

Abstract
Immunotherapy is currently recognized as the fourth modality in cancer therapy. CTL can detect cancer cells via complexes involving human leukocyte antigen (HLA) class I molecules and peptides derived from tumor antigens, resulting in antigen‐specific cancer rejection. The peptides may be predicted in silico using machine learning‐based algorithms. Neopeptides, derived from neoantigens encoded by somatic mutations in cancer cells, are putative immunotherapy targets, as they have high tumor specificity and immunogenicity. Here, we used our pipeline to select 278 neoepitopes with high predictive “SCORE” from the tumor tissues of 46 patients with hepatocellular carcinoma or metastasis of colorectal carcinoma. We validated peptide immunogenicity and specificity by in vivo vaccination with HLA‐A2, A24, B35, and B07 transgenic mice using ELISpot assay, in vitro and in vivo killing assays. We statistically evaluated the power of our prediction algorithm and demonstrated the capacity of our pipeline to predict neopeptides (area under the curve = 0.687, P < 0.0001). We also analyzed the potential of long peptides containing the predicted neoepitopes to induce CTLs. Our study indicated that the short peptides predicted using our algorithm may be intrinsically present in tumor cells as cleavage products of long peptides. Thus, we empirically demonstrated that the accuracy and specificity of our prediction tools may be potentially improved in vivo using the HLA transgenic mouse model. Our data will help to design feedback algorithms to improve in silico prediction, potentially allowing researchers to predict peptides for personalized immunotherapy.
Keywords: epitope peptide, HLA transgenic mice, HLA‐class I, neoantigen, prediction algorithm
Using HLA‐transgenic mice, we assessed in vivo immunogenicity of neoantigen peptides by our prediction pipeline from patient tissues. Our results demonstrated that our prediction pipeline could propose immunogenic candidates; however, the power prediction is still insufficient for clinical application. Assessment of immunogenicity in an in vivo model is essential for clinical efficiency of peptide vaccines.

1. INTRODUCTION
Immunotherapy is the fourth therapeutic modality for cancer after surgery, anticancer drugs, and radiotherapy. Immune checkpoint inhibitor (ICI) antibodies have strong and long‐lasting antitumor efficacy, 1 and the tumor mutational burden (TMB) correlates with ICI response. 2 , 3 , 4
Various immune cell groups can eliminate some types of cancer. For example, CD8+ T cells have high cytotoxicity and proliferative capacity and play major roles in cancer cell destruction. 5 , 6 They recognize peptides via the T‐cell receptor (TCR) that are derived from highly expressed cancer antigens degraded by the intracellular ubiquitin–proteasome complex and presented on the human leukocyte antigen (HLA) class I molecules. These peptides result from degradation of self or foreign antigens. The T cells are activated if the HLA/peptide complex and TCR match, following which they enter an expansion phase, migrate into tumor tissues, and induce tumor regression. 7 Contrary to the tumor‐associated antigens for which cancer vaccines were not as successful as expected, 8 the tumor neoantigens 9 belonging to the tumor‐specific antigen (TSA) family are believed to be more immunogenic and less tolerogenic. They consist of proteins bearing non‐synonymous insertions, deletions, point mutations, and fusions 10 They are sometimes of viral origin, the type and number of which vary with cancer type. 11 Those that are successfully presented on the HLA are treated as foreign bodies by the immune system and can activate neoantigen‐specific T cells.
In patients, each harbors a unique HLA haplotype, and the accumulated somatic mutations in tumors also vary with patients. Therefore, the development of personalized immunotherapy targeting on neoantigens is crucial. 12 To do that, it is also essential to identify neopeptides that can be presented on HLA to induce tumor rejection with the purpose of not only increasing the therapeutic performance but also for decreasing the associated expenses for peptide production. Now, neoantigens are mainly predicted using in silico algorithms based on information such as the amino acid sequences binding to the HLA, sequences recognized by the TCR, or the importance of specific amino acids in stabilizing the complex HLA–peptide–TCR. 13 In this manner, it is possible to prepare lists of candidates from protein sequences. The trained algorithm, such as netMHCpan, 14 predicts binding between peptides and the HLA. Recently, its efficiency has been improved, accompanied by the accumulation of the binding data with MHC class I molecules and many peptides. 15 However, the prediction power of this algorithm is based mainly on in vitro binding databases, and additional data are required to clarify the in vivo response. These include information regarding the number of peptides necessary for correct immunogenic responses, neoepitope presence and presentation in the tumor, TCR avidity, and cytokine background. 16 Here, we used an in vivo vaccination model to evaluate the immunogenicity of the predicted neopeptides derived from omics analyses of excised hepatocellular carcinoma (HCC) and metastasis of colorectal cancer (mCRC) tumors and normal tissues with the aim of using these data to potentially improve the accuracy of our prediction algorithm. We applied the HLA‐transgenic mouse (Tgm) model as it is stable and readily reproducible. It also enables high‐throughput evaluation of numerous peptides in vivo. We first determined the mutation background and expression profile of each patient. Then, based on the mutations, the peptides were predicted in silico, and those whose wild‐type sequence were identical with their corresponding murine counterparts were selected for in vivo HLA‐Tgm vaccination to determine their immunogenicity. This study is an important milestone for designing feedback algorithms and making them more powerful and precise for in silico prediction.
2. MATERIALS AND METHODS
2.1. Patients and samples
Paired fresh normal and tumor tissues were collected from surgically resected HCC and mCRC at the National Cancer Center Hospital East (NCCE) between 2017 and 2020. Written informed consent was acquired from all patients. Twenty‐eight patients with HCC and 23 with mCRC were enrolled in this study. Five of the 23 patients with mCRC were excluded from the analysis for reasons mentioned below. RNA was not recovered from three patients. Another patient harbored few mutations and was, hence, not included considering contamination with normal tissue. The last patient harbored a large number of mutations, which deviated significantly from the mean for the current population. The study design and protocol were approved by the Ethics Review Committee of NCCE (Permission Nos. 2016‐202 and G2010‐02). All experiments were performed in accordance with relevant committee guidelines and regulations and conformed to the Declaration of Helsinki.
