

Abstract
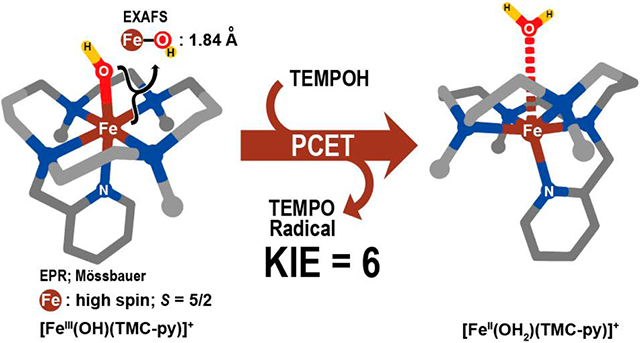
Nonheme mononuclear hydroxoiron(III) species are important intermediates in biological oxidations, but well-characterized examples of synthetic complexes are scarce due to their instability or tendency to form μ-oxodiiron(III) complexes, which are the thermodynamic sink for such chemistry. Herein, we report the successful stabilization and characterization of a mononuclear hydroxoiron(III) complex, [FeIII(OH)(TMC-py)]2+ (3; TMC-py = 1-(pyridyl-2′-methyl)-4,8,11-trimethyl-1,4,8,11-tetrazacyclotetradecane), which is directly generated from the reaction of [FeIV(O)(TMC-py)]2+ (2) with 1,4-cyclohexadiene at −40 °C by H-atom abstraction. Complex 3 exhibits a UV spectrum with a λmax at 335 nm (ε ≈ 3500 M−1 cm−1) and a molecular ion in its electrospray ionization mass spectrum at m/z 555 with an isotope distribution pattern consistent with its formulation. Electron paramagnetic resonance and Mössbauer spectroscopy show 3 to be a high-spin Fe(III) center that is formed in 85% yield. Extended X-ray absorption fine structure analysis reveals an Fe─OH bond distance of 1.84 Å, which is also found in [(TMC-py)FeIII─O─CrIII(OTf)2]+ (4) obtained from the reaction of 2 with Cr(OTf)2. The S = 5/2 spin ground state and the 1.84 Å Fe─OH bond distance are supported computationally. Complex 3 reacts with 1-hydroxy-2,2,6,6-tetramethylpiperidine (TEMPOH) at −40 °C with a second-order rate constant of 7.1 M−1 s−1 and an OH/OD kinetic isotope effect value of 6. On the basis of density functional theory calculations, the reaction between 3 and TEMPOH is classified as a proton-coupled electron transfer as opposed to a hydrogen-atom transfer.
Graphical Abstract
INTRODUCTION
Since 2003 high-valent iron(IV)-oxo intermediates have been trapped and characterized for a number of nonheme iron enzymes.1-4 Such high-valent intermediates serve various roles, most importantly the cleavage of C─H bonds for the functionalization of substrates. The first well-documented example of an enzymatic oxoiron(IV) species is intermediate J of taurine dioxygenase, which is shown to perform the oxidation of the substrate C─H bond. Transformations at high-valent oxoiron active sites are expected to proceed via hydrogen atom transfer (HAT) reactions, where the oxoiron(IV) moiety is initially reduced to a hydroxoiron(III) species.
The field of bioinorganic chemistry has provided great insight into the area of nonheme oxoiron(IV) chemistry, particularly with respect to the characterization of their structural, spectroscopic, and reactivity properties.5-9 Such studies to date have almost exclusively focused on the high-valent oxoiron(IV) intermediates with less emphasis on the properties of the corresponding hydroxoiron(III) complexes, which are frequently proposed to form immediately after the initial HAT.4,10 The small number of reports in this field may be attributed to the challenges posed by such complexes due to their (thermal) instability.
Masuda and Borovik have reported the only crystal structures of nonheme hydroxoiron(III) complexes.11-13 These complexes have been stabilized by the introduction of H-bonding moieties in the second coordination sphere in the design of the supporting ligand (Figure 1). In addition, our group has trapped the [FeIII(OH)(TMG3tren)]2+ species (TMG3tren = 1,1,1-tris{2-[N2-(1,1,3,3-tetramethylguanidino)]ethyl}amine) obtained from the reaction of [FeIV(O)(TMG3tren)]2+ with 1,4-cyclohexadiene (CHD) and characterized the hydroxoiron-(III) complex using electron paramagnetic resonance (EPR), Mössbauer, and X-ray absorption spectroscopy.14 The tripodal TMG3tren ligand does not provide H-bonding moieties in the second coordination sphere like the other two complexes mentioned above but instead has sterically bulky tetramethyl-guanidino substituents that shield the hydroxo ligand and prolong the lifetime of the complex.
Figure 1.

Crystal structures of hydroxoiron(III) complexes reported by Masuda11 (left) and Borovik13 (right).
The relatively high stability of nonheme oxoiron(IV) complexes supported by the macrocyclic tetramethylcyclam ligand (TMC) or a pentadentate variant bearing an appended pyridine group (TMC-py; see Chart 1)5,15,16 has led us to investigate whether the corresponding hydroxoiron(III) complexes might have sufficient lifetimes to be trapped and characterized. Indeed, on the one hand, Nam and co-workers have provided some evidence for the formation of the one-electron reduced oxoiron(III) complex, but it is not very stable.17,18 On the other hand, Borovik and Fout have been successful in crystallizing such oxoiron(III) complexes with suitable ligand design.12,19 In the present report, we demonstrate that the FeIII(OH)(TMC-py)(OTf)2 (3) complex can be generated from the reaction of [FeIV(O)(TMC-py)]2+ (2) with CHD at −40 °C and characterized by a variety of spectroscopic methods. In addition, we also show that 3 can oxidize 1-hydroxy-2,2,6,6-tetramethylpiperidine (TEMPO-H/D) and exhibits a moderately high kinetic isotope effect (KIE) for this reaction.
Chart 1.

EXPERIMENTAL SECTION
General Information.
All reagents and solvents were purchased from commercial sources and used as received unless specified. n-PrCN was further purified using Na2CO3 and KMnO4. The mixture was heated at 75 °C for several hours and distilled under Ar atmosphere.20 The preparations for Fe(OTf)2(MeCN)2 (naFe or 57Fe),21 Cr(OTf)2(MeCN)2,22 TEMPOH,23 TEMPOD,24 and TMC-py,16 as well as iodosylbenzene (C6H5IO) and its 18O isotopomer,25 were performed following published procedures. Stock solutions of FeIV(O)(TMC-py)(OTf)2 (2) and FeIV(18O)(TMC-py)(OTf)2 were kept in the −40 °C refrigerator in a N2-filled glovebox. Samples for EPR experiments on FeIII(OH)(TMC-py)(OTf)2 (3) were prepared in MeCN under N2 atmosphere at −40 °C and then transferred via gastight syringe to precooled EPR tubes and frozen immediately in liquid N2. Corresponding extended X-ray absorption fine structure (EXAFS) and Mössbauer samples were quickly loaded into sample cups and then frozen in liquid N2. All moisture- and oxygen-sensitive compounds and solvent were prepared using standard Schlenk-line techniques and N2-filled glovebox.
UV–vis absorption spectra were recorded on an HP 8453A diode array spectrometer. Low-temperature visible spectra were obtained using a cryostat from UNISOKU Scientific Instruments, Japan. Electrospray ionization mass spectrometry (ESI-MS) was performed on a Finnigan LCQ ion trap mass spectrometer. 1H NMR spectra were collected on a Bruker 400 MHz spectrometer. X-band EPR spectra were measured on a Bruker Elexsys E-500 spectrometer equipped with an Oxford ESP-910 cryostat. Mössbauer spectra were recorded with home-built spectrometers using Janis Research Super-Varitemp dewars. Mössbauer spectral simulations were performed using the WMOSS software package (SEE Co., Edina, MN) and SpinCount software.26 Isomer shifts are quoted relative to Fe metal at 298 K. All Mössbauer figures were prepared using SpinCount software. EPR simulations were performed by using the SpinCount software.26 The spin Hamiltonian used for the EPR and Mössbauer simulations in Figures 4, 5, and S10 is
| (1) |
where .
Figure 4.

X-band EPR spectrum of a sample containing 3. The effective g values are indicated. The signal from a minor impurity is labeled as an asterisk. Measurement conditions: microwave frequency, 9.64 GHz; microwave power, 20 μW; modulation frequency, 100 kHz; modulation amplitude, 1 mT; measurement temperature, 15 K. (inset) T vs signal × T plot of g = 4.3 EPR resonance recorded at various temperatures (black dots: 4 K, 6 K, 15 K) and fitting curve (red) with D = 1.1 cm−1, E/D = 0.3.
Figure 5.

