

Abstract
Agents that safely induce, enhance, or sustain multiple innate immune signaling pathways could be developed as potent vaccine adjuvants or co-adjuvants. Using high-throughput screens with cell-based nuclear factor kappa B (NF-κB) and interferon stimulating response element (ISRE) reporter assays, we identified a bis-aryl sulfonamide bearing compound 1 that demonstrated sustained NF-κB and ISRE activation after a primary stimulus with lipopolysaccharide or interferon-α, respectively. Here, we present systematic structure-activity relationship (SAR) studies on the two phenyl rings and amide nitrogen of the sulfonamide group of compound 1 focused towards identification of affinity probes. The murine vaccination studies showed that compounds 1 and 33 when used as co-adjuvants with monophosphoryl lipid A (MPLA) showed significant enhancement in antigen ovalbumin-specific immunoglobulin responses compared to MPLA alone. SAR studies pointed to the sites on the scaffold that can tolerate the introduction of aryl azide, biotin and fluorescent rhodamine substituents to obtain several affinity and photoaffinity probes which will be utilized in concert for future target identification and mechanism of action studies.
Keywords: Adjuvant, NF-κB, ISRE, LPS, Interferon, Toll-like receptor, Sulfonamide, Affinity probe, Biotin, Fluorescent, Photoaffinity, Vaccine, Immunization
Graphical Abstract
Introduction:
Vaccines consisting of antigen and adjuvant rely primarily on adjuvants for enhancement of immune stimuli.1 These adjuvants include ligands for pattern recognition receptors (PRRs) such as Toll-like receptors (TLR) −2, −4, −7, −8, and −9, nucleotide-binding oligomerization domain-like receptors (NLRs), RIG-I-like receptors (RLRs) and cytokines such as interferon-α (IFN-α), IFN-γ and IL-12.2–13 Some of these adjuvants have been approved for human use by the U.S. Food and Drug Administration (FDA) including the TLR-4 agonist monophosphoryl Lipid A (MPLA),14 the TLR-9 agonist CpG 1018,15 and other adjuvants with different mechanisms of action such as alum and squalene based adjuvants. Despite the availability of approved adjuvants, the need for co-adjuvants is evident since single adjuvant vaccines often do not generate long lasting protective immunity.16 Alum has been used as an effective single adjuvant for decades primarily due to its safety record and induction of increased humoral immunity;17, 18 however it induces only weak cellular immunity and predominantly a T helper (Th) type 2 associate response; whereas in some cases a T helper type 1 response would be more effective for protection. In addition, it is not always sufficient for vaccinating immunocompromised and elderly populations.17, 19 A co-adjuvant is a substance that may or may not be an adjuvant by itself but can work with a known adjuvant to offer synergistic effects such as enhanced antibody response. For example, IL-2 has been shown to be a co-adjuvant with alum-adsorbed hepatitis B vaccine.20 Similarly, combination adjuvants can be obtained using a PRR or NLR ligand, an immunogenic protein, a delivery system or another adjuvant with a complementary mechanism of action.16 One such combination AS04 (Adjuvant System 04), consisting of MPLA and alum, has been FDA approved in a hepatitis B vaccine Fendrix® and human papillomavirus vaccine Cervarix®.21–24 Alternative combinations involving approved adjuvants, TLR agonists, NOD agonists, and delivery systems are being explored.25–28
Our approach towards identifying novel co-adjuvants focused on small molecules that may not lead to immune activation by themselves but may enhance the primary immune activation such as nuclear factor kappa B (NF-κB) or IFN stimulating response element (ISRE) activation induced by a TLR-4 agonist (LPS or MPLA). The rationale behind the approach is as follows: Upon vaccine administration, local antigen presenting cells (APCs) at the site of injection, such as dendritic cells and Langerhans cells, are activated by the TLR-4 agonist. These APCs engulf antigen and travel to local draining lymph nodes where the antigen is presented to T cells.29 The activation levels of APCs induced by a TLR-4 agonist peaks at 2–6 hours and then decays due to negative feedback mechanisms.30–37 Because it takes approximately 12–24 hours for an APC to travel to the lymph node after vaccination,38 APCs are arriving during the decay phase of the activation. This rationale is well supported from a report that showed that the absence of interleukin-1 receptor associated kinase M (IRAK-M, a negative regulator of TLR signaling)39 increases NF-κB activation, improves migration of dendritic cells (DCs) to lymph nodes thereby increasing the lifespan of the activated DCs and secretion of Th1-skewed cytokines and chemokines.31 Thus, we hypothesize that prolonging or sustaining the activation of APCs induced by the TLR-4 agonist for 12–24 hours will lead to optimal presentation of antigen to the T cells which would enhance the initial immune response and potentially allow for a longer lasting response. Our hypothesis is supported by reports that enhanced responses to vaccinations were observed in mice with genetic disruption of either IRAK-M, an inhibitor of the NF-κB pathway,31 or of UBP43, a negative regulator of type 1 IFN signaling.40 Thus, to address this issue, we sought HTS methods directed towards identification of co-adjuvants that prolonged activation of an immune response induced by a primary stimulus.41, 42
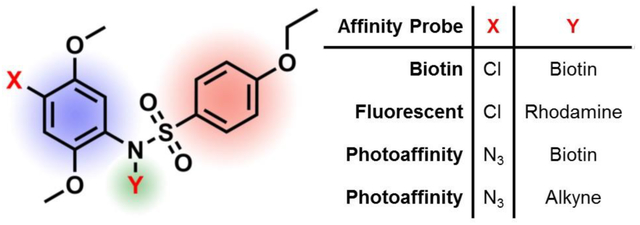
These cell based HTS campaigns tested protraction of a TLR-4 agonist lipopolysaccharide (LPS) stimulus through the NF-κB pathway41 or of IFN-α signaling via the interferon stimulating response element (ISRE)42 pathway. Compounds that prolonged LPS induced NF-κB signaling included a distinct set of pyrimido[5,4-b]indoles that were also found to be effective co-adjuvants with MPLA, an FDA approved adjuvant, in murine vaccination studies.41 In parallel, compounds that prolonged IFN-α induced ISRE signaling in vitro were also evaluated as co-adjuvants in vivo42 which led to identification of a potent bis-aryl sulfonamide compound 1 (Fig. 1) bearing 4-chloro-2,5-dimethoxy and 4-ethoxy substituted phenyl groups connected by a sulfonamide functional group. Compound 1 did not possess any NF-κB or ISRE activity when tested alone but it enhanced their activation when tested in presence of LPS or IFN-α respectively, compared to the stimulus alone. The further drug development of such hits identified through cell-based phenotypic assays and involved in cell signaling pathways is hampered without the knowledge of the target receptor or the compound’s mechanism of action.43
Figure 1.
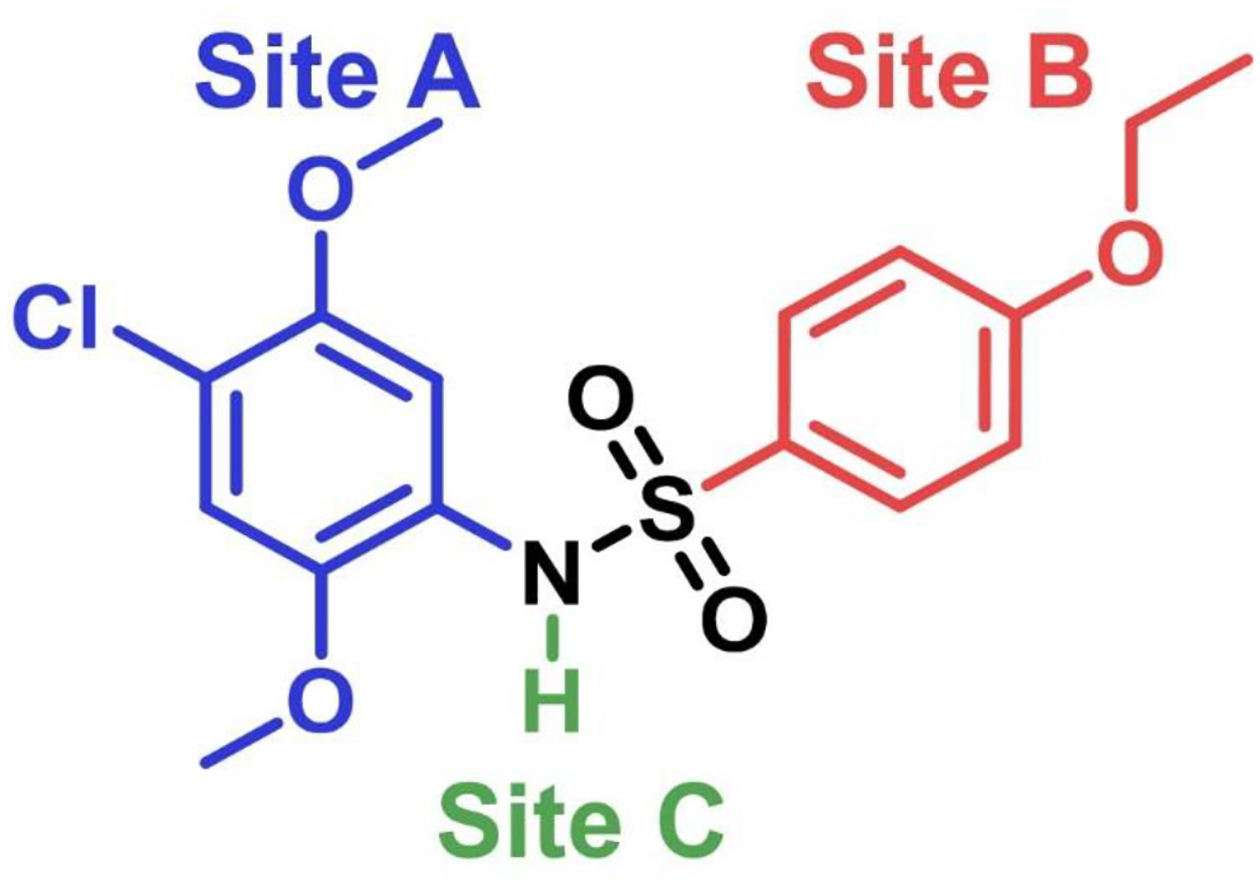
Structure of compound 1 and sites of modification on the scaffold. SAR studies on the bis-aryl sulfonamide compound 1 were approached by modifying one site at a time.
This necessitated SAR studies focused towards identification of affinity probes which involved evaluation of structural variations for compound 1 that were unexplored in the HTS with an aim to identify positions on the scaffold that can tolerate the introduction of small functional groups such as aryl azide or diazirine to make photoreactive probes or large substituents such as biotin and fluorescence moieties to generate affinity and fluorescent probes, respectively.44–50
Exploration of several different functional groups and substituents will allow us to systematically identify the position and size of the affinity probe as well as the reactive handle to be used for introducing these probes. These chemical probes would then be useful tools for future mechanistic and functional receptor studies. In addition, the chemical handle would allow one to covalently conjugate the small molecule to peptides or protein antigens to make self-adjuvanting vaccine constructs which are widely explored in vaccine development.51–56
Results and Discussion:
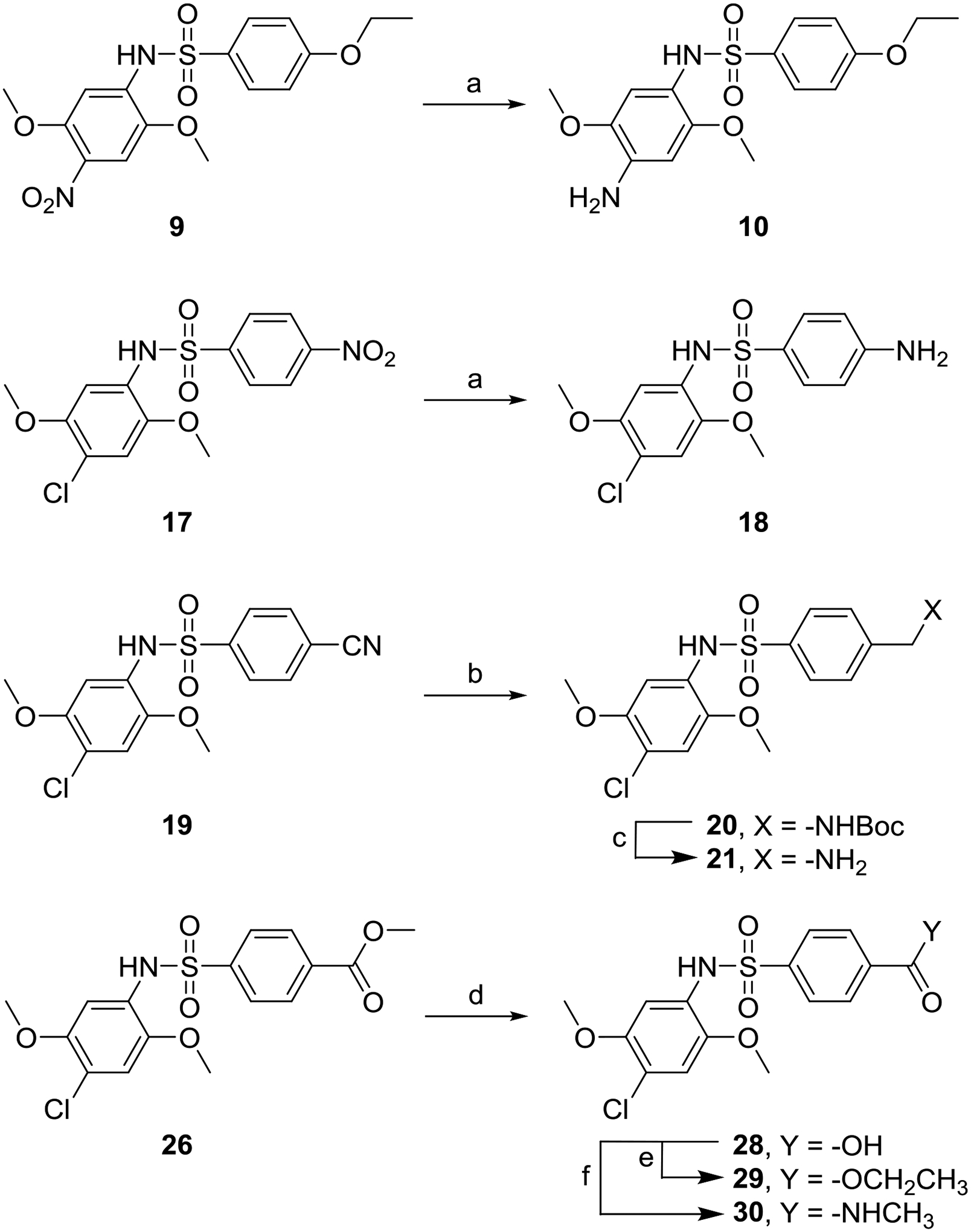
Approximately 3400 differently substituted bis-aryl sulfonamide compounds were screened in the original HTS libraries and a scatter plot showing activation data for these compounds in both cell-based NF-κB and ISRE assays is shown in Supporting Information Fig. S1. These results provided preliminary SAR analysis indicating the substituents on the two aryl rings necessary for activity and pointed to compound 1 as an advanced lead. Hence, we approached further SAR studies on compound 1 by first identifying three areas (sites A, B and C) of potential modification as shown in Fig. 1. To standardize the reaction, we began with synthesis of compound 1 by reaction of 4-ethoxysulfonyl chloride (3a) and 4-chloro-2,5-dimethoxy aniline (2a) in the presence of an organic base (Scheme 1). However, the reaction not only provided the desired compound 1, but also formed the bis-sulfonamide side-product in high yields. This undesired side-product was formed in situ by further reaction of compound 1 with another equivalent of 4-ethoxysulfonyl chloride (3a). We were able to isolate this bis-sulfonamide side-product but observed that it was somewhat unstable. Limited hydrolysis by lithium hydroxide facilitated the complete conversion of this bis-sulfonamide side-product to compound 1 without further hydrolysis of the mono-sulfonamide bond thereby improving reaction yields for compound 1 (Scheme 1). This reaction strategy was utilized for synthesis of several site A and site B modified compounds for SAR analysis.
