

Abstract
The volume-regulated anion channel (VRAC) is formed by LRRC8 proteins and responsible for the regulatory volume decrease (RVD) after hypotonic cell swelling. Besides chloride, VRAC transports other molecules including immunomodulatory cyclic dinucleotides (CDNs) including 2’3’cGAMP. Here we identify LRRC8C as a critical component of VRAC in T cells, where its deletion abolishes VRAC currents and RVD. T cells of Lrrc8c−/− mice have increased cell cycle progression, proliferation, survival, Ca2+ influx and cytokine production, a phenotype that was associated with downmodulation of p53 signaling. Mechanistically, LRRC8C mediates the transport of 2’3’cGAMP in T cells resulting in STING and p53 activation. Inhibition of STING recapitulates the phenotype of LRRC8C-deficient T cells whereas overexpression of p53 inhibits their enhanced T cell function. Lrrc8c−/− mice have exacerbated T cell-dependent immune responses including immunity to influenza A virus infection and experimental autoimmune encephalomyelitis. Our results identify cGAMP uptake through LRRC8C and STING-p53 signaling as a novel inhibitory signaling pathway in T cells and adaptive immunity.
Keywords: CD80, cGAMP, Ion channels, LRRC8A, LRRC8C, p53, SOCE, STING, T cells, VRAC
Introduction
The volume-regulated anion channel (VRAC) plays a critical role in regulating the volume of virtually all mammalian cells including immune cells upon osmotic swelling1,2. VRAC is activated by a reduction of ionic strength in the cytoplasm, but the mechanisms underlying channel gating remain unclear3,4. VRAC is a hexameric complex composed of paralogs of the leucine-rich repeat-containing protein 8 (LRRC8) family that contains 5 members, LRRC8A-E1–4. The best characterized homologue is LRRC8A5–8, which is an obligatory subunit of VRAC and forms heteromeric channel complexes with one or more of the other paralogs (LRRC8B-E) to constitute functional channels2–4. Compared to LRRC8A, the function of LRRC8B-E is much less understood.
In immune cells, LRRC8A has been reported to be required for the development and function of B cells5,9. Moreover, Lrrc8a−/− mice showed a severe defect in T cell development and function5,9. The role of LRRC8A in T cells appears to be independent of VRAC channel function since mice with a spontaneous Lrrc8a mutation (Lrrc8aebo/ebo) that abrogates Cl− currents have normal T cell development and function10. VRAC is not only required for cell volume regulation but also for the transport of metabolites, second messengers, anticancer drugs and antibiotics11–15. Heteromeric LRRC8A/E channels have recently been reported to mediate the paracrine transfer of the cyclic dinucleotide (CDN) second messenger 2’3’ cGMP-AMP (cGAMP) in macrophages, which results in activation of the STING pathway, production of type I interferons and antiviral immunity15. A similar 2’3’ cGAMP uptake by LRRC8A:C/E heteromeric channels was reported in human vasculature cells16. These findings indicated that several LRRC8 paralogs play important roles in innate immunity.
We here identify LRRC8C as an essential component of VRAC in T cells and a negative regulator of T cell function. By using a gene-expression based screening approach to identify ion channels and transporters (ICTs) that regulate ion transport in immune cells, we found that LRRC8C was highly and selectively expressed in T cells. T cells from Lrrc8c−/− mice completely lacked VRAC currents and regulatory volume decrease (RVD) upon osmotic swelling. Although T cell development was normal in Lrrc8c−/− mice, LRRC8C-deficient T cells had increased cell cycle progression, proliferation, survival, Ca2+ influx and cytokine production after TCR stimulation. Accordingly, Lrrc8c−/− mice displayed exacerbated T cell-mediated immunity characterized by increased CNS infiltration of T cells after induction of experimental autoimmune encephalomyelitis (EAE) and an augmented T cell-dependent humoral immune response to influenza infection. The enhanced function of Lrrc8c−/− T cells was due to reduced influx of 2’3’ cGAMP, which resulted in decreased activation of STING and expression as well as signaling of the tumor suppressor p53. Direct activation of STING or increasing p53 protein levels reversed the phenotype of Lrrc8c−/− T cells. Our study establishes a new inhibitory signaling pathway in T cells that links the uptake of 2’3’ cGAMP through LRRC8C-containing VRAC channels to the activation of STING and p53 and the suppression of Ca2+ influx, thereby suppressing T cell function and adaptive immunity.
Results
LRRC8C is expressed in T cells and regulated by IL-2 signaling
To identify ICTs that regulate immune cell function, we generated a database that contains 763 ICTs and analyzed their mRNA expression in myeloid and lymphoid cells using 3 publicly available transcriptomic databases (ImmGen17, Haemopedia18, Fantom519). A principal component analysis revealed distinct patterns of ICT expression in different immune cell subsets (Fig. 1a), suggesting that different immune cells utilize distinct ICTs for their function. Focusing on CD4+ T cells, we identified 148 ICTs with high mRNA expression in both mouse and human T cells (Extended Data Fig. 1a, Supplemental Table 1). Comparing the mRNA levels of these 148 ICTs to the average expression of all other immune cells, we found that 26 ICTs were highly and more selectively expressed in murine CD4+ T cells (Fig. 1b,c, Extended Data Fig. 1b–d). 6 of these ICTs were also enriched in human CD4+ T cells (Fig. 1b; Extended Data Fig. 1e). One of these 6 ICTs was LRRC8C. Because the function of LRRC8C in T cells is undefined, we investigated its role in T cell mediated immunity.
Figure 1. LRRC8C is selectively expressed in T cells.
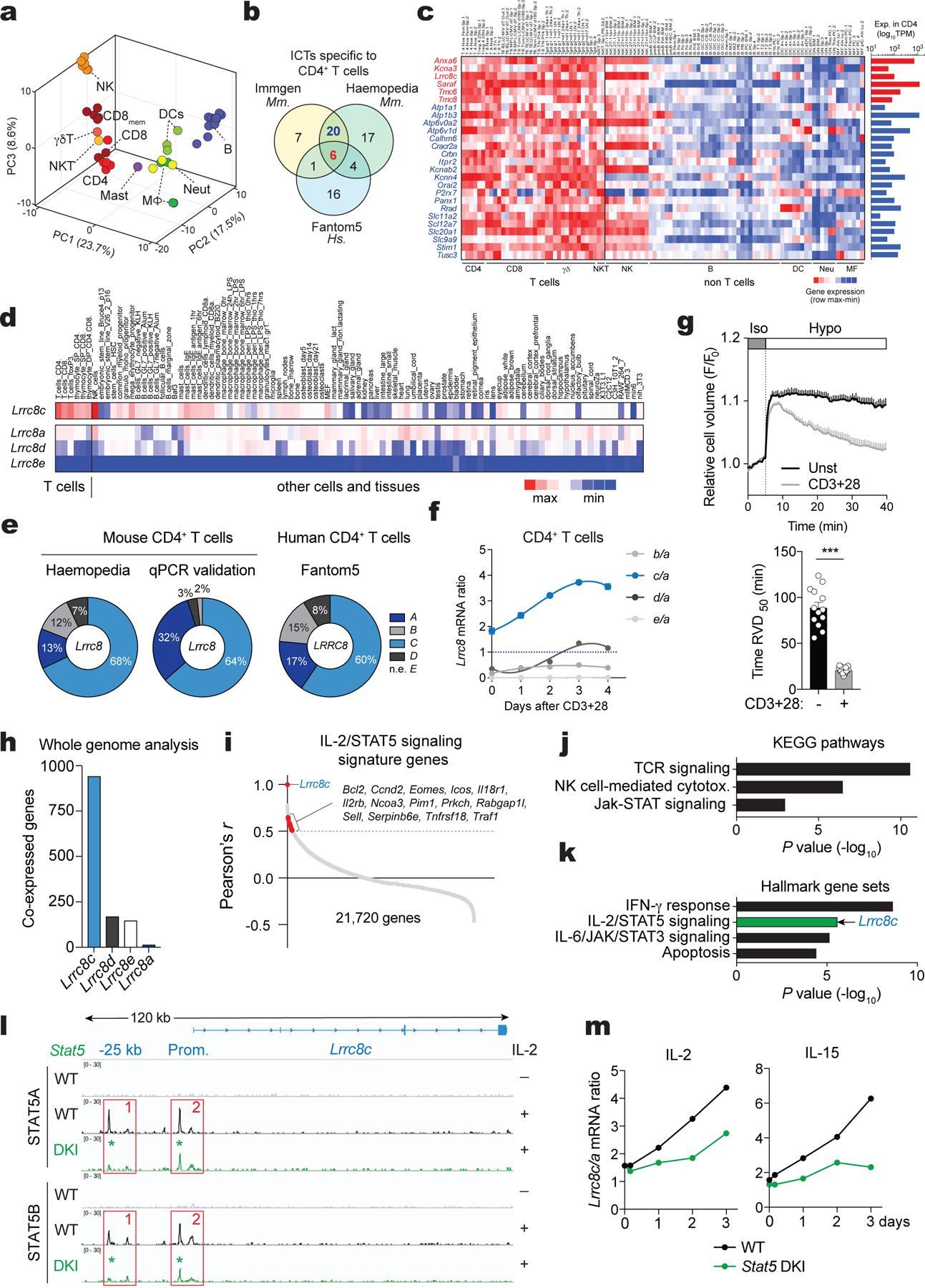
(a) 3D-PCA analysis of ion channels and transporters (ICT) and their regulators using murine immune cell RNA-Seq data (ImmGen). (b) Venn-diagram showing ICTs highly expressed in human and mouse CD4+ T cells. (c) Expression profile of the differentially expressed ICTs in murine CD4+ T cells (ImmGen). Heat map shows relative expression (red, high; blue, low). Bar graphs on the right show absolute gene expression. (d) Expression of Lrrc8a-e genes from different cell types and tissues (BioGPS). Heat map shows absolute mRNA expression (red, high; blue, low). Lrrc8b expression data was not available in BioGPS. (e) Proportion of Lrrc8a-e gene expression in mouse CD4+ T cells from Haemopedia RNA-Seq database (left) and validated by RT-qPCR (middle), and human CD4+ T cells from Fantom5 database (right). (f) Lrrc8b-e/Lrrc8a mRNA expression ratio from mouse CD4+ T cells upon stimulation with anti-CD3+CD28 and measured by RT-qPCR (n=5 independent experiments). (g) Cell volume measurements in unstimulated and stimulated CD4+ T cells upon challenge with hypotonic buffer (215 mOsm). Cell volume traces (top) are the mean ± s.e.m. from 13 and 15 independent experiments per unstimulated and stimulated conditions, respectively. The regulatory volume decrease (RVD) is quantified (bottom) as the time to reach half maximal cell volume (RVD50). Data are pooled from 5 independent experiments. Statistical analysis by two-tailed, unpaired Student’s t test. *** P<0.001. (h) Number of genes co-expressed with Lrrc8 genes (BioGPS). (i) Distribution of all murine genes based on their co-expression coefficient (Pearson’s r) relative to Lrrc8c (BioGPS). Genes linked to IL-2/STAT5 signaling highlighted in red are significantly enriched among the co-expressed genes. (j,k) Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis (j) and significant hallmark gene sets (Molecular Signatures Database, in k) of genes co-expressed with Lrrc8c from panel (h). Arrow indicates that Lrrc8c belongs to the IL-2/STAT5 signaling pathway. (l) In silico analysis of Lrrc8c gene locus for chromatin accessibility in activated T cells from wild-type and Stat5a-Stat5b double knock-in (DKI) mice measured by DNA binding of STAT5A and STAT5B before and after treatment with IL-2 for 1h23. (m) Lrrc8c/Lrrc8a mRNA expression ratio in wild-type and Stat5a-Stat5b DKI CD8+ T cells before and after re-stimulation with IL-2 or IL-15 (RNA-Seq data from ref.23).
Of all five LRRC8 family members, Lrrc8c mRNA was the most highly and selectively expressed in mouse T cells compared to 96 different cell types and tissues (BioGPS dataset20, Fig. 1d,e, Supplemental Fig.1a–c). Lrrc8a was the most ubiquitously expressed paralog in almost all tissues including T cells, whereas Lrrc8b, Lrrc8d and Lrrc8e expression was very low or undetectable in T cells. Stimulation of CD4+ T cells with anti-CD3/CD28 for 1–4 days resulted in a selective increase in Lrrc8c mRNA expression relative to Lrrc8a, whereas that of other Lrrc8 paralogs remained unchanged and low (Fig. 1f). This increase was associated with an enhanced RVD of activated compared to unstimulated T cells following exposure to hypotonic buffer and cell swelling (Fig. 1g), suggesting that LRRC8C contributes to VRAC channel function and RVD in T cells.
We hypothesized that the increase in LRRC8C expression following TCR stimulation is regulated by a T cell-specific signaling pathway. A whole-genome co-expression network analysis of Lrrc8c and its paralogs using BioGPS data20 identified 943 genes whose transcript expression correlated with those of Lrrc8c in all tissues (Fig. 1h,i, Pearson’s r > 0.5, P <0.001). By contrast, the numbers of genes co-expressed with other Lrrc8 genes were much lower (Lrrc8d, 172; Lrrc8e, 149; and Lrrc8a,16). Of note, the low number of genes co-regulated with Lrrc8a suggests that its expression is independent of a particular transcriptional program. Of the 943 genes co-regulated with Lrrc8c, 710 were known and analyzed further. Gene ontology and pathway analyses revealed an association with TCR signaling, NK cell-mediated cytotoxicity and JAK/STAT signaling (Fig. 1j, P < 0.001). Analysis of hallmark gene sets furthermore showed a strong association with the IFN-γ response, IL-2/STAT5 signaling, IL-6/JAK/STAT3 signaling and apoptosis (Fig. 1k, P < 10×10−4). Indeed, Lrrc8c itself is part of the IL-2/STAT5 signaling hallmark gene set (Fig. 1k, P = 2.7×10−6, FDR = 2.2×10−5)21,22. Because of the importance of IL-2/STAT5 signaling for T cell function, we next evaluated its role in the transcriptional regulation of Lrrc8 genes using genome-wide ChIP-seq data of IL-2 stimulated T cells23. This in silico analysis revealed strong binding of STAT5A and STAT5B to the promoter and putative regulatory regions of Lrrc8c in activated T cells upon IL-2 stimulation (Fig. 1l). By contrast, no STAT5A and STAT5B binding to other Lrrc8 genes was detectable (Supplemental Fig.2a). The expression of Lrrc8c, but not other Lrrc8 genes, was selectively upregulated in T cells stimulated with anti-CD3/CD28 and IL-2 for 3 days (Supplemental Fig.2b). Treatment of T cells with IL-2 or IL-15 resulted in a selective increase in the Lrrc8c/Lrrc8a ratio, which was blunted in T cells from mice lacking the ability to form STAT5 tetramers23 (Fig. 1m and Supplemental Fig.2c,d). Moreover, STAT5A and STAT5B binding to the Lrrc8c promoter was reduced in IL-2 treated T cells of Stat5a Stat5b double knock-in mice (Fig. 1l). Together these data demonstrate that the T cell-specific expression of Lrrc8c is controlled by IL-2/STAT5 signaling and STAT5 tetramerization.
LRRC8C is essential for VRAC function in T cells
We hypothesized that LRRC8C is critical for VRAC function and the RVD in T cells. We therefore measured Cl− currents in T cells from wild-type mice that were transduced with shLrrc8c or scrambled shRNA (control). Whereas shControl-transduced CD4+ T cells had robust swell-activated Cl− currents upon exposure to hypotonic solution (~215 mOsm) with properties characteristic of VRAC currents (IVRAC) including outward rectification, delayed activation, shLrrc8c-transduced T cells showed a substantial decrease in IVRAC (Extended Data Fig. 2a–e). Knockdown of Lrrc8a or both Lrrc8a/Lrrc8c abolished IVRAC (Extended Data Fig. 2a,b,d). As a consequence of reduced IVRAC in Lrrc8c-deficient CD4+ T cells, their RVD was strongly impaired (Extended Data Fig. 2f,g). Knockdown of Lrrc8a or both Lrrc8a/Lrrc8c in CD4+ T cells suppressed RVD to the same degree as shLrrc8c indicating that LRRC8A and LRRC8C are equally critical for VRAC-mediated RVD in CD4+ T cells (Extended Data Fig. 2f,g). To confirm these results, we evaluated VRAC function and RVD in T cells from Lrrc8c−/− mice, which were generated by deleting exon 4 of Lrrc8c that encodes 94% of the LRRC8C protein24. CD4+ T cells from Lrrc8c−/− mice lacked expression of exon 4 by RT-qPCR and RNA-Seq (Fig. 2a and Supplemental Fig.3a). Although mRNA corresponding to exons 1–3 was detectable, no LRRC8C protein was present in T cells of Lrrc8c−/− mice (Fig. 2b and Supplemental Fig.3a,b). We confirmed the lack of protein expression in cells transfected with a FLAG-tagged version of LRRC8C lacking the DNA encoded by exon 4 (Supplemental Fig.3c–e). Of note, deletion of Lrrc8c did not result in a compensatory upregulation of other Lrrc8 paralogs (Supplemental Fig.3f). As a consequence of abolished LRRC8C protein expression, Lrrc8c−/− T cells completely lacked IVRAC compared to wild-type T cells (Fig. 2c–e). As a consequence, Lrrc8c−/− T cells had significantly reduced RVD in hypotonic buffer (Fig. 2f).
Figure 2. LRRC8C is an essential component of the VRAC channel in T cells.
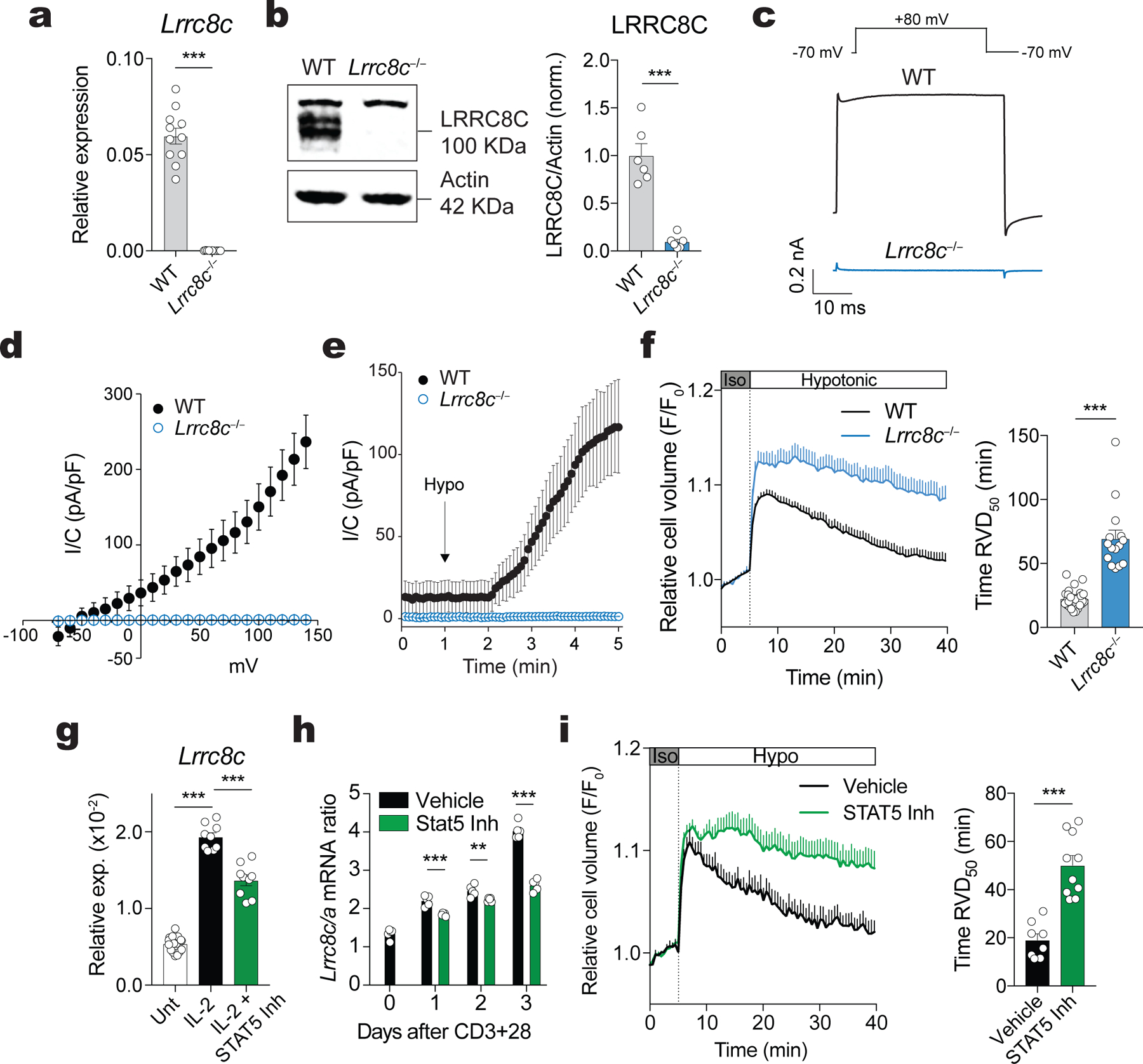
(a) Expression of Lrrc8c (exon 4) mRNA in unstimulated CD4+ T cells from wild-type and Lrrc8c−/− mice and measured by RT-qPCR (n=11 mice/genotype). Gapdh mRNA was used as housekeeping control. (b) Representative Western blot (left) and quantification of LRRC8C protein (right) in wild-type and Lrrc8c−/− CD4+ T cells (n=6 mice/genotype). Actin was used as loading control. (c) Representative volume-regulated anion current (IVRAC) traces from wild-type and Lrrc8c−/− CD4+ T cells stimulated with anti-CD3+CD28 and measured using whole-cell patch clamp recordings (n=10 wild-type and 33 Lrrc8c−/− T cells, 3 independent experiments). T cells were bathed in hypotonic solution (~215 mOsm) and held at −70 mV, followed by depolarization to +80mV every 5s. (d) Current density as a function of voltage at the end of each test pulse from experiments similar to that shown in (c). (e) Average current densities at −70 mV and +80 mV over time induced by hypotonic (Hypo) solution in wild-type and Lrrc8c−/− CD4+ T cells. Data in (d-e) are from 10 wild-type and 33 Lrrc8c−/− T cells from 3 independent experiments. (f) Cell volume measurements in wild-type and Lrrc8c−/− CD4+ T cells stimulated with anti-CD3+CD28 and challenged with hypotonic buffer (215 mOsm). Cell volume traces (left) are from 3 independent experiments and n=5 mice/genotype. The regulatory volume decrease (RVD) is quantified (right) as the time to reach half maximal cell volume (RVD50). (g) mRNA expression of Lrrc8c from activated CD4+ T cells and re-stimulated or not with 20 IU/ml IL-2 in the presence or absence of STAT5 inhibitor for 24h, and measured by RT-qPCR. Gapdh mRNA was used as housekeeping control (n=3 mice/treatment, done in triplicates and pooled from 3 independent experiments). (h) Lrrc8c/Lrrc8a mRNA expression ratio in CD4+ T cells stimulated with anti-CD3+CD28 in the presence or absence of STAT5 inhibitor (n=5 mice and 3 independent experiments). (i) RVD measurements (left) and quantification of RVD50 (right) in CD4+ T cells stimulated with anti-CD3+CD28 with and without STAT5 inhibitor as described in (f). Each dot represents one trace pooled from 5 mice and 3 independent experiments. All data are mean ± s.e.m. and were analyzed by two-tailed, unpaired Student’s t test. **P < 0.01 and ***P < 0.001.
Because Lrrc8c expression in T cells is regulated by IL-2/STAT5 signaling, we evaluated the effects of IL-2 and STAT5 inhibition on VRAC function. Treatment of CD4+ T cells with IL-2 resulted in increased Lrrc8c mRNA expression, which was attenuated by STAT5 inhibition (Fig. 2g). STAT5 inhibition also prevented the increase in the Lrrc8c/Lrrc8a mRNA ratio observed in activated T cells (Fig. 2h). Importantly, treatment of activated CD4+ T cells with a STAT5 inhibitor significantly impaired RVD in hypotonic buffer (~215 mOsm) compared to untreated T cells (Fig. 2i). The degree of RVD suppression following STAT5 inhibition was similar to that observed in Lrrc8c−/− T cells (Fig. 2f and Extended Data Fig. 2f,g). Collectively, these data demonstrate that LRRC8C is an essential component of VRAC in T cells.
