

Abstract
Despite IL-9 functioning as a pleiotropic cytokine in mucosal environments, the IL-9-responsive cell repertoire is still not well-defined. Here, we found IL-9 mediates pro-allergic activities in the lungs by targeting lung macrophages. IL-9 inhibits alveolar macrophage expansion and promotes recruitment of monocytes that develop into CD11c+ and CD11c− interstitial macrophage populations. Interstitial macrophages were required for IL-9-dependent allergic responses. Mechanistically, IL-9 affected the function of lung macrophages by inducing Arg1 activity. Compared to Arg1-deficient lung macrophages, Arg1-expressing macrophages expressed greater amounts of CCL5. Adoptive transfer of Arg1+ lung macrophages but not Arg1− lung macrophages promoted allergic inflammation that Il9r−/− mice were protected against. In parallel, the elevated expression of IL-9, IL-9R, Arg1 and CCL5 was correlated with disease in asthma patients. Thus, our study uncovers an IL-9/macrophage/Arg1 axis as a potential therapeutic target for allergic airway inflammation.
One Sentence Summary:
IL-9 promotes allergic inflammation by altering the heterogeneity and function of lung macrophages.
Introduction
Macrophages are the most abundant immune cells during homeostasis and a major antigen-presenting cell in the lung that bridges the innate and adaptive immune responses during disease development. Alveolar macrophages (AM) and interstitial macrophages (IM) are the two major heterogeneous lung macrophage populations (1). AMs reside in the alveoli, are directly in contact with the environment, and are derived from fetal liver and embryonic monocytes, maintaining a high level of self-proliferation. IMs are located in the interstitium and a represent small proportion of lung macrophages during homeostasis (1). Upon challenge, blood monocytes are recruited to the lung, where they can develop into IMs (1, 2). IMs were considered to be a uniform cell population but recent studies have challenged this paradigm and demonstrated that IMs can be divided into 2 or 3 distinct subpopulations (3, 4), including a CX3CR1+ SiglecF+ transitional macrophage identified by single cell RNA sequencing analysis and demonstrated to have profibrotic effects (5). These studies support the idea that lung IMs are a heterogeneous pool; however, our understanding of the heterogeneity of IMs under various disease states and their function during various immune responses is limited.
IL-9 plays a pleiotropic role in disorders including allergic disease and inflammatory bowel disease (6). It activates intracellular signaling by binding to IL-9 receptor (IL-9R) consisting of a unique alpha chain (IL-9Rα) and a common gamma chain shared with other family cytokine receptors including IL-2 and IL-4 (7, 8). IL-9R is expressed in multiple cell types including activated T cells, ILC2s, memory B cells and mast cells (6, 9–15). Numerous studies report that IL-9/IL-9R signaling can promote allergic inflammation characterized by the induction of chemokines and recruitment of eosinophils (16–18). Asthmatic patients also expressed high amounts of IL9 and IL9R (16, 17, 19). IL-9 enhances allergic responses by directly regulating mast cell function and also indirectly affecting other cells such as eosinophils, B cells and epithelial cells (6, 15). Nonetheless, the entire IL-9-responsive cell repertoire is still unclear.
As one of the most abundant immune cells in the lung, macrophages are key players in allergic responses. Previous studies showed that IL-9 affects the oxidative burst of human blood monocytes and AMs but little is known about how IL-9 affects the heterogeneity of lung macrophages and how that function might be linked to disease development (20, 21). In this study, we defined a comprehensive IL-9-responsive repertoire in allergic lung environment. We found that there are 3 distinct IL-9-responsive lung macrophage populations in the allergic lung. IL-9 inhibits the proliferation of AMs, promotes monocyte migration to the lung, and expands the IM population. More importantly, the induction of CD11c+ IM is mediated by IL-9 signaling in the context of allergic inflammation. Macrophages are the key drivers of IL-9-mediated immune response, with IL-9 regulating macrophage function by increasing Arg1 expression and arginase activity. Compared to Arg1− macrophages, Arg1+ macrophages express greater CCL5, a chemokine that promotes allergic airway inflammation through recruitment of eosinophils (22). Adoptive transfer of Arg1-expressing macrophages restores allergic inflammation in Il9r −/− mice. Importantly, evidence of the IL-9-macrophage-Arg1 axis was also found in asthma patients. Thus, our study advances the current understanding of IL-9-mediated allergic response and provides a rationale for targeting IL-9-responsive lung macrophages in therapeutic development.
Results
Deficiency of IL-9R inhibits allergic airway inflammation
Asthmatic patients express high amounts of IL-9 and IL-9R (16, 17, 19), and among all organs, the lung has one of the highest IL9R expression patterns in healthy donors (Fig. S1A). Although IL-9 can affect multiple cell types, the cells that directly respond to IL-9 in the lung are not well studied. To begin to explore this question in the context of a proallergic environment, we used a chronic house dust mite (HDM) challenge model (Fig. 1A). Compared to WT mice, Il9r−/− mice showed less lung inflammation demonstrated by decreased airway resistance (Fig. 1B), cellular infiltration of the lung (Fig. 1C, Fig. S1B), total bronchoalveolar lavage fluid (BALF) cell numbers (Fig. 1D), BALF eosinophils (Fig. 1E), lung eosinophils (Fig. 1F), HDM-specific IgE production (Fig. 1G) and mucus production (Fig. 1H and I). In our model, IL-9R-deficiency inhibited accumulation of mast cells (Fig. 1J and K, Fig. S1C), a well-known IL-9-responsive cell, and serum MCPT1 production (Fig. 1L). Serum MCPT1 was undetectable in PBS-challenged mice (Fig. 1L). The loss of IL-9R did not significantly affect the numbers of T cells, B cells, or cytokine production from CD4+ T cells (Fig. S1D and E). These results show that IL-9 signaling is critical in a chronic allergic lung inflammation model in vivo. Consistent with published reports (6, 15), CD4+ T cells were major IL-9 producers in the chronic allergic response (Fig. 1M and S1F).
Fig. 1. Deficiency of IL-9R inhibits allergic airway inflammation.
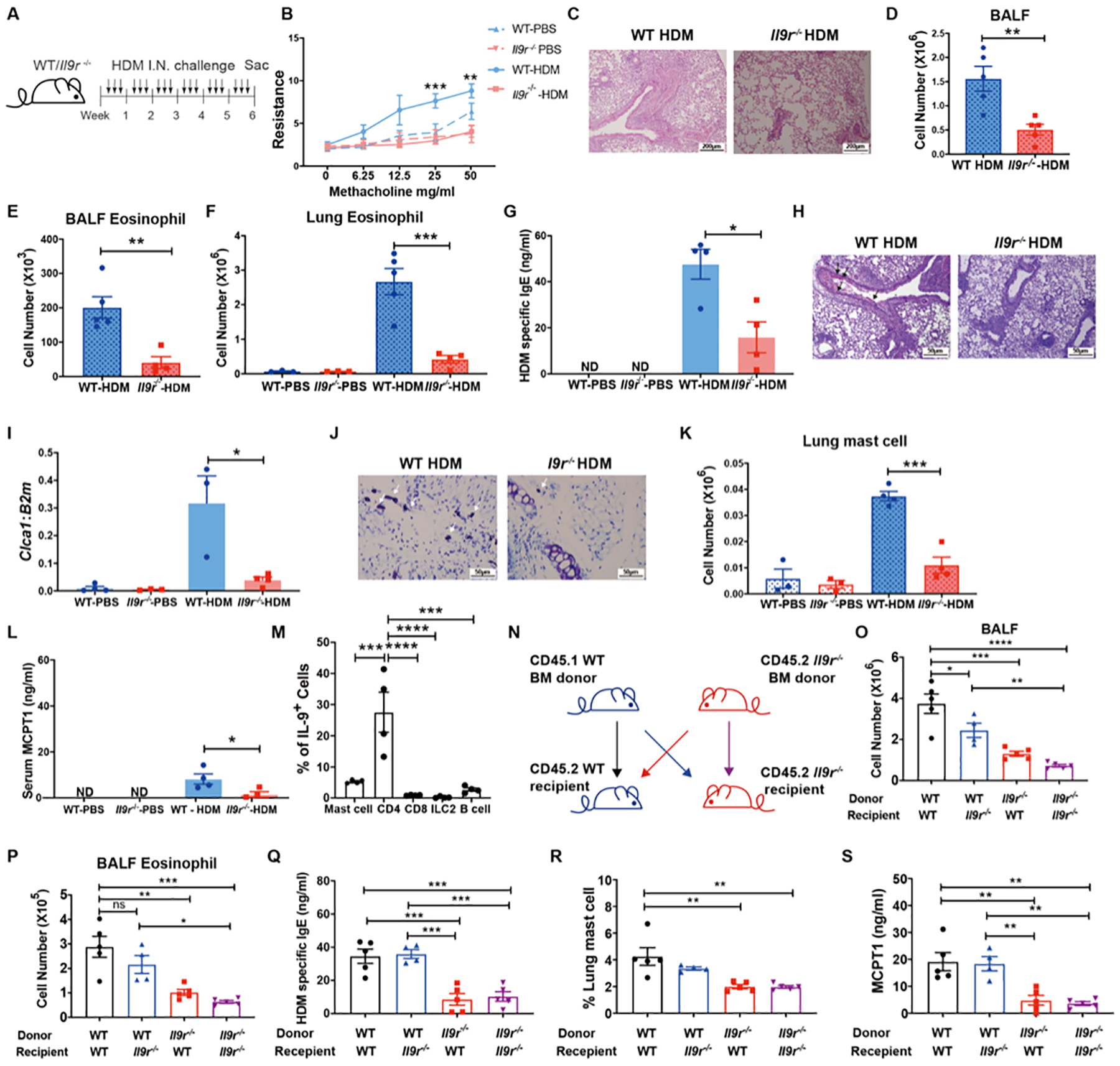
(A), Schematic of HDM extract–induced airway inflammation model. All measurements, unless indicated, are at the end of week 6.
(B), Airway resistance was measured in response to methacholine challenge one day after the last HDM treatment (n = 3–4 per group).
(C), Lung section with H&E staining.
(D–E), BALF total cell number and BALF eosinophil number were analyzed (n = 4–5 per group).
(F), Lung eosinophils were analyzed by flow cytometry (n = 3–5 per group).
(G), Serum HDM specific IgE level was analyzed by ELISA (n = 4 per group).
(H), Lung section with Periodic Acid-Schiff Stain (PAS) staining, arrows showed mucus production.
(I) Lung Clca1 mRNA expression was analyzed (n = 3–4 per group).
(J), Trachea section with toluidine blue staining, arrows show mast cells.
(K), Lung mast cells were analyzed by flow cytometry (n = 3–4 per group).
(L), Serum MCPT1 expression was analyzed by ELISA (n = 3–4 per group)
(M), IL-9+ cells were analyzed in HDM treated Il9 reporter (infer) mice (n=3–8).
(N-S), WT or Il9r−/− bone marrow cells were transferred to lethally irradiated WT or Il9r−/− mice. After reconstitution, the recipient mice were challenged with HDM as shown in (A). BALF total cell number (O), BALF eosinophils (P), Serum HDM specific IgE (Q), Lung mast cells (R) and serum MCPT1 level was analyzed (n = 4–5).
Data are presented as mean ± SEM from two independent experiments. Unpaired two-tailed Student t-test was used for comparison to generate p values in D–G, I, K–L. One-way ANOVA with Tukey’s multiple comparisons was used to generate p values in M, O-S. Two-way ANOVA with Sidak’s multiple comparisons was used to generate p values in B. ns p>0.05, *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001. ND: Undetected. See also Fig. S1.
To broadly determine the contribution of hematopoietic and non-hematopoietic cells to IL-9-dependent allergic responses, a bone marrow (BM) chimera experiment was performed (Fig. 1N). Compared to the WT bone marrow to WT recipient chimera group (WT → WT), the overall inflammation in both WT → Il9r−/− and Il9r−/− → WT chimeras was diminished (Fig. 1O–S), indicating both hematopoietic cells and non-hematopoietic cells responded to IL-9 and contributed to allergic inflammation. However, there was a greater impact of loss of IL-9R in hematopoietic cells (Fig. 1O–S) suggesting a stronger effect of IL-9/IL-9R signaling in hematopoietic cells during an allergic response.
Lung macrophages are key IL-9 responders in allergic pulmonary inflammation
We further observed lung F4/80+ CD206+ macrophages decreased in the Il9r−/− group compared to the WT group (Fig. S2A). To better understand how different pulmonary macrophage populations respond to IL-9 during allergic responses, we examined AM and IM separately to track their activities. Total lung macrophages were identified as CD64+ MerTK+ cells (Fig. 2A). AMs and IMs were distinguished by SiglecF and CD11c expression (Fig. 2A). In naïve mice, AM were the major pulmonary macrophage, while IM composed a small proportion of the total lung macrophages (Fig. 2A). During the allergic response, lung macrophages were present in three populations: AMs, CD11c− IMs and a third population that we termed CD11c+ IMs, which is supported in subsequent analyses (Fig. 2A and B). Among them, the CD11c+ IM population expanded considerably after HDM challenge (Fig. 2A and B). We did not observe a population of CX3CR1hi CD11c+ SiglecF− cells as previously noted in fibrosis models (Fig. S2B) (23–25). During development of allergic inflammation, the loss of Il9 or Il9r resulted in greater maintenance of the AM population and the reduced accumulation of CD11c− and CD11c+ IMs (Fig. 2A and B, S2C). Using a mixed bone marrow chimera approach to generate a model with WT and Il9r−/− cells in a uniform allergic inflammation (Fig. S2D), we defined intrinsic effects of IL-9R loss. Il9r−/− bone marrow cells had a decreased propensity to become IMs; the majority of AMs in the BAL have recipient mouse origin, and there was no significant difference in monocyte chimerism (Fig. 2C, Fig. S2E–F). To test whether these observations are consistent in other allergen-induced allergic disease model, we performed similar experiments with A. fumigatus (ASP)-induced allergic disease model. Consistent with results from the HDM model, IL-9 signaling inhibited AM and promoted CD11c− and CD11c+ IMs (Fig. S2G). Altogether, these data demonstrated that lung macrophage populations have discernable changes in response to IL-9 during allergic lung disease.