Scheme 1.
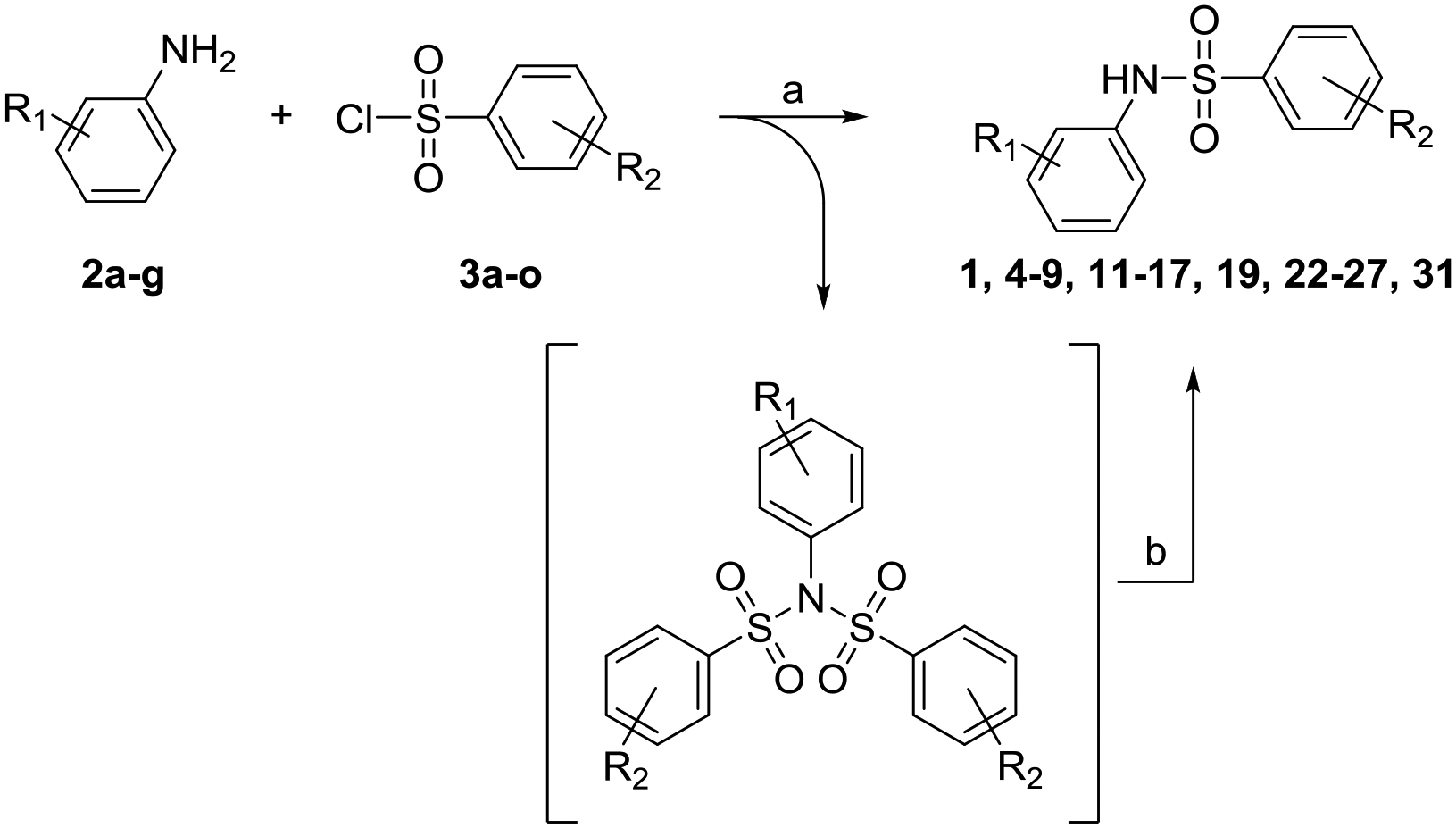
Syntheses of compound 1 and its site A and B modified analogs.
Reagents and conditions: a) Et3N, CH2Cl2; b) LiOH. MeOH, THF, H2O.
SAR studies were initiated by modifying the substituents at site A (Fig. 1). These compounds were synthesized according to Scheme 1 using different anilines (2a-g). We probed the removal of one aryl substituent at a time to obtain compounds 4, 5, and 6 lacking the 2-methoxy, 3-methoxy and 4-chloro substituent, respectively. Replacement of 4-chloro by a 4-bromo substituent gave compound 7 and migration of the 2-methoxy substituent to the 3-position gave compound 8. These compounds were evaluated for sustained activation of both NF-κB and ISRE pathways using LPS and IFN-α as primary stimuli, respectively. The SAR studies pointed to the importance of the methoxy substituents at the 2 and 5 positions of the aryl ring, because either removal of any one of the substituents as in compound 4 and 5 or its displacement to another position on the ring as in 8 led to complete loss of activity. Removal of the 4-chloro as in compound 6 or its replacement with a spatially larger bromo substituent as in compound 7 retained activity (Table 1). Thus, to further explore position 4 on the phenyl ring, we synthesized analogs with 4-nitro (9) substitution and its 4-amino (10) derivative. However, both these analogs were inactive suggesting that only hydrophobic substituents at this site are tolerated (Table 1).
Table 1.
Structure and bioactivity data for site A modified compounds.
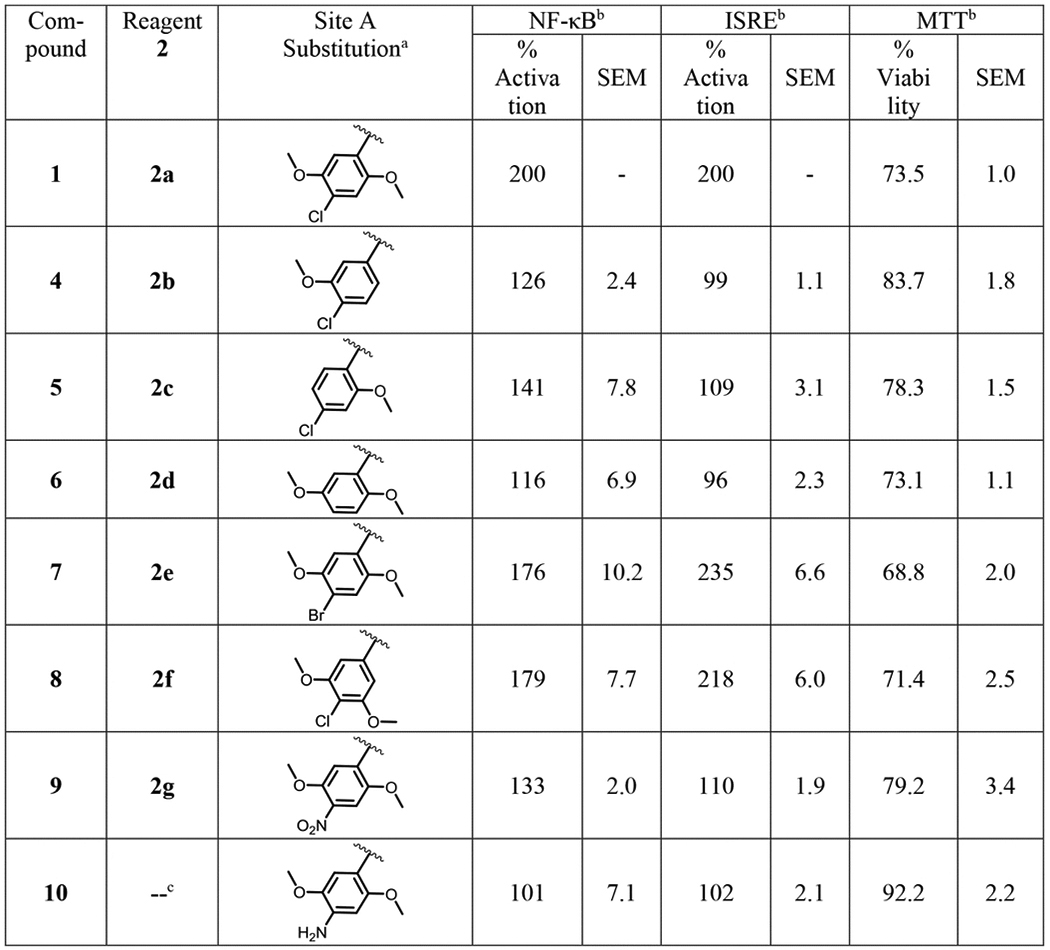
|
Compounds 1, 4–9 were obtained by reaction of reagent 2 with 4-ethoxybenzenesulfonyl chloride (3a) as shown in Scheme 1.
The % activation values in NF-ĸB and ISRE induction assays were two point normalized between compound 1 as 200% and LPS (10 ng/mL) for NF-κB or IFN-α (100 U/mL) for ISRE as 100%. The mean SEAP response in NF-ĸB assay for compound 1 + LPS and LPS alone was 3.44 ± 0.08 and 0.56 ± 0.06 μg/mL, respectively. The mean emission ratio in ISRE assay for compound 1 + IFN-α and IFN-α alone was 1.88 ± 0.04 and 0.69 ± 0.05 μg/mL, respectively. The % viability values for compounds in MTT assay were normalized to DMSO as 100%. The mean OD value at 405 nm for DMSO was 1.24 ± 0.03. All raw values used for normalization are represented as mean ± SEM.
Compound 10 was derived from compound 9 as shown in Scheme 2.
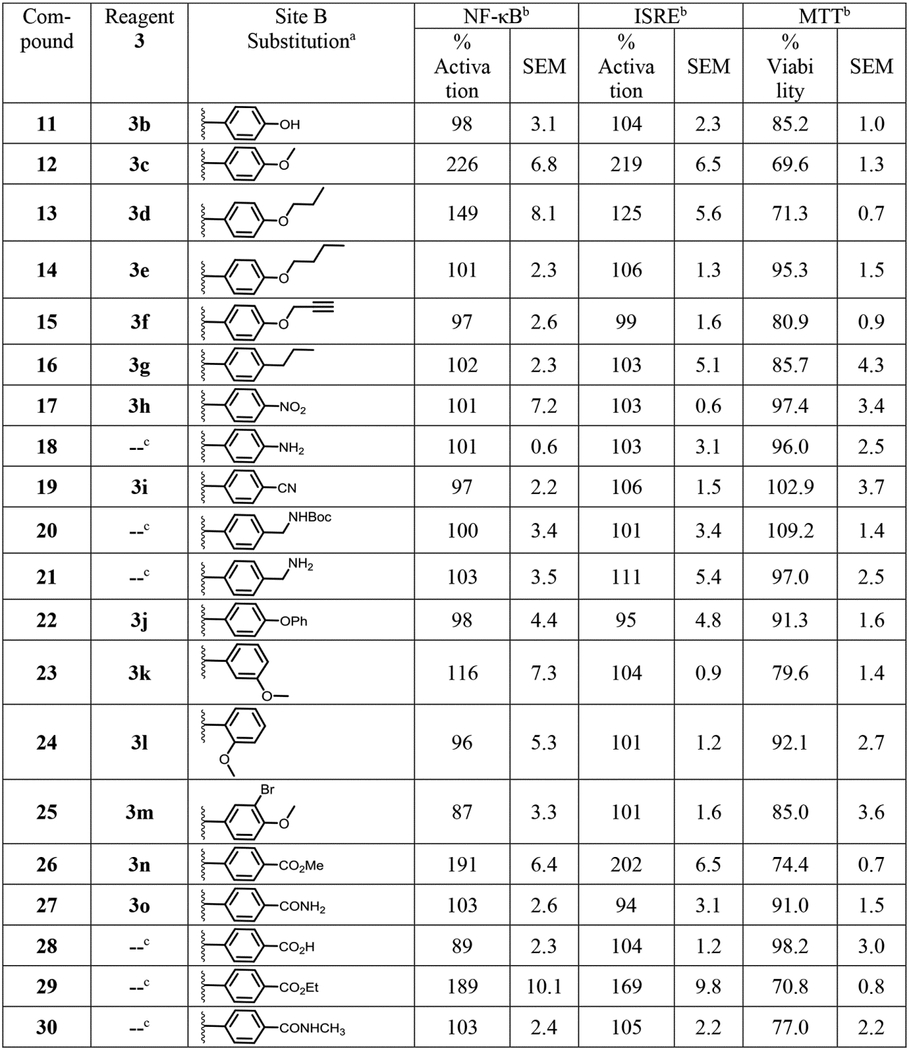
Next, we focused on site B as shown in Fig. 1. The compounds were synthesized as discussed earlier (Scheme 1) using different aryl sulfonyl chlorides (3a-p) and 4-chloro-2,5-dimethoxy aniline (2a). Some of the aryl sulfonyl chlorides were commercially available, while the others were synthesized as shown in Supporting Information Scheme S1. We first probed the homologous series of 4-O-alkylated compounds starting with 4-hydroxy analog 11, 4-methoxy analog 12, 4-propoxy analog 13 and 4-butoxy analog 14 compared to 4-ethoxy analog compound 1. Bioactivity evaluation of these compounds showed that only the smaller homolog as in 4-methoxy compound 12 was tolerated while the hydrophilic interaction with hydroxy group of 11 without any hydrophobic alkyl group was not tolerated. The higher 4-alkoxy chains showed gradual loss of activity (Table 2). While the 4-propoxy substituted compound was weakly active, the 4-propargyloxy compound 15, designed to use the alkyne as a handle for click chemistry reactions, was found to be inactive. Removal of the ether oxygen to obtain 4-propyl substituted compound 16 also led to loss of activity suggesting a crucial role of hydrogen bond interaction by the ether oxygen. Other functional groups that could be involved in such hydrogen bond interactions led to the syntheses of 4-nitro analog 17 and its amine bearing derivative 18 (Scheme 2) obtained by reduction of the nitro group. Also, the 4-nitrile analog 19, N-Boc methylamine derivative 20 obtained by in situ N-Boc protection during the reduction of the nitrile group and its free methylamine derivative 21 (Scheme 2) were synthesized. All these compounds were also evaluated but found to be either weakly active or completely inactive. A prior report indicated that analogs bearing a 4-O-phenyl substitution exhibited ubiquitin ligase inhibition activity,57 so we synthesized the 4-O-phenyl analog 22, but this compound was inactive. Encouraged by the activity of 4-methoxy substituted analog 12, we synthesized 3-methoxy and 2-methoxy substituted compounds 23 and 24, respectively. However, none of these molecules was active. In order to find an additional handle for modification, bromine was introduced to obtain a 3-bromo-4-methoxy substituted compound 25, which was also found to be inactive. Learning from the requirement of a hydrogen bonding functional group at site B for activity, we probed the addition of another oxo-containing group to obtain the 4-methylester analog 26 and an amide analog 27. Ester hydrolysis of compound 26 yielded the 4-carboxyl derivative 28 (Scheme 2). While the methyl ester bearing analog 26 was active, the hydrolyzed carboxylic acid analog 28 and the amide linked compound 27 lost activity (Table 2).
