

Abstract
To investigate influences on the topicity of perfluorinated halobenzenes as halogen bond (XB) donors in the solid state, we have conducted a database survey and prepared 18 novel cocrystals of potentially ditopic (13ditfb, 14ditfb) and tritopic (135titfb) XB donors with 15 monotopic pyridines. 135titfb shows high tendency to be mono- or ditopic, but with strong bases it can act as a tritopic XB donor. DFT calculations have shown that binding of a single acceptor molecule on one of the iodine atoms of the XB donor reduces the ESPmax on the remaining iodine atoms and dramatically decreases their potential for forming further halogen bonds, which explains both the high occurrence of crystal structures where the donors do not achieve their maximal topicity and the observed differences in halogen bond lengths. Despite the fact that this effect increases with the basicity of the acceptor, when the increase of halogen bond energy due to the basicity of the acceptor compensates its decrease due to the reduction of the acidity of the donor, it enables strong bases to form cocrystals in which a potentially polytopic XB donor achieves its maximal topicity.
Short abstract
Consecutive binding of halogen bond acceptors to donor molecules reduces electrostatic potential on free donor atoms, which can affect binding of further acceptor molecules and consequently the stoichiometry of the final product
Introduction
One of the most fascinating aspects of the study of intermolecular interactions is the effect one interaction can have on other interactions present in the same structure.1−4 This effect can be manifested through strengthening (cooperativity) or weakening (anticooperativity) of the interactions involved. This has particularly been studied in the case of hydrogen bonds5−7 where (anti)cooperativity and multiple hydrogen bonds have been found to have a profound effect on the properties of liquids,8−12 hydration of ions,13−16 the structures of biological macromolecules,17−20 etc. Cooperativity of hydrogen bonds is most commonly present in hydrogen bonded chains where the atom which is the donor of one hydrogen bond is an acceptor of another (sequence D–H···D–H···), while anticooperativity is most pronounced in systems with multiple hydrogen bonds involving the same donor or the same acceptor atom (sequences D–H···A···H–D and A···H–D–H···A). Anticooperativity of hydrogen bonds can also be observed when two or more different donor (or acceptor) sites are present in the same molecule (D–H···A–R–A···H–D and A···H–D–R–D–H···A), with the effect being reduced with the increase of the separator (R) between the two sites in the molecule.21
An interaction similar in many ways to a hydrogen bond is a halogen bond;22−25 both are strong and directional interactions with similar ranges of bond energies, and both can vary from purely electrostatic to largely covalent.26,27 Besides, in both hydrogen and halogen bonded systems, cooperativity of several bonds can lead to additional stabilization of the halogen (or hydrogen) bonded structure. In halogen bonded systems, this is commonly achieved in type II interhalogen contacts where the same halogen acts as a donor of one halogen bond and acceptor of an orthogonal one,22,28 although it has recently been shown that similar stabilization is mostly absent in the case of the triangular halogen bonded synthon.29
Anticooperativity in halogen bonded systems has also been demonstrated in the case of bifurcated halogen bonds with multiple acceptors interacting with the same donor (A···X···A) and less so when multiple donors interact with the same acceptor (D–X···A···X–D).30,31 Also, there have been strong indications that when the donor halogen atom is surrounded by additional electron density orthogonal to the halogen bond this does decrease the bond strength.25 However, the effect of multiple halogen bonds formed by different halogens on the same molecule (i.e., in structures comprising the A···X–D–R–D–X···A halogen bonded sequence) has remained an unresolved question.
In order to examine this point, a detailed study of (potentially) polytopic halogen bond donors (i.e., donors with multiple halogen atoms, which can potentially bind more than one XB acceptor molecule) was necessary, as here the anticooperativity of multiple halogen bonds formed by different halogens on the same molecule could prevent the formation of the maximum number of possible halogen bonds (i.e., prevent the XB donor molecule to achieve its maximal topicity). The obvious choice of compounds which can be used for such a study is perhalogenated hydrocarbons, the most commonly used organic halogen bond donors (in particular, the ortho-, meta-, and para-diiodotetrafluorobenzene—12ditfb, 13ditfb, and 14ditfb respectively). Of these, the ortho isomer is somewhat inappropriate because of possible steric hindrance upon binding of two Lewis base molecules on neighboring iodine atoms. The most commonly used halogen bond donor from this group is the 14ditfb originally introduced in 2000 by Metrangolo et al.32 which has shown to be a very reliable ditopic halogen bond donor, commonly forming two halogen bonds.33−38 As opposed to 12ditfb and 14ditfb, the third member of this group 13ditfb was introduced much later (2017).39 Although it also is a potentially ditopic halogen bond donor, it has often been found to form only a single halogen bond in crystal structures.33
Another especially interesting compound is a potentially tritopic halogen bond donor 1,3,5-triiodo-2,4,6-trifluorobenzene 135titfb. An early attempt to use 135titfb as a tritopic halogen bond donor was performed by van der Boom and co-workers40 who attempted to produce two-dimensional halogen bonded sheets by cocrystallizing it with ditopic bipyridyl acceptors. They observed that, rather than forming the expected 2:3 cocrystals, the molecules have assembled into halogen bonded chains of 1:1 stoichiometry, where each 135titfb formed only two halogen bonds. Consecutive binding of the pyridine molecules to the available donor atoms of 135tiftb was investigated also by computational methods, which have shown significant reduction in bond energies and increase in bond lengths as consecutive acceptor molecules were bonded to the 135titfb molecule (in the second and third step, the bond energy was reduced by 17% and 14% respectively, and the bond length increased by about 1% in each step). The observed reduction of the halogen bond donor potential of iodine atoms has led to the conclusion that 135titfb was unlikely to act as a tritopic halogen bond donor. However, in 2010, Roper et al. successfully obtained a 3:1 cocrystal of 4-N,N′-(dimethylamino)pyridine (dmap) with the 135titfb.41 In the same study, they also calculated changes in atomic charges on iodine atoms and halogen bond lengths during consecutive binding of ammonia (probe acceptor molecule) to the three donor atoms of 135titfb. The obtained results have shown that the halogen bond length increases with the number of bonded acceptors, but binding of the probe molecule caused only a negligible decrease in the partial charge on iodine atoms. Their results, contrary to those of van der Boom, seemed to indicate that 135titfb should quite easily act as a tritopic donor.
For the current study, we have decided to investigate the halogen bond donor and acceptor features which can affect the number of bonded acceptor molecules on a certain donor molecule, and how the binding of one acceptor molecule on the donor affects the binding of the second or the third molecule. To exclude both effects of crystal packing and formation of other noncovalent interactions in cocrystals, we have attempted to tackle this question by using 13ditfb, 14ditfb, and 135titfb as halogen bond donors with emphasis on simple, monotopic nitrogen heterocycles (pyridine derivatives, PDs) as halogen bond acceptors in a wide range of basicities (0.87 < pKa < 9.60; Scheme 1). In addition, quantum-chemical calculations were employed to rationalize the observed trends. This approach has enabled us to determine how both the topicity and basicity of the acceptor molecules influence the stoichiometry of halogen-bonded cocrystals.
Scheme 1. Molecular Diagrams of the Halogen Bond Donor and the Pyridine Derivatives Used in the Study.

