

Abstract
Despite the high mortality and disability associated with traumatic brain injury (TBI), effective pharmacologic treatments are lacking. Of emerging interest, bioactive lipids, including specialized pro-resolving lipid mediators of inflammation (SPMs), act to attenuate inflammation after injury resolution. The SPM lipidome may serve as a biomarker of disease and predictor of clinical outcomes, and the use of exogenous SPM administration represents a novel therapeutic strategy for TBI. This review article provides a comprehensive discussion of the current pre-clinical and clinical literature supporting the importance of bioactive lipids, including SPMs, in TBI recovery. We additionally propose a translational approach to answer important clinical and scientific questions to advance the study of bioactive lipids and SPMs towards clinical research. Given the morbidity and mortality associated with TBI with limited treatment options, novel approaches are needed.
Keywords: TBI, SPMs, lipids, inflammation, edema, pharmacologic treatment
Introduction
Traumatic brain injury (TBI) affects all age, ethnic and socioeconomic groups and is associated with high morbidity and mortality [1]. In survivors, chronic physical, cognitive, and mental health impairments are common. Despite its negative impact, there are no proven effective pharmacologic therapies that improve outcomes in mild, moderate, or severe TBI. Primary injury occurs at the time of impact or acceleration-deceleration and results in macro and microscopic injury to axons, brain parenchyma, and vasculature. The second phase of injury can occur immediately or be delayed for hours or days. It is characterized by ischemia, excitotoxicity, metabolic dysfunction, blood-brain barrier (BBB) breakdown, and subsequent cerebral edema [2]. Given the chronic effects and poor outcomes associated with TBI pathophysiology, novel therapeutic targets are needed to limit secondary brain injury.
Neuroinflammation as a mediator of secondary brain injury
Neuroinflammation is an important contributor to secondary cell death after TBI [3]. This is mediated by phenotypically activated microglia early after brain injury, which can persist for months [4,5]. Activated microglia release large amounts of pro-inflammatory signaling proteins that include tumor necrosis factor-α (TNF-α), interleukin (IL)-1β, and IL-6 [6,7]. After the primary TBI, a cascade of inflammatory events including further upregulation of pro-inflammatory mediators (e.g. cytokines, chemokines, and bioactive lipids) and matrix metalloproteinases (MMPs) target extracellular matrix proteins leading to BBB degradation. Pro-inflammatory signaling also results in recruitment of inflammatory cells, reduced integrity of tight junctions [8-10], upregulation of aquaporin-4 receptors [11], and oxidative injury secondary to mitochondrial dysfunction [12] that contribute to BBB permeability. Beyond promoting cerebral edema formation, neuroinflammation promotes secondary brain injury through metabolic crisis, mitochondrial dysfunction [13], alterations in central nervous system (CNS) glymphatics and lymphatics [14,15], increased pro-apoptotic signaling [16,17], and microthrombosis formation and coagulopathy, leading to ischemia [18-20] (Figure 1).
Figure 1.
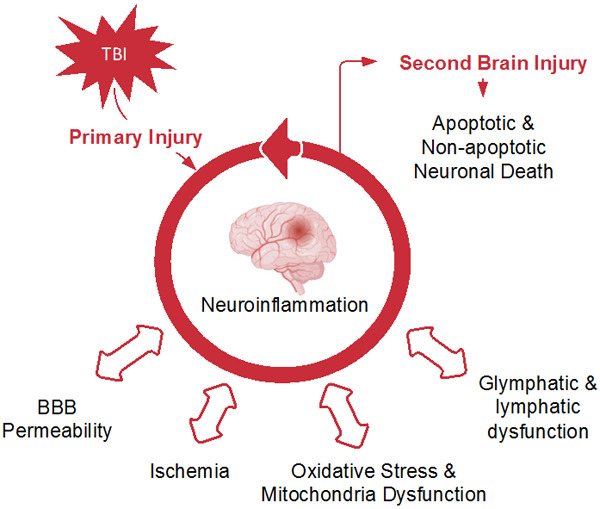
Conceptual schematic of the neuroinflammatory cascade promoting secondary brain injury.
Endogenous specialized pro-resolving lipid mediators of inflammation
While pro-inflammatory responses are well-established, less is known about the downregulation of inflammation after injury resolution. There is growing interest in the role of bioactive lipids in attenuating secondary brain injury. These endogenous lipids regulate cell growth, adhesion, migration, signaling, and death [21]. There is a diversity of bioactive lipids with pro-inflammatory or inflammatory-resolving functions. Those that downregulate inflammation represent a specific family called specialized pro-resolving lipid mediators (SPMs). This family of bioactive lipids has diverse biological activities, which include the ability to attenuate neuroinflammation and cell death in several neurologic diseases such as epilepsy [22], Alzheimer’s disease (AD) [23], subarachnoid hemorrhage [24], and ischemic stroke [25].
The most characterized SPMs include resolvins (e.g. RvD1, RvE1) and their aspirin-triggered stereoisomers (AT-RvD1, AT-RvE1), protectins (e.g. NPD1), maresins, and lipoxins (LXs). Derived from the metabolism of omega-3 and omega-6 fatty acid (FA) precursors, SPMs mitigate neurologic injury by acting as a molecular “stop signal” for neuroinflammation. At the injury site, SPMs promote killing and clearance of pathogens, reduce leukocyte infiltration, and stimulate macrophage-mediated efferocytosis of cellular debris and apoptotic-sequestered neutrophils. They also inhibit the expression of pro-inflammatory cytokines and chemokines while upregulating anti-inflammatory mediators [21]. However, despite what is known, their effect on TBI pathogenesis and recovery is not fully elucidated. In this review, we aim to summarize results from the literature supporting the use of SPMs as a novel therapeutic in TBI, identify remaining gaps in our knowledge, and provide a translational approach towards further research to improve outcomes in patients inflicted by TBI.
Role of specialized pro-resolving lipid mediators in traumatic brain injury
Rodent models of traumatic brain injury
Pre-clinical research investigating the use of pro-resolving lipid mediators of inflammation has been promising. Early studies in rodent models demonstrated that dietary supplementation of anti-inflammatory polyunsaturated fatty acids (PUFAs) prior to TBI reduced downstream effects of neuroinflammation. Pre-injury treatment in rats with dietary omega-3 PUFAS for 4 weeks prior to fluid percussion injury suggested reduced oxidative stress, decreased markers of abnormal cellular metabolism, and improved spatial learning [26,27]. In a rat impact acceleration model, Mills et al. also demonstrated that high-dose docosahexaenoic acid (DHA) supplementation prior to TBI was associated with reductions in CD-68+ cells and caspase-3 levels one week after injury, suggesting a neuroprotective effect against inflammatory cell recruitment and pro-apoptotic signaling [28].
Post-injury dietary supplementation of PUFAs also ameliorated cellular markers of secondary brain injury. In an impact acceleration injury model, rats given DHA for 30 days following TBI exhibited a dose-dependent decrease in axons positive for amyloid precursor protein [29]. Similarly, rats supplemented with an omega-3 FA preparation of eicosapentaenoic acid (EPA) and DHA in a post-injury model demonstrated decreased expression of pro-apoptotic caspase-3 [30]. Administered for 7-days after TBI, fish oil containing EPA and DHA in rats was associated with increased dopamine release, with a suggested beneficial effect on short and long-term cognitive and motor recovery [31].
