

Abstract
Histones are the primary protein component of chromatin and are involved in virtually all DNA-templated processes. Histones are abundantly post-translationally modified by a variety of chromatin modifying machinery. These post-translational modifications (PTMs) are recognized by a range of ‘reader’ proteins, which recruit additional proteins to specific locations on chromatin and impart precise and powerful effects on gene regulation. Each PTM typically exerts a positive or negative effect on transcription, and recent studies have shown that histone PTMs function in a combinatorial histone code: histone PTMs function in combination to exert precise DNA-templated regulation. Thus, there is a need to identify and understand proteoforms: unambiguously defined single protein molecules with all combinations of modifications. Top-down proteomics is currently the only viable approach for identifying and quantitating histone proteoforms, and mass spectrometry instruments have become sufficiently powerful to perform these quantitative analyses in a robust and high-throughput fashion. These recent innovations have enabled new experimental directions in chromatin research but have also introduced temporal and other constraints. This has led us to develop the protocol described here, which increases throughput, reduces sample requirements, and maintains robust quantitation. Although originally designed for high-throughput quantitative top-down proteomics, the protocols described here are useful for a wide range of chromatin biology applications. Starting with small amounts of cells or tissue, we describe two Basic Protocols for exceptionally rapid and efficient nuclei isolation, acid extraction of histones, and high-performance liquid chromatography fractionation of histones into histone families. We additionally describe the quantitative top-down proteomic analysis of histone H4 proteoforms.
Keywords: Histones, Proteomics, Proteoforms, Top-down, HPLC, mass spectrometry
Introduction
Proteins exist as ‘proteoforms’, which encompass all different molecular forms in which the product of a gene encoding a single primary amino acid sequence can be found, due, for instance, to the addition of post-translational modifications (PTMs) [1–2]. Physiologically, multiple PTMs are often present on each proteoform. These combinations are important for enzymatic functions, particularly in chromatin where PTM combinations are the norm (the majority of histone H4, for example, has more than one modification per molecule)[3]. However, there are limited approaches to studying such combinations of PTMs. Top-down proteomics is currently the only feasible method to characterize concurrent PTMs present on single protein molecules [4–5].
The physiological state of the eukaryotic genome is chromatin. The basic subunit of chromatin is the nucleosome: a histone octamer and approximately 147 base pairs of DNA [6–7]. There are five different families of histones: H1, H2A, H2B, H3, and H4 [8]. Among other functions, nucleosomes play a transcriptional role [8]. As regulators of transcription, histones can diminish or enhance access to DNA for transcription factors, polymerases, and other co-factors [10]. The many functions of histones are accomplished by the incorporation of different histone variants and post-translational modifications (PTMs) [11–12].
The resulting diversity allows for histones to play key roles in almost all transcriptional processes, and the dysregulation of such processes has been associated to various diseases. In particular, transcriptional alterations have been noted in cancer, where they have been associated with its progression, aggressiveness, and metastasis, and are useful in its prognosis [13]. A deep understanding of histone variants and proteoforms is thus needed to comprehend both normal cellular function and disease. A key issue, however, is that conventional PTM analysis methods do not capture proteoforms. A high-throughput middle-down or top-down proteomics approach is instead necessary to address these questions [3,14]. In these proteomic approaches, either the N-terminal tail or the entire protein are analyzed intact and the relationships between PTMs are maintained, and thus, proteoform level information is captured.
The sample preparation step has become a bottleneck for current strategies for the analysis of histone PTMs. Most prior protocols are not optimized for low sample amounts and are often quite inefficient with larger sample volumes. In addition, many have included substantially long incubation times with the expectation of improved efficiency with longer times; however, we have found that shorter times are both expeditious and equally or even more efficient. Speed also limits degradation processes, such as oxidation.
The protocol described here is an optimization and extension of previous histone extraction protocols progressively improved over more than 130 years [15–16]. We describe two protocols that have been streamlined and optimized for three different starting tissue culture cell amounts. In Basic Protocol 1, cells are lysed, nuclei are isolated, and histones are acid extracted. In Basic Protocol 2, histones are purified and separated by histone family with reversed phase HPLC, and Histone H4 is proteoform-resolved by HPLC and analyzed with top-down mass spectrometry. Additionally, we briefly describe protocols that are effective for common mammalian tissues, and endoproteinase Glu-C digestion of histones to obtain intact histone H3 tails for middle down proteomics (Support Protocol 1).
This protocol can be used together with multiple other proteomics protocols for the analysis of histones [17–28]. It also is useful for a variety of non-proteomics applications. Our goals in developing this protocol were: 1) to increase throughput, such that the entirety of the pipeline could be performed on a single day, 2) to decrease sample requirements, such that small tissue samples can be successfully analyzed, and 3) to not compromise data quality and reproducibility. The expediency of this protocol makes this an excellent choice for other chromatin biology approaches that are limited by sample size or throughput. Even in the absence of limiting sample and throughput constraints, the use of this protocol will substantially expedite and simplify histone extraction without any observed cost in quality.
Strategic Planning
Multiple solutions required throughout this protocol are best prepared in large batches for long term use. Solutions used at 4°C should be stored at 4°C for convenient use. Nuclei isolation buffer is conveniently made in large batches, aliquoted into 50–500 mL plastic bottles, and stored at −20°C. One of these should be brought to 4°C before starting the protocol.
Before performing the protocol, we recommend filling up an ice bucket and thawing all frozen solutions. All steps should be performed on ice unless otherwise specified. Basic PPE should be used at all times, in particular when handling acids. Trifluoracetic acid (TFA) is particularly volatile and hazardous and should be handled only with glass syringes or pipettes; it should only be added to solutions in a fume hood. Once TFA is in solution at relatively low concentrations, the buffers do not require special storage. Only use glass to transfer acetone; acetone dissolves most plastics.
This protocol begins with a set of cells or tissue that has already been harvested. The number of cells used is important for optimal efficiency. We suggest counting cells or estimating cell numbers based on volume: typically, there are roughly 8 million cells in a 100 μL cell pellet. For a smaller number of cells, we suggest skipping Basic protocol 1 steps 1–12 and starting at step 13, but instead of a 5:1 ratio of HCL to cells, a 10:1 ratio should be used.
For Basic Protocol 2, a high-performance liquid chromatography instrument to fractionate histones into histone families is required. Further, top-down Histone H4 analysis necessitates two key instruments: a high-performance liquid chromatography system connected to an electrospray ionization source and a high-resolution mass spectrometer that is capable of acquiring 60,000 full width at half maximum resolving power (FWHM) spectra 3 times a second and uses electron transfer dissociation for fragmentation. The specific instruments detailed in this protocol are not necessary, but we strongly suggest the use of an orbitrap mass spectrometer and to use HPLC systems that have compatible tubes/vials such that there are no liquid transfers. This protocol assumes that the reader is capable of operating these instruments and is familiar with the basic principles of chromatography and mass spectrometry.
For HPLC MS/MS, reverse chromatography with a C3 material designed for intact proteins (large pore size, small particle size) is preferred, and in our hands, yields superior results in separating histone H4 by the PTMs on the N-terminal tail.
Basic Protocol 1: Nuclei isolation and Histone Extraction
This protocol is for isolating nuclei from mammalian cells in culture or from tissues and extracting histones with acid precipitations. Cells are lysed with a detergent and nuclei are isolated by centrifugation. Histones are then acid extracted, precipitated, and washed. The final product of this protocol is a highly enriched mixture of histone proteins. While the protocol we designed for use with mammalian cells and tissues, we have also successfully used this protocol for Drosophila and C. elegans. The materials for this protocol are easy to obtain and relatively inexpensive, and the reagents are stable and easy to use.