2.2. Mice and cell lines
Male and female HLA‐A02:01‐HHD, B*07:02‐HHD, and B*35:01‐HHD Tgm were provided by the Département SIDA‐Retrovirus, Unite d’Immunite Cellulaire Antivirale, Institut Pasteur, Paris, France. HLA‐A24:02‐Tgm were provided by Satoru Senju of the Faculty of Life Sciences, Kumamoto University, Kumamoto, Japan. The mice were bred and maintained under specific‐pathogen‐free conditions at the animal facility of Sankyo Lab Service (Tokyo, Japan). Mice aged 6–20 weeks were maintained at the animal facility of the NCC Exploratory Oncology Research and Clinical Trial Center (EPOC), Chiba, Japan. All experiments were performed in accordance with the protocols approved by the Animal Care and Use Committee of NCCE (Chiba, Japan) (Permission No. M21‐011). RMA‐HHD stably transfected B cell lines expressing HLA‐A2.1‐HHD were kindly provided by Dr Masanori Matsui of Saitama Medical School, Saitama, Japan. The cells were cultured in 10% (v/v) FBS/RPMI‐1640 medium and used in the subsequent experiments. Data on HLA‐Tgm have been reported previously. 17 , 18 , 19 , 20
2.3. Next generation sequencing analysis
Tissue samples of 2–5 mm in diameter were harvested for DNA and RNA isolation. The samples were stored with RNAlater (Qiagen, Hilden, Germany) and/or frozen in liquid nitrogen. RNA and genomic DNA were isolated using RNeasy, DNeasy, or Allprep DNA/RNA kits (Qiagen) according to the manufacturer’s protocol. Precise methods and materials for DNA and RNAseq are described in supporting infromations. 21
2.4. Somatic variant identification in tumor tissues and peptide scoring
To identify germline/somatic variants for each patient, whole exome sequencing (WES) data of paired normal and tumor tissues were analyzed according to the Genome Analysis Toolkit (GATK) best practice workflow. 22 Briefly, the fastq read was mapped onto the human reference genome (hg38) using bwa‐mem (0.7.17) 23 and then converted to sorted bam files using samtools (v. 1.8). 24
GATK4 was used to remove duplicate reads, recalibrate the base quality score, and obtain analysis‐ready bam files. MSIsensor 22 was used to predict the microsatellite instability (MSI) status of each patient. Using the paired bam files of tumor and normal tissues, MSIsensor calculates the microsatellite length distribution at each site and statistically compares distributions between samples. 25 Germline and somatic variant calls were performed using HaplotypeCaller and Mutect2, respectively. The raw vcf files were filtered as described in the GATK best practice workflows. The filter‐passed variants were further checked based on variant allele frequency (VAF) and excluded if satisfying any of the following conditions: (1) allelic depth (depth of read with alternative base) less than three (both germline and somatic), (2) total depth less than 10 (germline only), (3) VAF less than 5% (somatic only), or (4) ratio of tumor VAF to normal VAF less than three times (somatic only). The germline/somatic variants were annotated with snpEff‐4.3. 26
According to the nonsynonymous somatic variant (nonsynonymous single/multiple‐nucleotide variant [SNV/MNV], frameshift insertion/deletion, and in‐frame insertion/deletion), the reference nucleotide coding sequences of all transcript isoforms bearing the variant site (subject variants) were modified to reflect it and other germline and somatic variants detected from the same coding sequence. The germline and somatic variants were locally phased with whatshap‐0.18 27 to reconstruct haplotype sequences. As the class I epitope is an 8–12 mer, an Illumina short read is sufficiently long for local physical phasing and coverage of this range or greater. Phasing information was applied if a subject variant had any confirmed phased‐in or phased‐out variants around it. Otherwise, all other unphased variants were left unconfirmed and were designated ambiguous nucleotides. All possible 9‐mer or 10‐mer (27 bp or 30 bp) nucleotide patterns, including a subject variant, were extracted from the modified coding sequence of each transcript isoform and translated into amino acid sequences to obtain a neoantigen peptide list. For nucleotide sequences, including ambiguous (unphased) bases, the peptide sequences were included in the list only if they exhibited the same amino acid translation. The N‐ and C‐terminal flank sequences were extracted from the same nucleotide template and translated into amino acid sequences required for downstream cleavage prediction and long peptide vaccine design. If a subject variant was a substitution mutation, the corresponding wild‐type peptides were recorded side by side.
To exclude self‐matching neoantigen peptides that are unfavorable in terms of both immunogenicity and safety, a patient‐oriented peptide database was generated from the reference protein sequences reflecting patient germline variants. The neoantigen peptides were then checked against it for segmental matching. First, the 8‐mer segments, including alternate residue(s), were listed from each 9‐mer or 10‐mer neoantigen peptide. Second, the segments were queried against the database for complete match. Finally, neoantigen peptides with at least one hit were filtered out of the list.
Transcriptome analysis was performed on the RNA‐seq data of the tumor samples to quantify mRNA expression in transcripts per million (TPM). The protocol used was GTEx/TOPMed RNA‐Seq pipeline. 28 Briefly, the STAR 29 index and RSEM 30 reference files were generated based on the foregoing GENCODE gene model. 31 Reads were mapped on the human reference genome (hg38) using STAR (v. 2.7.5b). All options and parameters were identical to those of the GTEx/TOPMed RNA‐Seq pipeline. Duplicates were marked with Picard v. 2.18.9. To calculate the TPM of each transcript isoform, RSEM v. 1.3.3 was run using the max 1000 fragment length, paired end, and rspd estimate options. Reads were also mapped using GSNAP (2018‐07‐04) 32 for increased robust alignment on the indel sites. Bam files generated by STAR and GSNAP were used downstream to investigate allelic expression of the somatic variants identified in the WES data of a corresponding patient (STAR bam for SNVs/MNVs; GSNAP bam for indels). The TPM values calculated using RSEM represent the combined expression levels of both alleles. However, mutated alleles are often downregulated. To reasonably quantify allelic expression of the mRNAs harboring somatic variants, the TPM was multiplied by the variant allele frequency in transcript molecules (VAFrna) that was defined as a proportion of altered reads over total reads overlapping on somatic variant sites. Reads were counted with bam‐readcount v. 0.0.8 33 on a STAR or GSNAP bam file if a somatic variant was a SNV/MNV or an indel, respectively. The adjusted TPM (TPMvar) was assigned to each neoantigen peptide in the foregoing list. As the neoantigen list at this point included redundancy derived from the multiplicity of gene transcript isoforms with various TPM and TPMvar, the redundancy was reduced by merging entries with amino acid sequences having identical N‐ and C‐flanks, following which the TPMvar values were summed to generate a TPM_SUMvar.
A series of predictions were performed to prioritize neoantigen peptides expected to be presented on patients’ HLA class I molecules. The EL‐score affinity %Rank and the IC50 affinity for each pair of neopeptide and HLA allele were calculated using NetMHCpan‐4.0 and MHCflurry‐1.6, respectively. 34 , 35 NetChop‐3.1 36 was used to calculate the C‐terminal cleavage probability score. The predicted values were plugged into a linear predictor function derived from the logistic regression of publicly available immunopeptidome data. 37 A linear predictor was converted using the softplus function to obtain a SCORE between 0 and a positive value:
| (1) |
where z is a linear predictor.
The linear predictor function was derived as follows. First, 9‐mer peptides in the immunopeptidome data in SysteMHC Atlas 37 were extracted. A total of 27,718 9‐mer peptides that were unambiguously identified for their consecutive C‐terminal flank sequences from reference human protein sequences were labeled as positive dataset. Next, the same number of random peptides that were not included in the positive dataset were derived from the human reference protein sequences together with their consecutive C‐terminal flank sequences and labeled as a negative dataset. The three types of predictive values mentioned above were calculated for the peptides in the positive and negative dataset. Finally, logistic regression was performed on these datasets using the glm function in R‐3.4.1 to optimize coefficients for the three predictive values to define the linear predictor function.
SCORE was adjusted according to mRNA expression such that neopeptides with lower TPM_SUMvar were ranked by attenuating SCORE or vice versa. This calculation was performed by weighting SCORE on an arctangent curve of TPM_SUMvar:
| (2) |
2.5. Peptide design for vaccination
The best SCORE and HLA allele were assigned to each neopeptide. Only those assigned with HLA‐A02:01, A24:02, B07:02, and B35:01 were selected. Only neopeptides with wild‐type counterparts 100% identical to the murine ortholog (mm10) were retained for the downstream processes, except for indel‐derived neoantigen peptides, the wild‐type sequences of which could not be clearly defined. Long peptides (Lp) were designed by extending the predicted epitopes with N‐ and C‐terminal flank sequences. The extended peptides were selected to contain either no or only one cysteine residue. When the short peptide contained a cysteine residue, an extended peptide was designed such that further cysteine residues were not present after the addition. Furthermore, if the lowest number of cysteine residue inclusions was equal for several extended patterns, the one with a mutant residue nearest to the center (14th position of 27‐mers) was selected. Those that did not extend to 27‐mers because of the presence of ≥2 cysteine residues were excluded. The identity of the extended parts of the 27‐mer Lp sequence were checked for identity with their murine counterpart sequences. No mismatch was permitted within a short epitope, but ≤3 mismatches were allowed within the flank sequences.