Apart from dietary supplementation, parenteral SPM delivery has also demonstrated benefits. Luo et al. investigated the use of LXA4 in a mouse model of TBI using a weight-drop impact method. A single dose of intrathecal (IT) drug given 10 minutes after TBI was associated with reductions in BBB permeability, brain water content, and pro-inflammatory cytokines TNF-α, IL-1β, and IL-6 at 6 H post-injury [32]. In a series of experiments by Chen et al. in a weight drop rat model, post-injury intraperitoneal (IP) administration of omega-3 FA for 7 days reduced inflammatory markers, including microglial activation and HMGB1 expression, and was associated with decreased neuronal apoptosis [33-35]. In a rat hemicerebellectomy model of focal injury, IP RvD1 promoted motor-behavioral recovery attributed to the expression of ALX/FPR2 receptor-regulated microRNAs [36]. Similarly, in adult mice treated with RvE1 or AT-RvD1 given IP for 7 days beginning 3 days before TBI, AT-RvD1 alone mitigated motor and cognitive deficits while RvE1 differentially showed reductions in activated microglia [37]. More recently, in a proof-of-concept study, single-dose intralesional administration of neuroprotection D1 (NPD1) given to rats after penetrating TBI was able to reduce lesion size at 72 h. No differences were detected in markers for inflammation, immune cells, or neuronal degeneration, suggesting that treatment induced a local biologic effect [38].
A summary of rodent studies is shown in Table 1. These data support the administration of SPMs or their precursors to mitigate secondary brain injury associated with TBI. Unfortunately, study design and disease models differ, and thus specific pathways important for these outcomes have not been clearly delineated yet. Further studies are necessary to fully understand the biochemical pathways associated with morbidity and the mechanistic role of bioactive lipids and SPMS after TBI induction. The use of these bioactive lipids has a variety of beneficial effects which is only partially explained by their anti-inflammatory activity.
Table 1.
Bioactive lipids studied in rodent models of TBI
| Administered Lipid | Model | Treatment Start | Dose and Frequency | Treatment Duration | Route | Outcomes |
|---|---|---|---|---|---|---|
| Resolvins [36] | Midline fluid percussion; mouse | 3 days pre-injury | RvE1 100 ng daily; | 7 days | IP | AT-RvD1 treatment associated with improved motor and cognitive scores. RvE1 treatment associated with reduced Iba-1+ microglia in cortical regions |
| AT-RvD1 100 ng daily | ||||||
| Resolvins [35] | Hemicerebellectomy; rat | Immediately post-injury | RVD1 0.4 μg/kg | Days 0, 3, 5, 7 | IP | Reduced activated Iba-1+ microglia and GFAP+ astrocytes |
| ALX/FPR2 50 μg/kg | Day 3 | IT | ||||
| LXA4 [31] | Weight drop; mouse | 10 min post-injury | LXA4 (0.3 nM) | Single Dose | IT | Decreases inflammatory cytokines TNF-α, IL-1β, IL-6, decreased BBB permeability and brain edema |
| DHA [28] | Impact acceleration; rat | 24 h post-injury | 10 mg/kg/d or 40 mg/kg/d | 30 days | PO | Decreased APP axons |
| EPA and DHA [29] | Impact acceleration; rat | 24 h post-injury | 10 mg/kg/d or 40 mg/kg/d | 30 days | PO | Decreased APP, reduced caspase-3 expression |
| Omega-3 FA [25] | Fluid percussion; rat | 4 weeks pre-injury | 8% fish oil (12.4% DHA and 13.5% EPA) | 5 weeks | PO | Reduced oxidative stress, increased BDNF levels, improved spatial learning |
| Omega-3 FA [26] | Fluid percussion; rat | 4 week pre-injury | 8% fish oil (12.4% DHA and 13.5% EPA) | 1, 7, or 14 days | PO | Reduced Sir2 expression, reduced oxidative stress, increased AMPK |
| Omega-3 FA [30] | Controlled cortical impact; rat | 6 min post-injury | 1.5 mL/d | 7 days | PO | Reduced dopamine release |
| Omega-3 FA [32,33] | Weight drop; rat | 30 min post-injury | 2 ml/kg | 7 days | IP | Reduced microglial activation, reduced HMGB1 expression and acetylation |
| Omega-3 FA [34] | Weight drop; rat | 30 min post-injury | 2 ml/kg | 7 days | IP | Increased SIRT1 expression, reduced apoptotic neurons |
| NPD1 [37] | Focal penetrating injury; rat | Immediately post-injury | 50 ng | Single dose | Intralesion | Decreased macroscopic lesion size at 72 H post injury, no change in neuronal degeneration or apoptosis |
LXA4: Lipoxin A4, DHA: docosahexanoic acid, EPA: eicosapentaenoic acid, FA: fatty acid, AT-RvD1: aspirin-triggered stereoisomer of resolving D1, RvE1: resolvin E1, GFAP: glial fibrillary acidic protein, APP: amyloid precursor protein, BDNF: brain-derived neurotrophic factor, Sir2: silent information regulator 2, AMPK: AMP-activated protein kinase, HMGB1: high mobility group box 1, SIRT1: sirtuin family of proteins 1, NPD1: neuroprotection D1.
Human experiences in acute brain injury
Reports on the impact of bioactive lipids in human patients after brain injury are limited (Table 2). Based on case reports and one case series, positive neurologic outcomes after acute brain injury have been partially attributed to therapy with large amounts of omega-3 FAs [39-41]. Dietary supplementation of PUFAs is considered a low-risk intervention, with future clinical trials being considered [41]. To our knowledge, no controlled randomized clinical trial has yet investigated the use of parenteral PUFA or SPM after TBI in human subjects. Enteral feeding formulations containing omega-3 FAs have found use in some intensive care units, but this is not widely implemented in clinical practice.
Table 2.
Human experiences using omega-3 fatty acid supplementation after acute brain injury
| Administered Lipid | Treatment Start | Dose and Frequency | Treatment Duration | Route | Type of Injury | Outcomes |
|---|---|---|---|---|---|---|
| Omega-3 FA [38] | 2 days post-injury | 5 g (3.4 g EPA/1.7 g DHA) And 16.2 g omega-3 FA, 10.8 g EPA/5.4 g DHA | Undefined | Enteral | Blast injury | Potential contributor to good neurologic outcome |
| 8 days post-injury | ||||||
| Omega-3 FA [39] | 10 days post-injury | 9,756 mg EPA, 6,756 mg DHA, and 19,212 mg total omega-3 FA 30 mL/day | Over 1 year | Enteral | Blunt-force motor vehicle accident | Potential contributor to good neurologic outcome |
| Omega-3 FA [40] | post-injury | 16.2 g (10.8 g of EPA and 5.4 g of DHA) | Variable duration: 7-35 days | Enteral | Blunt-force injury, primarily motor vehicle accident | Potential contributor to good neurologic outcome |
FA: fatty acid, EPA: eicosapentaenoic acid, DHA: docosahexanoic.
Bioactive lipids as an ideal biomarker and therapeutic candidate
Use as a biomarker of disease
Neuroinflammation plays a central role in mediating secondary brain injury after TBI; thus, the ability to modulate CNS inflammation may be an effective therapeutic approach. Several anti-inflammatory drugs have been studied in animal models of TBI (as recently reviewed by Bergold [42]); however, none have translated to widespread clinical use. Classes of drugs most extensively studied include corticosteroids, non-steroidal anti-inflammatory drugs, TNF-α and IL cytokine inhibitors, and HMG-CoA reductase inhibitors. Prior negative drug trials do not diminish the unique characteristics of bioactive lipids as an ideal biomarker of disease and therapeutic candidate.