LRRC8C expression is not required for T cell development
Given the abolished VRAC function and RVD in T cells of Lrrc8c−/− mice, we assessed the role of LRRC8C in T cell development and function. We did not observe significant defects in thymocyte numbers and subsets or the numbers of naïve, effector and memory CD4+ and CD8+ T cells (Extended Data Fig. 3). Moreover, the numbers of natural, thymic-derived CD4+CD25+FoxP3+ Treg and their suppressive function were normal in Lrrc8c−/− mice compared to wild-type littermates (Supplemental Fig.4a–d). Only the frequencies of induced Treg (iTreg) cells differentiated in vitro was increased in the absence of LRRC8C (Supplemental Fig.4e–g). Collectively, these data demonstrate that LRRC8C is not required for the development of conventional or regulatory T cells with the exception of a moderate effects on iTreg differentiation.
LRRC8C regulates p53 signaling in T cells
T cells rarely, if ever, encounter the hypotonic conditions that activate VRAC and RVD. To understand whether LRRC8C plays a role in T cell function, we investigated the effects of Lrrc8c deletion on gene expression before and after TCR stimulation. CD4+ T cells from Lrrc8c−/− and wild-type mice were left untreated or stimulated with anti-CD3/CD28 for 24h and 48h in vitro and analyzed by RNA-Seq. We identified 773, 160 and 337 differentially expressed genes (DEG ±1.5-FC, FDR <0.1) in Lrrc8c−/− vs. wild-type CD4+ T cells at 0, 24 and 48h after stimulation (Fig. 3a). Unbiased pathway enrichment analyses of DEG in TCR-stimulated CD4+ T cells using KEGG, IPA and GSEA revealed the dysregulation of several signaling pathways in the absence of LRRC8C. Of these, the p53 signaling pathway was most significantly and consistently downregulated (Fig. 3b–f). The validation of p53-related DEGs by RT-qPCR showed that Apaf1 and Bax as well as Ccng1 and Cdkn1a, which are involved in p53-dependent apoptosis and cell cycle arrest, were upregulated in wild-type but not in Lrrc8c-deficient T cells upon TCR stimulation (Fig. 3g). Moreover, we found that the expression of total and phosphorylated p53 protein was reduced in the absence of LRRC8C. TCR stimulation triggered a strong upregulation of total and phospho-p53 protein expression in wild-type and Lrrc8c−/− T cells after 24h, which decreased after 48 and 72h (Fig. 3h,i). This decrease was significantly more pronounced in the absence of LRRC8C. p53 is well established as a tumor suppressor that promotes apoptosis and inhibits cell cycle progression25. Consistent with their decreased p53 expression, Lrrc8c−/− T cells showed enhanced cell cycle progression with more cells in G2/M phase and proliferation, which was evident from increased CFSE dilution, total cell numbers and frequencies of Ki67 positive cells (Fig. 3j–m). Collectively, these results demonstrate that LRRC8C maintains p53 expression and signaling in T cells and thereby regulates cell cycle progression and proliferation.
Figure 3. LRRC8C regulates p53 expression in T cells.
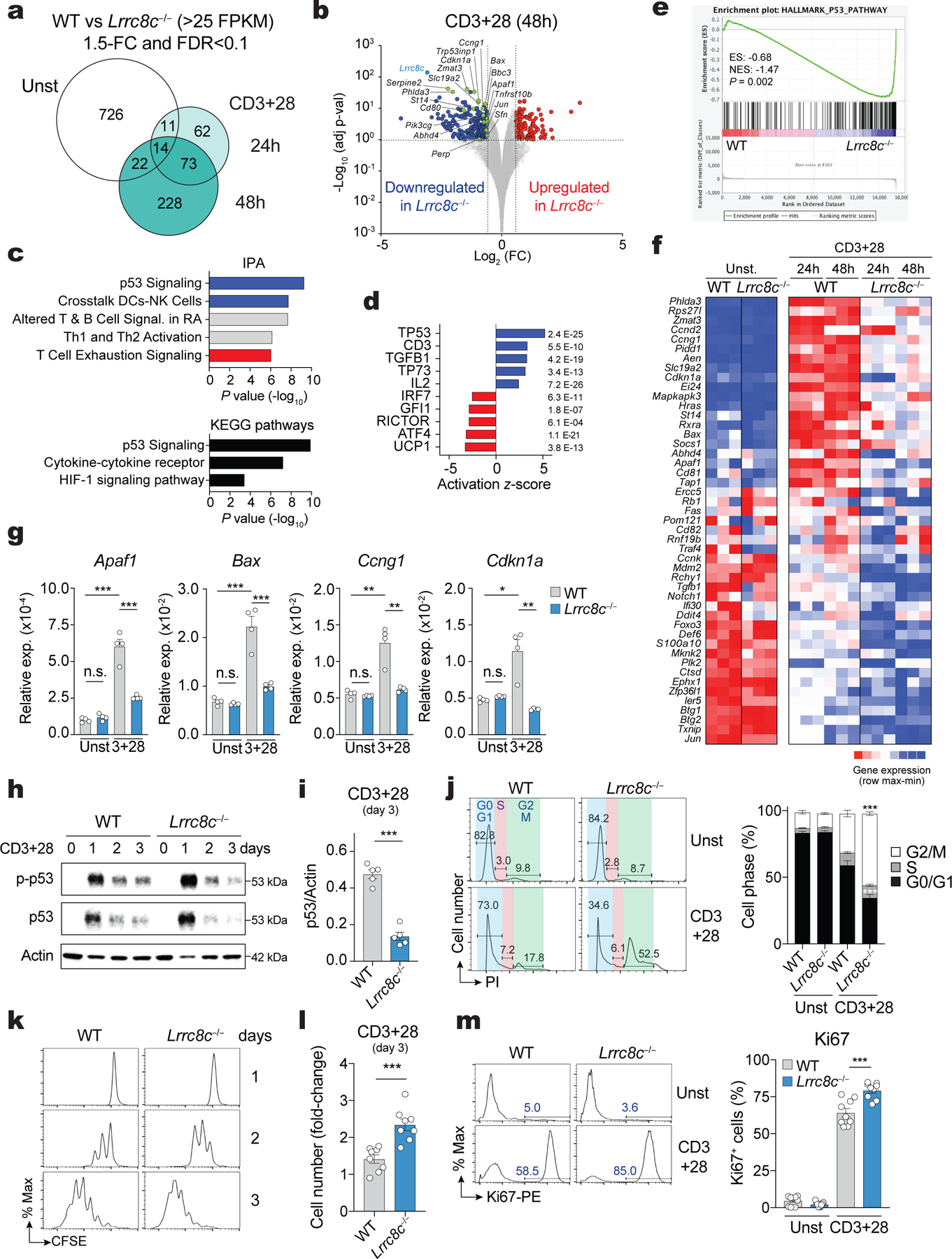
(a) Differentially expressed genes (DEG) between wild-type and Lrrc8c−/− CD4+ T cells before and after anti-CD3+CD28 stimulation and analyzed by RNA-seq. (b) Scatterplot of fold-change in gene expression (log2 FC) vs. adjusted p-value (−Log10) between wild-type and Lrrc8c−/− CD4+ T cells after anti-CD3+CD28 stimulation. Blue and red dots indicate downregulated and upregulated genes in Lrrc8c−/− T cells, respectively. Green dots indicate DEGs belonging to the p53 pathway. (c) IPA and KEGG pathways of DEGs in stimulated Lrrc8c−/− CD4+ T cells and ranked by P-value. (d) Top-5 upstream regulators that are inhibited (blue) or activated (red) in Lrrc8c−/− compared to wild-type CD4+ T cells after anti-CD3+CD28 stimulation and ranked by activation z-score. (e) GSEA showing enrichment in p53 pathway genes comparing transcriptomes of Lrrc8c−/− and wild-type CD4+ T cells after anti-CD3+CD28 stimulation. (f) Heat map of DEGs associated with the p53 pathway identified by GSEA in (e). Relative mRNA expression/row (red, high; blue, low). (g) mRNA expression of proapoptotic and cell cycle arrest genes in wild-type and Lrrc8c−/− CD4+ T cells before and after stimulation with anti-CD3+CD28 and measured by RT-qPCR. Rlp32 mRNA was used as housekeeping control (n=4 mice per genotype). (h) Immunoblot of total and phosphorylated p53 (p-p53 S15) in wild-type and Lrrc8c−/− CD4+ T cells before and after stimulation with anti-CD3+CD28. Actin was used as loading control. (i) Quantification of p53 expression in wild-type and Lrrc8c−/− CD4+ T cells stimulated for 3 days and normalized to actin loading control. Data are representative (h) and averaged (i) from n=5 mice/genotype and pooled from 2 independent experiments. (j) Cell cycle analysis of wild-type and Lrrc8c−/− CD4+ T cells before and after stimulation with anti-CD3+CD28. Representative flow cytometry plots (left) and quantification (right) of different cell cycle phases (n=6 and 8 mice/genotype for unstimulated and stimulated conditions, respectively). (k) CFSE dilution in wild-type and Lrrc8c−/− CD4+ T cells stimulated with anti-CD3+CD28. (l) Fold-change of CD4+ T cell numbers from (k) 3 days after anti-CD3+CD28 stimulation (compared to day 1, n=8 mice/genotype). (m) Ki67 expression in wild-type and Lrrc8c−/− CD4+ T cells before and after stimulation with anti-CD3+CD28. Representative flow cytometry plots (left) and quantification (right) of Ki67+ cells (n=9 mice/genotype, pooled from 3 independent experiments). All data are mean ± s.e.m. and were analyzed by two-tailed, unpaired Student’s t test. *P < 0.05, **P < 0.01 and ***P < 0.001.
LRRC8C controls T cell proliferation and apoptosis through p53
To investigate whether reduced p53 expression is responsible for the enhanced proliferation of Lrrc8c−/− T cells, we prevented the degradation p53 by treating activated Lrrc8c−/− T cells with the nutlin-derivative idasanutlin, an inhibitor of p53 interaction with the E3 ubiquitin ligase MDM226. Idasanutlin increased p53 protein expression in wild-type T cells and restored p53 levels in Lrrc8c−/− T cells to those observed in wild-type T cells (Fig. 4a). Moreover, idasanutlin suppressed the enhanced cell cycle progression and proliferation of Lrrc8c-deficient T cells (Fig. 4b,c). p53 signaling also regulates apoptosis26–28, and consistent with their decreased p53 expression, Lrrc8c−/− CD4+ T cells had reduced apoptosis compared to wild-type CD4+ T cells (Fig. 4d). Treatment of Lrrc8c−/− T cells with idasanutlin increased cell death to wild-type levels (Fig. 4d). A similar suppression of enhanced survival and proliferation of Lrrc8c−/− T cells was observed after overexpression of p53 (Extended Data Fig. 4a–c). To test whether p53 reduction is the main cause of the activated phenotype of Lrrc8c−/− T cells, we deleted Trp53 gene expression in activated wild-type T cells by CRISPR/Cas9 gene editing. Trp53 deletion enhanced T cell proliferation and abolished the suppressive effect of idasanutlin on T cell proliferation (Extended Data Fig. 4d–g). Collectively, these data suggest that the enhanced activation of Lrrc8c-deficient T cell is due to decreased p53 expression.
Figure 4. LRRC8C regulates T cell function through p53.
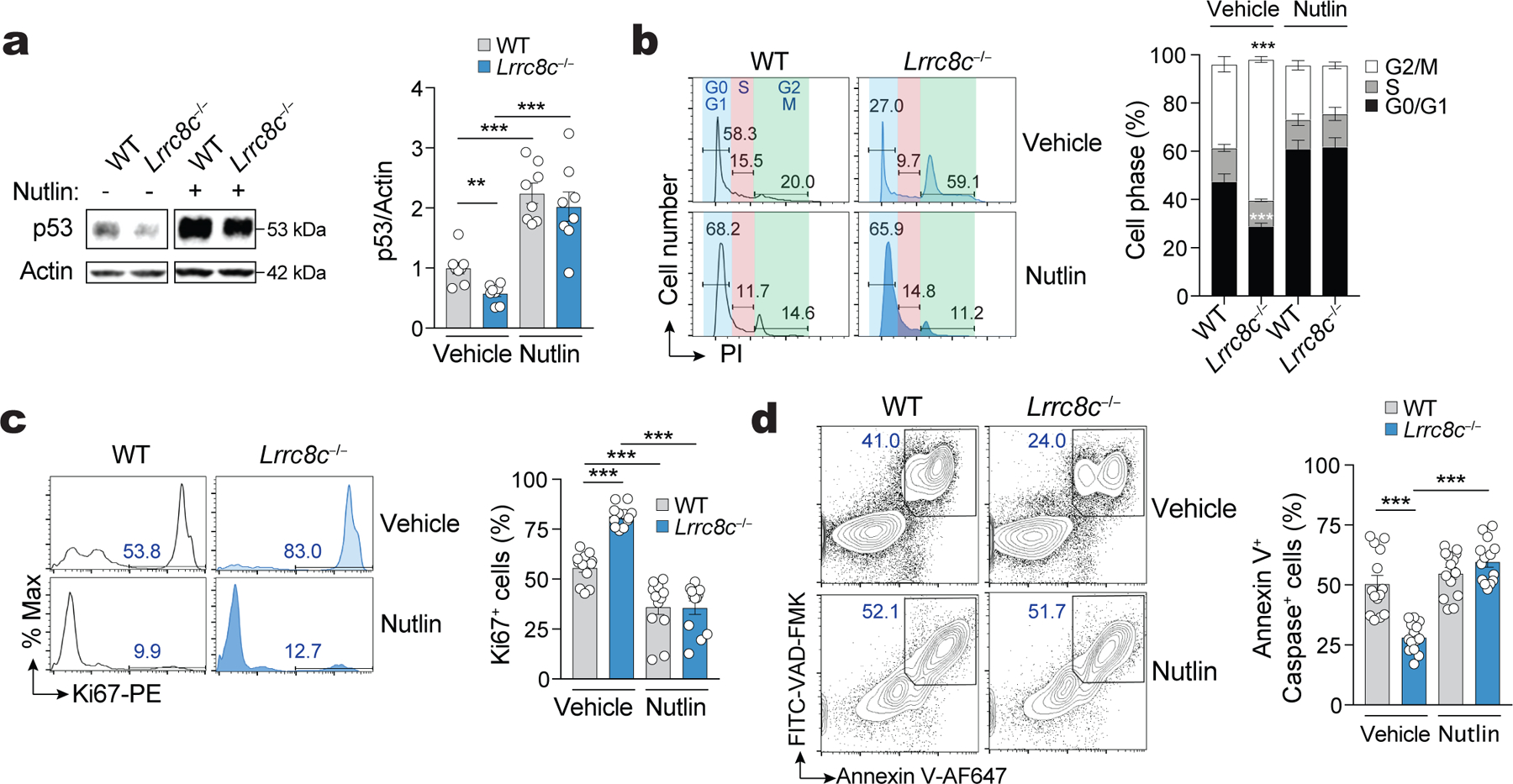
(a) Immunoblots of total p53 expression in wild-type and Lrrc8c−/− CD4+ T cells after stimulation with anti-CD3+CD28 for 3 days in the presence and absence of the MDM2 antagonist idasanutlin (abbreviated as nutlin). Actin was used as loading control. Representative blots (left) and quantification (right) from 4 independent experiments and 8 mice per genotype and treatment. (b) Cell cycle analysis of wild-type and Lrrc8c−/− CD4+ T cells after stimulation for 3 days in the presence or absence of nutlin. Representative flow cytometry plots (lefts) and quantification (right) of the different cell cycle phases. Data are from 12 mice per genotype and treatment, and pooled from 5 independent experiments. (c) Ki67 expression in wild-type and Lrrc8c−/− CD4+ T cells stimulated for 3 days and treated or not with nutlin. Representative flow cytometry plots (left) and quantification (right) of Ki67+ cells from 12 mice per genotype pooled from 5 independent experiments. (d) Apoptotic T cells from wild-type and Lrrc8c−/− mice measured by annexin V and active caspase (VAD-FMK) staining 3 days after stimulation and treated or not with nutlin. Representative contour plots (left) and quantification (right) of apoptotic cells from 14 mice per genotype pooled from 5 independent experiments. All data are mean ± s.e.m. and were analyzed by two-tailed, unpaired Student’s t test. **P < 0.01 and ***P < 0.001.
The effects of LRRC8C in T cells depend on its channel function
We next investigated if the ion channel function of LRRC8C is required for its effects on T cells. Deletion of Lrrc8a in T cells abolishes VRAC currents and blocks T cell development5. Likewise, the ébouriffé mutant of Lrrc8a, which results in the loss of 15 (of 17) C-terminal LRRs of LRRC8A, also abolishes VRAC currents in T cells but has no effects on T cell development, suggesting that the channel function of LRRC8A is dispensable for T cell development10. To investigate whether impaired p53 signaling and the aberrant function of Lrrc8c−/− T cells require the channel function of LRRC8C, we treated wild-type CD4+ T cells with the best-in-class VRAC inhibitor available, 4-(2-butyl-6,7-dichloro-2-cyclopentyl-indan-1-on-5-yl)oxy butyric acid (DCPIB)29. DCPIB blocked RVD and VRAC currents in CD4+ T cells exposed to hypotonic buffer (Extended Data Fig. 5a,b). DCPIB significantly suppressed p53 signaling in wild-type CD4+ T cells that were stimulated with anti-CD3/CD28 for 24h or 48h (Extended Data Fig. 5c). Notably, there was a large overlap between DEG in the p53 signaling pathway in DCPIB-treated wild-type T cells and Lrrc8c−/− T cells (Extended Data Fig. 5d,e). These results suggest that the VRAC channel function of LRRC8C is involved in the regulation of p53 signaling in activated T cells, although the nature of the physiologically relevant cargo of LRRC8C channels in T cells remains unclear.
LRRC8C mediates cyclic dinucleotide transport and p53 signaling
VRAC channels composed of LRRC8A:C and LRRC8A:E were recently reported to mediate the transport of cyclic dinucleotides (CDN) such as 2’3’ cGMP-AMP (cGAMP) and thereby regulate the activation of the stimulator of interferon genes (STING) and the production of type I interferons15,16. To elucidate how LRRC8C channels regulate p53 signaling and T cell function, we screened various CDNs, signaling molecules, amino acids and chemical compounds that had previously been reported to be transported by VRAC channels13–16,30 for their effects on T cell function. Of note, we had found that CD80 is a reliable marker for p53 activity in T cells. Nutlin-mediated stabilization of p53 enhanced p53 binding to the CD80 promoter and CD80 expression in T cells, which could be reversed by direct CRISPR-mediated deletion of p53 (Extended Data Fig. 6a–c). Moreover, CD80 mRNA and protein expression were reduced in Lrrc8c−/− CD4+ T cells and wild-type T cells treated with DCPIB (Extended Data Fig. 6d–f). Impaired CD80 expression in Lrrc8c-deficient T cells could be restored with nutlin or by overexpression of p53 (Extended Data Fig. 6g,h), suggesting that LRRC8C regulates CD80 expression through p53. We therefore used CD80 as a readout to identify cargo transported by the LRRC8C channel.
Treatment of anti-CD3/CD28 stimulated wild-type T cells with the CDNs 2’3’cGAMP, 3’3’c-GAMP, cdi-AMP and c-di-GMP resulted in significant suppression of proliferation (quantified by % CFSElow cells) and upregulation of CD80 expression, especially at the higher of two concentrations tested (Fig. 5a and Extended Data Fig. 7a,b). None of the other known or potential VRAC channel cargos had strong and consistent effects. Importantly, the CDN-mediated suppression of T cell proliferation and induction of CD80 expression was partially or completely abolished in Lrrc8c−/− CD4+ T cells, suggesting that CDN import across the plasma membrane depends on LRRC8C. To test this idea, we treated T cells with the cell-permeable STING agonist DMXAA. The proliferation of wild-type and Lrrc8c−/− CD4+ T cells was equally suppressed by DMXAA, and CD80 upregulation was induced to a similar extent in wild-type and Lrrc8c-deficient T cells (Fig. 5a and Extended Data Fig. 7a,b), confirming the potential role of LRRC8C in transporting CDNs across the plasma membrane. Titration experiments showed that the CDN effects were dose-dependent, with wild-type CD4+ T cells being more responsive to lower concentrations of CDNs (<10 μg/ml) than Lrrc8c−/− CD4+ T cells, which required considerably higher CDN concentrations to attain the same levels of proliferation arrest or CD80 expression (Fig. 5b,c and Extended Data Fig. 7c,d). Collectively these data suggest that in T cells LRRC8C mediates the influx of extracellular CDNs.
Figure 5. LRRC8C mediates cyclic dinucleotide transport, STING activation and p53 signaling.
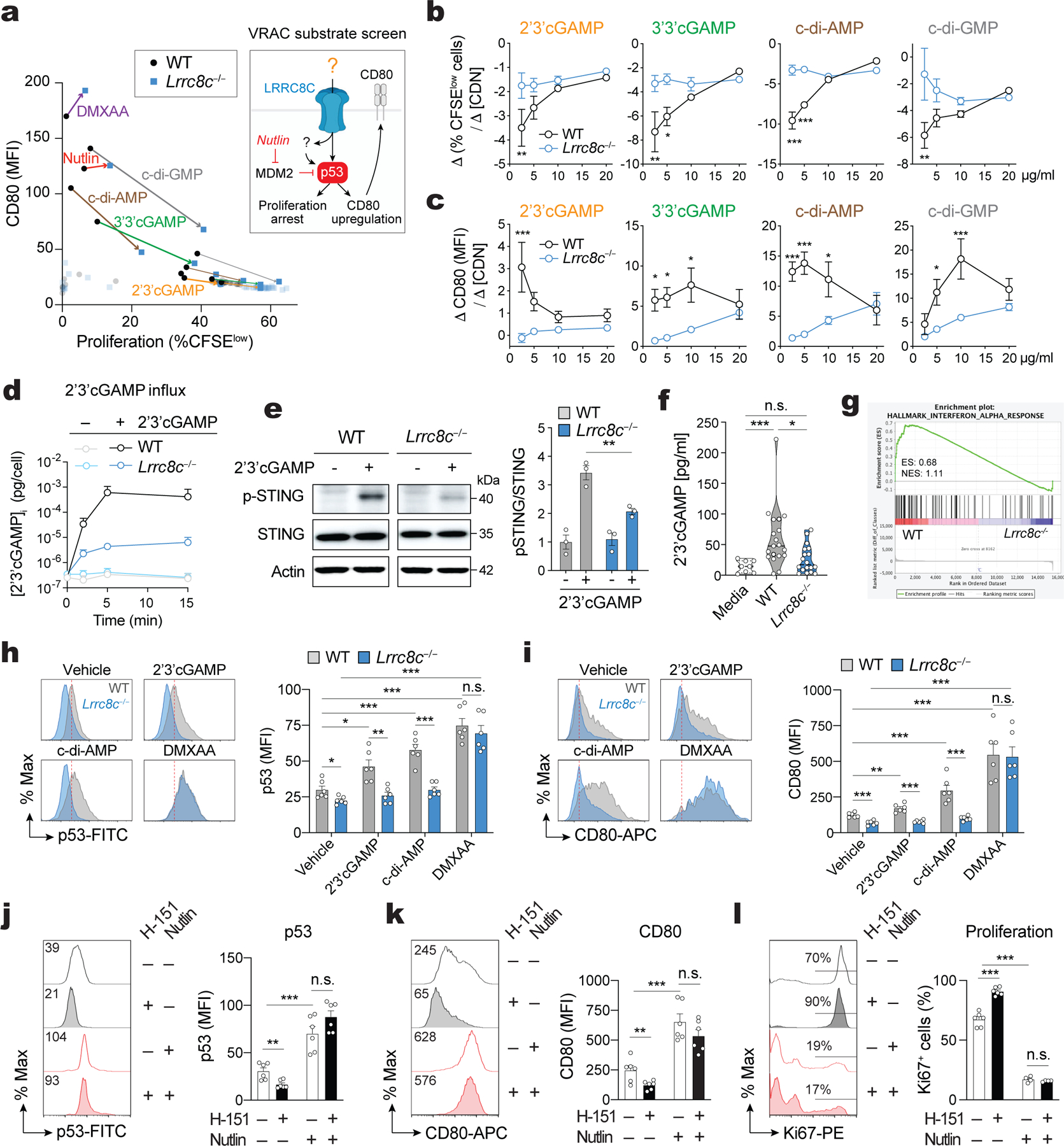
(a) Compound screening to identify substrates of LRRC8C in T cells (Created with BioRender.com). Correlation of CD80 expression and CFSE dilution in wild-type and Lrrc8c−/− CD4+ T cells stimulated for 2 days with anti-CD3/CD28 and treated with different substrates of VRAC channels. Arrows connect wild-type and Lrrc8c−/− T cell samples treated with the same compound at high (thick line) and low (thin line) compound concentrations (n= 4 mice/ genotype, pooled from 2 independent experiments). (b,c) Wild-type and Lrrc8c−/− CD4+ T cells stimulated with anti-CD3/CD28 were treated with increasing concentrations of CDNs and analyzed for cell proliferation and CD80 expression. Graphs show the differences in proliferation (Δ%CFSElow cells, in b) and the differences in CD80 expression (DMFI, in c) between at least three CDN concentrations (Δ[CDN]). Compare with Extended Data Fig. 7c,d (n=6 mice/genotype and treatment, pooled from 3 independent experiments). (d) Intracellular concentration of cGAMP in T cells exposed or not to 5 μg/ml 2’3’cGAMP in hypotonic buffer (~215 mOsm) for 15 min and measured by ELISA (n=10 mice/genotype, pooled from 2 independent experiments). (e) Immunoblots of total and phosphorylated STING (p-STING S366) in wild-type and Lrrc8c−/− CD4+ T cells after treatment with 10 μg/ml 2’3’cGAMP for 6h. Actin was used as loading control. Representative blots (left) and quantification (right) from n=3 mice/genotype and 2 independent experiments. (f) 2’3’cGAMP amount in culture media collected after in vitro stimulation of CD4+ T cells with anti-CD3+CD28 for 1–3 days measured by ELISA (n=6 mice/genotype, pooled from 5 independent experiments). (g) GSEA of RNA-Seq data identifies DEGs associated with IFN-α response in stimulated wild-type but not Lrrc8c−/− CD4+ T cells. (h,i) Flow cytometry analysis of p53 (h) and CD80 expression (i) in wild-type and Lrrc8c−/− CD4+ T cells stimulated with anti-CD3+CD28 and treated or not with STING agonists. Representative flow cytometry plots (left) and quantification (right). Data are from n=6 mice/genotype, pooled from 3 independent experiments. (j-l) Flow cytometry analysis of p53 (j), CD80 (k), and Ki67 (l) expression in wild-type CD4+ T cells stimulated for 3 days with anti-CD3+CD28 and treated or not with H-151 and idasanutlin (nutlin). Representative flow cytometry plots (left) and quantification (right) from n=6 mice/treatment, pooled from 3 independent experiments. All data are mean ± s.e.m. and were analyzed by two-tailed, unpaired Student’s t test. *P < 0.05, **P < 0.01 and ***P < 0.001.