Mössbauer spectra of the sample containing 3 measured under (a) 1.8 K, 45 mT; (b) 4.2 K, 0.1 T; (c) 10 K, 45 mT; (d) 4.2 K, 1.0 T; (e) 4.2 K, 2 T; (f) 4.2 K, 4.0 T; and (g) 4.2 K, 7.0 T. The magnetic fields were applied parallel to the γ-rays. The black vertical bars represent the experimental spectra, and the solid red lines represent the spectral simulation with the parameters of D = +1.1 cm−1, E/D = 0.3, σ(E/D) = 0.1, gx = gy = gz = 2, δ = 0.44 mm/s, ΔEQ = −0.85 mm/s, η = 0.3, Ax/gnβn = Ay/gnβn = Az/gnβn = −20.7 T. The solid arrows and the dashed arrows indicate the spectral features associated with the middle Kramers doublet and the upper Kramers doublet of the S = 5/2 system. The absorption areas that are not covered by the simulations belong to the minor species representing ~15% of the total iron in the sample. These simulations assumed slow relaxation behavior.
X-ray Absorption Spectroscopy (XAS).
XAS data were collected at Beamline 7–3 at the Stanford Synchrotron Radiation Lightsource (SSRL) of SLAC National Accelerator Laboratory. Fe K-edge XAS data were collected over the energy range of 6.9–8.0 keV on frozen samples maintained at 10 K. An Fe foil spectrum was measured simultaneously for internal energy calibration using the first inflection point of the K-edge energy (7112.0 eV). Data were obtained in the fluorescence mode using a solid-state germanium detector (Canberra).
Data reduction, averaging, and normalization were performed using the program EXAFSPAK.27 The coordination number of a given shell was a fixed parameter and was varied iteratively in integer steps, while the bond lengths (R) and mean-square deviation (σ2) were allowed to freely float. The amplitude reduction factor was fixed at 0.9, while the edge-shift parameter E0 was allowed to float as a single value for all shells. The pre-edge features were fitted using the Fityk program28 with pseudo-Voigt functions composed of 50:50 Gaussian/Lorentzian functions.
Preparation of FeII(TMC-py)(OTf)2 (1).
FeII(TMC-py)(OTf)2 (1) was synthesized by a published procedure.16 The 1H NMR spectrum of 1 at −40 °C showed well-resolved signals. The three methyl groups of 1 were assigned by peak integration29 (see Figure S1). 1H NMR (400 MHz, CD3CN, 233 K): δ 443.9 (1H), 440.2 (1H), 327.9 (1H), 264.3 (1H), 258.8 (1H), 239.3 (1H), 226.7 (3H, NMe), 207.5 (3H, NMe), 160.3 (1H), 143.3 (1H), 143.3 (1H), 126.5 (1H), 117.7 (3H, NMe), 95.1 (1H), 80.7 (2H), 47.1 (1H), 38.2 (1H), 32.2 (1H), 23.3 (1H), 14.1 (1H), 5.1 (1H), 2.4 (2H), −10.6 (1H), −19.2 (1H), −26.2 (1H), −37.5 (1H), −116.9 (1H). ESI-MS: Calcd for C20H35F3N5O3S1Fe1 {M-OTf}+, 537.9; Found: 538.2. 57Fe-labeled 1 was synthesized by the same procedure16 but was performed on a smaller scale using 22 mg of 57Fe(OTf)2(MeCN)2 as the starting material.
Generation of FeIV(O)(TMC-py)(OTf)2 (2).
FeIV(O)(TMC-py)-(OTf)2 (2) was generated by the reaction of 1 with 2 equiv of PhIO according to the published procedure.16 Solid 2 was obtained by adding 30 mL of anhydrous diethyl ether into a 48 mM solution of 2 in 3 mL of MeCN at −40 °C. The precipitate was washed with anhydrous diethyl ether several times, dried under vacuum for 2 min, and then stored at −40 °C. 18O-labeled 2 for the ESI-MS experiments was generated using C6H5I18O as the oxidant. 1H NMR (400 MHz, CD3CN, 298 K; Figure S2): δ 76.0 (1H), 59.1 (1H), 52.2 (1H), 43.1 (2H), 39.7 (1H), 29.8 (1H), 27.3 (1H), 20.1 (1H), 10.9 (1H), 6.3 (1H), −4.6 (1H), −19 (1H), −37.4, −38.2 (7H, NMe, NMe), −43.7 (3H, NMe), −71.4 (1H), −80 (1H), −149.5 (1H), −161.4 (1H), 166.8 (1H), 170.7 (1H). Six other proton signals were not found and may be significantly broadened by the paramagnetic center. ESI-MS: Calcd for C20H35F3N5O4S1Fe1 {M-OTf}+, 554.2; Found: 554.0. Calcd for C20H35F3N518O1O3S1Fe1 {M-OTf}+, 556.2; Found: 556.1.
Generation of FeIII(OH)(TMC-py)(OTf)2 (3).
FeIII(OH)(TMC-py)-(OTf)2 (3) was generated at −40 °C from the reaction of 2 (0.5 mM solution in MeCN) with 200 equiv of CHD under anaerobic conditions for 40 min. The reactions were monitored by the disappearance of the characteristic near-IR band of 2 at 834 nm and the appearance of a new absorbance band at 335 nm (ε ≈ 3500 M−1 cm−1), which was assigned to FeIII(OH)(TMC-py)(OTf)2 (3). The yield of 3 was estimated to be 80%, based on the amount of decamethylferrocenium ion produced in the reaction of 3 with 1 equiv of decamethylferrocene (relative to the amount of 2 used in its reaction with CHD), which was calibrated by the reaction of (NH4)2CeIV(NO3)6 with decamethylferrocene under the same conditions (see Figure S3).
Generation of (TMC-py)FeIII─O─CrIII(OTf)4(MeCN) (4).
(TMC-py)FeIII─O─CrIII(OTf)4(MeCN) (4) was generated at −40 °C by the reaction of 2 (0.45 mM solution in MeCN) with 1.25 equiv of CrII(OTf)2 under anaerobic conditions. The characteristic near-IR band of 2 at 834 nm decreased immediately, and this decrease was accompanied by the appearance of new bands at 382, 450, and 557 nm (ε ≈ 3800, 3200, and 920 M−1 cm−1, respectively), very similar to the spectral pattern recently reported for (MeCN)(TMC)FeIII─Oanti─CrIII(OTf)4(MeCN).22,30 18O-labeled 4 was generated similarly by using 18O-labeled 2 as starting material (64% 18O-labeled). ESI-MS: the molecular ion peak was observed at m/z 903.8, which shifted to m/z 905.8 upon 18O-labeling of the oxo atom in the precursor. Calcd for C22H35F9N5O10S3Fe1Cr1 {M-OTf-MeCN}+, 904.0 (see Figures S4-S8).
Reaction of FeIII(OH)(TMC-py)(OTf)2 (3) with TEMPOH/D.
FeIII(OH)(TMC-py)(OTf)2 (3) was generated in situ for each experiment at −40 °C as described above. Stock solutions of TEMPOH and TEMPOD were prepared in 1 mL of MeCN (2.1 × 10−1 and 2.54 × 10−1 M, respectively) within a glovebox and kept in a 0 °C ice bath outside of the glovebox. To the FeIII(OH)(TMC-py)(OTf)2 (3) solution in MeCN was introduced 10–50 equiv of TEMPOH or 10–100 equiv of TEMPOD at −40 °C. The reaction products were analyzed by ESI-MS and EPR spectroscopy, revealing a peak at m/z 538 corresponding to [FeII(TMC-py)(OTf)]+ and an EPR signal corresponding to the TEMPO radical (93% yield; see Figure S9).
Computational Details.
Geometry optimizations and single-point calculations were performed using the electronic structure code Turbomole v7.0.1.31-33 Geometry optimizations were performed using the M06-L functional34 in combination with the def2-TZVP basis set for Fe and the def2-SVP basis set for all other elements.35 Single-point energies were computed at these optimized structures using the def2-TZVPP basis set35 in combination with the M06-L and the M0636 functionals. In all calculations, acetonitrile solvation (ε = 35.8837 and n = 1.34438,39) was accounted for using the COSMO solvation model.40 Electronic energies reported include an outlying charge correction.41 Frequency calculations were performed to confirm the nature of stationary points resulting in no imaginary frequencies for minima and a single imaginary frequency for transition-state (TS) structures. To obtain thermal contributions to thermochemical quantities, frequencies were computed using frozen charges (NumForce –cosmo option), derivatives of quadrature weight, and small frequencies were raised to 100 cm−1.42,43 Frequencies were used without scaling. Free energies are referenced to a 1 M standard state in solution (233.15 K) and include a concentration-change term relative to a 1 bar gaseous standard state of RT ln(19.1) = 1.37 kcal mol−1.44 All calculations use grid m5 and were accelerated by the MARI-J45 approach using Weigend’s fitting basis sets.46 Because of the poor self-consistent field (SCF) convergence of some calculations, an initial level shift of 0.5 was introduced ($scforbitalshift automatic = 0.5) in all calculations. The VdW complexes before and after the transition state were obtained using the DRC tool in Turbomole (DRC –t 150 –f).47 Structural depictions and intrinsic bond orbital (IBO, iboexp was set to 2)48 analyses were made using IboView.49,50
RESULTS AND DISCUSSION
1. Generation of [FeIII(OH)(TMC-py)](OTf)2 (3).
The reaction of FeIV(O)(TMC-py)(OTf)2 (2) with 200 equiv of CHD (Scheme 1) in MeCN at −40 °C was monitored by UV–vis spectroscopy, and complete consumption of 2 was observed within 40 min. The light green solution of 2 turned light yellow, corresponding to the loss of its characteristic near-IR feature at 834 nm concomitant with an increase in absorbance at 335 nm (ε ≈ 3500 M−1 cm−1; Figure 2). As shown in the inset for Figure 2, there is an isosbestic point in the conversion of 2 to its product 3. We propose that 3 corresponds to [FeIII(OH)-(TMC-py)]2+, which would be formed by HAT from CHD by 2. The formulation for 3 was corroborated by the appearance of a dominant peak at m/z 555 in its ESI-MS (Figure 3) with a mass and isotope distribution pattern consistent with the [FeIII(OH)(TMC-py)(OTf)]+ ion. Moreover, the m/z 555 peak shifted to m/z 557 with the use of 18O-labeled 2 (64% 18O content). Furthermore, 3 was generated with a yield estimated to be ~80% based on the amount of decamethylferrocenium ion formed upon addition of decamethylferrocene to the reaction solution (see Figure S3).
Scheme 1.

