

Abstract
A functional variant on ALDH2 rs671 (G>A) confers a protective effect against alcohol‐induced carcinogenesis through an indirect pathway mediated by decreased alcohol consumption. Conversely, this variant also contributes to the accumulation of carcinogenic agents, resulting in a direct carcinogenic effect. This study aimed to separately quantify these two opposing effects of the rs671 A allele on pancreatic cancer risk and explore the impact of the rs671 A allele and alcohol consumption on pancreatic carcinogenesis. We included 426 cases and 1456 age‐ and sex‐matched controls. Odds ratio (OR) and 95% confidence interval (CI) for alcohol consumption were estimated using a conditional logistic regression model. By defining rs671 A allele and alcohol consumption as exposure and mediator, respectively, we used mediation analysis to decompose the total‐effect OR of the rs671 A allele into direct‐ and indirect‐effect ORs. Alcohol consumption (10 g/d) was associated with pancreatic cancer risk (OR, 1.05; 95% CI, 1.01‐1.10), but tests for interaction between the rs671 A allele and alcohol consumption were nonsignificant, indicating that the effect of alcohol consumption did not vary by genotype. Mediation analysis showed that the nonsignificant total effect (OR, 1.15; 95% CI, 0.92‐1.44) can be decomposed into the carcinogenic direct (OR, 1.34; 95% CI, 1.04‐1.72) and protective indirect effect (OR, 0.86; 95% CI, 0.77‐0.95). This study supports the association between alcohol consumption and pancreatic cancer risk and indicates the potential contribution of the rs671 A allele to pancreatic carcinogenesis through impaired metabolism of known or unknown ALDH2 substrates.
Keywords: alcohol consumption, aldehyde dehydrogenase 2, case‐control study, mediation analysis, pancreatic cancer
In this study, we undertook a mediation analysis with the aim of exploring the impact of alcohol consumption and ALDH2 rs671 A allele on pancreatic cancer risk in a case‐control study with 426 cases and 1456 controls. The results indicated an association between alcohol consumption and pancreatic cancer risk and the two opposing effects of the carcinogenic direct effect and the protective indirect effect of the rs671 A allele on pancreatic cancer risk, with odds ratios (95% confidence intervals) of 1.34 (1.04‐1.72) and 0.86 (0.77‐0.95), respectively.
Abbreviations
- 4‐HNE
4‐hydroxy‐2‐nonenal
- ALDH
aldehyde dehydrogenase
- BMI
body mass index
- CI
confidence interval
- HERPACC
Hospital‐based Epidemiologic Research Program at Aichi Cancer Center
- OR
odds ratio
1. INTRODUCTION
Pancreatic cancer remains a fatal type of cancer, with a 5‐year survival rate in Japan of only 8.5%. 1 , 2 Investigation of modifiable risk factors is therefore of critical importance. One candidate factor is alcohol consumption. Although alcohol consumption has been categorized as a risk factor with limited suggestive evidence, several epidemiological studies have shown a significantly increased pancreatic cancer risk with heavy alcohol consumption. 3 Nevertheless, the biological mechanism underlying an association between alcohol consumption and pancreatic cancer has not been fully elucidated. One important hypothesis among several possible mechanistic hypotheses is the involvement of ethanol metabolites, in particular acetaldehyde, which is considered one of the most likely carcinogens in alcohol. 4
Studies on the East Asian‐specific functional SNP of ALDH2 rs671 (c.1510G>A [p. Glu504Lys]) have revealed that acetaldehyde is a plausible carcinogen in some alcohol‐related cancers. 5 Ingested alcohol is metabolized predominantly to acetaldehyde through an oxidative pathway in the liver, and acetaldehyde is further oxidized to acetate by ALDH enzymes. 6 Aldehyde dehydrogenase 2 has the highest affinity for acetaldehyde (Km < 1 µM) among ALDH isoforms and is predominantly responsible for the oxidation of acetaldehyde. 6 , 7 The enzymatic activity of ALDH2 is dramatically reduced by the ALDH2 rs671 A allele, and individuals with this variant experience rapid accumulation of blood acetaldehyde after alcohol ingestion. 6 The rs671 variant‐drinking interaction observed in studies of alcohol‐related cancers (eg, head and neck and esophageal cancers) 8 is accordingly evidence for the possible involvement of accumulated ethanol‐derived acetaldehyde in alcohol‐induced carcinogenesis. 5 However, with regard to pancreatic cancer, although a few epidemiological studies 9 , 10 have reported a possible association of rs671 with pancreatic cancer risk, no clear evidence for the rs671 variant‐drinking interaction is yet available.
In intriguing contrast to its carcinogenic effect, the rs671 A allele also contributes to decreasing alcohol consumption. This effect is due to the occurrence of acetaldehyde‐related adverse effects (eg, flushing, headache, palpitation, and nausea), which induce the individual to decrease their alcohol intake 11 , 12 and thereby protect against alcohol‐induced carcinogenesis. 13 Due to these mutually opposing effects, the impact of the carcinogenic direct effect of the rs671 A allele appears attenuated by its indirect protective effect, mediated by reduced drinking intensity. Nevertheless, the conventional approach to risk using multivariate analysis focuses on simple adjustment for covariates, and cannot fully examine the mediation effects between rs671 and drinking intensity on the risk of pancreatic cancer.
Here, we undertook a pancreatic cancer case‐control study using mediation analysis 14 to decompose the total effect of the rs671 A allele on pancreatic cancer risk into direct and indirect effects. We previously used mediation analysis to successfully decompose the total effect of the rs671 A allele on digestive tract cancer risk into direct and indirect effects and determined the site‐specific impact of the carcinogenic direct effect as well as the site‐agnostic impact of the protective indirect effect. 13 In the present study, by quantifying the direct and indirect effects of the rs671 A allele on the risk of pancreatic cancer separately, we aimed to explore the impact of the ALDH2 rs671 variant in conjunction with alcohol consumption on pancreatic carcinogenesis.