LRRC8C controls STING activation
To directly assess if LRRC8C is required for CDN transport in T cells, we treated wild-type and Lrrc8c−/− CD4+ T cells with 2’3’cGAMP in hypotonic buffer (~215 mOsm) to activate VRAC and analyzed intracellular concentration of 2’3’cGAMP over time. Whereas significant uptake of 2’3’cGAMP was observable in wild-type CD4+ T cells, no such uptake was detected in Lrrc8c−/− T cells (Fig. 5d). Because CDNs activate STING, we treated wild-type and Lrrc8c−/− CD4+ T cells with 2’3’cGAMP and analyzed STING phosphorylation. LRRC8C deficient T cells showed significantly reduced STING phosphorylation compared to wild-type T cells (Fig. 5e). No differences in STING activation between wild-type and Lrrc8c-deficient T cells were observed using the cell permeable agonist DMXAA (Extended Data Fig. 7e). These data demonstrate that LRRC8C is required for 2’3’cGAMP influx and activation of STING in T cells. Because we had observed enhanced T cell proliferation and decreased p53 signaling in Lrrc8c−/− CD4+ T cells cultured in vitro in the absence of other cells and sources of CDNs, we speculated that T cell themselves may produce and secrete 2’3’cGAMP to activate STING in a paracrine manner as shown for macrophages15. Stimulation of wild-type T cells resulted in increased intracellular and extracellular concentration of 2’3’cGAMP (Fig. 5f, and Extended Data Fig. 7f). Compared to wild-type T cells, Lrrc8c−/− T cells had increased intracellular amount of 2’3’cGAMP but decreased extracellular concentration. Collectively, these data demonstrate that LRRC8C mediates the import as well as the export of 2’3’cGAMP in T cells.
In many immune and non-immune cells including macrophages and fibroblasts, the cGAS-STING pathway controls the production of type I interferons as an important innate immune response to cytosolic DNA31. To investigate if LRRC8C is required for STING activation in T cells, we analyzed the expression of genes associated with a type I IFN response in wild-type and Lrrc8c−/− CD4+ T cells. GSEA of DEGs in anti-CD3/CD28 stimulated T cells showed a reduction of IFN-α response genes in Lrrc8c−/− compared to wild-type CD4+ T cells consistent with impaired STING signaling (Fig. 5g). STING activates the TANK binding kinase 1 (TBK1), which in turn stimulates the interferon regulatory factor 3 (IRF3) and NF-kappa B, thereby inducing the expression of type I IFNs and cytokines such as TNF-α, respectively31–33. Consistent with a role of LRRC8C in STING activation and NF-κB dependent gene expression, we observed the downregulation of NF-κB-dependent TNF-α signaling genes in Lrrc8c−/− CD4+ T cells compared to wild-type T cells (Extended Data Fig. 7g,h). A similar defect was found in wild-type T cells treated with the VRAC channel blocker DCPIB, suggesting that NF-κB-dependent TNF-α signaling depends on the VRAC channel function of LRRC8C (Extended Data Fig. 7g,h).
STING is upstream of p53 activation in T cells
After having established that CDN transport through LRRC8C channels activates STING in T cells, we investigated if this pathway regulates the LRRC8C-dependent function of p53. STING activation was previously shown to result in the degradation of MDM2 and upregulation of p53 in T cells27, which may explain the cell cycle arrest and apoptosis observed in T cells after STING activation34,35. In our hands, TCR stimulation in the presence of 2’3’cGAMP and c-di-AMP resulted in the upregulation of p53 expression in wild-type T cells, and this response was significantly decreased in Lrrc8c−/− T cells (Fig. 5h). The cell permeable STING agonist DMXAA, however, caused a comparable upregulation of p53 in wild-type and LRRC8C-deficient T cells. These results are consistent with the similar effects of CDNs and DMXAA on the p53 regulated expression of CD80 (Fig. 5i). To further establish a direct connection between STING and p53 activation, we used the STING inhibitor H-15136. Treatment of anti-CD3/CD28 stimulated T cells with 2’3’cGAMP, c-di-AMP and DMXAA resulted in strong upregulation of STING phosphorylation, which was suppressed by H-151 (Extended Data Fig. 8a–c). Moreover, STING inhibition with H-151 increased the proliferation and viability of activated T cells, which was associated with significantly reduced expression of p53 and CD80 (Extended Data Fig. 8d–g). p53 and CD80 expression strongly correlated with the modulation of STING activation by various STING agonists and the inhibitor H-151 (Extended Data Fig. 8h).
To assess if p53 is downstream of STING signaling, we tested whether the effects of STING inhibition on T cell proliferation can be reversed by direct activation of p53. When H-151 treated T cells were incubated in addition with idasanutlin to inhibit MDM2 and stabilize p53, we observed increased p53 and CD80 expression and abolished T cell proliferation despite the presence of the STING inhibitor (Fig. 5j–l), suggesting that p53 is indeed downstream of STING. To exclude non-specific effects of idasanutlin unrelated to p53, we genetically deleted p53 and STING in activated T cells by CRISPR/Cas9 gene editing, which suppressed expression of the p53 target CD80 and increased T cell proliferation after 2’3’cGAMP, c-di-AMP or DMXAA treatment (Extended Data Fig. 9a–c). Of note, the inhibitory effects of idasanutlin on T cell proliferation could be reversed by the deletion of p53, whereas deletion of STING had no effects (Extended Data Fig. 9c). Collectively, these data demonstrate that p53 functions downstream of STING. Because STING activation depends on the transport of CDNs by LRRC8C, our data establish a new signaling pathway in T cells that links the uptake of CDNs through LRRC8C channels to the activation of STING and p53 signaling and thereby the regulation of T cell proliferation and survival (Supplemental Fig.5).
LRRC8C suppresses store-operated Ca2+ entry (SOCE) in T cells
Gain-of-function mutations of STING were shown to regulate Ca2+ homeostasis, ER stress and survival of T cells independent of STING-mediated regulation of type I IFN production37. To investigate a potential role of LRRC8C in the regulation of intracellular Ca2+ signals, we measured Ca2+ influx in T cells isolated from wild-type and Lrrc8c−/− mice. TCR crosslinking resulted in enhanced Ca2+ influx in Lrrc8c-deficient T cells compared to wild-type cells (Fig. 6a). Depletion of Ca2+ from the endoplasmic reticulum (ER) with ionomycin or by blocking the reuptake of Ca2+ into the ER with the sarco-endoplasmic reticulum Ca2+ ATPase (SERCA) inhibitor thapsigargin (TG) results in store-operated Ca2+ entry (SOCE) through Ca2+ release-activated Ca2+ (CRAC) channels38. We observed increased SOCE in Lrrc8c−/− T cells compared to wild-type controls in response to either ionomycin or TG stimulation (Fig. 6a, Supplemental Fig.6a) suggesting that LRRC8C suppresses SOCE in T cells. Because TG and ionomycin bypass proximal TCR signaling, LRRC8C likely regulates SOCE downstream of IP3 production and Ca2+ release from the ER through IP3 receptor channels. SOCE is mediated ORAI1 and its homologues ORAI2 and ORAI3, and activated by stromal interaction molecule 1 (STIM1) and STIM239,40. The mRNA and protein levels of these factors and mRNA amounts of other known regulators of SOCE 41 were unaltered in Lrrc8c−/− T cells (Supplemental Fig.6b–f), thus largely ruling out effects of LRRC8C on the expression of CRAC channel-related genes.
Figure 6. LRRC8C modulates Ca2+ influx in T cells via STING and p53 activation.
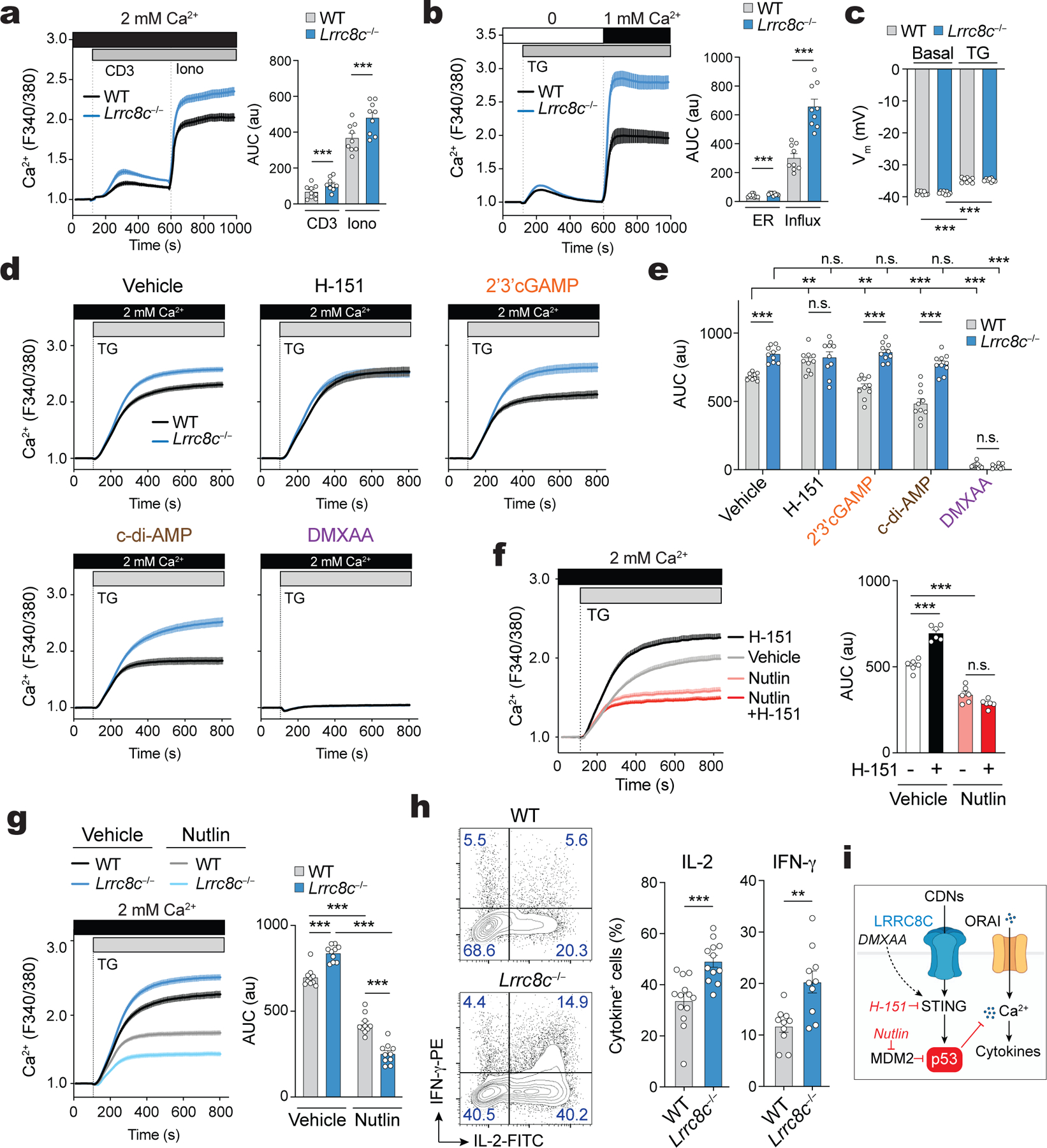
(a,b) Cytosolic Ca2+ signals in naïve (a) and activated (b) CD4+ T cells isolated from wild-type and Lrrc8c−/− mice. Fura-2-loaded T cells were stimulated by anti-CD3 cross-linking in 2 mM Ca2+ containing Ringer solution followed by ionomycin (Iono) stimulation (in a) or thapsigargin (TG) in Ca2+-free buffer followed by re-addition of extracellular Ca2+ (in b). Averaged Ca2+ traces (left) and quantification of the area under the curve (AUC, right) in the regions indicated by the dotted lines (n=9 mice/genotype, pooled from 3 independent experiments). (c) Plasma membrane potential (Vm) in wild-type and Lrrc8c−/− CD4+ T cells activated with CD3+CD28 and measured by patch-clamping. Gigaohm seals were stablished in isotonic solution in voltage-clamp configuration and cells were treated with TG for at least 5 min before recording Vm (n=8 cells/genotype, pooled from 2 independent experiments). (d,e) Cytosolic Ca2+ signals in wild-type and Lrrc8c−/− T cells stimulated with anti-CD3+CD28 and treated or not with STING agonists. T cells were stimulated with TG in Ca2+-containing Ringer buffer. Averaged Ca2+ traces (d) and quantification of the AUC (e) in the regions indicated by the dotted lines (n=10 mice/genotype, pooled from 5 independent experiments). (f) Cytosolic Ca2+ signals in T cells treated or not with STING inhibitor H-151 and idasanutlin (abbreviated as nutlin) using a similar protocol as in (d). Averaged Ca2+ traces (left) and quantification of the AUC (right) in the regions indicated by the dotted lines (n=6 mice/treatment, pooled from 3 independent experiments). (g) Cytosolic Ca2+ signals in wild-type and Lrrc8c−/− T cells treated or not with nutlin using a similar protocol as in (d and f). Averaged Ca2+ traces (left) and quantification of the AUC (right) in the regions indicated by the dotted lines (n=10 mice/treatment, pooled from 5 independent experiments). (h) IL-2 and IFN-γ production by wild-type and Lrrc8c−/− CD4+ T cells activated with CD3+CD28 and re-stimulated for 6h with PMA+Iono. Representative contour plots (left) and quantification (right) of IL-2+ and IFN-γ+ CD4+ T cells (n=10 mice per genotype, pooled from 6 independent experiments). (i) Schematic representation of LRRC8C regulating Ca2+ signals in T cells. CDNs influx via LRRC8C leads to STING activation and p53 stabilization, which in turn suppresses Ca2+ signals in T cells (Created with BioRender.com). All data are mean ± s.e.m. and were analyzed by two-tailed, unpaired Student’s t test. **P < 0.01 and ***P < 0.001.
SOCE depends on a negative membrane potential (Vm), which is generated by the concerted function of several K+, Na+ and Cl− channels39. The expression of K+ and Na+ channels that regulate Vm in T cells was normal in Lrrc8c−/− T cells (Supplemental Fig.6g)39, and measurements of Vm using the membrane potential-sensitive dye DiBAC4 did not reveal differences between Lrrc8c−/− and wild-type T cells at rest or after stimulation by TCR-crosslinking, ionomycin or TG (Supplemental Fig.6h). Additional direct measurements of Vm by patch clamp electrophysiology before and after TG stimulation did not show differences in the Vm of wild-type and Lrrc8c−/− T cells either (Fig. 6c). Of note, activation of VRAC channels by exposing T cells to hypotonic buffer resulted in an increase in baseline Ca2+ levels in both wild-type and Lrrc8c−/− T cells, which was, however, not associated with altered Vm (Supplemental Fig.6i,j). We conclude that LRRC8C controls the magnitude of SOCE in T cells through mechanisms unrelated to the regulation of Vm or CRAC channel gene expression.
LRRC8C regulates Ca2+ influx through STING and p53 signaling
To investigate if the regulation of SOCE by LRRC8C is linked to CDN transport and its effects on STING and p53 activation, we measured SOCE in the presence or absence of the STING inhibitor H-151 or different STING agonists. STING inhibition resulted in significantly enhanced SOCE in wild-type T cells to levels comparable to those in untreated Lrrc8c−/− T cells (Fig. 6d,e and Extended Data Fig. 10a,b). Conversely, STING activation with 2’3’ cGAMP and other CDNs strongly decreased SOCE in wild-type T cells, whereas Lrrc8c−/− T cells were resistant to the effects of CDNs on SOCE (Fig. 6d,e and Extended Data Fig. 10a,b). Only direct STING activation with DMXAA reduced Ca2+ influx in Lrrc8c−/− T cells. The suppressive effect of CDNs on SOCE in wild-type T cells could be partially rescued by STING inhibition with H-151 (Extended Data Fig. 10a,b). To test whether STING modulates SOCE in T cells by regulating p53 function, we enhanced p53 expression in wild-type CD4+ T cells with idasanutlin, which resulted in decreased TG-induced SOCE compared to untreated cells (Fig. 6f). The suppressive effect of idasanutlin on SOCE could not be rescued by inhibition of STING with H-151 (Fig. 6f). Increasing p53 expression with idasanutlin also suppressed the enhanced SOCE in Lrrc8c−/− T cells (Fig. 6g). Similar to idasanutlin, overexpression of p53 in T cells significantly decreased SOCE in both wild-type and Lrrc8c−/− T cells (Extended Data Fig. 10c). Deletion of p53, by contrast, enhanced SOCE (Extended Data Fig. 10d). Because SOCE is critical for the expression of many genes including cytokines in T cells40, we analyzed how deletion of LRRC8C and enhanced SOCE affect cytokine production. Compared to wild-type CD4+ T cells, stimulation of Lrrc8c−/− T cells resulted in the increased production of IL-2 and IFN-γ (Fig. 6h). Taken together, our findings demonstrate that LRRC8C suppresses SOCE and the production of Ca2+ dependent cytokines in T cells by mediating the uptake of CDNs, the activation of STING and p53 signaling (Fig. 6i and Supplemental Fig.5).
LRRC8C deletion enhances T cell-dependent CNS inflammation
Given the enhanced function of Lrrc8c−/− T cells in vitro, we sought to determine the role of LRRC8C in T cell-dependent immune responses in vivo. Lrrc8c−/− mice displayed no spontaneous immune phenotypes such as systemic inflammation. To test whether deletion of LRRC8C results in enhanced T cell-dependent autoimmunity, we induced experimental autoimmune encephalomyelitis (EAE), a murine model of multiple sclerosis (MS), in Lrrc8c−/− and wild-type mice by immunization with myelin oligodendrocyte glycoprotein (MOG35–55) peptide (Fig. 7a). The incidence and time to EAE onset in Lrrc8c−/− mice was comparable to wild-type control mice, but the severity of EAE, characterized by disease scores and body weight loss, was increased in Lrrc8c−/− mice (Fig. 7a and Supplemental Fig.7a). Exacerbated EAE was associated with significantly increased total cell numbers as well as numbers of CD3+ T cells, CD4+ (but not CD8+) T cells and MOG-specific CD4+ T cells in the spinal cords of Lrrc8c−/− mice compared to wild-type controls 23 days after MOG treatment (Fig. 7b,c and Supplemental Fig.7b–d). In addition, we observed elevated numbers of Lrrc8c−/− CD4+ T cells producing the proinflammatory cytokines IL-17A, IFN-γ and GM-CSF compared to wild-type controls (Fig. 7d). To test whether the increased presence of myelin-specific CD4+ T cells in the CNS of Lrrc8c−/− mice is due to their enhanced proliferative potential, we re-stimulated T cells isolated from the spinal cord of wild-type and Lrrc8c−/− mice with EAE ex vivo with MOG peptide. MOG-specific Lrrc8c-deficient T cells proliferated significantly more vigorously than wild-type cells (Fig. 7e). It is noteworthy that we did not detect functional defects in other immune cells of Lrrc8c−/− mice including B cells and macrophages (not shown). Overall, these data show that LRRC8C is essential to suppress the function of autoreactive T cells in autoimmune CNS inflammation in vivo.
Figure 7. LRRC8C suppresses T cell-dependent CNS inflammation.
