

Abstract
Cancer cells secrete large amounts of extracellular vesicles (EVs) originating from multivesicular bodies (MVBs). Mature MVBs fuse either with the plasma membrane for release as EVs, often referred as to exosomes or with lysosomes for degradation. However, the mechanisms regulating MVB fate remain unknown. Here, we investigated the regulators of MVB fate by analyzing the effects of signaling inhibitors on EV secretion from cancer cells engineered to secrete luciferase‐labeled EVs. Inhibition of the oncogenic MEK/ERK pathway suppressed EV release and activated lysosome formation. MEK/ERK‐mediated lysosomal inactivation impaired MVB degradation, resulting in increased EV secretion from cancer cells. Moreover, MEK/ERK inhibition prevented c‐MYC expression and induced the nuclear translocation of MiT/TFE transcription factors, thereby promoting the activation of lysosome‐related genes, including the gene encoding a subunit of vacuolar‐type H+‐ATPase, which is responsible for lysosomal acidification and function. Furthermore, c‐MYC upregulation was associated with lysosomal gene downregulation in MEK/ERK‐activated renal cancer cells/tissues. These findings suggest that the MEK/ERK/c‐MYC pathway controls MVB fate and promotes EV production in human cancers by inactivating lysosomal function.
Keywords: c‐MYC, extracellular vesicles, lysosome, MEK/ERK, renal cancer
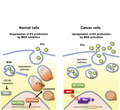
MEK/ERK‐mediated oncogenic signals promote EV secretion by lysosomal inactivation. MEK/ERK activation induces c‐MYC expression and suppresses nuclear localization of MiT/TFE transcription factors, leading to the downregulation of lysosome‐related genes critical for lysosome function.
1. INTRODUCTION
Cancer cells aberrantly secrete large amounts of extracellular vesicles (EVs) with cargo molecules different from those secreted by healthy cells 1 , 2 ; therefore, EVs have attracted widespread attention as a novel diagnostic marker that can be easily obtained through liquid biopsy. 3 EVs produced by cancer cells induce phenotypic changes in other cells in both the local and distant tumor microenvironment via the activity of their cargo, thereby supporting the establishment of the premetastatic niche and promoting organ‐specific metastasis. 4 , 5 , 6 Furthermore, EVs play a crucial role in the maintenance of cellular homeostasis as an additional mechanism for disposal of waste molecules. 7 Therefore, biogenesis of EV may be a promising target for cancer therapy; however, the mechanisms underlying the upregulation of EV biogenesis in cancer cells is largely unknown, and this lack of knowledge has hampered the development of EV‐targeting therapeutic strategy. 1 , 8 , 9
Endosome‐originated EVs known as exosomes are ~40–150 nm in diameter and contain various proteins, nucleic acids, and lipids. 7 , 10 During the maturation process of endosomes, invagination of endosomal membranes results in the formation of multivesicular bodies (MVBs) encapsulating a large number of intraluminal vesicles (ILVs). Matured MVBs face 2 alternative fates: fusion with lysosomes or autophagosomes that are driven to degradation within the cell, whereas fusion with the plasma membrane leads to the release of ILVs into the extracellular space as EVs. 8 , 11 , 12 The balance between these 2 fates is pivotal to regulate the secretion of MVB‐derived EVs in cancer cells; however, its mechanisms, such as the signaling pathways responsible for promoting MVB fusion with the plasma membrane for EV formation, still need to be defined.
In cancer cells, various signaling pathways are frequently deregulated, thereby contributing to cancer development, such is the case of the mitogen‐activated protein kinase kinase (MEK) and mitogen‐activated protein kinase (ERK) 13 , 14 that are aberrantly activated by the small G proteins Ras and Raf kinase, which are frequently mutated in several cancers. 15 , 16 The mechanisms underlying the regulation of the MEK/ERK pathway and the signaling circuits that drive the different features of cancers, including tumor growth, invasion, and metastasis, have been extensively studied since their discovery. 17 , 18 Nevertheless, very few studies have analyzed the effects of the MEK/ERK pathway on the phenotypic characteristics of cancer cells, including the upregulation of EVs. 19
In this study, the effects of known cellular signal inhibitors on EV secretion encouraged us to explore potential regulators of the fate of MVBs in cancer. Previously, we developed a system using engineered cancer cells that secrete luminescent EVs by the expression of a tetraspanin (such as CD63) fused with luciferase. 20 Detailed evaluation of these cells demonstrated that the intensity of luciferase luminescence in the cell culture medium correlated well with the number of EVs, demonstrating that this experimental system is convenient to estimate the quantity of secreted EVs including exosomes. Here, this system was used to study the molecular mechanisms underlying the upregulation of EV biogenesis in cancer cells. Based on the results, we propose a MYC‐MiT/TFE‐mediated mechanism by which MEK/ERK activation in cancer cells suppress lysosome function, causing the upregulation of EV secretion, which is, in turn, shown to be crucial for the growth of MEK/ERK‐activated and lysosome‐inhibited cancer cells. We further demonstrate that MEK/ERK activation is associated with the upregulation of MYC and downregulation of a subunit of the lysosomal vacuolar‐type H+ ATPase (V‐ATPase) proton pump, ATP6V1B1, in renal cancer tissues. These findings suggest that the MEK/ERK‐MYC‐MiT/TFE‐lysosomal pathway is pivotal for EV secretion and proliferation in human cancer cells in which the MEK/ERK oncogenic pathway is hyperactivated.
2. MATERIALS AND METHODS
2.1. Cell culture
Human colon cancer HT29 cells were obtained from the American Type Culture Collection. Mouse embryonic fibroblasts (MEFs) immortalized by large‐T antigen were kindly provided by Dr. Akira Imamoto. Human renal cancer Caki‐1, KMRC‐1, and VMRC‐RCW cells were obtained from the Japanese Collection of Research Bioresources Cell Bank, National Institutes of Biomedical Innovation, Health, and Nutrition (Osaka, Japan). Cells were cultured in the following media: HT29 in McCoy's 5A medium (Gibco), MEF and KMRC‐1 in Dulbecco’s modified Eagle's medium (DMEM; Sigma‐Aldrich), and Caki‐1 and VMRC‐RCW in Minimum Essential Medium Eagle (Sigma‐Aldrich). All media were supplemented with 10% fetal bovine serum (FBS; Gibco). The cells were cultured at 37°C in a humidified 5% CO2 chamber.
2.2. Chemicals
Chemicals were obtained commercially: U0126 (662005) from EMD/Calbiochem, GW4869 (D1692) from Millipore Sigma, MYCi975 (S8906) from Selleck Chemicals, trametinib (HY‐10999) from MedChemExpress, and bafilomycin A1 (BVT‐0252) from Adipogen AG.
2.3. Acidic lysosome detection
Cells on a glass‐bottomed dish were cultured in medium containing Lysotracker Red DND‐99 (50 nM) (Thermo Fisher Scientific) for 30 min, and then the medium was replaced with Lysotracker‐free medium, followed by observation of the cells using a ZEISS LSM 800 with Airyscan confocal microscope (Carl Zeiss).
2.4. Quantitative RT‐PCR
Quantitative RT‐PCR was performed as previously described. 21 Relative gene expression levels were calculated using 18S ribosomal RNA or GAPDH sequence as control, as per the method. Primer sequences are described in Table S1.