Reaction of FeIV(O)(TMC-py)(OTf)2 (2) with 1,4-Cyclohexadiene
Figure 2.

UV–vis spectral changes upon addition of 200 equiv of CHD into a 0.5 mM solution of FeIV(O)(TMC-py)(OTf)2 (2, black line) to form [FeIII(OH)(TMC-py)]2+ (3, red line) in MeCN at −40 °C. (inset) Expanded 280–400 nm region to show the isosbestic point associated with the reaction.
Figure 3.

ESI-MS spectrum of the solution after the reaction of FeIV(16O)(TMC-py)(OTf)2 (2) with CHD in MeCN at −40 °C. (inset) Isotopic pattern of the [FeIII(18OH)(TMC-py)(OTf)]+ ion with 64% isotope enrichment. ESI-MS spectra were obtained with the carrier gas set at 40 °C.
Complex 3 was further characterized by EPR and Mössbauer spectroscopy. The EPR spectrum measured on a sample containing 3 exhibits signals with observed g values at 4.3 and 9.0 (Figure 4), indicative of a rhombic (E/D ≈ 0.3) high-spin (S = 5/2) iron(III) species. The g = 4.3 resonance originates from the middle Kramers doublet of this S = 5/2 species, while the g = 9.03 resonance consists of signals from both the ground and upper Kramers doublets of the same S = 5/2 species. The broadness of the observed EPR signals from 3 likely originates, in part, from an E/D distribution (see the Supporting Information for more discussion), probably resulting from slight inhomogeneity of the structure of 3. The magnitude of the axial zero-field splitting parameter, D, was found to be 1.1 cm−1 from the temperature dependence of the intensity of the g = 4.3 resonance, but the sign of D could not be readily determined due to the high rhombicity of the spin system (E/D ≈ 0.3).
Mössbauer spectra on a sample containing 3 measured at different magnetic fields applied parallel to the γ-rays and at different temperatures further confirm the assignment of 3 as a typical high-spin ferric species with E/D ≈ 0.3, isomer shift δ = 0.44 mm/s, and an isotropic A tensor (Ax/gnβn = Ay/gnβn = Az/gnβn = −20.7 T). More specifically, the low-field (45 mT) spectrum measured at 1.8 K shows a six-line pattern (Figure 5a), which originates from the ground Kramers doublet of an S = 5/2 species having a uniaxial magnetic behavior (the spectrum is only sensitive to the y (D > 0) or z (D < 0) component of the internal field). Increasing the temperature to 4.2 K results in the appearance of spectral features (indicated by solid arrows in Figure 5b) belonging to the middle Kramers doublet of the same S = 5/2 species. Furthermore, the 10 K spectrum (Figure 5c) clearly shows the spectral features belonging to the upper Kramers doublet (indicated by dashed arrows), which also shows a uniaxial magnetic behavior. The relative intensities of the spectral features belonging to the different Kramers doublets provide an accurate measure of the magnitude of the axial zero-field splitting parameter D, which in this case equals 1.1 cm−1. However, the sign of D cannot be reliably determined for an S = 5/2 system with E/D ≈ 0.3. The magnetic splittings generated from the three Kramers doublets at low field are determined by E/D and the three principal components of the 57Fe hyperfine coupling tensor, Ax, Ay, and Az. At high field (>4 T), only the ground Kramers doublet is appreciably populated due to the strong magnetic Zeeman interactions; the magnetic splittings are thus largely determined by Ax, Ay, and Az but independent of E/D. Thus, by simultaneously simulating spectra obtained under low-field and high-field conditions, the full spin-Hamiltonian parameters can be determined reliably and are listed in the caption of Figure 5. (Although a simulation using D = +1.1 cm−1, ΔEQ = −0.85 mm/s, and η = 0.3 is presented in Figure 5, a satisfactory simulation can also be obtained using D = −1.1 cm−1, ΔEQ = +0.85 mm/s, and η = 2.) In addition, the broad line width of the spectra obtained under low-field conditions (Figure 5, top panel) can be best accounted for in a simulation with an E/D distribution of 0.1 centered at E/D = 0.3 (see Figure S10c for the simulation without E/D distribution), which is not observed in the spectra measured under high-field conditions due to the insensitivity of those spectral features to E/D. Overall, this S = 5/2 species revealed by the Mössbauer measurements accounts for ~85% of the total iron in the sample. The remaining 15% belongs to a minor impurity of unknown origin.
2. Comparison of [FeIII(OH)(TMC-py)]2+ (3) with [(TMC-py)FeIII─O─CrIII(OTf)4(NCMe)] (4).
To complement the data we collected thus far on 3, we also investigated the reaction of 2 with Cr(OTf)2 to generate the corresponding Fe─O─Cr adduct 4. As the feasibility of Cr adduct formation was recently demonstrated in the reactions of Cr(OTf)2 with syn or anti isomers of the [FeIV(O)(TMC)]2+ complex, we thought that 4 would be a suitable complex to compare, as the O─H group in 3 would be replaced by an O─Cr group in 4.22,30 Thus, the (TMC-py)FeIII─O─CrIII(OTf)4(NCMe) complex (4) was formed at −40 °C by the reaction of 2 (0.45 mM solution in MeCN) with 1.25 equiv of CrII(OTf)2 under anaerobic conditions. The characteristic near-IR band of 2 at 834 nm decreased immediately, concomitant with the appearance of new bands at 381, 451, and 558 nm (Figure 6). The latter chromophoric pattern closely resembles that for the (MeCN)-(TMC)FeIII─O─CrIII(OTf)4(NCMe) species.22 Formation of 4 was further supported by the observation of a peak at m/z at 903.8 in its ESI-MS spectrum, which is assigned to the [(TMC-py)Fe─O─Cr(OTf)3]+ ion based on its isotope distribution pattern. The formula was further confirmed by an 18O-labeling experiment, which resulted in the upshift of the molecular ion by 2 mass units to m/z 905.8 (see Figures S4 and S5).
Figure 6.

UV–vis spectra of 2 (black) and 4 (red) in MeCN at −40 °C.
The structures of 3 and 4 were further characterized by Fe K-edge XAS. The Fe K-edge energy of 3 is 7124.8 eV, which is a little higher compared with those of 4 (7124.2 eV) and (MeCN)(TMC)FeIII─Oanti─CrIII(OTf)4(NCMe) (7124.0 eV; see Figure 7). Complex 3 exhibits a pre-edge feature at ca. 7114 eV, which can be fitted with two peaks, providing a combined area of 9.4 units, a value that increases to 14.8 units in 4. Both pre-edge areas are comparable with that in (MeCN)(TMC)-FeIII-Oanti-CrIII(OTf)4(NCMe) (11 units), but much lower than those in (TMC)FeIII─Osyn─CrIII(OTf)4(NCMe) (30 units)22,30 and (TMC)FeIII─Osyn─ScIII(OTf)4(NCMe) (32 units),51 consistent with the presence of 6-coordinate Fe centers for both 3 and 4.
Figure 7.