Early studies, including those by Bazán et al. [43-45], demonstrated that FA precursors of SPMs are altered and can be reliably measured after brain injury. With the advent of validated high-performance liquid chromatography coupled with mass spectrometry (HPLC-MS), the lipidome can be rapidly and accurately characterized. Using HPLC-MS, endogenous lipids can be precisely identified and quantified, potentially serving as biomarkers. In a pilot study, Pilitsis et al. showed that free FA concentrations of AA and DHA are higher in cerebrospinal fluid (CSF) acutely after TBI compared to controls with chronic neurologic disease, with higher levels of AA correlated with TBI severity and worse clinical outcomes [46]. The proportional amounts of anti-inflammatory PUFAs may be similarly useful in detecting TBI and disease severity. The arachidonic acid (AA) to DHA ratio was decreased in the hippocampi, cortex, and plasma of brain-injured mice, while human studies suggest reduced ratios of AA:DHA more likely in patients exposed to TBI [47-49].
Lipid biomarkers of TBI hold advantages over proteomics and peptide biomarkers. Limitations of proteomics include a bias towards highly abundant proteins, challenges in identifying protein aggregations (e.g. tau), and difficulty in reliably assaying peptides generated in the CNS from plasma samples [50]. Levels of glial protein biomarkers are low acutely after TBI and non-specific, and have an inconsistent association with injury severity and outcomes [51]. Alternatively, lipids are abundant in the CNS, their metabolism is well-established, and TBI patients might possess a unique lipidome profile. These qualities provide an opportunity to characterize the lipidome in relation to disease severity and progression. Due to their ability to cross the BBB by active transport, lipids can easily be assayed from plasma, providing a “liquid biopsy” that might give insight into the biologic processes occurring at affected sites in the CNS [52]. Because of their high volume of distribution, lipids can be measured in other biofluids and biomatrices, including tears [53]; however, it is not yet known if lipidomes remain consistent when tested from different biofluids after brain injury. Correlation studies are ongoing to validate whether circulating lipidomics are accurate biomarkers in TBI.
Additional important lipid biomarkers are expected to be identified in the future with HPLC-MS techniques. In addition to identifying and quantifying analytes, new methods are also available to determine the metabolic fate of these analytes. When coupled with new technologies such as MALDI-TOF imaging, diagnostic tools used to characterize the post-TBI lipidome will also elucidate the molecular pathways by which SPMs are generated.
Therapeutic development
As a therapeutic agent, bioactive lipids are appealing due to the myriad of biological activities that promote tissue resolution after TBI. Their ability to attenuate inflammation without compromising immune activation is one of the positive aspects of developing SPMs. The pleiotropic impacts on multiple downstream mechanisms found in secondary brain injury are additional benefits [54]. While upstream targets modulating CNS lipid metabolism can be targeted [55], lipid formulations can be directly administered safely through intravenous (IV), subcutaneous (SC) or oral (PO) routes in human subjects, and additionally via IP injection in animal models of TBI. Because bioactive lipids of interest are naturally occurring, supplementation of PUFAs and other lipid agents and their analogs is considered safe and is thought to have a large therapeutic window. The low cost, wide availability and apparent safety of omega-3 FAs and other PUFAs warrant further investigation for therapeutic use in TBI. SPMs, including lipoxins, are not as readily available in clinical practice but might share many advantageous characteristics with other bioactive lipids.
A drawback associated with bioactive lipids includes metabolic instability resulting in a short systemic half-life. Despite this pharmacologic limitation, daily administration appears safe and leads to efficacy in various models, suggesting that SPM binding onto G-protein coupled receptors (GPCR) may activate an “on switch” where biological activity continues despite the absence of the SPM. More recently, our team has made mimics of the naturally occurring bioactive lipids like LXA4, with a systemic half-life beyond 6 hours [56]. Our compound library focused on bioactive lipids that were more stable as compared to the endogenous lipid without compromising GPCR binding. The ability to formulate these types of compounds is a continued challenge.
Research needs and translation to clinical care in traumatic brain injury
Given the importance of SPMs in mediating inflammation in disease animal models, and early human experiences supplementing PUFAs after brain injury, research efforts are urgently needed to translate therapeutic targets to clinical practice. Many biochemical and clinical questions remain. Mechanistically, the exact biological effects of SPMs on the neuroinflammatory cascade have not been fully elucidated. In clinical aspects, the optimal drug, dosage strategy, and clinical safety and efficacy have yet to be demonstrated in high-quality translational research. Here, we outline an analytic research approach to answer the clinical question of whether administration of SPMs can positively affect patient outcomes after TBI.
Establishing the importance of specialized pro-resolving lipid mediators of inflammation in human traumatic brain injury
As SPMs are naturally occurring endogenous lipids, they can be characterized before interventional clinical trials. Establishing their importance and mechanistic role in normal post-TBI repair and recovery would provide the rationale for SPMs to be utilized as a biomarker and therapeutic target in future clinical studies. Recently, they have been characterized in other human neurologic diseases that are mediated by chronic inflammation. In multiple sclerosis, SPM lipidome profiles carry unique signals that are associated with disease progression [57]. Investigators also found impaired gene expression of many biosynthetic enzymes and receptors important in the formation and biologic activity of SPMs, including reductions in COX-2 enzymes, 5-LOX enzymes, and GPCR receptors in progressive disease forms [57]. In vascular disease and stroke, Fredman et al. demonstrated that unstable areas of carotid plaques show reduced levels of SPMs, particularly RvD1, and reduced ratios of SPMs to pro-inflammatory leukotriene B4 (LTB4) [58]. In a middle cerebral artery occlusion model, Bazan et al. demonstrated that DHA administration results in increased NPD1 biosynthesis in the brain, suggesting the innate anti-inflammatory role of SPMs after stroke [25]. Lastly, in AD, levels of NPD1 and its precursor DHA are reduced in the hippocampi of dementia patients [59]. Wang et al. additionally demonstrated reduced LXA4 levels in the hippocampus of AD brains; notably, this profile was also seen when assayed from CSF [60].
Together, these studies demonstrate the value and feasibility of characterizing the SPM lipidome in human neurologic diseases. Identifying unique lipidomic signals compared to controls would not only confirm their importance in human disease but will also provide some insight into the specific pathophysiologic pathways by which these lipidomic signatures are produced. In clinical practice, it would be most relevant to determine the association between the SPM lipidome with disease severity, clinical outcomes, and advanced neuroimaging that includes perfusion and BBB permeability techniques. Imaging and lipidomics are both non-invasive techniques that may be used to predict outcomes after TBI.
Identifying similarities and differences between bioactive lipid signatures assayed from serum versus CSF samples could also be meaningful. If lipid signatures between sources show high agreement, it would suggest that SPMs measured from blood samples accurately reflect brain pathology without requiring invasive sampling of CSF. The TBI population is appropriate to use in confirming these correlations in an observational study, as the presence of a clinically indicated external ventricular drain to measure intracranial pressure allows access to CSF for analysis.
A validated rodent controlled-cortical impact model for pre-clinical research
Human observational trials are not sufficient to understand the full mechanistic role of SPMs. Validated animal models are also needed. While human TBI is a physiologically complex disease with many confounders impacting clinical outcomes, animal models allow investigators to perform controlled and reproducible experiments in which physiological and metabolic pathways are characterized. Despite SPM lipidomics being a relatively new field, much has been learned about the generation of SPMs and their role in attenuating a pro-inflammatory state in animal models [61].
The rodent-controlled cortical impact (CCI) model is a reasonable choice for both basic biochemical studies and for mechanistic translational research. It was first described in the 1980s and is now one of the most used TBI models. Compared to other methods of mechanical injury, namely lateral fluid percussion and drop-weight injury, CCI allows for more control over the force of injury, showing greater reproducibility, lower animal mortality, and reduced rebound injury [62]. Current CCI rodent models produce sufficient conditions for understanding the molecular, cellular, and biochemical mechanisms of secondary brain injury after focal blunt-force TBI [63]. The general methodology of CCI following craniotomy has been described in detail by Dean et al. [62]. Non-rodent gyrencephalic animal models of TBI have not been validated in the study of SPM lipidomics but may be necessary for additional safety outcomes prior to human research.