2. MATERIALS AND METHODS
2.1. Study population
The study population was selected from participants in HERPACC, which is described in detail elsewhere. 15 , 16 We undertook two case‐control studies of pancreatic cancer: HERPACC‐2 (2001‐2005), with 179 cases and 716 age‐ (±5 years) and sex‐matched controls; and HERPACC‐3 (2005‐2013), with 247 cases and 740 age‐ (±5 years) and sex‐matched controls. Cases were first‐visit outpatients diagnosed with pancreatic cancer at Aichi Cancer Center Hospital. Controls were first‐visit outpatients confirmed not to have cancer or a history of neoplasm. The most common reason for the visit to the Aichi Cancer Center Hospital among the control subjects was secondary screening after primary screening (31%), followed by patient discretion (25%), and referral from another clinic (24%). We obtained written informed consent and collected a self‐administered questionnaire and a peripheral blood sample from each participant. The study was approved by the Institutional Ethics Committee of Aichi Cancer Center and carried out in accordance with the Declaration of Helsinki.
2.2. Genotyping
DNA of each participant was extracted from the buffy coat fraction with a DNA Blood mini kit (Qiagen). Single nucleotide polymorphism genotyping was carried out using TaqMan Assays on the 7500 Real‐Time PCR System (Applied Biosystems). Genotype distributions in the controls were assessed for the Hardy‐Weinberg equilibrium using the χ2 test.
2.3. Environmental factors
Information on environmental factors was obtained using a self‐administered questionnaire that questioned participants on their exposure status before the development of the symptoms for which they first visited the hospital. Responses were checked by trained interviewers.
To measure drinking intensity, we adopted daily alcohol intake (g/d). Conversion from the information in the questionnaire to the total amount of daily alcohol consumption has been described in detail elsewhere. 13 In brief, the participants were first asked about their drinking status (never, including “almost never”, former, or current drinkers). We then required former and current drinkers to describe the frequency of alcohol drinking, type of beverage, and average consumption during each drinking session based on their average drinking behavior before stopping drinking (former drinkers) and during the year before the development of the symptoms or diagnosis (current drinkers). Each category of drinking frequency was assigned a score, as follows: 0 for never drinkers, 0.5 for <1 d/wk, 1.5 for 1‐2 d/wk, 3.5 for 3‐4 d/wk, and 6 for ≥5 d/wk in HERPACC‐2; and 0 for never drinkers, 0.5 for 1‐3 d/mo, 1.5 for 1‐2 d/wk, 3.5 for 3‐4 d/wk, 5.5 for 5‐6 d/wk, and 7 for every day in HERPACC‐3. Using the information on type of beverage and average consumption during each drinking session, we estimated the total amount of pure alcohol consumed per single drinking session. Based on the concentration of alcohol in each beverage, the HERPACC Study assumed that 23 g alcohol was contained in 180 mL (one “go”) of Japanese sake, 633 mL (one large bottle) of beer, 90 mL (half a “go”) of shochu (distilled spirit), 60 mL (double shot) of whiskey, and 200 mL (two and a half glasses) of wine. Finally, daily alcohol intake was estimated by multiplying the total amount of pure alcohol consumed per single drinking session by the frequency score/7. As a measure of cumulative smoking exposure, we used pack‐years, calculated by multiplying the number of packs consumed per day by the number of years of smoking. Body mass index (BMI) was estimated as the self‐reported weight (kg) divided by the square of the self‐reported height (m). Frequencies of meat (beef or pork) and processed meat intake were categorized into the three categories of <1 time/wk, 1‐4 times/wk, and ≥5 times/wk. A family history of pancreatic cancer in parents and siblings and history of diabetes were classified as “yes” or “no”.
2.4. Statistical analysis
Analyses were carried out for HERPACC‐2 and HERPACC‐3 separately. In order to obtain more accurate point estimates, a pooled analysis was further undertaken by combining the two studies. We used the t test or χ2 test to evaluate differences in the distribution of covariates between cases and controls. Odds ratios and their two‐sided 95% CIs for drinking intensity (0, 0< to <23, 23≤ to <46, and ≥46 g/d) were estimated using a conditional logistic regression model, with adjustment for age (continuous), pack‐years (continuous), BMI (kg/m2) (continuous), frequency of meat intake (<1, 1‐4, ≥5 times/wk), frequency of processed meat intake (<1, 1‐4, ≥5 times/wk), family history of pancreatic cancer (yes/no), history of diabetes (yes/no), and ALDH2 rs671 genotype. Trend analysis for drinking intensity (unit OR per 10 g daily alcohol intake) was also carried out with the same covariate adjustment. The interaction term that corresponded to multiplying a one‐allele change in rs671 by a 10‐g change in daily alcohol intake was further added into the model. We assessed the variant‐drinking interaction on both the additive and multiplicative scales using the relative excess risk due to interaction 17 and the Wald test of the coefficient of the interaction term, respectively.
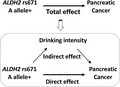
A mediation analysis was carried out to decompose the total‐effect OR of ALDH2 rs671 A allele on pancreatic cancer risk into direct‐ and indirect‐effect ORs with the paramed command of STATA. 18 In the mediation analysis, we assumed a hypothetical causal structure between rs671 A allele (exposure), drinking intensity (mediator), and pancreatic cancer risk (outcome) as depicted in the directed acyclic graph presented in Figure 1. Details of the methodology of this analysis are described elsewhere. 14 , 19 , 20 Briefly, we estimated direct‐ and indirect‐effect ORs by combining two models, namely: (a) the linear regression model for the mediator (drinking intensity) conditioned on the exposure (rs671 A allele) and covariates; and (b) the logistic regression model for the outcome (pancreatic cancer risk) conditioned on the exposure, the mediator, and covariates. As covariates, the model included age, sex, pack‐years, BMI, frequency of meat intake, frequency of processed meat intake, family history of pancreatic cancer, and history of diabetes. Daily alcohol intake (g/d) as a measure of drinking intensity was square‐root transformed and entered in the model. In the pooled analysis, we additionally included HERPACC version (2/3) as a covariate. The direct effect represents the effect of the rs671 A allele on pancreatic cancer risk through pathways that are independent of drinking intensity, whereas the indirect effect is interpretable as the effect controlled by drinking intensity. The direct‐effect OR multiplied by the indirect‐effect OR is equivalent to the total‐effect OR. 14 To preclude concerns about residual confounding by smoking, analyses restricted to never smokers were also carried out.
FIGURE 1.