(top) Pre-edge region of the Fe K-edge XAS spectrum of 3 (black squares): rising-edge fit (red line), pre-edge peak 1 (blue line), pre-edge peak 2 (magenta line), pre-edge fit (green line). (bottom) Pre-edge region of the Fe K-edge XAS spectrum of 4 (black squares): rising-edge fit (red line), pre-edge peak 1 (blue line), and pre-edge fit (green line).
The XANES data for 3 and 4 are compared with related complexes in Table 1. Interestingly, the table shows that K-edge energies of (TMC)FeIV(O) and (TMC)FeIII(O─X) complexes are not very different and suggests caution in using just K-energies to establish iron oxidation state. This challenge in interpreting K-edge energies has recently been discussed in detail by MacMillan and Lancaster.52 In our work, we found the K-edge energies of high-spin ferric centers to span a large range from 7123 to 7129 eV. The highest K-edge energy is associated with the aqueous ferric ion and is 3 eV higher than that for the corresponding FeIV(O) complex.53 In agreement with MacMillan and Lancaster,52 we believe that an interpretation of the K-edge energies must also consider the covalency of the iron─ligand bonds. Nevertheless, the oxidation state of the iron center in 3 was unambiguously established by the EPR and Mössbauer data we collected.
Table 1.
XANES Data for Iron Complexes of Interest
| K-edge energy (eV) |
pre-edge area (units) |
ref | |
|---|---|---|---|
| [FeIV(O)(TMC)(NCMe)]2+ | 7124.5 | 32.8 | 54 |
| [FeIV(O)(TMC-Py)]2+ (2) | 7124.0 | 34 | 29 |
| [FeIII(OH)(TMC-Py)]2+ (3) | 7124.8 | 9.4 | this work |
| (TMCPy)FeIII–O–CrIII(OTf)4 (4) | 7124.2 | 14.8 | this work |
| (TMC)FeIIIOCrIII(OTf)4 | 7124.0 | 11 | 22 |
| [FeIV(O)(OH2)5]2+ | 7126 | est 70 | 53 |
| [FeIII(OH2)6]3+ | 7129 | 53 |
Analysis of the EXAFS region provides structural information on the environment of the iron center. The best fit for the EXAFS data of 3 consists of one O/N scatterer at 1.84 Å, five O/N scatterers at 2.17 Å, four C scatterers at 2.92 Å, and four C scatterers at 3.07 Å (Figure 8, top; Table S1). Remarkably, 4 gives nearly identical results for the first coordination sphere with one O/N scatterer at 1.84 Å and five O/N scatterers at 2.16 Å (Figure 8, bottom; Table S2), but the outer sphere differs in having a strong contribution from a Cr scatterer at 3.65 Å. The FeIII─O/N bond distances of both 3 and 4 also fall in the range of the bond distances in other iron(III) complexes of TMC-based supporting ligands (see Table 2). Moreover, the Fe(III)─O(H) and Fe(III)─Nave bond distances in 3 agree with values found in the crystal structure of the high-spin FeIII(tnpa)(OH)(PhCOO)(ClO4) complex.11 In conclusion, the XAS data indicate the presence of 6-coordinate Fe(III) centers in both 3 and 4.
Figure 8.

(top) Fourier-transformed Fe K-edge EXAFS data for 3 (dotted black) and the corresponding best fit (solid red, fit No. 6 in Table S1). (inset) Unfiltered k-space data (dotted black) and its fit (solid red). (bottom) Fourier-transformed Fe K-edge EXAFS data for 4 (dotted black) and the corresponding best fit (solid red, fit No. 6 in Table S2). (inset) Unfiltered k-space data (dotted black) and its fit (solid red).
Table 2.
Select Bond Distances of Iron(III)–OX Complexes
| complexesa | Fe–Ob | Fe–Naveb | spin statec | ref |
|---|---|---|---|---|
| soybean lipoxygenase | 1.88 | 2.12 | 5/2 | 55 |
| [FeIII(OH)(TMC-py)](OTf)2 (3) | 1.84 | 2.17 | 5/2 | d |
| (TMC-py)FeIII–O–CrIII(OTf)4(NCMe) (4) | 1.84 | 2.16 | 1 | d |
| (Me3cyclam-acetate)FeIII–O–FeIIICl3 | 1.802(2) | 2.174 | 0 | 56 |
| (Cl)(TMC)FeIII–Oanti–FeIIICl3 | 1.8510(12) | 2.206 | 0 | 30 |
| (MeCN)(TMC)FeIII–Oanti–CrIII(OTf)4(NCMe) | 1.81 | 2.17 | 1 | 22 |
| (NCS)(TMC)FeIII–Oanti–CrIII(OTf)4(NCMe) | 1.85 | 2.17 | 1 | 22 |
| FeIII(OH)(tnpa)(O2CPh)(ClO4) | 1.876(2) | 2.188 | 5/2 | 11 |
| [FeIII(OH)(TMG3tren)](OTf)2 | 1.77 | 2.000 | 5/2 | 14 |
| K[FeIII(OH)(H3buea)] | 1.9264(17) | 2.016 | 5/2 | 13 |
Abbreviations used: H3buea = 1,1,1-tris[(N-tert-butylureaylato)-N-ethyl]aminate trianion; Me3cyclam-acetate = 1-(carboxymethyl)-4,8,11-trimethyl-1,4,8,11-tetrazacyclotetradecane anion; TMG3tren = 1,1,1-tris{2-[N2-(1,1,3,3-tetramethylguanidino)]ethyl}amine;. tnpa = tris(6-neopentylamino-2-pyridylmethyl)amine).
Bond distances from EXAFS analysis have uncertainties of ±0.02 Å, while those with higher precision derive from XRD.
Spin states listed for dinuclear complexes refer to the entire complex; individual iron(III) centers are all high-spin.
This work.
3. Density Functional Theory Calculations.
We next set out to compare the FeIII─OH bond distance obtained from EXAFS measurements with values predicted from density functional theory (DFT) calculations. For these calculations we considered two conformations of the TMC macrocycle derived from the X-ray crystal structures of [FeIV(O)(TMC-py)]2+, which has the two opposed N─CH2─CH2─N linkages oriented in a “parallel” fashion, and that of [FeII(TMC-py)]2+, which has these linkages oriented in a “crossed” fashion.16 At the M06-L/def2-SVP(Fe:def2-TZVP)/COSMO(MeCN) level of theory for the S = 5/2 spin state, we obtained Fe─OH bond distances of 1.837 and 1.850 Å for the [FeIII(OH)(TMC-py)]2+ complex with parallel and crossed conformations, respectively (Figure 9), and we found the parallel conformation to be energetically slightly favored ΔG233 = 2.7 kcal mol−1). These bond distances are in quantitative agreement with the experimental value of 1.84 Å as determined by EXAFS measurements. Single-point calculations at these geometries using the M06-L and M06 functionals in combination with the more complete def2-TZVPP basis set and COSMO(MeCN) predict the energetic difference between these conformers to be 2.6 and 3.0 kcal mol−1 (ΔG233), respectively. We also investigated intermediate (S = 3/2) and low (S = 1/2) spin states using the M06-L functional, which we recently demonstrated to be suitable for the determination of the spin ground state.57 We found these to be energetically less favorable and to provide poorer agreement with the experimentally determined Fe─OH bond distance (see Supporting Information Table S3).
Figure 9.

Structural depictions of the crossed and parallel conformations of [FeIII(OH)(TMC-py)]2+ at the M06-L/def2-SVP-(Fe:def2-TZVP)/COSMO(MeCN) level of theory for the S = 5/2 spin state.
4. Reactivity of FeIII(OH)(TMC-py)(OTf)2 (3) with TEMPOH.
[FeIII(OH)(TMC-py)](OTf)2 (3) is generated from 2 by reaction with excess CHD at −40 °C but is not a powerful enough oxidant to react further with excess CHD (bond dissociation energy (BDE) ≈ 77 kcal mol−1) indicating that the generated FeIII─OH complex 3 is only poorly reactive. This is in contrast to related FeIII complexes bearing the Py5 ligand (Py5 = (2,6-bis(bis(2-pyridyl)methoxymethane)-pyridine), which are capable of breaking relatively strong C─H bonds and readily oxidize CHD.58,59
However, 3 does decay at −40 °C over the course of 1 h, suggesting that it has the potential to react further. On the basis of the premise that 3 may be able to react with organic substrates having weaker X─H bonds, such as TEMPOH (DO─H ≈ 70 kcal/mol; Scheme 2),60,61 40 equiv of TEMPOH were added to a solution of 3, and the latter decayed completely within 1 min (Figure 10, top). A TEMPOH concentration dependence study afforded a k2 value of 7.1 M−1 s−1, which decreased sixfold to 1.2 M−1 s−1, when TEMPOD was used in place of TEMPOH (Figure 10, bottom). Analysis of the product solution by ESI-MS showed that [FeII(TMC-py)(OTf)]+ was formed as the major product, while EPR analysis revealed the formation of TEMPO radical in nearly quantitative yield (93%). Notably, the KIE value for TEMPOH/D oxidation by 3 is higher than those reported in the literature for O─H bond cleavage reactions by other FeIII─OH and MnIII─OH complexes (see Table 3). In addition, no reaction was observed at −40 °C for other potential substrates such as 2,4,6-tri-tert-butylphenol (O─H BDE ≈ 81 kcal/mol) and xanthene (C─H BDE ≈ 75.5 kcal/mol).61,62
Scheme 2.

Reaction of FeIII(OH)(TMC-py)(OTf)2 (3) with TEMPOH
Figure 10.