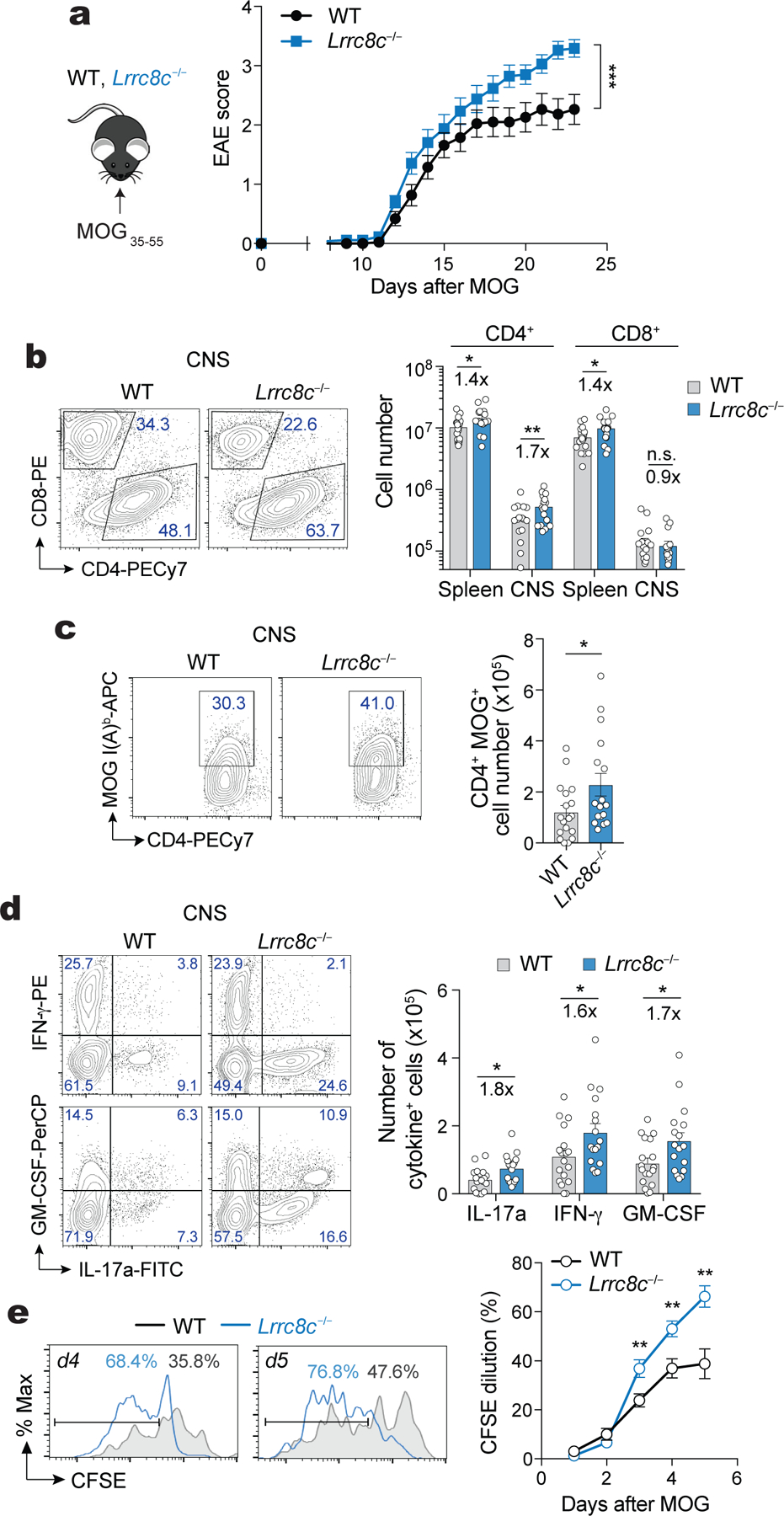
(a) Experimental design to actively induce experimental autoimmune encephalomyelitis (EAE) in mice (left) and EAE scores (right) in wild-type and Lrrc8c−/− mice at the indicated times after myelin oligodendrocyte glycoprotein (MOG) immunization. (b) Flow cytometry plots of T cells isolated from the spinal cord of mice with EAE on day 23. Representative contour plots (left) and absolute numbers of CD4+ and CD8+ T cells in the spleen and spinal cord (right) of wild-type and Lrrc8c−/− mice. (c) Representative contour plots (left) and absolute numbers (right) of MOG-specific CD4+ T cells from the spinal cord of mice. (d) Expression of IL-17a, IFN-γ and GM-CSF in CD4+ T cells isolated from the spinal cord. Representative contour plots (left) and total numbers (right) of cytokine-producing T cells after PMA+Iono re-stimulation for 6h. (e) CFSE dilution in wild-type and Lrrc8c−/− CD4+ T cells isolated from the spinal cord of mice with EAE and re-stimulated at the indicated time points with 50 μg/ml MOG35–55. Representative flow cytometry plots (left) and quantification (right) of proliferating cells. Data in (a-d) are pooled from 3 independent experiments using 19 wild-type and 17 Lrrc8c−/− mice. Data in (e) are pooled from 2 independent experiments using 15 wild-type and 12 Lrrc8c−/− mice. All data are mean ± s.e.m. and were analyzed by 2-way ANOVA with Sidak’s multiple comparison test (a) and two-tailed, unpaired Student’s t test (b-e). *P < 0.05, **P < 0.01, ***P < 0.001.
LRRC8C deficiency enhances antiviral immunity to influenza
To investigate whether LRRC8C also regulates T cell function in the context of T cell-dependent immunity to infection, we inoculated Lrrc8c−/− and wild-type mice with the A/Puerto Rico/8 (PR8) strain (H1N1) of influenza A virus (IAV). Lrrc8c−/− mice showed a similar mortality, but less pronounced weight loss and lower viral titers in their lungs compared to wild-type control mice after PR8 infection (Fig. 8a,b, and data not shown). The total numbers and frequencies of CD4+ T cells and B220+ B cells in the lungs of Lrrc8c−/− mice were significantly increased compared to wild-type mice (Supplemental Fig.8a,b). IAV-infected Lrrc8c−/− mice had significantly higher serum titers of IAV-specific IgM and IgG antibodies compared to wild-type mice (Fig. 8c and Supplemental Fig.8c). The production of high affinity IgG antibodies depends on T follicular helper (Tfh) cells that interact with germinal center (GC) B cells to induce class switch recombination and affinity maturation. Although the frequencies of CXCR5+PD-1+ Tfh cells and GL7+CD38− GC B cells were comparable in the spleen and mediastinal LNs of Lrrc8c−/− and wild-type mice, the total numbers of Tfh cells and GC B cells were significantly increased in Lrrc8c−/− mice (Fig. 8d,e). We did not observe differences in the numbers and frequencies of B220+ CD138+ plasma cells (Supplemental Fig.8d–f).
Figure 8. LRRC8C deficiency enhances T cell mediated antiviral immunity to influenza.
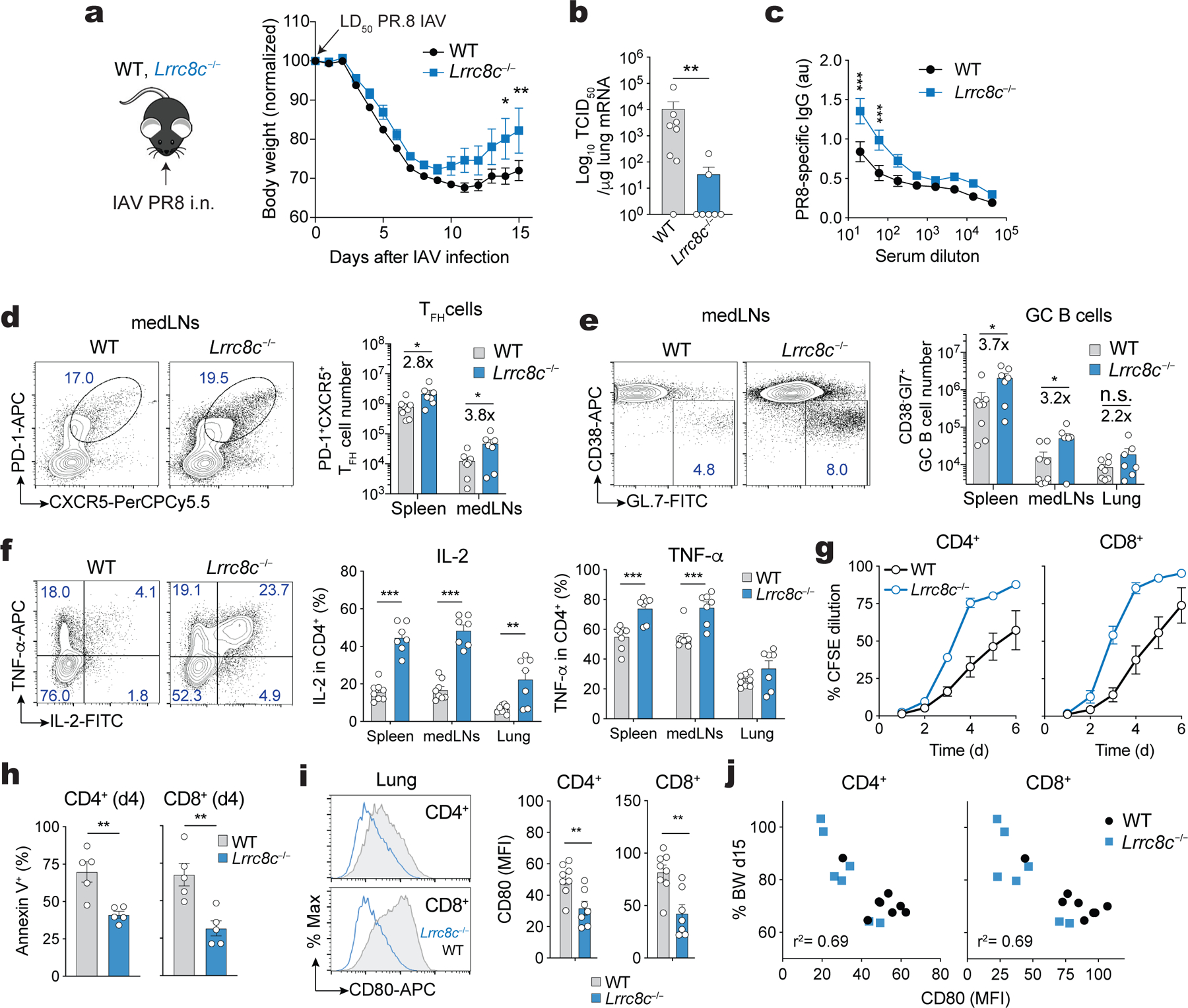
(a) Experimental design of influenza A virus (IAV) infection of wild-type and Lrrc8c−/− mice using the PR8 strain (H1N1, left), and body weight change (right) relative to the initial weight before infection. i.n., intranasal; LD, lethal dose. (b) Viral titers detected in the lung of IAV infected mice by measuring PR8 M2/M1 mRNA expression using RT-qPCR. TCID50, 50% tissue culture infective dose. TCID50 values where transform to TCID50+1 to plot titers on log10 scale. (c) anti-PR8 IgG antibodies in the sera of wild-type and Lrrc8c−/− mice detected by ELISA. (d) Representative flow cytometry plots and bar graphs showing the frequencies of T follicular helper (TFH) cells in the mediastinal lymph nodes (medLNs, left) and absolute number of TFH cells in spleen and medLNs (right). (e) Representative contour plots and bar graphs showing the frequencies of germinal center (GC) B cells in the medLNs (left) and absolute numbers of GC B cells in the spleen, medLNs and lungs (right). (f) Expression of IL-2 and TNF-α in CD4+ T cells isolated from IAV infected mice. Representative contour plots from lung-infiltrating T cells (left) and frequencies (right) of cytokine-producing T cells isolated from the spleen, medLNs and lungs after PMA+Iono re-stimulation for 6h. (g) T cell proliferation measured by CFSE dilution in wild-type and Lrrc8c−/− CD4+ T cells isolated from the lung of mice infected with IAV and re-stimulated with anti-CD3+CD28. (h) Apoptosis of CD4+ and CD8+ T cells from wild-type and Lrrc8c−/− mice shown in (g) measured by annexin V staining 4 days after stimulation with anti-CD3+CD28. (i) CD80 surface expression by CD4+ and CD8+ T cells in the lungs of wild-type and Lrrc8c−/− mice infected with IAV. Representative histogram plots (left) and quantification (right) of CD80 expression (MFI). (j) Correlation of CD80 expression by T cells in the lungs of IAV infected mice vs. body weight (% original BW of mice 15 days after IAV infection). Data in (a-j) are pooled from 2 independent experiments using 8 wild-type and 7 Lrrc8c−/− mice. Data in (g,h) are pooled from 2 independent experiments using 5 mice/genotype. All data are mean ± s.e.m. and were analyzed by 2-way ANOVA with Sidak’s multiple comparison test (a), two-tailed Mann Whitney test (b), and two-tailed, unpaired Student’s t test (b-f, h,i), *P < 0.05, **P < 0.01, ***P < 0.001.
Given the inhibitory role of LRRC8C on T cell function in vitro and the enhanced T cell and T-dependent antibody response to IAV of Lrrc8c−/− mice, we analyzed the function of Lrrc8c-deficient T cells isolated from IAV infected mice ex vivo. Stimulation of CD4+ T cells from the spleen, mediastinal LNs and lungs of IAV-infected mice showed increased production of IL-2 and TNF-α (Fig. 8f), but not IFN-γ and IL-21 (not shown), by Lrrc8c−/− T cells compared to wild-type controls. Furthermore, Lrrc8c-deficient CD4+ and CD8+ T cells proliferated significantly more and were more viable than wild-type T cells after restimulation in vitro (Fig. 8g,h). Because cell cycle progression and apoptosis are regulated by p53, we analyzed the expression of CD80 as a marker of p53 signaling. CD80 expression was significantly lower on CD4+ and CD8+ T cells isolated from the lungs of IAV-infected Lrrc8c−/− mice (Fig. 8i), and lower CD80 expression strongly correlated with protection from body weight loss after PR8 infection (Fig. 8j). Of note, LRRC8C deletion did not affect the expression of CD80 on other immune cells types that constitutively express CD80 such as macrophages and DCs; neither did CD80 expression on these cells correlate with IAV-induced body weight loss (Supplemental Fig.8g,h). Together these data demonstrate that deletion of LRRC8C enhances the function of CD4+ and CD8+ T cells during an antiviral immune response in vivo by downregulating p53 signaling.
Discussion
Here we show that LRRC8C is an essential component of the VRAC channel in T cells and negative regulator of T cell function. The canonical role of VRAC is to mediate Cl− secretion from cells and facilitate RVD after hypotonic cell swelling. Although we found that Lrrc8c-deficient T cells lack Cl− currents and efficient RVD when exposed to hypotonic conditions, T cells are rarely exposed to hypotonic environments in which RVD might be required. VRAC channels, however, are non-selective channels with a large pore that are permeable to many other molecules including amino acids, antibiotics (blasticidin S), chemotherapeutic drugs (cisplatin) and CDNs even under normotonic conditions12–14. Two recent studies showed that LRRC8A mediates the transport of CDNs in macrophages and non-immune cells to activate STING and the production of type I IFNs15,16. LRRC8A/C heteromeric channels were reported to transport cGAMP in human vascular epithelial cells16 and a similar role was shown for LRRC8A/E in murine macrophages, embryonic fibroblasts and L929 cells15. No such role of VRAC is known in T cells. Because the function of Lrrc8c−/− T cells was enhanced even under normotonic conditions, we speculated that the physiological role of VRAC in T cells might be related to CDN transport and STING signaling. Such a pathway would allow T cells to sense and respond to CDNs produced and secreted by other immune cells such as macrophages42.
We here provide several lines of evidence that establish LRRC8C as a CDN channel in T cells. (i) Stimulation of Lrrc8c−/− T cells resulted in decreased extracellular concentrations of 2’3’cGAMP compared to wild-type T cells, whereas intracellular amounts were increased, indicating that LRRC8C mediates CDN secretion. It is noteworthy that a recent study demonstrated the accumulation of double-stranded DNA in the cytosol of activated T cells, which may induce the production of 2’3’cGAMP by cGAS in T cells42. (ii) Incubation of T cells with exogenous 2’3’cGAMP results in increased cytosolic CDN concentration in wild-type but not Lrrc8c−/− T cells. (iii) Incubation of wild-type, but not Lrrc8c−/− T cells, with exogenous 2’3’cGAMP and other CDNs results in phosphorylation of STING, expression of CD80 and suppression of T cell proliferation, demonstrating that these T cell functions depend on LRRC8C-mediated influx of CDNs. Although these data are consistent with a role of LRRC8C in CDN transport, the increased intracellular concentration of 2’3’cGAMP and impaired STING phosphorylation in Lrrc8c−/− T cells appear at odds with one another. While we do not have a definite answer to resolve this conundrum, we speculate that a diffuse accumulation of 2’3’cGAMP in the cytoplasm of Lrrc8c−/− T cells is not sufficient to activate STING, presumably because 2’3’cGAMP concentrations are too low. We propose that STING activation may require the formation of microdomains in which LRRC8C as the 2’3’cGAMP influx channel in the plasma membrane comes into close proximity with STING in the ER membrane to achieve sufficiently high concentrations of 2’3’cGAMP in the intermembrane space. Such ER-plasma membrane junctions are well established in the Ca2+ signaling field as the site of CRAC channel activation that occurs following binding of STIM1 (located in the ER membrane) to ORAI1 (in the plasma membrane). Spatially confined Ca2+ microdomains in T cells are important signaling hubs that regulate specific signal transduction outcomes including the activation of transcription factors such as NFAT43,44. A similar microdomain formation may occur during cGAMP signaling to activate STING. It was recently reported that the activation of LRRC8 channels is facilitated by the recruitment of cGAMP synthase (cGAS) to the plasma membrane45. More generally speaking, the concept of signal transduction being initiated in microdomains is not new and a classical example is the immune synapse between T cells and APCs.
Lrrc8c−/− T cells have defective CDN influx, but are able to induce STING-p53 signaling at higher CDN concentrations (>10 μg/ml) than is required in wild-type T cells, suggesting that additional CDN transport mechanisms other than LRRC8C are present in T cells. One possible explanation is that Lrrc8c−/− T cells express LRRC8A which is capable of transporting CDNs. Indeed, as stated above, heteromeric VRAC channels composed of LRRC8A and either LRRC8C or LRRC8E can transport CDNs in vascular epithelial cells and murine macrophages, respectively15,16. Our RNA-seq data show that both LRRC8A and LRRC8C are expressed in murine T cells and deletion of either gene strongly suppressed VRAC currents and RVD after osmotic cell swelling. Another explanation might be the expression of additional transporters in T cells such as the folate carrier SLC19A1 that was shown to transport cGAMP in human monocytes46.
The mechanisms by which STING activation modulates T cell signaling and function are not well understood despite the fact that STING protein expression in T cells is higher than in myeloid cells27,37. In myeloid and non-immune cells, the main effect of CDN binding to STING is the activation of TBK1 and IRF3 resulting in the production of type I IFNs. Gain-of-function (GOF) mutations of STING are the cause of a type 1 interferonopathy called STING-associated vasculopathy with onset in infancy (SAVI) in human patients and severe T cell immunodeficiency in mice37,47–50. In contrast to innate immune cells, STING activation in T cells was shown to cause cell cycle arrest34,51 and cell death27,35,37,52, which are independent of type-I IFN production52. Whereas Sting−/− mice have normal lymphocyte development, their T cells show enhanced proliferation and survival upon TCR stimulation35,42,52. We made similar observations following pharmacological or genetic suppression of STING in wild-type T cells. Importantly, the effects of STING inhibition on T cell function and signaling were recapitulated in Lrrc8c−/− T cells suggesting that LRRC8C and STING are components of the same signaling pathway in T cells.
We here show that the downstream target of LRRC8C-STING signaling in T cells is p53. The expression of p53 was significantly lower in Lrrc8c−/− T cells 2–3 days post-stimulation, which was accompanied by downregulation of p53 dependent signaling pathways and genes such as CD80. Downregulation of p53 after TCR stimulation is critical for antigen-specific CD4+ T cell responses and preventing p53 degradation with MDM2 suppresses T cell proliferation26. We find that increasing p53 levels pharmacologically or by p53 overexpression reversed the enhanced proliferation and survival of Lrrc8c−/− T cells. Conversely, deletion of p53 in wild-type T cells reversed the suppressive effects of extracellular CDNs on T cell proliferation. Collectively, our data support a model in which LRRC8C regulates p53 expression and its function as a critical checkpoint of T cell activation and survival.
Deletion of LRRC8C in T cells, surprisingly, enhanced Ca2+ influx (SOCE) through CRAC channels following TCR stimulation or direct activation of CRAC channels with thapsigargin. Similarly, inhibition of STING in wild-type T cells increased SOCE, whereas activation of STING with DMXAA or CDNs decreased it, indicating that the LRRC8C-STING pathway regulates SOCE in T cells. These findings are consistent with observations in T cells from SAVI patients and StingN153S/+ mutant mice with GOF mutations of STING which showed altered Ca2+ homeostasis associated with ER stress, activation of the unfolded protein response and T cell death37. A recent study furthermore showed the reciprocal regulation of STING by another ER-resident protein, STIM1, that is the main activator of CRAC channels40,53. Deletion of STIM1 not only abolished SOCE but also enhanced STING-dependent production of type I IFNs in fibroblasts and HEK cells. Conversely, deletion of STING increased SOCE40,53. By contrast, we here show that the inhibitory effects of STING on SOCE in T cells are dependent on p53. Increasing p53 levels in T cells resulted in reduced SOCE that could not be rescued by inhibition of STING. We conclude that the suppression of SOCE by LRRC8C-STING signaling in T cells is dependent on the activation of p53 rather than effects of STING on STIM1 and CRAC channel activation. The mechanism by which the LRRC8C-STING-p53 pathway regulates SOCE is unclear. We excluded that the effects of LRRC8C deletion on SOCE are due to altered transcription of CRAC channel genes, or the membrane potential in T cells. Potential mechanisms of p53-mediated regulation of SOCE include direct effects on CRAC channel function at the protein level or the binding of p53 to the sarco/endoplasmic reticulum Ca2+ ATPase (SERCA), which was reported to increase SERCA activity and the reuptake of Ca2+ into the ER54, thereby shutting down STIM1 activation and SOCE. Regardless of the mechanisms underlying p53 control of SOCE, an important consequence of enhanced SOCE in Lrrc8c−/− T cells is the exacerbated production of IL-2, IFN-γ and potentially other Ca2+ dependent cytokines that mediate T cell function.
Our findings establish the LRRC8C-STING-p53 signaling axis as a novel inhibitory pathway in T cells that controls T cell mediated immune responses. This is evident from the enhanced proliferation, viability and cytokine production of Lrrc8c-deficient T cells in vitro, as well as exacerbated EAE and augmented immune responses to influenza A virus infection in vivo. The pharmacological suppression of LRRC8C may provide a novel approach to enhancing T cell function in the context of immunity to infection and antitumor immunity. Because LRRC8C is selectively enriched in T cells and its deletion is not lethal in mice (in contrast to the deletion of the obligatory VRAC channel subunit LRRC8A), targeting LRRC8C may represent a more specific and safer approach to modulating T cell function.
Methods
Mice.
Lrrc8c−/− (B6.129S2-Fad158<tm1Maim>/7Maim, RBRC05143, Riken)24, Stim1fl/flStim2fl/fl Cd4Cre 55 and Orai1fl/fl Cd4Cre 56 mice have been described previously. Cd4Cre (strain 017336) and Rosa26-LSL-Cas9 knock-in (strain 024857) mice were purchased from the Jackson laboratory (Bar Harbor, ME). Sperm from Lrrc8c+/− mice was used to in vitro fertilize female C57BL/6J mice (The Jackson Laboratory strain 000664) and F1 generation were further crossed with C57BL/6J mice. Mouse breeding was performed by crossing Lrrc8c+/− mice. Descendant Lrrc8c−/− mice and their Lrrc8c+/+ (wild-type) littermates were used for experiments. Genotyping of mice for the presence of the wild-type allele and inserted neoR gene allele was performed using mouse tail DNA and PCR using the following primer pairs: wild-type allele 5’-TCCAGGCAGAGGAAATGGAC-3’ and 5’-CTCGCTCGTAGCACATCTGG-3’ (product ~334 bp); neoR gene 5’-ACTCTGTTCTCACCCTTCTTAAC-3’ and 5’-ATGCCTGCTTGCCGAATA-3’ (product ~340bp). PCR conditions for all reactions were the following: denaturation at 94 °C for 4 min, followed by 37 cycles of 94 °C for 30 s, annealing at 60 °C for 30 s and elongation at 72 °C for 30 s with an additional 5 min final elongation step at 72 °C. All mice were maintained on a C57BL/6 genetic background and used between 6 and 20 weeks of age. Mice were maintained under specific pathogen-free conditions with a 12 hour dark/light cycle, at 22–25 °C, and 50–60% humidity with water and food provided ad libitum. All experiments were conducted in accordance with protocols approved by the Institutional Animal Care and Use Committee of at New York University School of Medicine.
Experimental autoimmune encephalomyelitis (EAE).
Active EAE was induced as described previously56. Briefly, mice were subcutaneously injected with 200 μg MOG35–55 peptide (Anaspec) emulsified in CFA (Difco). On days 1 and 3, mice were injected i.p. with 200 ng pertussis toxin (List Biological Laboratories). The severity of EAE was scored according to the following clinical scoring system: 0, no disease; 0.5, partially limp tail; 1, paralyzed tail; 2, hind limb weakness; 3, hind limb paralysis; 3.5, hind limb paralysis and hunched back; 4, hind and fore limb paralysis; and 5, moribundity and death56. All animals were supported with nutrigel and food on the floor. At the end of the experiment, cells were isolated from the spinal cord (CNS) and spleen of mice and analyzed by flow cytometry.
Influenza A Virus infection.