Results and Discussion
As shown in Table 1, out of the total number of crystal structures deposited in the CSD42 including 13ditfb, 14ditfb, and 135titfb as halogen bond donors (Table 1), a large proportion (98%, 81%, and 64%, respectively) are structures of organic compounds containing nitrogen atoms. In the majority of structures which contain the halogen bond donors above and the nitrogen atoms capable of acting as halogen bond acceptors, each donor molecule forms at least one I···N halogen bond (64% of structures with 13ditfb, 62% of structures with 14ditfb, and 56% of structures with 135titfb). However, in compounds in which I···N contact is present, there is a tendency among donors to create multiple halogen bonds. Formation of two halogen bonds is most prominent in structures with 14ditfb (56% of organic structures with 14ditfb and nitrogen bases), followed by 13ditfb (40%) and 135titfb (30%). Donor 135titfb can also form three halogen bonds, and this is found in 11% of structures. It follows therefore that out of these three XB donors, only 14ditfb tends to form the maximal number of I···N contacts in the majority of crystal structures in which such contacts are possible.
Table 1. Results of the CSD Survey of Crystal Structures Including 13ditfb, 14ditfb, and 135titfb as Halogen Bond Donorsa.
| 13ditfb | 14ditfb | 135titfb | |
|---|---|---|---|
| total number of structures | 51 | 490 | 153 |
| organic structures with N | 50 | 398 | 98 |
| one I···N contact | 12 (24%) | 22 (6%) | 15 (15%) |
| two I···N contacts | 20 (40%) | 223 (56%) | 29 (30%) |
| three I···N contacts | – | – | 11 (11%) |
| structures with Npy | 28 | 166 | 39 |
| structures with monotopic PD | 11 | 60 | 20 |
| one I···Npy contact | 8 (73%) | 14 (23%) | 14 (70%) |
| two I···Npy contacts | 3 (27%) | 46 (77%) | 5 (25%) |
| three I···Npy contacts | – | – | 1 (5%) |
| structures with polytopic PD | 17 | 106 | 19 |
| one I···Npy contact | 4 (24%) | 7 (7%) | 5 (26%) |
| two I···Npy contacts | 13 (76%) | 99 (93%) | 13 (69%) |
| three I···Npy contacts | – | – | 1 (5%) |
PD – Pyridine Derivative.
We have further performed a more specific CSD survey restricting nitrogen bases to pyridine derivatives. The results obtained with pyridine acceptors show somewhat different trends to those observed in the survey including all N-heterocycles. Among these cocrystals, both 13ditfb and 14ditfb mainly act as ditopic donors (in 57% and 87% of structures, respectively), and 135titfb is almost equally distributed as monotopic and ditopic (49% and 46%, respectively), while in only 5% of cases (i.e., two structures) it forms three halogen bonds. The reason for this discrepancy in the statistics lies in the relatively large number of structures comprising polytopic pyridine derivatives. This can be demonstrated by a further analysis of the data with respect to the number of the pyridine rings in a single molecule of the acceptor. Analysis has shown that there are many more structures with polypyridine acceptors in which 13ditfb, 14ditfb, and 135titfb are ditopic (76%, 93%, and 69%, respectively), than those in which they are monotopic. Conversely, in structures where pyridine is a simple monopyridine (molecule with a single pyridine ring), the probability for the halogen bond donor not to have the maximum possible topicity is much higher (73%, 23%, and 95% for 13ditfb, 14ditfb, and 135titfb, respectively). It is therefore evident that the presence of the polytopic acceptor molecules and polytopic donors has a significant effect on the topicity of the donor in the crystal structures of its cocrystals, favoring higher topicities of the donors. This is particularly pronounced in the case of donors of bent geometry, 13ditfb and 135titfb (both predominantly ditopic donors with polytopic acceptors, and monotopic donors with monotopic acceptors), and less so in the case of the linear 14ditfb. Therefore, in order to investigate the tendencies of the halogen bond donor toward different topicities per se (avoiding the effects of the topicity of the acceptor), simple pyridine derivatives should be used as acceptors. The best pyridine derivatives for such a study would be those which either have no other potential acceptor atoms or have such potential acceptors which are considerably less likely to participate in halogen bonding than the pyridine nitrogen (e.g., oxygen, sulfur, and halogen atoms, or certain nitrogen groups, e.g., cyanide, amide, or aliphatic tertiary amine). We have thus selected eight pyridine derivatives without any competing atoms, covering the pKa range of ca. 4.8–7.5. In order to further extend the range of basicities of pyridine derivatives used, we have also included four bases with heteroatoms which are less likely to compete with the pyridine nitrogen as halogen bond acceptors: a highly basic 4-(N,N′-dimethylamino)pyridine (pKa of 9.6) and three weak bases 4-cyanopyridine, 3-acetylpyridine, and 4-acetylpyridine, covering the 2.1–3.8 pKa range (Scheme 1, Table 2). While the majority of crystal structures of 13ditfb and 14ditfb cocrystals with the selected acceptors have already been determined,41,43,44 there were only three structures of cocrystals of 135titfb with simple pyridines published to date. For the purpose of this study, an additional eleven compounds were prepared in order to expand the data set needed for a more detailed analysis.
Table 2. pKa, Values of Used Acceptors and Donor:Acceptor Ratios in Studied Cocrystalsa.
| acceptor | pKa | 13ditfb | 14ditfb | 135titfb |
|---|---|---|---|---|
| 4cnpy | 2.10 | 1:1 (NUBTAI) | 1:1 (NUBSEL) | 1:1 |
| 4acpy | 3.50 | – | 1:2 | 1:2 |
| 3acpy | 3.82 | – | 1:2 | 1:1 |
| qin | 4.85 | 1:2 | 1:2 | 1:1 |
| iqin | 5.41 | 1:1 | 1:2 | 1:2 |
| acr | 5.58 | 1:1 | 1:2 (VOMHIP) | 1:1 (SAJDAL) |
| 2pic | 5.97 | – | 1:2 | 1:3 |
| 3pic | 5.68 | – | 1:2 | 1:3 |
| 4pic | 6.02 | – | 1:2 | – |
| 35lut | 6.24 | 1:1 | 1:2 | 1:3 |
| 34lut | 6.28 | 1:1 | 1:2 | 1:3 |
| 24lut | 6.46 | 1:1 | 1:2 | 1:2 |
| 26lut | 6.72 | 1:1 | – | – |
| 246col | 7.48 | 1:1 | 1:1 | 1:3 |
| dmap | 9.60 | 1:2 (RUYHOJ) | 1:2 (RUYHID) | 1:3 (RUYJAX) |
The tendency of ditopic (13ditfb, 14ditfb) and tritopic (135titfb) halogen bond donors to make cocrystals of a certain stoichiometry with the 13 chosen acceptors was investigated by grinding reaction mixtures in 1:2 (ditopic donors) or 1:3 (tritopic donor) stoichiometric ratios (Table 2). Based on the obtained XRPD patterns of the grinding products (Figures S25–S27 in Supporting Information), the formation of new phases has been observed in all the performed reactions. However, additional maxima corresponding to the pure acceptor have been observed in acr-13ditfb, acr-135titfb, 4cnpy-13ditfb, 4cnpy-14ditfb, and 4cnpy-135titfb reaction mixtures, which indicated the formation of cocrystals of lower stoichiometry than expected, and the presence of an excess of the acceptor in the reaction mixture. In those five cases, grinding experiments were repeated in 1:2 and 1:1 ratios, and they resulted in the formation of the pure 1:1 cocrystals. Cocrystal screening has also been performed by crystallization from solution, in 1:2 (ditopic donors) or 1:3 stoichiometric ratio (tritopic donor). In this way, we have prepared single crystals and determined crystal structures of seven novel compounds of the ditopic donors—(13ditfb)(24lut), (13ditfb)(26lut), (13ditfb)(34lut), (14ditfb)(3acp), (14ditfb)(4acp)2, (14ditfb)(24lut)2, and (14ditfb)(34lut)2 and eleven cocrystals of 135titfb (Table 2). It was found that both crystallization from solution and grinding experiments have resulted in the formation of identical crystal phases in all donor–acceptor combinations.