(top) UV–vis spectral changes upon addition of 40 equiv of TEMPOH into a solution of FeIII(OH)(TMC-py)(OTf)2 (3) in MeCN at —40 °C. (bottom) k2 plots for the reactions of 3 with varying amounts of TEMPOH (black ●) and TEMPOD (red ●) at −40 °C.
Table 3.
Second-Order Rate Constants for MIII–O(H/Me) Species Reactions with TEMPOH
| complexesa |
k2 (M− s−1, 25 °C) |
KIE | ref |
|---|---|---|---|
| [FeIII(OH)(TMC-py)]2+ (3) | 7.1 (−40 °C) | 6 (−40 °C) | b |
| [FeIII(OH)(PY5)]2+ | 4.3(3) × 10−4 | 6.3 (DHA) | 59 |
| [FeIII(OMe)(PY5)]2+ | 6.0(5) × 10−1 | 2.0 (4-t-BuArOH) | 58 |
| [FeIII(OH)(TMP)] | 7.6(5) × 101 | 63 | |
| [FeIII(OH)(OH2)(PyPz)]4+ | 2216(28) (20 °C) | 20.2(3) (xanthene) | 62 |
| [MnIII(OH)(dpaqH)]1+ | 1.3(1) × 10−1 | 1.8 | 61 |
| [MnIII(OMe)(dpaqH)]1+ | 8.0(1) × 10−2 | 1.8 | 64 |
| [MnIII(OH)(dpaq2Me)]1+ | 3.9(3) (−35 °C) | 2.7 (−35 °C) | 65 |
| [MnIII(OH) (SMe2N4(tren))]1+ | 2.1 × 103 | 3.1 | 66 |
| [MnIII(OMe) (SMe2N4(tren))]1+ | 3.6 × 102 | 2.1 | 66 |
Abbreviations used: dpaqH = 2-[bis(pyridin-2-ylmethyl)]amino-N-quinolin-8-yl-acetamidate anion; dpaq2Me = 2-[bis(pyridin-2-ylmethyl)]amino-N-2-methyl-quinolin-8-yl-acetamidate anion; ePy5 = (2,6-bis(bis(2-pyridyl)methoxymethane)pyridine; PyPz = quaternized tetra-2,3-pyridinoporphyrazine; SMe2N4(tren) = 3′-mercapto-3′,3′-dimethyl-2′-propylimino-tris(2-aminoethyl)amine; TMP = meso-tetra-mesitylporphyrinate.
This work.
The observed KIE of 6 for the reaction of 3 with TEMPOH at −40 °C is larger than all reactions listed in Table 3, except for the two cases involving C─H bond cleavage. This raises the question of whether the reaction mechanism involves HAT, where the proton and the electron both originate from the O─H bond and travel together to the same location, or a proton-coupled electron transfer (PCET), where the origin and destination of the proton and the electron transferred do not have to coincide, and thus both entities travel separately. We note here that the nomenclature for such processes is not always consistent in the literature. For the sake of simplicity we use the definition outlined above, which was also used by Usharani et al.67 (For more detailed definitions of different reaction scenarios associated with PCET reactions, see refs 68-70.) From the second-order rate constant of 7.1 M−1 s−1, we calculate an apparent free energy of activation of 12.6 kcal mol−1 as described in ref 71. To clarify the aspect outlined above, we optimized the reaction path at the M06-L/def2-SVP(Fe:def2-TZVP)/COSMO(MeCN) level of theory for the S = 5/2 spin state followed by single-point energy calculations at the M06-L/def2-TZVPP/COSMO(MeCN) and M06/def2-TZVPP/COSMO(MeCN) levels of theory. In the following, we will only refer to the given functional name reflecting the above outlined methodology. We again considered parallel and crossed conformations of the macrocycle. In Figure 11 Lewis structures of the studied path are depicted (for structural depictions see Figure S11). Computed free energies are shown in Table 4.
Figure 11.
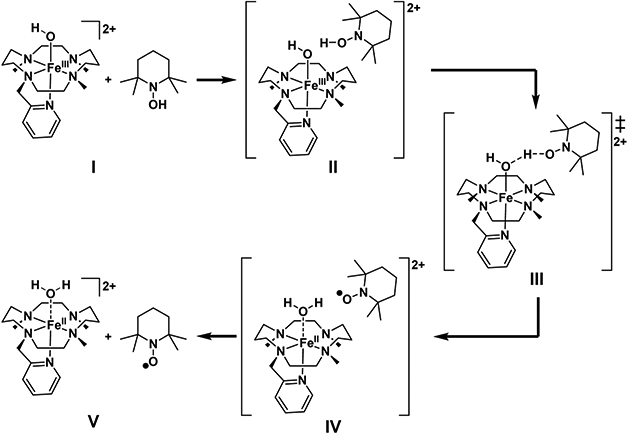
Lewis structure depictions of the studied reaction pathway.
Table 4.
Computed Free Energiesa (ΔG233 in kcal mol−1) for the Reaction between TEMPOH and [FeIII(OH)(TMC-py)]2+
| structure | I | II | III | IV | V |
|---|---|---|---|---|---|
| M06-L | 0.0 [2.6] | 3.5 [7.3] | 8.7 [10.4] | 0.6 [1.5] | −2.7 [−5.3] |
| M06 | 0.0 [3.0] | 5.0 [9.2] | 13.6 [16.2] | −3.7 [−3.1] | −8.2 [−12.2] |
Electronic energies were computed with the indicated functional using the def2-TZVPP basis set and COSMO(MeCN) on M06-L/def2-SVP(Fe:def2-TZVP)/COSMO(MeCN) geometries. Values refer to the “parallel” conformation, and values in brackets refer to the “crossed” conformation.
We obtained barriers of 8.7 and 10.4 kcal mol−1 for the parallel and crossed conformations. These values somewhat underestimate the experimentally observed values, which one might attribute to the local functional nature of M06-L. Computing energies with the hybrid functional M06 results in the prediction of slightly higher barriers of 13.6 and 16.2 kcal mol−1 for the parallel and crossed conformers (Table 4), which agree very well with the experimentally determined value of 12.6 kcal mol−1. We do note that we did not include any tunneling contribution, which would result in a reduction of the apparent barrier height.
One item we observe is that, once the +II oxidation state is reached, the crossed conformation becomes energetically favored, independent of the functional chosen. Furthermore, the FeII─OH2 distances in the resulting [FeII(OH2)(TMC-py)]2+ complexes with parallel and crossed conformations are rather long at 2.423 and 3.332 Å, respectively. We note that all FeII complexes supported by the TMC framework crystallographically characterized thus far are all five-coordinate and have a conformation consistent with our definition of crossed.16,29,72-76
To address the initially posed question of whether this reaction proceeds via HAT or PCET, we analyzed the changes in localized orbitals, more specifically, the intrinsic bond orbitals (IBOs),48 for intermediates II, III, and IV. It was previously demonstrated that this formalism can be used to analyze the electron flow of a given reaction.49 Notably, here we apply this methodology to an open-shell system for the first time. For this purpose, we treat α and β electrons separately, leading to two independent sets of localized orbitals. Consistent with the experimentally determined spin state of S = 5/2 for the FeIII─OH complex, we can identify five singly occupied d-orbitals in the α-spin manifold with no β counterparts on Fe for structure II. These are shown in Figure S12. As the FeIII center is reduced to FeII, one of the d-orbitals becomes occupied by a β electron derived from TEMPOH, leading to an S = 2 spin state on the reduced FeII center in IV. This leaves an α electron behind on the TEMPO radical that is ferromagnetically coupled to the FeII center (see spin density plot in Figure S13 and spin populations in Table S4). This scenario differs from the one described for the reaction between TEMPOH and a MnIII─OH complex, where an α electron is transferred, leading to an anti-ferromagnetic coupling of the resulting radical on TEMPO and the MnII─OH2 site.65 In the reaction of the FeIII─OH complex, we only need to inspect the changes that occur in the β spin manifold to identify which electron has been transferred from TEMPOH to the FeIII─OH complex. To be more specific, if the β electron transfers out of the O─H bond of TEMPOH and travels together with the proton in the form of a hydrogen radical, the reaction path would be classified as HAT. In contrast to this scenario, the transfer of an electron originating from some other orbital of TEMPOH to FeIII would more properly be described as a PCET. For a related analysis employing canonical orbital picture see ref 77.
In Figure 12 (top) we show the changes to the β electron of the O─H bond of the TEMPOH molecule. In the transformation from II to IV the electron associated with this localized orbital (IBO) is not transferred to the FeIII─OH complex. Instead, the changes to this IBO are modest in the TS structure III. After reduction of the FeIII center to FeII and generation of the TEMPO radical, the IBO becomes delocalized and is part of a π-bonding interaction between oxygen and nitrogen in structure IV. Further inspection of the IBOs associated with the TEMPOH molecule reveals that it is the β electron from the lone pair of the N atom that is transferred to the iron center, as shown in Figure 12 (bottom). This becomes particularly clear in the TS structure III and is consistent with the observed behavior of the β electron initially associated with the O─H bond of the TEMPOH molecule; that is, as the β electron is transferred from the N lone pair to the Fe center, a hole is generated, which is positioned such that in structure IV the β electron from the former O─H bond delocalizes as shown in Figure 12 (top). In this process, the proton is transferred to a lone pair on the FeIII─OH group. The IBO changes associated with the proton transfer are shown in Figure S14. As such, the proton transfer reaction resembles an acid/base reaction. Notably, acid/base reactions can exhibit nonclassical KIEs associated with proton transfer.78,79
Figure 12.