Table 2.
Structure and bioactivity data for site B modified compounds.
|
Compounds 11–17, 19, 22–27 were obtained by reaction of reagent 3 with 4-chloro-2,5-dimethoxyaniline (2a) as shown in Scheme 1.
The % activation values in NF-ĸB and ISRE induction assays were two point normalized between compound 1 as 200% and LPS (10 ng/mL) for NF-κB or IFN-α (100 U/mL) for ISRE as 100%. The mean SEAP response in NF-ĸB assay for compound 1 + LPS and LPS alone was 3.44 ± 0.08 and 0.56 ± 0.06 μg/mL, respectively. The mean emission ratio in ISRE assay for compound 1 + IFN-α and IFN-α alone was 1.88 ± 0.04 and 0.69 ± 0.05 μg/mL, respectively. The % viability values for compounds in MTT assay were normalized to DMSO as 100%. The mean OD value at 405 nm for DMSO was 1.24 ± 0.03. All raw values used for normalization are represented as mean ± SEM.
The compounds were synthesized as shown in Scheme 2.
Scheme 2.
Syntheses of site A and B modified derivatives.
Reagents and conditions: a) Pd/C, H2 (50psi), EtOAc b) (Boc)2O, Pd/C, H2 (50 psi), MeOH c) 4N HCl/dioxane d) LiOH, MeOH, THF, H2O e) TMSCl, EtOH f) EtNH2, HATU, Et3N, DMF.
Hypothesizing that the lack of hydrophobic alkyl group interaction could be a cause for the loss of activity, compound 28 was further derivatized to obtain the ethyl ester analog 29, and the N-methylamide analog 30 (Scheme 2). While analog 29 retained partial activity, compound 30 was completely inactive suggesting that only hydrogen bond accepting substituents were tolerated (Table 2). An additional analog (compound 31, Scheme 1) was synthesized by inversing the sulfonamide bond obtained by reaction of 2-ethoxyaniline and 4-chloro-2,5-dimethoxybenzenesulfonyl chloride, but the inactivity of this analog suggested that the positioning of the sulfonamide functional group was also critical for activity.
Moving forward, we wanted to probe the expansion at site C on the nitrogen of the sulfonamide function of compound 1. These compounds were synthesized by derivatization of compound 1 as shown in Scheme 3. The first extensive series of compounds were the N-alkylated derivatives including N-methyl (33), N-propyl (34), N-butyl (35), N-pentyl (36), N-hexyl (37), N-heptyl (38), and N-dodecyl (39). A clear correlation of bioactivity with the alkyl chain length was observed with potency gradually decreasing with increased alkyl chain length and compounds bearing alkyl chain lengths greater than N-pentyl were completely inactive (Table 3). We also probed the effect of steric bulk around the core structure by synthesizing N-isopropyl (40) and N-isobutyl (41) derivatives. Steric bulk closer to the core structure, as in compound 40, eliminated the NF-κB activity while retaining ISRE activity. In contrast, spacing the isopropyl group away by one methylene unit as in compound 41 regained the activity in both the NF-κB and ISRE assays. Encouraged by these results, we desired to synthesize alkyne bearing compounds with an additional aim to utilize the functional group as a biorthogonal reactive site. A homologous series of alkyne bearing molecules including N-propargyl (42), N-butynyl (43), and N-pentynyl (44) were synthesized (Scheme 3). Activity data showed that while N-alkyl derivatization with increasing alkyl chain length led to dramatic loss of activity, the corresponding N-alkynyl derivatives retained activity almost equivalent to that of compound 1 (Fig. 2, Table 3) for the corresponding alkyl chain length. As shown in Fig. 2, the retention of activity for the N-alkynyl compounds compared to loss in activity for the analogous N-alkyl derivatives for the same carbon unit chain length suggested the possible involvement of π-π interactions in near proximity with the target receptor(s). We were therefore interested in evaluation of a triethyleneglycol linked alkyne derivative (45) which would allow us to conveniently place the reactive functional group distant from the core. However, the 12-atom chain length equivalent to N-dodecyl compound 39 was too long to retain activity.
Scheme 3.
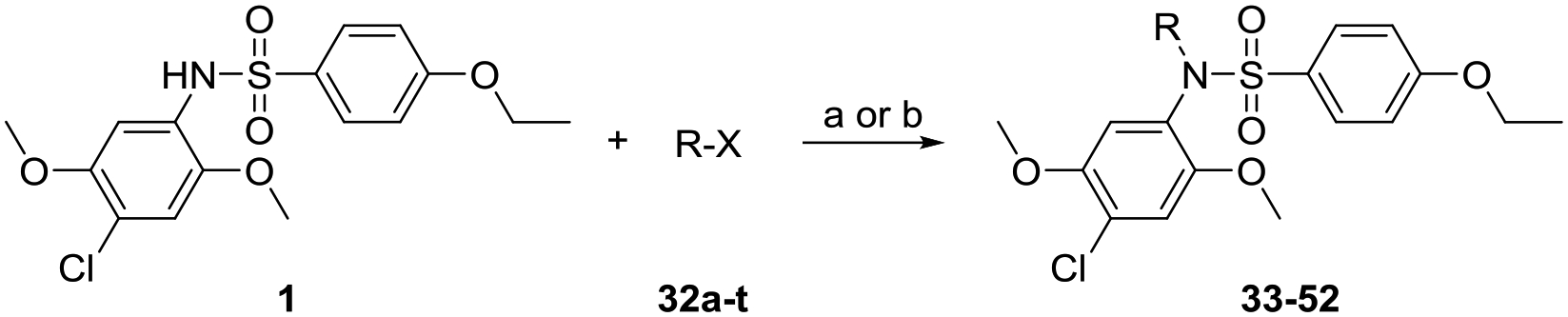
Syntheses of sulfonamide N-substituted derivatives of compound 1.
Reagents and conditions: a) K2CO3, DMF, 45 °C. For compound 48: b) Et3N, CH2Cl2.
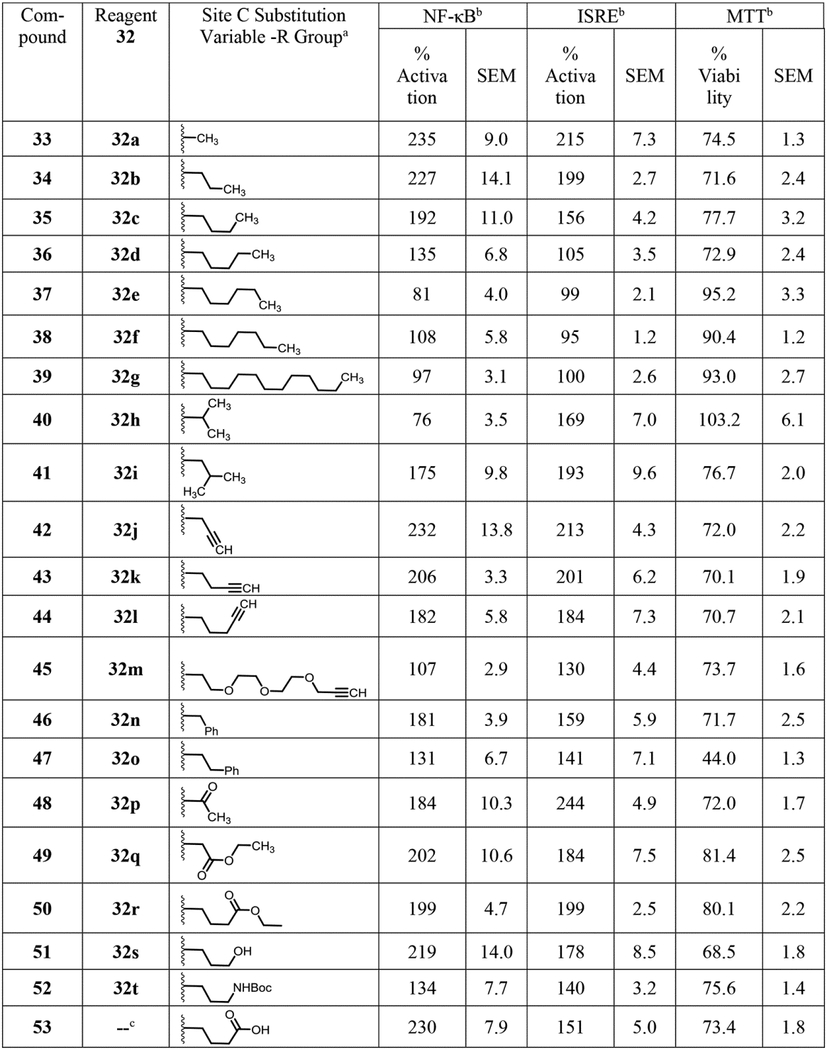
Table 3.
Structure and bioactivity data for site C modified compounds.
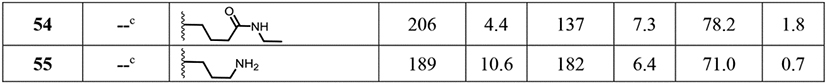
|
aCompounds 33–52 were obtained by reaction of reagent 32 with compound 1 as shown in Scheme 3. bThe % activation values in NF-ĸB and ISRE induction assays were two point normalized between compound 1 as 200% and LPS (10 ng/mL) for NF-κB or IFN-α (100 U/mL) for ISRE as 100%. The mean SEAP response in NF-ĸB assay for compound 1 + LPS and LPS alone was 3.44 ± 0.08 and 0.56 ± 0.06 μg/mL, respectively. The mean emission ratio in ISRE assay for compound 1 + IFN-α and IFN-α alone was 1.88 ± 0.04 and 0.69 ± 0.05 μg/mL, respectively. The % viability values for compounds in MTT assay were normalized to DMSO as 100%. The mean OD value at 405 nm for DMSO was 1.24 ± 0.03. All raw values used for normalization are represented as mean ± SEM. cThe compounds were derived from compounds 50 and 52 as shown in Scheme 4.
Figure 2.
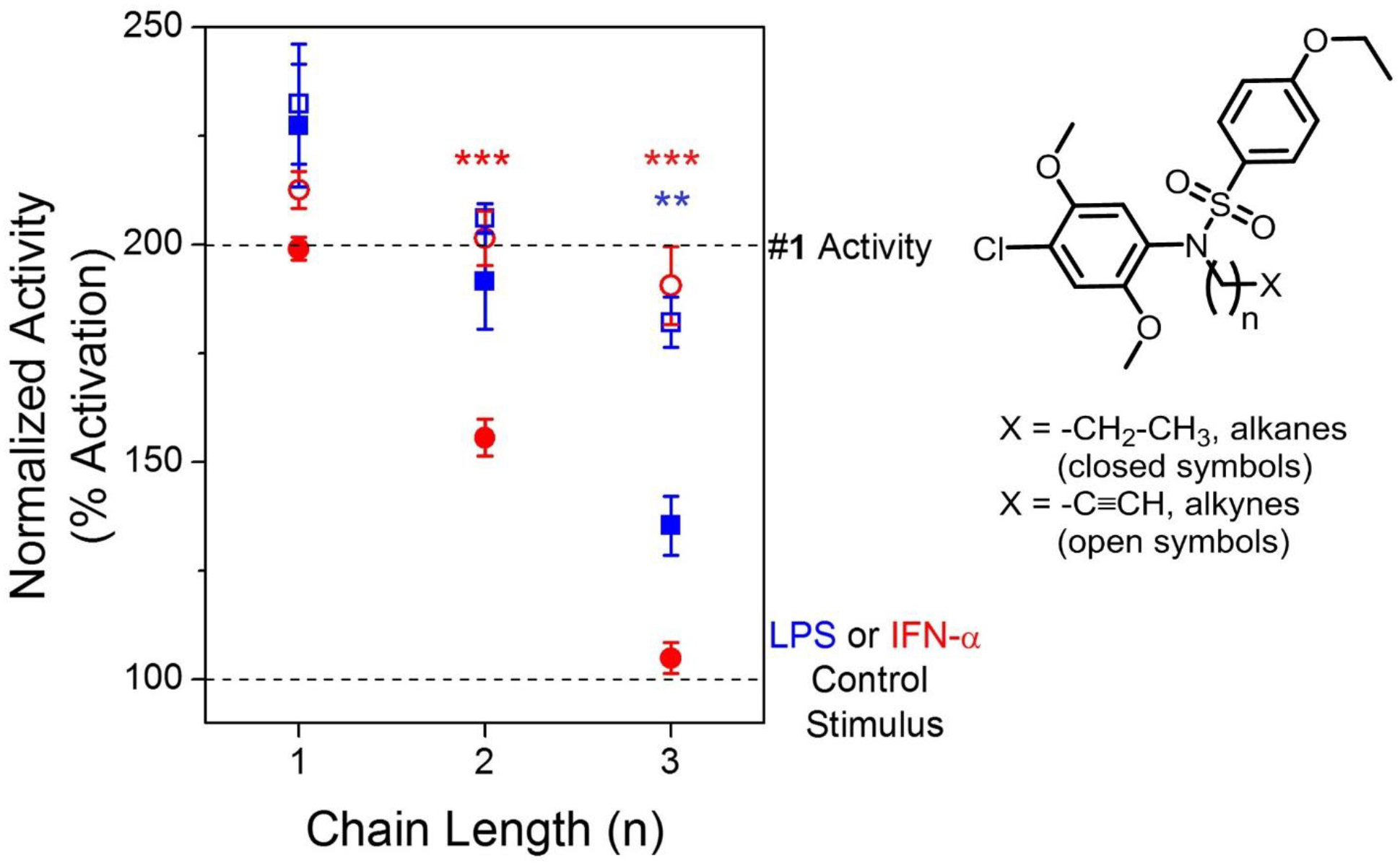
Differential activity profile for terminal alkane v/s terminal alkyne bearing derivatives of compound 1. Chain length (n) dependent reduction in activity was observed for site C modified terminal alkane (closed symbols) and terminal alkyne (open symbols) bearing compounds. The % activation values in NF-ĸB and ISRE induction assays were two point normalized between compound 1 as 200% (grey dotted line) and LPS (10ng/mL) for NF-κB or IFN-α (100 U/mL) for ISRE as 100% (grey dotted line). The relative reduction in the activity for terminal alkane compounds was significantly greater than the terminal alkynes for the same chain length suggesting involvement of π-π interactions. NF-κB activity is shown in blue squares and ISRE activity is shown in red circles. Structure of the compounds is shown to the right with variable chain length ‘n’ varying from 1 to 3. Data are presented as mean ± SEM. **p<0.01 and ***p<0.001 for alkyne bearing compounds compared to alkane bearing compounds for the same chain length using two-way ANOVA followed by Bonferroni post hoc analysis.
