

Abstract
Objective
Key augmented processes in atherosclerosis have been identified, whereas less is known about downregulated pathways. Here, we applied a systems biology approach to examine suppressed molecular signatures, with the hypothesis that they may provide insight into mechanisms contributing to plaque stability.
Approach and Results
Muscle contraction, muscle development, and actin cytoskeleton were the most downregulated pathways (false discovery rate=6.99e-21, 1.66e-6, 2.54e-10, respectively) in microarrays from human carotid plaques (n=177) versus healthy arteries (n=15). In addition to typical smooth muscle cell (SMC) markers, these pathways also encompassed cytoskeleton-related genes previously not associated with atherosclerosis. SYNPO2, SYNM, LMOD1, PDLIM7, and PLN expression positively correlated to typical SMC markers in plaques (Pearson r>0.6, P<0.0001) and in rat intimal hyperplasia (r>0.8, P<0.0001). By immunohistochemistry, the proteins were expressed in SMCs in normal vessels, but largely absent in human plaques and intimal hyperplasia. Subcellularly, most proteins localized to the cytoskeleton in cultured SMCs and were regulated by active enhancer histone modification H3K27ac by chromatin immunoprecipitation-sequencing. Functionally, the genes were downregulated by PDGFB (platelet-derived growth factor beta) and IFNg (interferron gamma), exposure to shear flow stress, and oxLDL (oxidized low-density lipoprotein) loading. Genetic variants in PDLIM7, PLN, and SYNPO2 loci associated with progression of carotid intima-media thickness in high-risk subjects without symptoms of cardiovascular disease (n=3378). By eQTL (expression quantitative trait locus), rs11746443 also associated with PDLIM7 expression in plaques. Mechanistically, silencing of PDLIM7 in vitro led to downregulation of SMC markers and disruption of the actin cytoskeleton, decreased cell spreading, and increased proliferation.
Conclusions
We identified a panel of genes that reflect the altered phenotype of SMCs in vascular disease and could be early sensitive markers of SMC dedifferentiation.
Keywords: actin cytoskeleton, atherosclerosis, downregulation, hyperplasia, smooth muscle cells
Graphical Abstract
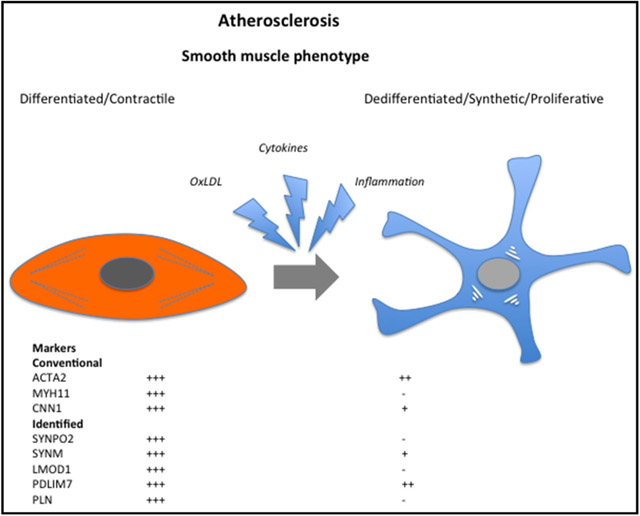
Unstable atherosclerosis in the carotid bifurcation is a common cause of stroke, and guidelines recommend treatment with stroke-preventive carotid endarterectomy in patients with signs of cerebral embolism.1 Stable, asymptomatic carotid lesions are generally rich in extracellular matrix and smooth muscle cells (SMCs), whereas unstable (symptomatic) plaques contain abundant inflammatory cells and a thin fibrous cap prone to rupture.2 Inflammation, cytokines, mitogens, extracellular matrix degradation, and altered cell–matrix interactions have been associated with intraplaque processes in atherogenesis, which promote activation of contractile SMCs in the media into a secretory and replicating phenotype that engage in intimal remodeling and formation of the fibrous cap.
Contractile SMCs are distinguished from other cell types by expression of a unique repertoire of markers, including smooth muscle actin (SMA, ACTA2 [actin alpha 2, smooth muscle, aorta]), CNN1 (calponin 1), TAGLN (transgelin), MYOCD (myocardin), and MYH11 (myosin heavy chain 11), mostly associated with the actomyosin cytoskeleton. These genes are downregulated in activated SMCs and may be undetectable using traditional immunohistochemical staining methods.3,4 However, it is unclear whether altered expression of these genes takes place concomitantly or successively during phenotypic modulation. This problem is notable in atherosclerotic lesions where SMA-positive (SMA+) cells define several distinct regions and can be found in the necrotic core and the fibrous cap (Figure 1A). Recently, SMC transdifferentiation into CD68+ macrophage-like cells has been demonstrated in atherosclerosis,5,6 which further emphasizes the complexity in characterizing SMC phenotypes in vascular disease. Apart from atherogenesis, activation of SMCs also dominates in healing reactions aimed to repair the vessel after injury, healing of ruptured atheromas,7 restenosis after arterial interventions,8 and in the failure of vein grafts and dialysis fistulas.9
Figure 1.

LMOD1, SYNPO2, SYNM, PDLIM7, and PLN were downregulated in carotid plaques. Microarray profiles comparing n=127 carotid plaques vs n=10 normal arteries (NAs) were used in discovery phase for analysis of downregulated genes and pathways. Arrowheads showing smooth muscle actin (SMA)+ smooth muscle cells (SMCs) in the media of NA, remaining media at the plaque periphery and fibrous cap. Images were taken with ×4 objective. A, Coexpression and cointeraction network of genes clustered in pathways muscle contraction, muscle development, actin cytoskeleton, and myofibril cytoskeleton show the presence of typical markers of SMCs in these categories (ie, CNN1, SMTN, TAGLN, ACTA2 [actin alpha 2, smooth muscle, aorta], and MYH11) and other potentially interesting genes of which some were selected for further investigations. Network weighted for closeness in biological function based on publically available data (B). PDLIM7, LMOD1, SYNPO 2, SYNM, and PLN were among the most downregulated genes in microarrays from plaques (CPs) vs NAs and symptomatic (S) vs asymptomatic (AS) patients (C) and strongly correlated to standard markers of SMCs (D). Plots showing log mean±SD in C and Pearson correlations in D. *P<0.05, **P<0.01, ****P<0.0001.
Understanding the molecular and cellular processes that convert asymptomatic plaques into symptomatic ones may facilitate the development of preventive pharmacotherapy with unprecedented impact on cardiovascular mortality and morbidity. For this purpose, intensive efforts have been dedicated to the identification of suitable targets through analysis of augmented pathways and molecules in vulnerable lesions.10,11 In contrast, less is known about pathways that are inhibited in atherogenesis and in the process of plaque instability. Because identification of such inherent functional changes within the vessel wall may give clues to therapies that can sustain arterial resistance to atherogenic stimuli or improve stability of established complex lesions, studies of downregulated genes and suppressed pathways may be equally important.12 Identification of ultimately translatable target molecules is bound to be more successful when generated directly from human disease, followed by clinical and experimental exploration.
Recently, we performed a comprehensive transcriptomic analysis of late-stage human carotid atherosclerosis based on defined clinical patient phenotypes.13 Our findings confirmed a central role for lipid accumulation, inflammation, and proteases in plaque instability and highlighted hemoglobin metabolism and bone resorption as important enriched pathways in plaques. Here, we instead analyzed downregulated molecular signatures in carotid plaques (CPs) by applying an integrative framework based on collaboration among 3 large human biobanks: initial discoveries were made using material from the Biobank of Karolinska Endarterectomies (BiKE, n=177 plaques from end-stage atherosclerosis patients and n=15 macroscopically healthy, normal arteries [NAs]), data were further validated using atheroprogression samples from the SOKRATES biobank (n=28 patients tissues), and genetic analyses were performed in the IMPROVE cohort (Carotid Intima Media Thickness [IMT] and IMT-PRogression as Predictors of Vascular Events in a High-Risk European Population; n=3378 high-risk patients without symptoms or signs of cardiovascular disease). We found that SMC-related functional categories were the most significantly affected in plaques and identified a set of downregulated SMC-related genes previously poorly studied in vascular disease. Temporal changes in the expression of these genes were followed in the rat carotid injury model and in primary SMCs in vitro in comparison with typical SMC markers. Genetic association with progression of carotid intima-media thickness (cIMT) as a surrogate marker of atherosclerosis was investigated in the large cohort of high-risk subjects, and mechanistic studies were performed for one of these genes in vitro. We report a panel of novel SMC markers that are suppressed in vascular disease in humans and may reflect the altered phenotype of SMCs during vascular remodeling.
Material and Methods
Material and Methods are available in the online-only Data Supplement.