2.6. In vivo vaccination
Five or six peptides (each 50 μg) with similar SCORE were pooled and subcutaneously injected along with 8 μg polyI:CLC (adjuvant) into HLA‐Tgm once weekly for 3 weeks. One week after the final vaccination, the mice were killed, and their spleens were excised. The splenocytes were treated with red blood cell lysis buffer (Sigma‐Aldrich), washed, and used in the subsequent assays.
2.7. ELISpot, intracellular staining assays, and cytotoxicity assay
Interferon (IFN)‐γ ELISpot assay was performed using a mouse IFN‐γ ELISpot set according to the manufacturer’s protocol (BD Biosciences, Franklin, Lakes, NJ, USA). In brief, 2 × 106 mouse splenocytes were seeded in each well containing 1 μg/mL of each peptide and the suspensions were incubated for 20 h. The plates were washed and stained with biotinylated secondary antibody and HRP‐conjugated streptavidin. IFN‐γ was detected as spots using a chromogenic substrate. The spots were enumerated in an Eliphoto system (Minerva Tech). The IFN‐γ intracellular staining (ICS) was performed according to standard methods. 38 In brief, splenocytes were cultured for 12 h in the presence of 1 μg of each peptide and for another 8 h with additional 2 μM monensin. After dead cell blocking and staining, the cell surface markers were stained with monoclonal antibodies against CD3 (2C11), CD8α (53‐6‐7), and CD4 (GK1.5) (BioLegend). The cells were fixed, permeabilized, stained with anti‐IFN‐γ mAb (XMG1.2), and analyzed using flow cytometry (Canto II; BD Pharmingen, San Diego, CA, USA) and the FACS Diva software (BD Biosciences, v.9). The protocols used were reported previously. 39 , 40
FCmct was performed in a 96‐well plate (Corning) on T cells co‐cultivated in the presence of CalceinAM‐labeled splenocytes or on RMA‐HDD target cells pulsed with the peptides. The cells were incubated at 37°C for 3–6 h and analyzed with Terascan VPC (Minerva Tech) to detect CalceinAM‐positive cells. The release rate in each well was calculated as the Cytotoxicity% (cytotoxicity rate). The difference between the spontaneous and maximum release of CalceinAM‐stained target cells was 100%. The protocols used were as reported previously. 6 , 40
2.8. In vivo killing assay
The protocols used were as described previously. 41 In brief, syngeneic splenocytes or RMA‐HHD cells were incubated with or without 10 μg/mL peptide at 37°C for 1 h, washed, and stained with various concentrations of carboxyfluorescein succinimidyl ester (CFSE; Dojindo Laboratories) to separate peptide‐pulsed and non‐pulsed cells. Ten million cells were injected into the tail veins of non‐immunized control and immunized (vaccinated) mice. Twenty‐four hours after injection, the mice were killed, their spleens were excised, and the percentages of CFSE‐labeled cells were evaluated using flow cytometry (Canto II; BD Pharmingen). The killing rates were calculated as described previously. 41
2.9. Statistical analysis
Statistical analyses were performed in GraphPad Prism v.9 (GraphPad Software). Differences in patient mutation background and SCORE between responder and non‐responder peptides were evaluated for statistical significance using Mann–Whitney U‐tests. Correlations between the number of nonsynonymous mutations and SCORE were assessed using Spearman’s correlation after normality testing. Spot counts disclosed by the IFN‐γ ELISpot assay and percentage killing in the in vitro killing assay were compared between the peptide and no‐peptide groups using paired multiple t‐tests.
When two mice vaccinated with the same peptide pool showed quite different results, the average spot number was used to test for significant differences. Moreover, additional experiments were conducted to confirm the reproducibility. Receiver operating characteristic (ROC) curve analysis and area under the curve (AUC) calculations were conducted to assess the performance of the prediction model.
3. RESULTS
3.1. Background of patient mutations and peptide selection
Samples were harvested from 28 patients with HCC, including one case of mixed type, intrahepatic cholangiocarcinoma (iCCC) and metastatic liver cancer, finally diagnosed by a pathologist, and 18 with mCRC. Their mutation backgrounds were comparable, and they all presented with microsatellite stable (MSS). Figure 1A shows the scheme of our pipeline for neoantigen prediction. The mutation background of each patient and the SCORE/SCOREadj predictions based on our prediction pipeline and correlations are shown in Table S1 and Figure 1B–D. Medians of 110 and 139 mutations were found in HCC and mCRC, respectively. The median SCORE and SCOREadj for the top 50 peptides in our prediction pipelines are shown in Figure 1C. Significant differences between tumor types were not detected for these factors (Table S2). In subsequent experiments, all peptides predicted from patients with HCC and mCRC were used together. According to our pipeline for the neoantigen prediction, as in Figure 1A, 25–50 neopeptides per patient were synthesized (Table S3), and we examined the reactivity of a total of 250 neoantigens peptides in six HCC patients with PBMC after in vitro stimulation. Unfortunately, although few patients showed a reactivity against neoantigens peptides, the positive rate was less than 3%, as shown in the Figure S1.
FIGURE 1.
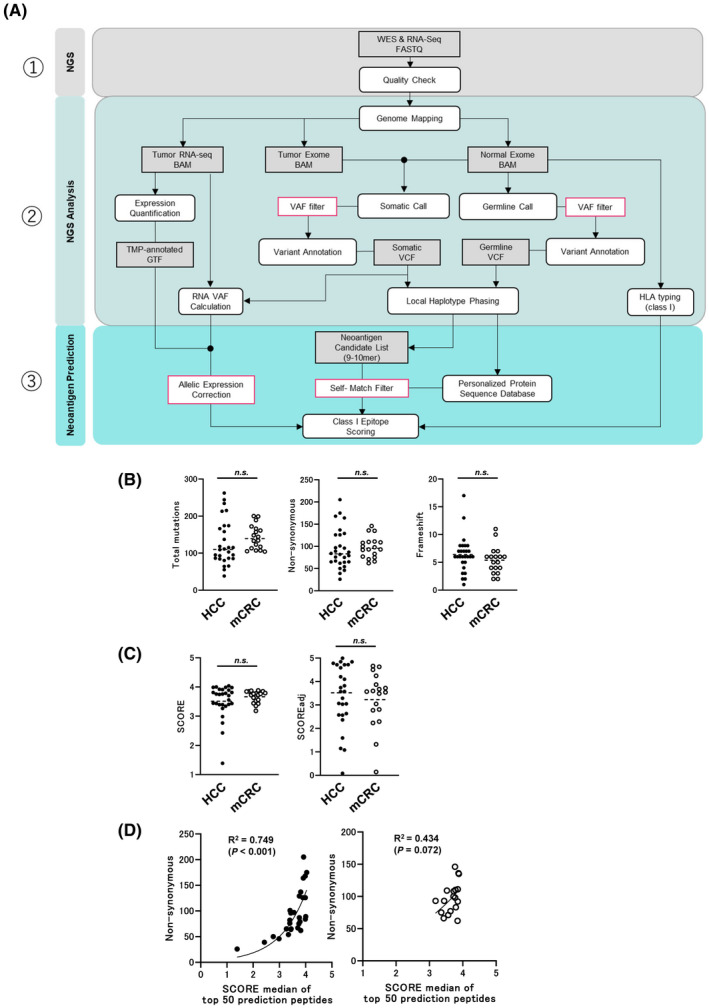
(A) Schematic of the workflow described in the Materials and Methods. (B) Total, non‐synonymous, and frameshift mutations summarized from data in Table S1. (C) SCORE and SCOREadj summarized from data in Table S1 and S2. (D) Correlations between median SCOREs of top 50 prediction peptides and nonsynonymous mutations in patients
For mouse experiments where the gene expression level in the original patients was irrelevant, peptide selection and evaluation were conducted based on SCORE rather than SCOREadj. To exclude the possibility that immunogenicity occurs due to difference in wild‐type sequence between humans and mice, only the neopeptides whose wild‐type sequences were 100% identical with murine counterparts were selected. Finally, 3–17 neopeptides per patient were chosen to be synthesized, resulting in 278 unique peptides in total. Several peptides with low SCORE were included as negative candidates.