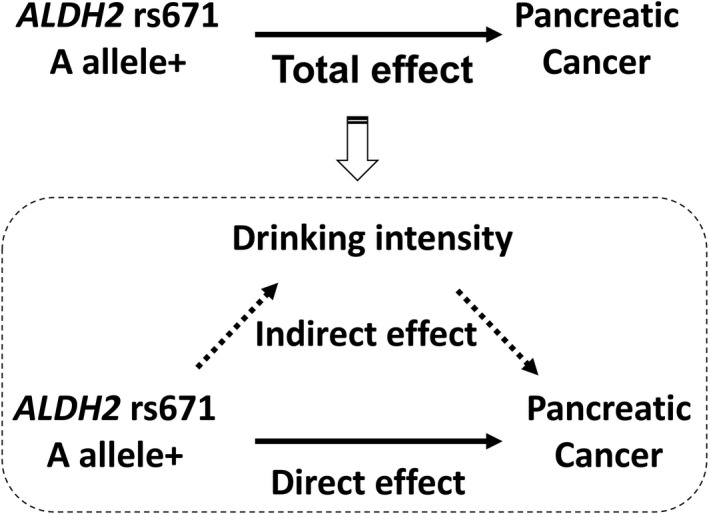
Directed acyclic graph illustrating assumed causal structure. A hypothetical causal structure between rs671 A allele (exposure), drinking intensity (mediator), and pancreatic cancer risk (outcome) is shown. We decomposed the total effect of rs671 A allele on pancreatic cancer risk into direct and indirect effects using mediation analysis. ALDH2, aldehyde dehydrogenase 2
Some data for daily alcohol intake, pack‐years, BMI, frequency of meat intake, frequency of processed meat intake, and history of diabetes were incomplete (Table 1). These missing values were handled by applying multiple imputation using chained equations. 21 The results from the 10 generated imputed datasets were combined using Rubin’s rules. 22 All analyses were carried out using STATA version 15 (Stata Corporation). We interpreted two‐sided P values of less than .05 as statistically significant.
TABLE 1.
Characteristics of pancreatic cancer cases and control subjects
| HERPACC‐2 (2001‐2005) | HERPACC‐3 (2005‐2013) | Pooled analysis | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Case (n = 179) | Control (n = 716) | P value a | Case (n = 247) | Control (n = 740) | P value a | Case (n = 426) | Control (n = 1456) | P value a | |
| Age (y) | .819 | .163 | .513 | ||||||
| <40 | 10 (5.6) | 40 (5.6) | 10 (4.0) | 24 (3.2) | 20 (4.7) | 64 (4.4) | |||
| 40‐<50 | 18 (10.1) | 79 (11.0) | 13 (5.3) | 63 (8.5) | 31 (7.3) | 142 (9.8) | |||
| 50‐<60 | 58 (32.4) | 208 (29.1) | 58 (23.5) | 183 (24.7) | 116 (27.2) | 391 (26.9) | |||
| 60‐<70 | 59 (33.0) | 265 (37.0) | 125 (50.6) | 320 (43.2) | 184 (43.2) | 585 (40.2) | |||
| 70‐ | 34 (19.0) | 124 (17.3) | 41 (16.6) | 150 (20.3) | 75 (17.6) | 274 (18.8) | |||
| Median (IQR) | 60 (53‐67) | 60 (53‐66.5) | 63 (57‐68) | 63 (56‐68) | 62 (55‐68) | 62 (54‐67) | |||
| Sex | 1.000 | .978 | .963 | ||||||
| Male | 125 (69.8) | 500 (69.8) | 170 (68.8) | 510 (68.9) | 295 (69.2) | 1010 (69.4) | |||
| Female | 54 (30.2) | 216 (30.2) | 77 (31.2) | 230 (31.1) | 131 (30.8) | 446 (30.6) | |||
| Alcohol intake (g/d) | .608 | .051 | .037 | ||||||
| 0 | 55 (30.7) | 242 (33.8) | 94 (38.1) | 284 (38.4) | 149 (35.0) | 526 (36.1) | |||
| <23 | 74 (41.3) | 294 (41.1) | 78 (31.6) | 286 (38.6) | 152 (35.7) | 580 (39.8) | |||
| <46 | 28 (15.6) | 103 (14.4) | 30 (12.1) | 89 (12.0) | 58 (13.6) | 192 (13.2) | |||
| ≥46 | 21 (11.7) | 63 (8.8) | 40 (16.2) | 77 (10.4) | 61 (14.3) | 140 (9.6) | |||
| Missing | 1 (0.6) | 14 (2.0) | 5 (2.0) | 4 (0.5) | 6 (1.4) | 18 (1.2) | |||
| Mean (SD) | 20.4 (50.5) | 14.8 (23.0) | 19.4 (33.3) | 15.1 (29.4) | 19.8 (41.4) | 15.0 (26.5) | |||
| Median (IQR) | 7.4 (0.0‐26.6) | 3.9 (0.0‐19.7) | 5.8 (0.0‐23.0) | 2.5 (0.0‐18.1) | 5.9 (0.0‐25.5) | 3.3 (0‐19.7) | |||
| Smoking pack‐years | .001 | .062 | .007 | ||||||
| 0 | 62 (34.6) | 311 (43.4) | 93 (37.7) | 333 (45.0) | 155 (36.4) | 644 (44.2) | |||
| <20 | 25 (14.0) | 104 (14.5) | 43 (17.4) | 127 (17.2) | 68 (16.0) | 231 (15.9) | |||
| <40 | 33 (18.4) | 155 (21.6) | 55 (22.3) | 115 (15.5) | 88 (20.7) | 270 (18.5) | |||
| <60 | 28 (15.6) | 85 (11.9) | 34 (13.8) | 76 (10.3) | 62 (14.6) | 161 (11.1) | |||
| ≥60 | 31 (17.3) | 57 (8.0) | 20 (8.1) | 62 (8.4) | 51 (12.0) | 119 (8.2) | |||
| Missing | 0 (0.0) | 4 (0.6) | 2 (0.8) | 27 (3.6) | 2 (0.5) | 31 (2.1) | |||
| BMI | .084 | 1.4 × 10−10 | 7.9 × 10−11 | ||||||
| <18.5 | 14 (7.8) | 32 (4.5) | 39 (15.8) | 34 (4.6) | 53 (12.4) | 66 (4.5) | |||
| 18.5‐<25 | 125 (69.8) | 479 (66.9) | 181 (73.3) | 526 (71.1) | 306 (71.8) | 1005 (69.0) | |||
| ≥25 | 40 (22.3) | 200 (27.9) | 27 (10.9) | 173 (23.4) | 67 (15.7) | 373 (25.6) | |||
| Missing | 0 (0.0) | 5 (0.7) | 0 (0.0) | 7 (0.9) | 0 (0.0) | 12 (0.8) | |||
| Mean (SD) | 22.7 (3.3) | 23.3 (3.1) | .026 | 21.6 (3.1) | 23.1 (3.1) | 2.9 × 10−10 | 22.1 (3.2) | 23.2 (3.1) | 1.7 × 10−10 |
| Meat intake | .319 | .929 | .468 | ||||||
| <1/wk | 50 (27.9) | 172 (24.0) | 61 (24.7) | 179 (24.2) | 111 (26.1) | 351 (24.1) | |||
| 1‐4/wk | 120 (67.0) | 499 (69.7) | 176 (71.3) | 523 (70.7) | 296 (69.5) | 1022 (70.2) | |||