Results
Genes and Pathways Associated With SMCs Are Repressed in Atherosclerosis
Pathways associated with muscle contraction, muscle development, and actin cytoskeleton were the most significantly downregulated in microarrays comparing late-stage human CPs versus NAs and in plaques extracted from symptomatic versus asymptomatic patients (eg, in CP versus NA comparison false discovery rate=6.99e-21, 1.66e-6, and 2.54e-10, respectively; Table I in the online-only Data Supplement). Genes clustered in these categories were the typical markers of SMCs and actomyosin cytoskeleton (Figure IA in the online-only Data Supplement). Among the most significantly downregulated genes appeared several whose function in SMCs was previously unexplored in the context of atherosclerosis: LMOD1 (leiomodin 1), SYNPO2 (synaptopodin 2), PLN (phospholamban), PDLIM7 (PDZ and LIM domain–containing 7), and SYNM (synemin). By constructing functional networks from their extended protein–protein interactions and expression profiles across tissues,14-16 we noted that these genes were coexpressed with actin and microtubule markers (Figure 1B) and some of them also cointeracted with the cytoskeleton based on available public data (ie, PDLIM7, SYNM, LMOD1, and SYNPO2; Figure IB in the online-only Data Supplement). Strong downregulation of these transcripts was found in 2 nonoverlapping microarray data sets comparing CPs to NAs (n=127 CP versus n=10 NA and n=50 CP versus n=5 NA, P<0.0001 for most transcripts) and downregulation was also noted in plaques from symptomatic versus asymptomatic patients (n=87 symptomatic versus 40 asymptomatic, P<0.01; Figure 1C, full list in Table II in the online-only Data Supplement). Moreover, the protein levels were also lower in plaques from symptomatic versus asymptomatic patients, as determined by mass spectrometry (n=9 symptomatic versus 9 asymptomatic, Figure II in the online-only Data Supplement). In addition, a trend toward downregulation of these genes was observed in publicly available microarray data sets comparing human CPs (n=12) versus NAs (n=9)10 and CPs (n=32) versus matched adjacent tissue (GSE43292). Strong positive correlations were seen between mRNA expression of LMOD1, SYNPO2, PLN, PDLIM7, and SYNM in CPs and typical SMC markers, such as ACTA2, MYH11, CNN1, and MYOCD (Pearson r>0.6, P<0.0001, representative examples in Figure 1D, full data in Table III in the online-only Data Supplement). Similarly, strong correlations between corresponding protein levels, as determined by mass spectrometry, were also demonstrated (Pearson r>0.8, P<0.0001, Table III in the online-only Data Supplement). To further investigate the association of the selected genes with SMCs, we analyzed a publicly available microarray data set (GSE23303) comparing microdissected SMC-rich subintimal regions of CPs with macrophage-rich regions from the necrotic core. We found that mRNA levels of these genes were strongly downregulated in macrophage-rich compared with SMC-rich regions, whereas no significant difference was seen in this comparison for ACTA2 (Table II in the online-only Data Supplement).
To experimentally corroborate our findings from human plaques, we analyzed the expression of the selected genes in an inducible plaque rupture model on apolipoprotein E background, where mice present atherothombotic events and morphological signs of plaque instability.17 Expression of all 5 genes of interest was strongly downregulated in ligated versus nonligated arteries (ie, mRNA mean fold change=−114, P<0.0001 for Synpo2; fold change=−45, P<0.0001 for Lmod1), and marginally also in comparison between ruptured versus stable plaques; thus, replicating results from the human data sets (Table IV in the online-only Data Supplement).
Collectively, these results demonstrated that we have identified a set of previously poorly characterized genes through transcriptomic profiling of late-stage human atherosclerosis, likely associated with the loss of contractile SMC features in the disease.
LMOD1, SYNPO2, PDLIM7, SYNM, and PLN Are Expressed by SMCs
The localization of selected genes and proteins in normal human vessels and CPs was performed by in situ RNA hybridization and immunohistochemistry and compared with typical SMC markers, such as MYH11, CNN1, and SMA, including proliferating cell nuclear antigen (PCNA) as a marker of cell proliferation. By immunohistochemistry, MYH11, considered to be an early marker of phenotypic changes in SMCs,3 was only detected in PCNA− SMCs in normal carotid artery media but was absent in late-stage plaques, whereas CNN1 was detectable in the NA and in the subintimal SMA+/PCNA− cells and in SMA+ cells in the fibrous cap of CPs (Figure 2A). By coimmunostaining, SMA, LMOD1, SYNPO2, PDLIM7, SYNM, and PLN were all abundantly expressed in SMCs in NAs (carotid artery stainings shown in Figure 2B). The stainings appeared mostly cytosolic, except in the case of PLN, which exhibited nuclear staining. In late-stage plaques, PDLIM7 was present in subintimal SMCs, and weak expression was also detected in stellate-shaped SMA+ cells in the fibrous cap. SYNM showed a similar staining pattern in subintimal SMCs but was absent from the fibrous cap. SYNPO2, LMOD1, and PLN were not detectable in these plaques by immunohistochemistry. RNA transcripts of MYH11, CNN1, ACTA2, and selected genes were all detectable in the NA media and to a lesser extent also in cells with elongated nuclei in the fibrous cap (except PLN, Figure III in the online-only Data Supplement).
Figure 2.

Selected candidates were localized to differentiated smooth muscle cells (SMCs) and reduced in late-stage plaques. By immunohistochemistry, Myh11 (red) was present only in normal carotid arteries, whereas calponin (red) and smooth muscle actin (SmA, green) were detectable in normal arteries, in plaques and in human intimal hyperplasia tissue with large proliferating cell nuclear antigen negative (PCNA)− areas (arrowheads, A). The identified SMCs markers (red) were all localized to SMCs in the normal carotid artery (left column panels, insets show higher magnification). Pdlim7 and Synm were also present in subintimal SMA+ cells at the plaque periphery and Pdlim7 was the only one still detectable in SMA+ cells in the fibrous cap. Signal for Lmod1, Synpo2, and Pln was lost in plaques. Abundant staining for Pdlim7 and Synm was seen in intimal hyperplasia tissues, and Synpo2, Lmod1, and Pln were also observed in PCNA− areas (B). Images were taken with ×10 objective, insets show ×40 magnification.
We further investigated the expression of these genes during atheroprogression, using human aortic lesions from different stages of disease graded according to the modified American Heart Association criteria,18 ranging from adaptive intimal thickening and xantomas (stages I and II), via pathological intimal thickening (stage III) to early and thin-cap fibroatheromas (stages IV and V). In these lesions, SMA and CNN1 were detectable in SMCs from early stages to advanced lesions, whereas MYH11 as expected was absent from PCNA+ SMCs already at stage I (Figure IV in the online-only Data Supplement). LMOD1 and SYNPO2 were mostly absent already from stage I, PLN was not detectable from stage III, whereas SYNM and PDLIM7 were present in subintimal SMCs but sparsely in cells that build the fibrous cap from stage III. Interestingly, in human intimal hyperplasia, we observed the reappearance of both typical SMC markers and the selected genes in SMA+/PCNA− areas (Figure 2). Abundant signal was observed for CNN1 and for PDLIM7 and SYNM, whereas LMOD1, SYNPO2, and PLN were sparsely present. Our results confirm that these genes localize to quiescent SMCs in NA media and undergo various degrees of downregulation at both transcript and protein levels in activated SMA+ cells of lesions, with reappearance in mature intimal hyperplasia. Of note, these proteins were also detected in SMA+ cells in several other smooth muscle–rich tissues (Figure V in the online-only Data Supplement).
Lmod1, Synpo2, Pdlim7, Synm, and Pln Are Downregulated Early in Reponse to Experimental Vascular Injury but Reappear in Mature Neointima
Time-dependent alterations in expression of SMC markers were examined by transcriptomic analysis from rat carotid arteries after balloon injury (Figure 3). Typical SMC genes along with Lmod1, Synpo2, Pdlim7, Synm, and Pln showed similar gene expression profiles with gradual downregulation in the early phases after injury, but upregulation after 2 to 12 weeks in the mature neointima. Expression correlations of Lmod1, Synpo2, Pln, Pdlim7, and Synm with typical SMC markers in this model were strongly positive (mostly Pearson r>0.8, P<0.0001; Figure 3C; Table III in the online-only Data Supplement) and negative with cytokines, such as Pdgfb, Igf1, and Tgfb1 (Figure 3C). By immunohistochemistry, we observed the loss of CNN1 from PCNA+ SMC layers closer to the lumen, whereas the staining was still present in deeper medial layers at day 5 and again abundant in the mature intima with reduced PCNA staining 12 weeks after injury (Figure 4A). Staining for LMOD1, SYNPO2, PDLIM7, SYNM, and PLN was absent at day 5 with gradual reappearance in medial SMCs in tissues with pronounced intimal hyperplasia 12 weeks after injury (Figure 4B). No similar changes in gene expression patterns were found in contralateral uninjured arteries (Figure VI in the online-only Data Supplement). These analyses indicated that downregulation of Lmod1, Synpo2, Pln, Pdlim7, and Synm might functionally relate to SMC activation in response to injury.