2.5. Western blotting
Western blotting was performed as previously described. 22 The following antibodies were used: anti‐Alix (ABC40) from Millipore Sigma; anti‐Tsg101 (C‐2), anti‐CD63 (MX‐49.129.5), anti‐ATP6V1B1/2 (F‐6) and anti‐GAPDH (6C5) from Santa Cruz Biotechnology; anti‐phospho‐ERK1/2 (D13.14.4E), anti‐ERK1/2 (137F5), anti‐phospho‐MEK1/2 (41G9), anti‐MEK1/2 (L38C12), and anti‐Ras (27H5) from Cell Signaling Technology; anti‐c‐MYC (Y69) from Abcam; anti‐TFEB (PA5‐34360) from Thermo Fisher Scientific; and anti‐TFE3 (HPA023881) from Sigma‐Aldrich65. All antibodies were used at a 1:1000 dilution.
2.6. Immunofluorescence staining
Immunocytochemistry was performed as described previously. 22 Fluorescence was observed using a ZEISS LSM 800 with Airyscan confocal microscope (Carl Zeiss).
2.7. Gene expression and gene silencing
Ectopic gene expression and site‐directed mutagenesis were performed using pCX4 retroviral vectors. 22 Each gene was PCR amplified and subcloned into the pCX4 vector. Virus production and infection were performed as previously reported. 23 Lentiviral vectors carrying each gene were purchased from Sigma‐Aldrich.
2.8. Soft‐agar colony formation assay
Soft‐agar colony formation assay was performed as previously described. 22 Colonies were stained with 3‐(4,5‐dimethylimidazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide (Sigma‐Aldrich), and micrographs were used to count the colony numbers.
2.9. Electron microscopy and lysosome/MVB quantification
Cells cultured on dishes were washed with phosphate‐buffered saline (PBS) and fixed for 1 h in 2.5% glutaraldehyde in 0.1 M phosphate buffer at room temperature. Then, the cells were slowly and gently scrapped and pelleted. Pellets were washed with phosphate buffer and incubated with 1% OsO4 for 90 min at 4°C. The samples were then dehydrated, embedded in Spurr's resin, and sectioned using a ultramicrotome (Leica Microsystems). Ultrathin sections (50–70 nm) were stained with 2% uranyl acetate for 10 min and with a lead‐staining solution for 5 min, and observed using a JEM‐1010 transmission electron microscope (JEOL) fitted with an Orius SC1000 (model 832; Gatan) digital camera. For the calibration of images, quantification, and analysis, ImageJ software (https://imagej.nih.gov/ij/) was used. MVBs were identified by morphology and counted; MVBs contain only discrete ILVs, whereas lysosomes contain multilamellar profiles. At least 14 MVBs were analyzed per experiment from separate cells. Data were analyzed from duplicate or triplicate independent experiments and 2‒4 grids were used for each condition. The minimum number of cells scored for each condition was 20.
2.10. Gene expression analysis of several cancer samples
Data analysis was performed using FireBrowse (http://firebrowse.org/), which provides access to data stored in The Cancer Genome Atlas (TCGA) database.
2.11. Quantification of EVs
NanoLuc luciferase assay was performed as described previously 20 using a Nivo multiplate reader (PerkinElmer). For EV preparation, cells were seeded onto a 150‐mm culture dish at the density of 5 × 106 cells, and cultured for 24 h. After washing with 20 ml PBS 2 times, the culture medium was replaced with 13 ml 1% EV‐depleted FBS containing medium. EV‐depleted FBS was prepared by ultracentrifuging at 110,000 g for 16 h at 4°C. After 48 h of culture, the culture supernatant was harvested and final cell amounts were counted to confirm no growth differences arose among the different conditions or cells. For nanoparticle tracking analysis (NTA), cell supernatant was centrifuged at 2000 g for 10 min at 4°C to remove cells and cellular debris, and then filtered through a 0.22‐μm filter (Millipore Sigma) and ultracentrifuged at 110,000 g for 70 min at 4°C (SW41Ti rotor; Beckman Coulter). Apolipoprotein A1, a non‐EV marker, in the sample was checked to be below the detection limit. The size distribution and concentration of the EVs were determined by NTA using a NanoSight LM10 (Malvern Panalytical). 24
2.12. Immunohistochemical analysis
Kidney carcinoma with matched kidney tissue array (KD244 and KD244a) was purchased from US Biomax. Briefly, tissue array slides were deparaffinized and treated at 120°C for 10 min with citrate buffer (pH 6.0). After washing with PBS, endogenous peroxidase was blocked with 0.3% H2O2 for 30 min. The slides were blocked with 5% bovine serum albumin in Tris‐buffered saline solution with 0.1% Tween 20 for 30 min, and then incubated with the primary antibody or IgG isotype control overnight at 4°C. After washing twice with PBS, the slides were incubated with Envision+Dual Link System‐HRP (Dako) for 30 min. The slides were developed using the Peroxidase Stain DAB Kit (Nacalai Tesque), a chromogenic substrate for peroxidase, and counterstained with hematoxylin. 21 The images were visualized under a BZ‐X710 fluorescence microscope (Keyence).
2.13. Gene expression data of kidney cancer samples
For gene expression data of kidney renal clear cell carcinoma patients from TCGA project, we downloaded text files containing fragments per kilobase of transcript per million mapped reads upper quartile (FPKM‐UQ) values from the Genomic Data Commons Data Portal (https://portal.gdc.cancer.gov/). For tumor samples, we selected a sample for each patient if it was annotated with “Primary Tumor” in a sample sheet from the portal site, which resulted in 530 samples. For normal samples, we selected a sample for each patient if it was annotated with “Solid Tumor Normal,” which resulted in 72 samples.
2.14. Gene set variation analysis
To estimate the variation of gene set enrichment through the samples of an expression data set, gene set variation analysis (GSVA), 25 a nonparametric unsupervised method, was performed on TCGA data sets using R software to obtain GSVA enrichment scores for lysosome‐related gene sets in MSigDB 7.1. 26 , 27 For GSVA, FPKM‐UQ gene expression values were transformed with log2 after addition of 1.
2.15. Statistical analysis
Data are presented as mean ± standard deviation. Statistical significance was calculated using Student t test or one‐way ANOVA with Dunnett's post hoc analysis using XLSTAT for Microsoft Excel. Test results were reported as two‐tailed p‐values, where p < 0.05 was considered statistically significant.
3. RESULTS
3.1. MEK/ERK pathway promotes EV secretion via suppression of lysosomal function
The effect of inhibitors on EV secretion in human colon cancer HT29 cells expressing CD63‐Nluc was evaluated (Figure 1A). U0126, a MEK inhibitor, decreased the luminescence of the culture medium of HT29/CD63Nluc cells in a dose‐dependent manner (Figures 1A and S1A), concurrent with the inhibition of downstream ERK activation (pERK) (Figure S1B). CD63‐positive EVs from HT29 cells were 100–150 nm in size and verified to contain MVB‐associated proteins, such as Alix and Tsg101, which were suppressed upon MEK inhibition (Figures 1B and S1C). An approved MEK‐targeting drug trametinib also inhibited EV secretion (Figure S1D). As inhibitors potentially have multiple effects, we confirmed the effect of MEK activation on EV secretion. Consistently, EV secretion significantly increased in transformed MEFs by constitutive activation of MEK, and this effect was almost reverted to basal levels by the addition of U0126 (Figure S1E). These findings suggest that the MEK/ERK pathway is involved in EV secretion.