These results for the alkyne bearing compounds led us to propose making compounds where substituents can form enhanced π-π interactions. Thus N-benzyl (46) and N-phenethyl (47) derivatives were synthesized and were also found to be potent analogs (Table 3). Since the N-isopropyl analog 40 was inactive, we desired to determine if steric bulk was the only reason for its inactivity and if that could be mitigated by some hydrogen bonding functional group such as acetyl. Thus, the N-acetyl derivative (48) was synthesized and the bioactivity assays showed that the compound was active. However, before proceeding with syntheses of additional acylated analogs, we wanted to evaluate its stability in stock solutions since during the assay this compound could behave as a prodrug by undergoing deacetylation to release active compound 1. While the stock of compound 48 in DMSO was stable, incubation of compound with assay media showed release of compound 1 (data not shown), suggesting that the bioactivity could be due to a prodrug effect and not true interaction with the receptor. Thus, syntheses of additional acylated analogs were not pursued.
Since the hydrophobic alkyl and alkynyl groups were well tolerated at site C, we examined if incorporating a hydrophilic group that could serve as a handle for further chemical modification would be acceptable for activity. A pair of compounds bearing a precursor to a reactive handle such as carboxylic esters were synthesized by alkylation of compound 1 to obtain the N-ethyl glycinate (49) and N-ethyl butanoate (50) analogs (Scheme 3). Our attempts to make a stable propionate analog failed after several attempts likely due to retro Michael type reaction, and despite isolating a few milligrams of the tert-butyl propionate ester derivative, activity studies were not pursued due to stability concerns. Both the ethyl ester substituted compounds 49 and 50 retained dual NF-κB and ISRE activities (Table 3). To avoid additional substitution closer to the core sulfonamide pharmacophore, we chose a propylene spacer for further analogs. A terminal hydroxy bearing analog as in N-propan-3-ol (51) and the N-Boc protected aminopropane analog (52) were then synthesized. The ethyl ester of compound 50 was de-esterified using lithium hydroxide to obtain its carboxylic acid analog 53, which was converted to the ethyl amide analog 54 (Scheme 4). Similarly, a free amine bearing molecule was obtained by N-Boc deprotection of 52 to obtain compound 55. Biological evaluation showed that the terminal hydroxy analog 51 retained activity in both the assays while the N-Boc protected compound 52 showed reduction in activity, which was recovered when the N-Boc group was removed as in compound 55. Both the free carboxylic acid and ethyl amide derivatives retained activity, which was more skewed towards the NF-κB pathway (Table 3).
Scheme 4.
Syntheses of chemically reactive handle bearing analogs of compound 1.
Reagents and conditions: a) LiOH, MeOH, THF, H2O b) EtNH2, HATU, Et3N, DMF; c) 4N HCl/dioxane.
All these compounds were evaluated for toxicity using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assays. All the active compounds showed viability between 69% and 81%. Some of the inactive compounds were completely non-toxic. Compound 47 with the N-phenethyl substitution was an exception showing somewhat higher toxicity (% viability = 44%) suggesting that an aryl group connected by an ethylene unit near the core sulfonamide structure may lead to toxicity (Tables 1–3).
The bioactivity data from both the assays for all the compounds were plotted to verify the correlation between the chemical structure and bioactivity. Most of the compounds were active in both NF-κB and ISRE bioassays and showed a good correlation (Pearson two-tailed, R2=0.6812, P<0.0001, Fig. 3). The SAR trends however varied depending on the site of modification. Site A modifications involving removal of the methoxy substituent (compounds 4 and 5) led to significant loss of activity (Fig. 3). On the other hand, nonpolar modifications at position 4 of site A (compounds 7 and 8) showed slightly skewed ISRE activity compared to compound 1, while hydrogen bond forming substituents at this position led to loss of activity (compounds 9 and 10). Most of the site B modified compounds were inactive suggesting restricted SAR tolerance due to limited spatial availability in the target receptor. Only short alkyl groups connected via ether-linkage as in compounds 1, 12 and 13 or carboxyl (ester)-linkage as in compounds 26 and 29 retained activity. A good correlation was seen, however, between the two assays for these compounds. In contrast, most of the site C modified compounds were active in both the bioassays suggesting that only a part of the substituent may be involved in receptor interaction and the rest of the group subtends out of the target receptor(s). A notable variation was observed in sterically hindered bulky groups close to the core structure as in compound 40 which led to a loss of NF-κB activity, while still retaining ISRE activity. On the other hand, another subset of compounds bearing a reactive handle such as carboxylic acid analog 53 and its amidated derivative 54, showed reduction in ISRE activity while retaining the NF-κB activity (Fig. 3). This suggested that a negative charge on the compound may be a deterrent for ISRE activity.
Figure 3.
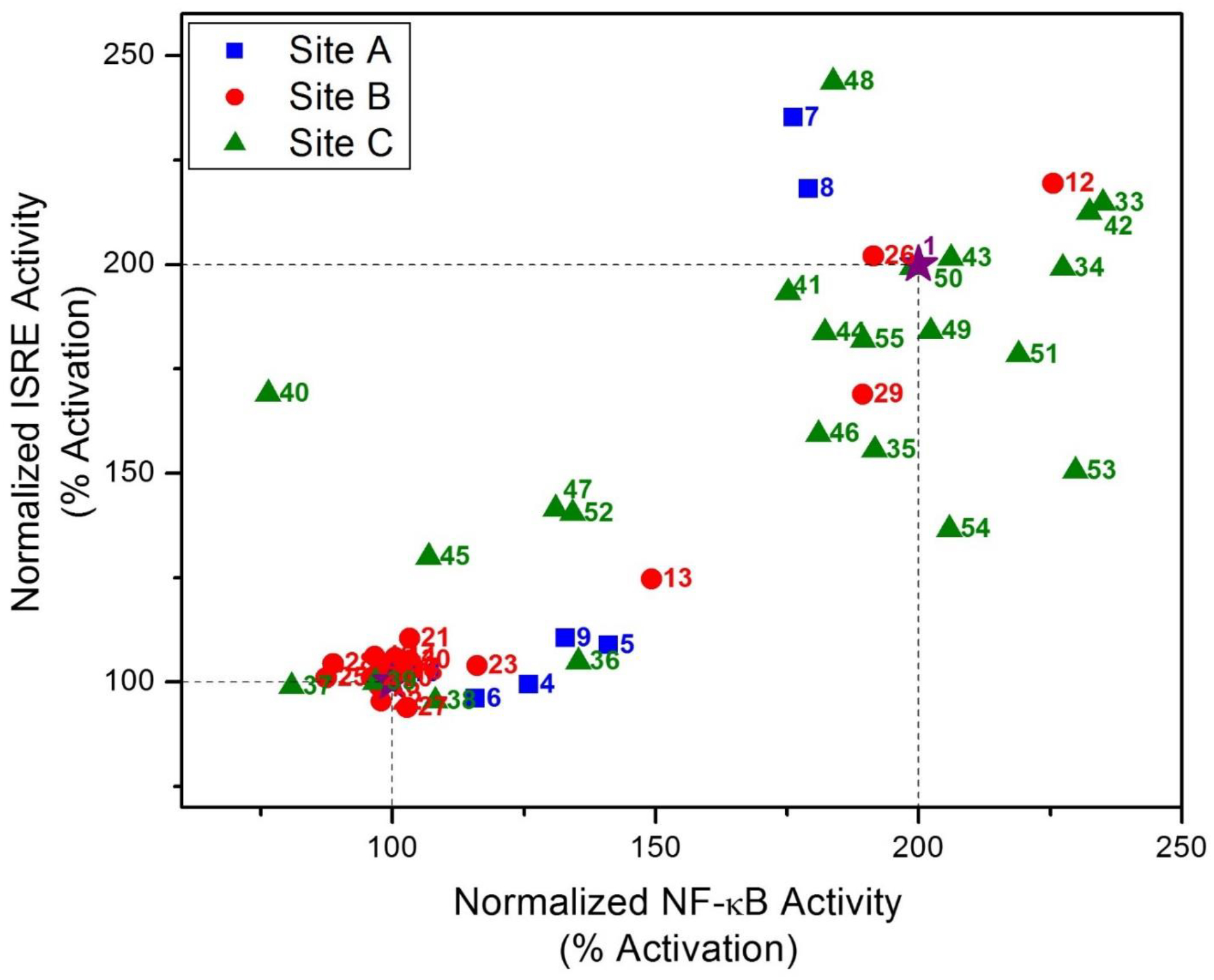
Bioactivity analysis for synthesized bis-aryl sulfonamide analogs. Scatter plot showing the ISRE activity on the Y-axis for cells treated with each compound and IFN-α and NF-κB activity on X-axis for cells treated with compound and LPS. Data are shown as a two-point normalization between the active hit compound 1 as 200% (purple star) and vehicle (0.1% LPS or IFN-α) as 100%. Site A, B, and C modified analogs are designated as blue, red, and green squares, circles and triangles, respectively. Pearson two-tailed correlation was significant (P<0.0001) for the two activities for all these compounds.
Continuing with the focus on compounds that retain dual NF-κB and ISRE activity similar to original hit compound 1, we selected site B modified compound 12, site C modified compound 33 and an aliphatic amine bearing compound 55 for dose response experiments and EC50 determination as these compounds are nearly equipotent in both the assays when evaluated at 5 μM concentration. As shown in Fig. 4, both compounds 12 and 33 showed relatively higher NF-κB activity at 5 μM concentration, but the activity of compound 12 decreased faster at lower concentrations which led to EC50 value of 1.85 μM. Compound 1 and 33 were almost equipotent with EC50 values of 0.60 μM and 0.69 μM, respectively. Compound 55 was relatively weaker with EC50 of 3.32 μM. The potency trends for these compounds remained the same in ISRE activity with compounds 1, 12 and 33 exhibiting EC50 of 0.66 μM, 1.4 μM, and 0.84 μM, respectively, and compound 55 with EC50 = 3.04 μM. Even though the activity of compound 55 was slightly attenuated, the amine handle can be utilized for derivatization to obtain affinity probes.
Figure 4.
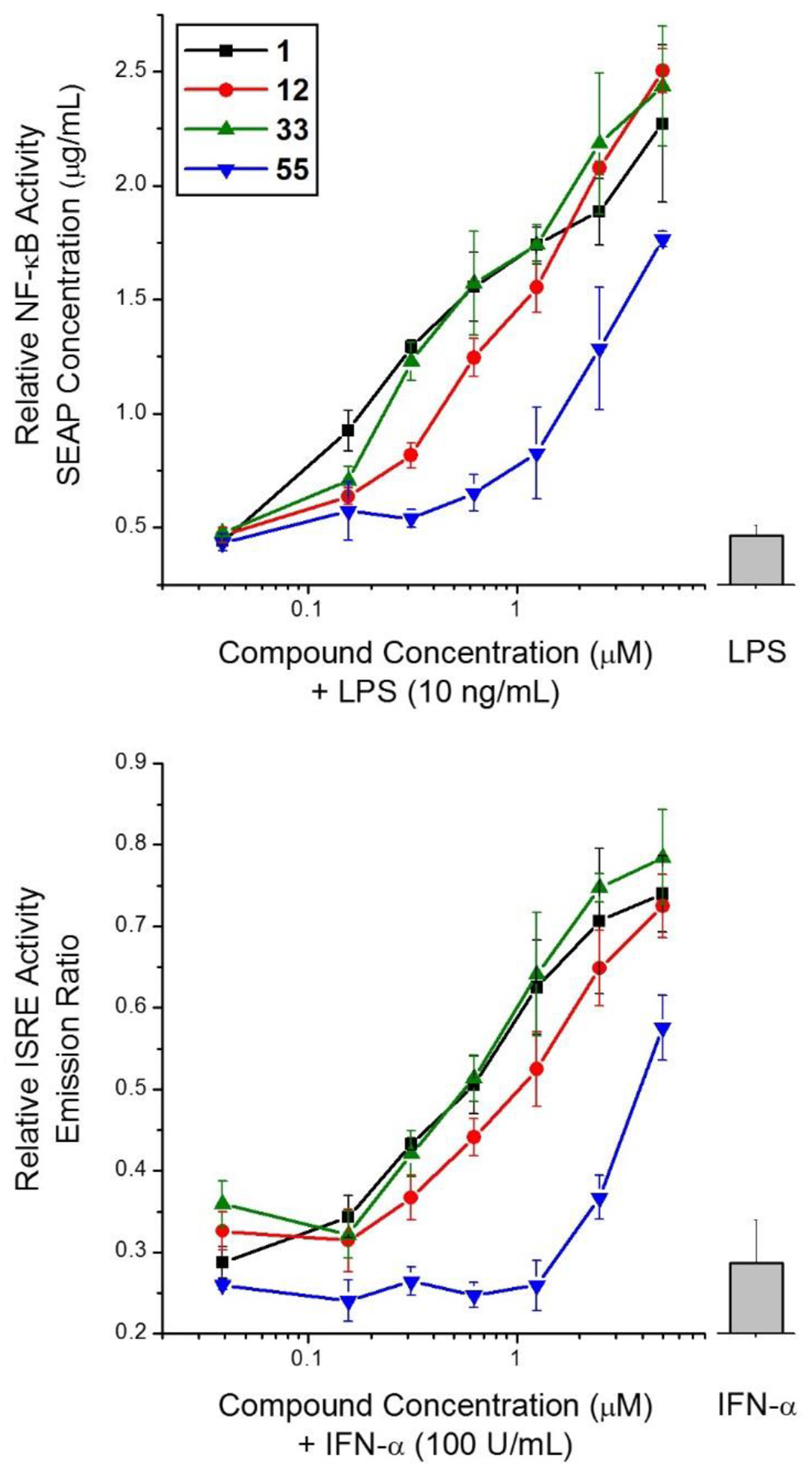
Dose response curves for selected active analogs. Compounds 1, 12, 33, and 55 were evaluated for enhancement of NF-κB and ISRE signaling at graded concentrations. Compound 33 was equipotent as compound 1 while chemically reactive handle (amino) bearing compound 55 retained activity even though slightly attenuated. The activity of stimulus alone is shown as grey bar. Data are presented as mean ± SEM. The NF-κB activity is measured as amount of SEAP induced, while ISRE activity is measured as emission ratio for the FRET based assay.
It was important to examine the adjuvanticity of the most potent compounds to verify if prolongation of immune stimulus by this chemotype leads to enhancement of in vivo antibody responses and if prolonged activation of the innate immune system could lead to systemic inflammation that may be harmful to the host.58, 59 In addition, it was also important to verify that the modifications on the scaffold that yielded potent compounds in vitro would retain the adjuvanticity in vivo as well. All the compounds administered to mice had low toxicity in the MTT assays. Since these vaccine co-adjuvants are designed to be administered locally (mostly intramuscularly) and show negligible toxicity (based on MTT data), we did not anticipate an excessive systemic inflammatory response. In the past, we utilized LPS as a widely recognized activator of the innate immune system and well characterized TLR-4 ligand to screen over 160,000 compounds for their ability to enhance APC activation.41, 42 However, to test these compounds for potency as co-adjuvants, we switched to the FDA approved TLR-4 adjuvant, MPLA for in vivo evaluation. Immunization experiments in mice (8 mice/group) were performed to evaluate the co-adjuvanticity of the lead compounds 1, 12 or 33 using ovalbumin (OVA) as a model antigen and MPLA as an adjuvant. Amine handle bearing compound 55 was not selected for immunization since it was designed for further derivatization as an intermediate to make probes as discussed below. Examination of OVA-specific IgG antibodies showed that co-immunization of MPLA with compounds 1 and 33 induced statistically significant increases in antigen-specific antibody titers when compared to mice immunized with MPLA alone (Fig. 5), without demonstrable systemic toxicity, as indicated by behavior change or weight loss. These results verified our approach that selected bis-aryl sulfonamide compounds that prolong immune stimulation could enhance the adjuvanticity of MPLA and that modified compounds that retained potency in vitro were equally potent in vivo as well.