Figure 3.

Expression of LMOD1, SYNPO2, SYNM, PLN, and PDLIM7 strongly correlated to typical markers of smooth muscle cells (SMCs) during the course of rat carotid artery injury and healing response. By microarray profiling, the identified SMC proteins and typical markers of SMCs were downregulated in early phases ≤day 5 after vessel injury and gradually upregulated in later phases in intimal hyperplasia after rat carotid balloon injury (A and B). Expression correlations of LMOD1, SYNPO2, SYNM, PLN, and PDLIM7 with typical SMC markers were significant and strongly positive in this model (mostly Pearson r>0.8), whereas they were negative with PDGFB (platelet-derived growth factor beta), IGF1 (insulin-like growth factor), and TGFB1 (transforming growth factor beta; C).
Figure 4.

Lmod1, Synpo2, Synm, Pln, and Pdlim7 were localized to smooth muscle cells (SMCs) in intact rat carotid artery and reduced in response to injury. By immunohistochemistry, the loss of calponin (red) from highly proliferative proliferating cell nuclear antigen positive (PCNA)+ SMCs layers in the injured rat artery closer to the lumen was observed (arrowheads), whereas staining was still present in deeper medial layers at day 5. At 12 weeks after injury, calponin was again abundant in the mature intima with less proliferative cells in the deeper layers but absent from luminal PCNA+ layers (arrowheads, A). The signal for identified SMC markers (red) was completely absent at day 5 with gradual reappearance in tissues with pronounced intimal hyperplasia at 12 weeks after injury (B). Images were taken with ×20 objective, insets show higher magnification (×100) of the media.
LMOD1, SYNPO2, SYNM, PDLIM7, and PLN Localize Mostly to Actin Cytoskeleton in SMCs and Relate To Phenotypic Changes In Vitro
To address the expression of the selected genes during the process of SMC phenotypic modulation, rat aortic SMCs were isolated by collagenase digestion, seeded on fibronectin and cultured in serum-free medium or medium supplemented with PDGFBB (platelet derived growth factor BB) for 7 days.19 Directly on isolation (day 0), almost 90% of the cells were SMA+ by flow cytometry, although lower SMA levels in a subgroup of cells were detectable (Figure 5A). After 7 days, 95% of the population cultured in serum-free medium uniformly expressed higher levels of SMA, whereas cells stimulated with PDGFBB showed the presence of 2 subpopulations of which one expressed lower signal for SMA (totally 77% SMA+ cells, Figure 5A, detailed analysis shown in Figure VII in the online-only Data Supplement). By quantitative polymerase chain reaction, mRNA levels of Acta2, Myocd, and Myh11 and Lmod1, Synpo2, Pdlim7, Synm, and Pln strongly decreased from day 1 to day 3 in culture, but on day 5 and 7, the expression of most of these genes (except Synm) gradually increased. At each reference time point, cells cultured in the presence of PDGFBB showed downregulation of the target genes compared with cells in serum-free medium (Figure 5B). By RNA-sequencing, downregulation of LMOD1, SYNPO2, PDLIM7, SYNM, and PLN was also observed in low-passage human SMCs cultured in serum-supplemented (dedifferentiation condition) versus serum-free medium (Table V in the online-only Data Supplement).
Figure 5.

LMOD1, SYNPO2, SYNM, PLN, and PDLIM7 were expressed by differentiated smooth muscle cells (SMCs) in vitro and localized to actin cytoskeleton. By flow cytometry, 90% of the primary rat aortic cells were smooth muscle actin (sMa)+ on day 0 (after overnight collagenase treatment, top, A) and 7 days after isolation when cultured in serum-free medium (second panel from top, A). A subpopulation of cells with lower sMa+ signal was identified in cultures at day 0, as well as 7 days after isolation when stimulated with PDGFB (platelet-derived growth factor beta; arrows, top and third panel, A). A, Bottom, Overlap of the upper 3 panels indicating the change in the SMA signal during 7 days of culture. By quantitative polymerase chain reaction analysis, rSMCs (rat smooth muscle cells) showed downregulation of conventional and identified SMC markers at day 3 on isolation and a trend toward upregulation after 5 days in culture. Graphs showing mean fold change±SEM, ANOVA P values, results representative of 3 independent primary cell isolations (B). In low-passage primary human carotid SMCs, Pdlim7, Synm, and Synpo2 colocalized with the actin cytoskeleton (as shown by phalloidin staining in red), Pln showed nuclear localization and the signal for LMOD1 was beyond detection. For comparison, calponin was localized to actin cytoskeleton (C). Images were taken with ×100 objective.
By chromatin immunoprecipitation-sequencing, we observed that all these genes were under the regulation of active enhancer histone modification H3K27ac (Table VI in the online-only Data Supplement). Prediction of putative-binding motifs in genomic sequences using MSigDB software searching a span of ±2000 basepairs around the transcription start site, we found 3 CArG motifs present upstream of human PDLIM7 (at positions +650, +654, +667) and 1 SRF (serum response factor)-binding site in the PLN gene, but no such motifs were found in either SYNPO2, LMOD1, or SYNM in this analysis. Another prediction program MotifMap, searching a wider region within 8000 basepairs around the transcription start, suggested regulation by several other transcription factors previously associated with SMCs or control of cell proliferation. Here, TEF1 (TEA domain transcription factor) and MAFA (V-Maf avian musculoaponeurotic fibrosarcoma oncogene homolog A) were predicted to regulate LMOD1; AP1 (activator protein) and SRF to regulate PDLIM7; TEF1 and SRF to regulate PLN; and CTCF (CCCTC-binding factor) and NEUROD (neuronal differentiation) to control SYNPO2 (full list in Table VII in the online-only Data Supplement).
Subcellular localization of SMC markers was also assessed in low-passage human SMCs (Figure 5C, additional images shown in Figure VIII in the online-only Data Supplement). CNN1, PDLIM7, and SYNPO2 were localized to the actin cytoskeleton by overlap with phalloidin staining; SYNM localized to cellular filopodia and to the cortical cytoskeleton; PLN exhibited nuclear staining, whereas MYH11 and LMOD1 could not be detected in these cells. Taken together, our data confirm that SMCs maintain phenotypic plasticity in vitro and show that the expression changes and cytoskeletal localization of the selected genes strongly correlate with those of typical SMC markers in vitro, as initially observed in situ.
Downregulation of LMOD1, SYNPO2, SYNM, PDLIM7, and PLN in Response to Inflammatory-, Hemodynamic-, and Lipid Stimuli
Next, we explored processes relevant in the environment of an atherosclerotic lesion that may influence expression of the genes identified in our study. The expression of standard SMC markers and LMOD1, SYNPO2, SYNM, PDLIM7, and PLN was rapidly downregulated in human SMCs in vitro by stimulation with IFNg (interferron gamma; Figure 6A). Downregulation of all genes was observed within 24 hours of IFNg treatment, whereas expression of PLN, PDLIM7, SYNM, and SYNPO2 was suppressed already after 2 hours. These genes were also downregulated in human SMCs after 48 to 72 hours of stimulation with oxLDL (oxidized low-density lipoprotein; in particular SYNPO2, LMOD1, and PLN, Figure 6B), which was validated by analyzing a public microarray data set comparing cholesterol-loaded primary mouse aortic SMCs with baseline controls (GSE47744, Figure IX in the online-only Data Supplement).20 In this model, the typical SMC markers ACTA2 and CNN1 were also downregulated, whereas the macrophage marker CD68 was upregulated. Finally, we analyzed the expression of these genes in an in vitro model of SMC exposure to laminar shear stress, mimicking the exposure of the injured vessel surface to the hemodynamic forces of the flowing blood.21 In microarrays comparing shear stress versus static conditions, we have previously observed apoptosis as an enriched pathway through the activation of CASP3 (data set accession no. GSE19909) and all genes (as well as other typical SMC markers) were also found to be downregulated in this model (Figure 6C).
Figure 6.