Mice were infected intranasally with influenza A/Puerto Rico/8 (PR8) virus strain (H1N1) at LD50 (250–500 TCID50) in PBS. Illness in mice was monitored by daily weighing after virus challenge. Mice were sacrificed at day 15 after IAV infection and mouse sera was collected for Ab detection by ELISA, and small pieces of lung tissue were collected in TRIZOL and stored at −80°C prior virus titters detection by RT-qPCR. PR8 M2/M1 mRNA expression was detected by RT-qPCR and calibrated to a standard curve using different median tissue culture infection doses (TCID50: 101–107). Cells isolated from the lungs, spleen and mediastinal LNs of mice were analyzed by flow cytometry.
T cell stimulation.
CD4+ T cells were isolated from the spleen using the MagniSort Mouse CD4+ T cell Enrichment Kit (Thermo Fisher Scientific). Purified CD4+ T cells were stimulated in flat bottom 12-well plates (1×106 cells/ml per well) with 1 μg/ml plate-bound anti-CD3 (clone 2C11) and 1 μg/ml anti-CD28 antibodies (clone 37.51; both Bio X cell) in complete RPMI media (Cellgro) containing 10% FBS, 1% L-glutamine, 1% penicillin-streptomycin and 0.1% β-mercaptoethanol. Alternatively, T cells were stimulated in round-U bottom 96-well plates (5×105 cells/ml per well) with Dynabeads Mouse T-Activator CD3/CD28 (Thermo Fisher Scientific) at 1:2 bead per cell ratio. For in vitro re-stimulation with MOG, total mononuclear cells isolated from the spinal cord of mice with EAE were stimulated with 50 μg/ml of MOG35–55 peptide. For differentiation into iTregs, CD4+ T cells were polarized for 3 days with anti-CD3+CD28 (1 μg/ml each) and 50 IU/ml rh-IL-2 (NIH), 2 ng/ml TGF-β (Peprotech), and 5 μg/ml anti-IL-4 (clone 11B11) and 5 μg/ml anti-IFN-γ (clone XMG1.2; both eBioscience). For cytokine expression, cells were re-stimulated with 1 μM ionomycin plus 20 nM phorbol myristate acetate (both Calbiochem) for 6h in the presence of brefeldin A (BioLegend) and analyzed by flow cytometry as described below. Additionally, activated T cells with anti-CD3+CD28 were treated in parallel with: 20 μM DCPIB, 5 μM Idasanutlin (abbreviated as nutlin) and 1 μM staurosporine (all from Tocris); 100 μM STAT5 inhibitor CAS 285986–31-4 (Millipore-Sigma), 10 μM H-151 (InvivoGen), and 3 μg/ml DMXAA (Selleckchem). For compound screening of VRAC substrates, the following compounds were used: cisplatin (Selleckchem), c-di-GMP (Cayman chemical), 2’3’cGAMP, 3’3’cGAMP, c-di-AMP, and Blasticidin S (all from InvivoGen); D-glutamic acid, taurine, D-lysine, D-aspartic acid, D-serine, D-sorbitol, sphingosine 1-phosphate (S1P), bradykinin acetate, folic acid, γ-Aminobutyric acid (GABA), myo-inositol, thiamine hydrochloride, and adenosine 5’-triphosphate (ATP) disodium salt (all from Sigma).
Recombinant DNA and retroviral transduction of T cells.
For RNA interference, shRNAs sequences targeting Lrrc8a and Lrrc8c were designed as described57 and cloned into the miR-E-based pLMPd retroviral vector which encodes mCherry or Ametrine (Amt) as fluorescent reporters58. For CRISPR/Cas9 gene editing, sgRNAs targeting STING and p53 were designed using the CRISPick sgRNA design tool (Broad Institute)59, and cloned into the pMRI-Amt retroviral vector60. shRNAs or sgRNAs targeting a non-mammalian gene (Renilla luciferase) or human VEGF were used as controls. Target sequences are listed in Supplemental Table 2. For overexpression of murine LRRC8C (Dharmacon, MMM1013–202767571) and p53 (Sino Biological, MG50534-UT), cDNAs were subcloned into the pMSCV retroviral vector (Addgene, 52114); empty pMSCV was used as control. Retroviral supernatant was produced in the Platinum-E retroviral packaging cell line following their transfection with retroviral expression plasmids and the ecotropic packaging vector pCL-Eco using lipofection (GeneJet, Fisher). Retroviral supernatant was collected 36 and 60h after transfection. For shRNA delivery, CD4+ T cells isolated from wild-type mice were transduced 24h after stimulation with anti-CD3+CD28 by spin-infection (2.500 rpm, 90 min, 32 °C) in the presence of retroviral supernatant and 8 μg/ml polybrene (SantaCruz). For CRISPR/Cas9 gene editing, CD4+ T cells isolated from Cd4Cre-Rosa26-LSL-Cas9 knock-in mice were infected twice at 24 and 48h after stimulation with anti-CD3+CD28. Retroviral supernatant from T cells was diluted 1:2 with full RPMI containing 20 IU/ml rh-IL-2 and 2.5 ng/ml IL-7 (Peprotech) 30 min after spin infection, and replaced with fresh full RPMI media containing IL-2 and IL-7 16h later. For overexpression of p53, T cells were spin infected twice at 24 and 48h after stimulation with anti-CD3+CD28 and cultured in full RPMI media without IL-2 and IL-7. For experiments that required RVD measurements and patch clamp electrophysiology, transduced Ametrine+ or mCherry+ T cells were sorted using a Sony SY3200 (HAPS1) cell sorter 2 days after spin infection and cultured in 20 IU/ml rh-IL-2 prior experiments.
Flow cytometry.
Cell from tissue culture or isolated from lymphoid organs (spleen, LNs and thymus) and tissues (lungs and spinal cord) were washed in cold PBS containing 3% FBS and 2 mM EDTA (FACS buffer). Staining on surface molecules with fluorescent labelled antibodies was performed at room temperature for 10 min in the dark. The complete list of antibodies used in this study can be found in Supplemental Table 3. Intracellular cytokine staining was performed using the IC staining buffer kit (eBioscience) according to the manufacturer’s protocol. For detection of Foxp3, Ki67, and p53 expression, cells were fixed and permeabilized with the Foxp3/Transcription Factor Staining Buffer Set (eBioscience). For cell cycle analysis, cells were fixed in 70% EtOH solution and washed once with FoxP3 permeabilization buffer before staining with propidium iodide (PI). For apoptosis measurements, cells were incubated for 20 min at 37°C with CaspACE FITC-VAD-FMK probe for active caspases (Promega) and co-stained with Annexin V in Annexin V Binding buffer (BioLegend) at room temperature. Samples were acquired on a LSR Fortessa cell analyzer (BD Biosciences) and analyzed using FlowJo software (FlowJo 10.5.3).
Proliferation assays.
CD4+ T cells were loaded with 2.5 μM CFSE according to the manufacturer’s instructions and stimulated with Dynabeads Mouse T-Activator CD3/CD28 at 1:2 bead per cell ratio. CFSE dilution was assessed by flow cytometry 24, 48 and 72h after stimulation. Alternatively, cell proliferation was assessed by flow cytometric measurement of Ki67 expression as described above.
In vitro Treg suppression assay.
Treg cells were isolated from the spleen and LNs of congenic CD45.2+ wild-type and Lrrc8c−/− mice using the EasySep Mouse CD4+CD25+ Regulatory T cell isolation Kit II (StemCell Technologies). CD4+ effector T cells were isolated from the spleens of congenic CD45.1+ wild-type mice, labeled with 2.5 μM CFSE (Invitrogen), and stimulated with Dynabeads Mouse T-Activator CD3/CD28 at 1:1 bead per cell ratio. Tregs and effector T cells were co-incubated at different ratios (1:8, 1:4, 1:2, 1:1) in round-U-bottom 96-well plates. Stimulated effector T cells cultured without Treg cells were used as positive control. Proliferation of CFSE labeled CD45.1+CD4+ effector T cells was assessed after 72h by flow cytometry.
ELISA.
For detection of PR8-specific anti-IgM and anti-IgG antibodies in mouse sera, ELISA plates (Corning Costar) were pre-coated with formaldehyde-inactivated PR.8 virus (1.6 × 106 TCID50) in PBS and incubated overnight at 4°C. After blocking, serial dilutions of mouse sera were tested for anti-IgM and anti-IgG antibodies using AP-conjugated goat-anti-mouse IgM and IgG as detection antibodies (both Southern Biotech). ELISAs using AP-conjugated detection antibodies were developed with a pNPP substrate (Thermo Fisher Scientific) and absorbance was analyzed at 405 nm using the FlexStation3 multi-mode microplate reader (Molecular Devices). For detection of 2’3’cGAMP in mouse T cell cultures, T cell media were collected at day 1, 2 and 3 after anti-CD3+CD28 stimulation from 12-well plates using a cell density of 1 × 106 cells/ml. Same media used for T cell culture at day 0 was store at −20°C as control. For intracellular detection of 2’3’cGAMP, T cells pellets were washed 3 times with ice cold PBS and lysed on ice using 200 μl of ice cold lysis buffer containing 80 % methanol (Fisher Scientific) and 0.1% trifluoroacetic acid (Sigma-Aldrich). Cell lysates were transferred to 2 ml screw cap tubes and subjected to mechanical lysis for 10 cycles (30s on/30s off) using a BeadBlaster homogenizer (D2400, Benchmark Scientific). Cell extracts were spun down at 21.000 × g and supernatants were transferred to 1.5 ml Eppendorf tubes and dry down using a SpeedVac Concentrator (SPD121P-115, Thermo Fisher Scientific). All samples were reconstituted in 50 μl of ultrapure HPLC grade water (Fisher Scientific). 2’3’cGAMP concentrations were measured using commercial ELISA kit (Cayman Chemical) and analyzed at 450 nm according to the manufacturers’ protocol using the FlexStation3 microplate reader. [2’3’cGAMP] was calculated from calibration curves generated using 2’3’cGAMP standards provided by the Kit (Cayman Chemical).
Quantitative PCR and IAV titers quantification.
Total RNA from cells was isolated using TRIZOL protocol. For total RNA extraction from lung, small pieces of lung tissue were collected in TRIZOL and grinded in a Precellys 24 Tissue Homogenizer using beads Precellys Lysing kits (both Bertin Instruments). Total mRNA was quantified in a Nanodrop 8000 spectrophotometer (Thermo Scientific) and cDNA was synthesized using the iScript cDNA synthesis kit (Bio-Rad). RT-qPCR was performed using the Maxima SYBR Green qPCR Master Mix (Thermo Fisher Scientific), acquired in a QuantStudio 3 PCR machine (Applied Biosystem-TermoFisher) and analyzed using the QuantStudio Design & Analysis Software (v 1.4.3). Transcripts amount was normalized to the expression of housekeeping genes using the 2−ΔCT method. The complete list of primers used in this study can be found in Supplemental Table 4.
Immunoblotting.
For p53 and phospho-p53 (S15) detection, total cell lysates were prepared in 1X Laemmli sample buffer (5% β-mercaptoethanol, 0.01% bromophenol blue, 10% glycerol, 2% SDS, 63 mM Tris-HCl, 1X PhosStop, and 1% protease inhibitor cocktail, pH 6.8, all from Sigma). Samples were sonicated for 10 min and heated at 95 °C for 10 min. For detection of LRRC8C, STIM2 and proteins from the STING pathway, total cell lysates were prepared in NP-40 lysis buffer (20 mM Tris-Cl, 150 mM NaCl, 5 mM EDTA, 1% Nonidet P40, 1 mM PMSF, 1 mM Na3VO4, 1% protease inhibitor cocktail, 1X PhosStop). Cell debris was removed by centrifugation at 15,000 × g for 5 min and protein extracts were treated with 4X Laemmli sample buffer (described above), heated at 95 °C for 5 min (except for LRRC8C protein detection) and spun down at 15,000 × g for 30s. Protein extracts were separated by SDS-PAGE using Novex™ WedgeWell™ 4 to 20% tris-glycine gel (Invitrogen) and transferred to polyvinylidene difluoride (PVDF) membranes. After blocking, membranes were incubated overnight at 4°C with the following primary antibodies: p53 rabbit mAb (D2H9O, 1:1000), phospho-p53-Ser15 rabbit polyclonal Ab (9284, 1:1000), STING rabbit mAb (D2P2F, 1:1000), phospho-STING-Ser365 rabbit mAb (D8F4W, 1:1000), STIM2 rabbit polyclonal Ab (4917S, 1:1000, all from Cell Signaling Technology), a custom-made rabbit anti-LRRC8C polyclonal antibody that recognizes the last 17aa at the C terminus of LRRC8C (1:1000, YenZym Antibodies), and a mouse anti-β-Actin monoclonal antibody (1:5000, 66009–1-Ig, proteintech) as loading control. For p53, phopsho-p53-Ser15, LRRC8C and actin, immunoreactive bands were detected after incubation with IRDye 800CW donkey anti-rabbit or IRDye 680RD donkey anti-mouse secondary antibodies (1:10,000, Licor) for 2h at room temperature, with an Odyssey Fc Western Blot Detection System (Licor Bioscience). For detection of proteins from the STING pathway, membranes were incubated with secondary antibodies coupled with horseradish peroxidase (A6154, Sigma-Aldrich, Cell Signaling Technology) and immunoreactive bands were visualized by enhanced chemiluminescence (Amersham 680-GE). Band densities were quantified and analyzed using ImageJ 1.52a.
VRAC current recordings.
T cells stimulated for 2 days with anti-CD3+CD28 were detached and cultured with 20 IU/ml hr-IL-2 prior patch clamp measurements. Cl− currents traces were acquired in the whole-cell patch clamp configuration using pClamp 9 and an Axopatch 200B amplifier (Molecular Devices, Sunnydale, CA). Only isolated individual cells were picked. Recordings were sampled at 2 kHz and filtered at 1 kHz. Pipette resistances were 2 MOhms and seal resistances were >1 GOhm. Cells were held at −70 mV and were depolarized to + 80 mV every 5s while washing in hypotonic solution. Voltage steps were from −70 mV to +140 mV at +10 mV voltage intervals for 40 ms. Voltage steps were recorded at 4 min after shift to hypotonic solution. All recordings were performed at room temperature (~23 °C). Hypotonic bath solution (~215 mOsm) contained 96 mM NaCl, 5.3 mM KCl, 0.8 mM MgSO4, 1 mM NaH2PO4, 1.8 mM Ca-acetate, and 5 mM HEPES (pH 7.4), and 100 mM mannitol was added to yield ~315 mOsm isotonic bath solution. Pipette solutions contained 120 mM CsASP, 10 mM Cs4BAPTA, 4 mM Mg-ATP, 2 mM MgCl2, 8 mM NaCl, and 10 mM HEPES, pH 7.2. VRAC inhibitor DCPIB (20 μM) was directly perfused onto individual cells using a multibarrel perfusion pipette.
Membrane potential (Vm) measurements.
For Vm measurements, cells were washed twice in isotonic solution and Gigaohm seals were stablished in isotonic buffer using the voltage clamp configuration with pClamp 9 software. Once seal was obtained, the amplifier was placed into track mode and Vm were recorded. T cells were allowed to dialyze for 2 min before recordings. Hypotonic solution and 1 μM thapsigargin were perfused onto the cells using a multibarrel pipette for 10 min before recording Vm. Isotonic and hypotonic bath solution composition were the same as described above for VRAC current recordings. Pipette solutions contained 62 mM K-gluconate, 38 mM KCl, 2 mM MgCl2, 8 mM NaCl, 145 μM CaCl2, HEDTA 100 μM, pH 7.2. The integrity of the Gigaohm seal was confirmed at the end of each experiment. Alternatively, Vm was measured in naïve and activated T cells using the Vm sensitive dye DiBAC4(3) (Thermo Fisher Scientific) in a FlexStation3. Briefly, naive CD4+ T cells were loaded with 2 μM DiBAC4(3) (Invitrogen) for 30 min in 2 mM Ca2+ Ringer solution (155 mM NaCl, 4.5 mM KCl, 2 mM CaCl2, 1 mM MgCl2, 10 mM D-glucose, and 5 mM HEPES) and plated on poly-L-lysine-coated translucent 96-well plates (BD Falcon) and incubated with 1 μg/ml biotin-conjugated anti-CD3ε Ab (145–2C11, BD Bioscience) for 15 min. Changes in Vm were induced either by TCR-crosslinking after addition of 1 μg/ml streptavidin (Invitrogen) or by 1 μM ionomycin stimulation (Invitrogen). For CD4+ T cells stimulated with anti-CD3+CD28 for 3 days, DiBAC4(3)-loaded cells were plated on poly-L-lysine-coated 96-well plates for 15 min and washed twice with Ca2+-free Ringer solution (155 mM NaCl, 4.5 mM KCl, 3 mM MgCl2, 10 mM D-glucose, and 5 mM HEPES ) containing 2 μM DiBAC4(3), and changes in Vm were detected after Ca2+ store depletion with 1 μM thapsigargin (EMD Millipore) stimulation or after activation of Ca2+ influx by addition of an equal volume of 2 mM Ca2+ Ringer solution to the cells to obtain a final extracellular [Ca2+] of 1 mM. DiBAC4(3) dye was kept at the same concentration of 2 μM throughout the experiment. Vm was measured as the ratio of fluorescence emission at 516 nm after excitation at 490 nm, and normalized to baseline DiBAC4(3) fluorescence in wild-type T cells.
Regulatory volume decrease.
Unstimulated and TCR-stimulated CD4+ T cells with anti-CD3+CD28 at the indicated time points were seeded onto UV-sterilized coverslips pre-coated with 0.01% poly-L-lysine (Sigma). T cells adhered on coverslips were loaded with 10 μM Calcein-AM (Life Technologies) for 30 min, and changes in calcein fluorescence (Ex: 488nm, Em: 510 nm) was analyzed using time-lapse imaging on an IX81 epifluorescence microscope (Olympus). Each individual cell was encircled by a region of interest. For measurements of RVD, baseline calcein fluorescence was acquired in isotonic solution (96 mM NaCl, 5.3 mM KCl, 0.8 mM MgSO4, 1 mM NaH2PO4, 1.8 mM Ca-acetate, 5 mM HEPES, and 100 mM mannitol, pH 7.4, ~315 mOsm) for 5 min followed by perfusion with hypotonic buffer (96 mM NaCl, 5.3 mM KCl, 0.8 mM MgSO4, 1 mM NaH2PO4, 1.8 mM Ca-acetate, and 5 mM HEPES, pH 7.4, ~215 mOsm) to induce hypotonic cell swelling and RVD for additional 35 min. Relative cell volume (F/F0) was measured as the ratio of fluorescence emission at 510 nm relative to baseline in isotonic buffer and analyzed using SlideBook imaging software v4.2 (Olympus). RVD measurements were represented as ΔPeak, area under the curve (AUC) and Time RVD50 (time to recover 50% of the volume upon hypoosmotic swelling) during the hypotonic challenge relative to baseline.
Intracellular Ca2+ measurements.
Naïve or stimulated CD4+ T cells for 3 days with anti-CD3+CD28 were loaded with 1 μM Fura-2-AM (Life Technologies) for 30 min in complete RPMI medium, and F340/380 ratios were measured with a FlexStation3. Briefly, Fura-2-AM-loaded CD4+ T cells were plated on poly-L-lysine-coated 96-well plates for 15 min, and incubated in Ca2+-free Ringer solution for 10 min before measurements of [Ca2+]i. Store depletion was induced by stimulating the cells with 1 μM thapsigargin (Invitrogen) in Ca2+-free Ringer solution and Ca2+ influx was induced after adding an equal volume of 2 mM Ca2+ Ringer solution to the cells. For stimulation by TCR cross-linking, T cells were incubated with 1 μg/ml biotin-conjugated anti-CD3ε Ab (145–2C11; BD Biosciences) at the time of Fura-2 loading and stimulated by addition of 1 μg/ml streptavidin (Invitrogen) in 2 mM Ca2+ Ringer buffer, followed by addition of 1 μM ionomycin. Alternatively, cells plated on poly-L-lysine-coated 96-well plates were incubated in either 2 mM Ca2+ Ringer, isotonic or hypotonic solutions described above, and Ca2+ signals were triggered by addition of 1 μM thapsigargin. [Ca2+]i, was represented as AUC for the store depletion and Ca2+ influx at the indicated time points.
ICT gene expression analysis.
Gene expression RNA-Seq datasets for the Immunological Genome Project17 and Haemopedia18 were downloaded from the Sequence Read Archive (SRA). FASTQ files were trimmed with Trimmomatic/0.36 and aligned with STAR/2.7.3a to the mouse GRCm38.85/mm10 genome assembly. Datasets for BioGPS20 and Fantom519 were downloaded from the NCBI Gene Expression Omnibus (GEO). Differential expression was performed on median-of-ratios-normalized datasets using DESeq261. Expression correlations were performed with TPM-normalized datasets. Information linking each gene to ICT function was extracted from GeneCards, UniProt KB, Swiss-Prot, OMIM and PubMed databases. A total of 763 ICT/ICT regulatory protein genes annotated in the mammalian genome based on ICT function were used to filter ICT genes from gene expression data sets. PCA analysis of the ImmGen RNA-Seq dataset was performed using MATLAB(R2020a). To identify specific ICTs in T cells, we screened for ICT genes whose expression is 1) 1.5-fold higher in T cells compared to the average of all other immune cells (adj-P value < 0.05) and 2) greater than the 50th percentile of expression among ICT genes in T cells. Heatmaps of selected ICT genes were created using the conditional formatting tool in Microsoft Excel. Highest and lowest expression for each gene (row min. and row max.) were displayed as red or blue color, respectively.
ChIP-Seq and whole-genome co-expression network analyses.
Reads from mouse23 and human62 ChIP-seq data were mapped to the mouse GRCm38.85/mm10 or human GRCh37/hg19 genome assembly, respectively, with Bowtie2 software package (v. 2.3.1). Binding peaks were identified using the model-based analysis of ChIP-Seq method (MACS, v2.1.2) and visualized using Integrative Genome Viewer (IGV_2.4.16) software. Whole-genome co-expression analysis for each Lrrc8 gene was performed by correlating their gene expression across 96 different cell types to the whole-genome using the BioGPS Affymetrix GeneChip Mouse Genome 430 2.0 array (Molecular Genomics Core, GSE10246). Genes with a Pearson’s r = 0.5 and P < 0.001 to Lrrc8 genes were considered as co-expressed and were subjected to pathway analyses using ingenuity’s pathway analysis (IPA, Qiagen), database for annotation, visualization, and integrated discovery (DAVID, https://david.ncifcrf.gov) and gene set enrichment analysis (GSEA, https://software.broadinstitute.org/gsea/index.jsp).
RNA sequencing.
Purified CD4+ T cells from three wild-type and three Lrrc8c−/− mice were isolated from the spleen and left unstimulated or stimulated with Ab-bound anti-CD3+CD28 in 12-well plates for 1 and 2 days. Additionally, wild-type CD4+ T cells were stimulated in the presence and absence of 20 μM DCPIB. Total RNA was extracted from 1 × 106 cells using TRIZOL reagent (Life Technologies), further purified with the RNeasy Micro RNA Isolation Kit (Qiagen) and analyzed as previously described63. Briefly, RNA quality was analyzed on a Bioanalyzer 2100 (Agilent) using a PICO chip. Samples with an RNA integrity number (RIN) of >9.0 were used for library preparation. RNA-seq libraries were prepared using the TruSeq RNA sample prep v2 kit (Illumina), starting from 100 ng of DNAse I (Qiagen) treated total RNA, following the manufacturer’s protocol with 15 PCR cycles. The amplified libraries were purified using AMPure beads (Beckman Coulter), quantified by Qubit 2.0 fluorometer (Life Technologies), and visualized in an Agilent Tapestation 2200. The libraries were pooled equimolarly, and loaded on the HiSeq 2500 Sequencing System (Illumina) and run as single 50 nucleotide reads63. For gene expression analysis, reads were aligned to the mouse reference genome (GRCm38.85/mm10) using STAR aligner (v2.5.0c)64. Alignments were guided by a Gene Transfer Format file (GTF GRCm38.85). The mean read insert sizes and their standard deviations were calculated using Picard tools (v.1.126) (http://broadinstitute.github.io/picard). The read count tables were generated using HTSeq (v0.6.0)65, normalized based on their library size factors using DEseq261, and differential expression analysis was performed. The Read Per Million (RPM) normalized BigWig files were generated using BEDTools (v2.17.0)66 and bedGraphToBigWig tool (v4). Differences in gene expression were considered significant if padj < 0.1. Gene expression signatures and canonical pathway analyses were performed using IPA, DAVID, and GSEA. Heatmaps of selected genes were created using the conditional formatting tool in Microsoft Excel. Highest and lowest expression for each gene (row min. and row max.) were displayed as red or blue color, respectively.