| 5‐/wk | 5 (2.8) | 34 (4.7) | 10 (4.0) | 34 (4.6) | 15 (3.5) | 68 (4.7) | |||
| Missing | 4 (2.2) | 11 (1.5) | 0 (0.0) | 4 (0.5) | 4 (0.9) | 15 (1.0) | |||
| Processed meat intake | .907 | .274 | .457 | ||||||
| <1/wk | 98 (54.7) | 372 (52.0) | 131 (53.0) | 377 (50.9) | 229 (53.8) | 749 (51.4) | |||
| 1‐4/wk | 66 (36.9) | 271 (37.8) | 106 (42.9) | 310 (41.9) | 172 (40.4) | 581 (39.9) | |||
| 5‐/wk | 13 (7.3) | 51 (7.1) | 9 (3.6) | 47 (6.4) | 22 (5.2) | 98 (6.7) | |||
| Missing | 2 (1.1) | 22 (3.1) | 1 (0.4) | 6 (0.8) | 3 (0.7) | 28 (1.9) | |||
| Family history of pancreatic cancer | .389 | .001 | .001 | ||||||
| No | 170 (95.0) | 690 (96.4) | 225 (91.1) | 714 (96.5) | 395 (92.7) | 1404 (96.4) | |||
| Yes | 9 (5.0) | 26 (3.6) | 22 (8.9) | 26 (3.5) | 31 (7.3) | 52 (3.6) | |||
| History of diabetes | 1.0 × 10−6 | .008 | 1.8 × 10−7 | ||||||
| No | 142 (79.3) | 658 (91.9) | 200 (81.0) | 648 (87.6) | 342 (80.3) | 1306 (89.7) | |||
| Yes | 37 (20.7) | 58 (8.1) | 47 (19.0) | 91 (12.3) | 84 (19.7) | 149 (10.2) | |||
| Missing | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (0.1) | 0 (0.0) | 1 (0.1) | |||
| ALDH2 rs671 | .121 | .706 | .212 | ||||||
| GG | 83 (46.4) | 354 (49.4) | .626 b | 117 (47.4) | 372 (50.3) | .428 b | 200 (46.9) | 726 (49.9) | .365 b |
| GA | 86 (48.0) | 295 (41.2) | 107 (43.3) | 299 (40.4) | 193 (45.3) | 594 (40.8) | |||
| AA | 10 (5.6) | 67 (9.4) | 23 (9.3) | 69 (9.3) | 33 (7.7) | 136 (9.3) | |||
Data are n (%) unless otherwise stated.
Abbreviations: ALDH2, aldehyde dehydrogenase 2; BMI, body mass index; HERPACC, Hospital‐based Epidemiologic Research Program at Aichi Cancer Center; IQR, interquartile range.
Differences between cases and controls were analyzed using the t test or χ2 test.
P values for the Hardy‐Weinberg equilibrium using the χ2 test.
3. RESULTS
The baseline characteristics of cases and controls are shown in Table 1. Genotype frequencies among controls did not deviate from the values predicted from the Hardy‐Weinberg equilibrium. Table 2 shows ORs for drinking intensity, along with tests for variant‐drinking interaction from the two studies and a pooled analysis. The pooled estimate showed a significantly increased pancreatic cancer risk among those with daily alcohol intake of 46 g or more (OR, 1.57; 95% CI, 1.00‐2.46). A significant dose‐dependent association between alcohol intake and pancreatic cancer risk (per 10‐g change in daily alcohol intake: OR, 1.05; 95% CI, 1.01‐1.10) was also shown. Individual results from the two studies showed the same tendency. Tests for interaction were not significant on either the additive or multiplicative risk scales, indicating that the effect of alcohol drinking on the risk of pancreatic cancer did not vary by rs671 genotype. We therefore carried out the mediation analysis without considering exposure‐mediator interaction.
TABLE 2.
Impact of alcohol intake on pancreatic cancer risk and assessment of variant‐drinking interaction
| Analysis | HERPACC‐2 (2001‐2005) | HERPACC‐3 (2005‐2013) | Pooled analysis | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| OR a | 95% CI | P value | OR a | 95% CI | P value | OR a | 95% CI | P value | ||||
| Alcohol intake (g/d) | ||||||||||||
| 0 | ref | ref | ref | |||||||||
| <23 | 1.16 | 0.73 | 1.86 | 0.532 | 0.81 | 0.54 | 1.22 | 0.321 | 0.99 | 0.73 | 1.34 | 0.953 |
| <46 | 1.27 | 0.67 | 2.39 | 0.460 | 1.22 | 0.69 | 2.15 | 0.502 | 1.26 | 0.83 | 1.90 | 0.283 |
| ≥46 | 1.34 | 0.65 | 2.77 | 0.433 | 1.75 | 0.97 | 3.17 | 0.065 | 1.57 | 1.00 | 2.46 | 0.050 |
| Trend (per 10 g/d) | 1.05 | 0.99 | 1.12 | 0.074 | 1.06 | 1.00 | 1.12 | 0.042 | 1.05 | 1.01 | 1.10 | 0.011 |
| Interaction analyses b | ||||||||||||
| Additive interaction | 0.067 | −0.076 | 0.209 | 0.360 | 0.046 | −0.088 | 0.179 | 0.501 | 0.068 | −0.027 | 0.164 | 0.161 |
| Multiplicative interaction | 1.06 | 0.93 | 1.20 | 0.406 | 1.03 | 0.92 | 1.16 | 0.577 | 1.06 | 0.97 | 1.15 | 0.196 |
Estimates in bold show statistical significance (P < .05).