3.2. Peptides predicted from somatic mutations in tumors induced significant immune response in vivo
The HLA‐Tgm models were used to evaluate the immune response of the predicted neopeptides derived from patients harboring somatic mutations. Each HLA‐Tgm was vaccinated thrice, with five to six peptides grouped by SCORE (50 μg/peptide) + poly‐I:CLC (8 μg) adjuvant. The immune responses of splenocytes to each peptide were assessed using the IFN‐γ ELISpot assay (Figure 2A). Figure 2B shows the representative results obtained after vaccinating the peptide mix into HLA‐A02‐Tgm and HLA‐A24‐Tgm; 126‐1‐01, 126‐2‐08, 126‐1‐21 and 126‐1‐31 were A02‐restricted peptides, 117‐1‐01, 117‐2‐07, 117‐1‐14, and 117‐2‐16 were A24‐restricted peptides, respectively. To confirm mutation specificity, mice were vaccinated with short neopeptides, and their immune responses were assessed for all neopeptides and their wild‐type counterparts. Figure 2C shows that IFN‐γ was produced against A02‐restricted neopeptides but not their wild‐type counterparts. Therefore, mutation‐specific immune responses were significantly induced after HLA‐Tgm were immunized with the predicted peptides derived from patients harboring somatic mutations.
FIGURE 2.
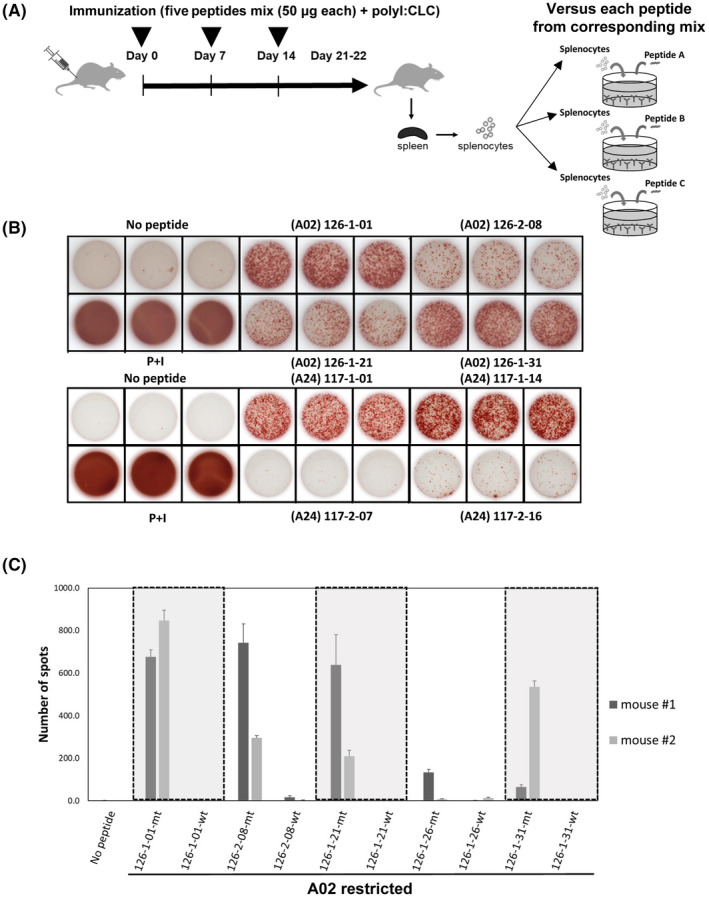
Screening of predicted neoepitopes in human leukocyte antigen (HLA) transgenic mice (Tgm). (A) Scheme of vaccinations and assays. (B) Representative pictures of ELISpot assays with A02‐restricted 126‐1‐01, 126‐2‐08, 126‐1‐21, and 126‐1‐31 peptides in vaccinated HLA‐A02 Tgm (upper pictures) and A24‐restricted 117‐1‐01, 117‐1‐14, 117‐2‐07 and 117‐2‐16 peptides in vaccinated HLA‐A24 Tgm (lower pictures). “No peptide” and “PMA + ionomycin” conditions were the negative and positive controls, respectively. (C) ELISpot assay showing mutation‐specific immune response and no immune response to corresponding wild‐type peptides in HLA‐A02 Tgm. Mt, neopeptides; wt, wild‐type peptides
3.3. Killing activity of CTL induced by vaccination with predicted neoantigen peptides
Figure 3A shows the scheme of the in vivo killing assay. The peptide immunogenicity used for in vivo killing assay were confirmed by ELISpot assay (Figure 3B); 145‐1‐21 and 145‐1‐30 were A02 restricted peptides, and 129‐3‐22 and 129‐2‐03 were A24 restricted peptides, respectively. The in vivo experiment using immunized mice showed strong killing activity (93.7% for 145‐1‐21 and 94.5% for 145‐1‐30 in HLA‐A2 Tgm; 93.63% for 129‐3‐22 and 85.46% for 129‐2‐03 in HLA‐A24 Tgm; Figure 3C). The killing activity was also confirmed when RMA‐HHD‐A2 was the target. Therefore, in the immunized HLA‐A2 Tgm, the killing rates were 26.3% and 64.5% for 145‐1‐21 and 30.8% and 49.1% for 145‐1‐30, demonstrating a modest killing activity against tumor cells (Figure 3D).
FIGURE 3.