Despite the small differences in ESPmax values on donor iodine atoms on 13ditfb (122 kJ mol–1) and 14ditfb (127 kJ mol–1), their tendency to form cocrystals as ditopic donors is quite different. The bent 13ditfb is generally a monotopic donor, with two exceptions—(13ditfb)(qin)2 and (13ditfb)(dmap)2. In both crystal structures the two halogen bonds formed are of different lengths and angles. Out of the three possible potentially ditopic acceptors (4cnpy, 3acpy, 4acpy) only in the case of 4cnpy does the second acceptor (cyano nitrogen) participate in an additional XB contact with 13ditfb (Figure 1a). This contact is considerably longer (d(I···NCN) = 3.144(2) Å) than the one between the other iodine and the pyridine nitrogen (d(I···Npy) = 2.998(1) Å;). Unlike 13ditfb, the linear 14ditfb is mostly ditopic, with two exceptions: (14ditfb)(4cnpy) and (14ditfb)(246col). In the former case, 4cnpy again acts as ditopic acceptor forming a short I···Npy halogen bond (d(I···N) = 2.946(8) Å) and a longer halogen bond with cyano nitrogen (d(I···N) = 3.094(9) Å; Figure 1b), and resulting in a halogen-bonded chain. In (14ditfb)(246col), the deviation from the expected topicity is the result of close packing of molecules, which was explained in more detail in our previous study.33 In all cocrystals of 1:2 stoichiometry, the 14ditfb molecule is positioned on the crystallographic inversion center making the two halogen bonds identical.
Figure 1.
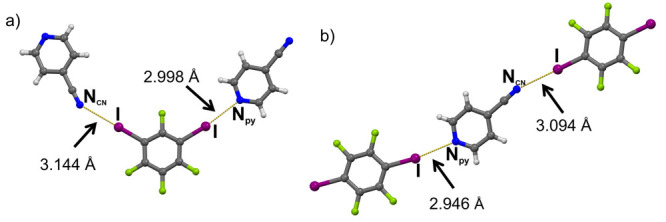
Two types of I···N halogen bonds and their lengths formed in (a) (13ditfb)(4cnpy) and (b) (14ditfb)(4cnpy).
A more complex situation has been found among cocrystals of 135titfb: in four compounds it was found to be monotopic, in three compounds ditopic, and in six of them it formed three I···Npy halogen bonds (Table 2, Figure 2). Additional contacts between iodine atoms and either cyano or keto groups (longer than I···Npy halogen bonds) have been noticed in crystal structures of (135titfb)(3acpy) and (135titfb)(4cnpy) (Figure 3). In (135titfb)(3pic)3 and (135titfb)(246col)3, where 135titfb has been found to be tritopic, the three I···Npy halogen bonds are of different lengths, while in other 1:3 cocrystals, there are also three I···Npy contacts of which two are related by symmetry. From the data represented in Table 2, one can notice an interrelation between acceptor basicity and donor topicity observed in the crystal structures of the prepared cocrystals. The vast majority of strong bases (2pic, 3pic, 35lut, 34lut, 246col, and dmap) form cocrystals of 1:3 stoichiometry, while weaker bases (4cnpy, 3acpy, 4acpy, qin, iqin, acr) form cocrystals either of 1:1 or 1:2 stoichiometries. This indicates that there is a significant effect of the basicity of the acceptor on the stoichiometry of the cocrystal formed with 135titfb (and therefore on the topicity of 135titfb). It has to be noted that this conclusion does not seem to be valid for 13ditfb and 14ditfb, which preferentially form 1:1 and 1:2 cocrystals, respectively, with almost all bases used.
Figure 2.
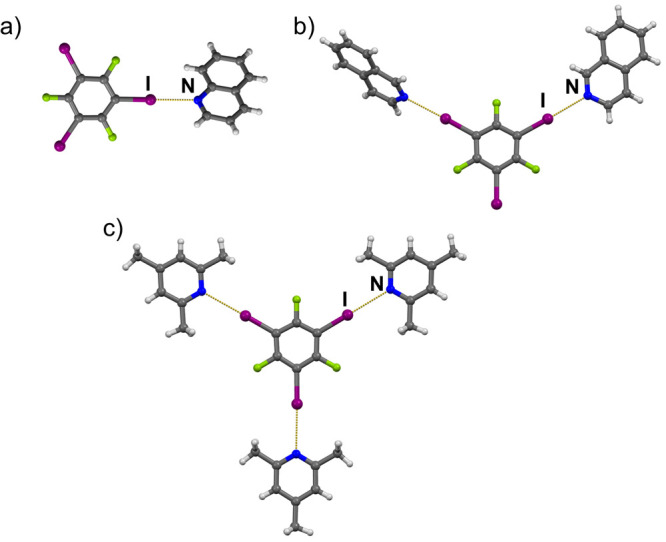
Halogen bonded molecular complexes in (a) (135titfb)(qin), (b) (135titfb)(iqin)2, and (c) (135titfb)(246col)3.
Figure 3.
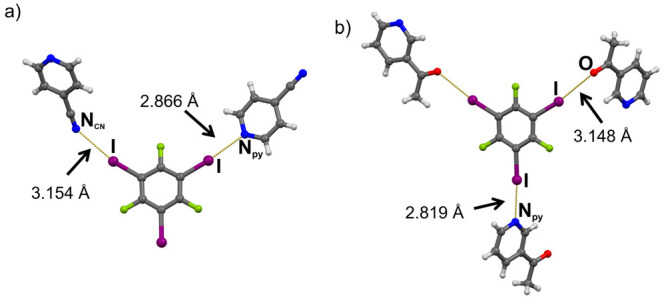
(a) Two types of I···N halogen bonds and their lengths formed in (135titfb)(4cnpy). (b) I···N and I···O halogen bonds formed in (135titfb)(3acpy).
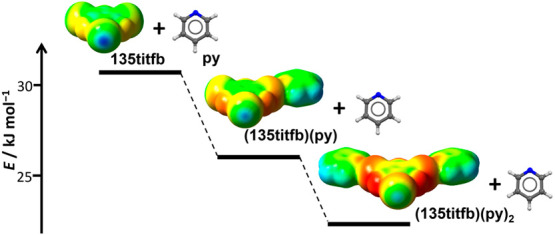
In order to ascertain whether the differences in the behavior of 13ditfb, 14ditfb, and 135titfb as halogen bond donors are due to their electronic structures, we have performed a series of quantum-chemical computations aimed at observing the differences in the effect of binding of base molecules on each of the studied halogen bond donors. Computations using pyridine as the probe acceptor molecule have shown that binding of a single pyridine molecule on one of the iodine atoms of the XB donor reduces the ESPmax (on 0.001 au electron density isosurface; Figure 4) on the remaining iodine atom(s) by 23.0, 23.4, and 21.9 kJ mol–1 e–1 in 14ditfb, 13ditfb, and 135titfb, respectively (on average by ca. 17%). Binding of a further pyridine molecule on a second iodine atom of 135titfb reduces the ESPmax on the remaining unbonded iodine atom by a further 19.3 kJ mol–1 e–1. This indicates a dramatic decrease in the potential of the nonhalogen-bonded iodine atom for forming a further halogen bond. This is mirrored by the reduction of halogen bond energies: the binding energies of the first pyridine molecule onto the three donors are 31.4, 30.8, and 30.1 kJ mol–1, while for the second pyridine they are 27.8, 26.8, and 26.4 kJ mol–1 (for 14ditfb, 13ditfb, and 135titfb, respectively), which corresponds to relative reductions of 11%, 13%, and 12%. Further binding energy of the third pyridine molecule onto 135titfb (23.4 kJ mol–1) is overall reduced by 22%. It should be noted, however, that the overall partial charge of the nonhalogen-bonded iodine atoms changes significantly less: binding of a pyridine molecule on one of the iodine atoms of either donor reduces the NBO charge of the other iodine atom(s) by ca. 0.015 e—a decrease of only 6%. The apparent discrepancy can be explained by referring to the electron density difference (EDD) plots (Figure 5).