Abbreviations: ALDH2, aldehyde dehydrogenase 2; CI, confidence interval; HERPACC, Hospital‐based Epidemiologic Research Program at Aichi Cancer Center; OR, odds ratio.
ORs were calculated by a conditional logistic regression model adjusted for age, pack‐years, body mass index, meat intake, processed meat intake, family history of pancreatic cancer, history of diabetes, and ALDH2 rs671 genotype.
Multiplicative interaction was assessed using a Wald test of the coefficient of the interaction term corresponding to a one‐allele change in the genetic variant multiplied by a 10‐g change in daily alcohol intake; the test for additive interaction was carried out using the relative excess risk due to interaction.
Table 3 shows the estimated direct‐, indirect‐, and total‐effect ORs for the ALDH2 rs671 A allele. A significant positive direct‐effect OR was observed in the pooled analysis (OR, 1.34; 95% CI, 1.04‐1.72), even when restricted to never smokers (OR, 1.50; 95% CI, 1.02‐2.20) (Table 3). With regard to indirect effect, we observed a significant protective indirect effect (OR, 0.86; 95% CI, 0.77‐0.95). In the analysis among never smokers, a marginally significant protective indirect effect (OR, 0.89; 95% CI, 0.79‐1.01) (Table 3) was observed. In contrast, the total effects were nonsignificant, with the pooled estimated OR (95% CI) of 1.15 (0.92‐1.44) (Table 3). Further analysis by treating rs671 GA and AA separately showed consistent results. Most of the individual results from the two studies were nonsignificant but showed a similar tendency (Table 3).
TABLE 3.
Direct and indirect effects of the ALDH2 rs671 A allele on pancreatic cancer
| Analysis (Ca/Co) | HERPACC‐2 (2001‐2005) | HERPACC‐3 (2005‐2013) | Pooled analysis b | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| OR a | 95% CI | P value | OR a | 95% CI | P value | OR a | 95% CI | P value | ||||
| GA/AA vs GG | (179/716) | (247/740) | (426/1456) | |||||||||
| Direct effect | 1.39 | 0.95 | 2.03 | 0.090 | 1.27 | 0.91 | 1.78 | 0.163 | 1.34 | 1.04 | 1.72 | 0.022 |
| Indirect effect | 0.87 | 0.74 | 1.01 | 0.068 | 0.86 | 0.74 | 1.00 | 0.043 | 0.86 | 0.77 | 0.95 | 0.004 |
| Total effect | 1.20 | 0.86 | 1.69 | 0.290 | 1.09 | 0.81 | 1.48 | 0.572 | 1.15 | 0.92 | 1.44 | 0.228 |
| Never smokers only | (62/311) | (93/333) | (155/644) | |||||||||
| Direct effect | 1.80 | 0.99 | 3.26 | 0.054 | 1.33 | 0.80 | 2.22 | 0.281 | 1.50 | 1.02 | 2.20 | 0.038 |
| Indirect effect | 1.00 | 0.82 | 1.22 | 0.967 | 0.84 | 0.71 | 0.99 | 0.034 | 0.89 | 0.79 | 1.01 | 0.071 |
| Total effect | 1.81 | 1.02 | 3.19 | 0.041 | 1.11 | 0.68 | 1.80 | 0.684 | 1.34 | 0.93 | 1.92 | 0.116 |
| GA vs GG | (169/649) | (224/671) | (393/1320) | |||||||||
| Direct effect | 1.42 | 0.97 | 2.08 | 0.074 | 1.28 | 0.91 | 1.80 | 0.158 | 1.36 | 1.06 | 1.76 | 0.017 |
| Indirect effect | 0.91 | 0.79 | 1.04 | 0.165 | 0.85 | 0.75 | 0.97 | 0.019 | 0.88 | 0.80 | 0.96 | 0.006 |
| Total effect | 1.29 | 0.91 | 1.83 | 0.159 | 1.09 | 0.80 | 1.50 | 0.594 | 1.20 | 0.95 | 1.51 | 0.135 |
| AA vs GG | (93/421) | (140/441) | (233/862) | |||||||||
| Direct effect | 1.00 | 0.44 | 2.29 | 0.996 | 1.54 | 0.80 | 2.96 | 0.199 | 1.23 | 0.74 | 2.03 | 0.429 |
| Indirect effect | 0.80 | 0.55 | 1.17 | 0.255 | 0.76 | 0.54 | 1.05 | 0.099 | 0.78 | 0.62 | 1.00 | 0.046 |
| Total effect | 0.81 | 0.39 | 1.68 | 0.575 | 1.16 | 0.67 | 2.02 | 0.601 | 0.96 | 0.63 | 1.48 | 0.874 |
Estimates in bold show statistical significance (P < .05).
Abbreviations: ALDH2, aldehyde dehydrogenase 2; Ca, case; CI, confidence interval; Co, control; HERPACC, Hospital‐based Epidemiologic Research Program at Aichi Cancer Center; OR, odds ratio.
Mediator variable: square root of alcohol intake (g/d). Covariates: age, sex, pack‐years, body mass index, meat intake, processed meat intake, family history of pancreatic cancer, and history of diabetes.
HERPACC version was additionally included as a covariate in the pooled analysis.
To explore the heterogeneous impact between sexes, we undertook a sex‐stratified analysis (Table S1). Results were consistent between the sexes, albeit that the estimates for women included only a small number of cases and are accordingly unstable. In the main analysis, we treated former drinkers (32 cases [7.5% of cases] and 55 controls [3.8% of controls]) as those exposed to alcohol to the extent of their former drinking. To obviate concerns about bias caused by misclassification of former drinkers, we additionally undertook a sensitivity analysis that omitted data of former drinkers. The result showed no substantial difference from the main analysis, namely a direct‐effect OR of 1.31 (95% CI, 1.02‐1.70), indirect‐effect OR of 0.89 (95% CI, 0.80‐0.99), and total‐effect OR of 1.17 (95% CI, 0.93‐1.47).