Figure 5.
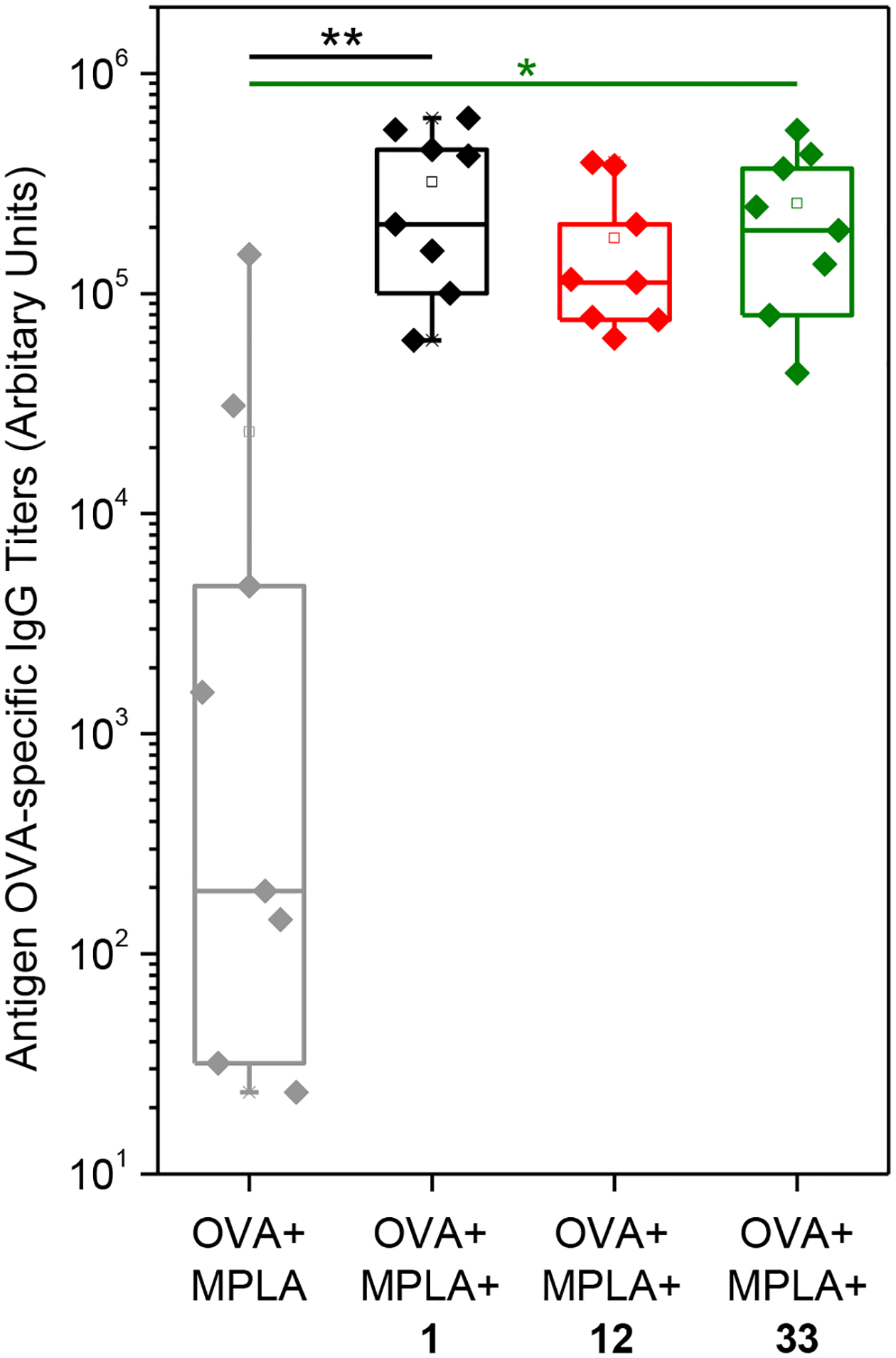
Co-adjuvanticity of potent bis-aryl sulfonamide compounds with MPLA. Mice (n=8 per group) were immunized on day 0 and day 21 with antigen (ovalbumin, 20 μg/animal), MPLA (10 μg/animal) and compound 1, 12 or 33 (100 nmol/animal). The immunized mice were bled on day 21 and OVA-specific IgG titers were measured using ELISA. Note, that the potent compounds 1 and 33 showed significant enhancement of antibody titers when co-adjuvanted with MPLA compared to MPLA alone. **p<0.01 and *p<0.05 compared to MPLA group using one-way ANOVA followed by Dunnett’s post hoc testing.
Now that we had confirmed the in vitro and in vivo potency of selected active compounds, we were further interested to utilize the SAR studies for designing affinity probes. The activity data guided us to utilize site C for the introduction of an identifiable tag by derivatizing compound 55. Although compound 55 was less potent than compound 1, the changes in the hydrophobic interaction after amine derivatization may improve the potency. Compound 55 was derivatized to obtain fluorescein labeled compound 56, rhodamine labeled compound 57 and biotin labeled compound 58 (Scheme 5). In primary screens, the biotin labeled compound 58 was equipotent to compound 1 and thus could serve as the affinity probe (Fig. 6, Table 4). The rhodamine analog 57 showed reduced activity compared to compound 1 in both the NF-κB and ISRE assays likely due to the presence of a fixed charge on the molecule similar to the amine bearing compound 55. In contrast, the fluorescein analog 56 was completely inactive in both the assays (Fig. 6, Table 4).
Scheme 5.
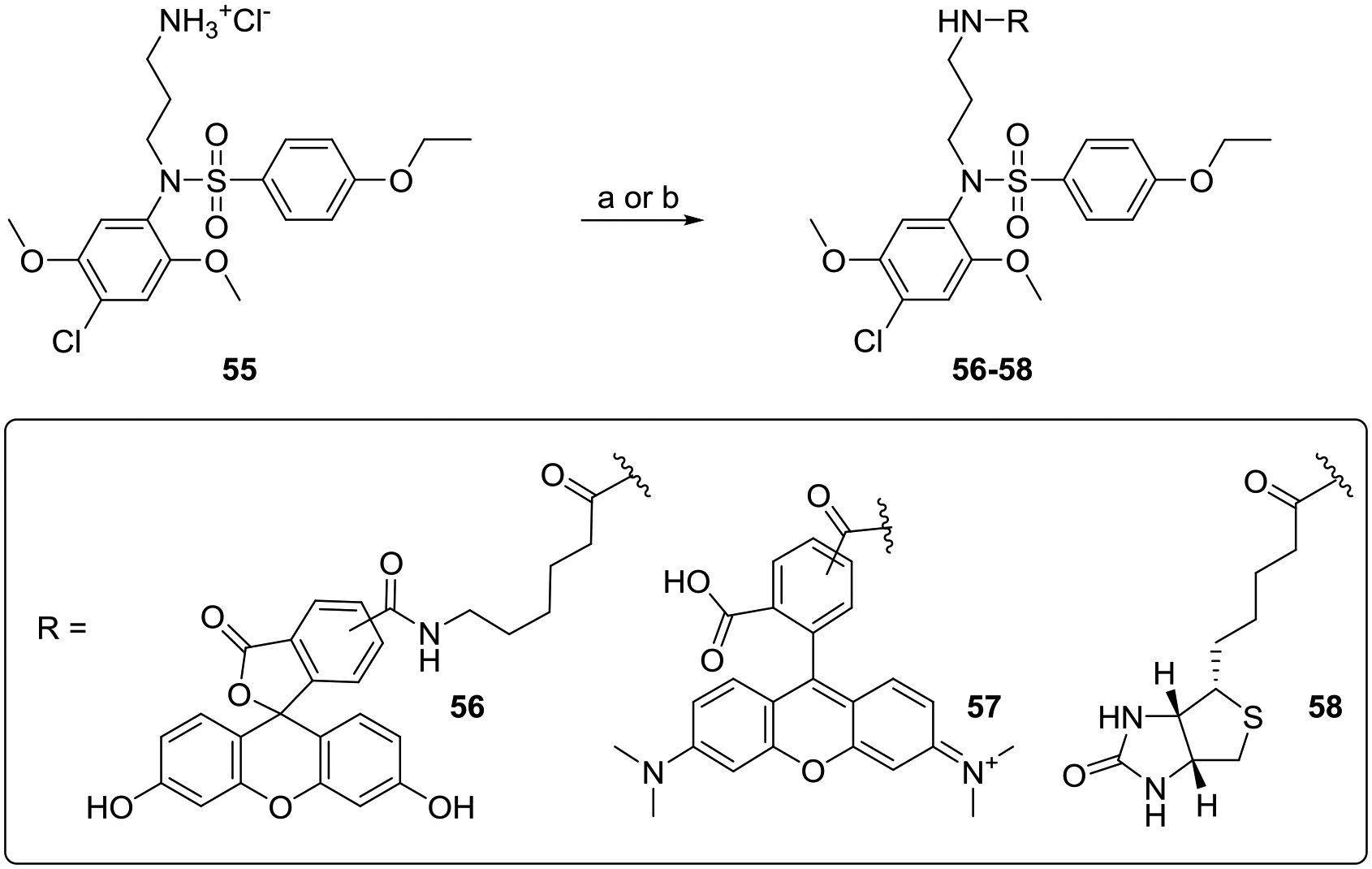
Syntheses of fluorescent and biotin labeled derivatives of compound 1.
Reagents and conditions: For compound 56: a) 5(6)-SFX SE, Et3N, DMF; For compound 57: b) NHS-Rhodamine, Et3N, DMF; For compound 58: b) biotin, HATU, Et3N, DMF.
Figure 6.
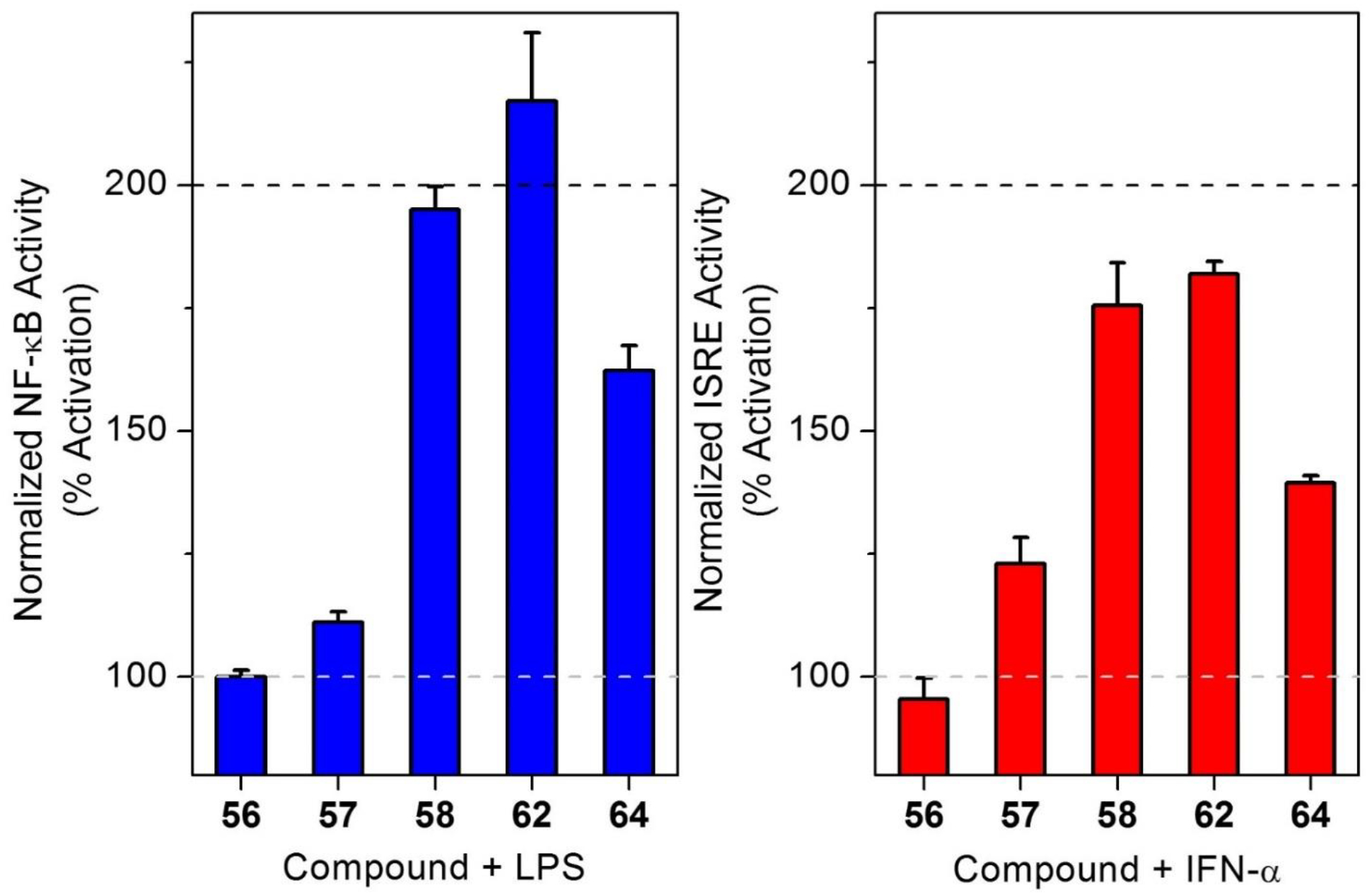
Bioactivities of different affinity probes for compound 1. NF-κB activity of affinity probes in presence of LPS is shown by blue bars (left) while ISRE activity in presence of IFN-α is shown by red bars (right). Reporter cells for NF-κB and ISRE activation were treated with LPS or IFN-α respectively with 5μM compounds 56, 57, 58, 62, and 64, in addition to compound 1 and respective vehicle (LPS of IFN-α) as controls. Data are presented as mean ± SEM after normalized to the activity of vehicle (100%, grey dotted lines) and compound 1 (200%, black dotted lines), respectively.
Table 4.
Bioactivity data for fluorescent, biotin and photoreactive analogs of compound 1.
| Compound | NF-κBa | ISREa | MTTa | |||
|---|---|---|---|---|---|---|
| % Activation | SEM | % Activation | SEM | % Viability | SEM | |
| 56 | 100 | 1.3 | 96 | 4.2 | 98.1 | 1.7 |
| 57 | 111 | 2.1 | 123 | 5.4 | 76.9 | 1.3 |
| 58 | 195 | 4.7 | 175 | 8.6 | 69.6 | 1.2 |
| 59 | 215 | 13.8 | 201 | 3.8 | 76.6 | 2.6 |
| 60 | 158 | 5.9 | 166 | 2.4 | 77.3 | 2.5 |
| 61 | 104 | 5.1 | 104 | 3.1 | 87.2 | 1.8 |
| 62 | 217 | 13.9 | 182 | 2.5 | 73.4 | 1.5 |
| 63 | 138 | 4.2 | 117 | 1.2 | 68.9 | 0.8 |
| 64 | 162 | 5.1 | 140 | 1.4 | 73.8 | 0.6 |
The % activation values in NF-ĸB and ISRE induction assays were two point normalized between compound 1 as 200% and LPS (10 ng/mL) for NF-κB or IFN-α (100 U/mL) for ISRE as 100%. The mean SEAP response in NF-ĸB assay for compound 1 + LPS and LPS alone was 3.44 ± 0.08 and 0.56 ± 0.06 μg/mL, respectively. The mean emission ratio in ISRE assay for compound 1 + IFN-α and IFN-α alone was 1.88 ± 0.04 and 0.69 ± 0.05 μg/mL, respectively. The % viability values for compounds in MTT assay were normalized to DMSO as 100%. The mean OD value at 405 nm for DMSO was 1.24 ± 0.03. All raw values used for normalization are represented as mean ± SEM.