Figure 4.
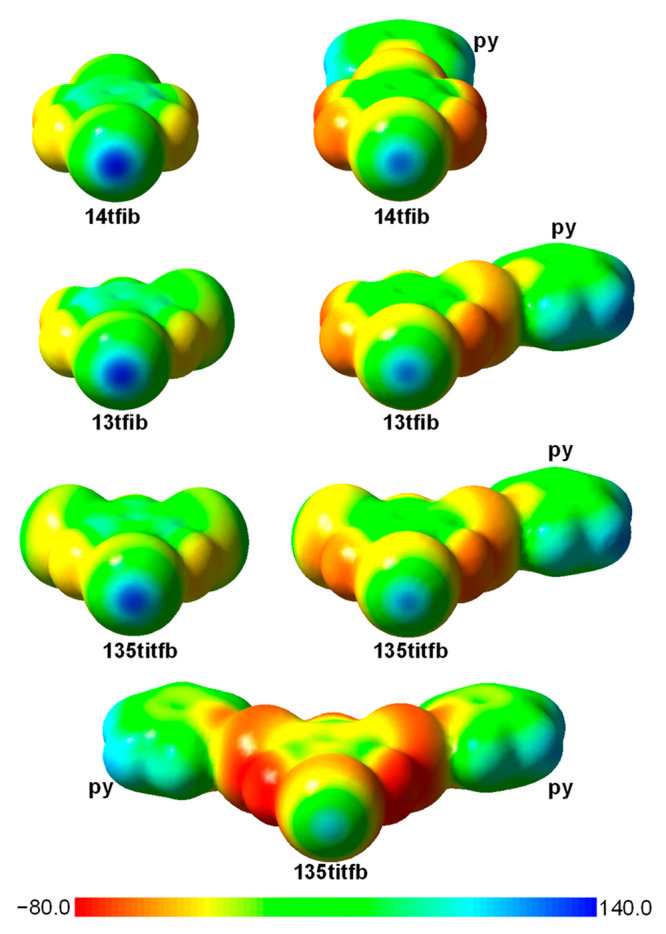
ESP mapped on the electron density isosurface (ρel = 0.001 au) in systems 14ditfb, 13ditfb, 135titfb, (14ditfb)•(py), (13ditfb)•(py), (135titfb)•(py), and (135titfb)•(py)2. Boundaries of ESP values are given in kJ mol–1 e–1.
Figure 5.

EDD isosurfaces (|Δρel| = 2 × 10–4 au) upon binding of a py molecule to 14ditfb, 13ditfb, and 135titfb, and upon binding of two py molecules to 135titfb. Blue parts of isosurfaces correspond to the positive and brown parts to the negative value of Δρel.
These reveal how electron densities in both the donor and the acceptor molecules are ostensibly perturbed by the formation of the halogen bond, primarily about the donor and acceptor atoms (as demonstrated in earlier studies by crystallographic charge density analysis).26 By concentrating however on the nonbonding iodine atom, a large increase of electron density can be seen in the σ-hole region of the atom, coupled with a slight decrease of electron density perpendicular to it—particularly visible in the 2:1 complex of 135titfb (Figure 4). Therefore, while there is a significant decrease of the (positive) ESP on the iodine atom, the change of the total charge is slighter, as the corresponding increase of electron density is partially compensated by a slight increase in the perpendicular direction.
The reduction of ESPmax (and the energies of binding of subsequent molecules) is in accord with the observed high occurrence of crystal structures where the donors do not achieve their maximal topicity. Another question requiring attention is the effect of the basicity of the acceptor on the topicity of the donor. As demonstrated by the crystal structures of cocrystals with 135titfb with simple heterocyclic acceptors, there is a definite increase in the probability of achieving higher topicities with the increase of the basicity of the acceptor. However, one would also expect stronger bases to exact a stronger influence on the donor molecule and to reduce the ESPmax on free iodine atoms (and subsequently the binding energy for subsequent molecules) more than the weaker ones. In order to elucidate this issue, we have performed additional computations with two extremes, 4-cyanopyridine and 4-(N,N-dimethylamino)pyridine, binding to 135titfb. To provide a more detailed view of the changes of the ESP with binding of base molecules, we have plotted ESP as the function of angle φ (Scheme 2) which corresponds to the deflection from linearity (XB angle −180°) in the plane of the donor molecule (Figure 6). The maxima of the obtained curves occur at φ = 0 and correspond to the ESPmax of the σ-hole, while the values of φ where ESP(φ) changes sign (ESP(φmax) = 0) indicate the angular width of the σ-hole (i.e., the ESP(φ) is positive in the region −φmax > φ > φmax).
Scheme 2. (a) Definition of the Parameters of the σ-Hole on the Halogen Atom (X); (b) Qualitative Representation of ESP on a Halogen Atom in the Plane Containing the D–X Bond As a Function of Angle φ.

Figure 6.

Angular dependence (see Scheme 2 for the definition of the φ angle) of molecular electrostatic potential in the donor-molecule plane on the σ-hole of the free iodine atom, evaluated on the 0.001 au electron density isosurface for (a) pure 14ditfb and its 1:1 complexes, (b) pure 13ditfb and its 1:1 complexes, (c) pure 135titfb and its 1:1 complexes, and (d) pure 135titfb and its 1:2 complexes. Black curves represent the pure donors, while the colored curves represent halogen-bonded complexes with corresponding acceptor (see legend).
As can be observed, the ESP(φ) is globaly lowered upon binding of the base molecules (however maintaining the shape), leading to the reduction of both ESPmax and φmax. As expected, the effect increases with the basicity of the base—binding of 4cnpy, py, and dmap reduces ESPmax by 10.7, 22.0, and 31.5 kJ mol–1 e–1 and φmax by 4.5°, 10.5°, and 14.5°, respectively. Also, the effect appears to be nearly proportional to the number of bound acceptor molecules—upon binding of two acceptor molecules, ESPmax on the third iodine atom is reduced by 20.0, 41.3, and 58.3 kJ mol–1 e–1, while φmax is reduced by 9.0°, 18.1°, and 24.4°. As can be seen, this effect can be (depending of the basicity and the number of acceptor molecules) significant—specifically, the binding of two dmap molecules on 135titfb reduces ESPmax on the third iodine atom by 46%, making it a much weaker Lewis acid as compared to free 135titfb.