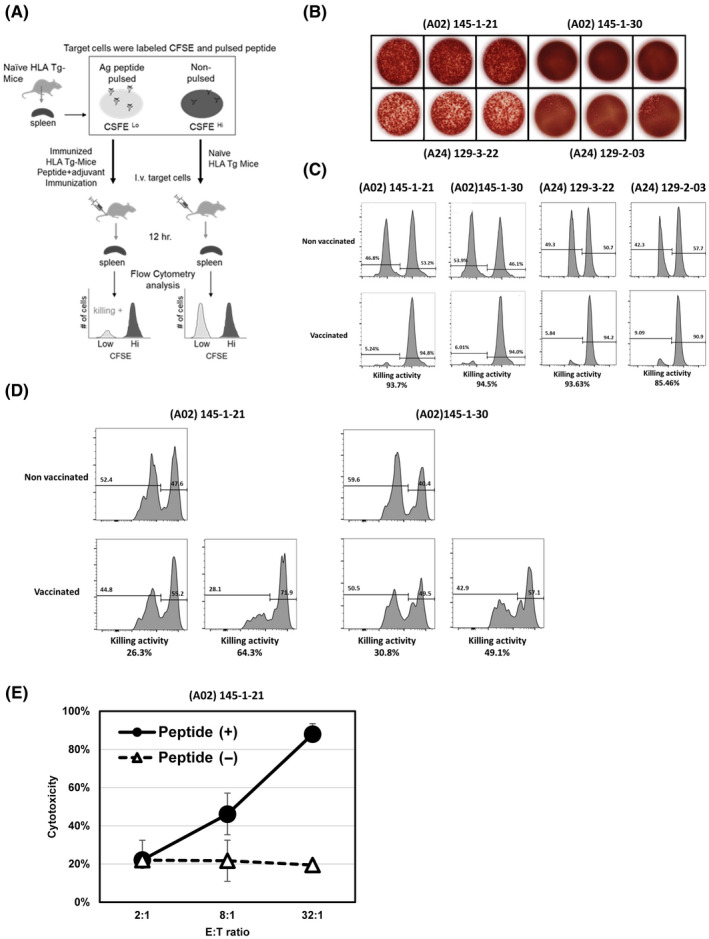
In vitro and in vivo killing assays of induced immune response. (A) Protocol for in vivo killing assay. (B) The results of ELISpot assays illustrating the immune reactivity of (A02) 145‐1‐21, (A02) 145‐1‐30, (A24) 129‐3‐22 and (A24) 129‐2‐03 peptides that used for in vivo killing assay. (C) In vivo cytotoxicity measured in A02‐Tgm treated with 145‐1‐21 and 145‐1‐30 peptides (histograms, left side) and in A24‐Tgm treated with 129‐3‐22 and 129‐2‐03 peptides (histograms, right side). The target cells were syngeneic splenocytes pulsed with each peptide, respectively. (D) In vivo cytotoxicity measured in A02‐Tgm with 145‐1‐21 and 145‐1‐30 peptides using RMA‐HHD myeloma cells as targets. (E) In vitro killing assay targeting T2 cells pulsed with A02‐restricted 145‐1‐21 peptide. Cytotoxicity is shown as a percentage. Effector and target ratio (E:T ratio) gradually increased from 2:1 to 8:1 and to 32:1
To confirm the cytotoxicity induced in effector cells by the neopeptide vaccine, we performed in vitro killing experiments using CTLs isolated from vaccinated HLA‐A02‐Tgm. Splenocytes harvested from mice vaccinated with the A02‐restricted 145‐1‐21 neopeptide were repeatedly stimulated in vitro for 4 weeks in the presence of the neopeptide. CD8+ cells (CTLs) were then purified from these splenocytes and co‐cultivated with RMA‐S‐HHD pulsed with or without the neopeptides. Neopeptide‐induced CTLs showed significant cytotoxic activity against A02‐restricted 145‐1‐21‐pulsed RMA‐HHD tumor cells in vitro (Figure 3E).
3.4. Evaluation of prediction model
We tested the immunogenicity of 100, 90, 42, and 46 neopeptides on A2‐, A24‐, B35‐, and B07‐Tgm, respectively. The response of each peptide (number of IFN‐γ spots/2 × 106 splenocytes/well) is shown along with its SCORE in Figure 4A and Table S4. For A02‐Tgm, A24‐Tgm, B07‐Tgm, and B35‐Tgm (responder), 50%, 51%, 33.3%, and 52.1% of the peptides showed positive responses, respectively (Figure 4B).
FIGURE 4.
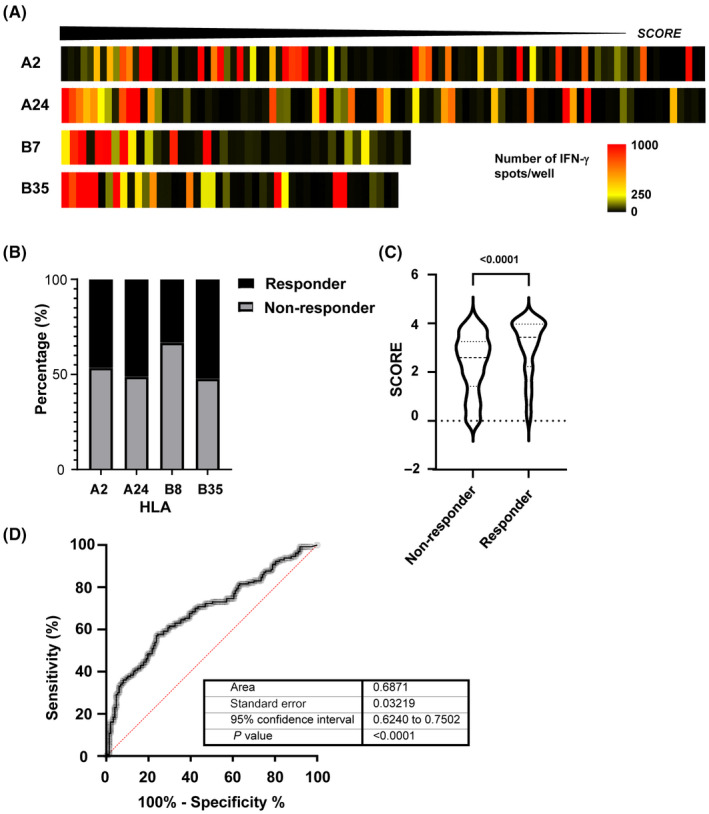
Pipeline prediction power. (A) Heat map showing each human leukocyte antigen (HLA) used in SCORE function. The highest values are shown on the left side. The color pattern shows the average number of spots per peptide. (B) Percentage of immunogenic peptides (responders) in total peptide for each HLA‐Tgm. (C) Score comparison between responders and non‐responders. (D) Area under the receiver operating characteristic (AUC ROC) curve = 0.6871, P < 0.0001
We examined the relationships between the prediction results and in vivo immune response after vaccination (Figure 4C). The responder peptides had relatively higher SCOREs than the non‐responder peptides. For the responder peptides (n = 130), the median SCORE was 3.427, while it was 2.596 for the non‐responder peptides (n = 147). ROC analysis of the SCORE and the immune responses to the 288 peptides confirmed the reasonable prediction performance of our pipeline (AUC = 0.687; Figures 4D and S2).
3.5. Induction of immune responses after long peptide vaccination
We then compared the efficacies of Lp (27‐mers) containing short neoepitopes with those of their short counterparts (Figure 5A). The long neopeptide sequences and their short counterparts are shown in Table S5. Peptide Pool1, Pool2, and Pool3 contained mixtures of seven Lp and were administered to A24‐Tgm. Results showed that immune responses were upregulated against each Lp when 129‐Long‐29, 129‐Long‐18, 129‐Long‐14, and 117‐Long‐21 were administered as vaccines (Figure 5B). Of these, 129‐Long‐18 and 117‐Long‐21 induced immune responses against their short peptide counterparts. The ICS of IFN‐γ confirmed immune responses in CD8+ T cells of mice immunized with 129‐Long‐18 and 117‐Long‐21 (Figure 5C). The long‐18 Lp induced an immune response against the 129‐3‐22 pulsed target, but the 129‐Long‐29 Lp did not.