Using animal models to further dissect mechanisms of action of specialized pro-resolving lipid mediators
As previously discussed, neuroinflammation mediates several pathologic processes that result in secondary brain injury [14,20,64,65]. We hypothesize that by acting as a “stop signal” for inflammation after TBI, SPMs can be safely administered as a therapeutic drug that will have a pleiotropic and beneficial effect on disease biomarkers. This has not been extensively tested in humans; however, as described, the rodent CCI model is reasonable to further elucidate the biochemical effect of SPMs. Their potential effect on BBB permeability and cerebral edema is a central clinical question, as cerebral edema is typically symptomatic and rapidly life-threatening; therefore, a priority for any target SPM drug should be in establishing the effect on these outcomes.
Using a validated rodent CCI model, the mechanistic effect of SPMs can be further dissected, and novel hypotheses can be tested. Of emerging interest, whether SPMs can promote the resolution of metabolic crisis and CNS glymphatic and lymphatic dysfunction after TBI can be investigated with this approach.
Given the close association between inflammation and cellular metabolism, research is needed to describe the effect of SPMs on energetics post-TBI. In a lung injury model, RvD1 was protective for mitochondria, showing increased Na+-K+-ATPase activity, improved energetics, and reduced apoptosis [66]. Similarly, oral administration of DHA protected against liver injury under metabolic stress in a rat model, with clinical applications in chronic metabolic disorders and liver transplant [67]. After TBI, neuronal metabolic crisis has been demonstrated in both mammalian models and human patients [68,69]. The full mechanism by which this occurs is not fully known, but excess glutamate release and oxidative stress is a mediator [70]. In a rat intracerebral hemorrhage model, glutamate receptor blockade attenuated glucose hypermetabolism in perihematomal tissue [71]. After TBI, glutamate toxicity might also trigger cortical spreading depressions-pathologic waves of cortical activation that increase anaerobic metabolism and energy substrate depletion [72].
There is emerging evidence about the importance of brain lymphatic drainage in maintaining homeostasis. Comprised of perivascular spaces, glymphatics, and nasal and meningeal lymphatics, it functions to promote CSF and interstitial fluid drainage, clearance of waste and water balance and represents a novel target in both acute and chronic neurologic disease (for a complete review, see Sun et al. [73]). Bocheng et al. showed that lymphatics discharged iron from the CNS after experimental intraventricular hemorrhage, reducing the development of hydrocephalus [74]. As an interface between the immune system and the brain, lymphatics likely also play an important role in neuroinflammation. In a rat model, meningeal lymphatic dysfunction was triggered by either TBI or elevated intracranial pressure and led to overexpression of pro-inflammatory mRNA at 24 h [14]. Lymphatic blockage also reduced regional cerebral blood flow in the cortex and exacerbated neuronal injury after middle cerebral artery occlusion [75,76]. As recently reviewed by Kraft et al. [15], the effect of SPMs on inflammation may be partially mediated by lymphatic function and can be further elucidated in a rodent model of TBI.
Establishing the optimal pharmacologic conditions to maximize therapeutic effect
The rat CCI model and other models are needed to define the optimal pharmacologic strategy for clinical research. The most important question is to identify the best candidate SPM or SPM-analog drug to use in translational research. Additional questions regarding the optimal timing, dosing, frequency, duration of treatment, and route of drug delivery can also be explored. A summary of pharmacologic conditions used for SPMs in rodent models of TBI is described in Table 1.
Although numerous SPMs continue to be identified, few have been studied as therapeutic agents. The most commonly used SPMs include RvD1 and its aspirin-triggered stereoisomer, RvE1 and LXA4, and its analogs. Dietary supplementation with SPM precursors DHA and EPA have also been studied. SPM and other FA compounds are considered relatively safe with no significant adverse effects reported in the literature; however, given the paucity of data in this field, no conclusions can be made about the relative efficacy of different agents. In a mouse model of TBI, Harrison et al. demonstrated that AT-RvD1 and E1 differentially impact microglial activation and motor-behavioral outcomes, suggesting that individual SPMs have distinct properties and effects [37]. Characterizing which metabolic pathways are up and down-regulated with each specific SPM or precursor would be useful in identifying a candidate drug that minimizes off-target effects and directly leads to clinical benefit.
Dose-escalation trials are needed to determine the therapeutic window of candidate drugs that minimizes off-target effects. Few studies in TBI models have compared drug efficacy at different doses or demonstrated a large dose-dependent response. In a series of rat experiments by Bailes and Mills et al., DHA or omega-3 FA at a dose of 40 mg/kg/day resulted in a greater reduction in amyloid precursor protein staining on axons and caspase-3 expression compared to a lower dose of 10 mg/kg/day [29,30]. LXA4, which is more extensively studied, appears to have a robust dose-dependent response in rodent models of inflammatory vascular and retinal disease [77,78]; however, dose-escalation trials are lacking in TBI. Pharmacokinetic studies are necessary to help determine the appropriate dosing frequency. Beyond dosing, the optimal timing and duration of treatment have not been established. In rodent models, both pre-injury neuroprotection [26,27,37] and post-injury administration [29-32,36] have been studied. As outcome measures in rat and mouse models are typically short-term, often within days of injury, there is no substantial evidence to suggest the optimal treatment duration.
Lastly, the route of drug delivery is essential for clinical trials. In human populations with critical illness, dietary supplementation has several practical limitations. Dosing variability and impaired gastrointestinal absorption and motility can affect drug bioavailability. Additionally, drug-drug interactions and increased metabolic rates in critical illness can result in low, subtherapeutic drug levels. Administration of SPMs by IT routes has only recently been used in TBI models. In a rodent brain ischemia-reperfusion model, Wu et al. administered intraventricular LXA4 analog LXA4ME following middle cerebral artery occlusion, demonstrating decreased Evans Blue extravasation, reduced MMP-9 expression, upregulation of tissue inhibitors of metalloproteinase-1, and decreased final infarct volume [79]. Similarly, intraventricular LXA4 has also been used in a mouse model of TBI by Luo et al. as previously described [32]. Although IT dosing is appealing, it is limited in clinical practice. In human patients, IT drug is exclusively given through an external ventricular drain or long-term ventricular shunt, both of which require neurosurgery and are not readily available in most patients. There is an additional concern for ventriculostomy-associated infection with repeated CSF accessing [80], with infections negatively impacting hospital metrics. Similar considerations apply to intralesional drug delivery [38]. IP injections are commonly used in rodent models and have been used in SPM TBI research [36]. To date, IV, SC, or intramuscular SPM formulations have not been studied in mammalian TBI models but are accepted methods of drug delivery in clinical practice.
Translation to clinical applications: prediction models and human safety and efficacy trials
Once the therapeutic window and mechanistic effects of SPMs have been established in mammalian models of TBI, clinical trials are warranted. In future clinical applications, SPM lipidomes could be integrated into prediction models of clinical outcomes while the safety and efficacy of SPM drug therapy are studied.
Prognostication after human TBI is challenging compared to other acute neurologic diseases such as ischemic stroke, spontaneous hemorrhage, or aneurysmal subarachnoid hemorrhage. The term “TBI” represents a heterogeneous group of injuries, both with and without intracranial hemorrhage, involving multiple CNS sites, and are often accompanied by non-neurologic injuries that impact outcomes. To date, over 60 prediction models have been proposed; yet, none of them have gained widespread use [81]. TBI models typically use clinical exams, imaging findings, physiologic derangements, or a combination of these elements; however, these are surrogate markers of disease severity rather than a description of the underlying pathophysiology. As discussed, the SPM lipidome might represent a novel biomarker that more accurately identifies mediators of secondary brain injury: neuroinflammation, ischemic injury, and metabolic crisis. Modern HPLC-MS SPM lipidomic assays are validated, feasible and produce relatively rapid results that could be implemented into future prediction models targeted at measuring these mediators. In combination, SPM lipidomics, general metabolomics, and neuroimaging would allow for a noninvasive and more complete picture of the structural and molecular impact TBI has on individual patients and would also allow for tailored prognostication.
