

A Perspective on “ATP synthase K+- and H+-fluxes drive ATP synthesis and enable mitochondrial K+-“uniporter” function: I. Characterization of ion fluxes” & “ATP synthase K+- and H+-fluxes drive ATP synthesis and enable mitochondrial K+-“uniporter” function: II. Ion and synthase flux regulation”
Mitochondria are the dominant source of energy in the form of adenosine triphosphate (ATP) in most cells. In the mitochondrial matrix, the Krebs cycle is fueled by nutrients to reduce nicotinamide- (NADH) and flavin adenine dinucleotide (FADH2)1, which donate electrons to the respiratory chain (Figure 1). The ensuing electron transfer along complexes I-IV of the chain and onto oxygen (O2) provides the energy to pump protons (H+) from the matrix to the intermembrane space, generating a chemical (∆pH) and an electrical potential (∆Ψm) across the inner mitochondrial membrane (IMM), which together constitute the protonmotive force (∆μH). According to the chemiosmotic theory developed by Peter D. Mitchell, ∆μH is the driving force for oxidative phosphorylation of adenosine diphosphate (ADP) to ATP at the F1Fo-ATP synthase (Figure 1)2. This concept, for which Mitchell was awarded the Nobel Prize for Chemistry in 1978, has been accepted for more than 50 years and can be found in literally every textbook of biology.
Figure 1.
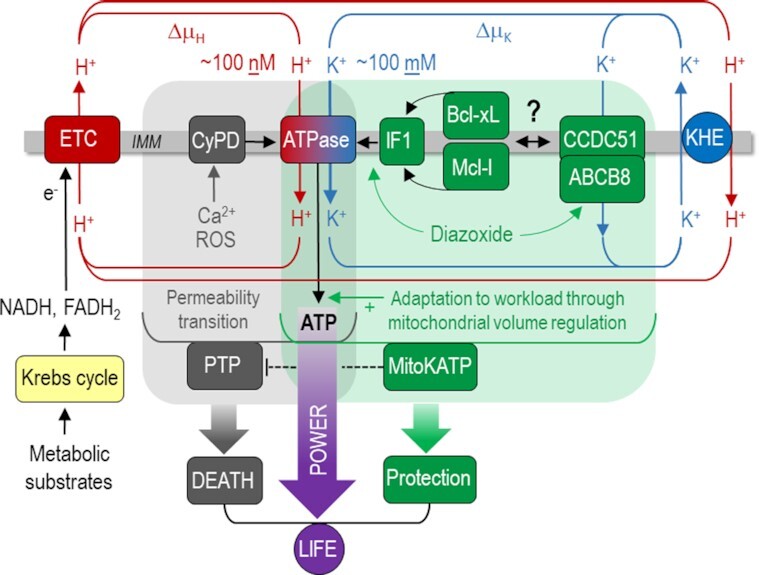
The Janus-faced mitochondrial F1Fo-ATP synthase as the master regulator of life and death. The electron transport chain (ETC) receives electrons from NADH and FADH2 to translocate protons (H+) across the inner mitochondrial membran (IMM) to provide the driving force for the F1Fo-ATP synthase to produce ATP. The current studies3,6 suggest that in addition to the H+ motive force (∆μH), the even greater K+-motive force (∆μK) is harnessed to drive ATP production at the ATPase. Through its impact on mitochondrial volume, this optimizes ATP production during increased ATP demand. Pathological concentrations of Ca2+ and/or reactive oxygen species (ROS) trigger cyclophilin D (CyPD) binding to the ATPase and thereby the formation of a permeability transition pore (PTP), which can induce cell death9. K+ flux via the ATPase is under the control of survival-related protein Inhibitory Factor 1 (IF1), which in turn is regulated by the Bcl-family proteins Bcl-xL and Mcl-1, to constitute a mitochondrial ATP-dependent K+ current (KATP) that protects against PTP opening during ischemia/reperfusion and other stress conditions. Organ protection during ischemia/reperfusion provided by the canonical mKATP activator diazoxide requires IF1. K+ influx via the ATPase is counterbalanced by K+ extrusion via the K+/H+-exchanger (KHE). In addition to the ATPase, also CCDC51 complexing with ABCB8 constitutes functional mKATP16, but without coupling to ATP production.
