IR, those with a positive WT1‐specific IR experienced significantly longer OS (P = .0034). Because patients with a longer OS were able to receive DSP‐7888 for a longer period of time, WT1‐specific IR may have been more common in such patients. Conversely, the WT1‐specific IR may have directly contributed to the longer OS observed.
The limitations of this study were inherent to phase 1/2 studies and included the small sample size, lack of a control arm, and use of historical controls to set the survival threshold. There may have been important differences in patient characteristics compared with historical data, which may have impacted the reliability of relative assessments of outcomes. For example, only 16.7% of patients with RAEB‐t were included in the efficacy analysis set in our study, compared to 26.0% and 23.0% of patients in the studies by Prébet et al (2011) and Garcia‐Manero et al (2016), respectively. 19 , 37 As the prognosis for patients with AML transformation is particularly poor, this discrepancy may have resulted in an overestimation of the benefits observed in our study. Nevertheless, the results of this phase 1/2 study showed the tolerability and clinical activity of DSP‐7888 in patients with higher‐risk MDS who had failed AZA, a population with few treatment options.
In summary, DSP‐7888 was well tolerated in patients with higher‐risk MDS who had failed prior AZA treatment. Further, the clinical activity of DSP‐7888 was shown, and WT1‐specific IR, including WT1 specific CD8+ T cell induction, was demonstrated. These findings indicate that further investigation is warranted, including the potential for DSP‐7888 to be combined with a wide range of other treatments or evaluated in patients with less aggressive disease.
DISCLOSURE
YU has no conflict of interest to disclose. KU receives lecture fees, honoraria, or other fees from Novartis Pharma, and research funds from Astellas Pharma, Alexion Pharmaceuticals, Inc, AbbVie, Gilead Sciences, Symbio Pharmaceuticals, Daiichi‐Sankyo, Sumitomo Dainippon Pharma, Chugai Pharmaceutical, Otsuka Pharmaceutical, Novartis Pharma, Bristol‐Myers Squibb, Takeda Pharmaceutical, Amgen Astellas BioPharma, Apellis Pharmaceuticals, and Nihon Shinyaku. JF has no conflict of interest to disclose. IM receives lecture fees, honoraria, or other fees from Bristol‐Myers, Novartis Pharma, Pfizer Japan, Daiichi‐Sankyo, Otsuka Pharmaceutical, Astellas Pharma, Amgen Astellas BioPharma, and Janssen Pharmaceutical, and research funds from Chugai Pharmaceutical, Eisai, Ono pharmaceutical, Kyowa Kirin, SHIONOGI, Sumitomo Dainippon Pharma, Asahi Kasei Pharma, Takeda Pharmaceutical, Nihon Shinyaku, Taiho Pharmaceutical, Mitsubishi Tanabe Pharma, MSD, Japan Blood Products Organization, and AbbVie. NA has no conflict of interest to disclose. NS receives research funds from Sumitomo Dainippon Pharma. TN has no conflict of interest to disclose. HI receives research funds from Kyowa Kirin. MTM, SS, and MG are employees of Sumitomo Dainippon Pharma, Osaka, Japan, who funded this study. TN receives research funds from Daiichi‐Sankyo, FUJIFILM, and Astellas Pharma. MK receives lecture fees, honoraria, or other fees from Celgene, Bristol‐Myers Squibb, Janssen Pharmaceutical, Ono Pharmaceutical, Novartis Pharma, and Sumitomo Dainippon Pharma, and research funds from Takeda Pharmaceutical, Ono Pharmaceutical, Chugai Pharmaceutical, and Kyowa Kirin. YM receives lecture fees, honoraria, or other fees from Astellas Pharma, Chugai Pharmaceutical, Novartis Pharma, Celgene, Sumitomo Dainippon Pharma, Nihon Shinyaku, Otsuka Pharmaceutical, and Kyowa Kirin, and research funds from Sumitomo Dainippon Pharma and Chugai Pharmaceutical. KA receives lecture fees, honoraria, or other fees from Abbvie, Eisai, Astellas Pharma, Bristol‐Myers Squibb, Celgene, Chugai Pharmaceutical, Janssen Pharmaceutical, Kyowa Kirin, and Novartis Pharma, and research funds from Abbvie, Asahi Kasei Pharma, Astellas Pharma, Chugai Pharmaceutical, Daiichi‐Sankyo, Eisai, Kyowa Kirin, Ono Pharmaceutical, Otsuka Pharmaceutical, Mochida Pharmaceutical, MSD, Mundhipharma, Nihon Shinyaku, Sanofi, Shin Nippon Biomedical Laboratories, SHIONOGI, Sumitomo Dainippon Pharma, Taiho Pharmaceutical, Takeda Pharmaceutical, Toyama Chemical, and Yakult Honsha.