Materials List
1–10 million cells (fresh or frozen) that have been harvested and are in a microcentrifuge tube, or fresh/frozen tissue (see below)
1 x phosphate-buffered saline (PBS, Sigma, cat P5493), 4°C
Nuclei Isolation Buffer (NIB) (see Reagents and Solutions, and Table 1), 4°C
Table 1.
NIB inhibitor recipe
| DTT | Protease | 1M | 1000x | 1mM | 50μL | 10μL | 1μL |
| AEBSF | Protease | 200mM | 400x | 0.5mM | 125μL | 25μL | 2.5μL |
| Microcystin-LR | Phosphatase | 2.5μM | 500x | 5nM | 100μL | 20μL | 2μL |
| in 100% EtOH | |||||||
| Sodium Butyrate | HDAC | 5M | 500x | 10mM | 100μL | 20μL | 2μL |
| NP-40 Alternative | Detergent | 10% | 33.3x | 0.30% | 1.5mL | 300μL | 30μL |
Inhibitors (see Reagents and Solutions), 4°C
10% NP-40 replacement (Sigma, cat 74385)
1.5 mL dolphin tubes (Costar, Cat 3213)
0.2M Hydrochloric acid (Fisher, Cat A144–212), 4°C
1g/mL Trichloroacetic acid solution (ACROS, Cat 152135000), 4°C
TCA to be used is at 1g/mL in H2O, which is often referred to as “100% TCA” in other protocols (1g/ml is 100% on a weight/volume basis but not on a w/w or v/v basis). The final concentration once this stock is added to the sample will be around 0.2 g/mL TCA.
Acetone with 0.1% v/v HCl, −20°C
Acetone, −20°C (Fisher, Cat. A929–4)
Protocol Steps
Nuclei Isolation
-
1
For small numbers of cells (less than 1 million cells), we suggest skipping nuclei isolation and starting with step 13. Despite evidence that nuclei isolation can be skipped and cells taken directly into acid extraction, these steps substantially remove lipids and other contaminants that are detrimental to the longevity of reversed phase columns and other downstream processes. If these issues are not a concern, proceeding directly to acid extraction comes at little cost
Thaw frozen cell pellets or tissue, put on ice, and let them come to ~4°C. Process the sample as follows:- If cells contain media, wash with PBS until no media is visible:
- To cell pellet, add 1 mL PBS
- Centrifuge 300 × g for 5 min, 4°C
- Remove PBS; be careful to leave the cell pellet intact
-
Repeat until no media is visibleThe end result should be a clean cell pellet
-
If using fresh soft tissues such as brains or livers, mince the organs with a razor blade, and Dounce homogenizeTougher tissues may require mechanical homogenization first and typically require more material for an equivalent nuclear pellet and histone yield.
-
Frozen tissues can be crushed with a mortar and pestleWe generally perform this isolation protocol for cell numbers from 1 to 10 million. Above 10 million cells, it is typically more efficient to split the cells and parallel-process aliquots of 5–10 million cells.
- If using fresh cells, skip straight to Step 2. For this protocol, cells should be in a 1.5 mL tube.
-
2
Visually estimate your cell pellet volumes
Either look for tick marks on your tube that corresponds to a particular volume, or pipette water into an empty tube to estimate a given volume.
100 μL is a typical volume from a 10–15 cm tissue culture plate and represents around 8 million cells depending on the cell type
-
3
Aliquot 40x the volume of the pellets (from the estimate in step 2) of NIB stock and add inhibitors (see Reagents and Solutions)
If you have two cell pellets, each with 100 μL volume of cells, prepare 8 mL of NIB with inhibitors. The inhibitors should be kept in small aliquots at −20°C and not in a premixed cocktail.
-
4
Take 1/4th of the NIB mixture made in Step 3 and add 10% NP-40 Alternative to a final concentration of 0.3% (see Reagents and Solutions)
For instance, for 2mL of NIB (which would be ¼ of the NIB mixture volume in the example in Step 3), add 60 μL of 10% NP-40
-
5
To each of your cell pellets, add NIB and inhibitors with 0.3% NP-40 alternative (mixture from Step 4) to a final ratio of 10:1
This means, you will need 1mL of NIB buffer from step 3 for every 100 μL cells.
-
6
Mix by pipetting gently until any clumps of cells have been dispersed, and incubate on ice for 5 min
We generally find that pipetting is more effective than vortexing for disrupting cell pellets and typically generates less heat. The 5-minute incubation time is sufficient; however, we have not observed any significant changes with incubations of up to 30 minutes.
-
7
Pellet the nuclei at 600 × g for 5 min, at 4°C
600 × g is sufficient to pellet nuclei and will not pellet other organelles.
-
8
Transfer supernatant to a new 1.5 mL tube
The supernatant corresponds to the cytoplasmic extract. If you will not keep or use the cytoplasm, simply discard.
-
9
Resuspend the nuclei pellet gently by adding a 10:1 volume ratio of NIB (without NP-40 detergent) to the cell pellet.
For instance, a 100 μL nuclei pellet would require 1 mL of NIB. Be sure to get the nuclei suspended and reasonably unclumped, but be gentle. This step is important in removing detergents which will affect subsequent steps.
-
10
Centrifuge nuclei at 600 × g for 5 min, at 4°C
-
11
Repeat steps 9–10 at least twice, or multiple times if necessary, until no NP-40 is left and only a clean nuclei pellet remains
Typically, 2 washes are sufficient for removing the detergent. The presence of detergent can be judged by the formation of soap bubbles.
-
12
Pellet nuclei at max speed (15,000–21,000 × g) for 5 min at 4°C and use promptly for acid extraction
These pellets can be flash-frozen, however, we recommend proceeding immediately to acid extraction
Acid Extraction
Acid extraction is a standard strategy for histone isolation.
-
13
Gently vortex the pellet of isolated nuclei obtained in Step 12 and add 0.2 M, 4°C HCl dropwise at a 5:1 volume ratio. (10:1 ration if fewer than 1 million cells in the starting material)
The bright white nuclear pellet derived from the nuclei isolation procedure above will be 40–50% smaller than the starting cell pellet. Be careful to reassess the volume of the nuclear pellet for the acid extraction procedure.
If volumes are too small to add dropwise, slowly pipette instead. Adding the HCl too quickly can precipitate some histones and decrease yield. The final volume added for 100 μL of nuclei is 500 μL of HCl. If a cell pellet is smaller than 40 μL, we recommend adding 200 μL of HCl. If the nuclei clump is not readily dissolved, gently pipette up and down to disrupt.
-
14
Put on ice for 5 minutes
This step was typically performed for hours or even overnight. We have found that 5 minutes is sufficient (Figure 1A). We observe no benefit from increasing the duration with regards to signal-to-noise nor are there any proteoform changes. Further, we have not noticed issues with subsequent HPLC fractionation. This precipitation is likely completed almost instantly. Alternatively, this step is a reasonable breakpoint as it can be extended without affecting the final results.
-
15
Spin down at max speed (15,000–21,000 × g), for 5 min at 4°C.
-
16
Transfer the supernatant to one 1.5 mL Eppendorf tube
This supernatant contains primarily histones and potentially other histone-like proteins, while the precipitate should contain the majority of other nuclear proteins
-
17
To the supernatant, add 1/4 of the supernatant volume of 1 g/mL TCA and fully mix
For instance, for 200 μL of supernatant, add 50 μL of TCA. This mixing can be done aggressively as the objective is to precipitate the histones. Ensure that the TCA is mixed, by vortexing.