With the foundational knowledge from animal models, SPM drug therapy would be studied for its safety and efficacy. The dosing and frequency of any candidate SPM would be informed by dose-escalation trials in animal models that identify potential off-target effects that might lead to adverse effects in clinical research. If SPM therapy is safe in animal and pilot human studies, longer treatment durations may be beneficial in attenuating early and delayed inflammatory responses. Although case reports of the possible beneficial effect of omega-3 FA supplementation after acute brain injury are described [39-41], controlled clinical trials are needed to establish the true safety and efficacy of treatment. The target SPM or SPM precursor may have an impact on the speed by which translation and implementation to clinical practice occurs. Omega-3 and 6 PUFAs are readily available, have a strong safety profile, and may be amenable to study without extensive pre-clinical research. In comparison, any novel SPM, SPM analog, or other new formulation would require thorough study and is described by the scientific approach outlined here.
Conclusions
There is increasing scientific interest in the role SPMs play in mediating the resolution of inflammation after TBI, with many potential clinical applications. The SPM lipidome may serve as a novel biomarker of disease and predictor of clinical outcomes, and the use of exogenous SPM administration as a therapeutic target after acute brain injury is promising. Given this potential, a careful and comprehensive analytic approach is necessary to answer the relevant remaining translational research questions. The greatest priorities should be in determining the mechanisms by which SPMs attenuate secondary brain injury, establishing the SPM lipidome as a useful biomarker of disease, identifying candidate SPMs for clinical research, and demonstrating their safety and efficacy in both animal models of TBI and human populations. Given the morbidity and mortality associated with TBI with limited treatment options, novel approaches are urgently needed.
Acknowledgements
This research is supported by an institutional award from the Southern California Clinical and Translational Science Institute (grant number: UL1TR001855). SGL is funded by NCI R01CA213129. PL is funded by the NINDS U24 NS113452. Figure 1 was created using BioRender.com.
Disclosure of conflict of interest
Dr. Poblete and Dr. Louie report grants from NIH; Dr. Louie reports non-financial support and other from Eyemedix, LLC, outside the submitted work; In addition, Dr. Louie has a patent for lipid-based compounds licensed to Eyemedix. Dr. Lyden receives compensation as a member of the “PORTICOTM Re-sheathable Transcatheter Aortic Valve System US IDE Trial (PORTICO)” DSMB, royalty income from Thromboltyic Therapy for Acute Stroke, 3rd Ed, as a consultant to APEX Innovations and to various law firms as an expert in medical malpractice litigation.
References
- 1.Miller GF, Daugherty J, Waltzman D, Sarmiento K. Predictors of traumatic brain injury morbidity and mortality: examination of data from the national trauma data bank: predictors of TBI morbidity & mortality. Injury. 2021;52:1138–1144. doi: 10.1016/j.injury.2021.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kaur P, Sharma S. Recent advances in pathophysiology of traumatic brain injury. Curr Neuropharmacol. 2018;16:1224–1238. doi: 10.2174/1570159X15666170613083606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hinson HE, Rowell S, Schreiber M. Clinical evidence of inflammation driving secondary brain injury: a systematic review. J Trauma Acute Care Surg. 2015;78:184–191. doi: 10.1097/TA.0000000000000468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Engel S, Schluesener H, Mittelbronn M, Seid K, Adjodah D, Wehner HD, Meyermann R. Dynamics of microglial activation after human traumatic brain injury are revealed by delayed expression of macrophage-related proteins MRP8 and MRP14. Acta Neuropathol. 2000;100:313–322. doi: 10.1007/s004019900172. [DOI] [PubMed] [Google Scholar]
- 5.Gentleman SM, Leclercq PD, Moyes L, Graham DI, Smith C, Griffin WS, Nicoll JA. Long-term intracerebral inflammatory response after traumatic brain injury. Forensic Sci Int. 2004;146:97–104. doi: 10.1016/j.forsciint.2004.06.027. [DOI] [PubMed] [Google Scholar]
- 6.Block ML, Hong JS. Microglia and inflammation-mediated neurodegeneration: multiple triggers with a common mechanism. Prog Neurobiol. 2005;76:77–98. doi: 10.1016/j.pneurobio.2005.06.004. [DOI] [PubMed] [Google Scholar]
- 7.Rodríguez-Gómez JA, Kavanagh E, Engskog-Vlachos P, Engskog MKR, Herrera AJ, Espinosa-Oliva AM, Joseph B, Hajji N, Venero JL, Burguillos MA. Microglia: agents of the CNS pro-inflammatory response. Cells. 2020;9:1717. doi: 10.3390/cells9071717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Banks WA, Gray AM, Erickson MA, Salameh TS, Damodarasamy M, Sheibani N, Meabon JS, Wing EE, Morofuji Y, Cook DG, Reed MJ. Lipopolysaccharide-induced blood-brain barrier disruption: roles of cyclooxygenase, oxidative stress, neuroinflammation, and elements of the neurovascular unit. J Neuroinflammation. 2015;12:223. doi: 10.1186/s12974-015-0434-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chodobski A, Chung I, Koźniewska E, Ivanenko T, Chang W, Harrington JF, Duncan JA, Szmydynger-Chodobska J. Early neutrophilic expression of vascular endothelial growth factor after traumatic brain injury. Neuroscience. 2003;122:853–867. doi: 10.1016/j.neuroscience.2003.08.055. [DOI] [PubMed] [Google Scholar]
- 10.Argaw AT, Gurfein BT, Zhang Y, Zameer A, John GR. VEGF-mediated disruption of endothelial CLN-5 promotes blood-brain barrier breakdown. Proc Natl Acad Sci U S A. 2009;106:1977–1982. doi: 10.1073/pnas.0808698106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Neri M, Frati A, Turillazzi E, Cantatore S, Cipolloni L, Di Paolo M, Frati P, La Russa R, Maiese A, Scopetti M, Santurro A, Sessa F, Zamparese R, Fineschi V. Immunohistochemical evaluation of aquaporin-4 and its correlation with CD68, IBA-1, HIF-1α, GFAP, and CD15 expressions in fatal traumatic brain injury. Int J Mol Sci. 2018;19:3544. doi: 10.3390/ijms19113544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kumar Sahel D, Kaira M, Raj K, Sharma S, Singh S. Mitochondrial dysfunctioning and neuroinflammation: recent highlights on the possible mechanisms involved in traumatic brain injury. Neurosci Lett. 2019;710:134347. doi: 10.1016/j.neulet.2019.134347. [DOI] [PubMed] [Google Scholar]