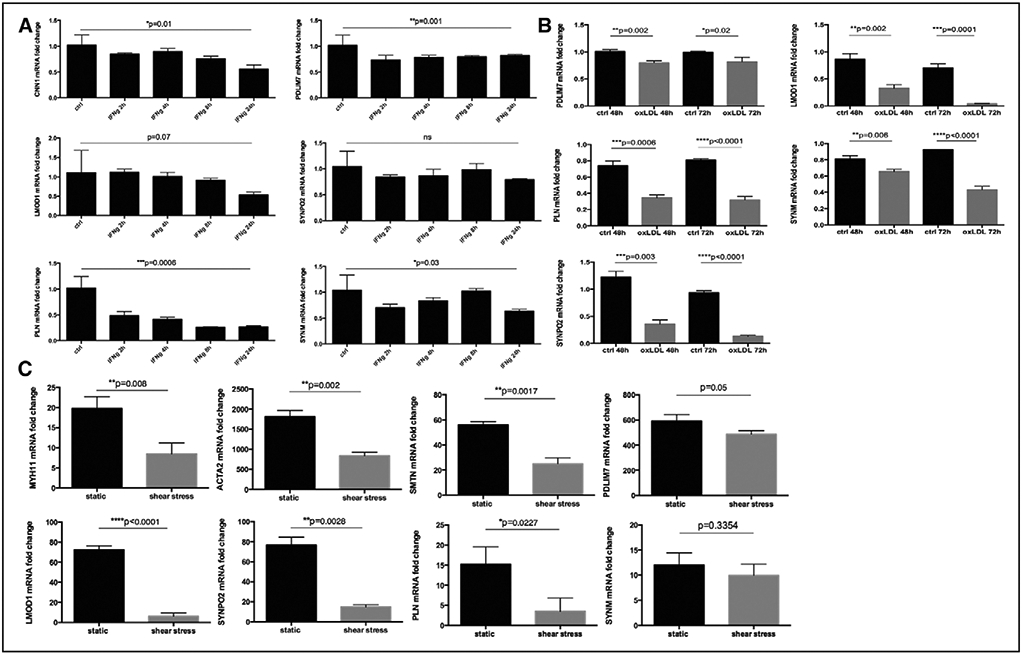
Smooth muscle cell (SMC) markers were downregulated in relation to inflammation, lipid loading, and hemodynamic stress. CNN1, PDLIM7, LMOD1, SYNPO2, PLN, and SYNM were rapidly downregulated by IFNg (interferron gamma) treatment of cultured human carotid SMCs (A) and stimulation with oxLDL (oxidized low-density lipoprotein) similarly resulted in downregulation of these genes (B). These SMC genes were also repressed in primary rat aortic SMCs by exposure to laminar shear stress (data set Gene Expression Omnibus [GEO] accession no. GSE19909, C). Plots show mean fold change±SEM, P values from t test or ANOVA when appropriate. Results are representative of 3 independent experiments.
Collectively, our results demonstrate that downregulation of LMOD1, SYNPO2, SYNM, PDLIM7, and PLN, along with standard SMC markers, functionally relates to clinical symptoms of plaque instability, vascular injury, and to key molecular processes in atherosclerosis, such as apoptosis, shear stress, inflammatory stimuli, and lipid uptake.
Polymorphisms in PDLIM7, SYNPO2, and PLN Associate With Surrogate Markers of Atherosclerosis
To investigate the involvement of LMOD1, SYNPO2, PDLIM7, SYNM, and PLN in early processes shown to be predictive of carotid and coronary artery disease in humans, we examined the association of genetic variants in these loci with severity and rate of cIMT progression. Several variants in the PDLIM7, SYNPO2, and PLN genomic regions were found to be associated with cIMT phenotypes in a large cohort of high-risk subjects (n=3378, IMPROVE)22 after adjustment for age, sex, and population stratification (Table VIII in the online-only Data Supplement). Variants rs11746443 and rs35716097 (PDLIM7) were associated with the maximum thickness of the common carotid artery (P=0.002) and the fastest cIMT progression (P=0.0009 and P=0.002, respectively), and variants rs67456868 (PLN) and rs4833611 (SYNPO2) were associated with the maximum common carotid artery thickness (P=0.00004 and P=0.0007, respectively). Full functional information obtained from Haploreg for these variants is presented in Tables IX and X in the online-only Data Supplement. The SYNPO2 variant rs4833611 was located in the intronic region of the USP53 gene and by eQTL (expression quantitative trait locus) analyses marginally linked to SYNPO2 (P=0.09) and USP53 (P=0.02) gene expression in plaques. Of particular interest, PDLIM7 variant rs35716097 was predicted to constitute a putative-binding site for the HNF4A transcription factor. The other PDLIM7 variant rs11746443 was localized in the genomic region of RGS14 (regulator of G-protein signaling) and predicted to constitute the binding site for the HEY1 transcription factor, whereas its proxy (rs4075958, R2=0.927, D′=0.963) was mapped within the putative-binding site for the ETS1 (avian erythroblastosis virus E26 [V-Ets] oncogene homolog-1) transcription factor. By eQTL analysis in plaques, rs4075958 was found to be significantly associated with the expression of both PDLIM7 and RGS14 (P=0.007 and P=0.0002, respectively; Figure 7A and 7B), and the expression levels of both genes were strongly correlated (Pearson r=0.61, P<0.0001; Figure 7C). PDLIM7 and RGS14 also appeared to be linked in a protein interaction network via actin cytoskeleton and markers of differentiated SMCs, SMTN, and CNN2 (Figure 7D). Altogether, our results underline the possibility that genetic variants associated with PDLIM7 may be causal to altered intima-media phenotypes and predisposition to atherosclerosis.
Figure 7.

Polymorphism in the PDLIM7 genomic region associated with carotid intima-media thickness affects its expression in plaque tissue. By eQTL (expression quantitative trait locus) analysis, variant rs4075958 was associated with the mRNA expression of both PDLIM7 and RGS14 in plaque tissue (A and B), and the expression levels of these 2 genes were strongly correlated (C). Functional network coupling based on protein–protein interactions links Pdlim7 and Rgs14 via actin cytoskeleton proteins (D). Plots in A and B show median with minimum and maximum.
Silencing of PDLIM7 Leads to Downregulation of Other SMC Markers and Increased SMC Proliferation
Of the 5 genes that were identified, PDLIM7 emerged as one of the hub genes in the interaction network with other signature genes in atherosclerosis.13 Because PDLIM7 was causally implicated in atherogenesis at the genetic level, localized to SMC actin cytoskeleton and in addition, interconnected with other cytoskeletal proteins, we decided to mechanistically investigate its role in SMCs. Silencing PDLIM7 expression in human carotid SMCs in vitro, resulted in downregulation of other SMC markers (ACTA2, MYH11, LMOD1, PLN, and particularly SYNPO2 by ≈70% on the mRNA level). Cell adhesion and spreading on fibronectin were defective compared with controls, and proliferation was significantly increased in these cells as evaluated by BrdU (bromodeoxyuridine) incorporation (P<0.0001; Figure 8; Figure X in the online-only Data Supplement). These findings add mechanistic support to the notion that PDLIM7 is an important structural molecule in the regulation of SMC phenotype.
Figure 8.

Silencing of PDLIM7 leads to downregulation of other smooth muscle cell (SMC) markers and increased SMC proliferation. PDLIM7 expression was silenced in human SMCs in vitro using siRNA (Crystal Violet staining, A), which resulted in downregulation of several other SMC markers (B), increased cell proliferation (as evaluated by BrdU [bromodeoxyuridine] incorporation) and impaired cell spreading/adhesion ability (C). Plots show mean±SEM.
Discussion
A large biobank of endarterectomies obtained from patients undergoing surgery for symptomatic or asymptomatic carotid stenosis was used to identify suppressed processes in atherosclerosis. We found molecular pathways related to SMC function and phenotype but also a panel of genes (SYNPO2, SYNM, LMOD1, PDLIM7, and PLN) previously not associated with vascular disease, not only to be the most repressed in end-stage atherosclerosis but also in relation to clinical symptoms of plaque instability, both on the transcriptomic and proteomic level. We hypothesized that some of these genes may show early expression variations and demarcate the initiation of SMC phenotypic switching. Most of these genes were linked to the SMC cytoskeleton, downregulated during neointima formation after rat carotid balloon injury, and polymorphisms in PDLIM7, PLN, and SYNPO2 genomic regions were associated with cIMT phenotypes in high-risk subjects. In addition, expression of these genes was sensitive to predominant processes in the atherosclerotic lesion, such as apoptosis, inflammation, hemodynamic stress, and lipid exposure. We propose that these SMC genes may improve definition of the phenotypic state of these cells in vascular disease and may be further explored in relation to the capacity of SMCs to contribute to plaque stabilization.