This result is apparently in contrast with the experimental observation that only the strongest bases (dmap in particularly) form 1:3 cocrystals, whereas the weakest bases (such as 4cnpy) often form 1:1 cocrystals. One should keep in mind, however, that the halogen bond energy is also highly influenced by the basicity of the acceptor: the difference in XB energies formed by N-halogenosuccinimides (halogen = chlorine, bromine, and iodine) with dmap and 4cnpy was found to be as much as 20 kJ mol–1.25 The differences between XB energies for binding of the first base molecule to 135titfb as the XB donor are somewhat less—36.0 kJ mol–1 for dmap and 24.7 kJ mol–1 for 4cnpy with an intermediary energy of 30.1 kJ mol–1 for py, it being an intermediary base (Figure 7). As noted above, the energy of binding the second molecule of py is by 3.7 kJ mol–1, and the third is by ca. 6.7 kJ mol–1 less than the first one. Upon binding of dmap, the reduction of binding energy is considerably more pronounced (ΔE of 6.8 kJ mol–1 and 12.1 kJ mol–1), while with 4cnpy it is considerably less (1.6 kJ mol–1 and 2.9 kJ mol–1), again mirroring the differences in the reduction of ESPmax upon binding of each of the three bases. However, in spite of the largest reduction of binding energies upon binding of subsequent molecules of dmap, the overall binding energies of dmap are still considerably higher than those of weaker bases. Indeed, the binding energy of the third molecule of dmap to 135titfb (23.9 kJ mol–1) is higher than the binding energy of the second molecule of 4cnpy (23.2 kJ mol–1). Therefore, although the reduction of the Lewis acidity of the free iodine atom(s) of the 135titfb molecule is most pronounced with the strongest bases as acceptors, the increase of halogen bond energy due to the basicity of the acceptor more than compensates its decrease due to the reduction of the acidity of the donor.
Figure 7.
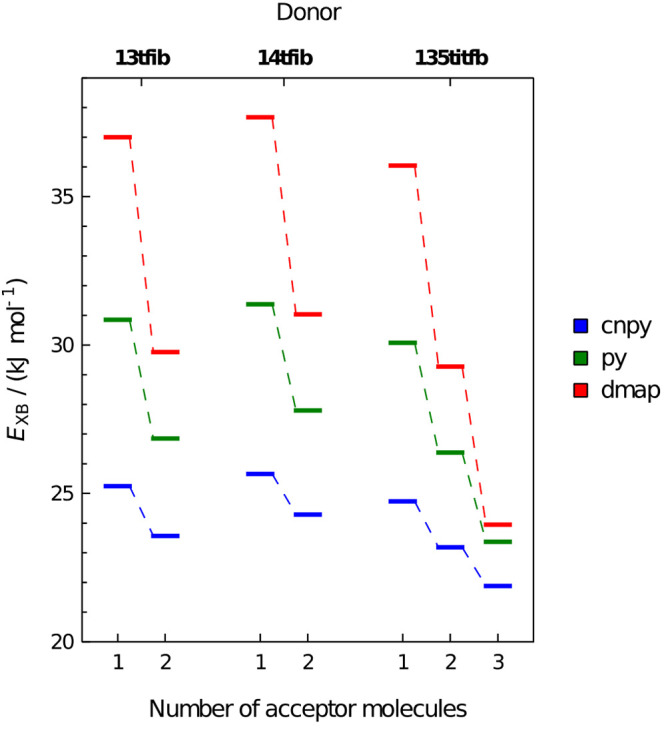
First, second, and (for the case of 135titfb) third binding energy of dmap, py, and 4cnpy to donors 14ditfb, 13ditfb, and 135titfb.
While the pKa-dependent topicity of 135titfb can be explained by the above considerations, the disparate behavior of the other two donors—the generally monotopic 13ditfb and generally ditopic 14ditfb—does not seem to stem from similar reasons. Although the ESPmax of 14ditfb is somewhat higher than that of 13ditfb, its reduction upon binding a single base molecule is smaller and the energies of binding of both base molecules are higher (for all three studied bases, Figure 7); these differences are minute and do not seem to be able to account for the observed difference in behavior. The binding energy of the second molecule of an intermediate base (py) is for both donors considerably higher than the energy of binding the first molecule of the weak base (4cnpy). Therefore, as both XB donors can form the 1:1 cocrystal with 4cnpy, they should both be expected to form 1:2 cocrystals with intermediate (and even more so with strong) bases. It is therefore probable that the main determining factor for the topicity of 13ditfb and 14ditfb is not the energy of the halogen bonds they form, but rather the ability of the resulting complexes to achieve close packing. When the linear 14ditfb binds a pair of base molecules, the resulting complex is also linear, and generally centrosymmetric, and therefore likely to efficiently fill the space in a crystal structure. The bent 2:1 complex resulting from binding two base molecules on 13ditfb is sterically more demanding, and less likely to be able to achieve close packing.
Conclusions
The structural and statistical study have pointed out significant differences in topicities of the 13ditfb (monotopic) and 14ditfb (ditopic) molecules as halogen bond donors in cocrystals with monotopic pyridines. It has been shown that the preferential cocrystal stoichiometry (and donor topicity) for both donor molecules is generally independent of the acceptor basicity but is rather determined by the overall crystal structure. Thus, 13ditfb will be the predominantly ditopic donor in cocrystals with polytopic acceptors, while the bent 2:1 complexes it forms with monotopic bases only exceptionally can efficiently pack in a crystal structure, resulting in the predominance of 1:1 cocrystals.
The topicity of 135titfb in cocrystals with monotopic pyridines highly depends on the basicity of the used acceptors—in combinations with more basic pyridines, 135titfb forms three halogen bonds, but with others, one or two bonds have formed. DFT calculations have shown that binding of the one acceptor molecule to the accessible iodine atoms leads to the reduction of the ESPmax values on free iodine atoms, and this effect increases with the basicity of the base. However, stronger bases form stronger halogen bonds, and the corresponding increase in bond energy can compensate the reduction of the Lewis acidity of the donors, thus allowing the formation of the three halogen bonds. Obtained results indicate that in addition to the donor molecule itself, the number of halogen bonds in the crystal structure is predetermined by the topicity and basicity of the acceptor molecule, which have been recognized as important factors in both the formation of the halogen-bonded molecular complexes and their packing in the solid state.
Experimental Section
Synthesis of Cocrystals
All the solvents and compounds used as halogen bond acceptors were procured from Sigma-Aldrich Chemie GmbH, Taufkirchen, Germany, and used without additional purification. Halogen bond donors 13tfib, 14tfib, and 135titfb were procured from Manchester Organics Ltd., Cheshire, UK and used without additional purification.
Cocrystals of 14ditfb and 135titfb and acceptors used have been prepared by both grinding and crystallization from solution. The grinding experiments were conducted in a Retsch MM200 ball mill using 10 mL stainless steel jars and two stainless steel balls (5 mm in diameter) for 15 min, under normal laboratory condition (40–60% relative humidity and temperature ca. 25 °C). Due to the large number of experiments performed, masses and volumes of the reactants used in the mechanochemical synthesis of cocrystals are given in Tables S5 and S6 in the Supporting Information. Single crystals of cocrystals with liquid acceptors were prepared by dissolving a halogen bond donor 135titfb or 14ditfb (50 mg) in hot ethanol (1.5 mL), after which a large excess of liquid acceptor was added (500 μL). The resulting solution was stirred and left at room temperature. Single crystals of 135tfib cocrystal with 4cnpy were prepared by dissolving donor m(135titfb) = 48 mg and acceptor m(4cnpy) = 53 mg in 1.00 mL of hot ethanol, after which solution was left at room temperature.
Cocrystals of 13ditfb and solid acceptors (acr, 4cnpy, and dmap) have been prepared by grinding as described above, while the cocrystals with liquid acceptors were synthesized by mixing of 13ditfb and corresponding acceptor in a 1:2 stoichiometric ratio on a microscope glass slide, after which crystallization of the product occurred. Masses and volumes of the reactants used in the synthesis of cocrystals are given in Table S7 in the Supporting Information. Single crystals of 13ditfb cocrystals were prepared by dissolving a halogen bond donor (40 μL) in hot ethanol (1.5 mL), after which a large excess of corresponding acceptor was added (500 μL).