FIGURE 1.
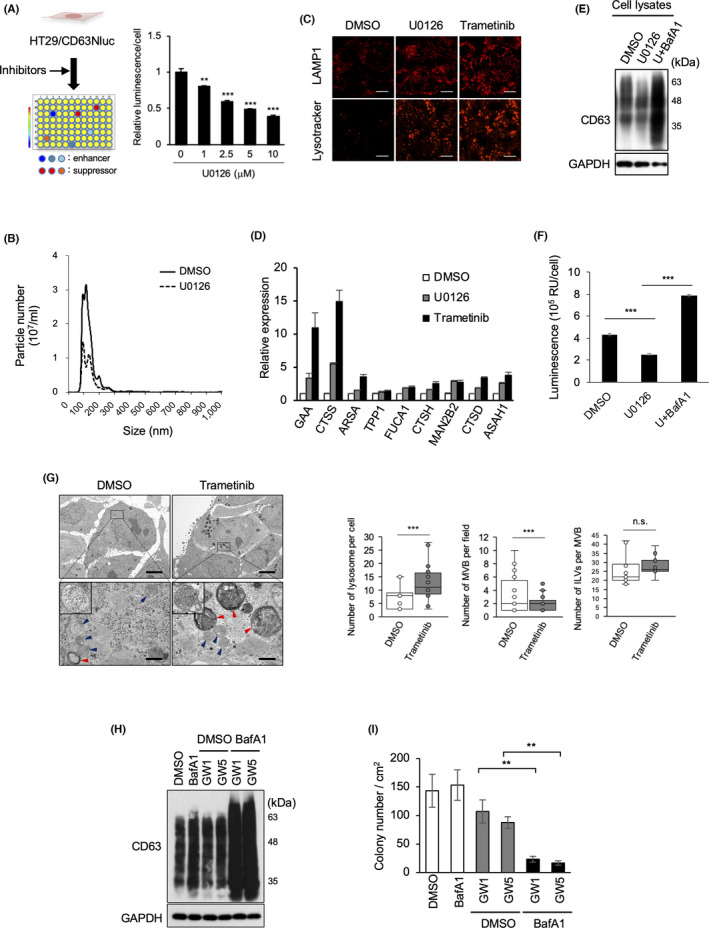
MEK/ERK activation promotes EV secretion and impairs lysosome function. (A) Schematic representation of the selection of EV inhibitors. After CD63Nluc‐expressing HT29 cells (HT29/CD63Nluc) were treated with signaling inhibitors, luminescence in the culture medium was analyzed. Relative luminescence normalized by cell viability in the presence for 24 h of the MEK inhibitor U0126 at the indicated concentrations. (B) HT29 cells treated with or without U0126 for 24 h were used for EV preparation followed by nanoparticle tracking analysis of isolated particles. (C) HT29 cells were treated with DMSO, 10 μM U0126, or 100 nM trametinib for 24 h, and then subjected to immunostaining for anti‐LAMP1 and Lysotracker Red DND‐99. Scale bars, 10 μm. (D) Expression analysis of lysosomal genes in cells used in (C). (E) Total cell lysates from HT29 cells treated with DMSO, 10 μM U0126, 10 μM U0126, or 50 nM bafilomycin A1 (BafA1) for 24 h were immunoblotted with the indicated antibodies. (F) HT29/CD63Nluc cells were treated with DMSO or 10 μM U0126, and 50 nM bafilomycin A1 for 24 h, luminescence in the culture medium was analyzed. (G) Electron microscopy images of MVBs (blue arrowheads) and lysosomes (red arrowheads) in HT29 cells treated with DMSO or trametinib (100 nM) for 24 h are presented in upper panels with a scale bars representing 5 μm. Boxed areas with dotted line are enlarged at lower panels with a scale bar of 500 nm. High magnification pictures of MVB are shown in insets of lower panels. Quantification of number of lysosomes per cell (n = 17: DMSO, n = 25: trametinib), MVBs in more than 21 fields per condition, and ILVs per MVB (n ≥ 14). Boxes represent the interquartile range and line inside the box represents median value. (H) Total cell lysates from HT29 cells treated for 48 h with 1 nM BafA1, and/or GW4869 (GW) at 1 or 5 μM were immunoblotted with the indicated antibodies. (I) HT29 cells treated with DMSO, or 1 nM BafA1, and/or GW at 1 or 5 μM were used for the soft‐agar colony formation assay. Relative colony numbers obtained from 3 independent experiments are shown. Data are represented as means ± standard deviation of 3 independent measurements. Statistical analysis was performed using one‐way ANOVA. **p < 0.01, ***p < 0.001; n.s., not significant
Next, the mechanism by which inhibition of MEK mediates the suppression of EV secretion was assessed by the reanalysis of the gene expression profile of HT29 cells treated with or without U0126 (available data set: GSE18232 28 ). Pairwise significance analysis of the microarray data revealed that 3070 and 899 genes were upregulated and downregulated, respectively, upon MEK inhibition. In gene ontology analysis using the DAVID bioinformatics resource, 29 extracellular exosome (139 genes) and lysosome‐related gene (24 genes) categories were enriched among the upregulated genes (Figure S1F). Therefore, the impact of MEK inhibition on lysosome function was evaluated next. Levels of LAMP1, a lysosomal biomarker, and lysosome acidity, visualized by Lysotracker staining, were found to be considerably increased in HT29 cells upon MEK inhibition (Figure 1C). Moreover, the expression of lysosome‐related genes was upregulated in HT29 cells treated with MEK inhibitors (Figure 1D).
To verify the significance of lysosome activation on suppression of EV release, we examined the effect of bafilomycin A1. The levels of cellular CD63 protein, as a marker of MVBs, were downregulated in U0126‐treated HT29 cells and restored by the addition of the lysosome inhibitor bafilomycin A1, although CD63 expression remained unaltered (Figures 1E and S1G). U0126‐induced suppression of EV secretion was significantly restored by addition of bafilomycin A1 (Figure 1F) without activation of ERK (Figure S1H). Therefore the upregulation of EV secretion by bafA1 treatment (Figure 1F) can be attributed to the suppression of lysosomal activity below basal level (Figure S1I). By MEK inhibition, the LC3‐positive foci were not emerged and the conversion of LC3 to LC3‐Ⅱ was not induced, suggesting that autophagic activity is ignorable and Bafilomycin A1 would mainly inhibit lysosome formation in our experimental conditions (Figure S1J,K). Altogether, these findings demonstrated that MEK inhibition promotes lysosome biogenesis and activity via upregulation of lysosomal gene expression, further suggesting that the upregulation of EV production in cancer cells is due to the inactivation of lysosomes downstream of the MEK/ERK pathway. Electron microscopy analysis of HT29 cells demonstrated that the number of lysosomes was significantly increased by trametinib treatment; conversely, the number of MVBs was markedly decreased (Figure 1G).
The apparent compensation between EV secretion and lysosome activation led us to a hypothesis that EV secretion acts as an alternative mechanism to avoid accumulation of MVBs. To examine the consequence of the accumulation of MVB in cancer cells, the effects of low concentrations of bafilomycin A1 and GW4869, an inhibitor of N‐sphingomyelinase and exosome secretion, on cancer cells were assessed. Neither inhibitor affected the cellular amount of the MVB biomarker CD63 (Figure 1H); bafilomycin A1 did not inhibit the growth of cancer cells, and GW4869 moderately suppressed cancer cell growth in a dose‐dependent manner (Figure 1I), as previously reported. 24 Surprisingly, the presence of both inhibitors synergistically attenuated the growth of the cancer cells (Figures 1I and S1L), with stronger band intensity of CD63 suggesting the retention of MVBs in the cells (Figure 1H).