- 13.Lamade AM, Anthonymuthu TS, Hier ZE, Gao Y, Kagan VE, Bayır H. Mitochondrial damage & lipid signaling in traumatic brain injury. Exp Neurol. 2020;329:113307. doi: 10.1016/j.expneurol.2020.113307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bolte AC, Dutta AB, Hurt ME, Smirnov I, Kovacs MA, McKee CA, Ennerfelt HE, Shapiro D, Nguyen BH, Frost EL, Lammert CR, Kipnis J, Lukens JR. Meningeal lymphatic dysfunction exacerbates traumatic brain injury pathogenesis. Nat Commun. 2020;11:4524. doi: 10.1038/s41467-020-18113-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kraft JD, Blomgran R, Lundgaard I, Quiding-Järbrink M, Bromberg JS, Börgeson E. Specialized pro-resolving mediators and the lymphatic system. Int J Mol Sci. 2021;22:2750. doi: 10.3390/ijms22052750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hiebert JB, Shen Q, Thimmesch AR, Pierce JD. Traumatic brain injury and mitochondrial dysfunction. Am J Med Sci. 2015;350:132–138. doi: 10.1097/MAJ.0000000000000506. [DOI] [PubMed] [Google Scholar]
- 17.Ola MS, Nawaz M, Ahsan H. Role of Bcl-2 family proteins and caspases in the regulation of apoptosis. Mol Cell Biochem. 2011;351:41–58. doi: 10.1007/s11010-010-0709-x. [DOI] [PubMed] [Google Scholar]
- 18.Ekdahl KN, Teramura Y, Hamad OA, Asif S, Duehrkop C, Fromell K, Gustafson E, Hong J, Kozarcanin H, Magnusson PU, Huber-Lang M, Garred P, Nilsson B. Dangerous liaisons: complement, coagulation, and kallikrein/kinin cross-talk act as a linchpin in the events leading to thromboinflammation. Immunol Rev. 2016;274:245–269. doi: 10.1111/imr.12471. [DOI] [PubMed] [Google Scholar]
- 19.Dietrich WD, Alonso O, Busto R, Prado R, Dewanjee S, Dewanjee MK, Ginsberg MD. Widespread hemodynamic depression and focal platelet accumulation after fluid percussion brain injury: a double-label autoradiographic study in rats. J Cereb Blood Flow Metab. 1996;16:481–489. doi: 10.1097/00004647-199605000-00015. [DOI] [PubMed] [Google Scholar]
- 20.Schwarzmaier SM, Kim SW, Trabold R, Plesnila N. Temporal profile of thrombogenesis in the cerebral microcirculation after traumatic brain injury in mice. J Neurotrauma. 2010;27:121–130. doi: 10.1089/neu.2009.1114. [DOI] [PubMed] [Google Scholar]
- 21.Shimizu T. Lipid mediators in health and disease: enzymes and receptors as therapeutic targets for the regulation of immunity and inflammation. Annu Rev Pharmacol Toxicol. 2009;49:123–150. doi: 10.1146/annurev.pharmtox.011008.145616. [DOI] [PubMed] [Google Scholar]
- 22.Musto AE, Walker CP, Petasis NA, Bazan NG. Hippocampal neuro-networks and dendritic spine perturbations in epileptogenesis are attenuated by neuroprotectin d1. PLoS One. 2015;10:e0116543. doi: 10.1371/journal.pone.0116543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Medeiros R, Kitazawa M, Passos GF, Baglietto-Vargas D, Cheng D, Cribbs DH, LaFerla FM. Aspirin-triggered lipoxin A4 stimulates alternative activation of microglia and reduces Alzheimer disease-like pathology in mice. Am J Pathol. 2013;182:1780–1789. doi: 10.1016/j.ajpath.2013.01.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guo Z, Hu Q, Xu L, Guo ZN, Ou Y, He Y, Yin C, Sun X, Tang J, Zhang JH. Lipoxin A4 reduces inflammation through formyl peptide receptor 2/p38 MAPK signaling pathway in subarachnoid hemorrhage rats. Stroke. 2016;47:490–497. doi: 10.1161/STROKEAHA.115.011223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bazan NG, Eady TN, Khoutorova L, Atkins KD, Hong S, Lu Y, Zhang C, Jun B, Obenaus A, Fredman G, Zhu M, Winkler JW, Petasis NA, Serhan CN, Belayev L. Novel aspirin-triggered neuroprotectin D1 attenuates cerebral ischemic injury after experimental stroke. Exp Neurol. 2012;236:122–130. doi: 10.1016/j.expneurol.2012.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wu A, Ying Z, Gomez-Pinilla F. Dietary omega-3 fatty acids normalize BDNF levels, reduce oxidative damage, and counteract learning disability after traumatic brain injury in rats. J Neurotrauma. 2004;21:1457–1467. doi: 10.1089/neu.2004.21.1457. [DOI] [PubMed] [Google Scholar]
- 27.Wu A, Ying Z, Gomez-Pinilla F. Omega-3 fatty acids supplementation restores mechanisms that maintain brain homeostasis in traumatic brain injury. J Neurotrauma. 2007;24:1587–1595. doi: 10.1089/neu.2007.0313. [DOI] [PubMed] [Google Scholar]
- 28.Mills JD, Hadley K, Bailes JE. Dietary supplementation with the omega-3 fatty acid docosahexaenoic acid in traumatic brain injury. Neurosurgery. 2011;68:474–481. doi: 10.1227/NEU.0b013e3181ff692b. discussion 481. [DOI] [PubMed] [Google Scholar]
- 29.Bailes JE, Mills JD. Docosahexaenoic acid reduces traumatic axonal injury in a rodent head injury model. J Neurotrauma. 2010;27:1617–1624. doi: 10.1089/neu.2009.1239. [DOI] [PubMed] [Google Scholar]
- 30.Mills JD, Bailes JE, Sedney CL, Hutchins H, Sears B. Omega-3 fatty acid supplementation and reduction of traumatic axonal injury in a rodent head injury model. J Neurosurg. 2011;114:77–84. doi: 10.3171/2010.5.JNS08914. [DOI] [PubMed] [Google Scholar]
- 31.Shin SS, Dixon CE. Oral fish oil restores striatal dopamine release after traumatic brain injury. Neurosci Lett. 2011;496:168–171. doi: 10.1016/j.neulet.2011.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Luo CL, Li QQ, Chen XP, Zhang XM, Li LL, Li BX, Zhao ZQ, Tao LY. Lipoxin A4 attenuates brain damage and downregulates the production of pro-inflammatory cytokines and phosphorylated mitogen-activated protein kinases in a mouse model of traumatic brain injury. Brain Res. 2013;1502:1–10. doi: 10.1016/j.brainres.2013.01.037. [DOI] [PubMed] [Google Scholar]
- 33.Chen X, Wu S, Chen C, Xie B, Fang Z, Hu W, Chen J, Fu H, He H. Omega-3 polyunsaturated fatty acid supplementation attenuates microglial-induced inflammation by inhibiting the HMGB1/TLR4/NF-κB pathway following experimental traumatic brain injury. J Neuroinflammation. 2017;14:143. doi: 10.1186/s12974-017-0917-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen X, Chen C, Fan S, Wu S, Yang F, Fang Z, Fu H, Li Y. Omega-3 polyunsaturated fatty acid attenuates the inflammatory response by modulating microglia polarization through SIRT1-mediated deacetylation of the HMGB1/NF-κB pathway following experimental traumatic brain injury. J Neuroinflammation. 2018;15:116. doi: 10.1186/s12974-018-1151-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen X, Pan Z, Fang Z, Lin W, Wu S, Yang F, Li Y, Fu H, Gao H, Li S. Omega-3 polyunsaturated fatty acid attenuates traumatic brain injury-induced neuronal apoptosis by inducing autophagy through the upregulation of SIRT1-mediated deacetylation of Beclin-1. J Neuroinflammation. 2018;15:310. doi: 10.1186/s12974-018-1345-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bisicchia E, Sasso V, Catanzaro G, Leuti A, Besharat ZM, Chiacchiarini M, Molinari M, Ferretti E, Viscomi MT, Chiurchiù V. Resolvin D1 halts remote neuroinflammation and improves functional recovery after focal brain damage via ALX/FPR2 receptor-regulated microRNAs. Mol Neurobiol. 2018;55:6894–6905. doi: 10.1007/s12035-018-0889-z. [DOI] [PubMed] [Google Scholar]