In the current issue of Function, Juhaszova and colleagues3 substantially challenge—or rather expand, but do not tumble—this concept in revealing that in addition to H+, potassium ion (K+) flux through the F1Fo-ATP synthase (working the same way as H+) provides the majority of energy to produce ATP (Figure 1). Why was this was overlooked for more than six decades? Presumably because the F1Fo-ATP synthase has a > 107-fold selectivity for H+ over other cations.4 But what was not sufficiently considered is that due to the 106-fold higher cytosolic concentration for K+ (∼100 mM) than for H+(∼100 nM), such that K+ flux—driven mostly by the same high electrical driving force (∆Ψm) - could be comparable to H+ flux via the ATP synthase.
Employing a variety of experimental systems, including proteoliposomes containing purified mammalian F1Fo-ATP synthase, planar lipid membranes, but also intact rat cardiac mitochondria, Juhaszova et al.3 elegantly demonstrate that for each H+, 2.7 K+ ions are transferred at the ATP synthase under physiological conditions. Since contraction of intramitochondrial volume hinders the activity of the respiratory chain, and K+ influx osmotically allows water to expand the matrix, such two-ion flux through the F1Fo-ATP synthase not only increases ATP synthesis, but also improves its efficiency: Compared to H+ flux, K+ flux exhibited a 3.5-fold higher ATP synthesis, but only a 2.6-fold higher O2 consumption rate.3 Although a “two-ion theory of energy coupling” was proposed previously by Nath,18 the models differ substantially: while Nath proposed a H+/K+antiport within the F1Fo-ATP synthase may maintain electroneutrality,18 the model presented here3 defines a H+/K+symport via the ATP synthase, where K+ extrusion is accounted for by the distinct K+/H+ exchanger (KHE; Figure 1). Importantly, this novel concept, which allows an optimized matching of energy supply to demand, was corroborated by a minimal computational model comprising the “core” mechanism constituted by ATP synthase, driven by both H+- and K+-motive force, respiratory chain, adenine nucleotide translocator, phosphate carrier, and the K+/H+ exchanger in a parallel study published elsewhere.5
As if this discovery was not enough of a scientific earthquake, in a second manuscript, the same authors6 uncover that by this previously unrecognized K+ flux, the F1Fo-ATP synthase is also a major candidate for the long-sought mitochondrial ATP-dependent K+ channel (mKATP), which is a central downstream effector of a phenomenon termed ischemic preconditioning, where repetitive brief episodes of ischemia and reperfusion of an organ reduce necrosis after a subsequent longer phase of ischemia with reperfusion by delaying the opening of the mitochondrial permeability transition pore (mPTP),7,8 an event that dissipates the mitochondrial membrane potential and—if nonreversible—induces cell death.9
KATP channels, located on the sarcolemma and the IMM, are controlled by the metabolic state of a cell: when the cellular ATP/ADP ratio drops, activation of sarcolemmal KATP channels hyperpolarizes the cell membrane, reducing its excitability to reduce ATP demand,10 whereas activation of mitochondrial KATP channels (mKATP) optimizes ATP production through mitochondrial volume regulation,11 as described above. Although after its first description in the early 1990s12, the electrophysiological and pharmacological properties of the mKATP were extensively characterized,11 its molecular identity has long remained elusive. It was initially proposed that, akin to its sarcolemmal counterpart, mKATP comprised K+-selective pore-forming subunits from the Kir6.x family; however, this model was discarded as genetic ablation of Kir6.x channels did not suppress mKATP responses.13 Subsequently, the renal outer medullary K+ channel (ROMK) evolved as a potential pore-forming subunit of mKATP based on a proteomic screen and in vitro evidence,14 but cardiac-specific knock-out of ROMK later revealed that it is dispensable for cardioprotection and mKATP responses.15 Recently, a protein with previously unknown function (CCDC51) was identified to form a channel with mKATP-like properties when associating with the ATP Binding Cassette protein 8 (ABCB8),16 which had already been shown to modulate mKATP activity.17 Since knock-out of CCDC51 in vivo confirmed its essential role to regulate mitochondrial function in unstressed conditions and protect from necrosis during ischemia/reperfusion,16 CCDC51 and ABCB8 are currently the most accepted candidates in the field to constitute the mKATP (Figure 1).