Data analysis and statistics.
Data are expressed as mean ± s.e.m. No statistical methods were used to predetermine sample sizes, but our sample sizes are similar to those reported in previous publications55,56. Data distribution was assumed to be normal, but this was not formally tested. Unless when restricted by the genotype, animals and cell plates were assigned randomly to experimental conditions. Data collection and analysis were not blinded to the conditions of the experiments. No data exclusion was performed. Statistical significance was determined by using two-tailed, unpaired Student’s t-test, paired t-test, two-tailed Mann Whitney test, one-way analysis of variance (ANOVA), and 2-way ANOVA corrected for Sidak’s or Dunnett’s multiple comparisons tests, as mentioned in the figure legends. Statistical analyses were performed using GraphPad Prism software (v 9.0.2). A value of two-tailed P < 0.05 was considered statistically significant.
Data availability.
The RNA-Seq data generated in this study have been deposited in the GEO database under accession number GSE163679. Additional available gene expression data sets and ChiP-Seq data used in this study were downloaded from GEO using the following accession numbers: GSE127267 for mouse RNA-seq data from the Immunological Genome Project (www.immgen.org) 17. GSE116177 for mouse RNA-Seq data from Haemopedia (www.haemosphere.org) 18. GSE10246 for mouse microarray data from BioGPS (www.biogps.org) 20. GSE49834 for human CAGE-Seq data from Fantom5 (https://fantom.gsc.riken.jp/5/) 19. GSE36890 for mouse RNA-Seq data and STAT5 ChIP-Seq data using T cells from Stat5a-Stat5b-DKI N-domain mutant mice in which STAT5 proteins form dimers but not tetramers23. GSE110370 for gene expression analysis and p53 ChIP-seq data from PHA-stimulated human T-lymphocytes treated with nutlin-3 62. Source data for each figure, extended data figure, and supplemental figure are provided with this paper.
Extended Data
Extended Data Figure 1. Transcriptomic analysis of the ion channelome in immune cells identifies LRRC8C and other specific ICTs in T cells.
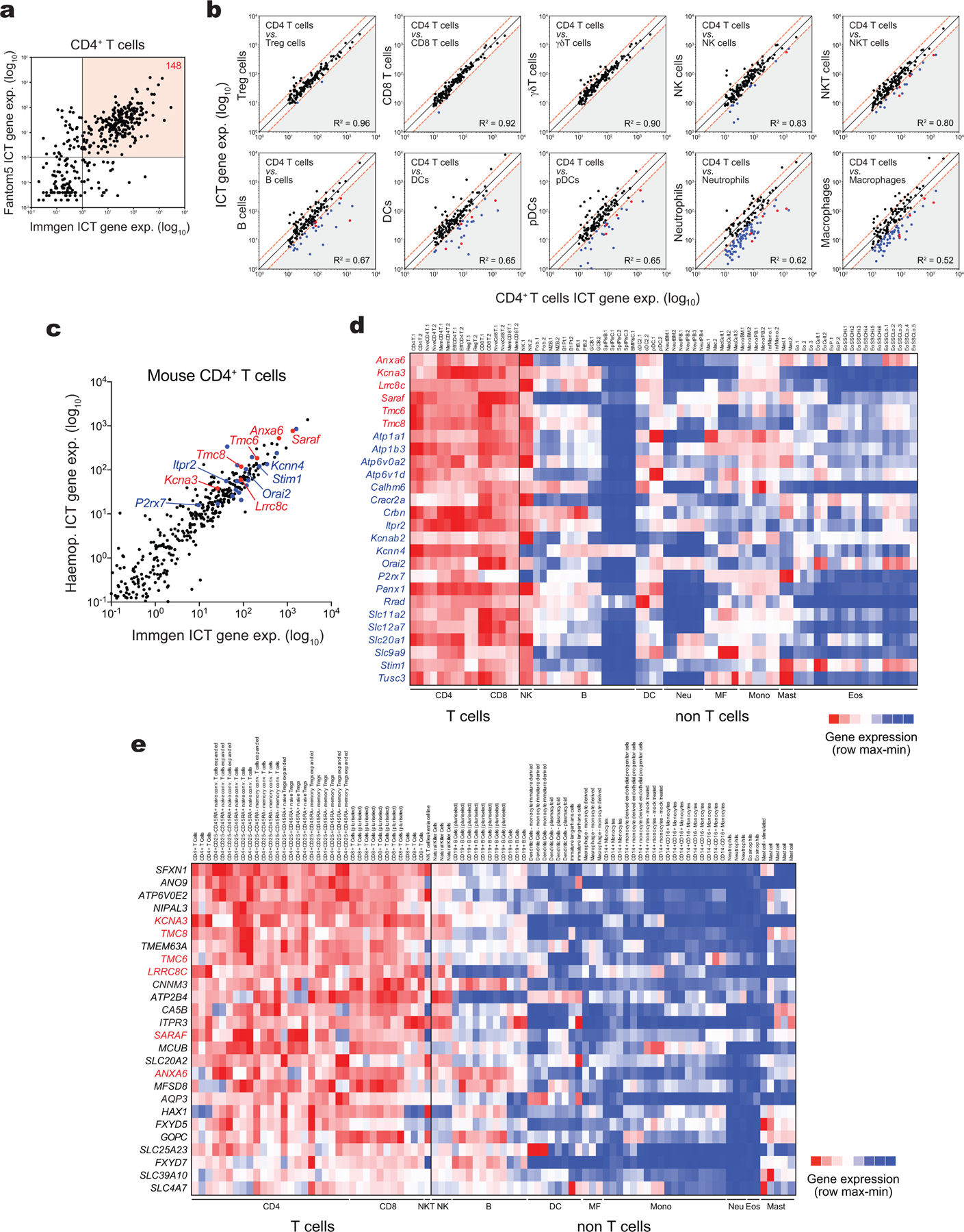
(a) Correlation analysis of ICT genes in CD4+ T cells from mouse ImmGen RNA-Seq and human Fantom5 CAGE-Seq databases. 148 ICTs are highly expressed in both human and mouse T cells (red quadrant). (b) Correlation analysis of 148 highly expressed ICT genes in CD4+ T cells and other immune cells from mouse ImmGen RNA-Seq. ICT genes on the bottom right (grey triangle) are 2-fold higher expressed in CD4+ T cells compared to other immune cells. CD4-specific ICT genes highlighted in blue are shared in both mouse ImmGen and Haemopedia datasets, and red are shared in ImmGen, Haemopedia, and human Fantom5 datasets. (c) Correlation analysis of ICT genes in mouse CD4+ T cells from ImmGen RNA-Seq and Haemopedia RNA-Seq databases. Most specific ICTs are highlighted in red and known ICTs to play a role in T cells shared in both mouse ImmGen and Haemopedia datasets are highlighted in blue. (d,e) Expression profile of differentially expressed ICTs in CD4+ T cells from (d) Haemopedia RNA-Seq and (e) Fantom5 CAGE-Seq databases. Color coding: high (red) and low (blue) relative mRNA expression per row.
Extended Data Figure 2. VRAC channels in mouse T cells are composed by LRRC8C and LRRC8A.
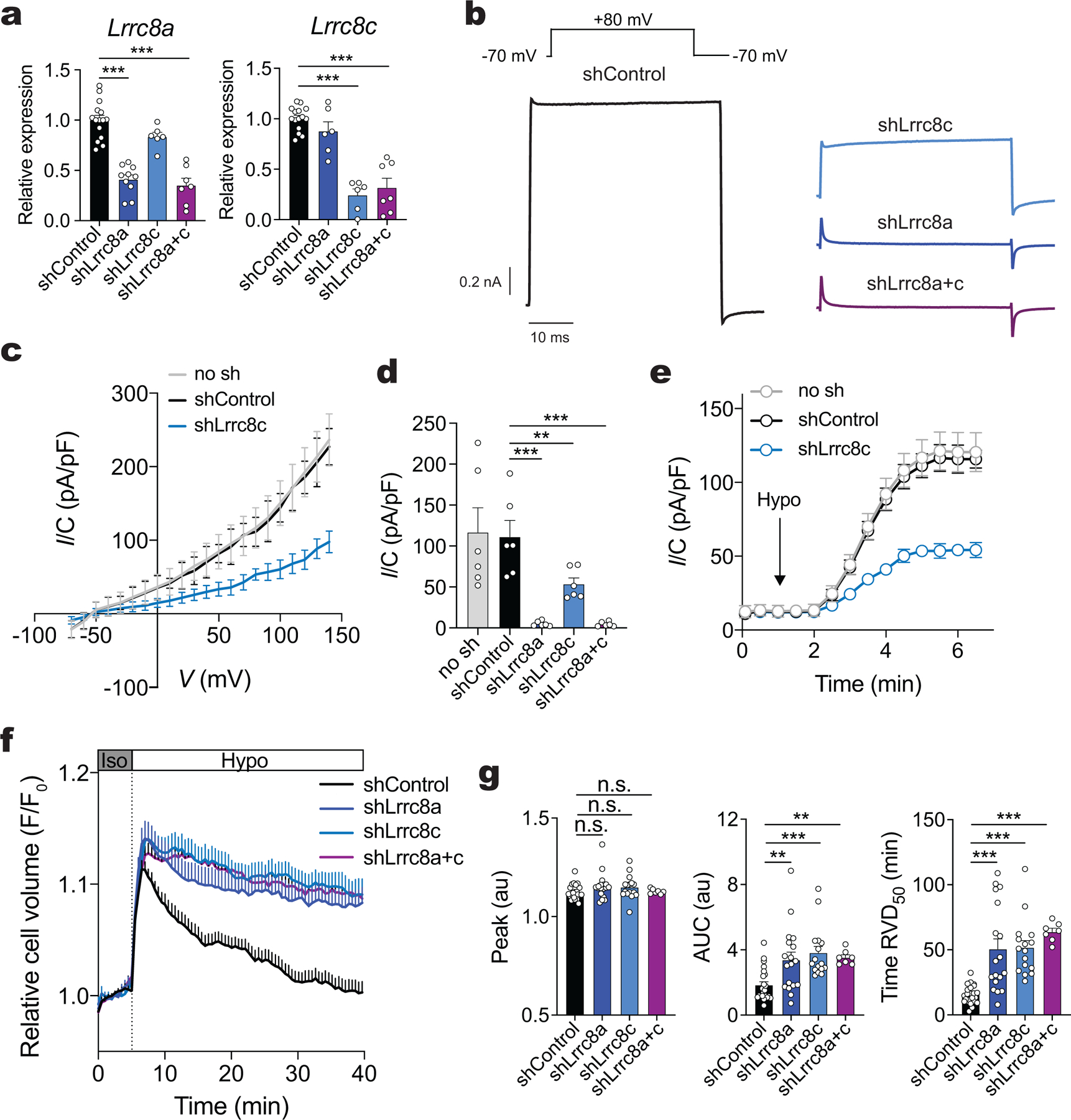
(a) Relative expression of Lrrc8a and Lrrc8c mRNA from CD4+ T cells transduced with shRNAs targeting Lrrc8a, Lrrc8c, or both Lrrc8a/c, and measured by RT-qPCR 3 days after transduction. shRNA targeting non-mammalian gene Renilla Luciferase (Ren.713) was used as shControl. Gapdh mRNA was used as housekeeping control and relative expression was normalized to shControl. Data are the mean ± s.e.m. from 6 independent experiments and mice (representing 15, 6, 6 and 7 transductions of T cells with shControl, shLrrc8a, shLrrc8c, and shLrrc8a/c, respectively). (b-e) Volume-regulated anion currents (IVRAC) from CD4+ T cells transduced with shRNAs. (b) IVRAC from T cells shown in (a) and measured by patch clamping in whole-cell configuration (representative traces from at least 6 cells per shRNA from 2 independent experiments). Recording protocol (top): T cells were held at −70 mV and were depolarized to +80mV every 5s in hypotonic solution (~215 mOsm). Current densities as a function of voltage (c) and extracted at +80 mV (d) at the end of each test pulse from experiment shown in (b). (e) Average current densities at −70 mV and +80 mV over time induced by hypotonic (Hypo) solution (applied at the arrow) in shRNA transduced CD4+ T cells (mean ± s.e.m., 6–10 cells per shRNA from 2 independent experiments). Unapparent error bars are smaller than symbols. (f,g) RVD traces (f) and quantification of maximum peak, area under the curve (AUC) and time to RVD50 (g) in CD4+ T cells transduced with shRNAs and subjected to hypoosmotic swelling. Data are the mean ± s.e.m. from 23, 18, 16 and 7 traces for shControl, shLrrc8a, shLrrc8c, and shLrrc8a/c, respectively, and pooled from 3 independent experiments. Statistical analysis by two-tailed, unpaired Student’s t test in (a and g), and one-way ANOVA with Dunnett’s multiple comparisons test in (d). Not significant (n.s.) P > 0.05, **P<0.01 and ***P<0.001.
Extended Data Figure 3. Lrrc8c−/− mice have normal T cell development.
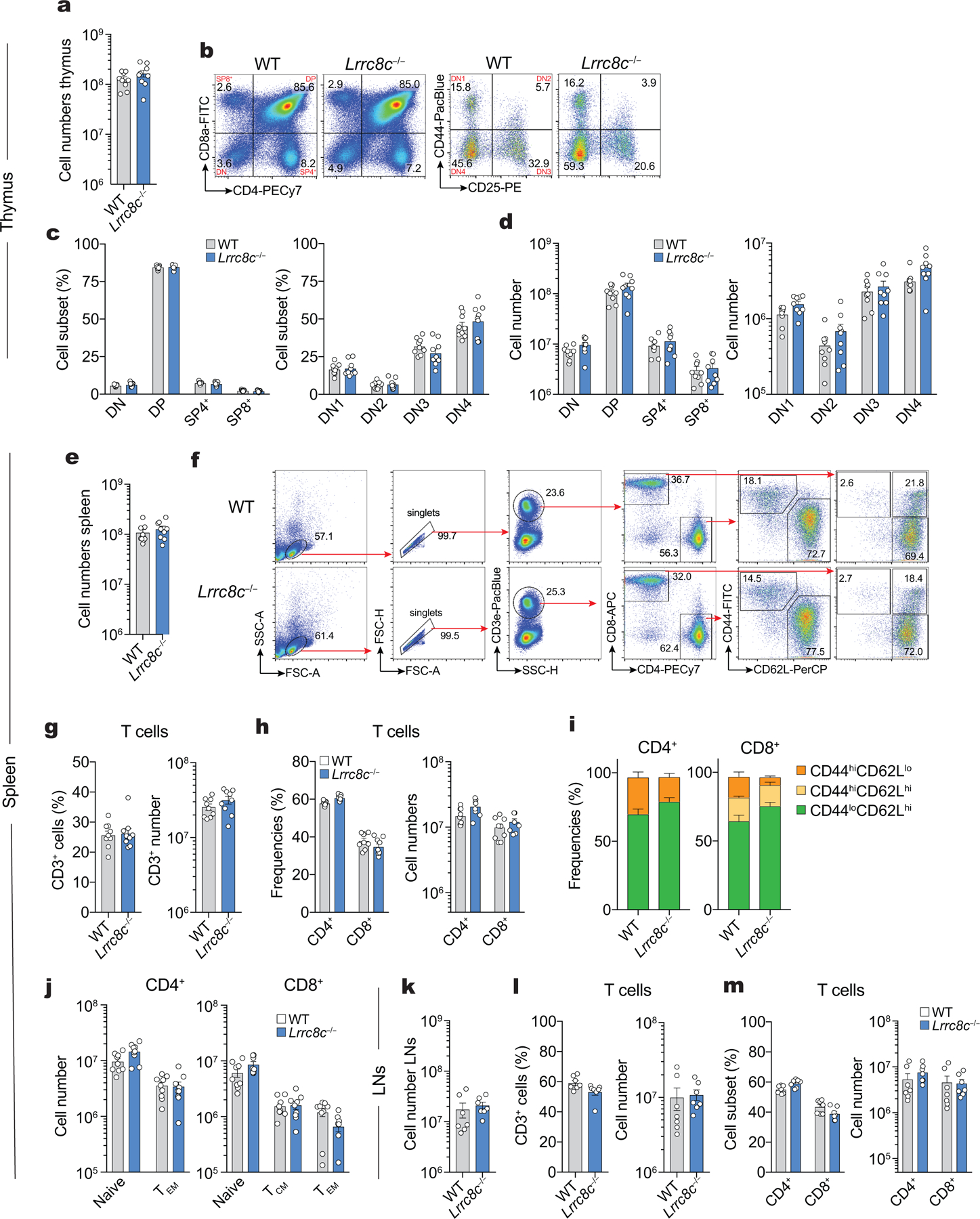
(a-d) Cell numbers and proportion of thymocytes from WT and Lrrc8c−/− mice. (a) Absolute cell numbers, (b) representative flow cytometry plots showing the proportion (c) and numbers (d) of thymocyte subsets DN1, DN2, DN3, DN4, DP, SP4+ and SP8+ cells. (e-j) Cell numbers and proportion of splenocytes from WT and Lrrc8c−/− mice. (e) Absolute cell numbers, (f) representative flow cytometry plots showing the gating strategy to identify the different subsets of T cells in the spleen from WT and Lrrc8c−/− mice. Proportion and absolute cell numbers of (g) CD3+ T cells, (h) CD4+ and CD8+ T cells, and (i,j) naïve CD44loCD62Lhi, T central memory (TCM) CD44hiCD62Lhi, and T effector memory (TEM) CD44hiCD62Llo T cell subsets in both CD4+ and CD8+ T cells. (k-m) Cell number and proportion of T cells in the lymph nodes (LNs) from WT and Lrrc8c−/− mice. (k) Absolute cell numbers, and proportion and absolute cell numbers of (l) CD3+ T cells, and (m) CD4+ and CD8+ T cells. Data represent mean ± s.e.m. of 9 mice per genotype (a-j) and 7 mice per genotype (k-m), pooled from at least 4 independent experiments.
Extended Data Figure 4. p53 control the proliferation and survival in T cells.
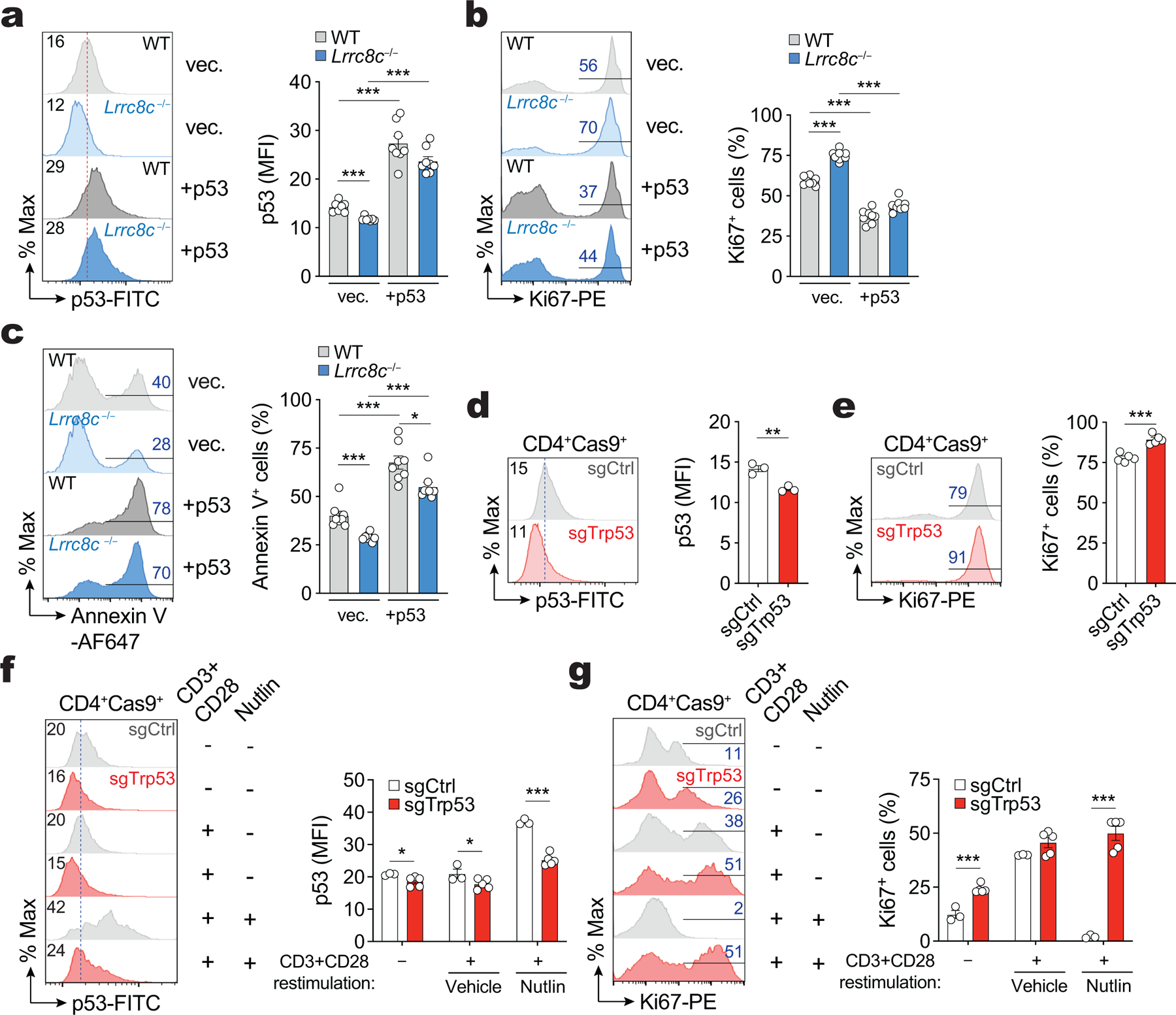
(a-c) Protein expression of p53 (a), Ki67 (b), and apoptosis determined by annexin V staining (c) in WT and Lrrc8c−/− CD4+ T cells after 3 days of retroviral transduction. Representative flow cytometry plots (left) and quantification (right). Data represent the mean ± s.e.m. of 8 mice per genotype, pooled from 2 independent experiments. (d,e) Protein expression of p53 (d) and Ki67 (e) in CD4+Cas9+ T cells transduced with sgRNAs targeting p53 (Trp53) after 3 days of retroviral infection. Representative flow cytometry plots (left) and quantification (right). Data represent the mean ± s.e.m. of 3 (in d) and 5 (in e) mice, pooled from 2 independents. (f,g) Protein expression of p53 (f) and Ki67 (g) in T cells shown in (d,e) after restimulation for additional 3 days with anti-CD3+28 dynabeads and treated or not with idasanutlin (nutlin). Representative flow cytometry plots (left) and quantification (right). Data represent the mean ± s.e.m. of 3 and 5 mouse donors for sgControl and sgTrp53, respectively, pooled from 2 independents. Statistical analysis by two-tailed, unpaired Student’s t test. *P<0.05 and ***P<0.001.
Extended Data Figure 5. VRAC inhibitor DCPIB suppresses p53 signaling in activated T cells.