-
18
Precipitate on ice for 5 min.
-
19
Centrifuge at max speed on a table-top centrifuge (15,000–21,000 × g), 4°C for 5 minutes
The TCA precipitates all histones and does not affect the majority of PTMs (there are limited exceptions, e.g. histidine phosphorylation). You may directly use this precipitate for HPLC purification or other purposes, but only if you do so within an hour. Otherwise, follow the remaining steps.
-
20
Gently add 0.1 mL cold (−20°C) acetone w/0.1% HCl to tube containing precipitate
Do not disturb the precipitate. Let it stay on the tube walls. The HCl is used to displace the TCA from proteins, and this is very important for further HPLC-MS analysis. Steps 18–20 are designed to remove extra TCA and water in the samples – too much residual TCA may degrade the samples. However, adding acetone to Eppendorf tubes can dissolve the plastic, so do these steps quickly, but carefully.
-
21
Centrifuge at max speed on a table-top centrifuge (15,000–21,000 × g), 4°C for 2 minutes and carefully remove acetone with a glass pipet.
Do not scrape the walls. Err on the side of leaving solvent behind rather than losing histone. A small amount of acetone will evaporate quickly. The use of a vacuum aspiration system is ideal. Evaporation can be accelerated by aspirating the air above the solvent.
-
22
Add 0.1 mL 4°C pure acetone (without HCl) and repeat step 21.
-
23
Air dry final pellet (or SpeedVac for 10 min). What is left is mostly histone and is ready to be fractionated by HPLC.
Samples can be stored in this state at RT for a few days (~5 days) but should not be stored long-term in this condition. Storage at −80°C may be used to extend this time if necessary. Perform HPLC fractionation and purification (Basic Protocol 2) for longer-term storage. HPLC purified histones are stable dry in sealed tubes at room temperature for years.
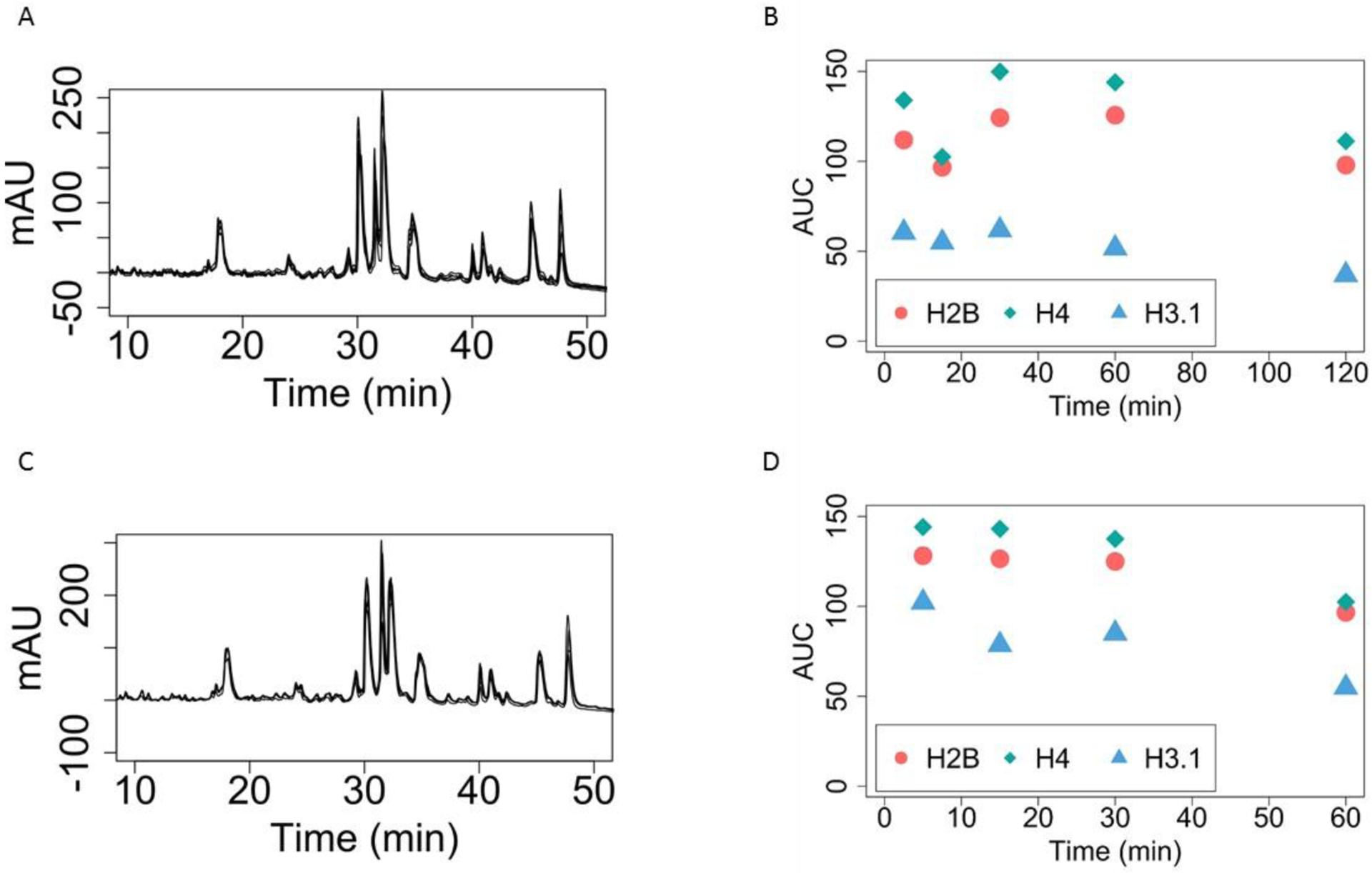
Figure 1. Five Minute HCl and TCA Precipitations are Sufficient
A) HPLC Chromatogram of SUM159 cell line histones with varying HCl precipitation times. Each black line represents a different HCl precipitation time: 5 min, 15min, 30min, 60 min and 120 min. Note that these are not readily distinguishable from each other and overlap almost perfectly. Thus, extensive HCl precipitations, such as overnight, are neither necessary nor beneficial.
B) Quantitation of Histones by area under the curve (AUC) with varying HCl precipitation times. The areas under the curve from Figure 1A are integrated for the different time points of HCl precipitation. There is no significant trend for increasing HCl times.
C) HPLC Chromatogram of SUM159 histones with varying TCA precipitation times. Each black line represents a different TCA precipitation time: 5 min, 15min, 30min and 60min. There is no difference in retention time or peak heights by increasing TCA precipitation time.
D) Quantitation of Histones by AUC with varying TCA precipitation times. The areas under the curve from Figure 1C are integrated for the different time points of TCA precipitation. There is no significant trend for increasing TCA times.
Basic Protocol 2: HPLC Fractionation of histones, and Histone H4 HPLC-MS/MS
This protocol focuses on reversed phase chromatography of histones into individual histone families, followed by Histone H4 HPLC-MS/MS.
First, purified histones from Basic Protocol 1 will be dissolved, injected onto a reversed phase column, eluted, and fractionated. These fractions will contain individual histone families. Performing HPLC fractionation (steps 1–7) is beneficial in providing relative abundances of histones and variants. In particular, the H3 variant peaks can be informative. Further, the HPLC step serves to clean and desalt the histones, yielding extremely pure samples that are both suitable for mass spectrometry and for long-term storage. Histone fractionation into histone families may be essential —or completely unnecessary—, depending on the biological question being asked. For intact H4 analysis alone, or for whole histone analysis, it can be skipped. For H3 middle-down methodologies, on the other hand, it is required if one wishes to distinguish between H3.1, H3.2, or H3.3. Support Protocol 1 describes subsequent steps for Glu-C digestion for middle-down H3 after histone family fractionation. Support protocol 1 is essential for future middle down H3 analysis.
While the histones obtained with the protocols described in this article can be used for multiple assays, here, we describe one such use, top down proteomics of histone H4. After fractionation, the histone H4 fraction is loaded onto another HPLC, separated by proteoforms, and analyzed by an orbitrap mass spectrometer. The final results are a UV trace of histone families from which abundances of histones can be determined, and an MS/MS raw file which can be analyzed to determine identity and quantity of histone proteoforms.
Basic Protocol 2 is specifically designed for use with a Thermo U3000 HPLC system with a 150× 2.1 mm Vydac 218TP 3 μm C18 column (Part Number: 218TP3215). Flow rates, pressure limits, buffer gradients, separation, and UV mAU signal intensity will be different if using a different setup. This specific HPLC system is not required, as discussed in Strategic Planning. The HPLC system used here for the Histone H4 HPLC MS/MS section has an autosampler tube system to is compatible with the one used in this Histone family fractionation. Ideally, no liquid transfers are performed, ensuring no additional losses after histone family fractionation. We have set up our inline HPLC for 1 μL pickup with a sample loop size of 3 μL.
Materials List
Precipitated histones from Basic Protocol 1.
A high-performance liquid chromatography instrument with a reversed phase column.
Compatible auto-sampler vials (We use 250 μL vials (Thermo Cat. 200 046) for both auto-sampler based injection and fraction collection)
Offline high-performance liquid chromatography Buffer A (5% acetonitrile, 0.2% trifluoracetic acid (TFA))
Offline high-performance liquid chromatography Buffer B (95% acetonitrile, 0.188% TFA)
SpeedVac capable of handling acetonitrile and TFA (Thermo Savant SPD131DDA)
Mass spectrometry Buffer A (see Reagents and Solutions)
Mass spectrometry Buffer B (see Reagents and Solutions)
A high-performance liquid chromatography instrument with a reversed phase column (Thermo U3000, Agilent Zorbax 300SB-C3).
A high-resolution mass spectrometer with electron transfer dissociation and an electrospray source (Thermo Orbitrap Fusion Lumos, Thermo nano-spray flex)
Protocol Steps
HPLC fractionation of Histone families
-
1
Dissolve histones from Basic Protocol 1 with 85 μL Buffer A by vortexing vigorously for 10 seconds.
Some pellets will not fully dissolve, in particular if one has skipped nuclei isolation in Basic Protocol 1. These pellets do not adversely affect HPLC fractionation and purification except that they may clog a column or filter. If no pellet is visible, you may use 80 μL instead of 85 μL.
-
2
Centrifuge at max speed (15,000–21,000 × g) for 15 min at 4°C.
This step is recommended to prevent any large particles from being injected into the HPLC
-
3
Transfer 80 μL into an HPLC compatible vial and place in the HPLC autosampler
Leave approximately ~5 μL of buffer in the tube to prevent transferring any large particles into your HPLC system
-
4
Run the sample in accordance with the parameters in Table 2.
From this run, a UV trace will be obtained from which quantification of histone families can be performed.
-
5
Collect and label the vials
Depending on the amount of buffer in each vial, it is possible to combine multiple tubes into a single one at this stage.
-
6
SpeedVac for 2 hours or until dry
We recommend a Speedvac that has a delayed and burst vacuum feature to prevent sample loss. If you wish, samples can be left in a running Speedvac overnight. After the samples are fully dry, they can be stored at RT for months to years, and in the −80 °C indefinitely.
-
7
Optional: If accurate quantitation of histones is needed, inject in-vitro purified histones with known concentrations, at varying concentrations to obtain a standard curve and interpolate your sample amounts.
Accurate quantification is needed for the following steps. We have included our standard curves for H4 and H3 (Figures 2A and 2B); however, these values may not be accurate for a different HPLC and we recommend performing this step for precise numbers. Purified histones can be purchased (Sigma cat. 14–697 and Sigma cat. 14–494). If you are interested in middle-down Histone H3, proceed to Support Protocol 1.
Table 2.
Offline HPLC Parameters
| Sample Volume | 80 μl |
| Temperature | 4.0 [°C] |
| Collection Tube Max Volume | 0.20 [ml] |
| Column Oven Temperature | 37.0 [°C] |
| Buffer A | 5% ACN 0.2% TFA |
| Buffer B | 95% ACN 0.188% TFA |
| UV wavelength | 214 [nm] |
| Equilibration | Duration = 10.000 [min] |
| Flow Rate | 0.250 [ml/min] |
| Column | 218TP52 |
| Time: | %B: |
| 0 | 25.0 [%] |
| 60 | 60.0 [%] |
| 75 | 100.0 [%] |
| 80 | 100.0 [%] |
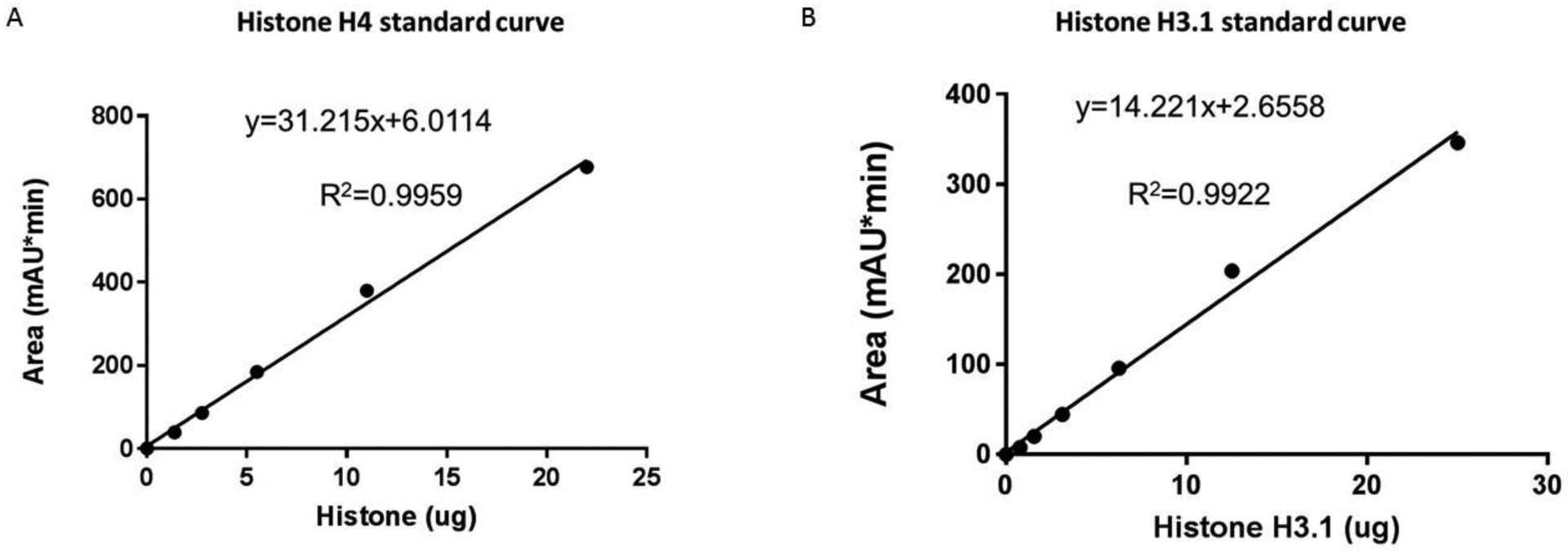
Figure 2 Histone H3 and H4 Standard Curves for Estimating Protein Amount
A) Histone H4 Standard Curve. Area of mAU obtained from HPLC analysis of purified histone H4.
B) Histone H3.1 Standard Curve. Area of mAU obtained from HPLC analysis of purified histone H3.1.
Top-Down HPLC-MS/MS of Histone H4
-
8
Quantitate histones from HPLC fractionation in step 7: integrate the area under the curve for histone H4 and plug this number into our provided standard curve equations (see Figure 2), or into a standard calculated in step 7.
-
9
Dissolve histone H4 with mass spectrometry buffer A to a final concentration of 55 ng/μL.
The volume used to dissolve histones is dependent on the amount determined in step 8 and the volume of your sample loop. Typically, from a 10cm plate (8 million cells), one can expect 40–60 runs of histone H4. Additionally, different mass spectrometers will have different sensitivities, and more sample may be necessary per run. The objective is to load the same amount of test samples for an unbiased comparison, which is equally important as maximizing signal.
Some samples will require pipetting of small volumes (<5 μL). When using such small amounts, scrape along the walls of the tube, vortex, and centrifuge. It is critical to not have any air bubbles especially at the bottom of the tube.
-
10
Run the sample according to Table 3
-
11
Samples can be stored at 4°C for 1 week without a significant increase in oxidation. For long term storage, Speedvac samples and store at −80°C indefinitely
Table 3.
LC-MS analysis parameters
| Equilibration time | 10 min | One advantage of reversed phase chromatography over hydrophilic interaction chromatography is ease in establishing reproducible gradients with short equilibration times |
| Equilibration %B | 27% | |
| Starting %B | 27% | |
| Linear Gradient | 70 min | |
| Final %B | 30% | |
| Flow Rate | 200 nL/min | |
| Electrospray Voltage | 1700 V | Generally, 10% more voltage than the minimum required for a stable spray is recommended |
| Ion funnel temperature | 320 °C | Histone H4 has no problems with higher temperatures if there are desolvation issues, other histones may require lower temperatures |
| Mode | Positive | |
| Ion funnel RF% | 80% | Higher RF% may be beneficial in the removal of adducts, however for routine running there is no observable benefit for increasing past 80% |
| Detector | Orbitrap | |
| Scan Window | 700–1400 m/z | This range contains the most abundant charge state and is generally low in interfering ions |
| Resolution | 60,000 | This resolution is sufficient to fully resolve isotopic peaks |
| Automatic Gain Control (AGC) | 5.00E+05 | In our system, increasing this past 5.00e5 results in reduced mass accuracy |
| Maximum Injection Time | 200 ms | For MS1 scans we usually achieve AGC in under 20 ms |
| Microscans | 5 | We observe diminishing returns after 3 microscans, with 5 microscans yielding our optimal results |
| Mode | Profile | |
| Mode | Top-20 | Top-20 may seem excessive, however in some instances we observe significant increases to H4 proteoforms and their precursor diversity, which warrants so many dependent scans |
| Mass List | see note | We have developed a mass list of H4 targeted masses. Refer to [10] Supp. Table 1 for the full list |
| Isolation | Quadrupole | |
| Isolation window | 1 m/z | This isolation window is critical, as our MS1 precursor peaks are slightly over 1 m/z apart |
| Fragmentation | ETD | |
| ETD reaction time | 14 ms | This parameter will likely have to be optimized for each instrument and over time |
| ETD reagent target | 5.00E+05 | |
| Maximum ETD reagent injection time | 200 ms | |
| Detector | Orbitrap | |
| Scan Window | ||
| Resolution | 60,000 | This resolution is more than is necessary for the majority of proteoforms, however we observe an increase in confident MS2 peak matches with increased resolution. This is true for higher resolutions as well, however the duty time increases do not justify going past 60,000 resolution |
| Automatic Gain Control (AGC) | 5.00E+05 | In our system, increasing this past 5.00e5 results in reduced mass accuracy, at this setting we typically observe <10 ppm matches |
| Maximum Injection Time | 200 ms | For MS2s, we often reach max injection time, 200 ms has been sufficient for full proteoform characterization for the top ~50% of proteoforms, while not sacrificing duty time |
| Microscans | 3 | |
| Mode | Profile | |
Support Protocol 1: Preparation of intact H3 histone tails by Glu-C Digestion
In this protocol, Histone H3 is digested with endoproteinase Glu-C. The histone fractions obtained from Basic Protocol 2 steps 1–7 are dissolved in digestion buffer, digested with endoproteinase Glu-C for 1 hour at 37°C, and SpeedVac dried. The result of this sample processing protocol are intact 1–50 aa N-terminal histone tails which are rich in PTMs and considerably easier to analyze than intact H3 with mass spectrometry. Perform this protocol if you wish to analyze histone H3 N-terminal tail PTMs.
Materials List
Histone H3 fraction from Basic Protocol 2, steps 1–8.
100 mM Ammonium acetate pH 4.0 (Fisher, A637–500), 37°C
Endoproteinase Glu-C (Thermo Cat. 90054)
Protocol Steps
Add 20 μL of 100 mM ammonium acetate to purified histone H3 from Basic Protocol 2 steps 1–7.
-
Add Glu-C to a final ratio of 10:1 protein to enzyme amounts
For example, for 10μg of protein, add 1μg of Glu-C
Incubate at 37°C for 1 hour
-
SpeedVac for 30 minutes or until dry
This sample is now ready for mass spectrometry.
Reagents and Solutions
NIB (Nuclei Isolation Buffer)
15 mM Tris-HCL (pH 7.5)
60 mM KCl
15 mM NaCl
5 mM MgCl2
1 mM CaCl2
250 mM Sucrose
Nuclei isolation buffer is conveniently made in large batches, aliquoted into 50–500 mL plastic bottles (see Table 1), and stored at −20°C. NIB can be stored at −20°C for a year, or stored at 4°C for a month
10% NP-40 Alternative:
Add 5 mL of NP-40 alternative to a 50 mL conical.
Add 45 mL of milli-pure water to the 50 mL graduation.
Shake, vortex, wait, repeat.
It will take up to several hours to completely dissolve. Be patient. It is best prepared days in advance.
DTT Stock Solution (1.0 M):
Use PPE: Gloves, Safety Glasses
Using a P1000 pipette, add 6.48 mL of milli-pure water to the 1 g bottle of DTT (Thermo, Cat. R0861)
Mix gently by vortexing
Aliquot 250 μL (used per 250 mL of NIB) into sterile 750 μL Eppendorf tubes (approx. 26) using 1 mL pipette.
Mark or label the Eppendorf tubes with “DTT” and store in a labeled box at −20°C
Stable for 1 year at −20°C; mark the expiration date on the box
AEBSF Stock Solution (200.0 mM):
Use PPE: Gloves, Safety Glasses
Add 20.86 mL per 1 g of AEBSF
Add an additional 860 μL of milli-pure water to the 1.0 g bottle of AEBSF
Mix gently by vortexing
Transfer the AEBSF solution to a larger container, e.g. 125 mL flask
Measure 20 mL of milli-pure water in a 25 mL graduated glass cylinder and use to rinse the AEBSF bottle in several aliquots to transfer and recover as much as possible.
Aliquot 625 μL of the solution into 750 μL sterile Eppendorf tubes (approx. 34) using a 1 mL pipette. One such aliquot will be used per 250 mL of NIB.
Mark or label the Eppendorf tubes with “AEBSF” and store in a labeled box in the −20°C
Stable for 2 months at −20°C, mark the expiration date on the box
Microcystin – LP Stock Solution (2.5 μM)
Use PPE: Gloves, Safety Glasses (Microcystin is a potent hepatotoxin)
Measure 43.95 mL (approx. 44 mL) of absolute ethanol in a glass 50 mL graduated cylinder.
Add the measured absolute ethanol (approx. 100%) to 100 μg of Microcystin – LP in a 100 mL glass beaker.
Aliquot 500 μL of the solution into 750 μL sterile Eppendorf tubes (approx. 88) using a 1 mL pipette. One such aliquot will be used per 250 mL of NIB.
Mark or label the Eppendorf tubes “MC”
Stable for 6 months at −20°C, mark the expiration date on the box.
5M Sodium Butyrate:
Use PPE – Gloves, Face shield, chemical hood. (Butyric acid is corrosive and smells like vomit/rancid butter/body odor. Sodium Butyrate is harmless and is a listed food additive.)
In a 250 mL bottle, add 250 mL of water. Precisely mark the 250 mL level for future use. Pour out approximately 150 mL of water to waste, leaving ~100 mL.
Add a stir bar to the bottle and put it on a magnetic stir plate in the chemical hood and stir at a moderate-fast rate.
In a clean, dry glass 100 mL graduated cylinder, measure out 97 mL of Butyric Acid
Slowly add the butyric acid to the water.
Using a pH meter, adjust the pH to 7.0 with solid sodium hydroxide. Approximately 50 g will be needed.
Add Milli-pure water to the 250 mL mark on the bottle.
Aliquot 1000 μL of the solution into 750 μL sterile Eppendorf tubes (approx. 88) using a 1 mL pipette. One such aliquot will be used per 500 mL of NIB.
Mark or label the Eppendorf tubes
Stable for 2 years at −20°C, mark the expiration date on the box.
Commentary
Background Information
Epigenetic studies are critical to understanding cell biology. Histones are a fundamental component of all epigenetic mechanisms and thus, understanding their characteristics and abundances is of great value. Scientific endeavors as a whole have begun transitioning into the “omics” and high-throughput era, and modern science necessitates methodologies that minimize sample requirements and maximize speed. The protocol described here, for histone extraction, is based on previous acid extraction protocols but with timing and sample size improvements to accommodate high-throughput proteomic analyses of histones.
With recent improvements in mass spectrometry methodology and instrumentation, high-throughput top-down and middle-down histone proteomics experiments are not only feasible but preferable. These methods deliver previously unattainable data: quantities of proteoforms. We have previously shown that this approach is robust and high-throughput [3]. It is a misconception that these approaches are time- or cost-prohibitive. At full speed, this protocol can be completed in a single eight-hour day. This is largely due to changes in the duration of acid-extraction steps. The acid extraction protocol described here is designed specifically for mass spectrometry but will generally yield highly efficient extraction of histones that exceed the purity requirements of other workflows. We avoid salt, detergents, and other chemicals that are not compatible with mass spectrometry. Other acid extraction protocols call for extended HCl and TCA precipitation times; we have found that there is no difference in performing these reactions in 5 minutes or 2 hours (Figure 1A). The precipitation reactions with the concentrations given in this protocol happen relatively quickly and efficiently. The TCA precipitation of histones has previously been performed for an hour or more. We, however, find no benefit for such extended precipitation time (Figure 1B), and given that precipitation for too long may degrade the samples due to the acidic conditions, this is an important consideration. Regarding cost, we estimate the total cost per proteomic analysis to be less than 200 USD (considering 5-year mass spectrometer depreciation, reagents, column lifetime, and labor).
Sample limits are a further consideration that we address here. By minimizing the number of tube transfers and overall steps, we have increased sensitivity sufficiently to enable analysis of ~1 million cells. It is important to note, that with 5–10 million cells, we routinely achieve over 20 replicates for histone H4, which makes sample handling easier, increases reproducibility, and allows for re-analysis, if necessary. At 1 million cells, H3 becomes challenging as different species and certain cell types have varying levels of H3 variants, and thus may not have sufficient material for even a single replicate. These concerns do not apply to histone H4, as there is only one variant. When not sample-limited, we recommend using 5–10 million cells to maximize success.
One key basis of our sensitivity improvement is minimizing sample transfers and total steps. The HPLC system that we use to fractionate histones into histone families is the same as our mass spectrometer HPLC, and, thus, we use the same tube for both instruments.
Lastly, which column you choose for histone fractionation is important. The Vydac 218TP 3 μm C18, 150 × 2.1 mm we use in this protocol offers greater sensitivity and sufficient capacity when compared to larger columns that are more commonly used.
Critical Parameters
Cells
Quantity and quality of the cells are critical parameters of this protocol. The optimizations we have performed are all centered on 10 cm tissue culture plates and an upper bound of 10 million cells. If more cells are to be processed, we suggest splitting into smaller fractions. A lower limit of cells is also important: anything less than 1 million cells will not be effectively processed with this protocol, and mass spectrometry analysis will be challenging and limited. Cells should have 80%+ viability and stable nuclei. We also suggest that cells be cleaned. For instance, large quantities of blood when processing mouse hearts will result in a large hemoglobin peak that obfuscates histones. Cells from tissues need to be effectively homogenized in basic protocol step 1.
HPLC
Effective chromatography is critical for Basic Protocol 2. It is important to have an instrument capable of handling acetonitrile, trifluoroacetic acid, and formic acid. Further, these instruments must deliver flow rates comparable to those described in Basic Protocol 2. We strongly suggest using compatible HPLC systems that allow for collection of offline purification fractions into the same vials that are used for online HPLC-MS/MS sample injection, thereby avoiding unnecessary liquid transfers. UV quantitation of histones is also necessary to ensure correct loading into the next stage of HPLC MS/MS. The columns we have discussed in this protocol are recommended for histone purification and analysis, but comparable C18 and C3 columns may be used. Ensure that columns are designed for intact proteins: large pore sizes (300 Å recommended) and small bead size (5 μm or smaller) are essential for best results.
Mass Spectrometry
It is not necessary to use an orbitrap instrument, however, only FT-ICR instruments have shown any advantages at usually a higher cost. The Fusion Lumos is our recommended mass spectrometer given the sensitivity, accuracy, resolution, and speed of the instrument. Electron transfer dissociation (ETD) has yielded more MS2 matches than collision-based fragmentation events, and we do not suggest attempting this approach without ETD. We have not tested top-down H4 with ultraviolet photodissociation; however, others have had success [29]. Electrospray ionization is necessary for top-down proteomics of Histone H4.
Troubleshooting
No nuclear pellet
For small cell numbers (<1 million), nuclear pellets are often very hard or impossible to see. Centrifuging longer and faster (30 minutes, 15,000 × g) may slightly increase yields. For some tissues, mechanical homogenization and doubling the incubation to 10 minutes may increase yields. Proceeding with the protocol without a visible pellet has yielded decent results in our experience.
No histone signal on offline
Lack of signal in the offline could be due to a multitude of issues. Follow your troubleshooting guide to rule out any mechanical HPLC issues such as leaks, blockages, or pump issues. In our experience, it is rare to not see any signal at all. We suggest purifying 5+ million cells to rule out any sample processing issues. Note that histones contain few aromatic amino acids and thus 280 nm UV wavelength detection (A280) is an inaccurate means of estimating protein concentration. Similarly, acidic residues are strongly underrepresented, making accurate Bradford assays for protein concentration difficult.
High noise or unexpected peaks in offline HPLC UV-trace
High noise and additional peaks are indicative of ineffective sample preparation or acid extraction. We have attempted to skip the HCl acid extraction step and our signal-to-noise ratio suffered due to the presence of other proteins. While histones were still time-resolved and represented the dominant peaks, additional peaks partially obfuscated and contaminated the histone peaks. We recommend performing Basic Protocol 1 in its entirety to increase the purity of histones. Before beginning Basic Protocol 1, wash your cells with PBS an additional time. Increase the ratio of HCl to nuclei pellet to 10:1. If peaks continue to interfere, increase the offline HPLC gradient time to better resolve overlapping peaks. Change the column temperature: decreasing to room temperature may shift peak order.
Insoluble pellet
In certain tissue samples, we observe white insoluble pellets post-acid-extraction. Histones are highly soluble, and we find that these pellets do not contain significant quantities of histones. Extensive vortexing and sonication solubilized our pellets, but no improvement in histone quantity was observed. We recommend centrifuging insoluble pellets and not disturbing them when removing dissolved histones.
Understanding Results
Basic Protocol 1 results in a purified histone fraction that is usable for a HPLC-MS/MS. This fraction is also suitable for typical western blot analysis. Basic Protocol 2 has two outputs. The first is a UV trace, from which histone family abundances can be quantitated by integrating the areas under their respective curves. The specific peaks which contain histones are reasonably consistent between C18 reversed phase columns, however, we strongly recommend verifying the composition of each chromatographic peak. This can be accomplished with western blotting or mass spectrometry. For quantitation, we have included standard curves for human H3.1 and H4.
The second output of Basic Protocol 2 is a top-down HPLC-MS/MS mass spectrometry raw file that contains intact H4, and specific precursor fragment spectra. For intact Histone H4, two predominant peaks are expected (Figure 3A) from the total ion current (TIC). The first peak, at 20min, contains H4 that is oxidized. This oxidation is mainly on M84. The second peak is un-oxidized H4, at around 25 min. Within each of these peaks, is a rich mixture of H4 proteoform sub-peaks, which are near Gaussian elutions. We primarily target the +15 charge state of H4 (Figure 3B) due to a lack of interferences, abundance, and efficient ETD fragmentation (Figure 3C). For maximum sequence coverage, optimization of ETD parameters will be required. To achieve this, maximize +2 and +3 charged fragment ions while maintaining +10 and larger fragment ions. The +1 fragment ions observed under ETD are easily identified and have few interferences. These ions typically do not cover much past the first 10 amino acids. The +2 and +3 ions will be critical in accurately characterizing the N-terminal tail of histone H4, in particular the modification state of K20.
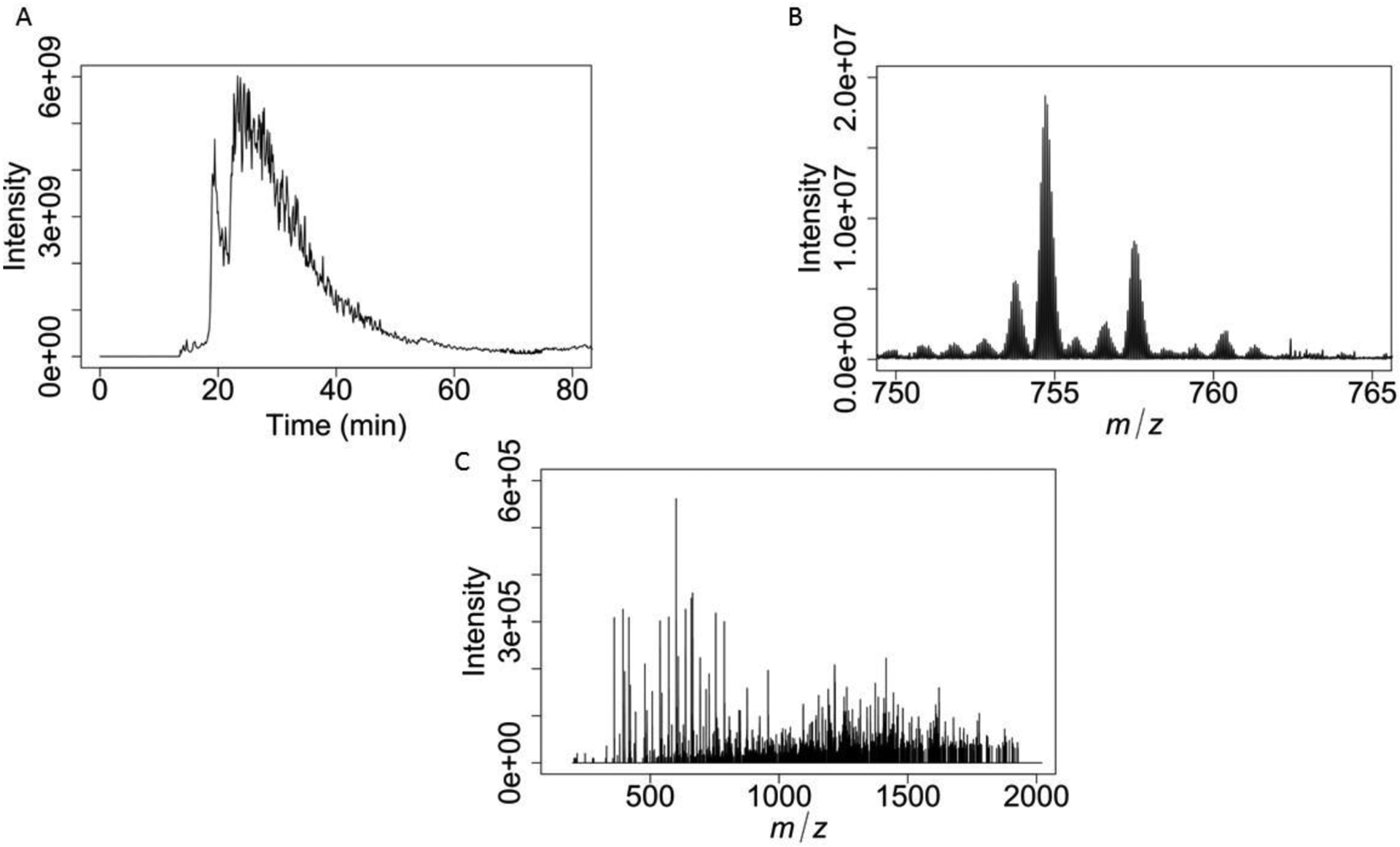
Figure 3 Quantitative Top Down Proteomic Analysis of Histone H4 Proteoforms
A) Histone H4 MS1 extracted ion chromatogram. The peak at 20 min corresponds to oxidized H4, while the dominant peak contains unoxidized H4 proteoforms.
B) MS1 scan of +15 charge state histone H4 precursors.
C) A single MS2 scan of Histone H4 K20me2.
Time Considerations
Basic Protocol 1 takes approximately 1.5 hours. General preparation such as thawing and preparing solutions may take 20 minutes. Nuclei isolation requires less than 40 minutes for 10 samples. Acid extraction requires less than 30 minutes for 10 samples.
Basic Protocol 2 takes approximately 4 hours. HPLC fractionation of histones requires approximately 1.5 hours for running a sample. The collection and drying of samples typically require 3 hours. HPLC-MS/MS requires approximately 1.5 hours for each sample.
Acknowledgments
This research was supported in part by the National Institute of General Medicine (R01GM139295-01). The authors thank Ms. Kala T. Pham and Mr. Jonathan C. Pickett for the critical reading of the manuscript for clarity.
Literature Cited
- 1.Smith LM, Kelleher NL, The Consortium for Top Down Proteomics Proteoform: a single term describing protein complexity. Nat Methods. 2013. Mar; 10(3): 186–187. doi: 10.1038/nmeth.2369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aebersold R, Agar JN, Amster IJ, et al. How Many Human Proteoforms are There? Nat Chem Biol. 2018. Feb 14;14(3):206–214 doi: 10.1038/nchembio.2576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Holt MV, Wang T & Young NL High-Throughput Quantitative Top-Down Proteomics: Histone H4. J. Am. Soc. Mass Spectrom 30, 2548–2560 (2019). [DOI] [PubMed] [Google Scholar]
- 4.Kelleher NL, Lin HY, Valaskovic GA, Aaserud David J., et al. Top Down versus Bottom Up Protein Characterization by Tandem High-Resolution Mass Spectrometry, J. Am. Chem. Soc 1999, 121, 4, 806–812. doi: 10.1021/ja973655h [DOI] [Google Scholar]
- 5.Cupp-Sutton KA, Wu S. High-throughput quantitative top-down proteomics. Mol Omics. 2020;16(2):91–99. doi: 10.1039/c9mo00154a [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Luger K, Mäder AW, Richmond RK, Sargent DF, and Richmond TJ: Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature 389, 251–260 (1997) [DOI] [PubMed] [Google Scholar]
- 7.Kornberg RD: Chromatin structure: a repeating unit of histones and DNA. Science 184, 868–871. (1974) [DOI] [PubMed] [Google Scholar]
- 8.Mariño-Ramírez Leonardo, Kann Maricel G, Shoemaker Benjamin A & Landsman David (2005) Histone structure and nucleosome stability, Expert Review of Proteomics, 2:5, 719–729, doi: 10.1586/14789450.2.5.719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Castillo J, López-Rodas G, Franco L. Histone Post-Translational Modifications and Nucleosome Organisation in Transcriptional Regulation: Some Open Questions. Adv Exp Med Biol. 2017. [DOI] [PubMed] [Google Scholar]
- 10.Allfrey VG, Faulkner R, Mirsky AE (1964) Acetylation and methylation of histones and their possible role in the regulation of RNA synthesis. Proc Natl Acad Sci U S A 315:786–794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Strahl BD, Allis CD. The language of covalent histone modifications. Nature. 2000;403:41–5 [DOI] [PubMed] [Google Scholar]
- 12.Talbert PB, Henikoff S. Histone variants on the move: substrates for chromatin dynamics. Nat Rev Mol Cell Biol. 2017. Feb;18(2):115–126. doi: 10.1038/nrm.2016.148. [DOI] [PubMed] [Google Scholar]
- 13.Dawson MA, Kouzarides T. Cancer epigenetics: from mechanism to therapy. Cell. 2012. Jul 6;150(1):12–27. doi: 10.1016/j.cell.2012.06.013. [DOI] [PubMed] [Google Scholar]
- 14.Wang T, Holt MV, Young NL. The histone H4 proteoform dynamics in response to SUV4–20 inhibition reveals single molecule mechanisms of inhibitor resistance. Epigenetics Chromatin. 2018;11(1):29. doi: 10.1186/s13072-018-0198-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kossel A Uber einen peptonartigen bestandteil des zellkerns. Z Physiol. Chem 1884. 8: 511–5 [Google Scholar]
- 16.Shechter D, Dormann HL, Allis CD, Hake SB. Extraction, purification and analysis of histones. Nat Protoc. 2007;2(6):1445–1457. doi: 10.1038/nprot.2007.202 [DOI] [PubMed] [Google Scholar]
- 17.Plazas-Mayorca MD, Zee BM, Young NL, Fingerman IM, Leroy G, Briggs SD, Garcia BA. One-Pot Shotgun Quantitative Mass Spectrometry Characterization of Histones. J Proteome Res. 2009. Nov;8(11):5367–74 doi: 10.1021/pr900777e [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sidoli S, Bhanu NV, Karch KR, Wang X, Garcia BA. Complete Workflow for Analysis of Histone Post-translational Modifications Using Bottom-up Mass Spectrometry: From Histone Extraction to Data Analysis. J Vis Exp. 2016;(111):54112. doi: 10.3791/54112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Young NL, DiMaggio PA, Plazas-Mayorca MD, Baliban RC, Floudas CA, Garcia BA, High-Throughput Characterization of Combinatorial Histone Codes, Mol Cell Proteomics. 8:2266–2284, 2009. doi: 10.1074/mcp.M900238-MCP200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sidoli S, Garcia BA. Characterization of Individual Histone Posttranslational Modifications and Their Combinatorial Patterns by Mass Spectrometry-Based Proteomics Strategies. Methods Mol Biol. 2017;1528:121–148. doi: 10.1007/978-1-4939-6630-1_8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen Y, Hoover ME, Dang X, et al. Quantitative Mass Spectrometry Reveals that Intact Histone H1 Phosphorylations are Variant Specific and Exhibit Single Molecule Hierarchical Dependence. Mol Cell Proteomics. 2016. Mar;15(3):818–33 doi: 10.1074/mcp.M114.046441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang T, Holt MV, Young NL, Early Butyrate Induced Acetylation of Histone H4 is Proteoform Specific and Linked to Methylation State Epigenetics. 2018;13(5):519–535. doi: 10.1080/15592294.2018.1475979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhou M, Malhan N, Ahkami AH, et al. Top-down mass spectrometry of histone modifications in sorghum reveals potential epigenetic markers for drought acclimation Methods. 2019. Oct 23;S1046–2023(19)30185–9. doi: 10.1016/j.ymeth.2019.10.007. [DOI] [PubMed] [Google Scholar]
- 24.Holt MV, Wang T, Young NL, One Pot Quantitative Top- and Middle-down Analysis of GluC Digested Histone H4. J Am Soc Mass Spectrom. 2019. Dec;30(12):2514–2525. doi: 10.1007/s13361-019-02219-1 [DOI] [PubMed] [Google Scholar]
- 25.Moradian A, Kalli A, Sweredoski MJ, Hess S. The top-down, middle-down, and bottom-up mass spectrometry approaches for characterization of histone variants and their post-translational modifications. Proteomics. 2014;14(4–5):489–497. doi: 10.1002/pmic.201300256 [DOI] [PubMed] [Google Scholar]
- 26.Dang X, Scotcher J, Wu S, et al. The first pilot project of the consortium for top-down proteomics: a status report. Proteomics. 2014;14(10):1130–1140. doi: 10.1002/pmic.201300438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gargano AFG, Shaw JB, Zhou M, et al. Increasing the Separation Capacity of Intact Histone Proteoforms Chromatography Coupling Online Weak Cation Exchange-HILIC to Reversed Phase LC UVPD-HRMS. J Proteome Res. 2018;17(11):3791–3800. doi: 10.1021/acs.jproteome.8b00458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dang X, Singh A, Spetman BD, et al. Label-Free Relative Quantitation of Isobaric and Isomeric Human Histone H2A and H2B Variants by Fourier Transform Ion Cyclotron Resonance Top-Down MS/MS. J. Proteome Res, 2016, 15 (9), pp 3196–3203. doi: 10.1021/acs.jproteome.6b00414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Greer SM, Sidoli S, Coradin M, Schack Jespersen M, Schwämmle V, Jensen ON, Garcia BA, Brodbelt JS. Extensive Characterization of Heavily Modified Histone Tails by 193 nm Ultraviolet Photodissociation Mass Spectrometry via a Middle-Down Strategy. Anal Chem. 2018. Sep 4;90(17):10425–10433. doi: 10.1021/acs.analchem.8b02320. [DOI] [PMC free article] [PubMed] [Google Scholar]