Changes of the intrinsic bond orbitals associated with the ß electrons of the O–H bond (top) and N lone pair (bottom) at the M06-L/def2-TZVPP/COSMO(MeCN) level of theory at M06-L/ def2-SVP(Fe:def2-TZVP)/COSMO(MeCN) geometries.
We compared our IBO-based results with respect to the HAT versus PCET nature of this reaction to an analysis of deformation energies proposed by Usharani et al.67 With the latter approach, we again obtain results more consistent with a PCET scenario (see Table S4). We therefore assign the reaction of our FeIII─OH complex with TEMPOH, to be PCET in nature. This reaction may thus be regarded as a model for the reactivity of soybean lipoxygenase, which has similarly been proposed to react with weak C─H bonds via PCET.80-88
CONCLUSIONS
To date, there are only two crystallographically characterized FeIII─OH complexes in nonheme ligand environments; in both examples, the FeIII─OH units are stabilized by hydrogen bonding groups designed into the supporting ligands.11,13 Here, we successfully characterized the thermally unstable high-spin FeIII(OH)(TMC-py)(OTf)2 (3) complex using UV–vis, EPR, EXAFS, and Mössbauer spectroscopies and ESI-MS. Structural parameters obtained from EXAFS measurements were supported computationally. The results not only shed new light into structural and electronic features of intermediates of this type but also provide evidence that the reaction of FeIV(O)(TMC-py)(OTf)2 with CHD forms an Fe(III)─OH complex as the initial product. Insight into the reactivity of such poorly understood iron(III)─hydroxide complexes has been obtained by studying the reaction of 3 with TEMPOH both experimentally and computationally. On the basis of computational analysis we classify the reaction as having PCET-like character, consistent with the proposed reaction channel for the FeIII─OH oxidant associated with soybean lipoxygenase.80-88
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the U.S. National Science Foundation (Grant Nos. CHE-1361773 to L.Q., CHE-1361595 to C.J.C., and CHE-1654060 to Y.G.) and the U.S. National Institutes of Health (Grant No. GM38767 to L.Q.). W.-M.C. acknowledges the Ministry of Science and Technology, Taiwan, for a postdoctoral fellowship, and J.E.M.N.K. thanks the Alexander von Humboldt Foundation for a Feodor Lynen Research Fellowship. XAS data were collected on Beamlines 7-3 and 9-3 at the Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, which is supported by the U.S. Department of Energy (DOE), Office of Science, Office of Basic Energy Sciences under Contract No. DE-AC02-76SF00515. Use of Beamlines 7-3 and 9-3 is supported by the DOE Office of Biological and Environmental Research, and by the National Institutes of Health, National Institute of General Medical Sciences (including P41GM103393). The authors acknowledge the Minnesota Supercomputing Institute (MSI) at the University of Minnesota for providing resources that contributed to the research results reported within this paper. R.F. and Y.G. also thank Prof. M. Hendrich for his assistance with the EPR instrument.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.inorgchem.7b01459.
Additional information on EXAFS analysis for complexes 3 and 4; NMR data for 1 and 2; mass spectrometric data for 4; EPR and Mössbauer data for 3; additional computational details and Cartesian coordinates (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Price JC; Barr EW; Tirupati B; Bollinger JM Jr.; Krebs C The First Direct Characterization of a High-Valent Iron Intermediate in the Reaction of an α-Ketoglutarate-Dependent Dioxygenase: A High-Spin Fe(IV) Complex in Taurine/α-Ketoglutarate Dioxygenase (TauD) from Escherichia coli. Biochemistry 2003, 42, 7497–7508. [DOI] [PubMed] [Google Scholar]
- (2).Bollinger JM; Krebs C Stalking intermediates in oxygen activation by iron enzymes: Motivation and method. J. Inorg. Biochem 2006, 100, 586–605. [DOI] [PubMed] [Google Scholar]
- (3).Krebs C; Galonić Fujimori D; Walsh CT; Bollinger JM Jr. Non-Heme Fe(IV)-Oxo Intermediates. Acc. Chem. Res 2007, 40, 484–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Bollinger JM; Price JC; Hoffart LM; Barr EW; Krebs C Mechanism of Taurine: α-Ketoglutarate Dioxygenase (TauD) from Escherichia coli. Eur. J. Inorg. Chem 2005, 2005, 4245–4254. [DOI] [PubMed] [Google Scholar]
- (5).Rohde J-U; In J-H; Lim MH; Brennessel WW; Bukowski MR; Stubna A; Münck E; Nam W; Que L Jr. Crystallographic and Spectroscopic Characterization of a Nonheme Fe(IV)=O Complex. Science 2003, 299, 1037–1039. [DOI] [PubMed] [Google Scholar]
- (6).Klein JEMN; Que L Jr. Biomimetic High-Valent Mononuclear Nonheme Iron-Oxo Chemistry in Encyclopedia of Inorganic and Bioinorganic Chemistry (EIBC); John Wiley & Sons, Ltd, 2016, DOI: 10.1002/9781119951438.eibc2344. [DOI] [Google Scholar]
- (7).McDonald AR; Que L Jr. High-valent nonheme iron-oxo complexes: Synthesis, structure, and spectroscopy. Coord. Chem. Rev 2013, 257, 414–428. [Google Scholar]
- (8).Ray K; Pfaff FF; Wang B; Nam W Status of Reactive Non-Heme Metal–Oxygen Intermediates in Chemical and Enzymatic Reactions. J. Am. Chem. Soc 2014, 136, 13942–13958. [DOI] [PubMed] [Google Scholar]
- (9).Puri M; Que L Jr. Toward the Synthesis of More Reactive S = 2 Non-Heme Oxoiron(IV) Complexes. Acc. Chem. Res 2015, 48, 2443–2452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Sastri CV; Lee J; Oh K; Lee YJ; Lee J; Jackson TA; Ray K; Hirao H; Shin W; Halfen JA; Kim J; Que L Jr.; Shaik S; Nam W Axial ligand tuning of a nonheme iron(IV)–oxo unit for hydrogen atom abstraction. Proc. Natl. Acad. Sci. U. S. A 2007, 104, 19181–19186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Ogo S; Wada S; Watanabe Y; Iwase M; Wada A; Harata M; Jitsukawa K; Masuda H; Einaga H Synthesis, Structure, and Spectroscopic Properties of [FeIII(tnpa)(OH)(PhCOO)]ClO4: A Model Complex for an Active Form of Soybean Lipoxygenase-1. Angew. Chem., Int. Ed 1998, 37, 2102–2104. [DOI] [PubMed] [Google Scholar]
- (12).MacBeth CE; Golombek AP; Young VG; Yang C; Kuczera K; Hendrich MP; Borovik AS O2 Activation by Nonheme Iron Complexes: A Monomeric Fe(III)-oxo Complex Derived from O2. Science 2000, 289, 938–941. [DOI] [PubMed] [Google Scholar]
- (13).MacBeth CE; Gupta R; Mitchell-Koch KR; Young VG; Lushington GH; Thompson WH; Hendrich MP; Borovik AS Utilization of Hydrogen Bonds To Stabilize M–O(H) Units: Synthesis and Properties of Monomeric Iron and Manganese Complexes with Terminal Oxo and Hydroxo Ligands. J. Am. Chem. Soc 2004, 126, 2556–2567. [DOI] [PubMed] [Google Scholar]
- (14).England J; Guo Y; Farquhar ER; Young VG Jr.; Münck E; Que L Jr. The Crystal Structure of a High-Spin Oxoiron(IV) Complex and Characterization of Its Self-Decay Pathway. J. Am. Chem. Soc 2010, 132, 8635–8644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Prakash J; Rohde GT; Meier KK; Münck E; Que L Jr. Upside Down! Crystallographic and Spectroscopic Characterization of an [FeIV(Osyn)(TMC)]2+ Complex. Inorg. Chem 2015, 54, 11055–11057. [DOI] [PubMed] [Google Scholar]
- (16).Thibon A; England J; Martinho M; Young VG Jr.; Frisch JR; Guillot R; Girerd J-J; Münck E; Que L Jr.; Banse F Proton- and Reductant-Assisted Dioxygen Activation by a Nonheme Iron(II) Complex to Form an Oxoiron(IV) Intermediate. Angew. Chem., Int. Ed 2008, 47, 7064–7067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Lee Y-M; Kotani H; Suenobu T; Nam W; Fukuzumi S Fundamental Electron-Transfer Properties of Non-heme Oxoiron(IV) Complexes. J. Am. Chem. Soc 2008, 130, 434–435. [DOI] [PubMed] [Google Scholar]
- (18).Fukuzumi S; Kotani H; Suenobu T; Hong S; Lee Y-M; Nam W Contrasting Effects of Axial Ligands on Electron-Transfer Versus Proton-Coupled Electron-Transfer Reactions of Nonheme Oxoiron(IV) Complexes. Chem. - Eur. J 2010, 16, 354–361. [DOI] [PubMed] [Google Scholar]