Fig. 2. Lung macrophages are key IL-9 responders in allergic response.
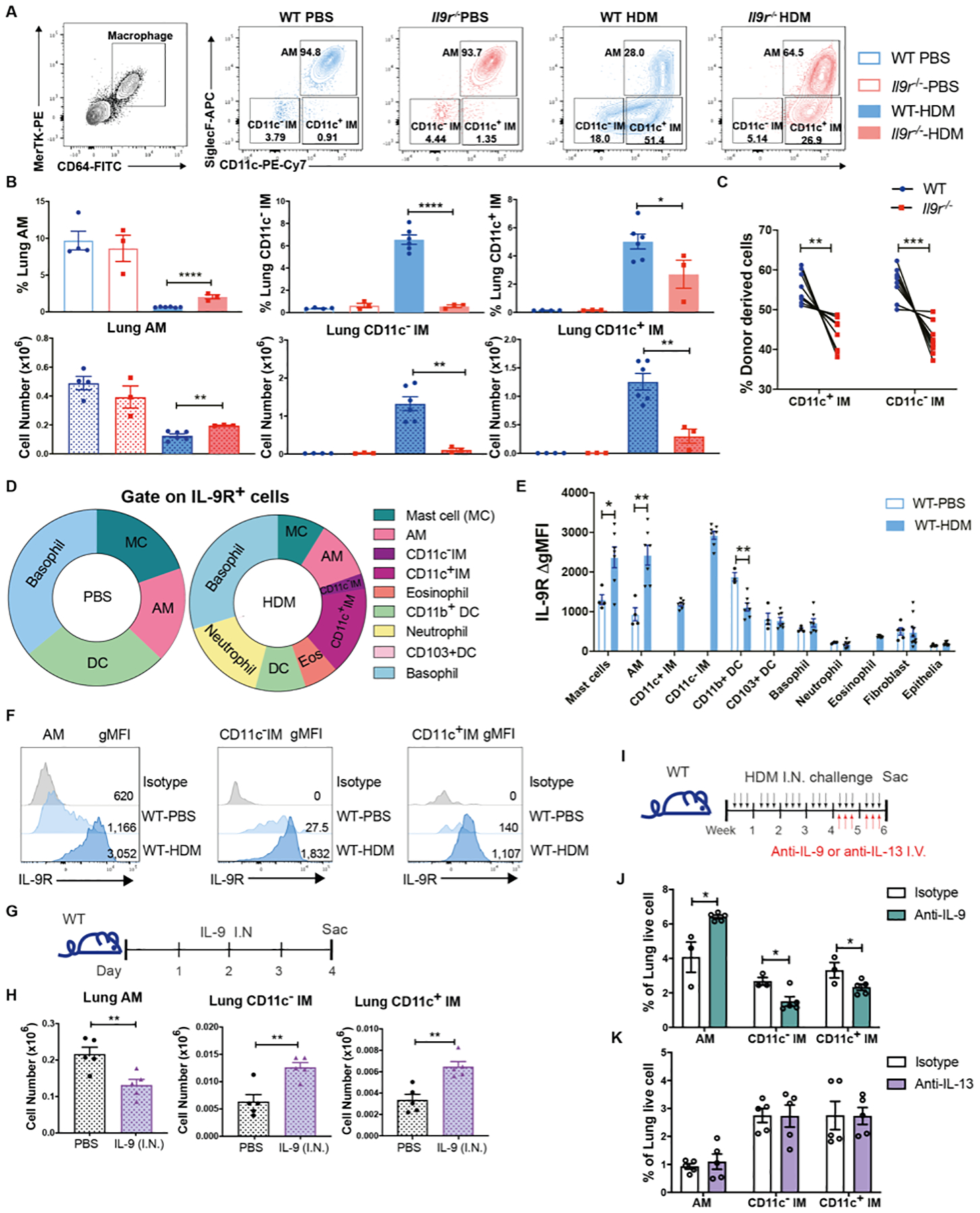
Mice were intranasally treated with HDM three times a week for six weeks.
(A and B), Flow cytometry analysis of lung macrophages (n = 3 for WT-PBS and Il9r−/−-PBS group, n = 6 for WT-HDM group, n = 3 for Il9r−/−-HDM group).
(C), WT (CD45.1) and Il9r−/− mice (CD45.2) bone marrow cells were mixed in 1:1 ratio and transferred to lethally irradiated recipient mice (CD45.1+CD45.2+ mice). After reconstitution, chimeric mice were treated with HDM, lung macrophages from donor cells were analyzed by flow cytometry (n = 9).
(D–F), Lung cells from PBS- or HDM-treated mice were stained with IL-9R and other surface markers for analysis by flow cytometry. (D), Pie chart analysis of IL-9R+ cells. (E), Fluoresence intensity of IL-9R among various populations were analyzed, ΔgMFI is the gMFI of each population minus the gMFI of isotype controls in that population; there were too few IMs for analysis in PBS-treated mice (n = 4–7). (F), Histogram of IL-9R in lung macrophage populations.
(G and H), Mice were intranasally treated with IL-9 for three days (G). (H), Lung macrophages were analyzed by flow cytometry (n = 5 per group).
(I-K), Mice were treated with HDM for six weeks, anti-IL-9, anti-IL-13 or isotype-matched control antibodies were intravenously injected every other day during the last two weeks (I). (J and K), Lung macrophages were analyzed by flow cytometry (n = 3–5).
Data are presented as mean ± SEM from three independent experiments. Unpaired two-tailed Student t-test was used for comparison to generate p values in B, E, H, and J. Paired two-tailed t-tests were used to generate p values in C. *p<0.05, **p<0.01, ****p<0.0001. See also Fig. S2.
To further define if lung macrophages directly responded to IL-9, we assessed IL-9R expression on lung cell populations (26). In PBS-treated mice, mast cells, basophils, DCs and AMs are the major IL-9R+ cells (Fig. 2D). During chronic allergic challenge, macrophages occupied an increased proportion of IL-9R+ cells (Fig. 2D). Moreover, macrophages expressed greater IL-9R than other populations (Fig. 2E and F, S2H). To assess if IL-9 is sufficient to control the redistribution of lung macrophages, IL-9 was intranasally administered to naïve mice for three days (Fig. 2G). Consistent with the results in allergic models, intranasal injection of IL-9 led to the redistribution of lung macrophages and increased mast cell numbers, but IL-9 alone did not alter IL-9Rα expression (Fig. 2H, S2I). IL-9 injection failed to alter type 2 cytokine expression in CD4+ T cells (Fig. S2J). Thus, macrophages are a prominent IL-9-responsive cell type during allergic lung inflammation.
Since we demonstrated that lung macrophages respond to IL-9 in allergic inflammation with altered dynamics, we next defined whether this is an IL-9-specific effect. Blocking antibodies against IL-9 or another type 2 cytokine, IL-13, were given to mice for the last two weeks of HDM challenges (Fig. 2I). Airway resistance was decreased in mice receiving anti-IL-9 treatment (Fig. S2K). Blocking IL-9 signaling increased AMs and inhibited the two IM populations, similar to the pattern observed in Il9r−/− mice (Fig. 2J and S2L). In contrast, anti-IL-13 antibody failed to alter the lung macrophage populations (Fig. 2K and S2M). Blocking IL-9 or IL-13 did not significantly affect reciprocal cytokine or other type 2 cytokine production in the lung (Fig. S2N–P). We further examined whether IL-9 alters macrophage populations by regulating IL-4R signaling. IL-4R expression in lung macrophages was not affected by IL-9 treatment in naïve mice or IL-9R deficiency in HDM model (Fig. S2Q and R). Il4−/− mice demonstrated decreased BAL total cell number (Fig. S2S), but comparable IL-9 detected in the BALF of WT mice and Il4−/− mice (Fig. S2T). In contrast to observations in Il9/Il9r-deficient mice or with blocking IL-9, IL-4R deficiency resulted in increases of both AMs and CD11c− IMs (Fig. S2U), while CD11c+ IMs were increased in Il4−/− mice receiving anti-IL-13 treatment (Fig. S2U). Together, these results suggest that IL-9 is distinct among type 2 cytokines in a broad scope of altering macrophage responses, and that blocking IL-9 at a late stage in the allergen challenge protocol results in similar changes to the macrophage populations as germ-line deletion of Il9 or Il9r.
Heterogeneity of pulmonary macrophages and blood monocytes in allergic inflammation
To better define the heterogeneity of pulmonary macrophages upon allergic response, a cellular indexing of transcriptomes and epitopes by sequencing (CITE-seq) with cell hashing experiment was performed. CD64+MerTK+ total lung macrophages and CD115+ blood monocytes were sorted from HDM-challenged mice, and further labelled by hashtag oligonucleotide (HTO) antibodies separately. Lung macrophages were stained with Antibody Derived Tag (ADT) antibodies against CD11c and SiglecF. Lung macrophages and blood monocytes were pooled for analysis with the 10X Genomics Chromium platform (Fig. 3A). Twenty-three clusters were identified from the transcriptome data and visualized by UMAP projection (Fig. 3B and S3A). As defined by HTO labeling of sorted cells, 6 clusters were blood monocytes and 14 clusters were lung macrophages. Some clusters were either dead cells or non-monocytes/macrophages and were excluded from further analysis (Fig. 3B and C, Fig.S3A). Based on the ADT-defined protein expression profiles and standard cell identifying gene profiles, we clustered cells as AM, CD11c− IM, and CD11c+ IM (Fig. 3D and E, Fig. S3B). Several small clusters were ADT-SiglecF+ but did not have Siglecf mRNA and were excluded from analysis.
Fig. 3. Heterogeneity of pulmonary macrophages and blood monocytes in allergic inflammation.
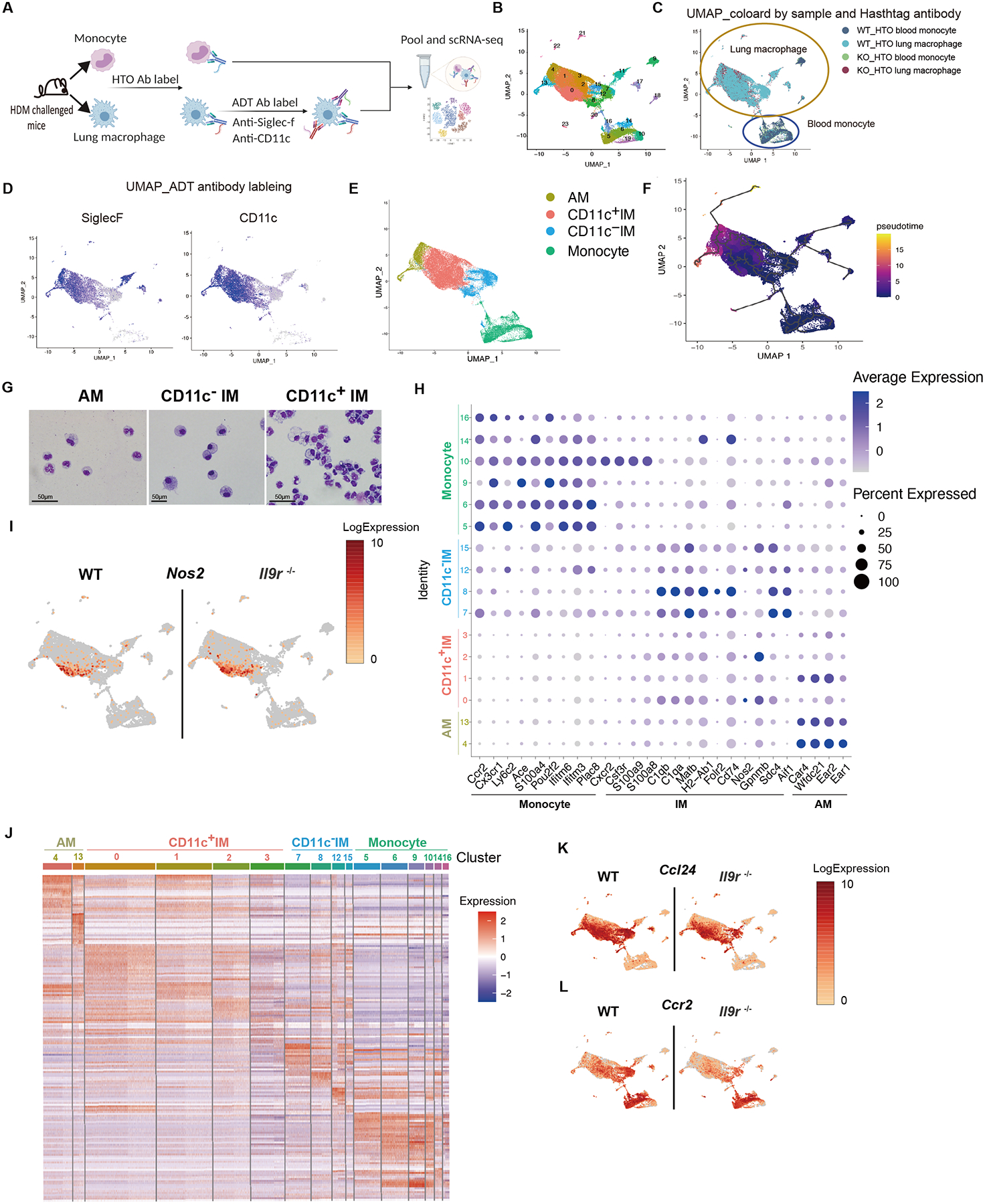
(A-G), CITE-seq combined with cell hashing analysis for blood monocytes and lung macrophages from HDM-challenged mice, 15 mice were pooled per group before FACS sorting (A). (B), UMAP showing clusters based on gene expression. (C), UMAP based on cell hashing antibodies. (D), UMAP based on ADT antibodies. (E), Annotation of the lung macrophages within the UMAP based on both transcriptome and surface proteome. (F), Pseudotime analysis using CCR2+ blood monocytes as the root cell type. (G), Lung macrophages were sorted from HDM-treated mice and morphology were analyzed by cytospin.
(H), Dot plot showing the gene expression in different clusters. The number on the y-axis are the cluster numbers shown in (B).
(I), UMAP showing Nos2 expression.
(J), Heatmap showing differential expressed genes in different clusters. The number on the top are the cluster numbers shown in (B).
(K-L), UMAP showing gene expression of the indicated genes.
See also Fig. S3.
To further identify the relationship among these clusters, a pseudotime analysis was performed using CCR2+ monocytes as the root cell type. We found that CD11c− IM were most closely related to blood monocytes, and AM shared the least similarity with either CD11c− IM or blood monocytes (Fig. 3F and S3C). Pseudotime relationships were not altered among cells from Il9r−/− mice, suggesting that IL-9 impacts effector recruitment and/or function but not differentiation (Fig. S3C). Consistent with the gene expression profile, the CD11c+ IM were more related to blood monocytes and CD11c− IM than AM (Fig. 3F). CD11c+ IM also shared similar morphology with CD11c− IM. Both populations displayed vacuoles in their cytoplasm and nucleus, whereas AM demonstrated more regular round or oval shape nuclei (Fig. 3G). Hence, we defined this population as CD11c+ IM. Although various pulmonary macrophage populations shared some common transcriptional profiles, they also exhibited distinct gene expression patterns (Fig. 3H–J and S3D). Moreover, even within each population, transcriptional heterogeneity further defined subsets of cells (Fig. 3J). Nos2 and Folr2 are exclusively expressed in CD11c+ IM clusters (Fig. 3I and S3E). Cd72 is preferentially expressed in CD11c− IM (Fig. S3E). Blood monocytes shared some common markers with IM including Tmem176a and Tmem176b (Fig. S3G), and also expressed unique genes such as Trem14 (Fig. S3E). The loss of Il9r caused alterations in gene expression in both blood monocyte and lung macrophage populations (Fig. 3K–L and S3F–G). IL-9 signaling significantly promoted chemokine expression in macrophage populations (Fig. 3L and S3F–G). Thus, lung macrophages not only act as IL-9 responders, but potentially contribute to the IL-9-dependent allergic response.
Lung macrophages amplify IL-9-mediated allergic lung inflammation
To further dissect if the altered lung macrophage dynamics are a critical component in the development of the allergic response, we first tested whether depleting macrophages would impact allergic airway inflammation. We depleted macrophages in the airway or IM and circulating monocytes by administering clodronate intranasally or intravenously, respectively (Fig. 4A). Consistent with previous studies, intranasal clodronate injection primarily depleted the AM without affecting the CD11c− IM (Fig. S4A and B) (27, 28). This treatment did not alter the CD11c+ IM (Fig. S4B), indicating the majority of this population does not reside in the airway. Intravenous injection of clodronate successfully depleted blood monocytes and CD11c− IM, but not the AM population (Fig. 4B, S4C and D). Strikingly, it significantly depleted the CD11c+ IM, further supporting that they are indeed residing in the tissue (Fig.4B and C, S4E). Importantly, WT mice developed reduced lung inflammation similar to Il9r−/− mice after depletion of monocytes and IMs (Fig. 4D–F, S4F–H), which supports the possibility that blood monocytes and IMs are required for the development of allergic airway inflammation.
Fig. 4. Lung macrophages amplify IL-9 mediated allergic lung inflammation.
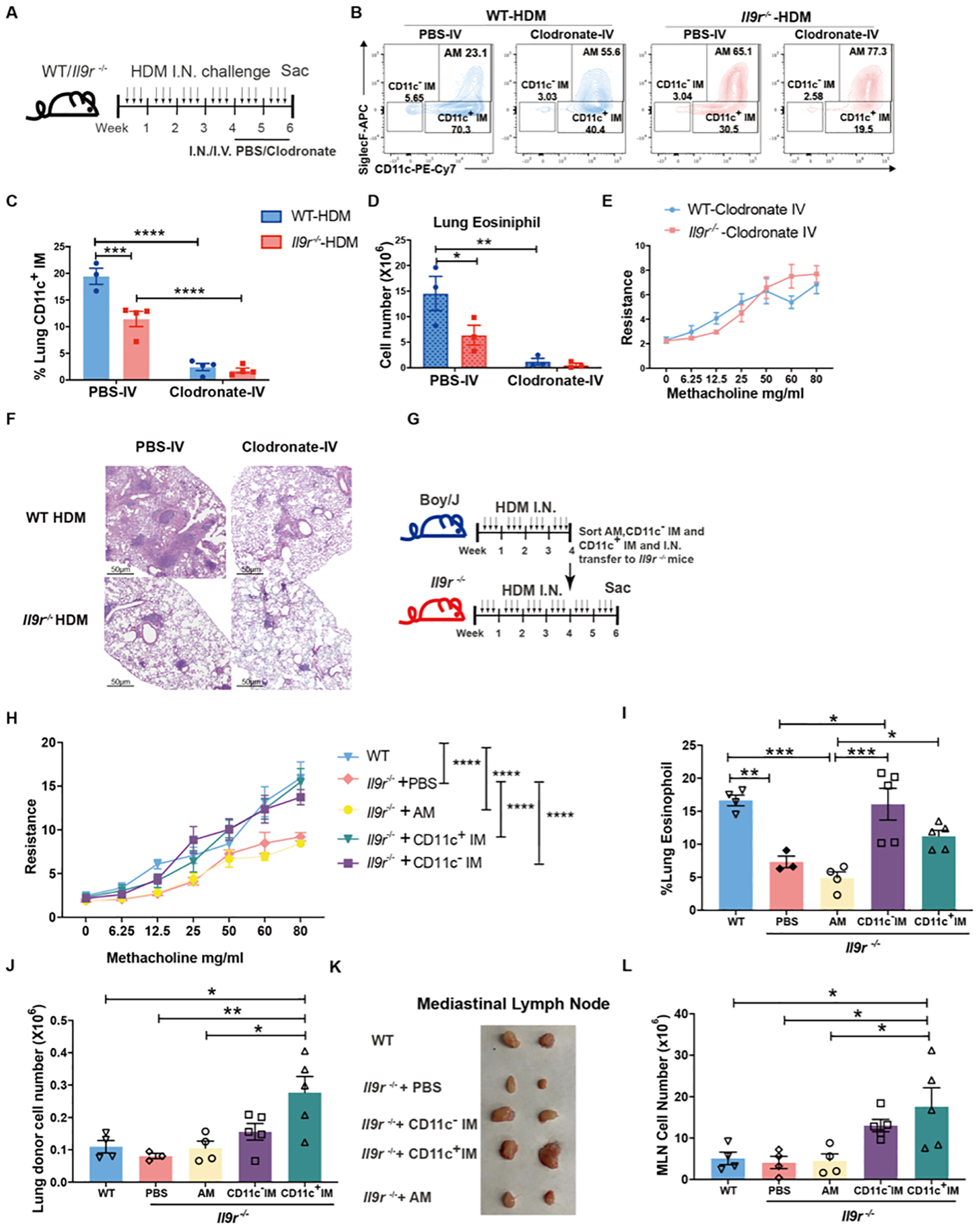
(A), Mice were intranasally treated with HDM three times a week for six weeks. Clodronate was intranasally or intravenously injected to the mice every other day for the last two weeks of the HDM model.
(B-F), Mice were treated as described in A. Lung macrophages (B-C) and eosinophils (D) were analyzed by flow cytometry from lavaged lung. Dot plots indicate the percentage of total lung macrophage populations. Bar graphs quantify live lung macrophage populations. (E), Airway resistance was measured in response to methacholine challenge one day after the last dose of HDM treatment. (F), H&E staining of lung sections (n = 3–4 per group).
(G-L), Boy/J and Il9r−/− mice were treated with HDM three times a week for 4 weeks. Macrophage populations were sorted from Boy/J mice one day after the last HDM treatment and intranasally transferred to Il9r−/− mice followed by HDM challenge every other day for another two weeks (G). (H), Airway resistance was measured in response to methacholine challenge one day after the last HDM treatment at the end of week 6. Statistical differences are indicated between groups in the panel key for clarity. Lung eosinophils (I) and lung donor cells (J) were analyzed by flow. MLN size (K) and cell numbers (L) were analyzed (n = 3–5 per group).
Data are presented as mean ± SEM from two independent experiments. One-way ANOVA with Tukey’s correction for multiple comparisons was used to generate p values in I, J and L. Two-way ANOVA with Sidak’s multiple comparisons was used to generate p values in C and D. Two-way ANOVA with Tukey’s multiple comparisons was used to generate p values in H to compare group effect by using mean of every group. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001. See also Fig. S4.
Since the clodronate injection might affect other cell populations, such as monocytes and DCs (Fig. S4C and S4I), we further defined the functional contribution of the macrophage populations by an adoptive transfer experiment (Fig. 4G). Intranasal adoptive transfer of WT CD11c− IM or CD11c+ IM increased airway resistance and lung eosinophil infiltration in Il9r−/− mice (Fig. 4H and I). Donor cells were successfully detected in the lung interstitium (Fig. 4J and S4J–K), and percentages of DCs were not affected by treatment (Fig. S4L). Donor DCs were not detected in mediastinal LN (MLN) (Fig. S4M). Compared to WT mice, the Il9r−/− mice that received PBS showed much smaller MLN and transferred AM did not affect the size of the lymph node (Fig. 4K). However, transfer of either CD11c− IM or CD11c+ IM resulted in enlarged MLN to a size comparable to WT mice (Fig. 4K). Mice that received CD11c+ IM, had significantly larger lymph nodes than the WT mice (Fig. 4K). This was consistent with increased total cell numbers in the MLN (Fig. 4L). Collectively, these findings suggest that lung macrophages, particularly CD11c+ IM and CD11c− IM, contribute to IL-9-induced allergic airway disease.
IL-9 enhances allergic inflammation via recruiting monocytes
To further explore the underlying mechanisms on how IL-9 alters the subpopulations of lung macrophages, we examined the proliferation and cell death of macrophages during the allergic response. Deficiency of Il9r increased the proliferation of AM indicated by higher amount of Ki67 but did not affect the rate of cell death (Fig. S5A and B). Both WT and Il9r−/− IM showed similar capacity for proliferation and cell death, suggesting other mechanisms were involved in the regulation of IM by IL-9 (Fig. S5A and B). The efferocytosis capacity of lung macrophages was not affected by the loss of Il9r (Fig. S5C). Since monocytes are a major source of increased IM in inflammatory environments (2), we hypothesized that IL-9 might affect monocyte recruitment to the lung. Compared to WT mice, the Il9r−/− mice treated with HDM retained more monocytes in blood and bone marrow (Fig. 5A and B). However, IL-9R expression was positively correlated with CCR2 expression, a key chemokine receptor required for classical monocyte migration (Fig. 5C and D). These data suggested that IL-9 might regulate classical monocyte migration to the lung via CCR2. To directly test this, we performed a migration assay, in which IL-9 and/or CCL2 were added to the lower chamber of a transwell and bone marrow-derived monocytes were placed in the upper chamber before cells were allowed to migrate for 3 hours. IL-9 exclusively promoted monocyte migration in the presence of CCL2 (Fig. 5E). Human monocytes from healthy donors also expressed IL-9R, which is consistent with previous studies (Fig. 5F) (21). IL-9 enhanced CCL2-stimulated human PBMC-derived monocyte migration, which likely results from the upregulation of CCR2 following IL-9 treatment (Fig. 5G and H). In parallel, we observed that peripheral blood CD14+ monocytes from children with a diagnosis with asthma showed increased IL-9R expression compared to monocytes from children that are atopic but did not have physician-diagnosed asthma (Fig. 5I) (29–31). Together, these results suggest that IL-9 promotes the capacity of classical monocytes to migrate to the allergic lung.
Fig. 5. IL-9 enhances allergic inflammation via recruiting monocytes.
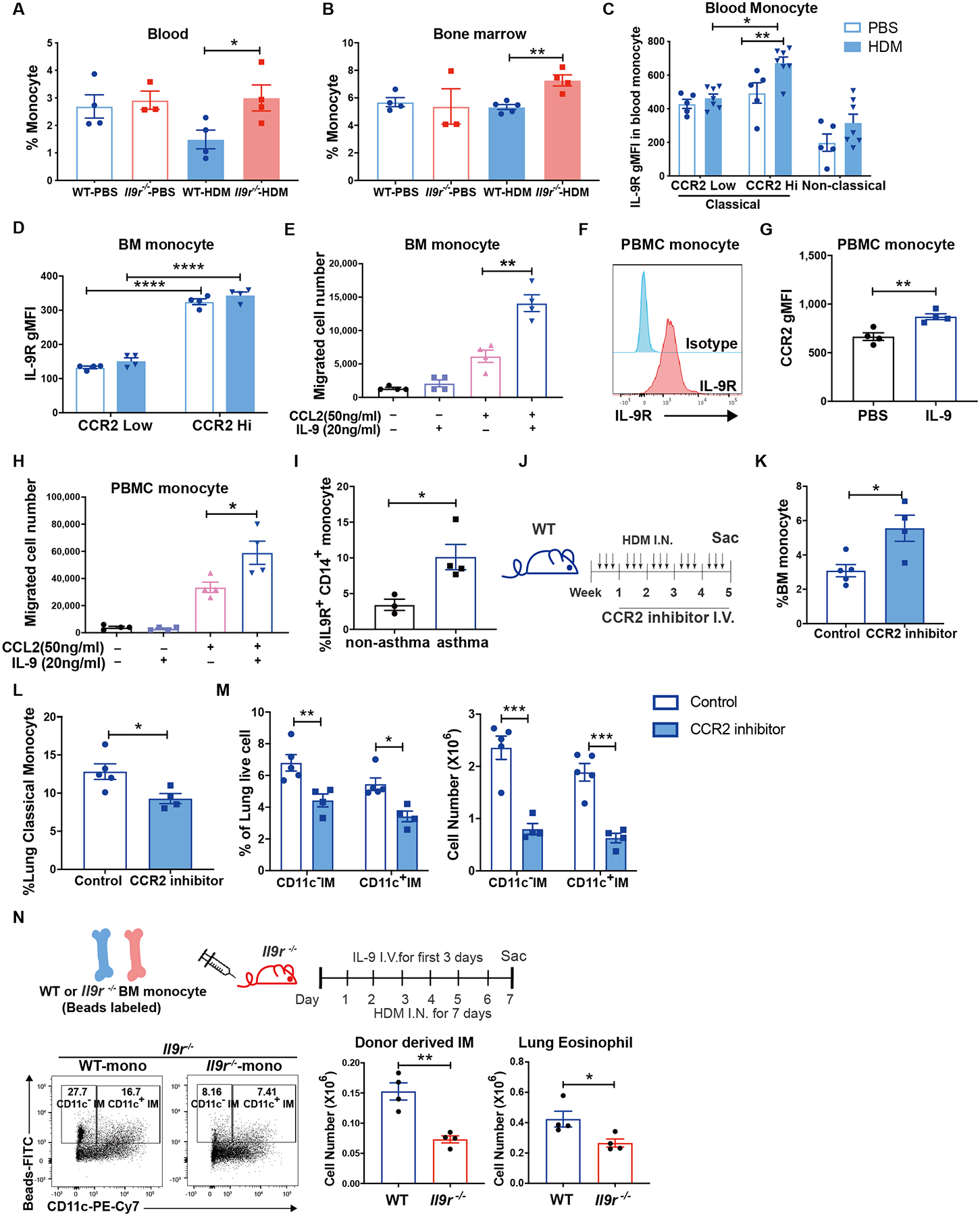
(A-D), Mice were intranasally treated with HDM three times a week for 6 weeks. Blood (A) and bone marrow (B) monocytes were analyzed (n = 3–5 per group). (C and D), CCR2lo and CCR2hi monocytes were gated and IL-9R expression was further analyzed (n = 4–7 per group).
(E), Bone marrow monocytes were isolated and plated on the top chamber of a transwell, IL-9 or CCL2 was added to the lower chamber, and cells were allowed to migrate for 3 hours before counting (n = 4 per group).
(F-H), Monocytes were isolated from human PBMCs, IL-9R (F) and CCR2 (G) expression was analyzed by flow, migration assay was performed as described in (E), migrated cells were analyzed (H) (n = 4 per group).
(I), IL-9R expression were analyzed in non-asthma or asthma donors’ blood CD14+ monocytes (n = 3 for non-asthma group, n = 4 for asthma group).
(J-M), Mice were treated with HDM three times a week for 5 weeks. CCR2 inhibitor was intravenously injected to the mice twice a week for 4 weeks (J). Bone marrow monocytes (K), lung monocytes (L) and lung macrophages (M) were analyzed by flow cytometry (n = 5 for control group, n = 4 for CCR2 inhibitor group).
(N), Fluorescent bead-labeled monocytes were transferred to Il9r−/− mice, recipient mice were treated with IL-9 and HDM, lung macrophages and eosinophils were analyzed on day 7, dot plots were gated on MerTK+ CD64+ SiglecF− live cells (n=4 per group).
Data are presented as mean ± SEM from two independent experiments. Unpaired two-tailed Student t-test was used for comparison to generate p values in A-B, E, G-I, K-L and N. Two-way ANOVA with Sidak’s multiple comparisons was used to generate p values in C-D and M. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001. See also Fig. S5.
Since the results above supported the idea that IL-9 promotes CCR2-dependent monocyte migration, we tested whether CCR2+ monocytes were required for the accumulation of CD11c− and CD11c+ IM. Following administration of a CCR2 inhibitor to mice in the HDM model (Fig. 5J), there were increased BM monocytes and reduced lung monocytes, compared to control mice (Fig. 5K and L, S5D). This confirmed that the inhibitor successfully arrested the monocytes in the bone marrow. Furthermore, the inhibitor significantly reduced CD11c− and CD11c+ IM but had no impact on AM (Fig. 5M and S5E), supporting the hypothesis that CD11c− and CD11c+ IM accumulation in the lung were dependent on CCR2+ monocytes. To further confirm this, we isolated BM monocytes from HDM-challenged mice and transferred them to WT CD45.1 recipient mice. IL-9 and HDM were given to the recipient mice for 7 days to establish an environment for monocytes to differentiate into IM (Fig. S5F). Mice that received WT monocytes showed slightly increased donor monocytes and CD11c− IM in the lung, and significantly more CD11c+ IM than mice that received Il9r−/− monocytes (Fig. S5G and H). The number of AM was not altered by the donor cells (Fig. S5I). In addition, the WT but not the Il9r−/− monocyte recipients showed strongly elevated BALF total cell numbers, lung eosinophils and lung inflammation (Fig. S5J–L). To assess the effect of IL-9 signaling on monocytes, fluorescent bead-labeled monocytes were transferred into Il9r−/− mice (Fig. 5N) (32). Flow cytometric analysis of bead-labeled monocytes showed that IL-9 signaling promotes monocytes to become lung IMs (Fig. 5N). Together, these results demonstrated that mechanistically, IL-9 inhibited the self-renewal capacity of AM and promoted monocyte recruitment to the lung for subsequent development into IM. Moreover, IL-9 signaling induces the pro-allergic function of monocytes and IMs.
IL-9 impacts lung macrophage function by regulating Arg1 expression
As IL-9 signaling is required for the pro-allergic effect of lung macrophages, we next wanted to define the underlying mechanism of IL-9-mediated lung macrophage function. We found that Arg1 was one of the most differentially expressed genes from our CITE-Seq data (Fig. 6A), and Arg1 has been shown to be a key factor related to the pathogenesis of asthma (33). Thus, we hypothesized that Arg1 might be a potential effector in the IL-9-macrophage axis. Consistent with our CITE-Seq data, Arg1 protein expression was substantially reduced in lung IM populations in Il9r−/− mice with allergic inflammation (Fig. 6B). IMs were the predominant Arg1+ macrophages in this model (Fig. S6A). In the mixed bone marrow chimeric mice described in Fig. S2D, the increased proportion of Arg1+ IMs, but not AMs, from WT donors further confirms that IL-9 signaling regulates Arg1 expression (Fig. 6C and S6B). Because Arg1 is also a marker for immunosuppressive macrophages and to confirm that differential expression is related to functional differences, arginase activity was measured in total lung macrophages and serum from HDM-treated mice (Fig. 6D and E). In accordance with the diminished expression of Arg1, Il9r−/− mice also showed significantly lower arginase activity than WT mice (Fig. 6D and E). Moreover, IL-9 promoted Arg1 expression from IMs both in vivo and after ex vivo stimulation (Fig. 6F and G). Loss of IL-9R did not impact Arg1 production of IMs in response to ex vivo stimulation of IL-4 and IL-13 (Fig. S6C). ILC2s have also been shown to be an important Arg1 producer (35), and while ILC2s did expand early during the chronic challenge (Fig. S6D), Arg1 expression from ILC2s in Il9r−/− mice was comparable to WT mice (Fig. S6E). Together, these data suggest that Arg1 is a downstream target of IL-9 signaling in lung macrophages.
Fig. 6. IL-9 impacts lung macrophage function by regulating Arg1 expression.
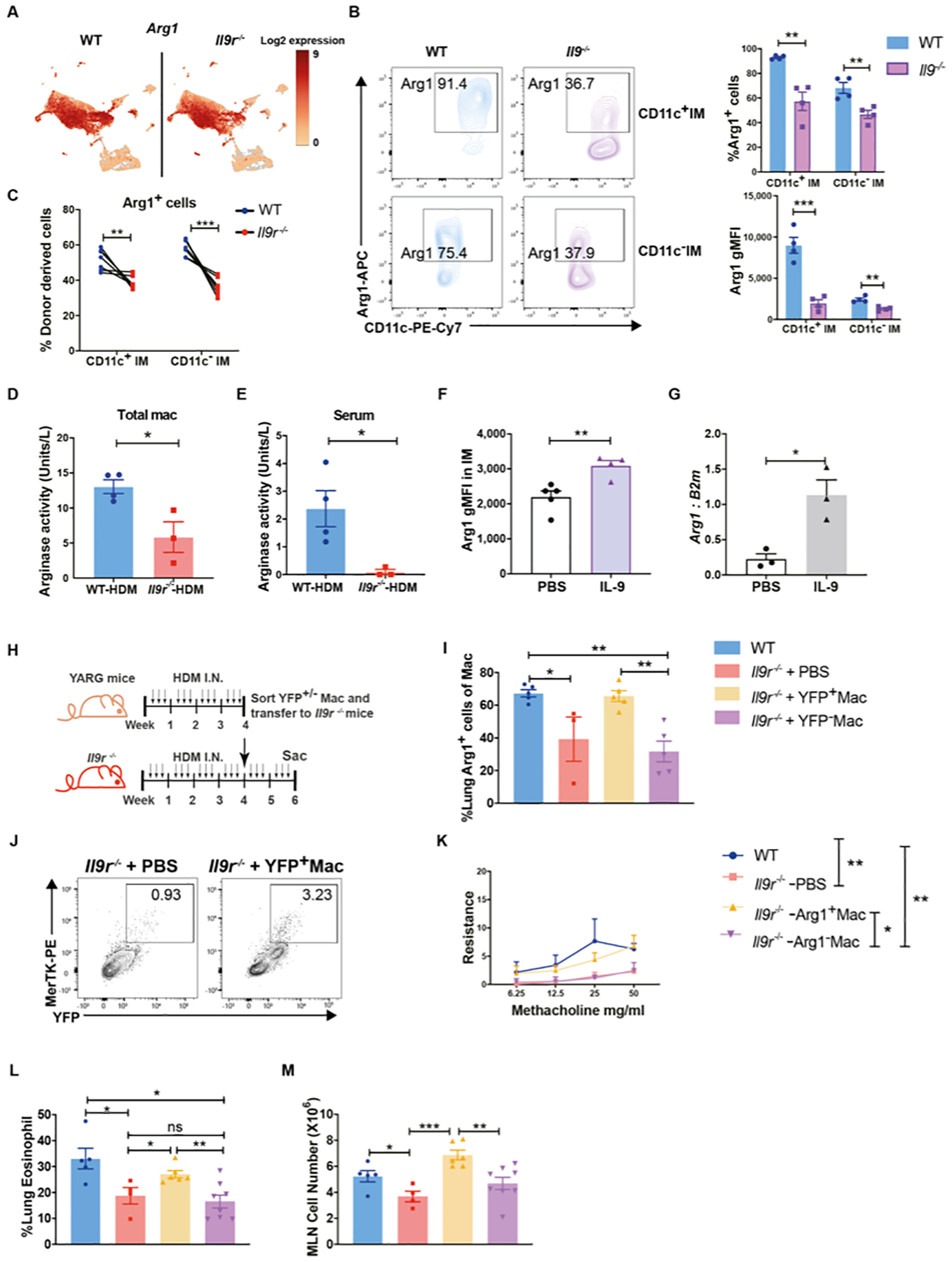
(A), UMAP showing Arg1 expression from CITE-seq experiment described in Fig. 3.
(B), Arg1+ cells from HDM-treated mice were analyzed by flow cytometry (n=4 per group). Dot plots were gated on lived IMs.
(C), Mixed bone marrow chimeric mice were generated and treated as described in Fig. S2D, donor derived Arg1+ IMs were analyzed by flow cytometry (n = 8 per group).
(D and E), Arginase activity was analyzed in lung macrophages and serum from HDM-treated mice (n=3–4 per group).
(F), Naïve mice were treated with IL-9 for three days, Arg1 production from IMs were analyzed (n=5 for PBS group, n=4 for IL-9 treated group).
(G), IMs were sorted from WT HDM-treated mice and stimulated with IL-9 for 24hs, Arg1 mRNA expression was analyzed (n=3).
(H-M), YARG and Il9r−/− mice were treated with HDM three times a week for 4 weeks. YFP+/−macrophage populations were sorted from YARG mice one day after the last HDM treatment and intranasally transferred to Il9r−/− mice followed by HDM challenge every other day for another two weeks (H). Lung Arg1+ macrophages (I) and YFP+ macrophages (J) were analyzed by flow cytometry. (K), Airway resistance was measured in response to methacholine challenge one day after the last HDM treatment at the end of week 6. Statistical differences are indicated between groups in the panel key for clarity. (L), Lung eosinophils were analyzed by flow. (M), MLN cell number was analyzed (n= 3–8 per group).
Data are presented as mean ± SEM from two independent experiments. Paired two-tailed t-tests were used to generate p values in B and C. Unpaired two-tailed Student t-test was used for comparison in D–G and L–M. One-way ANOVA with Tukey’s multiple comparisons was used to generate p values in I. Two-way ANOVA with Tukey’s multiple comparisons was used to generate p values in K to comparing group effect by using mean of every group. *p<0.05, **p<0.01, ***p<0.001, ****p<0.001. See also Fig. S6.
To test whether Arg1+ macrophages contribute to IL-9-mediated allergic responses, Arg1-expressing or non-expressing macrophages were sorted from Arg1 YFP reporter (YARG) mice that were subjected to HDM challenge, and subsequently transferred into Il9r−/− mice (Fig. 6H). YFP+ macrophages migrated to the lung (Fig. 6I) and restored the Arg1+ macrophage population in the lung of Il9r−/− mice (Fig. 6J). Strikingly, YFP+ (Arg1+) macrophages successfully rescued allergic lung inflammation in Il9r−/− mice, while YFP− macrophages did not (Fig. 6K–M, S6F).
To directly test the requirement for Arg1 in allergic lung inflammation, Arg1fl/flLysM-Cre+ or littermate control mice were challenged with HDM; Arg1fl/flLysM-Cre+ showed decreased Arg1 expression in IMs and lower serum arginase activity (Fig. 7A and S6G). Lung macrophages were the major Arg1 producers in vivo (Fig. S6H). Mice lacking Arg1 expression in myeloid cells showed significantly decreased allergic inflammation, indicated by decreased BALF total numbers, BALF eosinophils, lung eosinophils and MLN cell numbers (Fig. 7B–E). Interestingly, consistent with the decreased Ccl5 expression from Il9r−/− IMs in our CITE-Seq data (Fig. 7F), Arg1-deficient IMs also demonstrated lower CCL5 expression than control IMs (Fig. 7G), which is also consistent with the lower level of serum CCL5 in both Arg1fl/flLysM-Cre+ and Il9r−/− mice (Fig. 7H and I). In response to IL-9 stimulation in vitro, Arg1+ macrophages secreted more CCL5 than Arg1− macrophages (Fig. 7J). IL-9 also increased CCL5 expression from IMs in vivo (Fig. 7K). Furthermore, IL-9 expanded the Arg1+ CCL5+ IM population in naïve mice, and CCL5 is exclusively expressed in Arg1+ IMs (Fig. 7L). Moreover, in a uniform inflammatory environment, Arg1+ IMs from HDM-treated mice expressed more CCL5 than Arg1− IMs (Fig. 7M). These results suggest that IL-9 regulation of allergic airway inflammation requires Arg1 expression in lung macrophages, and Arg1+ IMs secrete more CCL5 to induce eosinophil-mediated allergic inflammation.
Fig. 7. IL-9 promotes allergic inflammation by inducing CCL5 production from lung macrophages.
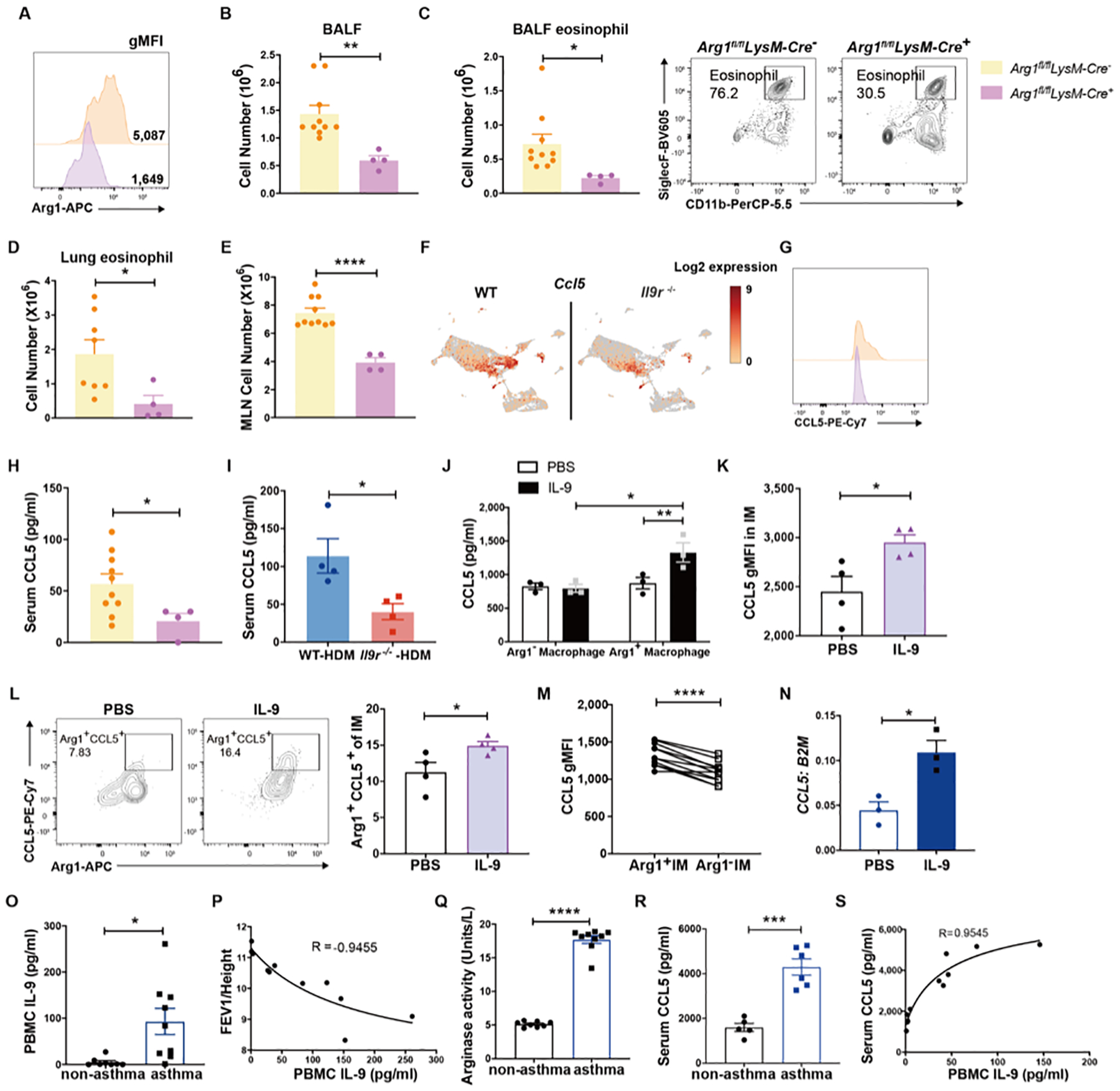
Arg1fl/fl LysM-Cre+/− mice were treated with HDM for 6 weeks.
(A), Arg1 expression in IMs was analyzed. BALF total cell number (B), BALF eosinophils (C), Lung eosinophils (D) and MLN cell number were analyzed (n = 4–10).
(F), UMAP showing Ccl5 expression from CITE-seq experiment described in Fig. 3.
(G), CCL5 expression in IMs was analyzed by flow cytometry (n = 4–10).
(H and I), Serum CCL5 level was analyzed by ELISA (n = 4–10).
(J), FACS sorted Arg1+/− macrophages were treated with IL-9 for 24 hours, CCL5 level was measured by ELISA (n=3).
(K-L), Naïve mice were treated with IL-9 for three days, CCL5 and Arg1 expression from IMs were analyzed (n=4).
(M), CCL5 expression was analyzed by gating on Arg1+ or Arg1− IMs from WT HDM treated mice (n=11).
(N), Human monocyte were cultured under M2 macrophage condition, CCL5 mRNA expression was analyzed (n = 3).
(O), IL-9 production from PBMCs was analyzed from non-asthma donors and asthma patients (n=7–9).
(P), Correlation between FEV1 corrected for height and IL-9 production from patient PBMCs.
(Q-R), Arginase activity (Q) and CCL5 level (R) was analyzed from non-asthma donors and asthma patients (n=5–9).
(S), Correlation of PBMC IL-9 and serum CCL5 concentration (n=11).
Data are presented as mean ± SEM from two independent experiments. Unpaired two-tailed Student t-test was used for comparison in B–C, D–E, H–I, K–L, N–O and Q–R. A paired two-tailed t-test was used to generate the p value in M. Two-way ANOVA with Sidak’s multiple comparisons was used for comparisons in J. Spearman correlation was performed for P and S, *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001. See also Fig. S6.
Consistent with the results from murine cells, IL-9 promotes CCL5 expression from human bone marrow-derived macrophages (Fig. 7N). To further detect if this paradigm is observed in patient samples, we analyzed IL-9 concentration and arginase activity from a well-defined population of atopic children (29, 30) and IL-9 produced from infant PBMCs was inversely correlated with parameters of lung function (36). Atopic children with asthma diagnosis at age 5 showed increased IL-9 production from anti-CD3-stimulated PBMCs that was inversely correlated with lung function and associated with increased serum arginase activity compared to children that are atopic but did not have physician-diagnosed asthma (Fig. 7P and Q). Compared to non-asthmatic patients, asthmatic patients also showed increased CCL5 expression (Fig. 7R). IL-9 production is positively corelated to serum CCL5 level (Fig. 7S). Collectively, these data suggest that IL-9 regulates allergic airway inflammation by increasing the expression of Arg1 and CCL5 in lung macrophages.
Discussion
Lung macrophages play an important role in maintaining immune homeostasis and fine-tuning immune responses. During an immune response, de novo monocytes migrate to inflammatory sites and develop into macrophages. Monocyte-derived macrophages had been regarded as a uniform interstitial macrophage population (1, 37). Recently, emerging studies showed that interstitial macrophages are actually heterogeneous and can be distinguished into Lyvello MHCIIhi CX3CR1hi and Lyvelhi MHCIIlo CX3CR1lo populations (4). Additional studies defined IM populations using expression of CD206, CD11c, and MHCII (3, 4, 38). However, these studies focused on the classification of IM in a steady state, and only some of them showed functional differences among the subsets in type 1 immune response (5, 39). Focusing on the heterogeneity and function of lung macrophages in allergic disease, this report demonstrates how lung macrophages contribute to IL-9-mediated allergic inflammation. Our results demonstrate that macrophage populations are among the cells that express the most IL-9R in the allergic lung. Interstitial macrophages are capable of conferring IL-9R-dependent allergic inflammation in Il9r−/− mice. Critical target genes of IL-9R signaling include Arg1 and Ccl5, and Arg1 is required for macrophages to promote allergic inflammation. We also found evidence that this pathway is conserved in human asthma patient samples.
IL-9 is known to enhance type 2 immune responses in allergic diseases (6), with mast cells being direct IL-9-responsive cells in asthma (15, 18). Additional studies have implicated IL-9 in the functions of multiple cell types, including Th2 cells, epithelial cells and B cells (9, 14, 16). However, knowledge about IL-9-targeted cells is still limited. In this study, we assessed IL-9R expression across various cell types to identify IL-9-responsive populations in allergic airway inflammation. Unexpectedly, lung macrophages occupied a significant proportion of the IL-9R+ cells. They also express relatively high amounts of IL-9R compared to other cell types, such as eosinophils and neutrophils. Currently, only a few studies have focused on the direct impact of IL-9 on macrophages, including identifying microglia as a downstream target of IL-9 in multiple sclerosis. Moreover, IL-9R expression has been detected in human blood monocytes and alveolar macrophages (20, 21, 40). Overall, IL-9 regulation of interstitial macrophages in diseases states is poorly understood. Our results suggest lung macrophages are not only an IL-9-responsive population, but also a key driver of the allergic response by secreting Arg1 and eosinophil-attracting chemokines.
A recent report showed the presence of the CD11c+ IM in OVA-induced allergic inflammation (41). Here, we found that the induction of CD11c+ IM is highly dependent on IL-9 signaling. This population acts as a key component for IL-9-mediated allergic responses, demonstrated by the restoration of disease progression in adoptive transfer experiments. Although this study has advanced the understanding of the functions of lung IM in the allergic environment, further studies are required to investigate the transition from monocytes to IM populations in various microenvironments, their biological functions in specific inflammatory responses, and their transcriptional programs.
Arg1 is one of the most important enzymes that regulates macrophage functions (42) and is a marker of macrophage alternative activation. Demonstration of Arg1 function in asthma models has been controversial and may vary with model or genetic background of the mice (43–51). In particular, Barron et al used Arg1 conditional mutant mice on a BALB/c background and with Cre expression that deleted in all hematopoietic cells and demonstrated a limited role for Arg1 in lung inflammation (52). Our studies deleted Arg1 more specifically in the myeloid compartment and were on a C57BL/6 background. It is possible that differences are observed due to compensation resulting from deletion in distinct populations between the models or from differences in protocols. It is also possible that in a genetic environment that is highly skewed towards type 2 inflammation as in the BALB/c mice, Arg1 is dispensable. In our studies however, Arg1 in macrophages is clearly required. In immune models where Arg1 function has been demonstrated, it appears to suppress inflammation potentially by depleting Arg in the environment(53, 54). This suppressive function is not consistent with pro-allergic function observed in our preliminary data and other studies, as well as patient samples where increased Arg1 expression and arginase activity are associated with progressive asthma development (33, 43–45). Together, these observations suggest that function of Arg1 in the lung may be dependent on the inflammatory milieu and the genetic environment.
The stimuli for Arg1 expression in lung macrophages during disease is still not clear. Previous studies have shown that IL-4 and IL-13 can induce Arg1 expression and promote an immunosuppressive macrophage phenotype (42). Here, we found IL-9 induced Arg1 expression in lung macrophages in both naïve and HDM-treated mice. Using mixed bone marrow chimeric mice, we found IL-9 signaling in IMs is intrinsically required for Arg1 expression. Il9r−/− IMs express IL-4R and responded to IL-4 or IL-13 to induce Arg1 indicating the requirement for IL-9 even in the presence of normal IL-4/IL-4R-induced Arg1 expression. Collectively, these results suggest that Arg1 is an important effector for IL-9-mediated macrophage function. Further investigations on how IL-9 promotes Arg1 expression in lung macrophages and whether this regulation is conserved in other diseases are required.
ILC2s have been shown to be important IL-9 producers and responders (11, 34), and are particularly important in acute allergic inflammation. ILC2s have also been shown to produce Arg1 in type2 inflammation (35), but Arg1 expression in ILC2s in the chronic allergic airway inflammation model was not impaired in the absence of IL-9 signaling. Moreover, the studies using clodronate depletion, macrophage adoptive transfer, and myeloid cell deletion of Arg1 argue against ILC2s as responders to IL-9 or as sources of Arg1. Thus, while ILC2s are important for the development of allergic inflammation in some contexts, there is no evidence that they contribute to the IL-9/macrophage/Arg1 axis described here that is critical for chronic airway inflammation.
Our CITE-Seq analysis showed expression of multiple chemokines was down-regulated in IL-9R-deficient macrophages. Among them, we found IL-9 signaling promotes CCL5 expression in both mouse and human macrophages. In asthmatic patients, CCL5 promotes disease progression by recruiting eosinophils to the airway, a function that has been recapitulated in mouse models as well (55, 56). Interestingly, our results suggest that IL-9 stimulates Arg1+ IMs to secrete CCL5. This is a potential explanation for why Arg1+ macrophages rescue the allergic response in IL-9R deficient mice and why the loss of IL-9 signaling hinders the pro-allergic function of lung macrophages. Thus, blocking IL-9 signaling in lung macrophages could be a feasible therapeutic strategy for asthma, and potentially other pulmonary inflammatory diseases.
MATERIALS AND METHODS
Study design
The aim of this study was to characterize the IL-9-responsive cells in allergic inflammation, and to uncover the mechanisms of the development of IL-9-mediated allergic diseases. Using flow cytometry we demonstrated that macrophage populations had high expression of IL-9R and that populations were significantly altered in the absence of IL-9 signaling. By performing CITE-Seq analysis, liposome clodronate mediated macrophage depletion assays, adoptive transfer experiments and morphology analysis, we demonstrated IL-9 signaling promotes monocyte migration and differentiation into CD11c+ IMs and CD11c− IMs. We used adoptive transfer experiments and IL-9/IL-9R-deficient mice to show the IMs are required for IL-9 mediated allergic inflammation, Moreover, by using Arg1 reporter mice and Arg1 conditional knock out mice, we found that Arg1 was required for IL-9-dependent pro-allergic activity in macrophages. Finally, we confirmed the IL-9-lung macrophage axis is conserved in human asthma patients using biobanked patient samples. The number of mice used in the studies is included in the figure legend.
Mice
All mice were on C57BL/6 background. Wild type mice (C57BL/6, 002014), Boy/J mice (C57BL/6, 002014), Il4−/− mice, YARG mice (C57BL/6, 015857)(57), Arg1fl/fl mice (C57BL/6, 008817) and LysM-Cre (C57BL/6, 004781) were purchased from The Jackson Laboratory. Il9r−/− mice (C57BL/6) were gift from Dr. Jean-Christophe Renauld (58). Il9−/− mice (C57BL/6) were provided by Dr. Andrew McKenzie (59), Dr. Alexander Kirsch and Dr. Sophie Paczesny. Infer mice were provided by Dr. Paula Licona-Limón and Dr. Richard A. Flavell. Both female and male mice were used between the age of 8 weeks to 16 weeks. All the mice were maintained in SPF animal facilities (ambient temperature 70–72°F, humidity 50%, light/dark cycle 12/12hr). All experiments were performed with the approval of the Indiana University Institutional Animal Care and Use Committee.
Patient Samples
Patients were recruited in infancy from pediatric allergy and dermatology clinics associated with IU Health and Riley Children’s Hospital based on a diagnosis of dermatitis. Subjects were excluded for a history of prior wheezing, lower respiratory tract illness, treatment with asthma medications, or congenital heart disease. The majority of the population (85%) had a family history of asthma or allergy, which was used as the criteria for high-risk. Samples in this study were a subset of patients that were physician-diagnosed with or without asthma at age 5 based on parameters including persistent wheezing, the requirement for asthma controlling medications, respiratory function and response to methacholine challenge (60). The primary outcome of this cohort was to analyze airway function in a high-risk population of infants (30, 31, 36). All studies with human subjects and samples were performed with approval from the Indiana University Institutional Review Board and required informed consent from a parent or guardian. All subjects were evaluated at James Whitcomb Riley Hospital for Children, Indianapolis, Indiana. Characteristics of the subset of patients selected for the presence or absence of physician-diagnosed asthma for this study are listed in Table S2. Patient sample collection and analysis were approved by the Institutional Review Board of Indiana University and required parental consent for samples from infants.
Induction of allergic airway inflammation
Mice were challenged intranasally with House dust mite (HDM; Greer Laboratories) or A. fumigatus (ASP; Greer Laboratories) extract three times a week for 6 weeks. HDM or ASP (25 μg) extract was diluted with PBS (25 μl) and administered into the nose. Mice were sacrificed 1 day after final intranasal challenge. Mice were euthanized and lungs were lavaged with cold PBS for two times. BAL fluid was collected and cells were centrifuged at 1500g for 5 minutes at 4°C for further surface staining. The supernatant was saved at −80°C for ELISA analysis. The rest of the lung tissue was digested in 0.5 mg/ml collagenase A (Roche) media for 1 hour at 37°C with rotation. After digestion, the lungs were filtered through mesh and red blood cells lysed with ACK lysis buffer for 3 mins (Lonza). One million cells were kept for RNA analysis. Cells were washed with FACS buffer and stained for granulocytes, mast cells and lymphocytes. Eosinophils were identified as live Ly6G− SiglecF+ CD11c− CD11b+ cells; Neutrophils were identified as live Ly6G+ CD11b+ cells; Mast cells were identified as CD49b− FcεR1+ c-Kit+ cells; Monocytes were identified as live Ly6c+ CD11b+ cells; DCs were identified as live MHCII+ CD11b+ or MHCII+ CD103+ cells; Total macrophages were identified as live CD64+ MerTK+ cells; AMs, CD11c− IMs and CD11c+ IMs were distinguished by the expression of Siglec-F and CD11c. Blood was collected by cardiac puncture. A portion of the blood was kept for FACS analysis, red blood cells were lysed with ACK lysis buffer for 3 mins (Lonza). The remaining blood samples were kept for serum. Serum were taken after centrifugation at 14000 rpm for 10 mins at 4°C. For some of the experiments, CCR2 inhibitor (2mg/kg, R&D systems, RS 504393) was intravenously injected to the mice for 3 weeks. Anti-IL-9 (BioXcell, 9C1), anti-IL-13(R&D systems, MAB413) antibodies and their isotype IgG antibodies were intravenously injected to the mice every other day for two weeks.
Airway resistance
For airway resistance, mice were subjected to increasing doses of methacholine and airway resistance was measured using a ventilator (Elan Series Mouse RC Site; Buxco Electronics) and BioSystem XA software (Buxco Electronics).
Macrophage depletion
Mice were challenged with 150 μl clodronate intranasally or 200 μl clodronate intravenously every other day for 2 weeks. Mice were sacrificed 1 day after the final challenge. The subsequent procedures for sample harvesting are the same as for the House dust mite extract or A. fumigatus extract -induced allergic airway inflammation experiment.
Cell migration assay
Monocyte migration assay was performed by using CytoSelectTM 24-well cell migration assay kit (Cell biolabs, INC). Mouse bone marrow derived monocytes were isolated by magnetic selection according to the company protocol (Miltenyi biotec, monocyte isolation kit 130-100-629). Monocytes (0.5 × 106 in 100 μl of serum free DMEM media containing 1% BSA) were added to the upper chamber of the insert. The lower chamber contained 400 μl of complete DMEM media with or without cytokine and chemokine as indicated. The plates were incubated at 37°C in 5% CO2 incubator for 3 h and cells that migrated into the lower chamber were counted.
IL-9 injection model
Mice were treated with 4 μg IL-9 (Biolgend, 556004) by intravenously or intraperitoneally for three days. Mice were sacrificed 1 day after final intranasal challenge. The subsequent procedures for sample harvesting are the same as for the House dust mite extract or A. fumigatus extract -induced allergic airway inflammation experiment.
Arginase activity assay
For measurements of arginase activity we harvested ~106 cells per sample and centrifuged at 1000xg at 4°C for 10 min. Cell pellets were lysed for 10 min in 100 μl of 10 mM Tris-HCL (pH 7.4) containing 1 μM pepstatin A, 1 μM leupeptin, and 0.4% (w/v) Triton X-100. The cell lysate was centrifuged at 14,000×g at 4°C for 10 min and use supernatant was assessed for arginase activity using QuantiChrom Arginase Assay Kit (BioAssay Systems) according to the manufacturer’s directions.
Mixed bone marrow chimera
The F1 generation of CD45.1 X CD45.2 mice were irradiated at 1000 rads. One day after the irradiation, 8 million bone marrow cells (4 million from Boy/J mice, 4 million from Il9r−/− mice) were injected to the recipient mice. Mice were used 12 weeks after donor cell injection.
Macrophage transfer
Lung macrophage populations were sorted from HDM-treated mice. Cells were transferred into Il9r−/− mice (0.3 million from Boy/J mice, 0.15 million from YARG mice). Recipient mice were harvested as indicated in the figures.
Mouse BM-derived macrophage cell polarization
Bone marrow cells were isolated and red blood cells were lysed by using ACK buffer. 0.5 million/ml cells were cultured with complement DMEM media containing 20% L929 supernatant (cell biologics, 3368) for 7 days. IFN-γ (50ng/ml, Peprotech) for M1 macrophages or IL-4 (20ng/ml, Peprotech) for M2 macrophages were added on day 7 for 24 hours.
Human macrophage polarization
De-identified buffy coat blood packs from healthy donors were purchased from Indiana Blood Center. Peripheral blood mononuclear cells (PBMCs) were isolated by density gradient centrifugation using Ficoll-Paque (GE Healthcare). Buffy coat cells (10 ml) was diluted with 10 ml DPBS and gently added to 15 ml Ficoll-paque. After spinning down at 400 × g for 30 mins at room temperature without the brake, the upper layer was removed. The mononuclear cell layer was collected and transferred to a new conical tube and filled with MACS up to 50 ml. After mixing, cells were centrifuged at 300 × g for 10 mins, repeating this washing step three times. Human naïve CD14+ monocytes were isolated from the PBMCs by using magnetic separation (Miltenyi Biotec). Cells were cultured in the presence of GM-CSF (50 ng/ml, Peprotech) or M-CSF (50 ng/ml, Peprotech) for differentiating to M1 or M2 macrophages for 7 days. IFN-γ (20ng/ ml, Peprotech) or IL-4 (20 ng/ml, Peprotech) was added for 48hs. Cells were harvested for further analysis.
Cell hashing and CITE-Seq
Total lung macrophages and blood monocytes were sorted from HDM treated mice. 10 mice were pooled for each group before sorting. Sorted cells were labeled with Cell Hashing antibodies and CITE-seq antibodies according to the CITE-seq & Cell Hashing Protocol version 2019-02-13 (61, 62)(cite-seq.com/protocols). In brief, blood monocytes were stained with TotalSeq™-A0302 anti-mouse Hashtag 1 antibody (Biolegend) for 30 mins at 4°C. Lung macrophages were stained with CD11c-TotalSeqA ((Biolegend), SiglecF biotin antibody and TotalSeq™-A0301 anti-mouse Hashtag 1 antibody (Biolegend) for 30 mins at 4°C. After washing three times, lung macrophages were stained with a secondary Anti-biotin-TotalSeq A antibody (Biolegend) for 30 mins. The final single cell suspension was washed for 3 times with 0.1% BSA in PBS. Each clean single cell suspension was counted with a hemocytometer under microscope for cell number and cell viability. Labeled lung macrophages and blood monocytes were pooled at the ratio of 3:1. Twenty thousand targeted cell recovery per sample pool were super-loaded to a single cell master mix with lysis buffer and reverse transcription reagents, following the Chromium Single Cell 3’ Reagent Kits V3 User Guide, CG000183 Rev A (10X Genomics, Inc). Along with the single cell gel beads and partitioning oil, the single cell master mixture containing the cells was dispensed onto a Single Cell Chip B in separate wells, and the chip loaded to the Chromium Controller for GEM generation and barcoding, followed by reverse transcription. During cDNA amplification, HTO (HashTag Oligonucleotide) and ADT (Antibody-Derived Tags) PCR additive oligonucleotides were spiked into the 10X cDNA amplification to increase the yield of HTO and ADT products. Following cDNA amplification, each sample was separated into ADT-derived (<180bp), HTO-derived (<180bp) and mRNA-derived cDNAs (>300bp). The mRNA-derived cDNAs were further processed with the standard 10X Genomics protocol. The ADT and HTO libraries were prepared using the Illumina TruSeq Small RNA adapter and Illumina TruSeq adapter sequences, respectively. At each step, the quality and quantity of cDNA and library were examined by Bioanalyzer and Qubit. The resulting libraries were pooled at a ratio of 5:10:85 of HTO, ADT and mRNA in molarity and sequenced on an Illumina NovaSeq 6000. About 6K antibody reads per cell of ADT and HTO and 40–50K reads per cell of mRNA were generated, with 28 bp read 1 of cell barcode and UMI and 91 bp read 2 of RNA or HTO/ADT.
Analysis of Cell hashing and CITE-Seq
CellRanger 3.1.0 (http://support.10xgenomics.com/) was utilized to process the raw sequence data generated. Briefly, CellRanger used bcl2fastq (https://support.illumina.com/) to demultiplex raw base sequence calls generated from the sequencer into sample-specific FASTQ files. The FASTQ files were then processed with the cellranger count pipeline with feature barcode analysis to generate matrices of gene expression counts alongside HTO and ADT quantifications for each cell barcode. Mouse reference genome GRCm38 was used in the sequence data alignment and counting processes. The filtered feature-cell barcode matrices generated from CellRanger were used for further analysis with the R package Seurat (63, 64)(Seurat 3.1.1) with Rstudio version 1.2.5001 and R version 3.5.1. To identify tissue origin of cells in the WT and KO samples, the HTO counts were normalized using centered log-ratio (CLR) transformation and cells were demultiplexed with the HTODemux function. Doublets and negative cells identified from demultiplexing were removed from further analysis. Cells with low number of detected genes/UMIs and high mitochondrial gene content were excluded as well. For gene expression data analysis, gene expression levels for each cell were log normalized with the NormalizeData function in Seurat. Highly variable genes were subsequently identified with FindVariableFeatures using the “vst” approach. To integrate the single cell data of the WT and Il9r−/− samples, functions FindIntegrationAnchors and IntegrateData from Seurat were applied. The integrated data was scaled and PCA was performed. Clusters were identified with the Seurat functions FindNeighbors and FindClusters. The FindConservedMarkers function was subsequently used to identify cell cluster specific marker genes. Cell cluster identities were manually defined with the cluster-specific marker genes or known marker genes and small populations that stained with ADT-SiglecF Abs but were negative for transcripts encoding SiglecF, or were obviously not macrophage/monocyte populations were excluded from analyses as indicated. The cell clusters were visualized using the t-Distributed Stochastic Neighbor Embedding (t-SNE) plots and Uniform Manifold Approximation and Projection UMAP) plots. Analysis of the cell surface epitope expression ADT data was performed following the standard Seurat workflow of multi-modal data (https://satijalab.org/seurat/v3.1/multimodal_vignette.html).
Real-time Quantitative PCR analysis
RNA was extracted by using TRIzol reagent (ThermoFisher Schientific) or RNeasy Plus Micro Kit (QIAGEN). cDNA synthesis was performed according to manufacturer’s instructions (qScript™ cDNA Synthesis Kits, Quantabio). Taqman real time PCR assay (ThermoFisher Scientific) was used for detecting gene expression (Table S3). The relative mRNA expression was normalized to housekeeping gene expression (β2-microglobulin).
Flow cytometry
Single cell suspensions were stained with a fixable viability dye (eBioscience) and antibodies for surface markers for 30 mins at 4 °C, before fixation with 4% formaldehyde for 10 mins dark at room temperature. After fixation, cells were permeabilized with permeabilization buffer (eBioscience) for 30 mins at 4°C and stained for cytokines for another 30 mins at 4°C. For transcription factor staining, after surface staining, cells were fixed with Fixation & Permeabilization Buffer (eBioscience) for 2 hours or overnight at 4 °C, and then permeabilized with permeabilization buffer (eBioscience). After intracellular staining, cells were washed with FACS buffer and analyzed by LSR4 or Fortessa (BD Biosciences) and analyzed with Flowjo 10.7.1 software (Tree Star).
Cell sorting
Mouse macrophage were stained with anti-CD64, anti-Mertk, anti-CD11c and anti-Siglec-F antibody and viability dye and further sorted with FACSAria or SORPAria (BD Bioscience) by gating on live cells. Human patient samples were stained with viability dye, anti-hCD14 antibody, followed by FACS sorting. Sorted cells were used for further experiments. Details of antibodies are listed in Table S1.
Enzyme linked immunosorbent assay
IL-9 (Biolgend), CCL5 (Invitrogen) and HDM specific IgE ELISA were performed according to the manufacturer’s instruction. Briefly, 96 well-plate were coated with coating antibody overnight at 4°C. After washing 3 times with the wash buffer, 300 μl ELISA buffer was added to the plate and incubate at room temperature for 2 hours. After washing 3 times with washing buffer, 100 μl samples were added to the plate and incubated at room temperature for 2 hours. After washing three times, 100 μl diluted detection antibody was added to the plate and incubated at room temperature for 1 hour. After washing the plate three times, 100 μl of diluted Avidin-HRP solution was added to the plate and incubated at room temperature for 30 mins at dark. After washing the plate 3 times, 100 μl substrate was added to the plate. Plates were read at absorbance 450nm and 570 nm.
Cytospin
Isolated lung macrophages were washed with cold PBS and resuspended with up to 200 μl of PBS in a density of 0.1 × 106 cells/ml. After the cytospin at 600 rpm for 5 minutes, slides were stained with a modified Wright-Giemsa stain on a Hema-Tek 2000 slide stainer (Bayer Corp, Diagnostics Division, Leverkusen, Germany). The morphology of isolated lung macrophages was evaluated under a microscope and photos were taken.
Immunofluorescence staining
Frozen tissue sections (5 um) were recovered at RT. Slides were washed with PBST for three times and blocked with donkey serum at RT for 1h. After blocking, sections were incubated with anti-CD45.1 antibody at 4°C overnight. After three times washing, sections were incubated with Goat Anti-Mouse IgG H&L (Alexa Fluor® 488) at RT for 1h. After 3 times washing with PBST, nuclear were stained with DAPI.
Histology
Lung tissue was fixed with 4% formalin for 24 hours at room temperature. Tissues were embedded in paraffin, sectioned, and further stained with H&E, periodic acid-Schiff (PAS) and toluidine blue staining.
Statistics analysis
Statistical was analyzed by using GraphPad Prism 8.0 (GraphPad Software) and presented as means ± SEM. Unpaired or paired Student t tests and one-way or two-way ANOVA analysis were used in data analysis. A p value <0.05 was considered statistically significant. ns p>0.05, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Supplementary Material
Acknowledgments:
We thank Dr Henrique Serezani for review of this manuscript. The authors thank the members of the Indiana University Melvin and Bren Simon Cancer Center Flow Cytometry Resource Facility for their outstanding technical support. Schematics in Fig. 3 were generated with BioRender.com.
Funding:
This work was supported by Public Health Service grants from the National Institutes of Health (R01 AI129241to M.H.K.). B.Z. was supported by Public Health Service grants from the National Institutes of Health (R01 AI085046). B.J.U. was supported by National Institutes of Health Grants T32 AI060519 and F30 HL147515. A.P. was supported by National Institutes of Health Grant T32 AI060519. J.S. was supported by Public Health Service grants from the National Institutes of Health (AG069264 and AI147394). The Indiana University Melvin and Bren Simon Comprehensive Cancer Center Flow Cytometry Resource Facility is funded in part by NIH, National Cancer Institute (NCI) grant P30 CA082709 and National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) grant U54 DK106846. The FCRF is supported in part by NIH instrumentation grant 1S10D012270. Support provided by the Herman B Wells Center for Pediatric Research was, in part, from the Riley Children’s Foundation.
Footnotes
Competing interests: The authors declare no competing interests with this work. RAF is an advisor to Glaxo Smith Kline, Zai labs and Ventus Therapeutics.
Data and Materials Availability:
CITE-seq data are available under GEO accession number GSE162898. All other data needed to support the conclusions of the paper are present in the paper or the Supplementary Materials. Il9r−/− mice are available from Jean-Christophe Renauld under a material transfer agreement with the Ludwig Institute for Cancer Research Ltd. The anti-IL-9R mAb was initially obtained from Dr. Daisuke Kitamura under a material transfer agreement with Tokyo University of Science. The anti-IL-9R mAb is now commercially available.
References
- 1.Hussell T, Bell TJ, Alveolar macrophages: plasticity in a tissue-specific context. Nature Reviews Immunology 14, 81–93 (2014). [DOI] [PubMed] [Google Scholar]
- 2.Epelman S, Lavine Kory J., Randolph Gwendalyn J., Origin and Functions of Tissue Macrophages. Immunity 41, 21–35 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gibbings SL, Thomas SM, Atif SM, McCubbrey AL, Desch AN, Danhorn T, Leach SM, Bratton DL, Henson PM, Janssen WJ, Jakubzick CV, Three Unique Interstitial Macrophages in the Murine Lung at Steady State. Am J Respir Cell Mol Biol 57, 66–76 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chakarov S, Lim HY, Tan L, Lim SY, See P, Lum J, Zhang X-M, Foo S, Nakamizo S, Duan K, Kong WT, Gentek R, Balachander A, Carbajo D, Bleriot C, Malleret B, Tam JKC, Baig S, Shabeer M, Toh S-AES, Schlitzer A, Larbi A, Marichal T, Malissen B, Chen J, Poidinger M, Kabashima K, Bajenoff M, Ng LG, Angeli V, Ginhoux F, Two distinct interstitial macrophage populations coexist across tissues in specific subtissular niches. 363, eaau0964 (2019). [DOI] [PubMed] [Google Scholar]
- 5.Aran D, Looney AP, Liu L, Wu E, Fong V, Hsu A, Chak S, Naikawadi RP, Wolters PJ, Abate AR, Butte AJ, Bhattacharya M, Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nature Immunology 20, 163–172 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kaplan MH, Hufford MM, Olson MR, The development and in vivo function of T helper 9 cells. Nat Rev Immunol 15, 295–307 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kimura Y, Takeshita T, Kondo M, Ishii N, Nakamura M, Van Snick J, Sugamura K, Sharing of the IL-2 receptor γ chain with the functional IL-9 receptor complex. International Immunology 7, 115–120 (1995). [DOI] [PubMed] [Google Scholar]
- 8.Russell SM, Johnston JA, Noguchi M, Kawamura M, Bacon CM, Friedmann M, Berg M, McVicar DW, Witthuhn BA, Silvennoinen O, et al. , Interaction of IL-2R beta and gamma c chains with Jak1 and Jak3: implications for XSCID and XCID. Science (New York, N.Y.) 266, 1042–1045 (1994). [DOI] [PubMed] [Google Scholar]
- 9.Takatsuka S, Yamada H, Haniuda K, Saruwatari H, Ichihashi M, Renauld J-C, Kitamura D, IL-9 receptor signaling in memory B cells regulates humoral recall responses. Nature Immunology 19, 1025–1034 (2018). [DOI] [PubMed] [Google Scholar]
- 10.Price AE, Liang HE, Sullivan BM, Reinhardt RL, Eisley CJ, Erle DJ, Locksley RM, Systemically dispersed innate IL-13-expressing cells in type 2 immunity. Proceedings of the National Academy of Sciences of the United States of America 107, 11489–11494 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wilhelm C, Hirota K, Stieglitz B, Van Snick J, Tolaini M, Lahl K, Sparwasser T, Helmby H, Stockinger B, An IL-9 fate reporter demonstrates the induction of an innate IL-9 response in lung inflammation. Nature immunology 12, 1071–1077 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nowak EC, Weaver CT, Turner H, Begum-Haque S, Becher B, Schreiner B, Coyle AJ, Kasper LH, Noelle RJ, IL-9 as a mediator of Th17-driven inflammatory disease. The Journal of experimental medicine 206, 1653–1660 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Elyaman W, Bradshaw EM, Uyttenhove C, Dardalhon V, Awasthi A, Imitola J, Bettelli E, Oukka M, van Snick J, Renauld JC, Kuchroo VK, Khoury SJ, IL-9 induces differentiation of TH17 cells and enhances function of FoxP3+ natural regulatory T cells. Proceedings of the National Academy of Sciences of the United States of America 106, 12885–12890 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang Y, Shi J, Yan J, Xiao Z, Hou X, Lu P, Hou S, Mao T, Liu W, Ma Y, Zhang L, Yang X, Qi H, Germinal-center development of memory B cells driven by IL-9 from follicular helper T cells. Nat Immunol 18, 921–930 (2017). [DOI] [PubMed] [Google Scholar]
- 15.Sehra S, Yao W, Nguyen ET, Glosson-Byers NL, Akhtar N, Zhou B, Kaplan MH, TH9 cells are required for tissue mast cell accumulation during allergic inflammation. The Journal of allergy and clinical immunology 136, 433–440.e431 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wambre E, Bajzik V, DeLong JH, O’Brien K, Nguyen QA, Speake C, Gersuk VH, DeBerg HA, Whalen E, Ni C, Farrington M, Jeong D, Robinson D, Linsley PS, Vickery BP, Kwok WW, A phenotypically and functionally distinct human T(H)2 cell subpopulation is associated with allergic disorders. Science translational medicine 9, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shimbara A, Christodoulopoulos P, Soussi-Gounni A, Olivenstein R, Nakamura Y, Levitt RC, Nicolaides NC, Holroyd KJ, Tsicopoulos A, Lafitte JJ, Wallaert B, Hamid QA, IL-9 and its receptor in allergic and nonallergic lung disease: increased expression in asthma. The Journal of allergy and clinical immunology 105, 108–115 (2000). [DOI] [PubMed] [Google Scholar]
- 18.Kearley J, Erjefalt JS, Andersson C, Benjamin E, Jones CP, Robichaud A, Pegorier S, Brewah Y, Burwell TJ, Bjermer L, Kiener PA, Kolbeck R, Lloyd CM, Coyle AJ, Humbles AA, IL-9 governs allergen-induced mast cell numbers in the lung and chronic remodeling of the airways. American journal of respiratory and critical care medicine 183, 865–875 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kauppi P, Laitinen T, Ollikainen V, Mannila H, Laitinen LA, Kere J, The IL9R region contribution in asthma is supported by genetic association in an isolated population. European Journal of Human Genetics 8, 788–792 (2000). [DOI] [PubMed] [Google Scholar]
- 20.Pilette C, Ouadrhiri Y, Van Snick J, Renauld J-C, Staquet P, Vaerman J-P, Sibille Y, Oxidative burst in lipopolysaccharide-activated human alveolar macrophages is inhibited by interleukin-9. 20, 1198–1205 (2002). [DOI] [PubMed] [Google Scholar]
- 21.Pilette C, Ouadrhiri Y, Van Snick J, Renauld JC, Staquet P, Vaerman JP, Sibille Y, IL-9 inhibits oxidative burst and TNF-alpha release in lipopolysaccharide-stimulated human monocytes through TGF-beta. Journal of immunology (Baltimore, Md. : 1950) 168, 4103–4111 (2002). [DOI] [PubMed] [Google Scholar]
- 22.Gonzalo J-A, Lloyd CM, Wen D, Albar JP, Wells TNC, Proudfoot A, Martinez-A C, Dorf M, Bjerke T, Coyle AJ, Gutierrez-Ramos J-C, The Coordinated Action of CC Chemokines in the Lung Orchestrates Allergic Inflammation and Airway Hyperresponsiveness. Journal of Experimental Medicine 188, 157–167 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Misharin AV, Morales-Nebreda L, Reyfman PA, Cuda CM, Walter JM, McQuattie-Pimentel AC, Chen CI, Anekalla KR, Joshi N, Williams KJN, Abdala-Valencia H, Yacoub TJ, Chi M, Chiu S, Gonzalez-Gonzalez FJ, Gates K, Lam AP, Nicholson TT, Homan PJ, Soberanes S, Dominguez S, Morgan VK, Saber R, Shaffer A, Hinchcliff M, Marshall SA, Bharat A, Berdnikovs S, Bhorade SM, Bartom ET, Morimoto RI, Balch WE, Sznajder JI, Chandel NS, Mutlu GM, Jain M, Gottardi CJ, Singer BD, Ridge KM, Bagheri N, Shilatifard A, Budinger GRS, Perlman H, Monocyte-derived alveolar macrophages drive lung fibrosis and persist in the lung over the life span. The Journal of experimental medicine 214, 2387–2404 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cui H, Jiang D, Banerjee S, Xie N, Kulkarni T, Liu RM, Duncan SR, Liu G, Monocyte-derived alveolar macrophage apolipoprotein E participates in pulmonary fibrosis resolution. JCI Insight 5, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ogger PP, Albers GJ, Hewitt RJ, O’Sullivan BJ, Powell JE, Calamita E, Ghai P, Walker SA, McErlean P, Saunders P, Kingston S, Molyneaux PL, Halket JM, Gray R, Chambers DC, Maher TM, Lloyd CM, Byrne AJ, Itaconate controls the severity of pulmonary fibrosis. Science immunology 5, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Takatsuka S, Yamada H, Haniuda K, Ichihashi M, Chiba J, Kitamura D, DNA Immunization Using in vivo Electroporation for Generating Monoclonal Antibodies Against Mouse IL-9R. Bio-protocol 9, e3174 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huang L, Nazarova EV, Tan S, Liu Y, Russell DG, Growth of Mycobacterium tuberculosis in vivo segregates with host macrophage metabolism and ontogeny. Journal of Experimental Medicine 215, 1135–1152 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zasłona Z, Przybranowski S, Wilke C, van Rooijen N, Teitz-Tennenbaum S, Osterholzer JJ, Wilkinson JE, Moore BB, Peters-Golden M, Resident Alveolar Macrophages Suppress, whereas Recruited Monocytes Promote, Allergic Lung Inflammation in Murine Models of Asthma. 193, 4245–4253 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yao W, Zhang Y, Jabeen R, Nguyen ET, Wilkes DS, Tepper RS, Kaplan MH, Zhou B, Interleukin-9 is required for allergic airway inflammation mediated by the cytokine TSLP. Immunity 38, 360–372 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tepper RS, Llapur CJ, Jones MH, Tiller C, Coates C, Kimmel R, Kisling J, Katz B, Ding Y, Swigonski N, Expired nitric oxide and airway reactivity in infants at risk for asthma. The Journal of allergy and clinical immunology 122, 760–765 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yao W, Barbé-Tuana FM, Llapur CJ, Jones MH, Tiller C, Kimmel R, Kisling J, Nguyen ET, Nguyen J, Yu Z, Kaplan MH, Tepper RS, Evaluation of airway reactivity and immune characteristics as risk factors for wheezing early in life. The Journal of allergy and clinical immunology 126, 483–488.e481 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tacke F, Ginhoux F, Jakubzick C, van Rooijen N, Merad M, Randolph GJ Immature monocytes acquire antigens from other cells in the bone marrow and present them to T cells after maturing in the periphery. Journal of Experimental Medicine 203, 583–597 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Litonjua AA, Lasky-Su J, Schneiter K, Tantisira KG, Lazarus R, Klanderman B, Lima JJ, Irvin CG, Peters SP, Hanrahan JP, Liggett SB, Hawkins GA, Meyers DA, Bleecker ER, Lange C, Weiss ST, ARG1 is a novel bronchodilator response gene: screening and replication in four asthma cohorts. American journal of respiratory and critical care medicine 178, 688–694 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Turner JE, Morrison PJ, Wilhelm C, Wilson M, Ahlfors H, Renauld JC, Panzer U, Helmby H, Stockinger B, IL-9-mediated survival of type 2 innate lymphoid cells promotes damage control in helminth-induced lung inflammation. The Journal of experimental medicine 210, 2951–2965 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Monticelli LA, Buck MD, Flamar AL, Saenz SA, Tait Wojno ED, Yudanin NA, Osborne LC, Hepworth MR, Tran SV, Rodewald HR, Shah H, Cross JR, Diamond JM, Cantu E, Christie JD, Pearce EL, Artis D, Arginase 1 is an innate lymphoid-cell-intrinsic metabolic checkpoint controlling type 2 inflammation. Nat Immunol 17, 656–665 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sarria EE, Mattiello R, Yao W, Chakr V, Tiller CJ, Kisling J, Tabbey R, Yu Z, Kaplan MH, Tepper RS, Atopy, cytokine production, and airway reactivity as predictors of pre-school asthma and airway responsiveness. Pediatr Pulmonol 49, 132–139 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wynn TA, Vannella KM, Macrophages in Tissue Repair, Regeneration, and Fibrosis. Immunity 44, 450–462 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schyns J, Bai Q, Ruscitti C, Radermecker C, De Schepper S, Chakarov S, Farnir F, Pirottin D, Ginhoux F, Boeckxstaens G, Bureau F, Marichal T, Non-classical tissue monocytes and two functionally distinct populations of interstitial macrophages populate the mouse lung. Nature communications 10, 3964 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ural BB, Yeung ST, Damani-Yokota P, Devlin JC, de Vries M, Vera-Licona P, Samji T, Sawai CM, Jang G, Perez OA, Pham Q, Maher L, Loke P, Dittmann M, Reizis B, Khanna KM, Identification of a nerve-associated, lung-resident interstitial macrophage subset with distinct localization and immunoregulatory properties. Science immunology 5, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Donninelli G, Saraf-Sinik I, Mazziotti V, Capone A, Grasso MG, Battistini L, Reynolds R, Magliozzi R, Volpe E, Interleukin-9 regulates macrophage activation in the progressive multiple sclerosis brain. Journal of Neuroinflammation 17, 149 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lai JF, Thompson LJ, Ziegler SF, TSLP drives acute T(H)2-cell differentiation in lungs. The Journal of allergy and clinical immunology, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Murray PJ, Macrophage Polarization. 79, 541–566 (2017). [DOI] [PubMed] [Google Scholar]
- 43.Cloots RH, Sankaranarayanan S, de Theije CC, Poynter ME, Terwindt E, van Dijk P, Hakvoort TB, Lamers WH, Köhler SE, Ablation of Arg1 in hematopoietic cells improves respiratory function of lung parenchyma, but not that of larger airways or inflammation in asthmatic mice. Am J Physiol Lung Cell Mol Physiol 305, L364–376 (2013). [DOI] [PubMed] [Google Scholar]
- 44.Cloots RHE, Poynter ME, Terwindt E, Lamers WH, Köhler SE, Hypoargininemia exacerbates airway hyperresponsiveness in a mouse model of asthma. Respir Res 19, 98 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cloots RHE, Sankaranarayanan S, Poynter ME, Terwindt E, van Dijk P, Lamers WH, Eleonore Köhler S, Arginase 1 deletion in myeloid cells affects the inflammatory response in allergic asthma, but not lung mechanics, in female mice. BMC Pulm Med 17, 158 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Niese KA, Collier AR, Hajek AR, Cederbaum SD, O’Brien WE, Wills-Karp M, Rothenberg ME, Zimmermann N, Bone marrow cell derived arginase I is the major source of allergen-induced lung arginase but is not required for airway hyperresponsiveness, remodeling and lung inflammatory responses in mice. BMC Immunol 10, 33 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Maarsingh H, Dekkers BG, Zuidhof AB, Bos IS, Menzen MH, Klein T, Flik G, Zaagsma J, Meurs H, Increased arginase activity contributes to airway remodelling in chronic allergic asthma. Eur Respir J 38, 318–328 (2011). [DOI] [PubMed] [Google Scholar]
- 48.Maarsingh H, Zuidhof AB, Bos IS, van Duin M, Boucher JL, Zaagsma J, Meurs H, Arginase inhibition protects against allergen-induced airway obstruction, hyperresponsiveness, and inflammation. American journal of respiratory and critical care medicine 178, 565–573 (2008). [DOI] [PubMed] [Google Scholar]
- 49.North ML, Khanna N, Marsden PA, Grasemann H, Scott JA, Functionally important role for arginase 1 in the airway hyperresponsiveness of asthma. Am J Physiol Lung Cell Mol Physiol 296, L911–920 (2009). [DOI] [PubMed] [Google Scholar]
- 50.Yang M, Rangasamy D, Matthaei KI, Frew AJ, Zimmmermann N, Mahalingam S, Webb DC, Tremethick DJ, Thompson PJ, Hogan SP, Rothenberg ME, Cowden WB, Foster PS, Inhibition of arginase I activity by RNA interference attenuates IL-13-induced airways hyperresponsiveness. Journal of immunology (Baltimore, Md.: 1950) 177, 5595–5603 (2006). [DOI] [PubMed] [Google Scholar]
- 51.Zimmermann N, King NE, Laporte J, Yang M, Mishra A, Pope SM, Muntel EE, Witte DP, Pegg AA, Foster PS, Hamid Q, Rothenberg ME, Dissection of experimental asthma with DNA microarray analysis identifies arginase in asthma pathogenesis. The Journal of clinical investigation 111, 1863–1874 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Barron L, Smith AM, El Kasmi KC, Qualls JE, Huang X, Cheever A, Borthwick LA, Wilson MS, Murray PJ, Wynn TA, Role of arginase 1 from myeloid cells in th2-dominated lung inflammation. PLoS One 8, e61961 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Herbert DR, Orekov T, Roloson A, Ilies M, Perkins C, O’Brien W, Cederbaum S, Christianson DW, Zimmermann N, Rothenberg ME, Finkelman FD, Arginase I suppresses IL-12/IL-23p40-driven intestinal inflammation during acute schistosomiasis. Journal of immunology (Baltimore, Md.: 1950) 184, 6438–6446 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pesce JT, Ramalingam TR, Mentink-Kane MM, Wilson MS, El Kasmi KC, Smith AM, Thompson RW, Cheever AW, Murray PJ, Wynn TA, Arginase-1-expressing macrophages suppress Th2 cytokine-driven inflammation and fibrosis. PLoS pathogens 5, e1000371 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Venge J, Lampinen M, Håkansson L, Rak S, Venge P, Identification of IL-5 and RANTES as the major eosinophil chemoattractants in the asthmatic lung. The Journal of allergy and clinical immunology 97, 1110–1115 (1996). [DOI] [PubMed] [Google Scholar]
- 56.Marques RE, Guabiraba R, Russo RC, Teixeira MM, Targeting CCL5 in inflammation. Expert Opin Ther Targets 17, 1439–1460 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Reese TA, Liang H-E, Tager AM, Luster AD, Van Rooijen N, Voehringer D, Locksley RM, Chitin induces accumulation in tissue of innate immune cells associated with allergy. Nature 447, 92–96 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Steenwinckel V, Louahed J, Orabona C, Huaux F, Warnier G, McKenzie A, Lison D, Levitt R, Renauld J-C, IL-13 Mediates In Vivo IL-9 Activities on Lung Epithelial Cells but Not on Hematopoietic Cells. The Journal of Immunology 178, 3244–3251 (2007). [DOI] [PubMed] [Google Scholar]
- 59.Townsend MJ, Fallon PG, Matthews DJ, Smith P, Jolin HE, McKenzie ANJ, IL-9-Deficient Mice Establish Fundamental Roles for IL-9 in Pulmonary Mastocytosis and Goblet Cell Hyperplasia but Not T Cell Development. Immunity 13, 573–583 (2000). [DOI] [PubMed] [Google Scholar]
- 60.Chang D, Yao W, Tiller CJ, Kisling J, Slaven JE, Yu Z, Kaplan MH, Tepper RS, Exhaled nitric oxide during infancy as a risk factor for asthma and airway hyperreactivity. Eur Respir J 45, 98–106 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Stoeckius M, Hafemeister C, Stephenson W, Houck-Loomis B, Chattopadhyay PK, Swerdlow H, Satija R, Smibert P, Simultaneous epitope and transcriptome measurement in single cells. Nat Methods 14, 865–868 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Stoeckius M, Zheng S, Houck-Loomis B, Hao S, Yeung BZ, Mauck WM 3rd, Smibert P, Satija R, Cell Hashing with barcoded antibodies enables multiplexing and doublet detection for single cell genomics. Genome Biol 19, 224 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Butler A, Hoffman P, Smibert P, Papalexi E, Satija R, Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol 36, 411–420 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM 3rd, Hao Y, Stoeckius M, Smibert P, Satija R, Comprehensive Integration of Single-Cell Data. Cell 177, 1888–1902.e1821 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
CITE-seq data are available under GEO accession number GSE162898. All other data needed to support the conclusions of the paper are present in the paper or the Supplementary Materials. Il9r−/− mice are available from Jean-Christophe Renauld under a material transfer agreement with the Ludwig Institute for Cancer Research Ltd. The anti-IL-9R mAb was initially obtained from Dr. Daisuke Kitamura under a material transfer agreement with Tokyo University of Science. The anti-IL-9R mAb is now commercially available.