Previously, transition of SMCs from a contractile and quiescent phenotype into synthetic, matrix-producing, and replicating cells has been widely accepted as a central feature in early atherogenesis and an essential part of lesion stability and repair. This process, where the typical contractile features of SMCs are lost, represents an example of disturbed vessel wall homeostasis in disease progression. However, it has become evident that these conclusions oversimplify the complexity of SMC function in vascular disease and that these phenotypes probably represent the extremes of a spectrum of intermediate phenotypes that may to various extents coexist in the vessel wall, as dictated by exposure to environmental cues affecting gene expression patterns.23 Recent studies have presented strong evidence that SMCs and macrophages can activate the same genes by demonstrating that 50% of foam cells within advanced human coronary artery lesions express the SMC marker SMA besides the macrophage marker CD68, whereas lineage tracing in mice confirmed that ≤80% of the lesion cells (including mesenchymal stem cells and macrophage-like cells) are SMC derived.5,6 Here, we demonstrated that several SMC markers remain repressed on the protein level in stellate-shaped SMA+ cells of the fibrous cap, whereas expression was still detectable on the transcript level in situ, as previously reported by others.24 Together, these observations highlight the problem of correct SMC identification with respect to our understanding of human disease. Other studies seeking to establish the earliest determinants of SMC phenotypic switch have shown that, for example, mitochondrial fragmentation represents an early mark of SMC activation.25 Currently, changes in histone modifications, novel SMC-enriched transcription factors, such as TCF21 (transcription factor) and TET2,26-29 and epigenetic regulation of SMC phenotype by noncoding RNAs30 are also being intensively investigated. Nevertheless, our study highlights that we have not yet fully explored the transcriptomic landscape in relation to the plethora of SMC phenotypes and adds to elucidation of molecular signatures that characterize their plasticity.
Here, we confirmed that muscle contraction, muscle development, and actomyosin cytoskeleton were some of the most repressed categories in atherosclerotic tissue, including typical markers of SMCs and several genes previously poorly characterized in the context of SMC function. Synemin (SYNM) is an intermediate filament protein whose knockdown in saphenous vein SMCs in vitro leads to increased collagen production, downregulation of typical SMC markers, and disassembly of actin fibers.31 Phospholamban (PLN) was previously immunolocalized to the nuclear envelope and sarcoplasmic reticulum of cardiac SMCs, where it has been proposed to regulate intracellular and intranuclear Ca2+ levels.32 It has also been shown that Ca2+ signaling pathways are altered in synthetic vascular SMCs, which possibly occurs in conjunction with PLN translocation from the nucleus to the other subcellular compartments. In a recent study, PLN mutations were linked to dilated cardiomyopathy, ventricular arrhythmias, and interstitial fibrosis.33 Nanda et al34 described LMOD1 as a new SMC-restricted, myofilament-related, SRF/MYOCD target gene enriched in SMCs in embryonic and adult mouse tissues. Earlier, LMOD1 was predicted to belong to a gene battery involved in SMC differentiation by a bioinformatics screen for regulators of conserved functional gene modules in mammals.35 Interestingly, SYNPO (synaptopodin) and PDLIM proteins have been discovered as neuronal components and also investigated as adaptor molecules orchestrating actin-cytoskeletal organization in foot processes of podocytes,36-39 but sparsely linked to SMCs.40-43 Apart from these few publications, limited information exists about these genes in the literature up to date, and to the best of our knowledge, this study is the first to systematically examine their implication in human atherosclerosis and vascular remodeling.
SMCs are currently considered to be the main cell type responsible for intimal repair after balloon injury in the rat carotid artery, although cells of mesenchymal origin may also contribute.44 In this model, intimal hyperplasia develops in 3 major stages45,46 with initial SMC activation, replication, and beginning of migration to the luminal surface during the first 2 days after injury. Between days 2 and 5, SMCs colonize the intimal surface after activity related to chemoattractants and extracellular matrix degradation. In the next few weeks, the number of SMCs in the neointima continues to increase, but from 1 month after injury, SMC proliferation ceases, the cells become quiescent and regain ultrastructural features typical for the contractile state.46 Our results show that expression profiles for both typical SMC markers and for Synm, Pln, Lmod1, Synpo2, and Pdlim7 reflect these stages by gradual downregulation until 5 days after injury, followed by subsequent upregulation later as SMCs become quiescent and regain contractile features. In a similar fashion, immunohistochemical staining for typical SMC markers and for SYNM, PLN, LMOD1, SYNPO2, and PDLIM7 was detected in human intimal hyperplasia, especially in large PCNA− areas. On the basis of these results, we hypothesize that SYNM, PLN, LMOD1, SYNPO2, and PDLIM7 functionally relate to the phenotypic state of SMCs.
Freshly isolated rat aortic SMCs seeded on fibronectin and cultured under serum-free conditions have previously been used to study the subcellular properties related to SMC phenotypic modulation in vitro. Under these conditions, interactions between fibronectin, integrin α5β1 and FAK (focal adhesion kinase)-dependent intracellular signaling promote cell cycle entry and dedifferentiation into a synthetic state, accompanied by structural reorganization and loss of myofilaments.19,47,48 Here, we observed that Synm, Pln, Lmod1, Synpo2, and Pdlim7 (as well as Acta2, Myocd, Myh11) were indeed downregulated in primary rat SMCs during the first day of culture on fibronectin, but re-expressed from about 5 days of culture, suggesting that SMCs retain their inherent plasticity in vitro. Several of the examined genes were localized to the actin cytoskeleton in human SMCs implying that they may be involved in reorganization of cytoskeletal structures. Interestingly, although plasticity of SMCs and re-expression of target genes and proteins was apparent in human and rat intimal hyperplasia, expression of the proteins remained repressed in stellate-shaped SMA+ cells of the fibrous cap in CPs. As discussed, this may be because of either a heterogeneous population of cells expressing SMA5,6 or a repression of these genes in SMCs by disease-specific factors, such as inflammatory, apoptotic, or lipid mediators.
Therefore, to explore whether dominant processes in the atherosclerotic environment and in vascular disease in general may influence the genes of interest in our study, we investigated the expression of SYNM, PLN, PDLIM7, LMOD1, and SYNPO2 and other typical SMC markers in SMCs exposed to disease-associated stimuli in vitro. To summarize, although we have not yet fully dissected which specific stimulus or combination of stimuli may repress the expression of SYNM, PLN, PDLIM7, LMOD1, and SYNPO2 in atherosclerosis and vascular remodeling, we showed that they were downregulated by inflammatory stimuli and cholesterol uptake, and in response to shear stress (and apoptosis). In support of these observations, exposure to lipids has previously been associated with dramatic effects on SMC phenotype and transdifferentiation into CD68+ macrophage-like foam cells, as also demonstrated in our study.20
Intima-media thickness of extracranial carotid arteries, measured by ultrasound is a commonly accepted noninvasive marker of subclinical atherosclerosis. Several studies have established that cIMT changes over time are associated with vascular risk factors22 and prediction of vascular events both in subjects with plaques at baseline and in those without. Here, genetic variants in PDLIM7, SYNPO2, and PLN showed association with cIMT measurements, suggesting that these genes could have a causal role in carotid disease. Of note, SYNPO2 variants were located in the intron of the USP53 gene and marginally linked to expression in BiKE atherosclerotic plaques. USP53 (ubiquitin-specific peptidase 53) is a poorly studied protein, highly expressed in the heart muscle and found to be genetically associated with the Cantu syndrome, a rare condition characterized clinically by hypertrichosis, cardiomegaly, and bone abnormalities.49 Of particular interest, PDLIM7 SNPs linked to fastest-IMT-max-progression were shown to influence the expression of PDLIM7 in plaques and predicted to constitute binding sites for transcription factors previously implicated in cardiovascular development, SMC migration, and SMC proliferation in response to cytokine stimulation.50,51 One of these SNPs was positioned in the intronic region of the RGS14 gene, and interestingly, the expression of RGS14 also strongly correlated with the expression of PDLIM7 in plaques. RGS14 has been shown to act as a positive modulator of microtubule polymerization and spindle organization during cell division by integrating G protein and MAPK (mitogen-activated protein kinase) signaling pathways.52,53 It inhibits platelet-derived growth factor–stimulated ERK1 (extracellular signal-regulated kinase)/ERK2 phosphorylation and may indirectly interact with PDLIM7 via the actin cytoskeleton.
The importance of PDLIM7 for SMC phenotype was confirmed by silencing experiments that resulted in perturbed cytoskeletal structure, adhesion, and spreading, as well as SMC proliferation. Previous studies in other cell types have shown similar effects of PDLIM7 knockdown on proliferation (ie, periodontal ligaments54), and studies of other PDLIM family members have shown that they can directly interact with actin–cytoskeleton proteins, such as α-actinin-4 to stabilize actin fibers.39 Similarly, missense mutations of ACTA2 in humans are associated with diminished gene expression, defective actin-filaments, and actin-based spreading in SMCs, and formation of occlusive lesions because of increased SMC proliferation and intimal hyperplasia.55,56 Overall, our findings suggest an important structural and mechanistic role for PDLIM7 in SMCs, with possible genetic influence on disease development.