The second study by Juhaszova et al.6 in this issue of Function delineates the endogenous and exogenous regulation of the F1Fo-ATP synthase in its function as a KATP channel. The survival-related protein Inhibitory Factor 1 (IF1) is regulated by Bcl-family proteins, in particular Bcl-xL and Mcl-1, but not Bcl-2, through interaction at a BH3-like domain, which increases chemo-mechanical efficiency of the F1Fo-ATP synthase to function as mKATP (Figure 1).6 Furthermore, the cardioprotective effect of diazoxide, the canonical mKATP activator, is shown to be mediated by IF1. By applying Bayesian phylogenetic analysis, the authors conclude that IF1 is likely an ancient Bcl family member that evolved from bacteria resident in eukaryotes and prevents excessive ATP consumption through the reversal of the ATP synthase to maintain the protonmotive force.6
The authors need to be applauded for providing groundbreaking results with fundamental implications for cellular bioenergetics and survival. First, these observations identify K+ import via the F1Fo-ATP synthase as one central mechanism by which the rate of ATP turnover in the cytosol is matched by ADP phosphorylation in mitochondria. Second, they assign the F1Fo-ATP synthase a central role in cardioprotection, where mitochondrial K+ influx via the F1Fo/K+ uniporter elevates the threshold to elicit ROS-induced permeability transition. Of note, by coupling mitochondrial K+ influx to ATP production, the subsequent K+ extrusion via the KHE at the expense of protonmotive force is energetically counterbalanced, which is not the case when K+ enters mitochondria via CCDC51/ABCB8 (Figure 1), thereby avoiding the production of futile heat through “uncoupled” K+ leak. This led the authors to suggest that CCDC51/ABCB8-related K+ flux may play a rather “fine-tuning” role compared with ATP synthase-dependent mKATP. However, since CCDC51 knock-out prevented most (but not all) of the cardioprotection provided by diazoxide,16 the herein suggested role of the ATP synthase as mKATP still needs to stand the in vivo test (of time), for instance in mice deficient of IF1.
In ancient Roman myth and religion, Janus is the god of beginnings, transitions, duality and endings, deciding over war and peace, or translated to biology—over life and death. Since in the past decade, the F1Fo-ATP synthase has already evolved as a central component of the mPTP under the control of cyclophilin D,9 the novel data presented in this issue of Function3,6 deservedly assign the mitochondrial F1Fo-ATP synthase a title as a Janus-faced enzyme, since in addition to its canonical role to produce ATP—utilizing both H+ and K+ flux—and its increasingly recognized role as a component of the mPTP,9 it also accounts for the protection from permeability transition through its novel function as an mKATP channel (Figure 1).
ACKNOWLEDGEMENTS
C. M. is supported by the German Research Foundation (DFG; Ma 2528/7–1; SFB 894; SFB 1525) and the Barth Syndrome Foundation. We thank Donald M. Bers for valuable input to the manuscript.
Contributor Information
Edoardo Bertero, Comprehensive Heart Failure Center (CHFC), University Clinic Würzburg, Würzburg, Germany; San Martino Policlinic Hospital, University of Genova, Genova, Italy.
Christoph Maack, Comprehensive Heart Failure Center (CHFC), University Clinic Würzburg, Würzburg, Germany; Department of Internal Medicine 1, University Clinic Würzburg, Würzburg, Germany.