FIGURE 5.
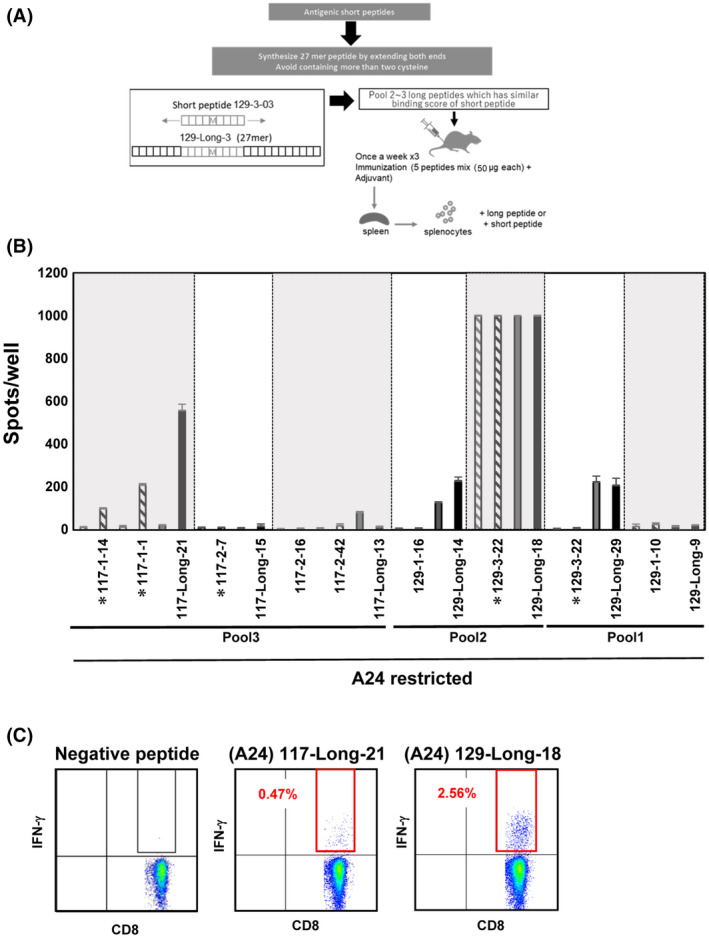
Immune response induction after vaccination with long peptide. (A) Protocol used to vaccinate mice with long or short peptides. Short peptides were lengthened at both extremities up to 27‐mers. (B) Number of spots per pool detected on ELISpot plates. The symbol * before peptide names represents P < 0.05. (C), Intracellular staining of IFN‐γ of CD8+ T cells for 117‐Long‐21 and 129‐Long‐18 including A24‐restricted neoepitopes
4. DISCUSSION
In the present study, we established a method of forecasting optimal peptide candidates based on a “SCORE” calculated by our prediction model from 46 patients with HCC and mCRC. The median SCORE of the top 50 predicted neopeptides from patients with HCC correlated strongly with the TMB of each patient. In the mCRC cohort, both TMB and SCORE were highly similar among patients.
Concerning the results from in vitro experiments with PBMC patients (Figure S1), it is important to mention that all patients in this study showed a lower accumulation of mutations and MSS status (Figure 1 and Table S1). Therefore, they may have weak tumor immunogenicity, 11 as well as a lower frequency of TCR repertoire that could recognize neoantigens. Moreover, in our phase I clinical trial of peptide vaccine targeting GPC3 or HSP105, we never found a response of CTL in PBMC against peptides before vaccination. However, after peptide vaccine, almost all patients showed reactivity against vaccinated peptides. 7 , 42 Recent reports about vaccination with long peptide of neoantigens have shown similar results such that before vaccination, the response of CD8+ T cells is rare. 43 , 44 , 45 To validate the efficacy of prediction algorithms for the development of personalized neoantigen vaccines, experiments with PBMC patients require great effort to obtain a sufficient amount of learning data. Therefore, in this study, we insisted on the using of HLA transgenic mice.
To establish the abilities of the predicted neopeptides to induce immune responses in vivo, the HLA‐restricted peptides were administered to HLA‐matched A24‐, A02‐, B07‐, and B35‐Tgm. Peptides inducing immune responses were designated responders and had higher SCOREs than the non‐responders. Hence, our prediction pipeline could extract antigenic neopeptides. However, the ROC analysis indicated relatively low predictive power, although the ROC AUC for B07‐Tgm, B35‐Tgm, A24‐Tgm, and A02‐Tgm were 0.8597, 0.7462, 0.6922, and 0.6259, respectively (Figure S1). The current prediction model was optimized based on an immunopeptidome training dataset and benefited from its relatively large data size. Consequently, the model partially considers factors related to peptide presentation. Thus, another model is required to predict immunogenicity in vivo and improve prediction performance for therapeutic applications. Nevertheless, construction of such a model is hindered by the lack of any appropriate large‐scale training dataset. We are attempting to overcome this issue by continuing in vivo vaccination assays and exporting the results to the algorithm. After administering vaccinations consisting of neopeptides and their wild‐type counterparts, we confirmed that the immune responses were mutation‐specific, none of the CTL induced by the neoantigen derived peptides showed a reactivity against the wild‐type counterpart, so we considered the off‐target effect as local. This specificity for the mutations underscores the importance of filtering out peptide sequences with high homology against germline sequences. In this manner, self‐matching neopeptides that are unfavorable in terms of immunogenicity and safety were excluded.
The limitations of our pipeline were revealed upon using Lp that included immunogenic short peptides in neoantigen prediction. We used NetChop to predict neopeptide production, although it only considers proteasome degradation 46 rather than endocytotic endosome cleavage. In vaccinations with Lp, the latter is more critical, as Lp cross‐presentation to MHC class I molecules occurs mainly via the phagosome‐to‐endosome and not the proteasome pathway. The observed differences in the response of A24‐Tgm to two Lp with the same short peptide sequence suggest that trimming of the flanking sequences in the endosome is vital for cross‐presentation and CTL activation. In the future, we will assess the improvement of the effect of Lp on CTL induction.
In 2020, HCC accounted for >75% of all liver cancers. 47 A recent study demonstrated an overall survival improvement compared to standard chemotherapy with sorafenib, consisting of a new drug combination in unresectable cases. 48 Tumor heterogeneity is a critical obstacle in immunotherapy‐based cancer treatment. 49 Use of insufficient neopeptides in cancer vaccines can increase selective pressure on tumor cells and increase the risk of immunoediting. Recent studies 50 , 51 have demonstrated the importance of frameshift mutations that enable neoantigen polyepitopes to provide adequate immunogenicity. As well as the importance of the quantity and quality of neoantigens, the function of tumor‐infiltrating cells must be considered in future research. 52 , 53 , 54 One research group studied the effects of a combination of neopeptide vaccine and ICI, in particular in patients with lower TMB. 55 Here, the prediction “SCORE” was not sufficiently accurate to detect all immunogenic neoantigens and neopeptides. Thus, in future, we will use these data to augment the prediction efficiency and speed of our algorithm. We will also determine whether the use of HLA‐Tgm in combination with the prediction algorithm can improve the outcomes of immunotherapies for HCC, mCRC, and other cancers.