Because the BiKE cohort comprises only late-stage lesions and cannot provide information about gene expression variations during atheroprogression, expression data were complemented with immunohistochemistry on aortic lesions collected from different stages of atherosclerotic disease. Of note, PCNA that was used as a proliferation marker in the immunohistochemical analysis has been reported to overestimate the number of replicating cells. Consensus is lacking regarding the selection of appropriate control tissues, and in the BiKE study, control vessels contained outer media that is not included in the endarterectomy samples. Furthermore, the discovery approach in our study was based on microarrays and the complexity of microarray data were reduced by pathway analyses and construction of functional networks where genes were clustered based on biological functions or protein interactions. Although this method is limited to semantic mining of existing knowledge from published literature and data-bases, it still permits for discovery of poorly explored genes in a certain context.
In conclusion, using a systems biology approach by integrating transcriptomic, in situ, in vivo, in vitro, and genetic studies we were able to overcome these limitations and discover several novel candidates that demarcate early phenotypic modulation of SMCs in vascular disease. In perspective, the full knowledge of key expression signatures is likely to help us derive a better definition of various SMC phenotypes that coexist in the vessel wall, and provide potential targets for prevention and therapy in vascular disease.
Supplementary Material
Highlights.
An integrative approach using large biobanks of carotid endarterectomies from patients undergoing surgery for symptomatic or asymptomatic stenosis was used to uncover mechanisms repressed in atherosclerosis.
A combination of transcriptomic and proteomic profiling revealed that smooth muscle cell–related molecular categories were the most downregulated in plaques and identified 5 genes previously not associated with atherosclerosis.
SYNPO2, SYNM, LMOD1, PDLIM7, and PLN were related to smooth muscle cell cytoskeleton, transiently downregulated during neointima formation after rat carotid balloon injury, by PDGFB (platelet-derived growth factor beta) and IFNg (interferron gamma), exposure to shear flow stress and oxLDL (oxidized low-density lipoprotein) loading.
Polymorphisms in PDLIM7 were associated with surrogate markers of atherosclerosis in high-risk subjects, whereas its silencing in vitro led to downregulation of smooth muscle cell markers, disruption of the actin cytoskeleton, decreased cell spreading, and increased proliferation.
The newly described smooth muscle cell genes may improve definition of the phenotypic state of these cells in vascular disease and relate to their capacity to contribute to stabilizing processes in atherosclerotic lesions.
Sources of Funding
The Biobank of Karolinska Endarterectomies (BiKE) study was conducted with support from the Swedish Heart and Lung Foundation, the Swedish Research Council, Uppdrag Besegra Stroke, the Strategic Cardiovascular Programs of Karolinska Institutet and Stockholm County Council, the Stockholm County Council, the Foundation for Strategic Research, and the European Commission (CarTarDis, AtheroRemo, VIA, and AtheroFlux projects). The SOKRATES study has been supported by the Foundation De Drie Lichten in The Netherlands. The IMPROVE study was supported by the European Commission (Contract number: QLG1-CT-2002-00896), Ministero della Salute Ricerca Corrente, Italy, the Swedish Heart-Lung Foundation, the Swedish Research Council (projects 8691 and 0593), the Foundation for Strategic Research, the Stockholm County Council (project 562183), the Foundation for Strategic Research, the Academy of Finland (Grant no. 110413), and the British Heart Foundation (RG2008/014). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the article.
Nonstandard Abbreviations and Acronyms
- BiKE
Biobank of Karolinska Endarterectomies
- CP
carotid plaque
- cIMT
carotid intima-media thickness
- MYH11
myosin heavy chain 11
- NA
normal artery
- PCNA
proliferating cell nuclear antigen
- PDLIM7
PDZ and LIM domain–containing 7
- SMA
smooth muscle actin
- SMC
smooth muscle cell
Footnotes
Disclosures
None.
The online-only Data Supplement is available with this article at http://atvb.ahajournals.org/lookup/suppl/doi:10.1161/ATVBAHA.116.307893/-/DC1.
Contributor Information
Ljubica Perisic Matic, Department of Molecular Medicine and Surgery, Karolinska Institutet, Solna, Sweden.
Urszula Rykaczewska, Department of Molecular Medicine and Surgery, Karolinska Institutet, Solna, Sweden.
Anton Razuvaev, Department of Molecular Medicine and Surgery, Karolinska Institutet, Solna, Sweden.
Maria Sabater-Lleal, Department of Medicine, Karolinska Institutet, Solna, Sweden.
Mariette Lengquist, Department of Molecular Medicine and Surgery, Karolinska Institutet, Solna, Sweden.
Clint L. Miller, Division of Vascular Surgery, Stanford University, CA
Ida Ericsson, Department of Molecular Medicine and Surgery, Karolinska Institutet, Solna, Sweden.
Samuel Röhl, Department of Molecular Medicine and Surgery, Karolinska Institutet, Solna, Sweden.
Malin Kronqvist, Department of Molecular Medicine and Surgery, Karolinska Institutet, Solna, Sweden.
Silvia Aldi, Department of Molecular Medicine and Surgery, Karolinska Institutet, Solna, Sweden.
Joelle Magné, Department of Medicine, Karolinska Institutet, Solna, Sweden.
Valentina Paloschi, Department of Medicine, Karolinska Institutet, Solna, Sweden.
Mattias Vesterlund, Science for Life Laboratory, Solna, Sweden.
Yuhuang Li, Department of Medicine, Karolinska Institutet, Solna, Sweden.
Hong Jin, Department of Medicine, Karolinska Institutet, Solna, Sweden.
Maria Gonzalez Diez, Department of Medicine, Karolinska Institutet, Solna, Sweden.
Joy Roy, Department of Molecular Medicine and Surgery, Karolinska Institutet, Solna, Sweden.
Damiano Baldassarre, Dipartimento di Scienze Farmacologiche e Biomolecolari, Università di Milano, Italy; Dipartimento di Scienze Cliniche e di Comunità, Centro Cardiologico Monzino, IRCCS, Milan, Italy.
Fabrizio Veglia, Dipartimento di Scienze Cliniche e di Comunità, Centro Cardiologico Monzino, IRCCS, Milan, Italy.
Steve E. Humphries, British Heart Foundation Laboratories, Department of Medicine, University College of London, United Kingdom
Ulf de Faire, Division of Cardiovascular Epidemiology, Institute of Environmental Medicine, Karolinska Institutet, Solna, Sweden; Department of Cardiology, Karolinska University Hospital Solna, Karolinska Institutet, Stockholm, Sweden.
Elena Tremoli, Dipartimento di Scienze Farmacologiche e Biomolecolari, Università di Milano, Italy; Dipartimento di Scienze Cliniche e di Comunità, Centro Cardiologico Monzino, IRCCS, Milan, Italy.
Jacob Odeberg, Science for Life Laboratory, Department of Proteomics, Stockholm, Sweden.
Vladana Vukojević, Department of Clinical Neuroscience, Center for Molecular Medicine, Karolinska Institutet, Solna, Sweden.
Janne Lehtiö, Science for Life Laboratory, Solna, Sweden.
Lars Maegdefessel, Department of Medicine, Karolinska Institutet, Solna, Sweden.
Ewa Ehrenborg, Department of Medicine, Karolinska Institutet, Solna, Sweden.
Gabrielle Paulsson-Berne, Department of Medicine, Karolinska Institutet, Solna, Sweden.
Göran K. Hansson, Department of Medicine, Karolinska Institutet, Solna, Sweden
Jan H.N. Lindeman, Department of Vascular Surgery, Leiden University Medical Center, The Netherlands
Per Eriksson, Department of Medicine, Karolinska Institutet, Solna, Sweden.
Thomas Quertermous, Division of Vascular Surgery, Stanford University, CA.
Anders Hamsten, Department of Medicine, Karolinska Institutet, Solna, Sweden.
Ulf Hedin, Department of Molecular Medicine and Surgery, Karolinska Institutet, Solna, Sweden.