4. DISCUSSION
In this study, we undertook a mediation analysis with the aim of exploring the impact of alcohol consumption and ALDH2 rs671 A allele on pancreatic cancer risk in a case‐control study with 426 cases and 1456 controls. The results showed: (a) an association between alcohol consumption and pancreatic cancer risk; and (b) the two opposing effects of the carcinogenic direct effect and the protective indirect effect of the rs671 A allele on pancreatic cancer risk, with ORs (95% CIs) of 1.34 (1.04‐1.72) and 0.86 (0.77‐0.95), respectively.
The results of this study emphasize that alcohol consumption, especially heavy consumption, is a preventable exposure associated with pancreatic cancer risk. This finding is consistent with the latest comprehensive meta‐analysis of 39 studies (18 cohort studies and 21 case‐control studies), which showed relative risks (95% CIs) of 0.95 (0.89‐1.01) for light, 1.03 (0.97‐1.09) for moderate, and 1.19 (1.11‐1.28) for heavy drinking compared with nondrinkers and occasional drinkers. 23 The protective indirect effect of the rs671 A allele also supported this association. Furthermore, we observed a lack of variant‐drinking interaction, consistent with previous studies. 10 , 24 This indicates that the accumulation of ethanol‐derived acetaldehyde with the rs671 A allele likely plays no major role in pancreatic cancer development because the absence of interaction indicates the absence of genetic susceptibility to the harmful effect of alcohol through acetaldehyde. Still, because the point estimates of the variant‐drinking interaction were positive in both the additive and multiplicative risk scales (Table 2), we cannot completely rule out the involvement of ethanol‐derived acetaldehyde in pancreatic carcinogenesis. Other mechanistic hypotheses for alcohol‐induced pancreatic carcinogenesis could include ethanol‐induced oxidative stress, 25 , 26 nonoxidative formation of fatty acid ethyl esters, 4 and pancreatitis related to chronic alcohol consumption. 27 , 28 The exact pathways by which alcohol causes pancreatic cancer remain unknown, and require further investigation.
In addition to ethanol‐derived acetaldehyde, ALDH2 is also capable of oxidating endogenous aldehydes and environmental aldehydes (present in tobacco smoke, car exhaust, etc.), 6 some of which are carcinogenic. For instance, formaldehyde has been classified as a group 1 carcinogen, 29 and endogenous aldehydes have been identified as a source of DNA damage. 30 , 31 Aldehyde dehydrogenase 2 therefore provides an important protective enzymatic function against several carcinogenic agents, and the rs671 A allele appears to contribute to carcinogenesis through the accumulation of these carcinogenic agents. The carcinogenic direct effect of the rs671 A allele therefore suggests that this allele might contribute to pancreatic carcinogenesis through impaired metabolism of ALDH2 substrates other than ethanol‐derived acetaldehyde. For example, formaldehyde is an environmental aldehyde to which humans are chronically exposed from environmental sources (eg, cigarette smoke), and is a possible candidate. Although the IARC evaluation did not support a causal role for formaldehyde and pancreatic cancer, 29 one meta‐analysis of 14 epidemiological studies showed a significant association. 32 In addition, an experimental study reported that oncogenic KRAS mutation, the major event in pancreatic carcinogenesis, mediated enhanced tolerance against formaldehyde. 33 This study further indicated that formaldehyde induces cytotoxicity in normal cells but represents a growth advantage for tumor cells carrying the oncogenic KRAS mutation. To take one more example, 4‐HNE, an endogenous aldehyde that arises from lipid peroxidation under oxidative stress that is suggested to play a role in the initiation and progression of cancer, 34 might also be a plausible candidate for pancreatic carcinogenesis. In fact, we observed a significant carcinogenic direct‐effect OR even when the analysis was restricted to never smokers (OR, 1.50; 95% CI, 1.02‐2.20) (Table 2). An experimental study that used 4‐HNE as a marker of mitochondrial oxidative stress showed that the acquisition of oncogenic KRAS mutation can initiate pancreatic cancer through increased mitochondrial oxidative stress. 35 A better understanding of the mechanism behind this carcinogenic effect of the rs671 A allele will deepen our understanding of pancreatic cancer development.
It is noteworthy that the direct and indirect effects mutually masked each other, resulting in a nonsignificant total effect of the rs671 A allele on pancreatic cancer risk. Mediation analysis enabled us to unveil this hidden relationship between ALDH2 rs671 and pancreatic cancer risk. Indeed, previous epidemiological studies, including genome‐wide association studies 36 , 37 , 38 of total effects, failed to detect any association between rs671 and pancreatic cancer. Given that the conventional approach simply identifies exposure‐outcome relationships, epidemiology has been criticized as adopting a “black box” approach to its subject matter. 39 However, by applying mediation analysis, we have been able to open the box slightly and shed new light on the mechanisms underlying the observed associations. In fact, the nonsignificant total‐effect OR of 1.15 (95% CI, 0.92‐1.44) can be decomposed into a direct‐effect OR of 1.34 (95% CI, 1.04‐1.72) and an indirect‐effect OR of 0.86 (95% CI, 0.77‐0.95), indicating the contribution of the rs671 A allele to pancreatic carcinogenesis through impaired metabolism of ALDH2 substrates as well as the association between alcohol consumption and pancreatic cancer risk.
Our study has several methodological strengths. First, we undertook two independent case‐control studies, with substantial numbers of participants and high response rates. As the HERPACC‐3 Study was carried out under an enrollment framework equivalent to that of the HERPACC‐2 Study, there should be no substantial differences in study characteristics between the two studies. However, as the HERPACC‐2 Study and the HERPACC‐3 Study were carried out between 2001 and 2005 and between 2005 and 2013, respectively, there was no overlap between the two studies in the participants they each included, and these two studies can accordingly be regarded as independent studies. By analyzing HERPACC‐2 and ‐3 separately, we successfully showed the consistency of the results between these two independent studies and ensured their robustness. Second, we had detailed information on potential confounding factors such as smoking, meat/processed meat intake, and BMI.