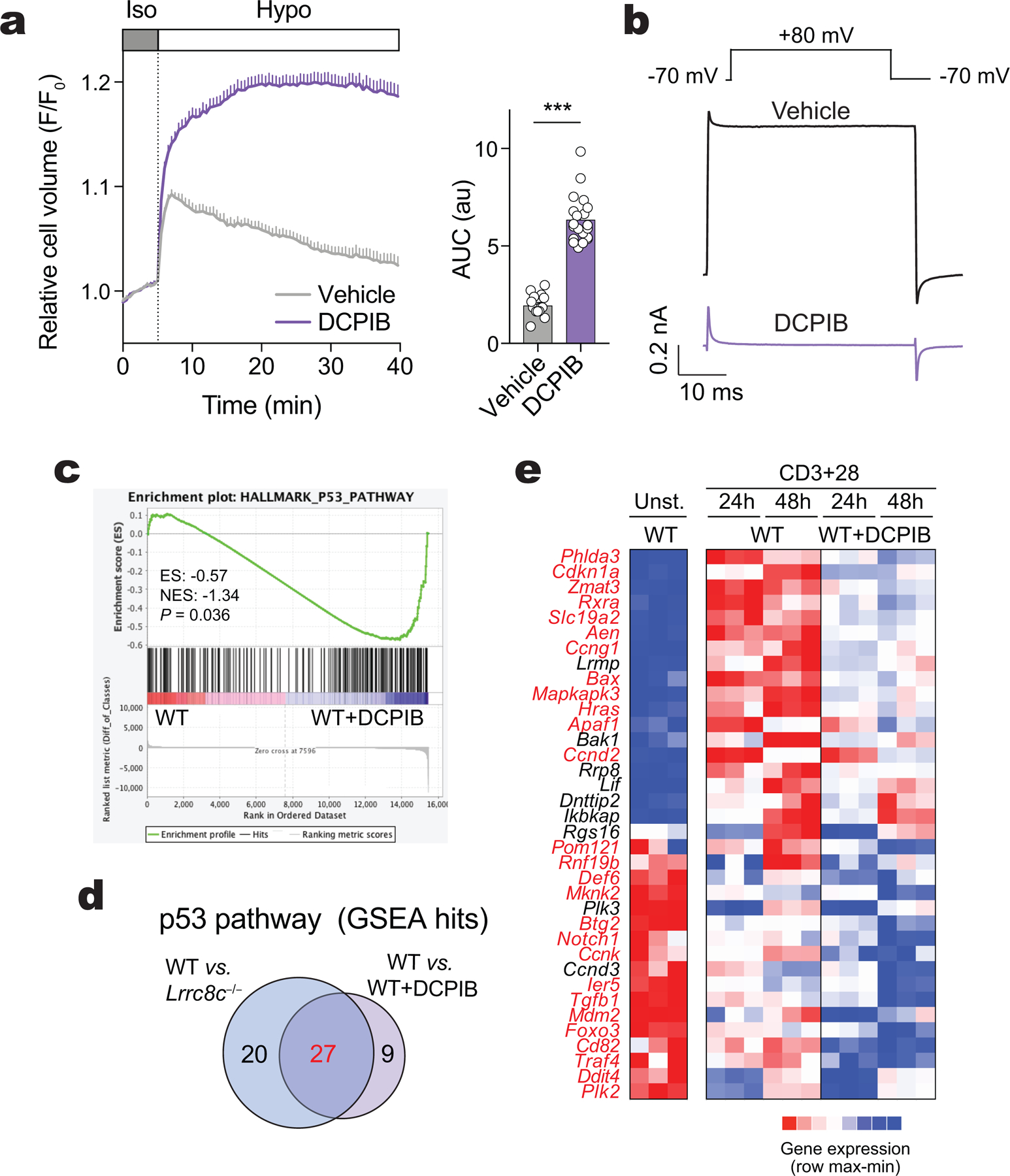
(a) Averaged traces (left) and quantification (right) of RVD (AUC) in CD4+ T cells stimulated with anti-CD3+28 for at least 3 days and subjected to hypotonic solution after treatment for 30 min with 20 μM DCPIB (data are the mean ± s.e.m. of 12 and 20 traces for vehicle and DCPIB treated cells, respectively, pooled from at least 5 independent experiments). (b) IVRAC traces from CD4+ T cells stimulated for 2d with anti-CD3+28 and pre-treated or not with 20 μM DCPIB, measured by patch clamping in whole-cell configuration. Representative traces from 10 cells (vehicle) and 5 cells (DCPIB) from at least 3 independent experiments. Recording protocol (top): T cells were held at −70 mV and were depolarized to +80mV every 5s in hypotonic solution (~215 mOsm). (c) GSEA of RNA-Seq data from WT CD4+ T cells treated or not with 20 μM DCPIB identifies DEGs associated with p53 pathway after anti-CD3+28 stimulation. (d) Venn-diagram showing number of DEGs related to the p53 pathway between WT vs Lrrc8c−/− and WT vs WT + DCPIB CD4+ T cells after anti-CD3+28 stimulation for 1 and 2 days. (e) Heat map of DEGs associated with p53 pathway identified by GSEA in (c). Color coding: high (red) and low (blue) relative mRNA expression per row. DEGs highlighted in red are shared between WT vs WT + DCPIB and WT vs Lrrc8c−/− CD4+ T cells. Statistical analysis in (a) by two-tailed, unpaired Student’s t test. ***P<0.001.
Extended Data Figure 6. p53 controls CD80 expression in T cells.
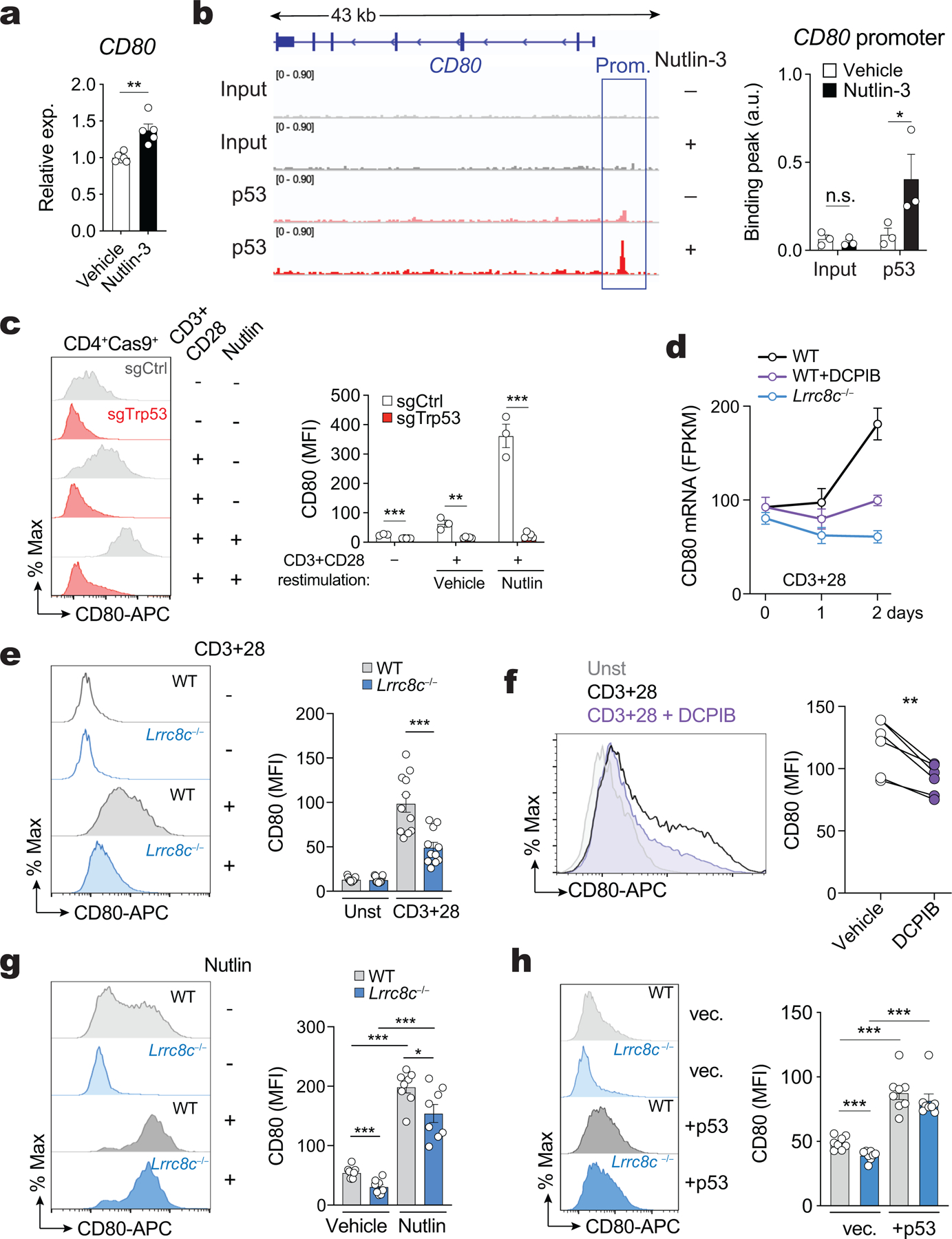
(a) CD80 mRNA expression in human T cells from healthy volunteers stimulated with phytohemagglutinin for 3 days and treated with nutlin-3 for 24h. CD80 mRNA expression based on microarray data from GSE110369 (Ref62). (b) p53 binding to the CD80 gene locus in human T cells treated as in (a) and analyzed by ChIP-Seq (data source: GSE110368)62. Representative binding peaks in the CD80 promoter (left) and quantification (right). (c) CD80 expression in CD4+Cas9+ T cells transduced with sgRNAs targeting p53 (Trp53) and restimulated for 3 days with anti-CD3+28 and treated or not with nutlin. Representative flow cytometry plots (left) and quantification (right) of mean fluorescence intensities (MFI) of CD80. Data are from 3 and 5 mice for sgControl and sgTrp53, respectively, pooled from 2 independent experiments. (d) Cd80 mRNA expression in CD4+ T cells of WT or Lrrc8c−/− mice stimulated with anti-CD3/28 for 1 and 2 days. T cells from WT mice were treated or not with DCPIB for the duration of T cell stimulation. mRNA expression based on RNA-Seq data (compare with Fig. 3). Data are from 3 mice per genotype and treatment. (e) CD80 expression in WT and Lrrc8c−/− CD4+ T cells before and after stimulation with anti-CD3+28 for 3 days. Representative flow cytometry plots (left) and quantification (right) from 11 mice per genotype, pooled from 4 independent experiments. (f) CD80 expression in WT CD4+ T cells treated with 20 μM DCPIB for 3 days following anti-CD3+28 stimulation. Representative overlay histograms (left) and quantification (right) of CD80 expression from 6 mice per condition. (g,h) CD80 cell expression in WT and Lrrc8c−/− CD4+ T cells upon stimulation with anti-CD3+28 for 3 days and treated or not with nutlin (in g), or after 3 days of retroviral transduction with empty vector or p53 (in h). Representative flow cytometry plots (left) and quantification (right) of 8 mice per genotype, pooled from 4 and 2 independent experiments in (g) and (h), respectively. All data are mean ± s.e.m. and were analyzed by two-tailed, unpaired Student’s t test (a-c,e,g,h) and by two-tailed, paired t test (f). Not significant (n.s.) P > 0.05, *P<0.05, **P<0.01 and ***P<0.001.
Extended Data Figure 7. Activation of STING in T cells depends on the channel function of LRRC8C.
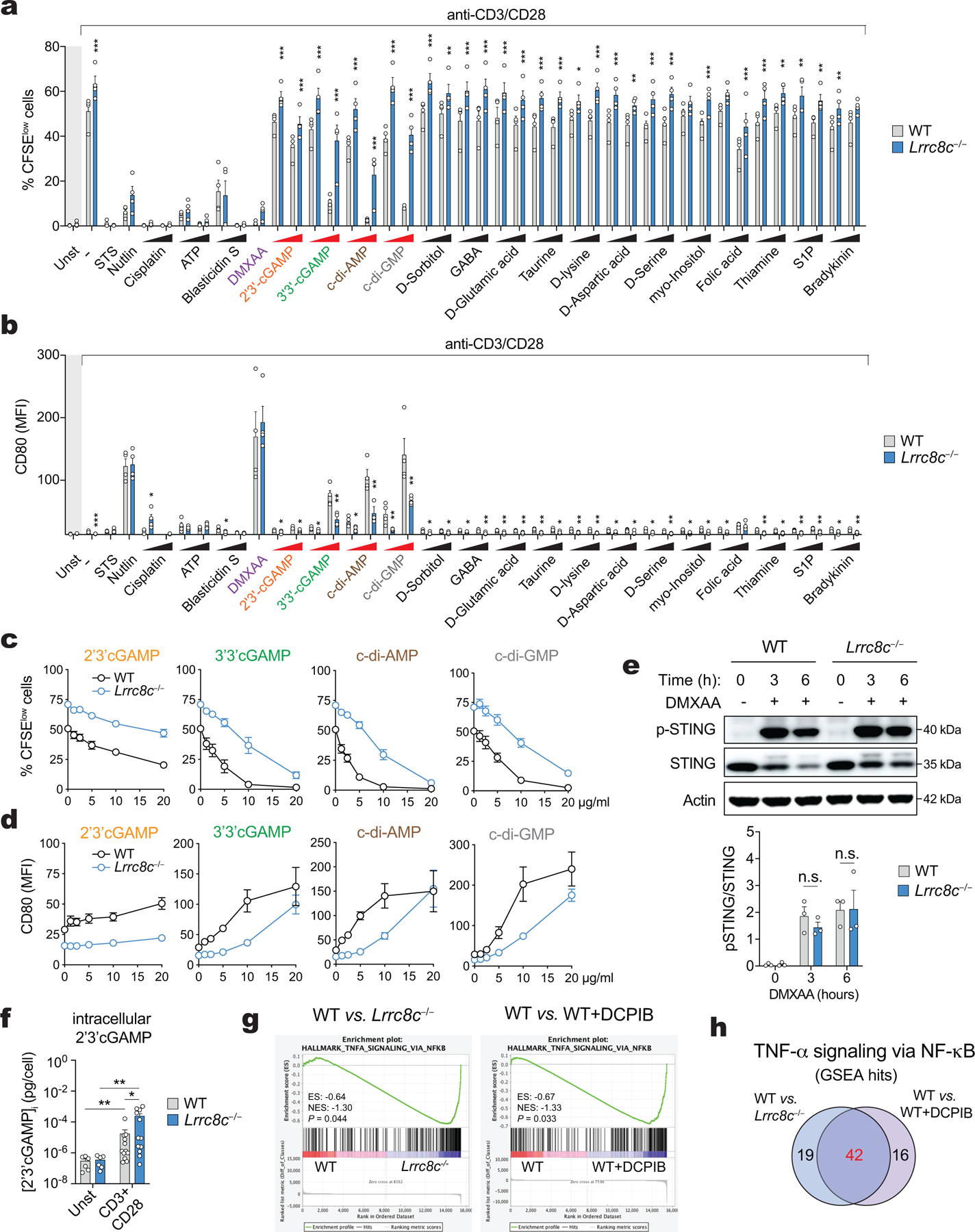
(a,b) Compound screening to identify substrates for LRRC8C in T cells. Flow cytometry analysis showing (a) the CFSE dilution and (b) CD80 surface expression of CD4+ T cells stimulated for 2 days with anti-CD3+28 dynabeads and treated with 19 different substrates known to be transported by VRAC channels. The concentration of tested compounds is: 3 and 10 μg/ml for 2’3’cGAMP, 3’3’cGAMP, c-di-AMP, and c-di-GMP; 5 and 10 μg/ml Blasticidin S, 0.1 and 1 μM sphingosine 1-phosphate (S1P), 1 and 10 μM bradykinin acetate, 50 and 100 μM cisplatin, 100 and 200 μM folic acid, 100 and 500 μM GABA, myo-inositol and thiamine hydrochloride; 250μM and 1 mM ATP, D-glutamic acid, taurine, D-lysine, D-aspartic acid, D-serine; 500μM and 1 mM D-sorbitol. The concentration of positive controls that lack requirement for VRAC transport are: 3 μg/ml DMXAA, 1 μM staurosporine (STS), and 5 μM idasanutlin (Nutlin). Data are from 4 mice per genotype, pooled from 2 independent experiments. Area highlighted in light grey represents unstimulated conditions. (c,d) Dose-dependent effects of CDNs on (c) proliferation measured by CFSE dilution and (d) CD80 expression in WT and Lrrc8c−/− CD4+ T cells stimulated for 2 days with anti-CD3+28. Data are from 6 mice per genotype and treatment, pooled from 3 independent experiments. (e) Immunoblots of total and p-STING (S366) protein in WT and Lrrc8c−/− CD4+ T cells activated for 2 days with anti-CD3+28 and treated with 3 μg/ml DMXAA at the indicated time points. Actin was used as loading control. Representative blot (top) and quantification (bottom) from at least 2 independent experiments. (f) Intracellular concentration of 2’3’ cGAMP in T cells stimulated with anti-CD3+28 for 12–72 hours and measured by ELISA. Data are from 6 (unstimulated) and 12 (stimulated) T cell samples, pooled from 12 mice per genotype and 2 independent experiments. (g) GSEA of RNA-Seq data from WT, Lrrc8c−/−, and WT + DCPIB-treated CD4+ T cells identifies DEGs associated with TNF-α signaling via NFκB pathway after anti-CD3+28 stimulation. (h) Venn-diagram showing number of DEGs related to the TNF-α signaling via NFκB pathway between WT vs. Lrrc8c−/− and WT vs. WT + DCPIB CD4+ T cells after anti-CD3+28 stimulation for 1 and 2 days. All data are the mean ± s.e.m. and were analyzed by two-tailed, unpaired Student’s t test. Not significant (n.s.) P > 0.05, *P<0.01, **P<0.01 and ***P<0.001.
Extended Data Figure 8. Inhibition of STING increases proliferation and decreases apoptosis, p53 and CD80 expression in T cells.
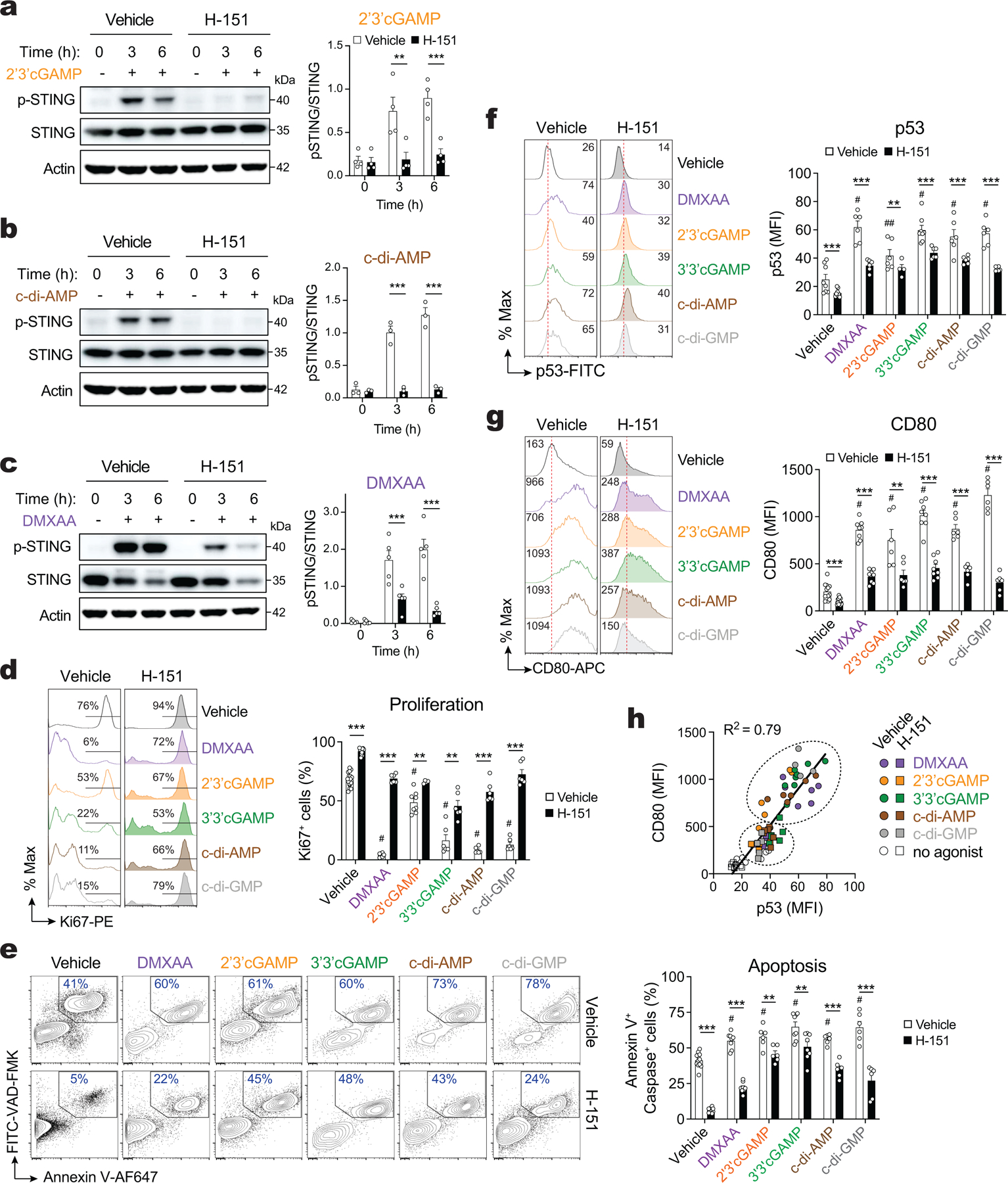
(a-c) Immunoblots of total and phospho-STING (S366) protein expression in WT CD4+ T cells activated for 2 days with anti-CD3+28 and treated or not with the STING inhibitor H-151. Cell were stimulated with 10 μg/ml 2’3’cGAMP (a), 5 μg/ml c-di-AMP (b) or 3 μg/ml DMXAA (c) for 3–6 hours. Actin was used as loading control. Representative blots (left) and quantification (right) of at least 3 independent experiments and 4, 3, and 5 mice for 2’3’cGAMP, c-di-AMP, and DMXAA treatment, respectively. (d-g) Flow cytometry analysis of Ki67 expression (d), apoptosis measured by annexin V and active caspase (e), p53 (f), and CD80 expression (g) in WT CD4+ T cells stimulated for 3 days with anti-CD3+28 and treated or not with STING agonists and pre-treated or not with the STING inhibitor H-151. Representative flow cytometry plots (left) and quantification (right) of at least 6 mice per treatment, pooled from 4–6 independent experiments and shown as mean ± s.e.m. (h) Correlation analysis of CD80 and p53 expression in T cells stimulated and treated as shown in (f,g). Statistical analysis in (a-g) by two-tailed, unpaired Student’s t test. **P<0.01 and ***P<0.001. ##P < 0.01 and #P<0.001 between STING agonists vs. vehicle untreated.
Extended Data Figure 9. Deletion of p53 protects T cells from STING-mediated proliferation arrest.