- (19).Matson EM; Park YJ; Fout AR Facile Nitrite Reduction in a Non-heme Iron System: Formation of an Iron(III)-Oxo. J. Am. Chem. Soc 2014, 136, 17398–17401. [DOI] [PubMed] [Google Scholar]
- (20).Armarego WLF; Chai C Chapter 4 - Purification of Organic Chemicals. In Purification of Laboratory Chemicals, 7th ed.; Butterworth-Heinemann: Boston, MA, 2013; pp 103–554. [Google Scholar]
- (21).Hagadorn JR; Que L Jr.; Tolman WB N-Donor Effects on Carboxylate Binding in Mononuclear Iron(II) Complexes of a Sterically Hindered Benzoate Ligand. Inorg. Chem 2000, 39, 6086–6090. [DOI] [PubMed] [Google Scholar]
- (22).Zhou A; Kleespies ST; Van Heuvelen KM; Que L Jr. Characterization of a heterobimetallic nonheme Fe(III)-O-Cr(III) species formed by O2 activation. Chem. Commun 2015, 51, 14326–14329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Mader EA; Davidson ER; Mayer JM Large Ground-State Entropy Changes for Hydrogen Atom Transfer Reactions of Iron Complexes. J. Am. Chem. Soc 2007, 129, 5153–5166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Wu A; Mader EA; Datta A; Hrovat DA; Borden WT; Mayer JM Nitroxyl Radical Plus Hydroxylamine Pseudo Self-Exchange Reactions: Tunneling in Hydrogen Atom Transfer. J. Am. Chem. Soc 2009, 131, 11985–11997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Havare N; Plattner DA Oxidative Cleavage of α-Aryl Aldehydes Using Iodosylbenzene. Org. Lett 2012, 14, 5078–5081. [DOI] [PubMed] [Google Scholar]
- (26).Petasis DT; Hendrich MP Chapter Eight-Quantitative Interpretation of Multifrequency Multimode EPR Spectra of Metal Containing Proteins, Enzymes, and Biomimetic Complexes. Methods Enzymol. 2015, 563, 171–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).George GN EXAFSPAK, Stanford Synchrotron Radiation Laboratory; Stanford Linear Accelerator Center: Stanford, CA, 2000. [Google Scholar]
- (28).Wojdyr M Fityk: a general-purpose peak fitting program. J. Appl. Crystallogr 2010, 43, 1126–1128. [Google Scholar]
- (29).England J; Bigelow JO; Van Heuvelen KM; Farquhar ER; Martinho M; Meier KK; Frisch JR; Münck E; Que L Jr. An ultra-stable oxoiron(IV) complex and its blue conjugate base. Chem. Sci 2014, 5, 1204–1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Zhou A; Prakash J; Rohde GT; Klein JEMN; Kleespies ST; Draksharapu A; Fan R; Guo Y; Cramer CJ; Que L Jr. The Two Faces of Tetramethylcyclam in Iron Chemistry: Distinct Fe–O–M Complexes Derived from [FeIV(Oanti/syn)(TMC)]2+ Isomers. Inorg. Chem 2017, 56, 518–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Ahlrichs R; Bär M; Häser M; Horn H; Kölmel C Electronic structure calculations on workstation computers: The program system turbomole. Chem. Phys. Lett 1989, 162, 165–169. [Google Scholar]
- (32).Furche F; Ahlrichs R; Hättig C; Klopper W; Sierka M; Weigend F Turbomole. WIREs Comput. Mol. Sci 2014, 4, 91–100. [Google Scholar]
- (33).TURBOMOLE, V7.0.1 2015, a development of University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 1989–2007; TURBOMOLE GmbH, since 2007; available from http://www.turbomole.com. [Google Scholar]
- (34).Zhao Y; Truhlar DG A new local density functional for main-group thermochemistry, transition metal bonding, thermochemical kinetics, and noncovalent interactions. J. Chem. Phys 2006, 125, 194101. [DOI] [PubMed] [Google Scholar]
- (35).Weigend F; Ahlrichs R Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys 2005, 7, 3297–3305. [DOI] [PubMed] [Google Scholar]
- (36).Zhao Y; Truhlar D The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc 2008, 120, 215–241. [Google Scholar]
- (37).Gagliardi LG; Castells CB; Ràfols C; Rosés M; Bosch E Static Dielectric Constants of Acetonitrile/Water Mixtures at Different Temperatures and Debye–Hückel A and a0B Parameters for Activity Coefficients. J. Chem. Eng. Data 2007, 52, 1103–1107. [Google Scholar]
- (38).Lowry TM; Henderson ST Molecular Structure and Physical Properties of Prussic Acid. Part I. Refractive Dispersion of Prussic Acid and Its Homologues. Proc. R. Soc. London, Ser. A 1932, 136, 471–487. [Google Scholar]
- (39).http://www.sigmaaldrich.com/chemistry/solvents/acetonitrile-center.html Accessed June 7, 2016.
- (40).Klamt A; Schüürmann G COSMO: A New Approach to Dielectric Screening in Solvents with Explicit Expressions for the Screening Energy and its Gradient. J. Chem. Soc., Perkin Trans. 2 1993, 2, 799–805. [Google Scholar]
- (41).Klamt A; Jonas V Treatment of the outlying charge in continuum solvation models. J. Chem. Phys 1996, 105, 9972–9981. [Google Scholar]
- (42).Averkiev BB; Truhlar DG Free energy of reaction by density functional theory: oxidative addition of ammonia by an iridium complex with PCP pincer ligands. Catal. Sci. Technol 2011, 1, 1526–1529. [Google Scholar]
- (43).Ribeiro RF; Marenich AV; Cramer CJ; Truhlar DG Use of Solution-Phase Vibrational Frequencies in Continuum Models for the Free Energy of Solvation. J. Phys. Chem. B 2011, 115, 14556–14562. [DOI] [PubMed] [Google Scholar]
- (44).Bryantsev VS; Diallo MS; Goddard WA III Calculation of Solvation Free Energies of Charged Solutes Using Mixed Cluster/Continuum Models. J. Phys. Chem. B 2008, 112, 9709–9719. [DOI] [PubMed] [Google Scholar]
- (45).Sierka M; Hogekamp A; Ahlrichs R Fast evaluation of the Coulomb potential for electron densities using multipole accelerated resolution of identity approximation. J. Chem. Phys 2003, 118, 9136–9148. [Google Scholar]
- (46).Weigend F Accurate Coulomb-fitting basis sets for H to Rn. Phys. Chem. Chem. Phys 2006, 8, 1057–1065. [DOI] [PubMed] [Google Scholar]
- (47).Hellweg A Heuristic control of kinetic energy in dynamic reaction coordinate calculations. J. Comput. Chem 2013, 34, 1835–1841. [DOI] [PubMed] [Google Scholar]
- (48).Knizia G Intrinsic Atomic Orbitals: An Unbiased Bridge between Quantum Theory and Chemical Concepts. J. Chem. Theory Comput 2013, 9, 4834–4843. [DOI] [PubMed] [Google Scholar]
- (49).Knizia G; Klein JEMN Electron Flow in Reaction Mechanisms—Revealed from First Principles. Angew. Chem., Int. Ed 2015, 54, 5518–5522. [DOI] [PubMed] [Google Scholar]
- (50).Knizia G. http://www.iboview.org/.
- (51).Prakash J; Rohde GT; Meier KK; Jasniewski AJ; Van Heuvelen KM; Münck E; Que L Jr. Spectroscopic Identification of an FeIII Center, not FeIV, in the Crystalline Sc–O–Fe Adduct Derived from [FeIV(O)(TMC)]2+. J. Am. Chem. Soc 2015, 137, 3478–3481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).MacMillan SN; Lancaster KM X-ray Spectroscopic Interrogation of Transition-Metal-Mediated Homogeneous Catalysis: Primer and Case Studies. ACS Catal. 2017, 7, 1776–1791. [Google Scholar]
- (53).Pestovsky O; Stoian S; Bominaar EL; Shan XP; Münck E; Que L Jr.; Bakac A Aqueous FeIV=O: Spectroscopic Identification and Oxo-Group Exchange. Angew. Chem., Int. Ed 2005, 44, 6871–6874. [DOI] [PubMed] [Google Scholar]
- (54).Jackson TA; Rohde J-U; Seo MS; Sastri CV; DeHont R; Stubna A; Ohta T; Kitagawa T; Münck E; Nam W; Que L Jr. Axial Ligand Effects on the Geometric and Electronic Structures of Nonheme Oxoiron(IV) Complexes. J. Am. Chem. Soc 2008, 130, 12394–12407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Scarrow RC; Trimitsis MG; Buck CP; Grove GN; Cowling RA; Nelson MJ X-ray Spectroscopy of the Iron Site in Soybean Lipoxygenase-1: Changes in Coordination upon Oxidation or Addition of Methanol. Biochemistry 1994, 33, 15023–15035. [DOI] [PubMed] [Google Scholar]
- (56).Berry JF; Bill E; Garcia-Serres R; Neese F; Weyhermuller T; Wieghardt K Effect of N-Methylation of Macrocyclic Amine Ligands on the Spin State of Iron(III): A Tale of Two Fluoro Complexes. Inorg. Chem 2006, 45, 2027–2037. [DOI] [PubMed] [Google Scholar]