Conflicts of interest
None declared in the context of this publication.
References
- 1. Krebs HA, Johnson WA. Metabolism of ketonic acids in animal tissues. Biochem J. 1937;31(4):645–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mitchell P. Coupling of phosphorylation to electron and hydrogen transfer by a chemi-osmotic type of mechanism. Nature. 1961;191(4784):144–148. [DOI] [PubMed] [Google Scholar]
- 3. Juhaszova M, Kobrinsky E, Zorov DBet al. ATP Synthase K+- and H+-Fluxes Drive ATP Synthesis and Enable Mitochondrial K+-“Uniporter” Function: I. Characterization of Ion Fluxes. Function. 2021;3(2), doi: 10.1093/function/zqab065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Feniouk BA, Kozlova MA, Knorre DA, Cherepanov DA, Mulkidjanian AY, Junge W.. The proton-driven rotor of ATP synthase: ohmic conductance (10 fS), and absence of voltage gating. Biophys J. 2004;86(6):4094–4109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cortassa S, Aon MA, Juhaszova M, Kobrinsky E, Zorov DB, Sollott SJ. Computational modeling of mitochondrial K+- and H+-driven ATP synthesis. J Mol Cell Cardiol. 2022;(165):9–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Juhaszova M, Kobrinsky E, Zorov DBet al. ATP Synthase K+- and H+-fluxes Drive ATP Synthesis and Enable Mitochondrial K+-“Uniporter” Function: II. Ion and ATP Synthase Flux Regulation. Function. 2022;3(2):zqac001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Juhaszova M, Zorov DB, Kim SHet al. Glycogen synthase kinase-3beta mediates convergence of protection signaling to inhibit the mitochondrial permeability transition pore. J Clin Invest. 2004;113(11):1535–1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Heusch G. Myocardial ischaemia-reperfusion injury and cardioprotection in perspective. Nat Rev Cardiol. 2020;17(12):773–789. [DOI] [PubMed] [Google Scholar]
- 9. Bernardi P, Carraro M, Lippe G.. The mitochondrial permeability transition: recent progress and open questions. FEBS J. 2021; doi: 10.1111/febs.16254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Flagg TP, Enkvetchakul D, Koster JC, Nichols CG.. Muscle KATP channels: recent insights to energy sensing and myoprotection. Physiol Rev. 2010;90(3):799–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. O'Rourke B. Evidence for mitochondrial K+ channels and their role in cardioprotection. Circ Res. 2004;94(4):420–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Inoue I, Nagase H, Kishi K, Higuti T.. ATP-sensitive K+ channel in the mitochondrial inner membrane. Nature. 1991;352(6332):244–247. [DOI] [PubMed] [Google Scholar]
- 13. Suzuki M, Sasaki N, Miki Tet al. Role of sarcolemmal K(ATP) channels in cardioprotection against ischemia/reperfusion injury in mice. J Clin Invest. 2002;109(4):509–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Foster DB, Ho AS, Rucker Jet al. Mitochondrial ROMK channel is a molecular component of mitoKATP. Circ Res. 2012;111(4):446–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Papanicolaou KN, Ashok D, Liu Tet al. Global knockout of ROMK potassium channel worsens cardiac ischemia-reperfusion injury but cardiomyocyte-specific knockout does not: implications for the identity of mitoKATP. J Mol Cell Cardiol. 2020;139:176–189., doi: 10.1016/j.yjmcc.2020.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Paggio A, Checchetto V, Campo Aet al. Identification of an ATP-sensitive potassium channel in mitochondria. Nature. 2019;572(7771):609–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ardehali H, O'Rourke B, Marbán E.. Cardioprotective role of the mitochondrial ATP-binding cassette protein 1. Circ Res. 2005;97(8):740–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nath S. Two-ion theory of energy coupling in ATP synthesis rectifies a fundamental flaw in the governing equations of the chemiosmotic theory. Biophys Chem. 2017;230:45–52. [DOI] [PubMed] [Google Scholar]