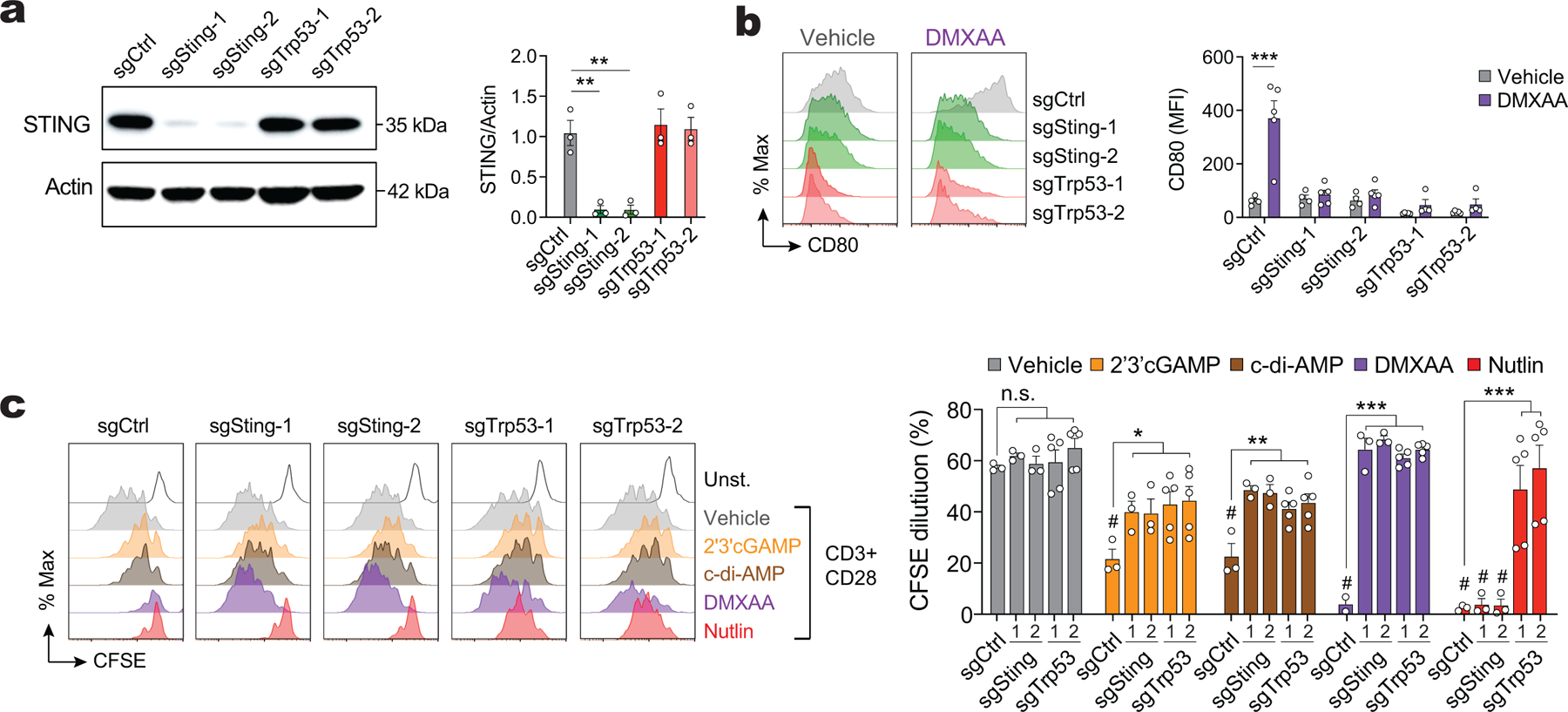
(a) Immunoblots of STING in CD4+Cas9+ T cells transduced with sgRNAs targeting STING and p53 at 4 days after retroviral infection. Actin was used as loading control. Representative blots (left) and quantification (right) represented as the mean ± s.e.m. of n = 3 mice and 2 independent experiments. (b) CD80 expression in CD4+Cas9+ T cells transduced with sgRNAs targeting STING and p53, restimulated for 2 days with anti-CD3+28 dynabeads and treated or not (vehicle) with 3 μg/ml DMXAA. Representative flow cytometry plots (left) and quantification (right). Data represent the mean ± s.e.m. of at least 3 mice, pooled from 2 independent experiments. (c) CFSE dilution in CD4+Cas9+ T cells transduced with sgRNAs targeting STING and p53, restimulated for 3 days with anti-CD3+28 dynabeads and treated or not (vehicle) with STING agonists and idasanutlin (nutlin). Representative flow cytometry plots (left) and quantification (right) of the frequency of proliferating cells. Data represent the mean ± s.e.m. of 3–5 mice, pooled from 2 independent experiments. Statistical analysis by two-tailed, unpaired Student’s t test (a,b) and 2-way ANOVA with Dunnett’s multiple comparisons test (c). Not significant (n.s.) P > 0.05, *P<0.05, **P<0.01 and ***P<0.001. #P<0.001 between treatment vs vehicle untreated.
Extended Data Figure 10. LRRC8C-STING-p53 signaling modulates Ca2+ influx in T cells.
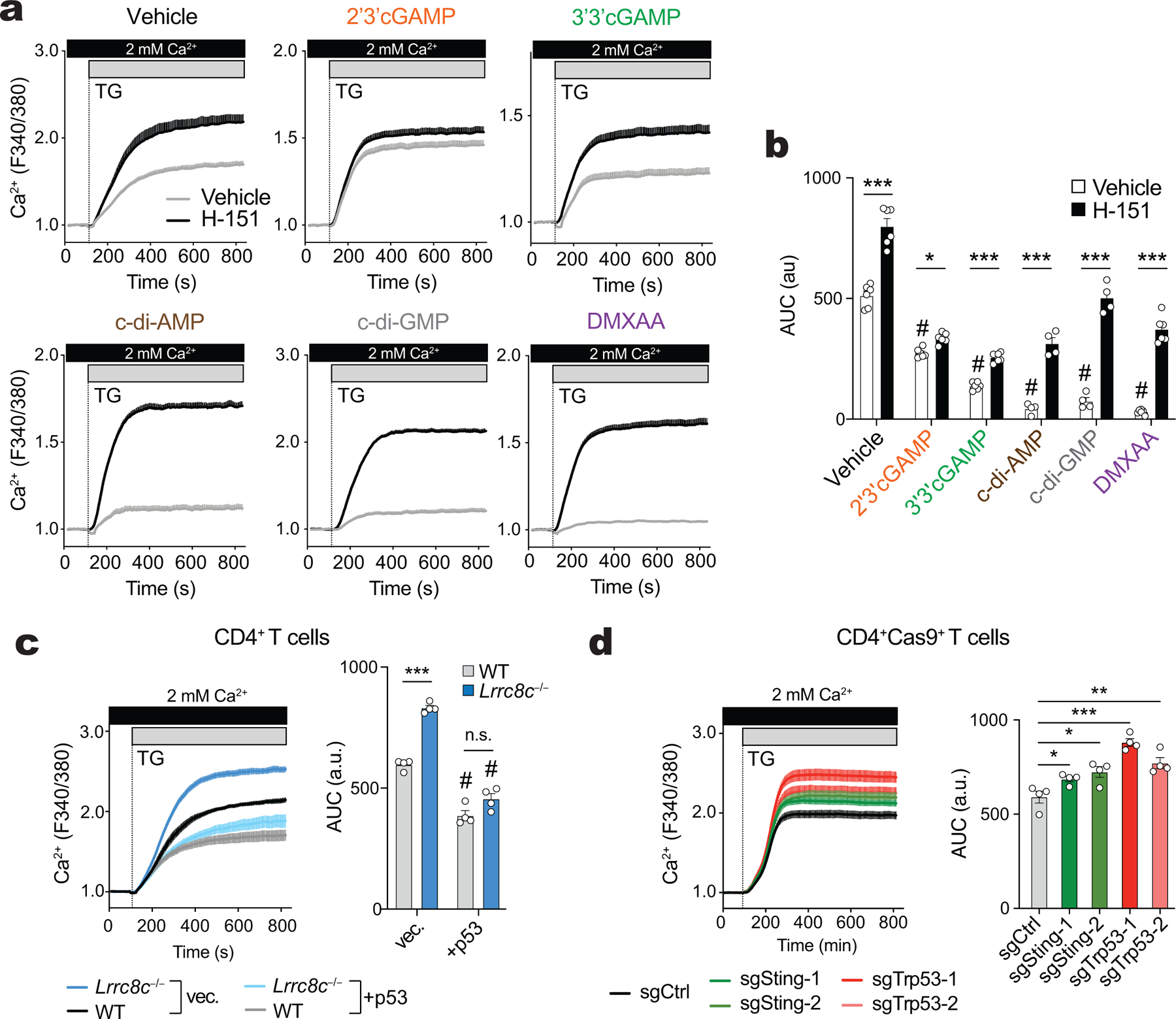
(a,b) Cytosolic Ca2+ signals in WT T cells stimulated for 3 days with anti-CD3+28 and treated or not with the STING inhibitor H-151 and STING agonists. T cells were stimulated with thapsigargin (TG) in Ca2+-containing Ringer buffer. Averaged Ca2+ traces (a) and area under the curve (AUC) following TG treatment (b). Data are the mean ± s.e.m. of 6 mice (vehicle, DMXAA, 2’3’cGAMP, 3’3cGAMP) or 4 mice (c-di-AMP, c-di-GMP) pooled from 2–3 independent experiments. (c) Cytosolic Ca2+ signals in WT and Lrrc8c−/− T cells 3 days after retroviral transduction with p53 using a similar protocol as in (a). Averaged Ca2+ traces (left) and quantification of the AUC (right) following TG treatment. Data are the mean ± s.e.m. of 4 mice per genotype and treatment, pooled from 2 independent experiments. (d) Cytosolic Ca2+ signals in CD4+Cas9+ T cells transduced with sgRNAs targeting STING and p53 three days after retroviral transduction using a similar protocol as in (a). Averaged Ca2+ traces (left) and quantification of the AUC (right) after TG treatment. Data are the mean ± s.e.m. of 3 mice pooled from 2 independent experiments. Statistical analysis by two-tailed, unpaired Student’s t test. Not significant (n.s.) P > 0.05, **P<0.01 and ***P<0.001. #P<0.001 between treatment vs vehicle untreated.
Supplementary Material
Acknowledgments
This work was funded by National Institutes of Health (NIH) grants AI097302, AI130143, AI137004 and AI125997 to S.F., and R01DE014756 to D.I.Y, an Irma T. Hirsch career development grant to S.F., a postdoctoral fellowship from the Alfonso Martin Escudero Foundation and the Bernard Levine postdoctoral fellowship in Immunology to A.R.C., and a pre-doctoral fellowship by the T32 training program in Immunology and Inflammation (AI100853) to A.Y.T. We thank M. Imagawa (Nagoya City University, Japan) for providing Lrrc8c−/− mice and P.G. Thomas (St. Jude Children’s Research Hospital) for the gift of influenza A/PR8 viral stocks. We thank P. Schwarzberg (NIH) and M. Pipkin (Scripps Research) for providing us with plasmid vectors for expression of sgRNA and shRNA delivery, respectively. We thank Dr. B. Desai (University of Virginia School of Medicine) for critical reading of the manuscript and members of the Feske laboratory for helpful discussions.
Footnotes
Competing interests
S.F. is a cofounder of CalciMedica; the other coauthors declare no conflict of interest.
References
- 1.Qiu Z et al. SWELL1, a plasma membrane protein, is an essential component of volume-regulated anion channel. Cell 157, 447–458, doi: 10.1016/j.cell.2014.03.024 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Voss FK et al. Identification of LRRC8 heteromers as an essential component of the volume-regulated anion channel VRAC. Science 344, 634–638, doi: 10.1126/science.1252826 (2014). [DOI] [PubMed] [Google Scholar]
- 3.Deneka D, Sawicka M, Lam AKM, Paulino C & Dutzler R Structure of a volume-regulated anion channel of the LRRC8 family. Nature 558, 254–259, doi: 10.1038/s41586-018-0134-y (2018). [DOI] [PubMed] [Google Scholar]
- 4.Syeda R et al. LRRC8 Proteins Form Volume-Regulated Anion Channels that Sense Ionic Strength. Cell 164, 499–511, doi: 10.1016/j.cell.2015.12.031 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kumar L et al. Leucine-rich repeat containing 8A (LRRC8A) is essential for T lymphocyte development and function. J Exp Med 211, 929–942, doi: 10.1084/jem.20131379 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Luck JC, Puchkov D, Ullrich F & Jentsch TJ LRRC8/VRAC anion channels are required for late stages of spermatid development in mice. J Biol Chem 293, 11796–11808, doi: 10.1074/jbc.RA118.003853 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stuhlmann T, Planells-Cases R & Jentsch TJ LRRC8/VRAC anion channels enhance β-cell glucose sensing and insulin secretion. Nat Commun 9, 1974, doi: 10.1038/s41467-018-04353-y (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang Y et al. SWELL1 is a regulator of adipocyte size, insulin signalling and glucose homeostasis. Nat Cell Biol 19, 504–517, doi: 10.1038/ncb3514 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sawada A et al. A congenital mutation of the novel gene LRRC8 causes agammaglobulinemia in humans. J Clin Invest 112, 1707–1713, doi: 10.1172/JCI18937 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Platt CD et al. Leucine-rich repeat containing 8A (LRRC8A)-dependent volume-regulated anion channel activity is dispensable for T-cell development and function. J Allergy Clin Immunol 140, 1651–1659 e1651, doi: 10.1016/j.jaci.2016.12.974 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Burow P, Klapperstuck M & Markwardt F Activation of ATP secretion via volume-regulated anion channels by sphingosine-1-phosphate in RAW macrophages. Pflugers Arch 467, 1215–1226, doi: 10.1007/s00424-014-1561-8 (2015). [DOI] [PubMed] [Google Scholar]
- 12.Lee CC, Freinkman E, Sabatini DM & Ploegh HL The protein synthesis inhibitor Blasticidin S enters mammalian cells via Leucine-Rich Repeat-Containing protein 8D. J Biol Chem 289, 17124–17131, doi: 10.1074/jbc.M114.571257 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lutter D, Ullrich F, Lueck JC, Kempa S & Jentsch TJ Selective transport of neurotransmitters and modulators by distinct volume-regulated LRRC8 anion channels. J Cell Sci 130, 1122–1133, doi: 10.1242/jcs.196253 (2017). [DOI] [PubMed] [Google Scholar]
- 14.Planells-Cases R et al. Subunit composition of VRAC channels determines substrate specificity and cellular resistance to Pt-based anti-cancer drugs. EMBO J 34, 2993–3008, doi: 10.15252/embj.201592409 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhou C et al. Transfer of cGAMP into Bystander Cells via LRRC8 Volume-Regulated Anion Channels Augments STING-Mediated Interferon Responses and Anti-viral Immunity. Immunity 52, 767–781 e766, doi: 10.1016/j.immuni.2020.03.016 (2020). [DOI] [PubMed] [Google Scholar]
- 16.Lahey LJ et al. LRRC8A:C/E Heteromeric Channels Are Ubiquitous Transporters of cGAMP. Mol Cell 80, 578–591 e575, doi: 10.1016/j.molcel.2020.10.021 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Heng TS, Painter MW & Immunological Genome Project C The Immunological Genome Project: networks of gene expression in immune cells. Nat Immunol 9, 1091–1094, doi: 10.1038/ni1008-1091 (2008). [DOI] [PubMed] [Google Scholar]
- 18.Choi J et al. Haemopedia RNA-seq: a database of gene expression during haematopoiesis in mice and humans. Nucleic Acids Res 47, D780–D785, doi: 10.1093/nar/gky1020 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Consortium F et al. A promoter-level mammalian expression atlas. Nature 507, 462–470, doi: 10.1038/nature13182 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wu C et al. BioGPS: an extensible and customizable portal for querying and organizing gene annotation resources. Genome Biol 10, R130, doi: 10.1186/gb-2009-10-11-r130 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liberzon A et al. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst 1, 417–425, doi: 10.1016/j.cels.2015.12.004 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Subramanian A et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102, 15545–15550, doi: 10.1073/pnas.0506580102 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lin JX et al. Critical Role of STAT5 transcription factor tetramerization for cytokine responses and normal immune function. Immunity 36, 586–599, doi: 10.1016/j.immuni.2012.02.017 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hayashi T et al. Factor for adipocyte differentiation 158 gene disruption prevents the body weight gain and insulin resistance induced by a high-fat diet. Biol Pharm Bull 34, 1257–1263, doi: 10.1248/bpb.34.1257 (2011). [DOI] [PubMed] [Google Scholar]
- 25.Joerger AC & Fersht AR The p53 Pathway: Origins, Inactivation in Cancer, and Emerging Therapeutic Approaches. Annu Rev Biochem 85, 375–404, doi: 10.1146/annurev-biochem-060815-014710 (2016). [DOI] [PubMed] [Google Scholar]
- 26.Watanabe M, Moon KD, Vacchio MS, Hathcock KS & Hodes RJ Downmodulation of tumor suppressor p53 by T cell receptor signaling is critical for antigen-specific CD4+ T cell responses. Immunity 40, 681–691, doi: 10.1016/j.immuni.2014.04.006 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gulen MF et al. Signalling strength determines proapoptotic functions of STING. Nat Commun 8, 427, doi: 10.1038/s41467-017-00573-w (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Madapura HS et al. p53 contributes to T cell homeostasis through the induction of pro-apoptotic SAP. Cell Cycle 11, 4563–4569, doi: 10.4161/cc.22810 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kern DM, Oh S, Hite RK & Brohawn SG Cryo-EM structures of the DCPIB-inhibited volume-regulated anion channel LRRC8A in lipid nanodiscs. Elife 8, doi: 10.7554/eLife.42636 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Konig B & Stauber T Biophysics and Structure-Function Relationships of LRRC8-Formed Volume-Regulated Anion Channels. Biophys J 116, 1185–1193, doi: 10.1016/j.bpj.2019.02.014 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ma Z, Ni G & Damania B Innate Sensing of DNA Virus Genomes. Annu Rev Virol 5, 341–362, doi: 10.1146/annurev-virology-092917-043244 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Abe T & Barber GN Cytosolic-DNA-mediated, STING-dependent proinflammatory gene induction necessitates canonical NF-κB activation through TBK1. J Virol 88, 5328–5341, doi: 10.1128/JVI.00037-14 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Balka KR et al. TBK1 and IKKε Act Redundantly to Mediate STING-Induced NF-κB Responses in Myeloid Cells. Cell Rep 31, 107492, doi: 10.1016/j.celrep.2020.03.056 (2020). [DOI] [PubMed] [Google Scholar]
- 34.Cerboni S et al. Intrinsic antiproliferative activity of the innate sensor STING in T lymphocytes. J Exp Med 214, 1769–1785, doi: 10.1084/jem.20161674 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Larkin B et al. Cutting Edge: Activation of STING in T Cells Induces Type I IFN Responses and Cell Death. J Immunol 199, 397–402, doi: 10.4049/jimmunol.1601999 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Haag SM et al. Targeting STING with covalent small-molecule inhibitors. Nature 559, 269–273, doi: 10.1038/s41586-018-0287-8 (2018). [DOI] [PubMed] [Google Scholar]
- 37.Wu J et al. STING-mediated disruption of calcium homeostasis chronically activates ER stress and primes T cell death. J Exp Med 216, 867–883, doi: 10.1084/jem.20182192 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Prakriya M & Lewis RS Store-Operated Calcium Channels. Physiol Rev 95, 1383–1436, doi: 10.1152/physrev.00020.2014 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Feske S, Wulff H & Skolnik EY Ion channels in innate and adaptive immunity. Annu Rev Immunol 33, 291–353, doi: 10.1146/annurev-immunol-032414-112212 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vaeth M, Kahlfuss S & Feske S CRAC Channels and Calcium Signaling in T Cell-Mediated Immunity. Trends Immunol 41, 878–901, doi: 10.1016/j.it.2020.06.012 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shaw PJ, Qu B, Hoth M & Feske S Molecular regulation of CRAC channels and their role in lymphocyte function. Cell Mol Life Sci 70, 2637–2656, doi: 10.1007/s00018-012-1175-2 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li W et al. cGAS-STING-mediated DNA sensing maintains CD8+ T cell stemness and promotes antitumor T cell therapy. Sci Transl Med 12, doi: 10.1126/scitranslmed.aay9013 (2020). [DOI] [PubMed] [Google Scholar]
- 43.Diercks BP et al. ORAI1, STIM1/2, and RYR1 shape subsecond Ca(2+) microdomains upon T cell activation. Sci Signal 11, doi: 10.1126/scisignal.aat0358 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kar P et al. Dynamic assembly of a membrane signaling complex enables selective activation of NFAT by Orai1. Curr Biol 24, 1361–1368, doi: 10.1016/j.cub.2014.04.046 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen X et al. Regulation of Anion Channel LRRC8 Volume-Regulated Anion Channels in Transport of 2’3’-Cyclic GMP-AMP and Cisplatin under Steady State and Inflammation. J Immunol 206, 2061–2074, doi: 10.4049/jimmunol.2000989 (2021). [DOI] [PubMed] [Google Scholar]
- 46.Luteijn RD et al. SLC19A1 transports immunoreactive cyclic dinucleotides. Nature 573, 434–438, doi: 10.1038/s41586-019-1553-0 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bouis D et al. Severe combined immunodeficiency in stimulator of interferon genes (STING) V154M/wild-type mice. J Allergy Clin Immunol 143, 712–725 e715, doi: 10.1016/j.jaci.2018.04.034 (2019). [DOI] [PubMed] [Google Scholar]
- 48.Liu Y et al. Activated STING in a vascular and pulmonary syndrome. N Engl J Med 371, 507–518, doi: 10.1056/NEJMoa1312625 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Motwani M et al. Hierarchy of clinical manifestations in SAVI N153S and V154M mouse models. Proc Natl Acad Sci U S A 116, 7941–7950, doi: 10.1073/pnas.1818281116 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Warner JD et al. STING-associated vasculopathy develops independently of IRF3 in mice. J Exp Med 214, 3279–3292, doi: 10.1084/jem.20171351 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Imanishi T et al. Reciprocal regulation of STING and TCR signaling by mTORC1 for T-cell activation and function. Life Sci Alliance 2, doi: 10.26508/lsa.201800282 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wu J, Dobbs N, Yang K & Yan N Interferon-Independent Activities of Mammalian STING Mediate Antiviral Response and Tumor Immune Evasion. Immunity 53, 115–126 e115, doi: 10.1016/j.immuni.2020.06.009 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Srikanth S et al. The Ca2+ sensor STIM1 regulates the type I interferon response by retaining the signaling adaptor STING at the endoplasmic reticulum. Nat Immunol 20, 152–162, doi: 10.1038/s41590-018-0287-8 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Giorgi C et al. p53 at the endoplasmic reticulum regulates apoptosis in a Ca2+-dependent manner. Proc Natl Acad Sci U S A 112, 1779–1784, doi: 10.1073/pnas.1410723112 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Oh-Hora M et al. Dual functions for the endoplasmic reticulum calcium sensors STIM1 and STIM2 in T cell activation and tolerance. Nat Immunol 9, 432–443, doi: 10.1038/ni1574 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kaufmann U et al. Selective ORAI1 Inhibition Ameliorates Autoimmune Central Nervous System Inflammation by Suppressing Effector but Not Regulatory T Cell Function. J Immunol 196, 573–585, doi: 10.4049/jimmunol.1501406 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fellmann C et al. An optimized microRNA backbone for effective single-copy RNAi. Cell Rep 5, 1704–1713, doi: 10.1016/j.celrep.2013.11.020 (2013). [DOI] [PubMed] [Google Scholar]
- 58.Chen R et al. In vivo RNA interference screens identify regulators of antiviral CD4(+) and CD8(+) T cell differentiation. Immunity 41, 325–338, doi: 10.1016/j.immuni.2014.08.002 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Doench JG et al. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat Biotechnol 34, 184–191, doi: 10.1038/nbt.3437 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Huang B, Johansen KH & Schwartzberg PL Efficient CRISPR/Cas9-Mediated Mutagenesis in Primary Murine T Lymphocytes. Curr Protoc Immunol 124, e62, doi: 10.1002/cpim.62 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Love MI, Huber W & Anders S Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550, doi: 10.1186/s13059-014-0550-8 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nguyen TT et al. Revealing a human p53 universe. Nucleic Acids Res 46, 8153–8167, doi: 10.1093/nar/gky720 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Vaeth M et al. Store-Operated Ca(2+) Entry in Follicular T Cells Controls Humoral Immune Responses and Autoimmunity. Immunity 44, 1350–1364, doi: 10.1016/j.immuni.2016.04.013 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dobin A et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21, doi: 10.1093/bioinformatics/bts635 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Anders S, Pyl PT & Huber W HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169, doi: 10.1093/bioinformatics/btu638 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Quinlan AR & Hall IM BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842, doi: 10.1093/bioinformatics/btq033 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The RNA-Seq data generated in this study have been deposited in the GEO database under accession number GSE163679. Additional available gene expression data sets and ChiP-Seq data used in this study were downloaded from GEO using the following accession numbers: GSE127267 for mouse RNA-seq data from the Immunological Genome Project (www.immgen.org) 17. GSE116177 for mouse RNA-Seq data from Haemopedia (www.haemosphere.org) 18. GSE10246 for mouse microarray data from BioGPS (www.biogps.org) 20. GSE49834 for human CAGE-Seq data from Fantom5 (https://fantom.gsc.riken.jp/5/) 19. GSE36890 for mouse RNA-Seq data and STAT5 ChIP-Seq data using T cells from Stat5a-Stat5b-DKI N-domain mutant mice in which STAT5 proteins form dimers but not tetramers23. GSE110370 for gene expression analysis and p53 ChIP-seq data from PHA-stimulated human T-lymphocytes treated with nutlin-3 62. Source data for each figure, extended data figure, and supplemental figure are provided with this paper.