- (57).Verma P; Varga Z; Klein JEMN; Cramer CJ; Que L; Truhlar DG Assessment of electronic structure methods for the determination of the ground spin states of Fe(II), Fe(III) and Fe(IV) complexes. Phys. Chem. Chem. Phys 2017, 19, 13049–13069. [DOI] [PubMed] [Google Scholar]
- (58).Goldsmith CR; Jonas RT; Stack TDP C–H Bond Activation by a Ferric Methoxide Complex: Modeling the Rate-Determining Step in the Mechanism of Lipoxygenase. J. Am. Chem. Soc 2002, 124, 83–96. [DOI] [PubMed] [Google Scholar]
- (59).Goldsmith CR; Stack TDP Hydrogen Atom Abstraction by a Mononuclear Ferric Hydroxide Complex: Insights into the Reactivity of Lipoxygenase. Inorg. Chem 2006, 45, 6048–6055. [DOI] [PubMed] [Google Scholar]
- (60).Wang C-C; Chang H-C; Lai Y-C; Fang H; Li C-C; Hsu H-K; Li Z-Y; Lin T-S; Kuo T-S; Neese F; Ye S; Chiang Y-W; Tsai M-L; Liaw W-F; Lee W-Z A Structurally Characterized Nonheme Cobalt–Hydroperoxo Complex Derived from Its Superoxo Intermediate via Hydrogen Atom Abstraction. J. Am. Chem. Soc 2016, 138, 14186–14189. [DOI] [PubMed] [Google Scholar]
- (61).Wijeratne GB; Corzine B; Day VW; Jackson TA Saturation Kinetics in Phenolic O–H Bond Oxidation by a Mononuclear Mn(III)–OH Complex Derived from Dioxygen. Inorg. Chem 2014, 53, 7622–7634. [DOI] [PubMed] [Google Scholar]
- (62).Gao H; Groves JT Fast Hydrogen Atom Abstraction by a Hydroxo Iron(III) Porphyrazine. J. Am. Chem. Soc 2017, 139, 3938–3941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Porter TR; Mayer JM Radical reactivity of the Fe(III)/(II) tetramesitylporphyrin couple: hydrogen atom transfer, oxyl radical dissociation, and catalytic disproportionation of a hydroxylamine. Chem. Sci 2014, 5, 372–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Wijeratne GB; Day VW; Jackson TA O-H bond oxidation by a monomeric MnIII-OMe complex. Dalton Trans. 2015, 44, 3295–3306. [DOI] [PubMed] [Google Scholar]
- (65).Rice DB; Wijeratne GB; Burr AD; Parham JD; Day VW; Jackson TA Steric and Electronic Influence on Proton-Coupled Electron-Transfer Reactivity of a Mononuclear Mn(III)-Hydroxo Complex. Inorg. Chem 2016, 55, 8110–8120. [DOI] [PubMed] [Google Scholar]
- (66).Coggins MK; Brines LM; Kovacs JA Synthesis and Structural Characterization of a Series of MnIIIOR Complexes, Including a Water-Soluble MnIIIOH That Promotes Aerobic Hydrogen-Atom Transfer. Inorg. Chem 2013, 52, 12383–12393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Usharani D; Lacy DC; Borovik AS; Shaik S Dichotomous Hydrogen Atom Transfer vs Proton-Coupled Electron Transfer During Activation of X–H Bonds (X = C, N, O) by Nonheme Iron–Oxo Complexes of Variable Basicity. J. Am. Chem. Soc 2013, 135, 17090–17104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Warren JJ; Tronic TA; Mayer JM Thermochemistry of Proton-Coupled Electron Transfer Reagents and its Implications. Chem. Rev 2010, 110, 6961–7001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Weinberg DR; Gagliardi CJ; Hull JF; Murphy CF; Kent CA; Westlake BC; Paul A; Ess DH; McCafferty DG; Meyer TJ Proton-Coupled Electron Transfer. Chem. Rev 2012, 112, 4016–4093. [DOI] [PubMed] [Google Scholar]
- (70).Hammes-Schiffer S Proton-Coupled Electron Transfer: Moving Together and Charging Forward. J. Am. Chem. Soc 2015, 137, 8860–8871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Cho K-B; Kim EJ; Seo MS; Shaik S; Nam W Correlating DFT-Calculated Energy Barriers to Experiments in Nonheme Octahedral FeIVO Species. Chem. - Eur. J 2012, 18, 10444–10453. [DOI] [PubMed] [Google Scholar]
- (72).Fiedler AT; Halfen HL; Halfen JA; Brunold TC Synthesis, Structure Determination, and Spectroscopic/Computational Characterization of a Series of Fe(II)–Thiolate Model Complexes: Implications for Fe–S Bonding in Superoxide Reductases. J. Am. Chem. Soc 2005, 127, 1675–1689. [DOI] [PubMed] [Google Scholar]
- (73).McDonald AR; Bukowski MR; Farquhar ER; Jackson TA; Koehntop KD; Seo MS; De Hont RF; Stubna A; Halfen JA; Münck E; Nam W; Que L Jr. Sulfur versus Iron Oxidation in an Iron-Thiolate Model Complex. J. Am. Chem. Soc 2010, 132, 17118–17129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Sastri CV; Park MJ; Ohta T; Jackson TA; Stubna A; Seo MS; Lee J; Kim J; Kitagawa T; Munck E; Que L Jr.; Nam W Axial Ligand Substituted Nonheme FeIV=O Complexes: Observation of Near-UV LMCT Bands and Fe = O Raman Vibrations. J. Am. Chem. Soc 2005, 127, 12494–12495. [DOI] [PubMed] [Google Scholar]
- (75).Hodges KD; Wollmann RG; Kessel SL; Hendrickson DN; Van Derveer DG; Barefield EK Preparations and Properties of Nitrosyl Complexes of Iron Tetramethylcyclam. X-ray Structures of [Fe(C14H32N4)NO](BF4)2, a S = 3/2–1/2 Spin-Equilibrium Complex, and [Fe(C14H32N4)(NO)(OH)](ClO4)2.CH3CN. J. Am. Chem. Soc 1979, 101, 906–917. [Google Scholar]
- (76).Wilson SA; Chen J; Hong S; Lee Y-M; Clemancey M; Garcia-Serres R; Nomura T; Ogura T; Latour J-M; Hedman B; Hodgson KO; Nam W; Solomon EI [FeIV=O(TBC)-(CH3CN)]2+: Comparative Reactivity of Iron(IV)-Oxo Species with Constrained Equatorial Cyclam Ligation. J. Am. Chem. Soc 2012, 134, 11791–11806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (77).Fang H; Jing H; Ge H; Brothers PJ; Fu X; Ye S The Mechanism of E–H (E = N, O) Bond Activation by a Germanium Corrole Complex: A Combined Experimental and Computational Study. J. Am. Chem. Soc 2015, 137, 7122–7127. [DOI] [PubMed] [Google Scholar]
- (78).Anslyn EV; Dougherty DA Modern Physical Organic Chemistry; University Science Books: Sausalito, CA, 2006. [Google Scholar]
- (79).Watt CIF Primary kinetic hydrogen isotope effects in deprotonations of carbon acids. J. Phys. Org. Chem 2010, 23, 561–571. [Google Scholar]
- (80).Lehnert N; Solomon EI Density-functional investigation on the mechanism of H-atom abstraction by lipoxygenase. J. Biol. Inorg. Chem 2003, 8, 294–305. [DOI] [PubMed] [Google Scholar]
- (81).Fukuzumi S Proton-Coupled Electron Transfer of Unsaturated Fatty Acids and Mechanistic Insight into Lipoxygenase. Helv. Chim. Acta 2006, 89, 2425–2440. [Google Scholar]
- (82).Soudackov AV; Hammes-Schiffer S Proton-coupled electron transfer reactions: analytical rate constants and case study of kinetic isotope effects in lipoxygenase. Faraday Discuss. 2016, 195, 171–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (83).Yu T; Soudackov AV; Hammes-Schiffer S Computational Insights into Five- versus Six-Coordinate Iron Center in Ferrous Soybean Lipoxygenase. J. Phys. Chem. Lett 2016, 7, 3429–3433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (84).Soudackov AV; Hammes-Schiffer S Probing Nonadiabaticity in the Proton-Coupled Electron Transfer Reaction Catalyzed by Soybean Lipoxygenase. J. Phys. Chem. Lett 2014, 5, 3274–3278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (85).Hatcher E; Soudackov AV; Hammes-Schiffer S Proton-Coupled Electron Transfer in Soybean Lipoxygenase: Dynamical Behavior and Temperature Dependence of Kinetic Isotope Effects. J. Am. Chem. Soc 2007, 129, 187–196. [DOI] [PubMed] [Google Scholar]
- (86).Hatcher E; Soudackov AV; Hammes-Schiffer S Proton-Coupled Electron Transfer in Soybean Lipoxygenase. J. Am. Chem. Soc 2004, 126, 5763–5775. [DOI] [PubMed] [Google Scholar]
- (87).Offenbacher AR; Hu S; Poss EM; Carr CAM; Scouras AD; Prigozhin DM; Iavarone AT; Palla A; Alber T; Fraser JS; Klinman JP Hydrogen–Deuterium Exchange of Lipoxygenase Uncovers a Relationship between Distal, Solvent Exposed Protein Motions and the Thermal Activation Barrier for Catalytic Proton-Coupled Electron Tunneling. ACS Cent. Sci 2017, 3, 570–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (88).Hu S; Soudackov AV; Hammes-Schiffer S; Klinman JP Enhanced Rigidification within a Double Mutant of Soybean Lipoxygenase Provides Experimental Support for Vibronically Non-adiabatic Proton-Coupled Electron Transfer Models. ACS Catal. 2017, 7, 3569–3574. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.