3.2. Nuclear localization of TFE3 regulates ATP6V1B1‐mediated lysosomal acidification under MEK/ERK pathway
As alteration of V‐ATPases expression might impair the ability of lysosomes to maintain low pH, 30 , 31 , 32 the expression of genes encoding V‐ATPase subunits was evaluated upon MEK inhibition. Analysis of a microarray data set (GSE18232) revealed that MEK inhibition upregulated several lysosomal V‐ATPase component genes (data not shown), including ATP6V1B1, which encodes subunit B of V1‐ATPase. Upregulation of ATP6V1B1 was confirmed by real‐time quantitative reverse transcription polymerase chain reaction (qRT‐PCR) (Figure S2A). Moreover, MEK inhibition increased ATP6V1B1 levels in HT29 cells, as determined by western blot analysis (Figure S2B). Lysotracker staining showed that lysosome acidification was increased upon ATP6V1B1 overexpression in HT29 cells (Figure S2C,D), whereas EV secretion was significantly downregulated (Figure S2E). Conversely, ATP6V1B1 was knocked down in nontransformed MEFs, lysosome acidification was suppressed, and EV secretion was increased (Figure S2F–H). Together, these findings suggest that MEK inhibition induces ATP6V1B1 expression and subsequently enhances lysosome activity, leading to the suppression of EV secretion.
Next, we evaluated the effects of MEK inhibition on the microphthalmia family of basic helix‐loop‐helix‐leucine‐zipper transcription factors (MiT/TFE), including TFEB, TFEC, TFE3, and MITF, given their reported contribution for the regulation of lysosomal gene expression. 33 , 34 Immunofluorescence analysis revealed that TFEB and TFE3 were distributed in the cytoplasm of HT29 cells. After addition of MEK inhibitors, most TFE3 translocated into the nucleus without a change in expression (Figures 2A and S3A,B), whereas TFEB showed partial nuclear localization. The expression of MITF was observed in the cytoplasm at quite low levels before and after the treatment with MEK inhibitors (Figure 2A). Similar MEK/ERK activity‐dependent TFE3 localization was observed in MEK‐transformed MEFs (Figure S3C,D).
FIGURE 2.
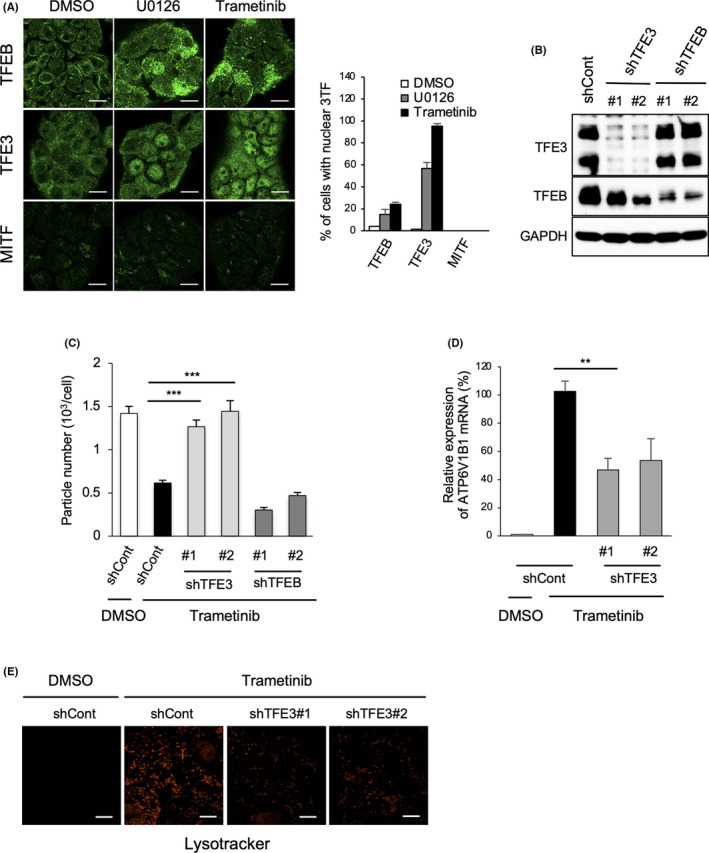
MEK/ERK inhibition induces nuclear localization of MiT/TFE transcription factor family members. (A) TFEB, TFE3, and MITF immunostaining using HT29 cells treated with DMSO, U0126 (10 μM), and trametinib (100 nM) for 24 h. The cells harboring nuclear TFEB, TFE3, and MITF colocalized with DAPI were counted as positive cells, and the ratio (%) of cells with nuclear localization of the 3 transcription factors was calculated by dividing positive cell number with total cell number (n > 50 cells, pooled from 3 independent experiments). Quantification of the ratio (%) of cells with nuclear localization of the 3 transcription factors (n > 50 cells, pooled from 3 independent experiments). (B) HT29 cells expressing control (shCont), TFE3 (shTFE3), or TFEB (shTFEB) shRNAs were immunoblotted with the indicated antibodies. (C) Cells indicated in (B) were treated with or without trametinib (100 nM) for 48 h and subjected to nanoparticle tracking analysis for the quantitative measurement of isolated particles. (D) ATP6V1B1 expression was assessed by real‐time quantitative reverse transcription polymerase chain reaction in the cells indicated in (C). (E) Cells indicated in (C) were stained with Lysotracker red. Scale bars, 10 μm (A,E). Data are represented as mean ± standard deviation of 3 independent measurements. Statistical analysis was performed using one‐way ANOVA. **p < 0.01, ***p < 0.001
To examine whether the MEK/ERK‐TFE3 axis could lead to lysosomal inactivation and upregulation of EV secretion, knockdown experiments were performed next (Figure 2B). TFE3 knockdown restored the trametinib‐induced downregulation of EV secretion in HT29 cells without trametinib, whereas TFEB knockdown did not (Figure 2C). Under the same conditions, knockdown of TFE3 markedly suppressed trametinib‐induced upregulation of ATP6V1B1 expression and lysosome acidification (Figure 2D,E). These results indicated that MEK/ERK‐mediated exclusion of TFE3 from the nucleus suppresses lysosome activity, accompanied by the upregulation of EV secretion from cancer cells.