ETHICAL APPROVAL
This study was conducted in accordance with ethical principles of the Declaration of Helsinki, the International Council for Harmonization Harmonized Tripartite Guideline for Good Clinical Practice, and the Institutional Review Board (IRB) regulations. The clinical study protocol, the investigator’s brochure, and other study‐related documents were reviewed and approved by the local or central IRB of study sites. Each investigator conducted the study according to applicable local or regional regulatory requirements and in accordance with the responsibilities listed in the protocol.
Supporting information
Fig S1
ACKNOWLEDGMENTS
The authors thank all clinicians for their involvement and contribution to the study. The authors also thank Jordana Campbell, BSc, CMPP of Science Communications, Springer Healthcare for writing the outline and the first draft of the manuscript. This medical writing assistance was funded by Sumitomo Dainippon Pharma Co., Ltd.
Ueda Y, Usuki K, Fujita J, et al. Phase 1/2 study evaluating the safety and efficacy of DSP‐7888 dosing emulsion in myelodysplastic syndromes. Cancer Sci. 2022;113:1377–1392. doi: 10.1111/cas.15245
Funding Information
This study was funded by Sumitomo Dainippon Pharma Co., Ltd.
REFERENCES
- 1. Steensma DP. Myelodysplastic syndromes current treatment algorithm 2018. Blood Cancer J. 2018;8:47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Efficace F, Gaidano G, Breccia M, et al. Prevalence, severity and correlates of fatigue in newly diagnosed patients with myelodysplastic syndromes. Br J Haematol. 2015;168:361‐370. [DOI] [PubMed] [Google Scholar]
- 3. Parylo S, Vennepurredy A, Terjanian T. Rapidly progressing myelodysplastic syndrome initially presenting as acute leukemia. Cureus. 2017;9:e1096–e1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chihara D, Ito H, Katanoda K, et al. Incidence of myelodysplastic syndrome in Japan. J Epidemiol. 2014;24:469‐473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Greenberg P, Cox C, LeBeau MM, et al. International scoring system for evaluating prognosis in myelodysplastic syndromes. Blood. 1997;89:2079‐2088. [PubMed] [Google Scholar]
- 6. Greenberg PL, Tuechler H, Schanz J, et al. Revised international prognostic scoring system for myelodysplastic syndromes. Blood. 2012;120:2454‐2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Platzbecker U. Treatment of MDS. Blood. 2019;133:1096‐1107. [DOI] [PubMed] [Google Scholar]
- 8. Leitch HA, Vickars LM. Supportive care and chelation therapy in MDS: are we saving lives or just lowering iron? Hematology Am Soc Hematol Educ Program. 2009;2009:664‐672. [DOI] [PubMed] [Google Scholar]
- 9. Shah SR, Tran TM. Lenalidomide in myelodysplastic syndrome and multiple myeloma. Drugs. 2007;67:1869‐1881. [DOI] [PubMed] [Google Scholar]
- 10. Blum W. How much? How frequent? How long? A clinical guide to new therapies in myelodysplastic syndromes. Hematol Am Soc Hematol Educ Program. 2010;2010:314‐321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chan O, Komrokji RS. Luspatercept in the treatment of lower‐risk myelodysplastic syndromes. Future Oncol. 2021;17:1473‐1481. [DOI] [PubMed] [Google Scholar]
- 12. Silverman LR, Demakos EP, Peterson BL, et al. Randomized controlled trial of azacitidine in patients with the myelodysplastic syndrome: a study of the cancer and leukemia group B. J Clin Oncol. 2002;20:2429‐2440. [DOI] [PubMed] [Google Scholar]
- 13. Fenaux P, Mufti GJ, Hellstrom‐Lindberg E, et al. Efficacy of azacitidine compared with that of conventional care regimens in the treatment of higher‐risk myelodysplastic syndromes: a randomised, open‐label, phase III study. Lancet Oncol. 2009;10:223‐232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kantarjian H, Issa JP, Rosenfeld CS, et al. Decitabine improves patient outcomes in myelodysplastic syndromes: results of a phase III randomized study. Cancer. 2006;106:1794‐1803. [DOI] [PubMed] [Google Scholar]
- 15. VIDAZA [prescribing information]. Celgene. 2020. https://packageinserts.bms.com/pi/pi_vidaza.pdf. Accessed September 24, 2020.
- 16. DACOGEN [prescribing information]. Otsuka America Pharmaceutical, Inc. 2018. https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/021790s021lbl.pdf. Accessed September 24, 2020.