CONFLICT OF INTEREST
This study was financially supported by the collaboration between the corresponding author and BrightPath Biotherapeutics (Tokyo, Japan). Tetsuya Nakatsura, corresponding author for this study, is Associate Editor of Cancer Science.
Supporting information
Supplementary Material
ACKNOWLEDGMENTS
The authors thank the members of Hepatobiliary Surgery at NCCE and Motohiro Kojima, Head of the Division of Pathology at NCCE, for their assistance with patient registration and tissue collection.
Charneau J, Suzuki T, Shimomura M, et al. Development of antigen‐prediction algorithm for personalized neoantigen vaccine using human leukocyte antigen transgenic mouse. Cancer Sci. 2022;113:1113–1124. doi: 10.1111/cas.15291
DATA AVAILABILITY STATEMENT
The DNA‐seq and RNA‐seq data generated in the present study were uploaded to the Sequence Read Archive of National Bioscience Database Center for human genome data (NBDC; https://humandbs.biosciencedbc.jp/data‐use/all‐researches) under accession No. JGAS000507. All other data are available from the corresponding author upon reasonable request.
REFERENCES
- 1. Robert C. A decade of immune‐checkpoint inhibitors in cancer therapy. Nat Commun. 2020;11:3801. 10.1038/s41467-020-17670-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Marabelle A, Fakih M, Lopez J, et al. Association of tumour mutational burden with outcomes in patients with advanced solid tumours treated with pembrolizumab: prospective biomarker analysis of the multicohort, open‐label, phase 2 KEYNOTE‐158 study. Lancet Oncol. 2020;21:1353‐1365. 10.1016/S1470-2045(20)30445-9 [DOI] [PubMed] [Google Scholar]
- 3. Yarchoan M, Hopkins A, Jaffee EM. Tumor mutational burden and response rate to PD‐1 inhibition. N Engl J Med. 2017;377:2500‐2501. 10.1056/NEJMc1713444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wu Y, Xu J, Du C, et al. The predictive value of tumor mutation burden on efficacy of immune checkpoint inhibitors in vancers: A systematic review and meta‐analysis. Front Oncol. 2019;9:1161. 10.3389/fonc.2019.01161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Waldman AD, Fritz JM, Lenardo MJ. A guide to cancer immunotherapy: From T cell basic science to clinical practice. Nat Rev Immunol. 2020;20:651‐668. 10.1038/s41577-020-0306-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yoshikawa T, Nakatsugawa M, Suzuki S, et al. HLA‐A2‐restricted glypican‐3 peptide‐specific CTL clones induced by peptide vaccine show high avidity and antigen‐specific killing activity against tumor cells. Cancer Sci. 2011;102:918‐925. 10.1111/j.1349-7006.2011.01896.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sawada Y, Yoshikawa T, Nobuoka D, et al. Phase I trial of a glypican‐3–derived peptide vaccine for advanced hepatocellular carcinoma: immunologic evidence and potential for improving overall survival. Clin Cancer Res. 2012;18:3686‐3696. 10.1158/1078-0432.CCR-11-3044 [DOI] [PubMed] [Google Scholar]
- 8. Hollingsworth RE, Jansen K. Turning the corner on therapeutic cancer vaccines. Npj Vaccines. 2019;4:7. 10.1038/s41541-019-0103-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Jiang T, Shi T, Zhang H, et al. Tumor neoantigens: From basic research to clinical applications. J Hematol OncolJ Hematol Oncol. 2019;12:93. 10.1186/s13045-019-0787-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Turajlic S, Litchfield K, Xu H, et al. Insertion‐and‐deletion‐derived tumour‐specific neoantigens and the immunogenic phenotype: a pan‐cancer analysis. Lancet Oncol. 2017;18:1009‐1021. 10.1016/S1470-2045(17)30516-8 [DOI] [PubMed] [Google Scholar]
- 11. Alexandrov LB, Nik‐Zainal S, Wedge DC, et al. Signatures of mutational processes in human cancer. Nature. 2013;500:415‐421. 10.1038/nature12477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Akazawa Y, Saito Y, Yoshikawa T, et al. Efficacy of immunotherapy targeting the neoantigen derived from epidermal growth factor receptor T790M/C797S mutation in non–small cell lung cancer. Cancer Sci. 2020;111:2736‐2746. 10.1111/cas.14451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wells DK, van Buuren MM, Dang KK, et al. Key parameters of tumor epitope immunogenicity revealed through a consortium approach improve neoantigen prediction. Cell. 2020;183:818‐834.e13. 10.1016/j.cell.2020.09.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. http://www.cbs.dtu.dk/services/netmhcpan/
- 15. Reynisson B, Alvarez B, Paul S, Peters B, Nielsen M. NetMHCpan‐4.1 and NetMHCIIpan‐4.0: improved predictions of MHC antigen presentation by concurrent motif deconvolution and integration of MS MHC eluted ligand data. Nucleic Acids Res. 2020;48:W449‐W454. 10.1093/nar/gkaa379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sarkizova S, Klaeger S, Le PM, et al. A large peptidome dataset improves HLA class I epitope prediction across most of the human population. Nat Biotechnol. 2020;38:199‐209. 10.1038/s41587-019-0322-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Pascolo S, Bervas N, Ure JM, Smith AG, Lemonnier FA, Pérarnau B. HLA‐A2.1–restricted Education and cytolytic activity of CD8+ T lymphocytes from β2 microglobulin (β2m) HLA‐A2.1 monochain transgenic H‐2Db β2m double knockout mice. J Exp Med. 1997;185:2043‐2051. 10.1084/jem.185.12.2043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Boucherma R, Kridane‐Miledi H, Bouziat R, et al. HLA‐A*01:03, HLA‐A*24:02, HLA‐B*08:01, HLA‐B*27:05, HLA‐B*35:01, HLA‐B*44:02, and HLA‐C*07:01 Monochain Transgenic/H‐2 Class I null mice: Novel versatile preclinical models of human T cell responses. J Immunol. 2013;191:583. 10.4049/jimmunol.1300483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rohrlich P, Cardinaud S, Firat H, et al. HLA‐B*0702 transgenic, H‐2KbDb double‐knockout mice: phenotypical and functional characterization in response to influenza virus. Int Immunol. 2003;15:765‐772. 10.1093/intimm/dxg073 [DOI] [PubMed] [Google Scholar]
- 20. Keun‐Ok J, Khan AM, Liang TBY, et al. West Nile virus T‐cell ligand sequences shared with other Flaviviruses: A multitude of variant sequences as potential altered peptide ligands. J Virol. 2012;86:7616‐7624. 10.1128/JVI.00166-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Andrews S. FastQC: A quality control tool for high throughput sequence data. Published online. 2010. https://www.bioinformatics.babraham.ac.uk/projects/fastqc/
- 22. Van der Auwera GA. Genomics in the Cloud: Using Docker, GATK, and WDL in Terra. Vol O’Reilly Media. 1st edition. 2020. [Google Scholar]
- 23. Li H, Durbin R. Fast and accurate short read alignment with Burrows‐Wheeler transform. Bioinformatics. 2009;25:1754‐1760. 10.1093/bioinformatics/btp324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Li H, Handsaker B, Wysoker A, et al. The sequence alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078‐2079. 10.1093/bioinformatics/btp352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Niu B, Ye K, Zhang Q, et al. MSIsensor: Microsatellite instability detection using paired tumor‐normal sequence data. Bioinformatics. 2014;30:1015‐1016. 10.1093/bioinformatics/btt755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cingolani P, Platts A, Wang LL, et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff. Fly (Austin). 2012;6:80‐92. 10.4161/fly.19695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Martin M, Patterson M, Garg S, et al. WhatsHap: Fast and accurate read‐based phasing. bioRxiv. Published online January 1, 2016:085050. 10.1101/085050 [DOI]