3.3. MEK/ERK pathway regulates MYC expression to control the localization of TFE3
The subcellular localization and activity of the MiT/TFE transcription factor family is known to be regulated through phosphorylation by the mammalian target of rapamycin (mTOR) or ERK2 35 , 36 : phosphorylated MiT/TFE is retained in the cytoplasm, whereas dephosphorylated MiT/TFE is translocated into the nucleus. 34 , 37 In addition, c‐MYC was recently reported to be involved in regulating lysosomal genes. 38 The presence of the mTOR inhibitor rapamycin did not induce lysosome acidification unlike U0126 or trametinib (Figure S3E). Moreover, TFE3 mutants with putative phosphorylation sites recognized by ERK or mTOR 39 (Figure S3F) also failed to show nuclear localization; this result contrasted those obtained upon treatment with the MEK inhibitor (Figure S3G–I). These observations suggest that the translocation of TFE3 into the nucleus induced by MEK inhibitor was not due to the inhibition of phosphorylation by ERK or mTOR. Alternatively, shRNA‐mediated MYC knockdown in HT29 cells was found to induce the nuclear localization of TFE3 and concomitant lysosome acidification, as well as upregulation of lysosome‐related genes (Figure 3A,B). More importantly, MYC knockdown significantly decreased EV secretion (Figure 3C) and the number of lysosomes and MVBs were changed (Figure S4A) as shown similarly by trametinib treatment (Figure 1G). The importance of c‐MYC in lysosome function and EV secretion was also confirmed by an inverse correlation between Lysotracker signal (in cells) and CD63Nluc luminescence (in culture medium) in HT29/CD63Nluc cells upon treatment with c‐MYC inhibitor MYCi975 40 (Figures 3D and S4B). Drastic reduction in c‐MYC expression by the treatment of trametinib demonstrated that MEK/ERK signaling regulates MYC expression (Figures 3E and S4C,D). To further verify the significance of MYC expression on MEK/ERK‐mediated regulation of TFE3 localization, we performed rescue experiments by exogenous expression of c‐MYC. Although the expression of MYC protein in the MYC‐introduced cell after treatment of trametinib was not marked, it was sufficient to restore cytoplasmic localization of TFE3 in these cells (Figure 3F,G). To confirm the contribution of TFE3 translocation to the reduction of EV secretion in MYC‐downregulated cancer cells, EV secretion was compared between MYC knockdown and MYC/TFE3 knockdown HT29 cells. As shown in Figure S4E,F, decrease of EV secretion by MYC knockdown was completely restored by TFE3 knockdown, suggesting that the regulation of EV secretion by MYC is dominantly caused via TFE3. These findings suggest that MEK/ERK‐mediated MYC expression plays a crucial role in the regulation of EV secretion from cancer cells through controlling TFE3 translocation.
FIGURE 3.
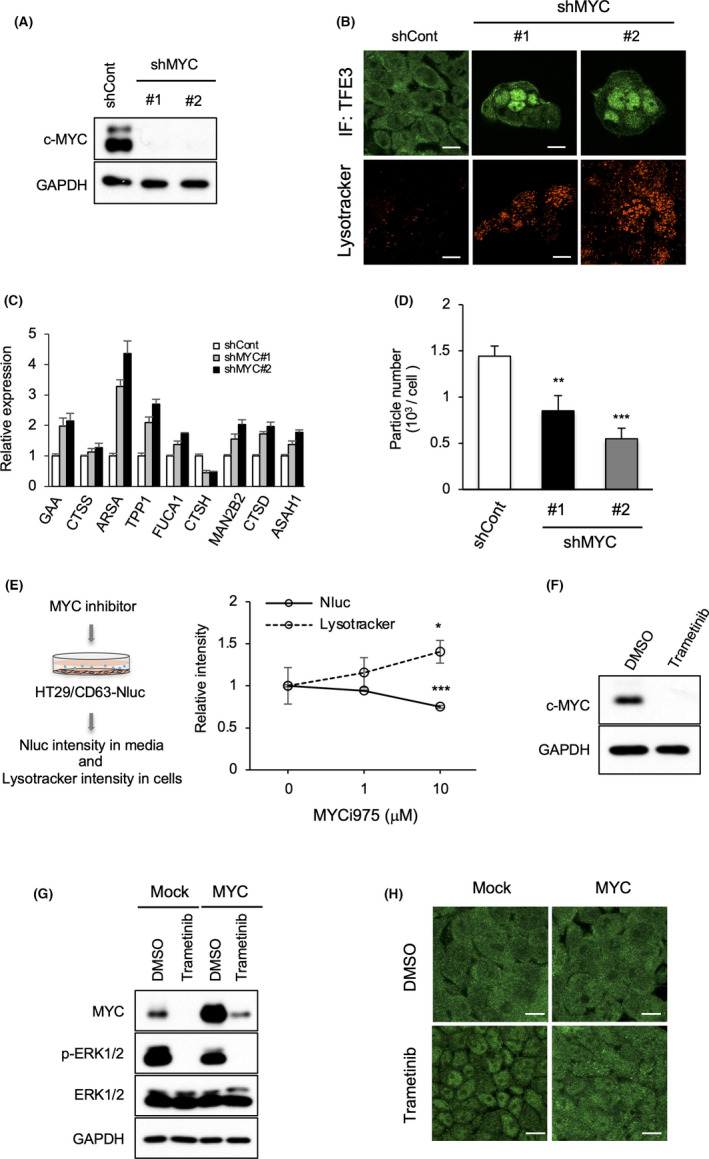
MEK/ERK‐mediated MYC upregulation controls lysosome function and EV secretion. (A) Cell lysates from HT29 cells expressing control (shCont) or MYC (shMYC) shRNAs were immunoblotted with the indicated antibodies. (B) Cells indicated in (A) were stained for anti‐TFE3 (green) or Lysotracker (red). Scale bars, 10 μm. (C) Nanoparticle tracking analysis for the isolated particles was performed using cells indicated in (A). (D) HT29 cells expressing CD63Nluc (HT29/CD63Nluc) were treated with c‐MYC inhibitor (MYCi975) at different concentrations for 24 h. (E) Relative luminescence intensity of CD63Nluc (solid lines) in the culture medium and Lysotracker‐positive intensity (dotted lines) in these cells are shown. (F) Total cell lysates from HT29 cells treated with or without trametinib (100 nM) for 24 h were immunoblotted with anti‐MYC antibody. (G) HT29 cells transfected with control (mock) or MYC were treated with DMSO or trametinib (100 nM) for 24 h, and the total cell lysates were immunoblotted with the indicated antibodies. (H) Cells indicated in (G) were stained for anti‐TFE3 (green). Scale bar, 10 μm (B, H). Data are represented as mean ± standard deviation of 3 independent measurements. Statistical analysis was performed using one‐way ANOVA. *p < 0.05, **p < 0.01, ***p < 0.001
3.4. MEK/ERK‐MYC‐lysosomal genes axis in human cancer tissues
To address the functional relevance of the MEK/ERK‐MYC‐lysosomal genes pathway to human cancers, expression of MYC and ATP6V1B1 was examined in various types of cancer and match‐paired adjacent normal tissues using publicly available data from TCGA database. MYC was highly expressed in several cancers, including colon, rectum, and colorectal adenocarcinoma, glioblastoma, and glioma, kidney cancers, lung squamous cell carcinoma, prostate adenocarcinoma, and skin cutaneous melanoma (Figure S5A). Analysis of ATP6V1B1 expression in these particular cancers revealed that it was much lower in renal tumors than in the match‐paired normal tissues (Figure S5B). Reassessment of renal cancer carcinoma (RCC) from the TCGA RNA‐seq data confirmed that MYC expression was significantly higher and ATP6V1B1 expression was lower in tumor tissues than in their normal counterparts (Figure 4A). Similarly, other RCC data sets from the Gene Expression Omnibus database also showed an inverse correlation between MYC and ATP6V1B1 expression (Figure S5C–E). These findings highlighted a potential correlation between MYC expression and lysosome function; therefore, a GSVA was performed in RCC samples. Consistently, an inverse correlation between the expression of MYC and lysosome‐related genes was observed when analyzing a large data set of renal cancers from the Kyoto Encyclopedia of Genes and Genomes database (p = 0.000323; Reactome, p = 0.0003729; Figure 4B). Furthermore, immunohistochemical analysis of the 24 primary tumors revealed that pERK (indicative of MEK/ERK activation) was greatly increased in most of the 24 primary tumor regions compared with adjacent normal tissues in agreement with previous findings (Figure 4C). 41 Notably, MiT/TFE proteins, including TFEB, TFE3, or MITF, were prominently localized in the nucleus and cytoplasm in normal tissues and tumor samples, respectively. Moreover, ATP6V1B1 and LAMP1 expressions were markedly downregulated in primary tumor tissues. These observations suggested a correlation between pERK/MYC expression and nuclear localization of MiT/TFEs, and the expression of lysosomal proteins in human cancer tissues.