Having validated specific site C modifications that tolerated the introduction of a trackable tag, we were interested to find a position where a photoreactive group such as aryl azide could be introduced to make photoaffinity probes. This prompted us to derivatize compounds 10 and 25, even though these were inactive but surmising that a change in the hydrogen bonding properties may have an opposite effect. The aromatic amine on position 4 at site A of compound 10 was converted to aryl azide using diazotization reaction to obtain compound 59 (Scheme 6). In parallel, the 3-bromo substitution at site B of compound 25 was reacted with sodium azide using copper catalyzed reaction. However, the major product of this reaction was aromatic amine analog 60, which was further converted to azide using the earlier described diazotization chemistry to obtain compound 61 (Scheme 6). The photoreactive aryl azide bearing compounds 59 and 61 and the aromatic amine analog 60 were then evaluated in the primary screens. While compound 61 was inactive just like its precursor bromo analog 25, the reversal of hydrogen bonding capacity in compound 60 led to resurgence of activity in both the assays possibly due to hydrophilic interaction with the aromatic amine (Fig. 6, Table 4). In contrast, the reversal of hydrogen bonding capacity of compound 10 led us to a potent aryl azide bearing analog 59 which was then utilized for making photoaffinity probes (Fig. 6, Table 4).
Scheme 6.
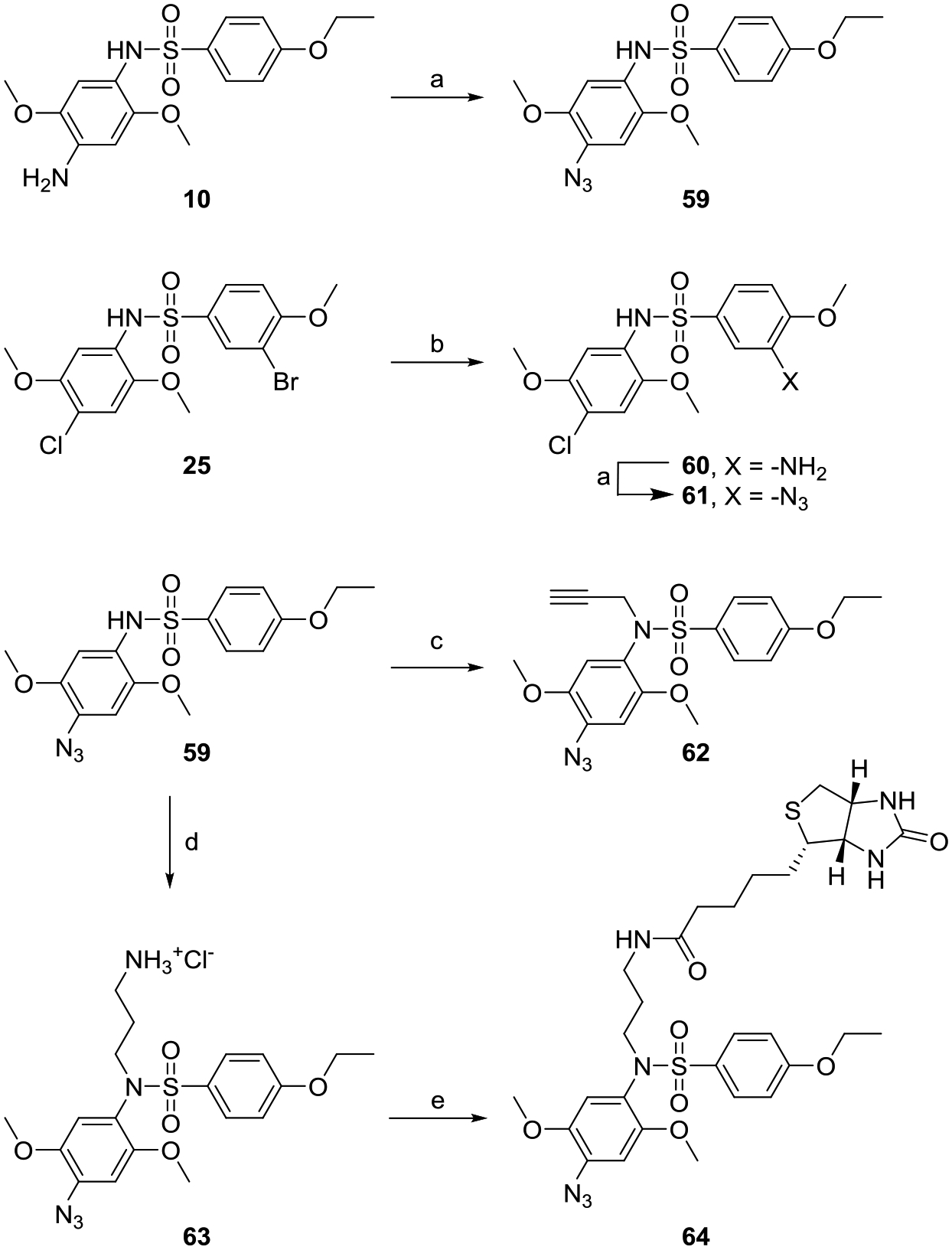
Syntheses of photoreactive analogs of compound 1.
Reagents and conditions: a) tert-butyl nitrite, trimethylsilyl azide, CH3CN, 40 °C b) Cul, NaN3, N,N′-dimethylethane-1,2-diamine, sodium ascorbate, EtOH, H2O c) propargyl bromide, K2CO3, DMF d) i. tert-butyl (3-bromopropyl)carbamate, K2CO3, DMF ii. 4N HCl/dioxane e) biotin, HATU, Et3N, DMF.
Using the methods utilized earlier, compound 59 was derivatized to obtain an alkyne analog 62, and a biotin analog 64 was obtained via an aliphatic amine derivative 63 (Scheme 6). Evaluation of these compounds in our primary screens showed that the alkyne probe 62 was very potent while the biotin probe 64 showed relatively weak activity in both the NF-κB and ISRE assays (Fig. 5, Table 4). Also, all the affinity probes had viability in the same range as the potent compounds in this series making them ideal candidates for future studies.
Our systematic SAR studies on bis-aryl sulfonamides that sustain NF-κB and ISRE activation have led to the identification of not only rhodamine labeled affinity fluorescent probe 57 and biotin-tagged affinity probe 58, but also alkyne and biotin labeled photoaffinity probes 62 and 64, respectively. These affinity probes will be utilized in concert for target identification and cell trafficking experiments.
Conclusions
Compound 1 was identified from HTS campaigns, that screened for agents capable of prolonging immune signaling, and was shown to be a potent co-adjuvant with MPLA in vivo. Here, we presented systematic SAR studies consisting of design, syntheses and evaluation of analogs of compound 1 to identify sites on the scaffold that can tolerate modification while still retaining dual NF-κB and ISRE enhancing activities in order to obtain affinity and photoaffinity probes. SAR studies pointed to key substitutions at site B and site C that retain potency in vitro and in vivo, while site A allowed the introduction of photoreactive aryl azide functionality. In addition, observed SAR trends at site C allowed the introduction of trackable tags such as rhodamine or biotin. This led to syntheses of several affinity probes which will be utilized to determine the mechanism of action and receptor target for this bis-aryl sulfonamide series of compounds that sustain NF-κB and ISRE activation.
Experimental Section:
Chemistry
Materials.
Reagents were purchased as at least reagent grade from commercial vendors unless otherwise specified and used without further purification. Solvents were purchased from Fischer Scientific (Pittsburgh, PA) and were either used as purchased or redistilled with an appropriate drying agent. All the reagents 2a-g and 3g-o were purchased from commercially available vendors while reagents 3a-f were synthesized from commercially available reagents as shown in Supporting Information. Compounds used for structure-activity studies were synthesized according to methods described below and all the compounds were identified to be least 95% pure using HPLC.
Instrumentation.
Analytical TLC was performed using precoated TLC silica gel 60 F254 aluminum sheets purchased from EMD (Gibbstown, NJ) and visualized using UV light. Flash chromatography was carried out using with a Biotage Isolera One (Charlotte, NC) system using the specified solvent. Microwave reaction was performed using Biotage Initiator+ (Charlotte, NC). Reaction monitoring and purity analysis were done using an Agilent 1260 LC/6420 Triple Quad mass spectrometer (Santa Clara, CA) with Onyx Monolithic C18 (Phenomenex, Torrance, CA) column. Purity of all final compounds was above 95% (also see LC-MS spectra in Supporting Information for all final compounds). All final compounds were analyzed by high resolution MS (HRMS) using an Agilent 6230 ESI-TOFMS (Santa Clara, CA). 1H and 13C NMR spectra were obtained on a Varian 500 with XSens probe (Varian, Inc., Palo Alto, CA). The chemical shifts are expressed in parts per million (ppm) using suitable deuterated NMR solvents.
General procedure A for the syntheses of select site A and site B modified compounds.
To a solution of a substituted phenyl sulfonyl chloride (reagent 3, 1 eq.) in anhydrous CH2Cl2 were added, triethylamine (2 eq.) and a solution of substituted aniline (reagent 2, 2 eq.) in CH2Cl2. The reaction mixture was stirred at room temperature overnight and then poured into water and acidified with 3N HCl followed by extraction with EtOAc. The EtOAc fraction was then dried over MgSO4, and solvent was removed under vacuum. The resultant residue was dissolved in MeOH and THF, followed by the addition of lithium hydroxide monohydrate (15 eq.) in water and stirred at room temperature until bis-sulfonamide side product is converted to the desired product. The solvent was then removed, dissolved in EtOAc, washed with water and brine, dried under vacuum to obtain the residue which was purified by column chromatography to obtain the final product.
Compound 1 and site A modified compounds 4–9 were synthesized using general procedure A described above.
N-(4-chloro-2,5-dimethoxyphenyl)-4-ethoxybenzenesulfonamide (1).
Compound 1 was synthesized using 4-chloro-2,5-dimethoxyaniline (2a, 1.7 g, 5.3 mmol) and 4-ethoxybenzenesulfonyl chloride (3a, 1g, 4.5 mmol) after recrystallization in EtOH as pink crystals (1.2 g, yield = 71%). 1H NMR (500 MHz, CHLOROFORM-d) δ 7.66 (d, J = 8.80 Hz, 2H), 7.24 (s, 1H), 6.91 (s, 1H), 6.86 (d, J = 8.80 Hz, 2H), 6.77 (s, 1H), 4.04 (q, J = 6.93 Hz, 2H), 3.87 (s, 3H), 3.60 (s, 3H), 1.42 (t, J = 6.97 Hz, 3H). 13C NMR (126 MHz, CHLOROFORM-d) δ 162.6, 149.2, 143.6, 130.0, 129.4, 125.2, 117.8, 114.4, 113.1, 106.3, 64.0, 56.8, 56.4, 14.6. HRMS for C16H17ClNO5S [M – H−] calculated 370.0521, found 370.0523.
N-(4-chloro-3-methoxyphenyl)-4-ethoxybenzenesulfonamide (4).
Compound 4 was synthesized using 4-chloro-3-methoxyaniline (2b, 142.84 mg, 0.92 mmol) and 4-ethoxybenzenesulfonyl chloride (3a, 100 mg, 0.46 mmol) as off-white solid (83 mg, yield = 54%). 1H NMR (500 MHz, CHLOROFORM-d) δ 7.70 (d, J = 8.80 Hz, 2H), 7.17 (d, J = 8.56 Hz, 1H), 7.01 (br. s., 1H), 6.89 (d, J = 9.05 Hz, 2H), 6.80 (d, J = 2.20 Hz, 1H), 6.51 (dd, J = 2.20, 8.56 Hz, 1H), 4.05 (q, J = 7.09 Hz, 2H), 3.83 (s, 3H), 1.42 (t, J = 6.97 Hz, 3H). 13C NMR (126 MHz, CHLOROFORM-d) δ 162.7, 155.3, 136.3, 130.4, 129.6, 129.4, 119.0, 114.6, 113.9, 105.8, 64.0, 56.2, 14.6. HRMS for C15H15ClNO4S [M − H]− calculated 340.0416, found 340.0416.
N-(4-chloro-2-methoxyphenyl)-4-ethoxybenzenesulfonamide (5).
Compound 5 was synthesized using 4-chloro-2-methoxyaniline (2c, 142.84 mg, 0.92 mmol) and 4-ethoxybenzenesulfonyl chloride (3a, 100 mg, 0.46 mmol) as tan solid (100 mg, yield = 64%). 1H NMR (500 MHz, CHLOROFORM-d) δ 7.66 (d, J = 8.80 Hz, 2H), 7.45 (d, J = 8.56 Hz, 1H), 6.83 – 6.91 (m, 2H), 6.85 (d, J = 8.80 Hz, 2H), 6.72 (d, J = 1.96 Hz, 1H), 4.04 (q, J = 7.09 Hz, 2H), 3.65 (s, 3H), 1.41 (t, J = 6.97 Hz, 3H). 13C NMR (126 MHz, CHLOROFORM-d) δ 162.5, 150.0, 130.4, 130.2, 129.4, 124.8, 121.9, 121.0, 114.3, 111.3, 63.9, 55.9, 14.6. HRMS for C15H16ClNO4SNa [M + Na+] calculated 364.0381, found 364.0382.
N-(2,5-dimethoxyphenyl)-4-ethoxybenzenesulfonamide (6).
Compound 6 was synthesized using 2,5-dimethoxyaniline (2d, 138.8 mg, 0.92 mmol) and 4-ethoxybenzenesulfonyl chloride (3a, 100 mg, 0.46 mmol) as off-white solid (117 mg, yield = 76%). 1H NMR (500 MHz, CHLOROFORM-d) δ 7.70 (d, J = 8.80 Hz, 2H), 7.14 (d, J = 2.93 Hz, 1H), 7.01 (s, 1H), 6.85 (d, J = 8.80 Hz, 2H), 6.65 (d, J = 8.80 Hz, 1H), 6.53 (dd, J = 2.93, 9.05 Hz, 1H), 4.03 (q, J = 6.85 Hz, 2H), 3.75 (s, 3H), 3.62 (s, 3H), 1.40 (t, J = 7.09 Hz, 3H). 13C NMR (126 MHz, CHLOROFORM-d) δ 162.4, 153.8, 143.4, 130.4, 129.4, 126.8, 114.3, 111.4, 109.5, 106.8, 63.9, 56.2, 55.8, 14.6. HRMS for C16H19NO5SNa [M + Na+] calculated 360.0876, found 360.0877.