X-ray Diffraction Experiments
Single crystal X-ray diffraction experiments were performed using an Oxford Diffraction Xcalibur Kappa CCD X-ray diffractometer with graphite-monochromated Mo Kα (λ = 0.71073 Å) radiation. The data sets were collected using the ω-scan mode over the 2θ range up to 54°. Programs CrysAlis CCD and CrysAlis RED were employed for data collection, cell refinement, and data reduction.46,47 The structures were solved by direct methods and refined using the SHELXS and SHELXL programs, respectively.48,49 The structural refinement was performed on F2 using all data. The hydrogen atoms were placed in calculated positions and treated as riding on their parent atoms [C–H = 0.93 Å and Uiso(H) = 1.2 Ueq(C); C–H = 0.97 Å and Uiso(H) = 1.2 Ueq(C)]. All calculations were performed using the WinGX crystallographic suite of programs.50 The figures were prepared using Mercury.51
Powder X-ray diffraction experiments on the samples were performed on an Aeris X-ray diffractometer (Malvern Panalytical, Malvern Worcestershire, UK) with Cu Kα1 (λ = 1.54056 Å) radiation. The scattered intensities were measured with a PIXcel-1D-Medipix3 detector. The angular range was from 5° to 40° (2θ) with a continuous step size of 0.02° and measuring a time of 0.5 s per step. Data collection methods were created using the program package START XRDMP CREATOR (Malvern Panalytical, Malvern Worcestershire, UK), while the data were analyzed using X’Pert HighScore Plus (Version 2.2, Malvern Panalytical, Malvern Worcestershire, UK).52
Thermal Analysis
Differential scanning calorimetry (DSC) and thermogravimetric (TG) measurements were performed simultaneously on a Mettler-Toledo TGA/DSC 3+ module (Mettler Toledo, Greifensee, Switzerland). Samples were placed in alumina crucibles (40 μL) and heated 25 to 300 °C, at a heating rate of 10 °C min–1 under nitrogen flow of 150 mL min–1.
Data collection and analysis were performed using the program package STARe Software (Version 15.00, Mettler Toledo, Greifensee, Switzerland).53 TG and DSC thermograms of the prepared compounds are shown in Figures S11–S15 in Supporting Information.
Calculation Details
All calculations were performed with Gaussian 09 (Rev. D.01) program suite.54 Geometry optimization of all donors, acceptors, and halogen-bonded complexes has been performed using B3LYP functional55 with Grimme’s GD3 dispersion correction56 and def2-TZVP basis set57 with effective core potential (ECP) for iodine atoms. Parameters of basis set and ECP for iodine were taken from the EMSL website.58 The same level of theory was used to calculate the binding energies on optimized geometries, employing the Boys–Bernardi counterpoise scheme59 to account for basis set superposition error and neglecting the relaxation energies of monomers, due to the high rigidity of all donors and acceptors. For overall population analysis (electron density, electrostatic potential and NBO analysis), single-point calculations on CAM-B3LYP60/def2-QZVP (with ECP for I) level were performed on optimized geometries.
Electron density and electrostatic potential were computed in a box around the halogen atom with length of 12.0 au using the cubegen utility of Gaussian. The number of points used was 150 in each dimension in the plane of the halogen bond donor molecule. Obtained densities were fitted with linear regression using basis of the type:
where Bi,k(x) is the ith B-spline function of the order k. B-spline functions were defined on the same box, and the order used was k = 4. The number of the B-spline function used was 30 in each dimension, so that there are 25 data points per coefficient that are fitted. An analogous procedure was used to fit the electrostatic potential in the plane of the donor molecule.
Fitted electron density was used to compute the isodensity curve in the plane of interest. This was done by defining a line in the plane which is perpendicular to the halogen bond and taking 1000 equidistant points along it. From each point, a linear search was performed in the direction perpendicular to the line (and parallel to the halogen bond) until the point with the desired density was found. For each point found, the value of electrostatic density was computed from the fit.
Database Survey
A data survey has been performed on the CSD database, version 5.42 (May 2021) with three updates using ConQuest Version 2020.3.0. For the halogen-bonded contacts, the upper limit of the distance between the donor atom (iodine) and the acceptors was defined as the sum of their van der Waals radii. In order to ascertain the frequency of halogen bonding, for each donor a number of searches were made: search for the total number of structures including perfluorinated halobenzenes as halogen bond donors; search which included the structures of the corresponding donor and nitrogen-containing molecule (defined as “N” fragment in ConQuest) which either can or cannot participate in halogen bonding; search for the structures in which donor and nitrogen-containing molecule participate in one or more I···N halogen bonds; search for structures of perfluorinated halobenzenes and pyridine derivatives from which structures with mono- and polytopic pyridines participating in one or more halogen bonds are manually extracted.
Acknowledgments
We acknowledge the support of the project CIuK cofinanced by the Croatian Government and the European Union through the European Regional Development Fund-Competitiveness and Cohesion Operational Programme (Grant KK.01.1.1.02.0016).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.cgd.2c00077.
ORTEP representations of formula units, PXRD patterns, DSC curves, crystallographic data, synthetic details and crystal structure descriptions (PDF)
Accession Codes
CCDC 2128222–2128241 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif, or by emailing data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336033.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
This research was supported by the Croatian Science Foundation under the project IP-2019–04–1868.
The authors declare no competing financial interest.
Supplementary Material
References
- Arman H. D.; Gieseking R. L.; Hanks T. W.; Pennington W. T. Complementary halogen and hydrogen bonding: sulfur···iodine interactions and thioamide ribbons. Chem. Commun. 2010, 46, 1854–1856. 10.1039/B925710A. [DOI] [PubMed] [Google Scholar]
- Berger G.; Soubhye J.; van der Lee A.; Vande Velde C.; Wintjens R.; Dubois P.; Clement S.; Meyer F. Interplay between Halogen Bonding and Lone Pair-π Interactions: A Computational and Crystal Packing Study. ChemPlusChem. 2014, 79, 552–558. 10.1002/cplu.201400005. [DOI] [PubMed] [Google Scholar]
- Nagels N.; Geboes Y.; Pinter B.; De Proft F.; Herrebout W. A. Tuning the Halogen/Hydrogen Bond Competition: A Spectroscopic and Conceptual DFT Study of Some Model Complexes Involving CHF2I. Chem.—Eur. J. 2014, 20, 8433–8443. 10.1002/chem.201402116. [DOI] [PubMed] [Google Scholar]
- Reid S. A.; Nyambo S.; Muzangwa L.; Uhler B. J. π-Stacking, C–H/π, and Halogen Bonding Interactions in Bromobenzene and Mixed Bromobenzene–Benzene Clusters. Phys. Chem. 2013, A117, 13556–13563. 10.1021/jp407544c. [DOI] [PubMed] [Google Scholar]
- Nochebuena J.; Cuautli C.; Ireta J. Origin of cooperativity in hydrogen bonding. Phys. Chem. Chem. Phys. 2017, 19, 15256–15263. 10.1039/C7CP01695F. [DOI] [PubMed] [Google Scholar]
- Masella M.; Flament J.-P. Influence of Cooperativity on Hydrogen Bond Networks. Molecular Modeling 2000, 24, 131–156. 10.1080/08927020008024192. [DOI] [Google Scholar]
- Patkar D.; Ahirwar M. B.; Gadre S. R.; Deshmukh M. M. Unusually Large Hydrogen-Bond Cooperativity in Hydrogen Fluoride Clusters, (HF)n, n = 3 to 8, Revealed by the Molecular Tailoring Approach. J. Phys. Chem. A 2021, 125, 8836–8845. 10.1021/acs.jpca.1c06478. [DOI] [PubMed] [Google Scholar]
- Cruzan J. D.; Braly L. B.; Liu K.; Brown M. G.; Loeser J. G.; Saykally R. J. Quantifying Hydrogen Bond Cooperativity in Water: VRT Spectroscopy of the Water Tetramer. Science 1996, 271, 59–62. 10.1126/science.271.5245.59. [DOI] [PubMed] [Google Scholar]
- Dashnau J. L.; Sharp K. A.; Vanderkooi J. M. Carbohydrate Intramolecular Hydrogen Bonding Cooperativity and Its Effect on Water Structure. J. Phys. Chem. B 2005, 109, 24152–24159. 10.1021/jp0543072. [DOI] [PubMed] [Google Scholar]
- Stokely K.; Mazza M. G.; Stanley H. E.; Franzese G. Effect of hydrogen bond cooperativity on the behavior of water. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 1301–1306. 10.1073/pnas.0912756107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanco S.; Pinacho P.; López J. C. Hydrogen-Bond Cooperativity in Formamide2–Water: A Model for Water-Mediated Interactions. Angew. Chem., Int. Ed. 2016, 128, 9477–9481. 10.1002/ange.201603319. [DOI] [PubMed] [Google Scholar]
- Guevara-Vela J. M.; Romero-Montalvo E.; Mora Gómez V. A.; Chávez-Calvillo R.; García-Revilla M.; Francisco E.; Pendása Á. M.; Rocha-Rinza T. Hydrogen bond cooperativity and anticooperativity within the water hexamer. Phys. Chem. Chem. Phys. 2016, 18, 19557–19566. 10.1039/C6CP00763E. [DOI] [PubMed] [Google Scholar]
- Sauza de la Vega A.; Rocha-Rinza T.; Guevara-Vela J. M. Cooperativity and anticooperativity in ion-water interactions. Implications for the aqueous solvation of ions. ChemPhysChem 2021, 22, 1269–1285. 10.1002/cphc.202000981. [DOI] [PubMed] [Google Scholar]
- Tielrooij K. J.; Garcia-Araez N.; Bonn M.; Bakker H. J. Cooperativity in ion hydration. Science 2010, 328, 1006–1009. 10.1126/science.1183512. [DOI] [PubMed] [Google Scholar]
- Vijay D.; Zipse H.; Sastry G. N. On the Cooperativity of Cation−π and Hydrogen Bonding Interactions. J. Phys. Chem. B 2008, 112, 8863–8867. 10.1021/jp804219e. [DOI] [PubMed] [Google Scholar]
- Broomsgrove A. E. J.; Addy D. A.; Di Paolo A.; Morgan I. R.; Bresner C.; Chislet V.; Fallis I. A.; Thompson A. L.; Vidovic D.; Aldridge S. Evaluation of Electronics, Electrostatics and Hydrogen Bond Cooperativity in the Binding of Cyanide and Fluoride by Lewis Acidic Ferrocenylboranes. Inorg. Chem. 2010, 49, 157–173. 10.1021/ic901673u. [DOI] [PubMed] [Google Scholar]
- Sheridan R. P.; Lee R. H.; Peters N.; Allen L. C. Hydrogen-bond cooperativity in protein secondary structure. Biopolymers 1979, 18, 2451–2458. 10.1002/bip.1979.360181006. [DOI] [Google Scholar]
- Li J.; Wang Y.; Chen J.; Liu Z.; Bax A.; Yao L. Observation of α-Helical Hydrogen-Bond Cooperativity in an Intact Protein. J. Am. Chem. Soc. 2016, 138, 1824–1827. 10.1021/jacs.5b13140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker L. L.; Houk A. R.; Jensen J. H. Cooperative Hydrogen Bonding Effects Are Key Determinants of Backbone Amide Proton Chemical Shifts in Proteins. J. Am. Chem. Soc. 2006, 128, 9863–9872. 10.1021/ja0617901. [DOI] [PubMed] [Google Scholar]
- DeChancie J.; Houk K. N. The Origins of Femtomolar Protein–Ligand Binding: Hydrogen Bond Cooperativity and Desolvation Energetics in the Biotin–(Strept)Avidin Binding Site. J. Am. Chem. Soc. 2007, 129, 5419–5429. 10.1021/ja066950n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffrey G. A.An Introduction to Hydrogen Bonding; Oxford University Press: Oxford, 1997. [Google Scholar]
- Cavallo G.; Metrangolo P.; Milani R.; Pilati T.; Priimagi A.; Resnati G.; Terraneo G. The Halogen Bond. Chem. Rev. 2016, 116, 2478–2601. 10.1021/acs.chemrev.5b00484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aakeröy C. B.; Fasulo M.; Schultheiss N.; Desper J.; Moore C. Structural Competition between Hydrogen Bonds and Halogen Bonds. J. Am. Chem. Soc. 2007, 129, 13772–13773. 10.1021/ja073201c. [DOI] [PubMed] [Google Scholar]
- Aakeröy C. B.; Panikkattu S.; Chopade P. D.; Desper J. Competing hydrogen-bond and halogen-bond donors in crystal engineering. CrystEngComm 2013, 15, 3125–3136. 10.1039/C2CE26747K. [DOI] [Google Scholar]
- Stilinović V.; Horvat G.; Hrenar T.; Nemec V.; Cinčić D. Halogen and Hydrogen Bonding between (N-Halogeno)-succinimides and Pyridine Derivatives in Solution, the Solid State and In Silico. Chem.—Eur. J. 2017, 23, 5244–5257. 10.1002/chem.201605686. [DOI] [PubMed] [Google Scholar]
- Eraković M.; Cinčić D.; Molčanov K.; Stilinović V. A Crystallographic Charge Density Study of the Partial Covalent Nature of Strong N··· Br Halogen Bonds. Angew. Chem., Int. Ed. 2019, 131, 15849–15853. 10.1002/ange.201908875. [DOI] [PubMed] [Google Scholar]
- Weinberger C.; Hines R.; Zeller M.; Rosokha S. V. Continuum of covalent to intermolecular bonding in the halogen-bonded complexes of 1,4-diazabicyclo[2.2.2]octane with bromine-containing electrophiles. Chem. Commun. 2018, 54, 8060–8063. 10.1039/C8CC04629H. [DOI] [PubMed] [Google Scholar]
- Desiraju G. R.; Ho P. S.; Kloo L.; Legon A. C.; Marquardt R.; Metrangolo P.; Politzer P.; Resnati G.; Rissanen K. Definition of the halogen bond (IUPAC Recommendations 2013). Pure Appl. Chem. 2013, 85, 1711–1713. 10.1351/PAC-REC-12-05-10. [DOI] [Google Scholar]