- 17. Santini V, Prebet T, Fenaux P, et al. Minimizing risk of hypomethylating agent failure in patients with higher‐risk MDS and practical management recommendations. Leuk Res. 2014;38:1381‐1391. [DOI] [PubMed] [Google Scholar]
- 18. Jabbour E, Garcia‐Manero G, Batty N, et al. Outcome of patients with myelodysplastic syndrome after failure of decitabine therapy. Cancer. 2010;116:3830‐3834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Prébet T, Gore SD, Esterni B, et al. Outcome of high‐risk myelodysplastic syndrome after azacitidine treatment failures. J Clin Oncol. 2011;29:3322‐3327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Call KM, Glaser T, Ito CY, et al. Isolation and characterization of a zinc finger polypeptide gene at the human chromosome 11 Wilms’ tumor locus. Cell. 1990;60:509‐520. [DOI] [PubMed] [Google Scholar]
- 21. Inoue K, Tamaki H, Ogawa H, et al. Wilms’ tumor gene (WT1) competes with differentiation‐inducing signal in hematopoietic progenitor cells. Blood. 1998;91:2969‐2976. [PubMed] [Google Scholar]
- 22. Ito K, Oji Y, Tatsumi N, et al. Antiapoptotic function of 17AA(+)WT1 (Wilms’ tumor gene) isoforms on the intrinsic apoptosis pathway. Oncogene. 2006;25:4217‐4229. [DOI] [PubMed] [Google Scholar]
- 23. Wagner KD, Cherfils‐Vicini J, Hosen N, et al. The Wilms’ tumour suppressor Wt1 is a major regulator of tumour angiogenesis and progression. Nat Commun. 2014;5:5852. [DOI] [PubMed] [Google Scholar]
- 24. Sugiyama H. WT1 (Wilms’ tumor gene 1): biology and cancer immunotherapy. Jpn J Clin Oncol. 2010;40:377‐387. [DOI] [PubMed] [Google Scholar]
- 25. Hastie ND. Wilms’ tumour 1 (WT1) in development, homeostasis and disease. Development. 2017;144:2862‐2872. [DOI] [PubMed] [Google Scholar]
- 26. Ohminami H, Yasukawa M, Fujita S. HLA class I‐restricted lysis of leukemia cells by a CD8+ cytotoxic T‐lymphocyte clone specific for WT1 peptide. Blood. 2000;95:286‐293. [PubMed] [Google Scholar]
- 27. Azuma T, Makita M, Ninomiya K, Fujita S, Harada M, Yasukawa M. Identification of a novel WT1‐derived peptide which induces human leucocyte antigen‐A24‐restricted anti–leukaemia cytotoxic T lymphocytes. Br J Haematol. 2002;116:601‐603. [DOI] [PubMed] [Google Scholar]
- 28. Tsuboi A, Oka Y, Udaka K, et al. Enhanced induction of human WT1‐specific cytotoxic T lymphocytes with a 9‐mer WT1 peptide modified at HLA‐A*2402‐binding residues. Cancer Immunol Immunother. 2002;51:614‐620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Oka Y, Tsuboi A, Taguchi T, et al. Induction of WT1 (Wilms’ tumor gene)‐specific cytotoxic T lymphocytes by WT1 peptide vaccine and the resultant cancer regression. Proc Nat Acad Sci USA. 2004;101:13885‐13890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Oka Y, Tsuboi A, Oji Y, Kawase I, Sugiyama H. WT1 peptide vaccine for the treatment of cancer. Curr Opin Immunol. 2008;20:211‐220. [DOI] [PubMed] [Google Scholar]
- 31. Sugiyama H, inventor; International Institute of Cancer Immunology Inc , assignee. Cancer antigen helper peptide. European patent EP. 2010. International filing date: April 22.
- 32. Ueda Y, Ogura M, Miyakoshi S, et al. Phase 1/2 study of the WT1 peptide cancer vaccine WT4869 in patients with myelodysplastic syndrome. Cancer Sci. 2017;108:2445‐2453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Swerdlow SH. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. WHO classification of tumours. 2008;2:439. [PubMed] [Google Scholar]
- 34. Bennett JM, Catovsky D, Daniel MT, et al. Proposed revised criteria for the classification of acute myeloid leukemia. A report of the French‐American‐British cooperative group. Ann Intern Med. 1985;103:620‐625. [DOI] [PubMed] [Google Scholar]
- 35. Cheson BD, Greenberg PL, Bennett JM, et al. Clinical application and proposal for modification of the international working group (IWG) response criteria in myelodysplasia. Blood. 2006;108:419‐425. [DOI] [PubMed] [Google Scholar]
- 36. Blauwet LA, Cooper LT. Myocarditis. Prog Cardiovasc Dis. 2010;52:274‐288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Garcia‐Manero G, Fenaux P, Al‐Kali A, et al. Rigosertib versus best supportive care for patients with high‐risk myelodysplastic syndromes after failure of hypomethylating drugs (ONTIME): a randomised, controlled, phase 3 trial. Lancet Oncol. 2016;17:496‐508. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1