N-(4-bromo-2,5-dimethoxyphenyl)-4-ethoxybenzenesulfonamide (7).
Compound 7 was synthesized using 4-bromo-2,5-dimethoxyaniline (2e, 105.2 mg, 0.45 mmol) and 4-ethoxybenzenesulfonyl chloride (3a, 50 mg, 0.23 mmol) as purple solid (59 mg, yield = 62%). 1H NMR (500 MHz, HLOROFORM-d) δ 7.67 (d, J = 8.80 Hz, 2H), 7.21 (s, 1H), 6.93 (s, 1H), 6.92 (s, 1H), 6.85 (d, J = 9.05 Hz, 2H), 4.04 (q, J = 7.09 Hz, 2H), 3.86 (s, 3H), 3.61 (s, 3H), 1.41 (t, J = 6.97 Hz, 3H). 13C NMR (126 MHz, CHLOROFORM-d) δ 162.6, 150.2, 143.7, 130.0, 129.4, 126.0, 115.8, 114.4, 106.2, 105.8, 64.0, 56.9, 56.4, 14.6. HRMS for C16H18BrNO5SNa [M + Na+] calculated 437.9981, found 437.9979.
N-(4-chloro-3,5-dimethoxyphenyl)-4-ethoxybenzenesulfonamide (8).
Compound 8 was synthesized using 4-chloro-3,5-dimethoxyaniline (2f, 50 mg, 0.27 mmol) and 4-ethoxybenzenesulfonyl chloride (3a, 29 mg, 0.13 mmol) as white solid (30 mg, yield = 61%). 1H NMR (500 MHz, CHLOROFORM-d) δ 7.71 (d, J = 8.80 Hz, 2H), 6.89 (d, J = 9.05 Hz, 2H), 6.76 (br. s., 1H), 6.35 (s, 2H), 4.06 (q, J = 7.01 Hz, 2H), 3.80 (s, 6H), 1.43 (t, J = 6.97 Hz, 3H). 13C NMR (126 MHz, CHLOROFORM-d) δ 162.8, 156.3, 136.1, 129.7, 129.5, 114.6, 107.2, 98.2, 64.0, 56.4, 14.6. HRMS for C16H17ClNO5S [M − H]− calculated 370.0521, found 370.0519.
N-(2,5-dimethoxy-4-nitrophenyl)-4-ethoxybenzenesulfonamide (9).
Compound 9 was synthesized using 3,5-dimethoxy-4-nitroaniline (2g, 50 mg, 0.27 mmol) and 4-ethoxybenzenesulfonyl chloride (3a, 29 mg, 0.13 mmol) as yellow solid (30 mg, yield = 61%). 1H NMR (500 MHz, CHLOROFORM-d) δ 7.69 – 7.86 (m, J = 8.80 Hz, 2H), 7.44 (s, 1H), 7.40 (s, 1H), 7.31 (s, 1H), 6.90 – 6.95 (m, 2H), 4.07 (q, J = 6.93 Hz, 2H), 3.93 (s, 3H), 3.82 (s, 3H), 1.43 (t, J = 6.97 Hz, 3H). 13C NMR (126 MHz, CHLOROFORM-d) δ 163.1, 149.4, 140.9, 133.1, 132.8, 129.5, 129.4, 114.8, 108.3, 103.2, 64.1, 57.0, 56.5, 14.5. HRMS for C16H19N2O7S [M + H+] calculated 383.0907, found 383.091.
N-(4-amino-2,5-dimethoxyphenyl)-4-ethoxybenzenesulfonamide (10).
To a solution of compound 9 (128 mg, 0.33 mmol) in EtOAc were added, a catalytic amount of palladium on carbon and some sodium sulfate. The reaction was subjected to Parr hydrogenation apparatus using hydrogen gas at 50 psi pressure for 6 hours. The solvent was then removed, and the residue was purified using silica gel column chromatography (5% MeOH/CH2Cl2) to obtain 84 mg of compound 10 as tan solid (yield = 72%). 1H NMR (500 MHz, METHANOL-d4) δ 7.55 (d, J = 8.80 Hz, 2H), 6.91 (d, J = 8.80 Hz, 2H), 6.87 (s, 1H), 6.26 (s, 1H), 4.06 (q, J = 7.01 Hz, 2H), 3.78 (s, 3H), 3.33 (s, 3H), 1.38 (t, J = 6.97 Hz, 3H). 13C NMR (126 MHz, METHANOL-d4) δ 162.2, 147.8, 140.9, 135.9, 131.2, 129.2, 114.4, 113.5, 110.3, 99.0, 63.6, 55.2, 54.7, 13.5. HRMS for C16H20N2O5SNa [M + Na+] calculated 375.0985, found 375.0988.
Site B modified compounds 11–17, 19, 22–27 were synthesized using general procedure A described above.
N-(4-chloro-2,5-dimethoxyphenyl)-4-hydroxybenzenesulfonamide (11).
Compound 11 was synthesized using 4-chloro-2,5-dimethoxyaniline (2a, 383 mg, 2.03 mmol) and 4-hydroxybenzenesulfonyl chloride (3b, 195 mg, 1.02 mmol) as dark brown solid (237 mg, yield = 68%). 1H NMR (500 MHz, DMSO-d6) δ 10.44 (s, 1H), 9.39 (s, 1H), 7.54 (d, J = 8.80 Hz, 2H), 7.02 (s, 1H), 6.97 (s, 1H), 6.83 (d, J = 8.80 Hz, 2H), 3.66 – 3.78 (m, 3H), 3.48 (s, 3H). 13C NMR (126 MHz, DMSO-d6) δ 161.2, 148.1, 146.0, 129.9, 129.2, 125.5, 117.3, 115.3, 113.9, 109.2, 56.5, 56.4. HRMS for C14H13ClNO5S [M − H]− calculated 342.0208, found 342.0205.
N-(4-chloro-2,5-dimethoxyphenyl)-4-methoxybenzenesulfonamide (12).
Compound 11 was synthesized using 4-chloro-2,5-dimethoxyaniline (2a, 450 mg, 2.40 mmol) and 4-methoxybenzenesulfonyl chloride (3c, 248 mg, 1.20 mmol) as light brown solid (189 mg, yield = 44%). 1H NMR (500 MHz, CHLOROFORM-d) δ 7.68 (d, J = 8.80 Hz, 2H), 7.24 (s, 1H), 6.92 (s, 1H), 6.88 (d, J = 9.05 Hz, 2H), 6.77 (s, 1H), 3.87 (s, 3H), 3.83 (s, 3H), 3.61 (s, 3H). 13C NMR (126 MHz, CHLOROFORM-d) δ 163.1, 149.2, 143.6, 130.3, 129.4, 125.2, 117.9, 114.0, 113.1, 106.3, 56.8, 56.4, 55.6. HRMS for C15H16ClNO5SNa [M + Na+] calculated 380.033, found 380.0326.
N-(4-chloro-2,5-dimethoxyphenyl)-4-propoxybenzenesulfonamide (13).
Compound 13 was synthesized using 4-chloro-2,5-dimethoxyaniline (2a, 106 mg, 0.57 mmol) and 4-propoxybenzenesulfonyl chloride (3d, 66 mg, 0.28 mmol) as off-white solid (65 mg, yield = 59%). 1H NMR (500 MHz, CHLOROFORM-d) δ 7.66 (d, J = 8.80 Hz, 2H), 7.23 (s, 1H), 6.92 (s, 1H), 6.86 (d, J = 9.05 Hz, 2H), 6.76 (s, 1H), 3.92 (t, J = 6.60 Hz, 2H), 3.87 (s, 3H), 3.60 (s, 3H), 1.76 – 1.85 (m, 2H), 1.02 (t, J = 7.46 Hz, 3H). 13C NMR (126 MHz, CHLOROFORM-d) δ 162.8, 149.2, 143.5, 130.0, 129.3, 125.2, 117.8, 114.4, 113.1, 106.2, 69.9, 56.8, 56.4, 22.3, 10.4. HRMS for C17H20ClNO5SNa [M + Na+] calculated 408.0643, found 408.0641.
4-Butoxy-N-(4-chloro-2,5-dimethoxyphenyl)benzenesulfonamide (14).
Compound 14 was synthesized using 4-chloro-2,5-dimethoxyaniline (2a, 191 mg, 1.02 mmol) and 4-butoxybenzenesulfonyl chloride (3e, 127 mg, 0.51 mmol) as off-white solid (95 mg, yield = 47%). 1H NMR (500 MHz, CHLOROFORM-d) δ 7.66 (d, J = 8.80 Hz, 2H), 7.23 (s, 1H), 6.91 (s, 1H), 6.85 (d, J = 8.80 Hz, 2H), 6.76 (s, 1H), 3.96 (t, J = 6.48 Hz, 2H), 3.87 (s, 3H), 3.60 (s, 3H), 1.76 (quin, J = 7.20 Hz, 2H), 1.47 (sxt, J = 7.40 Hz, 2H), 0.97 (t, J = 7.46 Hz, 3H). 13C NMR (126 MHz, CHLOROFORM-d) δ 162.8, 149.2, 143.5, 130.0, 129.3, 125.3, 117.8, 114.4, 113.1, 106.2, 68.1, 56.8, 56.4, 31.0, 19.1, 13.8. HRMS for C18H22ClNO5SNa [M + Na+] calculated 422.0799, found 422.0802.
N-(4-chloro-2,5-dimethoxyphenyl)-4-(prop-2-yn-1-yloxy)benzenesulfonamide (15).
Compound 15 was synthesized using 4-chloro-2,5-dimethoxyaniline (2a, 66 mg, 0.35 mmol) and 4-(prop-2-yn-1-yloxy)benzenesulfonyl chloride (3f, 40.7 mg, 0.18 mmol) as off-white solid (24 mg, yield = 32%). 1H NMR (500 MHz, CHLOROFORM-d) δ 7.69 (d, J = 9.05 Hz, 2H), 7.23 (s, 1H), 6.96 (d, J = 8.80 Hz, 2H), 6.90 (s, 1H), 6.76 (s, 1H), 4.72 (d, J = 2.20 Hz, 2H), 3.87 (s, 3H), 3.59 (s, 3H), 2.55 (t, J = 2.45 Hz, 1H). 13C NMR (126 MHz, DMSO-d6) δ 160.2, 148.1, 146.4, 132.5, 128.9, 125.1, 117.7, 114.9, 113.9, 109.9, 78.8, 78.5, 56.4, 55.8. HRMS for C17H15ClNO5S [M − H]− calculated 380.0365, found 380.0365.
N-(4-chloro-2,5-dimethoxyphenyl)-4-propylbenzenesulfonamide (16).
Compound 11 was synthesized using 4-chloro-2,5-dimethoxyaniline (2a, 210 mg, 1.12 mmol) and 4-propylbenzenesulfonyl chloride (3g, 100 μL, 0.56 mmol) as white solid (121 mg, yield = 59%). 1H NMR (500 MHz, CHLOROFORM-d) δ 7.64 (d, J = 8.31 Hz, 2H), 7.23 (s, 1H), 7.21 (d, J = 8.31 Hz, 2H), 6.92 (s, 1H), 6.76 (s, 1H), 3.87 (s, 3H), 3.53 – 3.58 (m, 3H), 2.60 (t, J = 7.70 Hz, 2H), 1.61 (sxt, J = 7.60 Hz, 2H), 0.91 (t, J = 7.34 Hz, 3H). 13C NMR (126 MHz, CHLOROFORM-d) δ 149.2, 148.6, 143.6, 136.0, 128.9, 127.2, 125.1, 117.9, 113.1, 106.4, 56.8, 56.3, 37.8, 24.1, 13.7. HRMS for C17H20ClNO4SNa [M + Na+] calculated 392.0694, found 392.0695.
N-(4-chloro-2,5-dimethoxyphenyl)-4-nitrobenzenesulfonamide (17).
Compound 17 was synthesized using 4-chloro-2,5-dimethoxyaniline (2a, 847 mg, 4.51 mmol) and 4-nitrobenzenesulfonyl chloride (3h, 500 mg, 2.25 mmol) as yellow solid (182 mg, yield = 22%). 1H NMR (500 MHz, DMSO-d6) δ 10.15 (s, 1H), 8.37 (d, J = 8.80 Hz, 2H), 7.93 (d, J = 8.80 Hz, 2H), 7.04 (s, 1H), 7.01 (s, 1H), 3.76 (s, 3H), 3.35 (s, 3H). 13C NMR (126 MHz, CHLOROFORM-d) δ 150.2, 149.4, 144.5, 144.0, 128.4, 124.1, 123.5, 119.6, 113.2, 107.4, 56.9, 56.3. HRMS for C14H12ClN2O6S [M − H]− calculated 371.011, found 371.0104.
4-Amino-N-(4-chloro-2,5-dimethoxyphenyl)benzenesulfonamide (18).
To a solution of compound 17 (150 mg, 0.4 mmol) in EtOAc were added, a catalytic amount of palladium on carbon and some sodium sulfate. The reaction was subjected to hydrogenation on Parr hydrogenation apparatus using hydrogen gas at 50 psi pressure for 6 hours. The solvent was then removed, and the residue was purified using silica gel column chromatography (9% MeOH/CH2Cl2) to obtain 79 mg of compound 18 as tan solid (yield = 58%). 1H NMR (500 MHz, DMSO-d6) δ 9.07 (s, 1H), 7.31 – 7.41 (m, J = 8.56 Hz, 2H), 6.97 (s, 1H), 7.01 (s, 1H), 6.47 – 6.56 (m, J = 8.80 Hz, 2H), 5.98 (s, 2H), 3.70 (s, 3H), 3.54 (s, 3H). 13C NMR (126 MHz, DMSO-d6) δ 152.9, 148.1, 145.5, 128.9, 126.1, 124.6, 116.4, 113.9, 112.4, 108.0, 56.6, 56.4. HRMS for C14H15ClN2O4SNa [M + Na+] calculated 365.0333, found 365.0335.
N-(4-chloro-2,5-dimethoxyphenyl)-4-cyanobenzenesulfonamide (19).
Compound 19 was synthesized using 4-chloro-2,5-dimethoxyaniline (2a, 372 mg, 1.98 mmol) and 4-cyanobenzenesulfonyl chloride (3i, 100 mg, 0.50 mmol) as white solid (40 mg, yield = 23%). 1H NMR (500 MHz, CHLOROFORM-d) δ 7.83 (d, J = 8.56 Hz, 2H), 7.73 (d, J = 8.31 Hz, 2H), 7.25 (s, 1H), 6.92 (s, 1H), 6.79 (s, 1H), 3.90 (s, 3H), 3.57 (s, 3H). 13C NMR (126 MHz, CHLOROFORM-d) δ 149.4, 144.0, 142.9, 132.6, 127.8, 123.5, 119.5, 117.1, 116.7, 113.1, 107.4, 56.8, 56.2. HRMS for C15H12ClN2O4S [M − H]− calculated 351.0212, found 351.021.
tert-butyl (4-(N-(4-chloro-2,5-dimethoxyphenyl)sulfamoyl)benzyl)carbamate (20).