FIGURE 4.
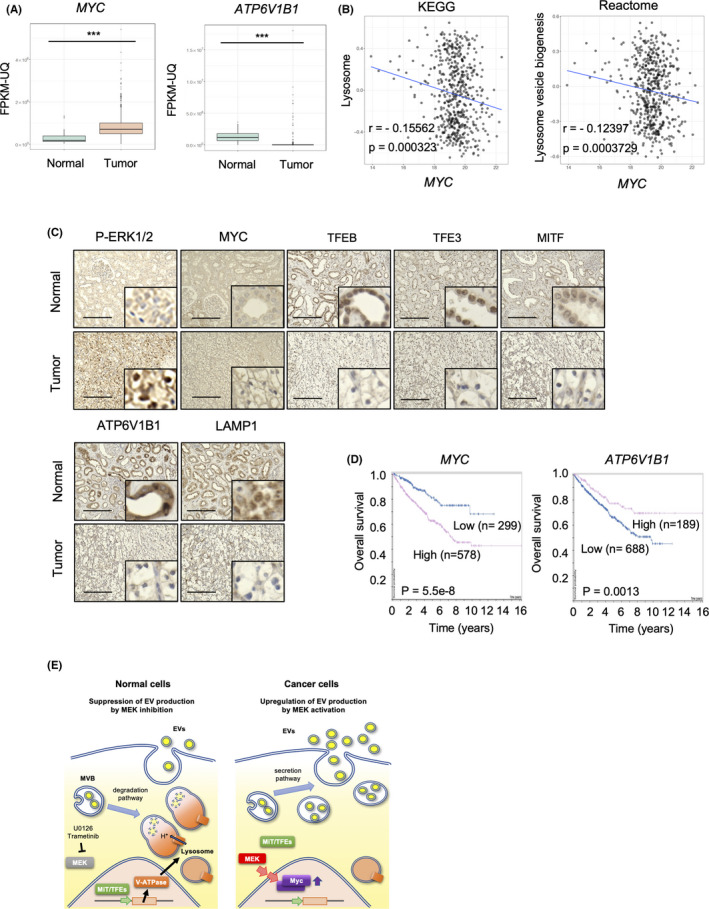
MEK/ERK‐MYC‐MiT/TFE‐lysosomal gene axis in renal cell carcinomas (RCCs). (A) MYC and ATP6V1B1 levels in several types of renal tumors and adjacent normal tissues. The expression of each gene in primary renal tumor (red) and normal tissue (blue) was analyzed (TCGA data set). Boxes represent the interquartile range and line inside the box represents median value. (B) Gene set variation analysis of MYC and lysosome‐related gene expression in MSigDB 7.1. (C) Immunohistochemistry for pERK, c‐MYC, TFEB, TFE3, MITF, ATP6V1B1, and LAMP1 in RCC and matched‐adjacent normal renal tissues (magnification: ×20). Inset: ×160 magnified view. Representative images from 24 samples are shown. Scale bars, 100 μm. (D) Kaplan‐Meier plots of MYC and ATP6V1B1 in renal cancer (TCGA data set). (E) Schematic model of MEK/ERK pathway role in regulating the fate of MVBs and control EV secretion. In normal cells, the MEK/ERK pathway is inactivated, presumably preserving the expression of lysosomal genes, including vacuolar‐type H+‐ATPase, and maintaining lysosome function. MVBs are degraded and therefore the number of MVBs fusing with the plasma membrane is low, leading to suppression of EV secretion. In cancer cells, when the MEK/ERK pathway is activated, MYC expression is increased and MiT/TFEs transcription factors are retained in the cytoplasm, inactivating the transcription of lysosomal genes and disrupting lysosome function. A large amount of MVBs can therefore fuse with the plasma membrane, promoting EV secretion. Statistical analysis was performed using one‐way ANOVA. ***p < 0.001
Next, the impact of MYC and ATP6V1B1 expression on the overall survival rate of RCC patients was investigated (Figure 4D). A cohort of 877 patients was classified into 2 groups based on their expression of each gene. At 10 y after diagnosis, 70% of the “low expression” group of MYC were alive, whereas 40% of the “high expression” group of MYC survived (p = 5.5 × 10−8, Figure 4D [left panel]). By contrast, 70% of the “high expression” group of ATP6V1B1 survived until 10‐year after diagnosis, whereas 40% of the “low expression” group of ATP6V1B1 survived for the same time (p = 0.0013, Figure 4D [right panel]). These results indicate a strong correlation between high and low expression of MYC and ATP6V1B1, respectively, and the poor prognosis of RCC patients. Overall, the present findings suggest that upregulation of MYC is induced by the activation of the MEK/ERK oncogenic pathway in various human cancers and is associated with the promotion of EV secretion via MiT/TFE‐mediated downregulation of lysosome biogenesis and activity.
4. DISCUSSION
This study revealed that the oncogenic MEK/ERK pathway affects the fate of MVBs by inactivating lysosome activity and the consequential upregulation of EV secretion. A schematic model for the MEK/ERK‐mediated regulation of the balance between the degradative and secretory fate of MVBs is depicted in Figure 4E. In normal cells, MEK/ERK activity is low, MYC expression is suppressed, thereby allowing MiT/TFE family members to localize into the nucleus and retain the transcription of lysosome‐related genes, such as ATP6V1B1. Expression of lysosomal genes in turn induces lysosome formation and promotes MVB degradation. Therefore, the secretion of endosome‐originated EVs is suppressed due to the depletion of MVBs (Figure 4E [left panel]). In cancer cells harboring MEK/ERK activation, MYC expression is upregulated and MiT/TFE members are localized in the cytoplasm, resulting in the repression of lysosome‐related genes and in the disruption of the lysosome pathway. Therefore, MVBs accumulate in cells and are destined to fuse with the plasma membrane for release of the inclusions as EVs (Figure 4E [right panel]). Currently, it is difficult to experimentally determine the origin of the secreted EVs and, therefore, whether they are so‐called exosomes or do not need to be investigated in the future. Therefore, the general term EV is used in this article in accordance with the recommendation of the international society for EVs. 42 It is noted that we present here the simplest hypothesis in which the observed changes in the secretion of EVs are directly attributable to changes in MVB.