References
- 1.Abbott AL, Paraskevas KI, Kakkos SK, et al. Systematic review of guidelines for the management of asymptomatic and symptomatic carotid stenosis. Stroke. 2015;46:3288–3301. doi: 10.1161/STROKEAHA.115.003390. [DOI] [PubMed] [Google Scholar]
- 2.Finn AV, Nakano M, Narula J, Kolodgie FD, Virmani R. Concept of vulnerable/unstable plaque. Arterioscler Thromb Vasc Biol. 2010;30:1282–1292. doi: 10.1161/ATVBAHA.108.179739. [DOI] [PubMed] [Google Scholar]
- 3.Gomez D, Owens GK. Smooth muscle cell phenotypic switching in atherosclerosis. Cardiovasc Res. 2012;95:156–164. doi: 10.1093/cvr/cvs115.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Alexander MR, Owens GK. Epigenetic control of smooth muscle cell differentiation and phenotypic switching in vascular development and disease. Annu Rev Physiol. 2012;74:13–40. doi: 10.1146/annurev-physiol-012110-142315. [DOI] [PubMed] [Google Scholar]
- 5.Allahverdian S, Chehroudi AC, McManus BM, Abraham T, Francis GA. Contribution of intimal smooth muscle cells to cholesterol accumulation and macrophage-like cells in human atherosclerosis. Circulation. 2014;129:1551–1559. doi: 10.1161/CIRCULATIONAHA.113.005015. [DOI] [PubMed] [Google Scholar]
- 6.Shankman LS, Gomez D, Cherepanova OA, Salmon M, Alencar GF, Haskins RM, Swiatlowska P, Newman AA, Greene ES, Straub AC, Isakson B, Randolph GJ, Owens GK. KLF4-dependent phenotypic modulation of smooth muscle cells has a key role in atherosclerotic plaque pathogenesis. Nat Med. 2015;21:628–637. doi: 10.1038/nm.3866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schwartz SM, Virmani R, Rosenfeld ME. The good smooth muscle cells in atherosclerosis. Curr Atheroscler Rep. 2000;2:422–429. [DOI] [PubMed] [Google Scholar]
- 8.Weintraub WS. The pathophysiology and burden of restenosis. Am J Cardiol. 2007;100(5A):3K–9K. doi: 10.1016/j.amjcard.2007.06.002. [DOI] [PubMed] [Google Scholar]
- 9.Hedin U, Roy J, Tran PK. Control of smooth muscle cell proliferation in vascular disease. Curr Opin Lipidol. 2004;15:559–565. [DOI] [PubMed] [Google Scholar]
- 10.Saksi J, Ijäs P, Nuotio K, Sonninen R, Soinne L, Salonen O, Saimanen E, Tuimala J, Lehtonen-Smeds EM, Kaste M, Kovanen PT, Lindsberg PJ. Gene expression differences between stroke-associated and asymptomatic carotid plaques. J Mol Med (Berl). 2011;89:1015–1026. doi: 10.1007/s00109-011-0773-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sluimer JC, Kisters N, Cleutjens KB, Volger OL, Horrevoets AJ, van den Akker LH, Bijnens AP, Daemen MJ. Dead or alive: gene expression profiles of advanced atherosclerotic plaques from autopsy and surgery. Physiol Genomics. 2007;30:335–341. doi: 10.1152/physiolgenomics.00076.2007. [DOI] [PubMed] [Google Scholar]
- 12.Clarke MC, Figg N, Maguire JJ, Davenport AP, Goddard M, Littlewood TD, Bennett MR. Apoptosis of vascular smooth muscle cells induces features of plaque vulnerability in atherosclerosis. Nat Med. 2006;12:1075–1080. doi: 10.1038/nm1459. [DOI] [PubMed] [Google Scholar]
- 13.Perisic L, Aldi S, Sun Y, Folkersen L, Razuvaev A, Roy J, Lengquist M, Åkesson S, Wheelock CE, Maegdefessel L, Gabrielsen A, Odeberg J, Hansson GK, Paulsson-Berne G, Hedin U. Gene expression signatures, pathways and networks in carotid atherosclerosis. J Intern Med. 2016;279:293–308. doi: 10.1111/joim.12448. [DOI] [PubMed] [Google Scholar]
- 14.Schmitt T, Ogris C, Sonnhammer EL. FunCoup 3.0: database of genome-wide functional coupling networks. Nucleic Acids Res. 2014;42(Database issue):D380–D388. doi: 10.1093/nar/gkt984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Alexeyenko A, Sonnhammer EL. Global networks of functional coupling in eukaryotes from comprehensive data integration. Genome Res. 2009;19:1107–1116. doi: 10.1101/gr.087528.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Warde-Farley D, Donaldson SL, Comes O, et al. The GeneMANIA prediction server: biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res. 2010;38(Web Server issue):W214–W220. doi: 10.1093/nar/gkq537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sasaki T, Nakamura K, Kuzuya M. Plaque rupture model in mice. Methods Mol Med. 2007;139:67–75. [DOI] [PubMed] [Google Scholar]
- 18.van Dijk RA, Virmani R, von der Thüsen JH, Schaapherder AF, Lindeman JH. The natural history of aortic atherosclerosis: a systematic histopathological evaluation of the peri-renal region. Atherosclerosis. 2010;210:100–106. doi: 10.1016/j.atherosclerosis.2009.11.016. [DOI] [PubMed] [Google Scholar]
- 19.Hedin U, Bottger BA, Forsberg E, Johansson S, Thyberg J. Diverse effects of fibronectin and laminin on phenotypic properties of cultured arterial smooth muscle cells. J Cell Biol. 1988;107:307–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vengrenyuk Y, Nishi H, Long X, Ouimet M, Savji N, Martinez FO, Cassella CP, Moore KJ, Ramsey SA, Miano JM, Fisher EA. Cholesterol loading reprograms the microRNA-143/145-myocardin axis to convert aortic smooth muscle cells to a dysfunctional macrophage-like phenotype. Arterioscler Thromb Vasc Biol. 2015;35:535–546. doi: 10.1161/ATVBAHA.114.304029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ekstrand J, Razuvaev A, Folkersen L, Roy J, Hedin U. Tissue factor pathway inhibitor-2 is induced by fluid shear stress in vascular smooth muscle cells and affects cell proliferation and survival. J Vasc Surg. 2010;52:167–175. doi: 10.1016/j.jvs.2010.02.282. [DOI] [PubMed] [Google Scholar]
- 22.Baldassarre D, Veglia F, Hamsten A, Humphries SE, Rauramaa R, de Faire U, Smit AJ, Giral P, Kurl S, Mannarino E, Grossi E, Paoletti R, Tremoli E; IMPROVE Study Group. Progression of carotid intima-media thickness as predictor of vascular events: results from the IMPROVE study. Arterioscler Thromb Vasc Biol. 2013;33:2273–2279. doi: 10.1161/ATVBAHA.113.301844. [DOI] [PubMed] [Google Scholar]
- 23.Shanahan CM, Weissberg PL. Smooth muscle cell heterogeneity: patterns of gene expression in vascular smooth muscle cells in vitro and in vivo. Arterioscler Thromb Vasc Biol. 1998;18:333–338. [DOI] [PubMed] [Google Scholar]
- 24.Gomez D, Shankman LS, Nguyen AT, Owens GK. Detection of histone modifications at specific gene loci in single cells in histological sections. Nat Methods. 2013;10:171–177. doi: 10.1038/nmeth.2332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang L, Yu T, Lee H, O’Brien DK, Sesaki H, Yoon Y Decreasing mitochondrial fission diminishes vascular smooth muscle cell migration and ameliorates intimal hyperplasia. Cardiovasc Res. 2015;106:272–283. doi: 10.1093/cvr/cvv005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu R, Jin Y, Tang WH, Qin L, Zhang X, Tellides G, Hwa J, Yu J, Martin KA. Ten-eleven translocation-2 (TET2) is a master regulator of smooth muscle cell plasticity. Circulation. 2013;128:2047–2057. doi: 10.1161/CIRCULATIONAHA.113.002887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Miller CL, Haas U, Diaz R, Leeper NJ, Kundu RK, Patlolla B, Assimes TL, Kaiser FJ, Perisic L, Hedin U, Maegdefessel L, Schunkert H, Erdmann J, Quertermous T, Sczakiel G. Coronary heart disease-associated variation in TCF21 disrupts a miR-224 binding site and miRNA-mediated regulation. PLoS Genet. 2014;10:e1004263. doi: 10.1371/journal.pgen.1004263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nurnberg ST, Cheng K, Raiesdana A, et al. Coronary artery disease associated transcription factor TCF21 regulates smooth muscle precursor cells that contribute to the fibrous cap. PLoS Genet. 2015;11:e1005155. doi: 10.1371/journal.pgen.1005155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sazonova O, Zhao Y, Nürnberg S, Miller C, Pjanic M, Castano VG, Kim JB, Salfati EL, Kundaje AB, Bejerano G, Assimes T, Yang X, Quertermous T. Characterization of TCF21 downstream target regions identifies a transcriptional network linking multiple independent coronary artery disease loci. PLoS Genet. 2015;11:e1005202. doi: 10.1371/journal.pgen.1005202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Miano JM, Long X. The short and long of noncoding sequences in the control of vascular cell phenotypes. Cell Mol Life Sci. 2015;72:3457–3488. doi: 10.1007/s00018-015-1936-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xiao Y, Huang Z, Yin H, Zhang H, Wang S. Desmuslin gene knockdown causes altered expression of phenotype markers and differentiation of saphenous vein smooth muscle cells. J Vasc Surg. 2010;52:684–690. doi: 10.1016/j.jvs.2010.03.069. [DOI] [PubMed] [Google Scholar]