- 28. https://github.com/broadinstitute/gtex‐pipeline/blob/master/topmed_rnaseq_pipeline.md
- 29. Dobin A, Davis CA, Schlesinger F, et al. STAR: Ultrafast universal RNA‐seq aligner. Bioinformatics. 2013;29:15‐21. 10.1093/bioinformatics/bts635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Li B, Dewey CN. RSEM: Accurate transcript quantification from RNA‐Seq data with or without a reference genome. BMC Bioinform. 2011;12:323. 10.1186/1471-2105-12-323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Frankish A, Diekhans M, Ferreira AM, et al. GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res. 2019;47:D766‐D773. 10.1093/nar/gky955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wu TD, Nacu S. Fast and SNP‐tolerant detection of complex variants and splicing in short reads. Bioinformatics. 2010;26:873‐881. 10.1093/bioinformatics/btq057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. https://github.com/genome/bam‐readcount
- 34. Jurtz V, Paul S, Andreatta M, Marcatili P, Peters B, Nielsen M. NetMHCpan‐4.0: improved peptide–MHC class I interaction predictions integrating eluted ligand and peptide binding affinity data. J Immunol. 2017;199:3360‐3368. 10.4049/jimmunol.1700893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. O’Donnell TJ, Rubinsteyn A, Bonsack M, Riemer AB, Laserson U, Hammerbacher J. MHCflurry: open‐source class I MHC binding affinity prediction. Cell Syst. 2018;7:129‐132.e4. 10.1016/j.cels.2018.05.014 [DOI] [PubMed] [Google Scholar]
- 36. Nielsen M, Lundegaard C, Lund O, Keşmir C. The role of the proteasome in generating cytotoxic T‐cell epitopes: insights obtained from improved predictions of proteasomal cleavage. Immunogenetics. 2005;57:33‐41. 10.1007/s00251-005-0781-7 [DOI] [PubMed] [Google Scholar]
- 37. Shao W, Pedrioli PGA, Wolski W, et al. The SysteMHC Atlas project. Nucleic Acids Res. 2018;46:D1237‐D1247. 10.1093/nar/gkx664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Suzuki T, Kishimoto H, Abe R. Requirement of interleukin 7 signaling for anti‐tumor immune response under lymphopenic conditions in a murine lung carcinoma model. Cancer Immunol Immunother. 2016;65:341‐354. 10.1007/s00262-016-1808-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Fujinami N, Yoshikawa T, Sawada Y, et al. Enhancement of antitumor effect by peptide vaccine therapy in combination with anti‐CD4 antibody: Study in a murine model. Biochem Biophys Rep. 2016;5:482‐491. 10.1016/j.bbrep.2016.02.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ueda T, Kumagai A, Iriguchi S, et al. Non–clinical efficacy, safety and stable clinical cell processing of induced pluripotent stem cell‐derived anti–glypican‐3 chimeric antigen receptor‐expressing natural killer/innate lymphoid cells. Cancer Sci. 2020;111:1478‐1490. 10.1111/cas.14374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Barber DL, Wherry EJ, Ahmed R. Cutting edge: rapid in vivo killing by memory CD8 T cells. J Immunol. 2003;171:27. 10.4049/jimmunol.171.1.27 [DOI] [PubMed] [Google Scholar]
- 42. Shimizu Y, Yoshikawa T, Kojima T, et al. Heat shock protein 105 peptide vaccine could induce antitumor immune reactions in a phase I clinical trial. Cancer Sci. 2019;110:3049‐3060. 10.1111/cas.14165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kloor M, Reuschenbach M, Pauligk C, et al. A frameshift peptide neoantigen‐based vaccine for mismatch repair‐deficient cancers: a phase I/IIa clinical trial. Clin Cancer Res. 2020;26:4503. 10.1158/1078-0432.CCR-19-3517 [DOI] [PubMed] [Google Scholar]
- 44. Ott PA, Hu Z, Keskin DB, et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature. 2017;547:217‐221. 10.1038/nature22991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sahin U, Derhovanessian E, Miller M, et al. Personalized RNA mutanome vaccines mobilize poly‐specific therapeutic immunity against cancer. Nature. 2017;547:222‐226. 10.1038/nature23003 [DOI] [PubMed] [Google Scholar]
- 46. Keşmir C, Nussbaum AK, Schild H, Detours V, Brunak S. Prediction of proteasome cleavage motifs by neural networks. Protein Eng Des Sel. 2002;15:287‐296. 10.1093/protein/15.4.287 [DOI] [PubMed] [Google Scholar]
- 47. Sung H, Ferlay J, Siegel RL, et al. Global Cancer Statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 vancers in 185 countries. CA Cancer J Clin. 2021;71:209‐249. 10.3322/caac.21660 [DOI] [PubMed] [Google Scholar]
- 48. Finn RS, Qin S, Ikeda M, et al. Atezolizumab plus Bevacizumab in unresectable hepatocellular carcinoma. N Engl J Med. 2020;382:1894‐1905. 10.1056/NEJMoa1915745 [DOI] [PubMed] [Google Scholar]
- 49. Dagogo‐Jack I, Shaw AT. Tumour heterogeneity and resistance to cancer therapies. Nat Rev Clin Oncol. 2018;15:81‐94. 10.1038/nrclinonc.2017.166 [DOI] [PubMed] [Google Scholar]
- 50. Roudko V, Bozkus CC, Orfanelli T, et al. Shared immunogenic poly‐epitope frameshift mutations in microsatellite unstable tumors. Cell. 2020;183:1634‐1649.e17. 10.1016/j.cell.2020.11.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Koster J, Plasterk RHA. A library of neo‐open reading frame peptides (NOPs) as a sustainable resource of common neoantigens in up to 50% of cancer patients. Sci Rep. 2019;9:6577. 10.1038/s41598-019-42729-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Balachandran VP, Łuksza M, Zhao JN, et al. Identification of unique neoantigen qualities in long‐term survivors of pancreatic cancer. Nature. 2017;551:512‐516. 10.1038/nature24462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Zhang J, Caruso FP, Sa JK, et al. The combination of neoantigen quality and T lymphocyte infiltrates identifies glioblastomas with the longest survival. Commun Biol. 2019;2:135. 10.1038/s42003-019-0369-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Hashimoto S, Noguchi E, Bando H, et al. Neoantigen prediction in human breast cancer using RNA sequencing data. Cancer Sci. 2021;112:465‐475. 10.1111/cas.14720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Subudhi SK, Luis V, Hao Z, et al. Neoantigen responses, immune correlates, and favorable outcomes after ipilimumab treatment of patients with prostate cancer. Sci Transl Med. 2020;12:eaaz3577. 10.1126/scitranslmed.aaz3577 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material
Data Availability Statement
The DNA‐seq and RNA‐seq data generated in the present study were uploaded to the Sequence Read Archive of National Bioscience Database Center for human genome data (NBDC; https://humandbs.biosciencedbc.jp/data‐use/all‐researches) under accession No. JGAS000507. All other data are available from the corresponding author upon reasonable request.