Considering that the MEK/ERK pathway is activated under the control of various oncogenic signaling pathways, including Ras and EGFR, 15 , 16 , 43 c‐MYC‐mediated upregulation of EV in cancer cells might be explained by similar mechanisms. Indeed, MEK/ERK‐mediated upregulation of c‐MYC and EV secretion in HRas‐ and KRas‐transformed cells was observed (Figure S6A–C). A previous report indicated that the stability of c‐MYC protein is regulated by ERK‐mediated phosphorylation 44 ; however, in the present study, MYC expression was regulated at the mRNA level (Figure S4C). Further extensive analysis will be necessary to elucidate the detailed mechanism of MEK/ERK‐mediated MYC expression; whereas the apparent positive correlation between MYC expression and MEK/ERK activity in human cancers would provide a missing link between c‐MYC, MEK/ERK, and EV secretion. Mechanisms driving the upregulation of EV secretion, which are activated by Src oncogenic signals, were also demonstrated to promote ILV formation in cancer cells. 24 , 45 Here it was shown that the MEK/ERK pathway does not change the number of ILV in MVBs (Figure 1G) and inhibition of Src does not induce activation of lysosome function (Figure S3E), suggesting that the mechanisms underlying the upregulation of EV secretion would differ between oncogenic signals. Very recently, it has been reported that EV secretion is enhanced by MYC overexpression, but can be attenuated by MEK in triple‐negative breast cancer (TNBC) cells. 46 We found that MEK inhibition decreased MYC expression in in a type of TNBC cells, MDA‐MB‐231 (Figure S7D). A MYC‐induced increase in EV secretion and decrease in the expression of lysosomal genes were observed in these cells as well. These results support the idea that the activation of the MEK/ERK‐MYC‐MiT/TFE‐lysosomal pathway can promote EV secretion in MDA‐MB‐231 cells. Therefore, although MEK activation induces the pathway driving the increase of EV secretion, it can be reduced by other cell type‐dependent pathways downstream of MEK.
We found that c‐MYC prevents the localization of MiT/TFE proteins into the nucleus (Figure 3B). A recent study demonstrated that cyclin‐dependent kinase CDK4/6 phosphorylates MiT/TFE in the nucleus, promoting their shuttling to the cytoplasm. 47 Considering that CDK4/6 is a transcriptional target of c‐MYC, CDK4/6 downregulation by MYC disruption might prevent MiT/TFE phosphorylation, thereby retaining them in the nucleus and resulting in the MiT/TFE‐mediated transcription of lysosomal genes. 48 Additional extensive analysis will be necessary to elucidate the precise mechanism underlying regulation of MYC‐mediated MiT/TFE subcellular localization. The present data demonstrating that repression of MYC suppresses EV secretion via upregulation of lysosomal genes provide the first firm evidence of MYC‐mediated EV secretion. As MYC overexpression is frequently observed in human cancers, 49 , 50 this mechanism is expected to contribute to EV upregulation in several cancers. Our findings also suggest that targeting MYC expression/function would effectively prevent EV secretion in several types of cancers, regardless of the status of the MEK/ERK pathway (Figures 3 and S7A–C).
Of note, overaccumulation of MVBs in cells was found to suppress the growth of cancer cells (Figure 1H,I). In agreement, it was reported that reduction of EV secretion by targeting Alix or Rab27b, or in the presence of GW4869, suppresses cell transformation, suggesting that overaccumulation of MVBs due to dysfunctional EV secretion suppresses the growth of cancer cells. 24 Several studies have shown that cancer cells use EVs to excrete waste molecules, such as unfolded proteins and damaged DNA. 1 , 51 These lines of evidence also suggest that the turnover of MVBs is important to support the growth of cancer cells, possibly by discarding harmful molecules via lysosomal digestion or extracellular excretion.
The inverse correlation observed between MEK/ERK activation and V‐ATPase expression in RCC tissues suggests that the MEK/ERK pathway has a crucial role in the upregulation of EV biogenesis in these cancers. Indeed, RCC cells, in which MEK/ERK inhibition induced the downregulation of MYC, exhibited activation of lysosome function and subsequent suppression of EV secretion (Figure S6D–F). The poor prognosis of RCC patients with high MYC expression associated with low ATP6V1B1 expression might suggest that the excretion pathway via EV is dominant in RCC as a mechanism for the disposal of waste molecules and is important for tumor progression.
In conclusion, this study demonstrates that the MEK/ERK‐MYC‐MiT/TFE‐lysosomal protein axis has a crucial role in the regulation of MVB fate for secretion or degradation, and consequently on EV secretion in cancer cells and cancer cell growth. Although the cellular context‐specific contribution of this axis should be further investigated, our findings suggest that the regulatory machinery of lysosome activity may offer new opportunities for therapeutic interventions targeting EV secretion in cancer.
DISCLOSURE
The authors have no conflict of interest.
Supporting information
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the Japan Science and Technology Agency, PRESTO program (JP1005457 to CO) and Grant‐in‐Aid Scientific Research (B) (20H03456 to CO).
Hikita T, Uehara R, Itoh RE, et al. MEK/ERK‐mediated oncogenic signals promote secretion of extracellular vesicles by controlling lysosome function. Cancer Sci. 2022;113:1264–1276. doi: 10.1111/cas.15288
REFERENCES
- 1. Kalluri R. The biology and function of exosomes in cancer. J Clin Invest. 2016;126(4):1208‐1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bebelman MP, Smit MJ, Pegtel DM, Baglio SR. Biogenesis and function of extracellular vesicles in cancer. Pharmacol Ther. 2018;188:1‐11. [DOI] [PubMed] [Google Scholar]
- 3. Torrano V, Royo F, Peinado H, et al. Vesicle‐MaNiA: extracellular vesicles in liquid biopsy and cancer. Curr Opin Pharmacol. 2016;29:47‐53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Feng Q, Zhang C, Lum D, et al. A class of extracellular vesicles from breast cancer cells activates VEGF receptors and tumour angiogenesis. Nat Commun. 2017;8:14450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hoshino A, Costa‐Silva B, Shen TL, et al. Tumour exosome integrins determine organotropic metastasis. Nature. 2015;527(7578):329‐335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Costa‐Silva B, Aiello NM, Ocean AJ, et al. Pancreatic cancer exosomes initiate pre‐metastatic niche formation in the liver. Nat Cell Biol. 2015;17(6):816‐826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Harding C, Heuser J, Stahl P. Receptor‐mediated endocytosis of transferrin and recycling of the transferrin receptor in rat reticulocytes. J Cell Biol. 1983;97(2):329‐339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. van Niel G, D'Angelo G, Raposo G. Shedding light on the cell biology of extracellular vesicles. Nat Rev Mol Cell Biol. 2018;19(4):213‐228. [DOI] [PubMed] [Google Scholar]
- 9. Hessvik NP, Llorente A. Current knowledge on exosome biogenesis and release. Cell Mol Life Sci. 2018;75(2):193‐208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Desrochers LM, Antonyak MA, Cerione RA. Extracellular vesicles: satellites of information transfer in cancer and stem cell biology. Dev Cell. 2016;37(4):301‐309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Blanc L, Vidal M. New insights into the function of Rab GTPases in the context of exosomal secretion. Small GTPases. 2018;9(1–2):95‐106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Vietri M, Radulovic M, Stenmark H. The many functions of ESCRTs. Nat Rev Mol Cell Biol. 2020;21(1):25‐42. [DOI] [PubMed] [Google Scholar]