To a crude solution of compound 19 (280 mg, 0.75 mmol) in methanol was added, a catalytic amount of palladium on carbon and di-tert-butyl dicarbonate (326 mg, 1.5 mmol). The reaction was subjected to hydrogenation on Parr hydrogenation apparatus using hydrogen gas at 50psi pressure overnight. The solvent was then removed, and the residue was purified using silica gel column chromatography (9% MeOH/CH2Cl2) to obtain 65 mg of compound 20 as white solid (yield = 20%). 1H NMR (500 MHz, CHLOROFORM-d) δ 7.70 (d, J = 8.07 Hz, 2H), 7.33 (d, J = 8.07 Hz, 2H), 7.25 (s, 1H), 6.93 (br. s., 1H), 6.76 (s, 1H), 4.94 (br. s., 1H), 4.34 (d, J = 5.87 Hz, 2H), 3.87 (s, 3H), 3.57 (s, 3H), 1.46 (s, 9H). 13C NMR (126 MHz, CHLOROFORM-d) δ 155.8, 149.3, 144.9, 143.7, 137.5, 127.5, 127.5, 124.8, 118.2, 113.1, 106.5, 80.0, 56.8, 56.3, 44.0, 28.3 HRMS for C20H25ClN2O6SNa [M + Na+] calculated 479.1014, found 479.1018.
4-(aminomethyl)-N-(4-chloro-2,5-dimethoxyphenyl)benzenesulfonamide (21).
Compound 20 (11 mg, mmol) was stirred in a solution of 4N HCl in dioxane for 1h. The solvent was then removed to obtain compound 21 in quantitative yield as hydrochloride salt (grey solid). 1H NMR (500 MHz, METHANOL-d4) δ 7.71 – 7.88 (m, J = 8.31 Hz, 2H), 7.51 – 7.67 (m, J = 8.31 Hz, 2H), 7.23 (s, 1H), 6.87 (s, 1H), 4.17 (s, 2H), 3.82 (s, 3H), 3.51 (s, 3H). 13C NMR (126 MHz, METHANOL-d4) δ 150.5, 147.2, 142.1, 139.5, 130.5, 129.3, 126.3, 120.3, 114.7, 110.4, 57.3, 57.0, 43.7. HRMS for C15H18ClN2O4S [M + H+] calculated 357.067, found 357.0674.
N-(4-chloro-2,5-dimethoxyphenyl)-4-phenoxybenzenesulfonamide (22).
Compound 22 was synthesized using 4-chloro-2,5-dimethoxyaniline (2a, 140 mg, 0.74 mmol) and 4-phenoxybenzenesulfonyl chloride (3j, 100 mg, 0.37 mmol) as light yellow solid (51 mg, yield = 33%). 1H NMR (500 MHz, CHLOROFORM-d) δ 7.69 (d, J = 8.80 Hz, 1H), 7.41 (t, J = 7.95 Hz, 1H), 7.25 (s, 2H), 7.23 (t, J = 7.60 Hz, 2H), 7.03 (d, J = 7.58 Hz, 2H), 6.95 (s, 1H), 6.94 (d, J = 8.80 Hz, 2H), 6.80 (s, 1H), 3.87 (s, 3H), 3.64 (s, 3H). 13C NMR (126 MHz, CHLOROFORM-d) δ 161.9, 154.8, 149.3, 143.6, 132.2, 130.2, 129.4, 125.1, 125.0, 120.3, 118.0, 117.2, 113.1, 106.3, 56.8, 56.4. HRMS for C20H18ClNO5SNa [M + Na+] calculated 442.0486, found 442.0489.
N-(4-chloro-2,5-dimethoxyphenyl)-3-methoxybenzenesulfonamide (23).
Compound 23 was synthesized using 4-chloro-2,5-dimethoxyaniline (2a, 182 mg, 0.97 mmol) and 3-methoxybenzenesulfonyl chloride (3k, 100 mg, 0.48 mmol) as brown solid (189 mg, yield = 54%). 1H NMR (500 MHz, CHLOROFORM-d) δ 7.29 – 7.35 (m, 2H), 7.21 – 7.26 (m, 2H), 7.00 – 7.09 (m, 1H), 6.94 (s, 1H), 6.78 (s, 1H), 3.87 (s, 3H), 3.77 (s, 3H), 3.58 (s, 3H). 13C NMR (126 MHz, CHLOROFORM-d) δ 159.6, 149.3, 143.7, 139.8, 129.9, 124.9, 119.5, 119.3, 118.2, 113.1, 111.7, 106.5, 56.8, 56.4, 55.6. HRMS for C15H16ClNO5SNa [M + Na+] calculated 380.033, found 380.0329.
N-(4-chloro-2,5-dimethoxyphenyl)-2-methoxybenzenesulfonamide (24).
Compound 24 was synthesized using 4-chloro-2,5-dimethoxyaniline (2a, 182 mg, 0.97 mmol) and 2-methoxybenzenesulfonyl chloride (3l, 100 mg, 0.48 mmol) as tan solid (121 mg, yield = 70%). 1H NMR (500 MHz, CHLOROFORM-d) δ 7.87 (td, J = 1.70, 7.70 Hz, 1H), 7.58 (s, 1H), 7.49 (tt, J = 1.50, 7.70 Hz, 1H), 7.22 (s, 1H), 6.99 (t, J = 7.70 Hz, 1H), 6.95 (d, J = 8.31 Hz, 1H), 6.78 (s, 1H), 3.95 (s, 3H), 3.80 (s, 3H), 3.73 (s, 3H). 13C NMR (126 MHz, CHLOROFORM-d) δ 156.4, 149.2, 142.9, 135.1, 130.9, 126.1, 125.7, 120.3, 116.8, 113.0, 111.8, 104.9, 56.7, 56.6, 56.1. HRMS for C15H16ClNO5SNa [M + Na+] calculated 380.033, found 380.0329.
3-Bromo-N-(4-chloro-2,5-dimethoxyphenyl)-4-ethoxybenzenesulfonamide (25).
Compound 25 was synthesized using 4-chloro-2,5-dimethoxyaniline (2a, 180 mg, 0.70 mmol) and 3-bromo-4-methoxybenzenesulfonyl chloride (3m, 100 mg, 0.35 mmol) as brown solid (68 mg, yield = 58%). 1H NMR (500 MHz, CHLOROFORM-d) δ 7.99 (d, J = 2.20 Hz, 1H), 7.64 (dd, J = 1.96, 8.56 Hz, 1H), 7.22 (s, 1H), 6.93 (s, 1H), 6.85 (d, J = 8.56 Hz, 1H), 6.79 (s, 1H), 3.93 (s, 3H), 3.89 (s, 3H), 3.65 (s, 3H). 13C NMR (126 MHz, CHLOROFORM-d) δ 159.4, 149.3, 143.7, 132.4, 131.4, 128.5, 124.6, 118.4, 113.1, 112.0, 111.0, 106.6, 56.8, 56.6, 56.4. HRMS for C15H14BrClNO5S [M − H]− calculated 433.947, found 433.9469.
Methyl 4-(N-(4-chloro-2,5-dimethoxyphenyl)sulfamoyl)benzoate (26).
Compound 26 was synthesized using 4-chloro-2,5-dimethoxyaniline (2a, 80 mg, 0.42 mmol) and of methyl 4-(chlorosulfonyl)benzoate (3n, 50 mg, 0.21 mmol) as tan solid (31 mg, yield = 38%). 1H NMR (500 MHz, CHLOROFORM-d) δ 8.08 (d, J = 8.31 Hz, 2H), 7.80 (d, J = 8.31 Hz, 2H), 7.25 (s, 1H), 6.94 (s, 1H), 6.76 (s, 1H), 3.94 (s, 3H), 3.89 (s, 3H), 3.55 (s, 3H). 13C NMR (126 MHz, CHLOROFORM-d) δ 165.5, 149.3, 143.8, 142.5, 134.1, 130.0, 127.2, 124.1, 118.9, 113.1, 107.0, 56.8, 56.2, 52.7. HRMS for C16H15ClNO6S [M − H]− calculated 384.0314, found 384.0308.
4-(N-(4-chloro-2,5-dimethoxyphenyl)sulfamoyl)benzamide (27).
Compound 27 was synthesized using 4-chloro-2,5-dimethoxyaniline (2a, 171 mg, 0.77 mmol) and 4-carbamoylbenzenesulfonyl chloride (3o, 100 mg, 0.46 mmol) as white solid (14 mg, yield = 8%). 1H NMR (500 MHz, DMSO-d6) δ 9.83 (br. s., 1H), 8.13 (br. s., 1H), 7.96 (d, J = 8.31 Hz, 2H), 7.76 (d, J = 8.31 Hz, 2H), 7.60 (br. s., 1H), 7.02 (s, 1H), 6.99 (s, 1H), 3.74 (s, 3H), 3.38 (br. s., 3H). 13C NMR (126 MHz, DMSO-d6) δ 166.7, 148.2, 146.7, 142.4, 138.0, 128.0, 126.7, 124.5, 118.3, 114.0, 110.7, 56.5, 56.3. HRMS for C15H14ClN2O5S [M − H]− calculated 369.0317, found 369.0315.
4-(N-(4-chloro-2,5-dimethoxyphenyl)sulfamoyl)benzoic acid (28).
To a solution of compound 26 (25.0 mg, 0.06 mmol) in MeOH and THF was added a solution of LiOH (40.1 mg, 0.97 mmol) in water. The reaction was stirred overnight and the solvent was then removed under vacuum. The residue was mixed with acidified water (3N aq. HCl) extracted with EtOAc, dried over MgSO4 and concentrated to dryness under vacuum to obtain compounds 28 as white solid (21 mg, yield = 87%). 1H NMR (500 MHz, DMSO-d6) δ 10.44 (s, 1H), 9.39 (s, 1H), 7.54 (d, J = 8.80 Hz, 2H), 7.02 (s, 1H), 6.97 (s, 1H), 6.83 (d, J = 8.80 Hz, 2H), 3.66 – 3.78 (m, 3H), 3.48 (s, 3H). 13C NMR (126 MHz, DMSO-d6) δ 166.4, 148.3, 147.0, 143.9, 134.5, 129.9, 127.1, 124.4, 118.7, 114.0, 111.1, 56.6, 56.3. HRMS for C15H13ClNO6S [M − H]− calculated 370.0158, found 370.0152.
Ethyl 4-(N-(4-chloro-2,5-dimethoxyphenyl)sulfamoyl)benzoate (29).
To a solution of compound 28 (20 mg, 0.05 mmol) in anhydrous EtOH was added trimethylsilyl chloride (68.3 μL, 0.54 mmol) and the reaction was stirred at room temperature until completion. The reaction mixture was poured into water and extracted with EtOAc. The organic layer was dried over MgSO4 and concentrated under vacuum to obtain the residue which was purified by silica gel column chromatography (25% EtOAc/hexanes) to obtain compound 29 as white solid. (17.6 mg, yield = 82%). 1H NMR (500 MHz, CHLOROFORM-d) δ 8.09 (d, J = 8.31 Hz, 2H), 7.80 (d, J = 8.31 Hz, 2H), 7.26 (s, 1H), 6.95 (s, 1H), 6.76 (s, 1H), 4.39 (q, J = 7.17 Hz, 2H), 3.89 (s, 3H), 3.56 (s, 3H), 1.40 (t, J = 7.09 Hz, 3H). 13C NMR (126 MHz, CHLOROFORM-d) δ 165.0, 149.3, 143.8, 142.4, 134.5, 130.0, 127.2, 124.2, 118.8, 113.1, 106.9, 61.8, 56.8, 56.3, 14.2. HRMS for C17H17ClNO6S [M − H]− calculated 398.0471, found 398.0467.
4-(N-(4-chloro-2,5-dimethoxyphenyl)sulfamoyl)-N-methylbenzamide (30).
To a solution of compound 23 (25 mg, 0.07 mmol) in anhydrous DMF were added, 2M methyl amine solution in THF (67 μL, 0.13 mmol), triethylamine (38 μL, 0.27 mmol) and HATU (31 mg, 0.08 mmol). The reaction was stirred at room temperature until completion. The reaction mixture was poured into water and extracted with EtOAc. The organic layer was dried over MgSO4 and concentrated under vacuum to obtain the residue which was purified by reverse-phase C18 column chromatography (56% MeOH/H2O with 0.1% CF3CO2H) to yield compound 30 as white solid (3 mg, yield = 12%). 1H NMR (500 MHz, CHLOROFORM-d) δ 7.79 (s, 4H), 7.26 (s, 1H), 6.94 (s, 1H), 6.76 (s, 1H), 6.14 (s, 1H), 3.89 (s, 3H), 3.56 (s, 3H), 3.03 (d, J = 4.89 Hz, 3H). 13C NMR (126 MHz, CHLOROFORM-d) δ 166.4, 149.3, 143.8, 141.2, 138.8, 127.5, 127.4, 124.2, 118.8, 113.1, 106.9, 56.8, 56.3, 27.0. HRMS for C16H16ClN2O5S [M − H]− calculated 383.0474, found 383.0468.
4-Chloro-N-(4-ethoxyphenyl)-2,5-dimethoxybenzenesulfonamide (31).
Compound 31 was synthesized using the general procedure A using p-phenetidine (2h, 166.04 μL, 1.21 mmol) and 4-Cl-2,5-dimethoxybenzenesulfonyl chloride (3p, 100 mg, 0.61 mmol) as brown solid (98 mg, yield = 43.6%). 1H NMR (500 MHz, DMSO-d6) δ 9.74 (br. s., 1H), 7.35 (s, 1H), 7.28 (s, 1H), 6.97 (d, J = 8.80 Hz, 2H), 6.75 (d, J = 9.05 Hz, 2H), 3.88 (q, J = 7.10 Hz, 2H), 3.86 (s, 3H), 3.77 (s, 3H), 1.24 (t, J = 6.85 Hz, 3H). 13C NMR (126 MHz, CHLOROFORM-d) δ 157.5, 149.7, 149.0, 128.4, 128.4, 125.4, 125.1, 114.9, 114.9, 113.7, 63.6, 57.3, 56.8, 14.8. HRMS for C16H18ClNO5SNa [M + Na+] calculated 394.0486, found 394.0489.
General procedure B for the syntheses of site C modified compounds 33–47 and 49–52.
To a solution of compound 1 (1 eq.) in anhydrous DMF were added, potassium carbonate (2 eq.), and reagent 32 (1.1 eq.). The reaction was then heated at 45°C with stirring until completion. The suspension was extracted with EtOAc and brine. Then the organic layer was isolated, dried over MgSO4 and concentrated in vacuo. The crude material was purified by chromatography to obtain the final compounds.
N-(4-chloro-2,5-dimethoxyphenyl)-4-ethoxy-N-methylbenzenesulfonamide (33).
Compound 33 was synthesized using compound 1 (10 mg, 0.03 mmol) and iodomethane (32a, 1.85 μL, 0.03 mmol) as white solid (10 mg, yield = 95%). 1H NMR (500 MHz, CHLOROFORM-d) δ 7.61 (d, J = 8.80 Hz, 2H), 6.96 (s, 1H), 6.92 (d, J = 8.80 Hz, 2H), 6.83 (s, 1H), 4.09 (d, J = 6.85 Hz, 2H), 3.85 (s, 3H), 3.39 (s, 3H), 3.19 (s, 3H), 1.45 (t, J = 6.97 Hz, 3H). 13C NMR (126 MHz, dichloroethane) δ 162.1, 150.4, 148.6, 130.6, 129.7, 128.0, 122.5, 116.1, 114.0, 113.8, 63.9, 56.7, 55.6, 37.8, 14.6. HRMS for C17H20ClNO5SNa [M + