

Abstract
BACKGROUND
The genetic determinants of heart failure (HF) and response to medical therapy remain unknown. We hypothesized that identifying genetic variants of HF that associate with response to medical therapy would elucidate the genetic basis of cardiac function.
OBJECTIVES
This study sought to identify genetic variations associated with response to HF therapy.
METHODS
This study compared extremes of response to medical therapy in 866 HF patients using a genome-wide approach that informed the systems-based design of a customized single nucleotide variant array. The effect of genotype on gene expression was measured using allele-specific luciferase reporter assays. Candidate gene transcription-deficient mice underwent echocardiography and treadmill exercise. The ability of the target gene agonist to rescue mice from chemically-induced HF was assessed with echocardiography.
RESULTS
Of 866 HF patients, 136 had an ejection fraction improvement of 20% attributed to resynchronization(n = 83), revascularization (n = 7), tachycardia resolution (n = 2), alcohol cessation (n = 1), or medications (n = 43). Those with the minor allele for rs7767652, upstream of hypocretin (orexin) receptor-2 (HCRTR2), were less likely to have improved left ventricular function (odds ratio: 0.40 per minor allele; p = 3.29 × 10−5). In a replication cohort of 798 patients, those with a minor allele for rs7767652 had a lower prevalence of ejection fraction >35% (odds ratio:0.769 per minor allele; p = 0.021). In an HF model, HCRTR2-deficient mice exhibited poorer cardiac function, worse treadmill exercise capacity, and greater myocardial scarring. Orexin, an HCRTR2 agonist, rescued function in this HF mouse model.
CONCLUSIONS
A systems approach identified a novel genetic contribution to human HF and a promising therapeutic agent efficacious in an HF model.
Keywords: genome-wide association study, HCRTR2, pharmacogenomics
Heart failure (HF), a syndrome characterized by impaired function and high filling pressures, affects more than 5 million people in the United States and is expected to touch 3.5% of the population over the next 20 years (1). A wide range of conditions can lead to HF, such as coronary artery disease and hypertension (2). Although heritable (3), few studies have explored the genetic basis for HF. Candidate gene studies identified associations between common variants in HSPB7 and FRMD4B and dilated cardiomyopathy (4) or advanced HF (5). Targeted genotyping of common variants in ADRB1 and GRK5, members of the β-adrenergic receptor signaling pathway, demonstrated associations with survival (6,7). In the limited genome-wide studies of HF (8–11), only 1 common variant associated with dilated cardiomyopathy of genome-wide significance has been replicated (11).
Systems approaches to studying the genetics of complex traits have been successful in uncovering promising gene targets and identifying fundamental disease patterns. Genome-wide association analyses have been combined with gene expression and metabolic data, for example, to identify AGPAT5 as an important effector of insulin resistance (12). Using gene coexpression network analyses, we previously identified patterns of gene expression found in common between diseased and developing myocardium(13). Gene expression patterns can also distinguish patients with HF and ischemic heart disease from those with dilated cardiomyopathy (14).
A phenomenon long recognized by HF physicians is that some patients dramatically respond to HF therapy with large increases in ejection fraction (EF) associated with positive remodeling of the left ventricle, whereas others deteriorate seemingly in spite of optimal medical management. Few studies have addressed even the clinical associations of such responders, a group for whom long-term survival is predictably better (15).
In this study, our primary hypothesis was that there are genomic variants associated with a dramatic response to HF medical therapy. We used a systems approach to design a genetic discovery platform optimized for HF, then evaluated whether there was a functional role for the gene target of the variant association.
METHODS
STUDY POPULATIONS.
Patients were recruited from Stanford University Medical Center, Stanford, California, and the Palo Alto Veterans Hospital, Palo Alto, California; patients were included who had clinically diagnosed HF, were referred for subspecialty care between 2005 and 2009, had an echocardiogram performed, and had an EF <55%. Patients were excluded if they could not be contacted by telephone or if they had insufficient clinical data. Also excluded were patients with congenital heart disease or cardiomyopathy due to infiltrative disease, a peripartum state, infection, or chemo-therapy; with a myocardial biopsy suggestive of viral cardiomyopathy who responded to medical therapy within 30 days of administration; who experienced an acute myocardial infarction and showed subsequent improvement in their EF within 3 months of infarction; or with a history of substance abuse (illicit drugs or alcohol) within 6 months before the study. Patients whose EF improved after surgical or percutaneous revascularization, resynchronization therapy using biventricular pacing, alcohol cessation, or cardioversion were excluded from the genomic analyses. The EF was measured using routine transthoracic echocardiography obtained by trained echocardiographers using 2-dimensional scanning in the parasternal long axis, parasternal short axis, and apical views. Change in EF was measured as maximal difference between lowest recorded EF and the highest subsequent recorded EF. Written informed consent was obtained from study participants in accordance with the Stanford University Internal Review Board policy.
For the informative genome-wide association studies (GWAS), case subjects (n = 29) were patients whose EF had improved by >20% while on medical therapy. Patients with poor sample quality were excluded. Control patients (n = 37) were selected from those followed in the Stanford University transplant clinic who had demonstrated lack of clinical improvement before transplant despite medical therapy. The case and control patients were matched by age, sex, race, medical therapy received, baseline EF on echocardiography, type of cardiomyopathy, and duration of HF. The remaining HF patients underwent genotyping with the custom genotyping array. Patient charts were retrospectively reviewed and their baseline demographics, clinical characteristics, and medication use were recorded. Serial measurements from clinically available echocardiograms were also recorded. Outcome data were obtained through detailed phone interviews.
REPLICATION.
For the independent replication study, patients with HF were recruited from the University of Pennsylvania as previously described (5). Briefly, Caucasian patients (n = 798) were recruited from the Penn Heart Failure Study, specifically those participating in an ongoing prospective observational study of patients with advanced HF referred for subspecialty care at the University of Pennsylvania Health System, Philadelphia, Pennsylvania. The primary inclusion criterion was a clinical diagnosis of HF with abnormal left ventricular function. Extensive clinical data were collected at enrollment. Written informed consent was obtained from study participants in accordance with the University of Pennsylvania Internal Review Board policy.
GENOTYPING.
Informative GWAS.
Genomic deoxyribonucleic acid (DNA) was isolated from whole blood using a commercial DNA extraction kit (Gentra Purgene Kit, Qiagen Inc., Valencia, California). The samples were genotyped using a 550K single nucleotide polymorphism (SNP) platform (Illumina, Inc., San Diego, California) at the Hudson Alpha Institute for Biotechnology (Huntsville, Alabama) (16). Additionally, genomic DNA was isolated from peripheral blood mononuclear cells and DNA quality was assessed utilizing optical absorbance and minigels.
A target of 1,536 SNPs was chosen to create a customized gene array. The overall strategy was to assign approximately one-third of the array to intergenic SNPs, one-third to genic SNPs, and one-third to SNPs ascertained from the network analyses and curated lists (Figure 1). Details of the informative GWAS analysis, including a Manhattan plot (Online Figure 1) and QQ plot (Online Figure 2), which used an additive logistic regression model, as well as the network analyses are included in the Online Appendix, which also includes details related to the animal portion of this study.
FIGURE 1. Custom Array: Primary Data Sources and Bioinformatic Analyses.
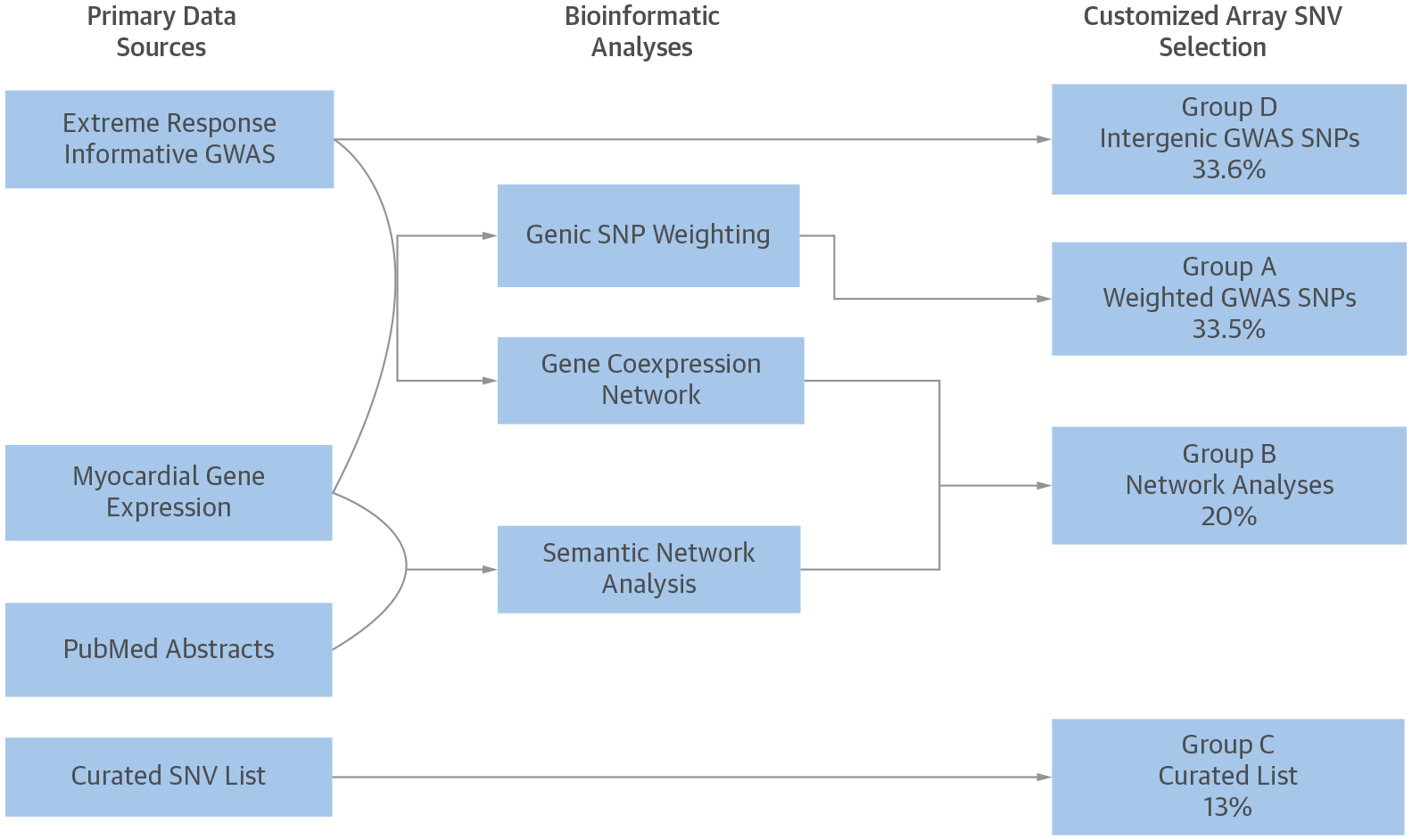
Approximately one-third of the 1,536 single nucleotide variants (SNVs) were derived from the gene expression-weighted analysis of extreme response genome-wide association studies (GWAS); one-third from the most statistically significant intergenic single nucleotide polymorphisms (SNPs) from the extreme response GWAS; and one-third from the GWAS-informed analyses of gene-coexpression networks, semantic published data networks, or a curated set of SNPs manually selected from the published data.
Custom array analysis was performed in PLINK (16) using a multivariate additive logistic regression model to measure the association between each SNP and an improvement in EF >10% compared with those without documentation of such an improvement. This association was adjusted for age, sex, and race. A Manhattan plot of −log10P was generated using Hap-loview (Figure 2A). Values for a quantile-quantile plot were generated using PLINK to evaluate the potential effect of population stratification (Online Figure 3). Odds ratio (OR) values were measured as OR per minor allele of each SNP. The threshold for statistical significance was estimated at the Bonferroni-corrected value of 0.05/1,402 = 3.56 × 10−5.
FIGURE 2. Significance of Association of EF Improvement.
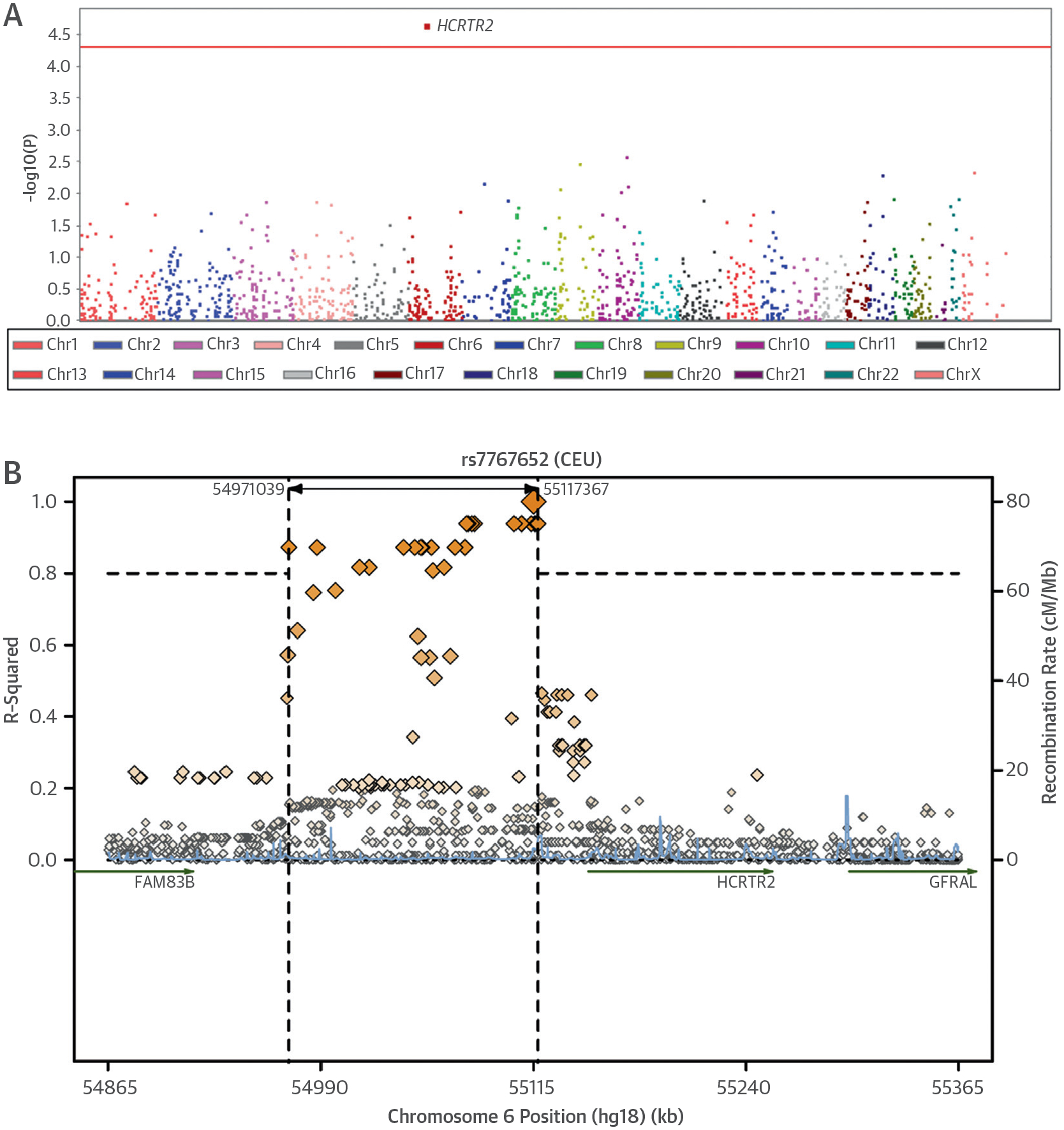
This Manhattan plot shows the significance of association with a 10% improvement in ejection fraction (EF) for single nucleotide polymorphisms (SNPs) in the customized heart failure array analysis of Phase II individuals and regional linkage disequilibrium (LD) plot of the statistically significant locus. (A) SNPs are plotted on the x-axis according to their position on each chromosome against association with >10% improvement in EF on the y-axis. The locus near hypocretin receptor 2 (HCRTR2) reached an association significance of p = 3.29 × 10−5 in the Phase II analysis. Red line = threshold for Bonferroni-adjusted significance (p < 3.6 × 10−5). (B) The linkage disequilibrium plot of the region surrounding rs7767652 was drawn according to 1,000 Genomes Pilot 1 CEU individuals. CEU = Utah residents with Northern and Western European Ancestry; other abbreviations as in Figure 1.
The p values from the informative GWAS and custom gene array were meta-analyzed with a weighted Z-method (17) using Stouffer’s Z trend, which considers sample size and effect directions, in MetaP.
Association analysis.
Analysis of the replication genotyping in the University of Pennsylvania cohort was performed in SAS version 9.1 (SAS Institute Inc., Cary, North Carolina) using a multivariate additive logistic regression model to measure association between the rs7767652 genotype and a baseline EF ≥35% compared with a baseline EF <35%. The association was adjusted for age, sex, hypertension, diabetes, renal function (glomerular filtration rate), and body mass index. The OR was measured as OR per minor allele. The threshold for statistical significance in this single replication association was 0.05.
For differences in relative luciferase activity, degree of hypocretin receptor-2 (HCRTR2) expression, E/E’ ratios, and peak VO2 difference where continuous values were compared between 2 groups, the Student t test was used. For differences in trichrome staining, blue/red ratios were calculated using mean values from the RGB histogram in Photoshop version12.0 (Adobe, San Jose, California) and the ratios were compared using Student t test. For EF differences in the chemical stress model, EF differences between the treatment and control groups were compared only at the 4-week time point using the Student t test. A p value <0.05 was considered statistically significant.
RESULTS
DRAMATIC RESPONSE TO HF THERAPY.
Of 866 patients with HF recruited at Stanford, 136 were found to have an absolute improvement in EF ≥20%. Dramatic improvement was attributed to resynchronization therapy in 83 (61%), revascularization therapy in 7 (5.1%), resolution of tachycardia in 2 (1.5%), alcohol cessation in 1 (0.7%), and standard HF medical therapy in 43 (32%) patients. Patients with a dramatic improvement in heart function (absolute increase of 20% in EF) attributed to medical therapy were on average 62 years of age, 41% were female, and 21% had ischemic cardiomyopathy. Compared with age- and sex-matched patients who ultimately underwent heart transplant, the patients with a dramatic improvement in heart function were more likely to have hypertension (37.9% vs. 16.2%; p = 0.045) or diabetes (48.2% vs. 13.5%; p = 0.002) (Table 1). There were no significant differences in beta-blocker or angiotensin-converting enzyme inhibitor use.
TABLE 1.
Baseline Demographic and Clinical Characteristics
| Phase I | Phase II | |||||||
|---|---|---|---|---|---|---|---|---|
| Overall (n = 66) | ΔEF ≥20%* (n = 29) | Transplant† (n = 37) | p Value‡ | Overall (n = 591) | ΔEF ≥10%* (n = 137) | ΔEF <10% (n = 454) | p Value§ | |
| Age, yrs | 60.6 ± 11.0 | 62.1 ± 13.1 | 59.4 ± 9.1 | 0.35 | 59.0 ± 15.5 | 56.9 ± 14.3 | 59.6 ± 15.8 | 0.071 |
| Female | 25 (37.9) | 12 (41.4) | 13 (35.1) | 0.60 | 169 (28.6) | 48 (35.0) | 121 (26.7) | 0.059 |
| African American | — | — | — | — | 43 (7.3) | 10 (7.3) | 33 (7.2) | 0.99 |
| Hispanic | — | — | — | — | 12 (2.0) | 4 (2.9) | 8 (1.8) | 0.40 |
| Asian | — | — | — | — | 26 (4.4) | 5 (3.6) | 21 (4.6) | 0.63 |
| Baseline EF, % | 27.8 ± 11.3 | 29.2 ± 10.7 | 26.7 ± 11.8 | 0.40 | 34.2 ± 14 | 29.0 ± 11.5 | 36.4 ± 14.6 | <0.0001 |
| NYHA functional class III-IV at baseline | 29 (43.9) | 16 (55.2) | 13 (35.1) | 0.10 | 199 (42.1) | 53 (42.0) | 146 (42.2) | 0.16 |
| Coronary disease | 18 (27.2) | 6 (20.7) | 12 (32.4) | 0.29 | 222(37.6) | 35 (25.5) | 187 (41.2) | 0.0009 |
| CABG | 9 (13.6) | 5 (17.2) | 4 (10.8) | 0.45 | 123 (20.8) | 20 (14.6) | 103 (22.7) | 0.041 |
| Atrial fibrillation | 18 (27.2) | 8 (27.6) | 10 (27.0) | 0.96 | 173 (29.3) | 40 (29.2) | 133 (29.3) | 0.98 |
| Diabetes | 19 (28.8) | 14 (48.2) | 5 (13.5) | 0.002 | 126 (21.3) | 36 (26.3) | 90 (19.8) | 0.11 |
| Hypertension | 17 (25.8) | 11 (37.9) | 6 (16.2) | 0.045 | 234 (39.6) | 55 (40.1) | 179 (39.4) | 0.88 |
| Device implant | 25 (37.9) | 9 (31.0) | 16 (43.2) | 0.31 | 305 (51.6) | 80 (58.4) | 225 (49.6) | 0.070 |
| Medications | ||||||||
| Beta-blocker | 48 (72.7) | 22 (75.9) | 26 (70.2) | 0.61 | 410 (69.4) | 127 (92.7) | 283 (62.3) | <0.0001 |
| ACE-I | 38 (57.6) | 17 (58.6) | 21 (56.8) | 0.88 | 274 (46.4) | 70 (51.1) | 204 (45.0) | 0.21 |
| ARB | 12 (18.2) | 4 (13.8) | 8 (21.6) | 0.41 | 81 (13.7) | 22 (16.1) | 59 (13.0) | 0.36 |
| Antiarrhythmic drug | 8 (12.1) | 2 (6.9) | 6 (16.2) | 0.25 | 105 (17.8) | 19 (13.9) | 86 (18.9) | 0.17 |
| CCB | 0 | 0 | 0 | — | 10 (1.7) | 5 (3.7) | 5 (1.1) | 0.0427 |
Values are mean ± SD or n (%).
ΔEF ≥20% and ≥EF >10% represent patients whose EF improved by an absolute value >20 or >10 percentage points, respectively.
Transplant patients are those with decompensation in their clinical status despite medical intervention and required heart transplantation.
p values represent significance from the Student t test for continuous variables and chi-square test for categorical variables between those with an improvement in EF by 20 percentage points and those who underwent transplant.
p value for differences between those whose EF improved by 10 percentage points and those without such an improvement.
ACE-I = angiotensin-converting enzyme inhibitor; ARB = angiotensin receptor blocker; CABG = coronary artery bypass grafting; CCB = calcium-channel blocker; EF = ejection fraction; NYHA = New York Heart Association.
DESIGN OF CUSTOMIZED ARRAY.
We first designed a customized single nucleotide variation (SNV) array, generating posterior probabilities by compiling publicly available myocardial gene expression data and combining them with a GWAS of extreme response to HF therapy and other resources (Figure 1). We evaluated 866 patients with HF at Stanford University. Patients at the extreme of the distribution of dynamic change were genotyped using a 561,464 SNV genotype chip that was used to inform the design of a customized genotyping array.
All publicly available microarray gene expression data from human cardiac tissue (13) was collected from the Gene Expression Omnibus (GEO) database and normalized using a median-absolute-deviation algorithm (18). Differences in expression levels between failing and normal human myocardium were measured using significance analysis of microarrays (19). The significance value (d-score) was used to weight (20,21) p values from the informative GWAS analysis. The top-ranking genic SNPs were included in the customized array (Online Table 1). The top intergenic SNPs from the informative GWAS analysis, ranked by unweighted p values, were also included in the customized array (Online Table 2).
Next, gene coexpression networks with scale-free topology were created from a collated file of all human myocardial gene expression data (n = 340 expression arrays) (13). Genes were clustered using average linkage hierarchical clustering and adaptively assigned to modules using a dynamic tree-cutting algorithm (22). Modules with topology shared between normal and failing myocardium were ranked according to differential gene expression (quantified by average d-score for modular gene expression differences between normal and failing myocardium) to identify sets of genes associated with adaptation to HF. Common variants from the hub genes, defined as the genes in each module with maximum intramodular connectivity, from the highest-ranking modules were included in the customized array (Online Table 3). The top-ranking module with SLC25A30 as its hub is depicted in Online Figure 4. Next, PubMed abstracts were retrieved and semantic mining was used to create a gene–gene interaction network (23,24). The top common variants of the hub genes, ranked by average d-scores (19), were included in the customized array (Online Table 4). Finally, a curated set of SNVs derived from multiple sources including significant SNPs from prior cardiovascular GWAS (Online Table 5), micro RNA, and coding SNVs in genes known to be targets of pharmacological agents used in HF were included. The selected SNVs were placed on a customized GoldenGate platform (Illumina, Inc.).
CUSTOMIZED ARRAY GENOTYPING AND REPLICATION.
After excluding patients with insufficient clinical data and failure to pass quality control measures, there were a total of 591 patients successfully genotyped with a custom array. Baseline characteristics of these HF patients, 23% of whom had an absolute improvement of 10% in their EF, are shown in Table 1. An additive logistic regression model, adjusted for age, sex, and race, was used to test the association between each SNV on the customized array and improved EF (Manhattan plot [Figure 2A], quantile-quantile plot [Online Figure 3]). Patients with the minor allele for rs7767652 were less likely to have a response >10% in EF (OR: 0.394 per minor allele; p = 3.29 × 10−5), with statistical significance below the Bonferroni cutoff of 0.05/1,402 = 3.56 × 10−5. This variant was chosen for the custom array as 1 of the intergenic SNVs. In the informative GWAS, patients with the minor allele for rs7767652 were less likely to have a response >20% in EF compared with transplanted patients (OR: 0.279 per minor allele; p = 0.0040). The meta-analysis p value was 9.04 × 10−6. The rs7767652 is 2,705 base pairs upstream from HCRTR2 splice variant 1A, within a haplotype block that encompasses several alternative splice sites (25) and the 5’ untranslated region (UTR) of HCRTR2 (Figure 2B). Top results from each of the custom array selection categories are presented in Table 2 and Online Tables 1 to 6. We sought replication in 798 HF patients from the cross-sectional Penn Heart Failure Study (5). After multivariate adjustment, HF patients with the minor allele for rs7767652 were less likely to have an EF >35% (OR: 0.769 per minor allele; p = 0.021).
TABLE 2.
Association Results for Ejection Fraction Response
| Phase I* | Phase II† | Combined | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SNP | Minor Allele | Candidate Gene | Source‡ | ΔEF ≥20%§ (n = 29) MAF | Transplant (n = 37)‖ MAF | OR | p Value | ΔEF ≥10%§ (n = 137) MAF | ΔEF <10% (n = 454) MAF | OR | p Value | p Value |
| Top Overall SNPs ¶ | ||||||||||||
| rs7767652 | T# | HCRTR2 | D | 0.14 | 0.36 | 0.28 | 0.0040 | 0.11 | 0.24 | 0.394 | 3.29 × 10−5 | 9.04 × 10−6 |
| rs2527366 | G | GTF2I | A | 0.45 | 0.26 | 2.35 | 0.016 | 0.39 | 0.29 | 1.728 | 0.00065 | 0.0003 |
| rs4901426 | A | DDHD1 | D | 0.31 | 0.63 | 0.27 | 0.0025 | 0.52 | 0.43 | 1.54 | 0.00383 | 0.0085 |
| rs1936602 | A | HTR7 | D | 0.60 | 0.30 | 3.60 | 0.0020 | 0.38 | 0.49 | 0.67 | 0.00650 | 0.0177 |
| rs3813089 | T | MRO | B | 0.22 | 0.18 | 1.36 | 0.465 | 0.31 | 0.23 | 1.60 | 0.00748 | 0.0062 |
| rs1546120 | T | MAD2L1 | A | 0.12 | 0.30 | 0.32 | 0.0130 | 0.19 | 0.27 | 0.61 | 0.00799 | 0.0037 |
| Top Curated SNPs ** | ||||||||||||
| rs8096199 | G | DLGAP1 | C-Can | — | — | — | — | 0.220 | 0.161 | 1.616 | 0.00902 | 0.0090 |
| rs1799983 | T | NOS3 | C-Can | — | — | — | — | 0.359 | 0.277 | 1.449 | 0.01504 | 0.01504 |
| rs767757 | G | PRKCH | C-Path | 0.362 | 0.216 | 2.057 | 0.0485 | 0.448 | 0.385 | 1.367 | 0.042 | 0.0254 |
| rs7220007 | A | PRKCA | C-Can | 0.603 | 0.378 | 2.5 | 0.0151 | 0.528 | 0.444 | 1.362 | 0.04335 | 0.0263 |
| rs12623467 | T | NRXN1 | C-GWA | — | — | — | — | 0.171 | 0.128 | 1.612 | 0.0267 | 0.0267 |
| rs7903146 | T | TCF7L2 | C-GWA | 0.362 | 0.284 | 1.432 | 0.364 | 0.354 | 0.292 | 1.377 | 0.04691 | 0.0381 |
| rs10511311 | T | CD200 | C-GWA | — | — | — | — | 0.409 | 0.336 | 1.342 | 0.04834 | 0.04834 |
| Replication†† Penn Heart Failure Study | ||||||||||||
| EF ≥35% (n = 333) MAF | EF <35% (n = 464) MAF | OR | p Value | |||||||||
| rs7767652 | T‡‡ | HCRTR2 | Penn | .209 | 0.247 | 0.769 | 0.021 | |||||
Phase I cohort was age-, sex-, and race-matched.
Phase II analyses were adjusted for age, sex, and race.
Source = group from which the variant was selected for placement on the customized array: D = top intergenic SNPs from phase I GWAS; A = top weighted SNPs from phase I GWAS; B-Hub = coexpression network analysis gene hub; C-can = candidate SNPs from prior studies; C-Path = variant in gene responsible for important disease pathway, C-GWA = SNPs associated with relevant phenotypes in prior studies.
ΔEF ≥20% and ΔEF ≥10% represent patients whose EF improved by an absolute value >20 or 10 percentage points, respectively.
Transplant patients are those with decompensation in their clinical status despite medical intervention and required heart transplantation.
Top Overall SNPs = the common variants from the customized heart failure gene array with the lowest p values.
21 of 591 Stanford Phase II heart failure patients were homozygous for the minor allele.
Top Curated SNPs = the variants with the lowest p values from a subset of variants on the customized array that had been manually pre-selected.
The Penn Heart Failure replication analysis was adjusted for age, sex, hypertension, diabetes, renal function (glomerular filtration rate), and body mass index.
59 of 767 Penn Heart Failure patients were homozygous for the minor allele.
GWAS = genome-wide association study; MAF = minor allele frequency; OR = odds ratio per minor allele; SNP = single nucleotide polymorphism; other abbreviations as in Table 1.
FUNCTIONAL VALIDATION OF HCRTR2.
To assess the potential regulatory role of the lead variant rs7767652 upstream of HCRTR2, we first mapped putative transcription factor binding sites in silico using various bioinformatics tools (Online Table 6). The minor allele (T) predicted disruption of a transcription factor 4 (TCF4) binding site containing the motif ATCAAAG. We then measured allele-specific gene regulation using luciferase reporters containing the predicted binding site in transfected C2C12 myoblast cells (Figure 3A). At baseline, transfection with the minor allele (T) construct of rs7767652 resulted in lower luciferase activity compared with the major allele (C) construct (p < 0.05) (Figure 3B). We also observed that the minor allele significantly disrupted β-catenin/TCF4-mediated transactivation in cells overexpressing human β-catenin and TCF4 cofactors (p < 0.01). These effects were abolished in the presence of a TCF4 transactivation mutant (p < 0.005).
FIGURE 3. Functional Roles of the rs7767652 Allele and HCRTR2.
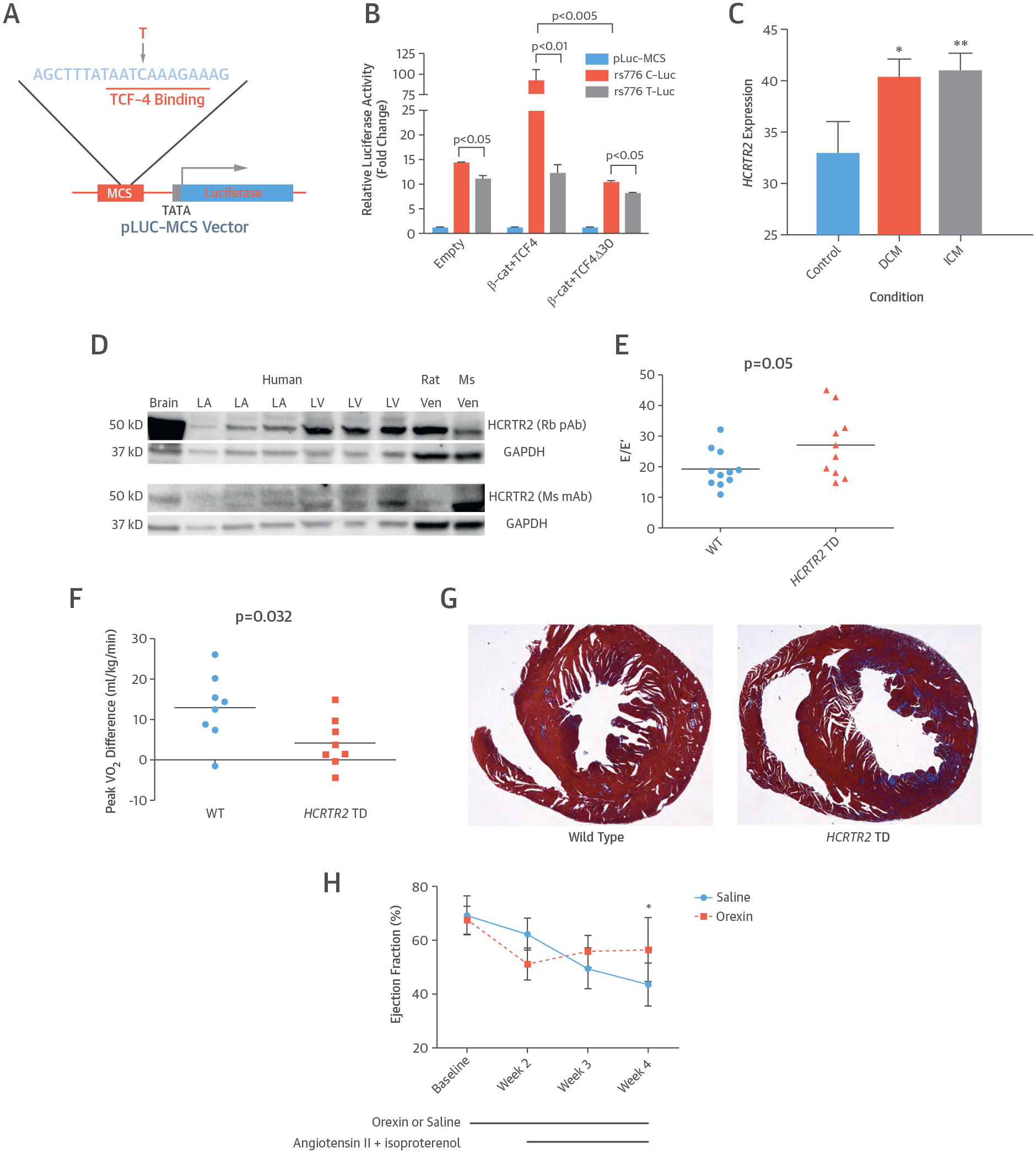
(A) Diagram of luciferase reporter with the rs7767652 locus, which contains a transcription factor-4 (TCF4) binding site. This SNP with its flanking sequence is inserted into the multiple cloning site (MCS) of a pLuc-MCS vector, which is driven by a minimal promoter upstream from the luciferase gene. (B) Allele-specific luciferase reporter assays at rs7767652 in C2C12 cells. The reporter construct carrying the minor allele (rs776 T-Luc) had slightly lower activity at baseline compared to the construct with the major allele (rs776 C-Luc). The major allele was highly responsive to β-catenin (β-cat)/TCF4 overexpression, whereas the minor allele disrupted β-catenin/TCF4 mediated transactivation. The TCF4 transactivation mutant (TCF4 Δ30) blocked β-catenin effects. (C) Human HCRTR2 gene expression measurements from microarray experiments in myocardial tissue of patients with dilated cardiomyopathy (DCM) (n = 162), ischemic cardiomyopathy (ICM) (n =143), and control subjects (n = 45) were obtained from the Gene Expression Omnibus repository. Gene expression values are graphed as mean fluorescence intensity ratios with 95% confidence intervals normalized across experiments. Compared with controls, HCRTR2 expression is higher in DCM tissue (*p = 0.00017) and in ICM tissue (**p = 0.000044). (D) Expression of HCRTR2 protein was examined in human brain, left atrium (LA), left ventricle (LV), rat ventricle (Ven), and mouse ventricle tissues using 2 different antibodies. (E) Diastolic function in C57BL/6 wild-type (WT) (n = 11) and HCRTR2 transcription-deficient (TD) mice (n = 10). The HCRTR2 TD mice had worse diastolic function as measured by a higher E/E’ ratio compared with WT mice (p = 0.05). (F) Change in exercise capacity before and after angiotensin II + isoproterenol chemical stress in C57BL/6 WT (n = 8) compared with HCRTR2 TD (n = 8) mice. The HCRTR2 TD mice had a smaller improvement in peak VO2 (13.0 ± 3.0 ml/kg/min vs. 4.2 ± 2.2 ml/kg/min; p = 0.032) after 2 weeks of chemical stress. (G) Representative examples of trichrome-stained myocardial sections from 5 C57BL/6 WT and 5 HCRTR2 TD mice after 2 weeks of angiotensin II + isoproterenol chemical stress. The HCRTR2 TD mouse hearts had a greater degree of trichrome stain (blue/red ratio 0.82 ± 0.1 vs. 0.85 ± 0.01; p = 0.022). (H) Systolic function in WT mice infused with either saline (n = 6) or orexin A (n = 7) for 4 weeks and stressed with angiotensin II + isoproterenol infusion during the latter 2 weeks. Mice who underwent infusion with orexin A had better systolic function after chemical stress at week 4 (ejection fraction 56.6 ± 4.5% vs. 43.6 ± 3.3%). *p = 0.045.
To assess a myocardial role for this neuropeptide receptor, we compared HCRTR2 expression in diseased human hearts (dilated n = 162; ischemic n = 143) with control tissue (n = 45). Expression of HCRTR2 was greater in dilated cardiomyopathy (p = 0.00017) and ischemic cardiomyopathy samples (p = 0.000044) compared with control samples (Figure 3C). We also confirmed that the HCRTR2 protein product was present in diseased human myocardial tissue and found that it was present in a greater concentration in the left ventricle compared with the left atrium (Figure 3D).
To investigate a causal role for HCRTR2 in HF pathogenesis, we performed heart function testing via ultrasound in HCRTR2 transcription-disrupted (TD) mice (n = 10) compared with wild-type mice (n = 11) (26). There was greater diastolic dysfunction in HCRTR2 TD mice compared with wild-type mice (p = 0.05) (Figure 3E), but no significant difference in systolic function. Similar studies in a small number of HCRTR2 knockout mice (27) (n = 5) compared with control subjects (n = 5) also demonstrated a trend toward greater diastolic dysfunction. HCRTR2 TD mice that underwent chemical stress with 2 weeks of angiotensin II and isoproterenol infusion had a smaller increase in treadmill exercise capacity (Figure 3F) and greater evidence of myocardial fibrosis (Figure 3G) compared with wild-type mice.
Finally, to assess the potential of HCRTR2 as a novel therapeutic target for HF, wild-type mice were infused with saline (n = 6) or the HCRTR2 agonist orexin A (n = 7) for 4 weeks. At week 2, all mice underwent infusion of angiotensin II and isoproterenol, neurohormones that mimic human HF. Echocardiography was performed blinded to drug infusion status at baseline and 2, 3, and 4 weeks (Figure 3H). Mice with orexin A had better systolic function compared with control subjects (p = 0.045).
DISCUSSION
Starting from a clinical observation of a dramatic response to HF medical therapy in a subset of patients, we categorized dramatic responders then studied the extremes of response using a customized gene array. Our primary hypothesis was that there are common genomic variants that associate with response to HF therapy. After identifying a genomic variant in the regulatory region of HCRTR2 associated with improved left ventricular function, we sought to validate this finding by performing a replication study in the University of Pennsylvania cohort and several functional experiments. Allele-specific reporter assays at this locus further suggested a role for this variant in regulating nearby gene transcription, and functional characterization of mice with HCRTR2 transcription deficiency demonstrated that HCRTR2 itself plays an important role in the regulation of cardiac function (Central Illustration).
CENTRAL ILLUSTRATION. HCRTR2 and Heart Failure: Therapy Through Genomic Variation and Functional Validation.
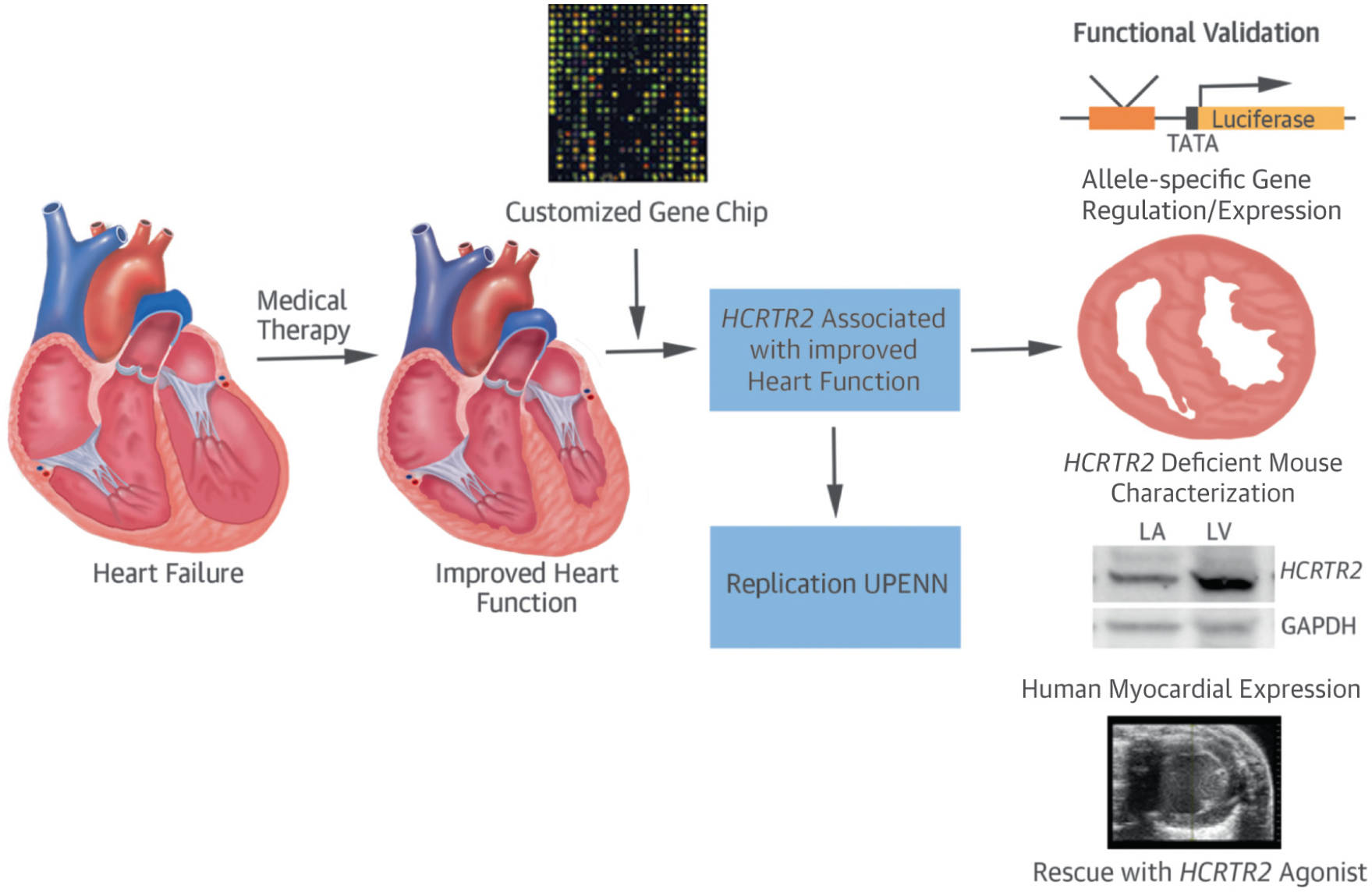
A systems approach was used in the design of a customized gene chip to study heart failure patients who had a dramatic response in heart function with medical therapy. A genetic variant near hypocretin receptor-2 (HCRTR2) was associated with improved heart function and was replicated in an independent cohort from the University of Pennsylvania. Finally, a series of functional studies were performed to validate the role of the genetic variant and the HCRTR2 gene product in heart failure. GAPDH = glyceraldehyde 3-phosphate dehydrogenase; LA = left atrium; LV = left ventricle; UPENN = University of Pennsylvania.
The HCRTR2 gene encodes a G protein-coupled receptor that binds hypocretin (orexin) A and B, neuropeptides involved in regulating appetite (28,29) and sleep (30). Although highly expressed in the hypothalamus (29), its expression has been documented in several tissues including the gut (31) and adrenal glands (32). Alternative splice variants of HCRTR2 were also previously expressed in heart tissue (25). Orexins provoke increases in blood pressure and heart rate via a centrally-mediated response (33–35).
The effect of these neurohormonal peptides on myocardial function, however, has not been studied. Yet, there are well-established links between sleep disorders and heart failure (36). This link is thought to be mediated by oxidative, inflammatory, and vascular endothelial mechanisms (37). Whether or not orexins mediate this association will need to be studied further.
The HCRTR2 gene has been best studied in context of its link to narcolepsy, which was first reported in Doberman canines (30). This later led to the discovery of the role of orexins in sleep regulation and, ultimately, a role for antagonists of HCRTR2 (such as suvorexant) in treating insomnia (38). Our observation that agonism of HCRTR2 leads to beneficial effects on myocardial function, however, suggests that patients using long-term HCRTR2 antagonists may benefit from monitoring for signs and symptoms of HF. Similarly, future studies to evaluate heart function in patients with narcolepsy are warranted.
Our finding that mice infused with an HCRTR2 agonist were protected from chemical stress-induced ventricular dysfunction suggests a promising role for HCRTR2 in regulation of ventricular function. The observation in the myocardial gene expression arrays that diseased myocardial tissue from the human left ventricle has a higher degree of HCRTR2 gene expression further implies that HCRTR2 may be involved in a protective response in the setting of myocardial insult. Additionally, the observation from the luciferase reporter assays that the minor allele results in lower gene expression suggests that minor allele carriers may lack the capacity to sufficiently up-regulate HCRTR2 in response to development of myocardial disease.
These findings may point to a novel G-protein-coupled receptor-mediated pathway that is distinct from, and could be complementary to, the well-established axis of the renin-angiotensin-aldosterone system. The effects of HCRTR2 agonists on myocardial function have not previously been characterized, but may play a role in regulating myocardial remodeling as indicated by our finding that myocardial fibrosis was more pronounced in HCRTR2-deficient mice.
Finally, we have identified a few clinical characteristics that are associated with the most dramatic response to HF therapy (Phase I [Table 1]), including the presence of diabetes along with HF. Although these were unadjusted comparisons in a relatively small number of subjects, it is worth validating in future studies whether or not these comorbid conditions may result in a more treatable form of cardiomyopathy compared with those with a primary form of HF.
STUDY LIMITATIONS.
Because no other cohort exists that has been studied from the perspective of dramatic response to therapy, our replication cohort relied on a single EF measurement. We used a neurohormonal model for HF, which produces a less-severe phenotype than models such as left anterior descending ligation. Despite this, we found significant changes in fibrosis and diastolic function. Future work will include more severe HF models as well as specific small molecule agonists. Additional studies to examine the central versus peripheral effects of orexin are beyond the scope of this work but will inform future therapeutic development.
CONCLUSIONS
We used a systems approach to identify a promising therapeutic target, HCRTR2, for the treatment of HF. Functional studies in several mouse models validated the role of HCRTR in regulating cardiac function, possibly via effects on heart remodeling. Further studies are needed to better elucidate the novel role of this neurohormonal pathway in regulating heart function.
Supplementary Material
PERSPECTIVES.
COMPETENCY IN PATIENT CARE AND PROCEDURAL SKILLS:
Some patients with systolic HF improve during treatment with neurohormonal blockade and other medical interventions, whereas others worsen despite similar management; genetic factors might explain some of this disparity.
TRANSLATIONAL OUTLOOK:
Combining genomic data with network-based analyses might not only isolate genetic variants associated with treatment failure but also identify others with the potential to rescue impaired myocardium.
Acknowledgments
Funding for this study was provided by the Robert Wood Johnson Foundation, a Harold Amos Medical Faculty Development Award, and the Stanford University Cardiovascular Institute (to Dr. Perez); by a National Institutes of Health NS-079940 award (to Dr. Peter); and by the Breetwor Foundation (to Dr. Ashley). Dr. Wheeler has modest ownership interest in Personalis, Inc. Dr. Dewey is an employee of Regeneron Genetics Center. Ms. Salisbury is a patient education consultant for Myokardia. Dr. Cappola has served on the data safety monitoring board for Novartis; and has received laboratory assays from BG Medicine. Dr. Ashley is cofounder of and scientific advisor to Personalis. All other authors have reported that they have no relationships relevant to the contents of this paper to disclose.
ABBREVIATIONS AND ACRONYMS
- GWAS
genome-wide association study
- HCRTR2
hypocretin receptor-2
- HF
heart failure
- SNP
single nucleotide polymorphism
- SNV
single nucleotide variant
Footnotes
APPENDIX For a supplemental Methods section as well as figures and tables, please see the online version of this article.
REFERENCES
- 1.Heidenreich PA, Trogdon JG, Khavjou OA, et al. Forecasting the future of cardiovascular disease in the United States: a policy statement from the American Heart Association. Circulation 2012;123: 933–44. [DOI] [PubMed] [Google Scholar]
- 2.Ho KK, Pinsky JL, Kannel WB, Levy D. The epidemiology of heart failure: the Framingham Study. J Am Coll Cardiol 1993;22:6A–13A. [DOI] [PubMed] [Google Scholar]
- 3.Lee DS, Pencina MJ, Benjamin EJ, et al. Association of parental heart failure with risk of heart failure in offspring. N Engl J Med 2006;355: 138–47. [DOI] [PubMed] [Google Scholar]
- 4.Stark K, Esslinger UB, Reinhard W, et al. Genetic association study identifies HSPB7 as a risk gene for idiopathic dilated cardiomyopathy. PLoS Genet 2010;6:e1001167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cappola TP, Li M, He J, et al. Common variants in HSPB7 and FRMD4B associated with advanced heart failure. Circ Cardiovasc Genet 2010;3: 147–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cresci S, Kelly RJ, Cappola TP, et al. Clinical and genetic modifiers of long-term survival in heart failure. J Am Coll Cardiol 2009;54:432–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liggett SB, Cresci S, Kelly RJ, et al. A GRK5 polymorphism that inhibits beta-adrenergic receptor signaling is protective in heart failure. Nat Med 2008;14:510–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Larson MG, Atwood LD, Benjamin EJ, et al. Framingham Heart Study 100K project: genome-wide associations for cardiovascular disease outcomes. BMC Med Genet 2007;8 Suppl 1:S5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Smith NL, Felix JF, Morrison AC, et al. Association of genome-wide variation with the risk of incident heart failure in adults of European and African ancestry: a prospective meta-analysis from the cohorts for heart and aging research in genomic epidemiology (CHARGE) consortium. Circ Cardiovasc Genet 2010;3:256–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Villard E, Perret C, Gary F, et al. A genome-wide association study identifies two loci associated with heart failure due to dilated cardiomyopathy. Eur Heart J 2011;32:1065–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Meder B, Ruhle F, Weis T, et al. A genome-wide association study identifies 6p21 as novel risk locus for dilated cardiomyopathy. Eur Heart J 2014; 35:1069–77. [DOI] [PubMed] [Google Scholar]
- 12.Parks BW, Sallam T, Mehrabian M, et al. Genetic architecture of insulin resistance in the mouse. Cell Metab 2015;21:334–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dewey FE, Perez MV, Wheeler MT, et al. Gene coexpression network topology of cardiac development, hypertrophy, and failure. Circ Cardiovasc Genet 2011;4:26–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu Y, Morley M, Brandimarto J, et al. RNA-Seq identifies novel myocardial gene expression signatures of heart failure. Genomics 2015;105: 83–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.de Groote P, Fertin M, Duva Pentiah A, Goeminne C, Lamblin N, Bauters C. Long-term functional and clinical follow-up of patients with heart failure with recovered left ventricular ejection fraction after beta-blocker therapy. Circ Heart Fail 2014;7:434–9. [DOI] [PubMed] [Google Scholar]
- 16.Purcell S, Neale B, Todd-Brown K, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 2007;81:559–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Whitlock MC. Combining probability from independent tests: the weighted Z-method is superior to Fisher’s approach. J Evol Biol 2005;18: 1368–73. [DOI] [PubMed] [Google Scholar]
- 18.Smyth GK, Speed T. Normalization of cDNA microarray data. Methods 2003;31:265–73. [DOI] [PubMed] [Google Scholar]
- 19.Storey JD, Tibshirani R. Statistical methods for identifying differentially expressed genes in DNA microarrays. Methods Mol Biol 2003;224: 149–57. [DOI] [PubMed] [Google Scholar]
- 20.Genovese CR, Roeder K, Wasswerman L. False discovery control with p-value weighting. Bio-metrika 2006;93:509–24. [Google Scholar]
- 21.Roeder K, Devlin B, Wasserman L. Improving power in genome-wide association studies: weights tip the scale. Genet Epidemiol 2007;31: 741–7. [DOI] [PubMed] [Google Scholar]
- 22.Langfelder P, Zhang B, Horvath S. Defining clusters from a hierarchical cluster tree: the Dynamic Tree Cut package for R. BMC Bioinformatics 2008;24:719–20. [DOI] [PubMed] [Google Scholar]
- 23.Ashley EA, Ferrara R, King JY, et al. Network analysis of human in-stent restenosis. Circulation 2006;114:2644–54. [DOI] [PubMed] [Google Scholar]
- 24.King JY, Ferrara R, Tabibiazar R, et al. Pathway analysis of coronary atherosclerosis. Physiol Genomics 2005;23:103–18. [DOI] [PubMed] [Google Scholar]
- 25.Chen J, Randeva HS. Genomic organization and regulation of the human orexin (hypocretin) receptor 2 gene: identification of alternative promoters. Biochem J 2010;427:377–90. [DOI] [PubMed] [Google Scholar]
- 26.Mochizuki T, Arrigoni E, Marcus JN, et al. Orexin receptor 2 expression in the posterior hypothalamus rescues sleepiness in narcoleptic mice. Proc Natl Acad Sci U S A 2011;108:4471–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chemelli RM, Willie JT, Sinton CM, et al. Narcolepsy in orexin knockout mice: molecular genetics of sleep regulation. Cell 1999;98: 437–51. [DOI] [PubMed] [Google Scholar]
- 28.Lubkin M, Stricker-Krongrad A. Independent feeding and metabolic actions of orexins in mice. Biochem Biophys Res Commun 1998;253: 241–5. [DOI] [PubMed] [Google Scholar]
- 29.Sakurai T, Amemiya A, Ishii M, et al. Orexins and orexin receptors: a family of hypothalamic neuropeptides and G protein-coupled receptors that regulate feeding behavior. Cell 1998;92: 1 page following 696. [DOI] [PubMed] [Google Scholar]
- 30.Lin L, Faraco J, Li R, et al. The sleep disorder canine narcolepsy is caused by a mutation in the hypocretin (orexin) receptor 2 gene. Cell 1999;98: 365–76. [DOI] [PubMed] [Google Scholar]
- 31.Kirchgessner AL, Liu M. Orexin synthesis and response in the gut. Neuron 1999;24:941–51. [DOI] [PubMed] [Google Scholar]
- 32.Lopez M, Senaris R, Gallego R, et al. Orexin receptors are expressed in the adrenal medulla of the rat. Endocrinology 1999;140:5991–4. [DOI] [PubMed] [Google Scholar]
- 33.Shirasaka T, Nakazato M, Matsukura S, et al. Sympathetic and cardiovascular actions of orexins in conscious rats. Am J Physiol 1999;277: R1780–5. [DOI] [PubMed] [Google Scholar]
- 34.Samson WK, Gosnell B, Chang JK, et al. Cardiovascular regulatory actions of the hypocretins in brain. Brain Res 1999;831:248–53. [DOI] [PubMed] [Google Scholar]
- 35.Matsumura K, Tsuchihashi T, Abe I. Central orexin-A augments sympathoadrenal outflow in conscious rabbits. Hypertension 2001;37: 1382–7. [DOI] [PubMed] [Google Scholar]
- 36.Shahar E, Whitney CW, Redline S, et al. Sleep-disordered breathing and cardiovascular disease: cross-sectional results of the Sleep Heart Health Study. Am J Respir Crit Care Med 2001;163:19–25. [DOI] [PubMed] [Google Scholar]
- 37.Kasai T, Bradley TD. Obstructive sleep apnea and heart failure: pathophysiologic and therapeutic implications. J Am Coll Cardiol 2011;57:119–27. [DOI] [PubMed] [Google Scholar]
- 38.Patel KV, Aspesi AV, Evoy KE. Suvorexant: A dual orexin receptor antagonist for the treatment of sleep onset and sleep maintenance insomnia. Ann Pharmacother 2015;49: 477–83. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.