- 32.Ferguson DG, Young EF, Raeymaekers L, Kranias EG. Localization of phospholamban in smooth muscle using immunogold electron microscopy. J Cell Biol. 1988;107:555–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.van der Zwaag PA, van Rijsingen IA, Asimaki A, et al. Phospholamban R14del mutation in patients diagnosed with dilated cardiomyopathy or arrhythmogenic right ventricular cardiomyopathy: evidence supporting the concept of arrhythmogenic cardiomyopathy. Eur J Heart Fail. 2012;14:1199–1207. doi: 10.1093/eurjhf/hfs119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nanda V, Miano JM. Leiomodin 1, a new serum response factor-dependent target gene expressed preferentially in differentiated smooth muscle cells. J Biol Chem. 2012;287:2459–2467. doi: 10.1074/jbc.M111.302224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nelander S, Larsson E, Kristiansson E, Månsson R, Nerman O, Sigvardsson M, Mostad P, Lindahl P. Predictive screening for regulators of conserved functional gene modules (gene batteries) in mammals. BMC Genomics. 2005;6:68. doi: 10.1186/1471-2164-6-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mundel P, Heid HW, Mundel TM, Krüger M, Reiser J, Kriz W. Synaptopodin: an actin-associated protein in telencephalic dendrites and renal podocytes. J Cell Biol. 1997;139:193–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Asanuma K, Yanagida-Asanuma E, Faul C, Tomino Y, Kim K, Mundel P. Synaptopodin orchestrates actin organization and cell motility via regulation of RhoA signalling. Nat Cell Biol. 2006;8:485–91. doi: 10.1038/ncb1400. [DOI] [PubMed] [Google Scholar]
- 38.Sistani L, Rodriguez PQ, Hultenby K, Uhlen M, Betsholtz C, Jalanko H, Tryggvason K, Wernerson A, Patrakka J. Neuronal proteins are novel components of podocyte major processes and their expression in glomerular crescents supports their role in crescent formation. Kidney Int. 2013;83:63–71. doi: 10.1038/ki.2012.321. [DOI] [PubMed] [Google Scholar]
- 39.Sistani L, Dunér F, Udumala S, Hultenby K, Uhlen M, Betsholtz C, Tryggvason K, Wernerson A, Patrakka J. Pdlim2 is a novel actin-regulating protein of podocyte foot processes. Kidney Int. 2011;80:1045–1054. doi: 10.1038/ki.2011.231. [DOI] [PubMed] [Google Scholar]
- 40.Watanabe T, Akishita M, Nakaoka T, He H, Miyahara Y, Yamashita N, Wada Y, Aburatani H, Yoshizumi M, Kozaki K, Ouchi Y Caveolin-1, Id3a and two LIM protein genes are upregulated by estrogen in vascular smooth muscle cells. Life Sci. 2004;75:1219–1229. doi: 10.1016/j.lfs.2004.03.004. [DOI] [PubMed] [Google Scholar]
- 41.Krcmery J, Gupta R, Sadleir RW, Ahrens MJ, Misener S, Kamide C, Fitchev P, Losordo DW, Crawford SE, Simon HG. Loss of the cytoskeletal protein Pdlim7 predisposes mice to heart defects and hemostatic dysfunction. PLoS One. 2013;8:e80809. doi: 10.1371/journal.pone.0080809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Weins A, Schwarz K, Faul C, Barisoni L, Linke WA, Mundel P. Differentiation- and stress-dependent nuclear cytoplasmic redistribution of myopodin, a novel actin-bundling protein. J Cell Biol. 2001;155:393–404. doi: 10.1083/jcb.200012039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Turczyńska KM, Swärd K, Hien TT, Wohlfahrt J, Mattisson IY, Ekman M, Nilsson J, Sjögren J, Murugesan V, Hultgårdh-Nilsson A, Cidad P, Hellstrand P, Pérez-García MT, Albinsson S. Regulation of smooth muscle dystrophin and synaptopodin 2 expression by actin polymerization and vascular injury. Arterioscler Thromb Vasc Biol. 2015;35:1489–1497. doi: 10.1161/ATVBAHA.114.305065. [DOI] [PubMed] [Google Scholar]
- 44.Zhao W, Wang C, Liu R, et al. Effect of TGF-β1 on the Migration and Recruitment of Mesenchymal Stem Cells after Vascular Balloon Injury: Involvement of Matrix Metalloproteinase-14. Sci Rep. 2016;6:21176. doi: 10.1038/srep21176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Majesky MW, Schwartz SM, Clowes MM, Clowes AW. Heparin regulates smooth muscle S phase entry in the injured rat carotid artery. Circ Res. 1987;61:296–300. [DOI] [PubMed] [Google Scholar]
- 46.Thyberg J, Blomgren K, Hedin U, Dryjski M. Phenotypic modulation of smooth muscle cells during the formation of neointimal thickenings in the rat carotid artery after balloon injury: an electron-microscopic and stereological study. Cell Tissue Res. 1995;281:421–433. [DOI] [PubMed] [Google Scholar]
- 47.Roy J, Tran PK, Religa P, Kazi M, Henderson B, Lundmark K, Hedin U. Fibronectin promotes cell cycle entry in smooth muscle cells in primary culture. Exp Cell Res. 2002;273:169–177. doi: 10.1006/excr.2001.5427. [DOI] [PubMed] [Google Scholar]
- 48.Roy J, Kazi M, Hedin U, Thyberg J. Phenotypic modulation of arterial smooth muscle cells is associated with prolonged activation of ERK1/2. Differentiation. 2001;67:50–58. doi: 10.1046/j.1432-0436.2001.067001050.x. [DOI] [PubMed] [Google Scholar]
- 49.Kurban M, Kim CA, Kiuru M, Fantauzzo K, Cabral R, Abbas O, Levy B, Christiano AM. Copy number variations on chromosome 4q26-27 are associated with Cantu syndrome. Dermatology. 2011;223:316–320. doi: 10.1159/000333800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fischer A, Schumacher N, Maier M, Sendtner M, Gessler M. The Notch target genes Hey1 and Hey2 are required for embryonic vascular development. Genes Dev. 2004;18:901–911. doi: 10.1101/gad.291004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jiang H, Huang S, Li X, Li X, Zhang Y, Chen ZY. Tyrosine kinase receptor B protects against coronary artery disease and promotes adult vasculature integrity by regulating Ets1-mediated VE-cadherin expression. Arterioscler Thromb Vasc Biol. 2015;35:580–588. doi: 10.1161/ATVBAHA.114.304405. [DOI] [PubMed] [Google Scholar]
- 52.Wieland T, Lutz S, Chidiac P. Regulators of G protein signalling: a spotlight on emerging functions in the cardiovascular system. Curr Opin Pharmacol. 2007;7:201–207. doi: 10.1016/j.coph.2006.11.007. [DOI] [PubMed] [Google Scholar]
- 53.Vellano CP, Brown NE, Blumer JB, Hepler JR. Assembly and function of the regulator of G protein signaling 14 (RGS14)·H-Ras signaling complex in live cells are regulated by Gαil and Gαi-linked G protein-coupled receptors. J Biol Chem. 2013;288:3620–3631. doi: 10.1074/jbc.M112.440057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lin Z, Navarro VP, Kempeinen KM, Franco LM, Jin Q, Sugai JV, Giannobile WV. LMP1 regulates periodontal ligament progenitor cell proliferation and differentiation. Bone. 2010;47:55–64. doi: 10.1016/j.bone.2010.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Guo DC, Pannu H, Tran-Fadulu V, et al. Mutations in smooth muscle alpha-actin (ACTA2) lead to thoracic aortic aneurysms and dissections. Nat Genet. 2007;39:1488–1493. doi: 10.1038/ng.2007.6. [DOI] [PubMed] [Google Scholar]
- 56.Papke CL, Cao J, Kwartler CS, et al. Smooth muscle hyperplasia due to loss of smooth muscle α-actin is driven by activation of focal adhesion kinase, altered p53 localization and increased levels of platelet-derived growth factor receptor-β. Hum Mol Genet. 2013;22:3123–3137. doi: 10.1093/hmg/ddt167. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.