- 37.Harrison JL, Rowe RK, Ellis TW, Yee NS, O’Hara BF, Adelson PD, Lifshitz J. Resolvins AT-D1 and E1 differentially impact functional outcome, post-traumatic sleep, and microglial activation following diffuse brain injury in the mouse. Brain Behav Immun. 2015;47:131–140. doi: 10.1016/j.bbi.2015.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Berg RWV, Davidsson J, Lidin E, Angéria M, Risling M, Günther M. Brain tissue saving effects by single-dose intralesional administration of Neuroprotectin D1 on experimental focal penetrating brain injury in rats. J Clin Neurosci. 2019;64:227–233. doi: 10.1016/j.jocn.2019.03.032. [DOI] [PubMed] [Google Scholar]
- 39.Roberts L, Bailes J, Dedhia H, Zikos A, Singh A, McDowell D, Failinger C, Biundo R, Petrick J, Carpenter J. Surviving a mine explosion. J Am Coll Surg. 2008;207:276–283. doi: 10.1016/j.jamcollsurg.2008.02.015. [DOI] [PubMed] [Google Scholar]
- 40.Lewis M, Ghassemi P, Hibbeln J. Therapeutic use of omega-3 fatty acids in severe head trauma. Am J Emerg Med. 2013;31:273, e275–278. doi: 10.1016/j.ajem.2012.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bailes JE, Abusuwwa R, Arshad M, Chowdhry SA, Schleicher D, Hempeck N, Gandhi YN, Jaffa Z, Bokhari F, Karahalios D, Barkley J, Patel V, Sears B. Omega-3 fatty acid supplementation in severe brain trauma: case for a large multicenter trial. J Neurosurg. 2020 doi: 10.3171/2020.3.JNS20183. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 42.Bergold PJ. Treatment of traumatic brain injury with anti-inflammatory drugs. Exp Neurol. 2016;275:367–380. doi: 10.1016/j.expneurol.2015.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bazán NG. Effects of ischemia and electroconvulsive shock on free fatty acid pool in the brain. Biochim Biophys Acta. 1970;218:1–10. doi: 10.1016/0005-2760(70)90086-x. [DOI] [PubMed] [Google Scholar]
- 44.Bazan NG, Rakowski H. Increased levels of brain free fatty acids after electroconvulsive shock. Life Sci. 1970;9:501–507. doi: 10.1016/0024-3205(70)90205-5. [DOI] [PubMed] [Google Scholar]
- 45.Aveldano MI, Bazán NG. Rapid production of diacylglycerols enriched in arachidonate and stearate during early brain ischemia. J Neurochem. 1975;25:919–920. doi: 10.1111/j.1471-4159.1975.tb04432.x. [DOI] [PubMed] [Google Scholar]
- 46.Pilitsis JG, Coplin WM, O’Regan MH, Wellwood JM, Diaz FG, Fairfax MR, Michael DB, Phillis JW. Free fatty acids in cerebrospinal fluids from patients with traumatic brain injury. Neurosci Lett. 2003;349:136–138. doi: 10.1016/s0304-3940(03)00803-6. [DOI] [PubMed] [Google Scholar]
- 47.Abdullah L, Evans JE, Ferguson S, Mouzon B, Montague H, Reed J, Crynen G, Emmerich T, Crocker M, Pelot R, Mullan M, Crawford F. Lipidomic analyses identify injury-specific phospholipid changes 3 mo after traumatic brain injury. FASEB J. 2014;28:5311–5321. doi: 10.1096/fj.14-258228. [DOI] [PubMed] [Google Scholar]
- 48.Emmerich T, Abdullah L, Crynen G, Dretsch M, Evans J, Ait-Ghezala G, Reed J, Montague H, Chaytow H, Mathura V, Martin J, Pelot R, Ferguson S, Bishop A, Phillips J, Mullan M, Crawford F. Plasma lipidomic profiling in a military population of mild traumatic brain injury and post-traumatic stress disorder with apolipoprotein E ε4-dependent effect. J Neurotrauma. 2016;33:1331–1348. doi: 10.1089/neu.2015.4061. [DOI] [PubMed] [Google Scholar]
- 49.Emmerich T, Zakirova Z, Klimas N, Sullivan K, Shetty AK, Evans JE, Ait-Ghezala G, Laco GS, Hattiangady B, Shetty GA, Mullan M, Crynen G, Abdullah L, Crawford F. Phospholipid profiling of plasma from GW veterans and rodent models to identify potential biomarkers of Gulf War Illness. PLoS One. 2017;12:e0176634. doi: 10.1371/journal.pone.0176634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sowers JL, Wu P, Zhang K, DeWitt DS, Prough DS. Proteomic changes in traumatic brain injury: experimental approaches. Curr Opin Neurol. 2018;31:709–717. doi: 10.1097/WCO.0000000000000613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dadas A, Washington J, Diaz-Arrastia R, Janigro D. Biomarkers in traumatic brain injury (TBI): a review. Neuropsychiatr Dis Treat. 2018;14:2989–3000. doi: 10.2147/NDT.S125620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hogan SR, Phan JH, Alvarado-Velez M, Wang MD, Bellamkonda RV, Fernández FM, LaPlaca MC. Discovery of lipidome alterations following traumatic brain injury via high-resolution metabolomics. J Proteome Res. 2018;17:2131–2143. doi: 10.1021/acs.jproteome.8b00068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.English JT, Norris PC, Hodges RR, Dartt DA, Serhan CN. Identification and profiling of specialized pro-resolving mediators in human tears by lipid mediator metabolomics. Prostaglandins Leukot Essent Fatty Acids. 2017;117:17–27. doi: 10.1016/j.plefa.2017.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Poblete RA, Arenas M, Sanossian N, Freeman WD, Louie SG. The role of bioactive lipids in attenuating the neuroinflammatory cascade in traumatic brain injury. Ann Clin Transl Neurol. 2020;7:2524–2534. [Google Scholar]
- 55.Wu Y, Pang J, Peng J, Cao F, Vitek MP, Li F, Jiang Y, Sun X. An apoE-derived mimic peptide, COG1410, alleviates early brain injury via reducing apoptosis and neuroinflammation in a mouse model of subarachnoid hemorrhage. Neurosci Lett. 2016;627:92–99. doi: 10.1016/j.neulet.2016.05.058. [DOI] [PubMed] [Google Scholar]
- 56.Dong T, Dave P, Yoo E, Ebright B, Ahluwalia K, Zhou E, Asante I, Salimova M, Pei H, Lin T, Mead A, Li Z, Humayun M, Petasis NA, Epstein AL, Louie SG. NAP1051, a lipoxin A4 biomimetic analogue, demonstrates antitumor activity against the tumor microenvironment. Mol Cancer Ther. 2021;20:2384–2397. doi: 10.1158/1535-7163.MCT-21-0414. [DOI] [PubMed] [Google Scholar]
- 57.Kooij G, Troletti CD, Leuti A, Norris PC, Riley I, Albanese M, Ruggieri S, Libreros S, van der Pol SMA, van Het Hof B, Schell Y, Guerrera G, Buttari F, Mercuri NB, Centonze D, Gasperini C, Battistini L, de Vries HE, Serhan CN, Chiurchiù V. Specialized pro-resolving lipid mediators are differentially altered in peripheral blood of patients with multiple sclerosis and attenuate monocyte and blood-brain barrier dysfunction. Haematologica. 2020;105:2056–2070. doi: 10.3324/haematol.2019.219519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fredman G, Hellmann J, Proto JD, Kuriakose G, Colas RA, Dorweiler B, Connolly ES, Solomon R, Jones DM, Heyer EJ, Spite M, Tabas I. An imbalance between specialized pro-resolving lipid mediators and pro-inflammatory leukotrienes promotes instability of atherosclerotic plaques. Nat Commun. 2016;7:12859. doi: 10.1038/ncomms12859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lukiw WJ, Cui JG, Marcheselli VL, Bodker M, Botkjaer A, Gotlinger K, Serhan CN, Bazan NG. A role for docosahexaenoic acid-derived neuroprotectin D1 in neural cell survival and Alzheimer disease. J Clin Invest. 2005;115:2774–2783. doi: 10.1172/JCI25420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang X, Zhu M, Hjorth E, Cortés-Toro V, Eyjolfsdottir H, Graff C, Nennesmo I, Palmblad J, Eriksdotter M, Sambamurti K, Fitzgerald JM, Serhan CN, Granholm AC, Schultzberg M. Resolution of inflammation is altered in Alzheimer’s disease. Alzheimers Dement. 2015;11:40–50. e41–42. doi: 10.1016/j.jalz.2013.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Serhan CN. Pro-resolving lipid mediators are leads for resolution physiology. Nature. 2014;510:92–101. doi: 10.1038/nature13479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dean DD, Frank JA, Turtzo LC. Controlled cortical impact in the rat. Curr Protoc Neurosci. 2017;81:9.62.1–9.62.12. doi: 10.1002/cpns.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ma X, Aravind A, Pfister BJ, Chandra N, Haorah J. Animal models of traumatic brain injury and assessment of injury severity. Mol Neurobiol. 2019;56:5332–5345. doi: 10.1007/s12035-018-1454-5. [DOI] [PubMed] [Google Scholar]
- 64.Beaumont A, Marmarou A, Hayasaki K, Barzo P, Fatouros P, Corwin F, Marmarou C, Dunbar J. The permissive nature of blood brain barrier (BBB) opening in edema formation following traumatic brain injury. Acta Neurochir Suppl. 2000;76:125–129. doi: 10.1007/978-3-7091-6346-7_26. [DOI] [PubMed] [Google Scholar]
- 65.Carre E, Ogier M, Boret H, Montcriol A, Bourdon L, Jean-Jacques R. Metabolic crisis in severely head-injured patients: is ischemia just the tip of the iceberg? Front Neurol. 2013;4:146. doi: 10.3389/fneur.2013.00146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhao Q, Wu J, Hua Q, Lin Z, Ye L, Zhang W, Wu G, Du J, Xia J, Chu M, Hu X. Resolvin D1 mitigates energy metabolism disorder after ischemia-reperfusion of the rat lung. J Transl Med. 2016;14:81. doi: 10.1186/s12967-016-0835-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Vargas R, Riquelme B, Fernández J, Álvarez D, Pérez IF, Cornejo P, Fernández V, Videla LA. Docosahexaenoic acid-thyroid hormone combined protocol as a novel approach to metabolic stress disorders: relation to mitochondrial adaptation via liver PGC-1α and sirtuin1 activation. Biofactors. 2019;45:271–278. doi: 10.1002/biof.1483. [DOI] [PubMed] [Google Scholar]
- 68.Zheng F, Zhou YT, Li PF, Hu E, Li T, Tang T, Luo JK, Zhang W, Ding CS, Wang Y. Metabolomics analysis of hippocampus and cortex in a rat model of traumatic brain injury in the subacute phase. Front Neurosci. 2020;14:876. doi: 10.3389/fnins.2020.00876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Timofeev I, Carpenter KL, Nortje J, Al-Rawi PG, O’Connell MT, Czosnyka M, Smielewski P, Pickard JD, Menon DK, Kirkpatrick PJ, Gupta AK, Hutchinson PJ. Cerebral extracellular chemistry and outcome following traumatic brain injury: a microdialysis study of 223 patients. Brain. 2011;134:484–494. doi: 10.1093/brain/awq353. [DOI] [PubMed] [Google Scholar]
- 70.Mendes Arent A, de Souza LF, Walz R, Dafre AL. Perspectives on molecular biomarkers of oxidative stress and antioxidant strategies in traumatic brain injury. Biomed Res Int. 2014;2014:723060. doi: 10.1155/2014/723060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ardizzone TD, Lu A, Wagner KR, Tang Y, Ran R, Sharp FR. Glutamate receptor blockade attenuates glucose hypermetabolism in perihematomal brain after experimental intracerebral hemorrhage in rat. Stroke. 2004;35:2587–2591. doi: 10.1161/01.STR.0000143451.14228.ff. [DOI] [PubMed] [Google Scholar]
- 72.Hinzman JM, Wilson JA, Mazzeo AT, Bullock MR, Hartings JA. Excitotoxicity and metabolic crisis are associated with spreading depolarizations in severe traumatic brain injury patients. J Neurotrauma. 2016;33:1775–1783. doi: 10.1089/neu.2015.4226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sun BL, Wang LH, Yang T, Sun JY, Mao LL, Yang MF, Yuan H, Colvin RA, Yang XY. Lymphatic drainage system of the brain: a novel target for intervention of neurological diseases. Prog Neurobiol. 2018;163-164:118–143. doi: 10.1016/j.pneurobio.2017.08.007. [DOI] [PubMed] [Google Scholar]
- 74.Bocheng W, Chaofeng L, Chuan C, Haiyong H, Tengchao H, Qun G, Ying G. Intracranial lymphatic drainage discharges iron from the ventricles and reduce the occurrence of chronic hydrocephalus after intraventricular hemorrhage in rats. Int J Neurosci. 2020;130:130–135. doi: 10.1080/00207454.2019.1667780. [DOI] [PubMed] [Google Scholar]
- 75.Xia ZL, Sun BL, Yang MF, Yuan H, Qiu PM, Chen YS. The effect of cerebral lymphatic blockage on cortex regional cerebral blood flow and somatosensory evoked potential. Clin Hemorheol Microcirc. 2003;29:345–349. [PubMed] [Google Scholar]
- 76.Sun BL, Xia ZL, Yan ZW, Chen YS, Yang MF. Effects of blockade of cerebral lymphatic drainage on cerebral ischemia after middle cerebral artery occlusion in rats. Clin Hemorheol Microcirc. 2000;23:321–325. [PubMed] [Google Scholar]
- 77.von der Weid PY, Hollenberg MD, Fiorucci S, Wallace JL. Aspirin-triggered, cyclooxygenase-2-dependent lipoxin synthesis modulates vascular tone. Circulation. 2004;110:1320–1325. doi: 10.1161/01.CIR.0000140985.89766.CB. [DOI] [PubMed] [Google Scholar]
- 78.Wei J, Mattapallil MJ, Horai R, Jittayasothorn Y, Modi AP, Sen HN, Gronert K, Caspi RR. A novel role for lipoxin A. Elife. 2020;9:e51102. doi: 10.7554/eLife.51102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wu Y, Wang YP, Guo P, Ye XH, Wang J, Yuan SY, Yao SL, Shang Y. A lipoxin A4 analog ameliorates blood-brain barrier dysfunction and reduces MMP-9 expression in a rat model of focal cerebral ischemia-reperfusion injury. J Mol Neurosci. 2012;46:483–491. doi: 10.1007/s12031-011-9620-5. [DOI] [PubMed] [Google Scholar]
- 80.Poblete R, Zheng L, Raghavan R, Cen S, Amar A, Sanossian N, Mack W, Kim-Tenser M. Trends in ventriculostomy-associated infections and mortality in aneurysmal subarachnoid hemorrhage: data from the nationwide inpatient sample. World Neurosurg. 2017;99:599–604. doi: 10.1016/j.wneu.2016.12.073. [DOI] [PubMed] [Google Scholar]
- 81.Dijkland SA, Foks KA, Polinder S, Dippel DWJ, Maas AIR, Lingsma HF, Steyerberg EW. Prognosis in moderate and severe traumatic brain injury: a systematic review of contemporary models and validation studies. J Neurotrauma. 2020;37:1–13. doi: 10.1089/neu.2019.6401. [DOI] [PubMed] [Google Scholar]