- Dominikowska J.; Rybarczyk-Pirek A. J.; Fonseca Guerra C. Lack of Cooperativity in the Triangular X3 Halogen-Bonded Synthon?. Cryst. Growth Des. 2021, 21, 597–607. 10.1021/acs.cgd.0c01410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheiner S. Comparison of Bifurcated Halogen with Hydrogen Bonds Comparison of Bifurcated Halogen with Hydrogen Bonds. Molecules 2021, 26, 350–358. 10.3390/molecules26020350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cinčić D.; Friščić T.; Jones W. Experimental and database studies of three-centered halogen bonds with bifurcated acceptors present in molecular crystals, cocrystals and salts. CrystEngComm 2011, 13, 3224–3231. 10.1039/c0ce00699h. [DOI] [Google Scholar]
- Corradi E.; Meille S. V.; Messina M. T.; Metrangolo P.; Resnati G. Halogen bonding versus hydrogen bonding in driving self-assembly processes. Angew. Chem., Int. Ed. 2000, 39, 1782–1786. . [DOI] [PubMed] [Google Scholar]
- Bedeković N.; Stilinović V.; Friščić T.; Cinčić D. Comparison of isomeric meta-and para-diiodotetrafluorobenzene as halogen bond donors in crystal engineering. New J. Chem. 2018, 42, 10584–10591. 10.1039/C8NJ01368C. [DOI] [Google Scholar]
- Ding X.-H.; Chang Y.-Z.; Ou C.-J.; Lin J.-Y.; Xie L.-H.; Huang W. Halogen bonding in the co-crystallization of potentially ditopic diiodotetrafluorobenzene: a powerful tool for constructing multicomponent supramolecular assemblies. National Science Review 2020, 7, 1906–1932. 10.1093/nsr/nwaa170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liantonio R.; Luzzati S.; Metrangolo P.; Pilati T.; Resnati G. Perfluorocarbon–hydrocarbon self-assembly. Part 16: Anilines as new electron donor modules for halogen bonded infinite chain formation. Tetrahedron 2002, 58, 4023–4029. 10.1016/S0040-4020(02)00264-8. [DOI] [Google Scholar]
- Aakeröy C. B.; Desper J.; Helfrich B. A.; Metrangolo P.; Pilati T.; Resnati G.; Stevenazzi A. Combining halogen bonds and hydrogen bonds in the modular assembly of heteromeric infinite 1-D chains. ChemComm 2007, 41, 4236–4238. 10.1039/b707458a. [DOI] [PubMed] [Google Scholar]
- Cinčić D.; Friščić T.; Jones W. Isostructural Materials Achieved by Using Structurally Equivalent Donors and Acceptors in Halogen-Bonded Cocrystals. Chem.—Eur. J. 2008, 14, 747–753. 10.1002/chem.200701184. [DOI] [PubMed] [Google Scholar]
- Nemec V.; Cinčić D. Uncommon halogen bond motifs in cocrystals of aromatic amines and 1,4-diiodotetrafluorobenzene. CrystEngComm 2016, 18, 7425–7429. 10.1039/C6CE01703G. [DOI] [Google Scholar]
- Metrangolo P.; Meyer F.; Pilati T.; Resnati G.; Terraneo G. 4,4’-Bipyridine-2,4,5,6-tetrafluoro-1,3-diiodobenzene (1/1). Acta Crystallogr. 2007, E63, o4243–4248. 10.1107/S1600536807047630. [DOI] [Google Scholar]
- Lucassen A. C. B.; Karton A.; Leitus G.; Shimon L. J. W.; Martin J. M. L.; van der Boom M. E. Co-Crystallization of Sym-Triiodo-Trifluorobenzene with Bipyridyl Donors: Consistent Formation of Two Instead of Anticipated Three N···I Halogen Bonds. Cryst. Growth Des. 2007, 7, 386–392. 10.1021/cg0607250. [DOI] [Google Scholar]
- Roper L. C.; Präsang C.; Kozhevnikov V. N.; Whitwood A. C.; Karadakov P. B.; Bruce D. W. Experimental and Theoretical Study of Halogen-Bonded Complexes of DMAP with Di- and Triiodofluorobenzenes. A Complex with a Very Short N···I Halogen Bond. Cryst. Growth Des. 2010, 10, 3710–3720. 10.1021/cg100549u. [DOI] [Google Scholar]
- Groom C. R.; Bruno I. J.; Lightfoot M. P.; Ward S. C. The Cambridge Structural Database. Acta Crystallogr., Sect. B 2016, 72, 171–179. 10.1107/S2052520616003954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torubaev Y. V.; Skabitsky I. V. The energy frameworks of aufbau synthon modules in 4-cyanopyridine co-crystals. CrystEngComm 2019, 21, 7057–7068. 10.1039/C9CE01174A. [DOI] [Google Scholar]
- Cinčić D.; Friščić T.; Jones W. Structural Equivalence of Br and I Halogen Bonds: A Route to Isostructural Materials with Controllable Properties. Chem. Mater. 2008, 20, 6623–6626. 10.1021/cm800923r. [DOI] [Google Scholar]
- Szell P. M. J.; Gabriel S. A.; Gill R. D. D.; Wan S. Y. H.; Gabidullin B.; Bryce D. L. 13C and 19F solid-state NMR and X-ray crystallographic study of halogen-bonded frameworks featuring nitrogen-containing heterocycles. Acta Crystallogr. 2017, C73, 157–167. 10.1107/S2053229616015023. [DOI] [PubMed] [Google Scholar]
- CrysAlis CCD V171.34; Oxford Diffraction Ltd.: Abingdon, Oxfordshire, UK, 2003.
- CrysAlis RED V171.34; Oxford Diffraction Ltd.: Abingdon, Oxfordshire, UK, 2003.
- Sheldrick G. M. A short history of SHELX. Acta Crystallogr. 2008, A64, 112–122. 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
- Sheldrick G. M. SHELXT - Integrated space-group and crystal-structure determination. Acta Crystallogr., Sect. A 2015, 71, 3–8. 10.1107/S2053273314026370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrugia L. J. WinGX suite for small-molecule single-crystal crystallography. J. Appl. Crystallogr. 1999, 32, 837–838. 10.1107/S0021889899006020. [DOI] [Google Scholar]
- Macrae C. F.; Bruno I. J.; Chisholm J. A.; Edgington P. R.; McCabe P.; Pidcock E.; Rodriguez-Monge L.; Taylor R.; van de Streek J.; Wood P. A. Mercury CSD 2.0 - new features for the visualization and investigation of crystal structures. J. Appl. Crystallogr. 2008, 41, 466–470. 10.1107/S0021889807067908. [DOI] [Google Scholar]
- Degen T.; Sadki M.; Bron E.; König U.; Nénert G. The HighScore suite. Powder Diffr. 2014, 29, S13–S18. 10.1017/S0885715614000840. [DOI] [Google Scholar]
- STARe Software; Mettler Toledo: Greifensee, Switzerland, 2016.
- Frisch M. J., et al. Gaussian 09, Revision D.01; Gaussian Inc.: Wallingford, CT, 2009.
- Becke A. D. Density-functional thermochemistry. I. The effect of the exchange-only gradient correction. J. Chem. Phys. 1993, 98, 5648. 10.1063/1.464913. [DOI] [Google Scholar]
- Grimme S.; Antony J.; Ehrlich S.; Krieg H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. 10.1063/1.3382344. [DOI] [PubMed] [Google Scholar]
- Weigend F.; Ahlrichs R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. 10.1039/b508541a. [DOI] [PubMed] [Google Scholar]
- Schuchardt K. L.; Didier B. T.; Elsethagen T.; Sun L.; Gurumoorthi V.; Chase J.; Li J.; Windus T. L. Basis Set Exchange: A Community Database for Computational Sciences. J. Chem. Inf. Model. 2007, 47, 1045–1052. 10.1021/ci600510j. [DOI] [PubMed] [Google Scholar]
- Boys S. F.; Bernardi F. The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol. Phys. 1970, 19, 553. 10.1080/00268977000101561. [DOI] [Google Scholar]
- Yanai T.; Tew D.; Handy N. A new hybrid exchange–correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. 10.1016/j.cplett.2004.06.011. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.