Several potential limitations also warrant mention. First, the information on potential confounding factors might have been affected by recall bias because the values were collected with a self‐administered questionnaire. However, information on environmental factors was collected prior to the first medical examination of participants, which possibly limited this bias, if it were present at all. Second, other unmeasured covariates might also have biased the results. Mediation analyses assume that conditioning on the covariates, there are: (a) no exposure‐outcome confounders; (b) no mediator‐outcome confounders; (c) no exposure‐mediator confounders; and (d) no mediator‐outcome confounders that are affected by the exposure. 40 Because exposure was a genetic variant occurring in a single ethnic group, our study conformed with assumptions (a) and (c). Given the specific functions of rs671 in drinking intensity, assumption (d) was also likely to hold. However, it is unclear whether assumption (b) was upheld. Nevertheless, given that we controlled for major potential confounders, we accordingly consider that assumption (b) is also likely upheld, to some degree at least. Finally, although we confirmed the consistency of the results between the two individual studies, validation of these observed associations requires replication in a larger sample size and in other populations.
In conclusion, we showed that alcohol consumption, especially heavy alcohol consumption, is a preventable exposure factor for pancreatic cancer and that the total effect of the ALDH2 rs671 A allele on pancreatic cancer can be decomposed into a carcinogenic direct effect and a protective indirect effect. This epidemiological study using mediation analysis highlighted the impact of alcohol consumption and the possible contribution of impaired metabolism of ALDH2 substrates on pancreatic cancer, and thereby provides additional evidence on the pathogenesis of pancreatic carcinogenesis.
DISCLOSURE
The authors have no conflict of interest.
Supporting information
Table S1
ACKNOWLEDGMENTS
This work was supported by Grants‐in‐Aid for Scientific Research from the Ministry of Education, Science, Sports, Culture and Technology of Japan, consisting of Priority Areas of Cancer (No. 17015018), Innovative Areas (No. 221S0001), and JSPS KAKENHI Grant (JP26253041, JP15H02524, JP16H06277, JP18H03045, JP19K19425) and a Grant‐in‐Aid for the Third Term Comprehensive 10‐year Strategy for Cancer Control from the Ministry of Health, Labour and Welfare of Japan.
Koyanagi YN, Oze I, Kasugai Y, et al. New insights into the genetic contribution of ALDH2 rs671 in pancreatic carcinogenesis: Evaluation by mediation analysis. Cancer Sci. 2022;113:1441–1450. doi: 10.1111/cas.15286
Issei Imoto and Keitaro Matsuo are editorial board members of Cancer Science (as of January 2022).
Funding information
Grants‐in‐Aid for Scientific Research from the Ministry of Education, Science, Sports, Culture and Technology of Japan: Priority Areas of Cancer, Grant/Award Number: 17015018; Innovative Areas, Grant/Award Number: 221S0001; and Japan Society for the Promotion of Science KAKENHI, Grant/Award Number: JP26253041, JP15H02524, JP16H06277, JP18H03045, and JP19K19425; Grant‐in‐Aid for the Third Term Comprehensive 10‐year Strategy for Cancer Control from the Ministry of Health, Labour and Welfare of Japan
REFERENCES
- 1. Matsuda T, Ajiki W, Marugame T, et al. Population‐based survival of cancer patients diagnosed between 1993 and 1999 in Japan: a chronological and international comparative study. Jpn J Clin Oncol. 2011;41:40‐51. [DOI] [PubMed] [Google Scholar]
- 2. Monitoring of Cancer Incidence in Japan ‐ Survival 2009‐2011 Report. Center for Cancer Control and Information Services, National Cancer Center, Japan; 2020.
- 3. World Cancer Research Fund/American Institute for Cancer Research . Continuous Update Project Expert Report 2018. Diet, nutrition, physical activity and pancreatic cancer. Available at dietandcancerreport.org
- 4. Duell EJ. Epidemiology and potential mechanisms of tobacco smoking and heavy alcohol consumption in pancreatic cancer. Mol Carcinog. 2012;51:40‐52. [DOI] [PubMed] [Google Scholar]
- 5. Cogliano VJ, Baan R, Straif K, et al. Preventable exposures associated with human cancers. J Natl Cancer Inst. 2011;103:1827‐1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chen CH, Ferreira JC, Gross ER, Mochly‐Rosen D. Targeting aldehyde dehydrogenase 2: new therapeutic opportunities. Physiol Rev. 2014;94:1‐34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Marchitti SA, Brocker C, Stagos D, Vasiliou V. Non‐P450 aldehyde oxidizing enzymes: the aldehyde dehydrogenase superfamily. Expert Opin Drug Metab Toxicol. 2008;4:697‐720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Druesne‐Pecollo N, Tehard B, Mallet Y, et al. Alcohol and genetic polymorphisms: effect on risk of alcohol‐related cancer. Lancet Oncol. 2009;10:173‐180. [DOI] [PubMed] [Google Scholar]
- 9. Miyasaka K, Hosoya H, Tanaka Y, et al. Association of aldehyde dehydrogenase 2 gene polymorphism with pancreatic cancer but not colon cancer. Geriatr Gerontol Int. 2010;10(Suppl 1):S120‐126. [DOI] [PubMed] [Google Scholar]
- 10. Kanda J, Matsuo K, Suzuki T, et al. Impact of alcohol consumption with polymorphisms in alcohol‐metabolizing enzymes on pancreatic cancer risk in Japanese. Cancer Sci. 2009;100:296‐302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chen CC, Lu RB, Chen YC, et al. Interaction between the functional polymorphisms of the alcohol‐metabolism genes in protection against alcoholism. Am J Hum Genet. 1999;65:795‐807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Matsuo K, Wakai K, Hirose K, Ito H, Saito T, Tajima K. Alcohol dehydrogenase 2 His47Arg polymorphism influences drinking habit independently of aldehyde dehydrogenase 2 Glu487Lys polymorphism: analysis of 2,299 Japanese subjects. Cancer Epidemiol Biomarkers Prev. 2006;15:1009‐1013. [DOI] [PubMed] [Google Scholar]
- 13. Koyanagi YN, Suzuki E, Imoto I, et al. Across‐site differences in the mechanism of alcohol‐induced digestive tract carcinogenesis: an evaluation by mediation analysis. Cancer Res. 2020;80:1601‐1610. [DOI] [PubMed] [Google Scholar]
- 14. Vanderweele TJ, Vansteelandt S. Odds ratios for mediation analysis for a dichotomous outcome. Am J Epidemiol. 2010;172:1339‐1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Matsuo K, Wakai K, Hirose K, et al. A gene‐gene interaction between ALDH2 Glu487Lys and ADH2 His47Arg polymorphisms regarding the risk of colorectal cancer in Japan. Carcinogenesis. 2006;27:1018‐1023. [DOI] [PubMed] [Google Scholar]
- 16. Hosono S, Ito H, Oze I, et al. A risk prediction model for colorectal cancer using genome‐wide association study‐identified polymorphisms and established risk factors among Japanese: results from two independent case‐control studies. Eur J Cancer Prev. 2016;25:500‐507. [DOI] [PubMed] [Google Scholar]
- 17. VanderWeele T, Knol M. A tutorial on interaction. Epidemiol Method. 2014;3:33‐72. [Google Scholar]
- 18. Emsley R, Liu H. PARAMED: Stata module to perform causal mediation analysis using parametric regression models. Statistical Software Components S457581, Boston College Department of Economics, revised 26 Apr 2013; 2013.