- 13. Chang L, Karin M. Mammalian MAP kinase signalling cascades. Nature. 2001;410(6824):37‐40. [DOI] [PubMed] [Google Scholar]
- 14. Robinson MJ, Cobb MH. Mitogen‐activated protein kinase pathways. Curr Opin Cell Biol. 1997;9(2):180‐186. [DOI] [PubMed] [Google Scholar]
- 15. Leevers SJ, Paterson HF, Marshall CJ. Requirement for Ras in Raf activation is overcome by targeting Raf to the plasma membrane. Nature. 1994;369(6479):411‐414. [DOI] [PubMed] [Google Scholar]
- 16. Prior IA, Lewis PD, Mattos C. A comprehensive survey of Ras mutations in cancer. Cancer Res. 2012;72(10):2457‐2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Samatar AA, Poulikakos PI. Targeting RAS‐ERK signalling in cancer: promises and challenges. Nat Rev Drug Discov. 2014;13(12):928‐942. [DOI] [PubMed] [Google Scholar]
- 18. Guo YJ, Pan WW, Liu SB, Shen ZF, Xu Y, Hu LL. ERK/MAPK signalling pathway and tumorigenesis. Exp Ther Med. 2020;19(3):1997‐2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Logozzi M, Spugnini E, Mizzoni D, Di Raimo R, Fais S. Extracellular acidity and increased exosome release as key phenotypes of malignant tumors. Cancer Metastasis Rev. 2019;38(1–2):93‐101. [DOI] [PubMed] [Google Scholar]
- 20. Hikita T, Miyata M, Watanabe R, Oneyama C. Sensitive and rapid quantification of exosomes by fusing luciferase to exosome marker proteins. Sci Rep. 2018;8(1):14035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Oneyama C, Ikeda J, Okuzaki D, et al. MicroRNA‐mediated downregulation of mTOR/FGFR3 controls tumor growth induced by Src‐related oncogenic pathways. Oncogene. 2011;30(32):3489‐3501. [DOI] [PubMed] [Google Scholar]
- 22. Oneyama C, Hikita T, Enya K, et al. The lipid raft‐anchored adaptor protein Cbp controls the oncogenic potential of c‐Src. Mol Cell. 2008;30(4):426‐436. [DOI] [PubMed] [Google Scholar]
- 23. Oneyama C, Hikita T, Nada S, Okada M. Functional dissection of transformation by c‐Src and v‐Src. Genes Cells. 2008;13(1):1‐12. [DOI] [PubMed] [Google Scholar]
- 24. Hikita T, Kuwahara A, Watanabe R, Miyata M, Oneyama C. Src in endosomal membranes promotes exosome secretion and tumor progression. Sci Rep. 2019;9(1):3265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hänzelmann S, Castelo R, Guinney J. GSVA: gene set variation analysis for microarray and RNA‐seq data. BMC Bioinformatics. 2013;14:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge‐based approach for interpreting genome‐wide expression profiles. Proc Natl Acad Sci USA. 2005;102(43):15545‐15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Liberzon A, Subramanian A, Pinchback R, Thorvaldsdottir H, Tamayo P, Mesirov JP. Molecular signatures database (MSigDB) 3.0. Bioinformatics. 2011;27(12):1739‐1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jürchott K, Kuban RJ, Krech T, et al. Identification of Y‐box binding protein 1 as a core regulator of MEK/ERK pathway‐dependent gene signatures in colorectal cancer cells. PLoS Genet. 2010;6(12):e1001231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. da Huang W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4(1):44‐57. [DOI] [PubMed] [Google Scholar]
- 30. Settembre C, Fraldi A, Medina DL, Ballabio A. Signals from the lysosome: a control centre for cellular clearance and energy metabolism. Nat Rev Mol Cell Biol. 2013;14(5):283‐296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lee JH, Yu WH, Kumar A, et al. Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer‐related PS1 mutations. Cell. 2010;141(7):1146‐1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Meo‐Evoli N, Almacellas E, Massucci FA, et al. V‐ATPase: a master effector of E2F1‐mediated lysosomal trafficking, mTORC1 activation and autophagy. Oncotarget. 2015;6(29):28057‐28070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Steingrimsson E, Copeland NG, Jenkins NA. Melanocytes and the microphthalmia transcription factor network. Annu Rev Genet. 2004;38:365‐411. [DOI] [PubMed] [Google Scholar]
- 34. Sardiello M, Palmieri M, di Ronza A, et al. A gene network regulating lysosomal biogenesis and function. Science. 2009;325(5939):473‐477. [DOI] [PubMed] [Google Scholar]
- 35. Settembre C, Di Malta C, Polito VA, et al. TFEB links autophagy to lysosomal biogenesis. Science. 2011;332(6036):1429‐1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Napolitano G, Ballabio A. TFEB at a glance. J Cell Sci. 2016;129(13):2475‐2481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Palmieri M, Impey S, Kang H, et al. Characterization of the CLEAR network reveals an integrated control of cellular clearance pathways. Hum Mol Genet. 2011;20(19):3852‐3866. [DOI] [PubMed] [Google Scholar]
- 38. Annunziata I, van de Vlekkert D, Wolf E, et al. MYC competes with MiT/TFE in regulating lysosomal biogenesis and autophagy through an epigenetic rheostat. Nat Commun. 2019;10(1):3623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ünal EB, Uhlitz F, Blüthgen N. A compendium of ERK targets. FEBS Lett. 2017;591(17):2607‐2615. [DOI] [PubMed] [Google Scholar]
- 40. Han H, Jain AD, Truica MI, et al. Small‐molecule MYC inhibitors suppress tumor growth and enhance immunotherapy. Cancer Cell. 2019;36(5):483‐497.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Oka H, Chatani Y, Hoshino R, et al. Constitutive activation of mitogen‐activated protein (MAP) kinases in human renal cell carcinoma. Cancer Res. 1995;55(18):4182‐4187. [PubMed] [Google Scholar]
- 42. Théry C, Witwer KW, Aikawa E, et al. Minimal Information for Studies of Extracellular Vesicles 2018 (MISEV2018): a position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J Extracell Vesicles. 2018;7(1):1535750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Daub H, Weiss FU, Wallasch C, Ullrich A. Role of transactivation of the EGF receptor in signalling by G‐protein‐coupled receptors. Nature. 1996;379(6565):557‐560. [DOI] [PubMed] [Google Scholar]
- 44. Thomas LR, Tansey WP. Proteolytic control of the oncoprotein transcription factor Myc. Adv Cancer Res. 2011;110:77‐106. [DOI] [PubMed] [Google Scholar]
- 45. Imjeti NS, Menck K, Egea‐Jimenez AL, et al. Syntenin mediates SRC function in exosomal cell‐to‐cell communication. Proc Natl Acad Sci USA. 2017;114(47):12495‐12500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kilinc S, Paisner R, Camarda R, et al. Oncogene‐regulated release of extracellular vesicles. Dev Cell. 2021;56(13):1989‐2006 e1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Yin Q, Jian Y, Xu M, et al. CDK4/6 regulate lysosome biogenesis through TFEB/TFE3. J Cell Biol. 2020;219(8):e201911036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Meyer N, Penn LZ. Reflecting on 25 years with MYC. Nat Rev Cancer. 2008;8(12):976‐990. [DOI] [PubMed] [Google Scholar]
- 49. Dang CV. MYC on the path to cancer. Cell. 2012;149(1):22‐35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Stine ZE, Walton ZE, Altman BJ, Hsieh AL, Dang CV. MYC, metabolism, and cancer. Cancer Discov. 2015;5(10):1024‐1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Vidal M. Exosomes: revisiting their role as "garbage bags". Traffic. 2019;20(11):815‐828. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material