- 19. Richiardi L, Bellocco R, Zugna D. Mediation analysis in epidemiology: methods, interpretation and bias. Int J Epidemiol. 2013;42:1511‐1519. [DOI] [PubMed] [Google Scholar]
- 20. VanderWeele TJ, Tchetgen Tchetgen EJ. Mediation analysis with matched case‐control study designs. Am J Epidemiol. 2016;183:869‐870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Stata Multiple‐Imputation Reference Manual. Release 13. StataCorp LP; 2013. [Google Scholar]
- 22. Carlin JB, Li N, Greenwood P, Coffey C. Tools for analyzing multiple imputed datasets. Stata J. 2003;3:226‐244. [Google Scholar]
- 23. Bagnardi V, Rota M, Botteri E, et al. Alcohol consumption and site‐specific cancer risk: a comprehensive dose‐response meta‐analysis. Br J Cancer. 2015;112:580‐593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Shan YS, Chen LT, Wu CH, et al. No association between alcohol consumption and pancreatic cancer even among individuals genetically susceptible to the carcinogenicity of alcohol. Sci Rep. 2021;11:14567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Norton ID, Apte MV, Haber PS, McCaughan GW, Pirola RC, Wilson JS. Cytochrome P4502E1 is present in rat pancreas and is induced by chronic ethanol administration. Gut. 1998;42:426‐430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Seitz HK, Stickel F. Molecular mechanisms of alcohol‐mediated carcinogenesis. Nat Rev Cancer. 2007;7:599‐612. [DOI] [PubMed] [Google Scholar]
- 27. Duell EJ, Lucenteforte E, Olson SH, et al. Pancreatitis and pancreatic cancer risk: a pooled analysis in the International Pancreatic Cancer Case‐Control Consortium (PanC4). Ann Oncol. 2012;23:2964‐2970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Apte MV, Phillips PA, Fahmy RG, et al. Does alcohol directly stimulate pancreatic fibrogenesis? Studies with rat pancreatic stellate cells. Gastroenterology. 2000;118:780‐794. [DOI] [PubMed] [Google Scholar]
- 29. International Agency for Research on Cancer . IARC Monographs on the Evaluation of Carcinogenic Risks to Humans. Formaldehyde, 2‐Butoxyethanol and 1‐tert‐Butoxypropan‐2‐ol, Vol.88: Lyon, France: IARC; 2006. [PMC free article] [PubMed] [Google Scholar]
- 30. Hira A, Yabe H, Yoshida K, et al. Variant ALDH2 is associated with accelerated progression of bone marrow failure in Japanese Fanconi anemia patients. Blood. 2013;122:3206‐3209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Garaycoechea JI, Crossan GP, Langevin F, et al. Alcohol and endogenous aldehydes damage chromosomes and mutate stem cells. Nature. 2018;553:171‐177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Collins JJ, Esmen NA, Hall TA. A review and meta‐analysis of formaldehyde exposure and pancreatic cancer. Am J Ind Med. 2001;39:336‐345. [DOI] [PubMed] [Google Scholar]
- 33. Recktenwald CV, Kellner R, Lichtenfels R, Seliger B. Altered detoxification status and increased resistance to oxidative stress by K‐ras transformation. Cancer Res. 2008;68:10086‐10093. [DOI] [PubMed] [Google Scholar]
- 34. Zhong H, Yin H. Role of lipid peroxidation derived 4‐hydroxynonenal (4‐HNE) in cancer: focusing on mitochondria. Redox Biol. 2015;4:193‐199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Liou GY, Doppler H, DelGiorno KE, et al. Mutant KRas‐induced mitochondrial oxidative stress in acinar cells upregulates EGFR signaling to drive formation of pancreatic precancerous lesions. Cell Rep. 2016;14:2325‐2336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Low SK, Kuchiba A, Zembutsu H, et al. Genome‐wide association study of pancreatic cancer in Japanese population. PLoS One. 2010;5:e11824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wu C, Miao X, Huang L, et al. Genome‐wide association study identifies five loci associated with susceptibility to pancreatic cancer in Chinese populations. Nat Genet. 2011;44:62‐66. [DOI] [PubMed] [Google Scholar]
- 38. Lin Y, Nakatochi M, Hosono Y, et al. Genome‐wide association meta‐analysis identifies GP2 gene risk variants for pancreatic cancer. Nat Commun. 2020;11:3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hafeman DM, Schwartz S. Opening the Black Box: a motivation for the assessment of mediation. Int J Epidemiol. 2009;38:838‐845. [DOI] [PubMed] [Google Scholar]
- 40. VanderWeele TJ, Asomaning K, Tchetgen Tchetgen EJ, et al. Genetic variants on 15q25.1, smoking, and lung cancer: an assessment of mediation and interaction. Am J Epidemiol. 2012;175:1013‐1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1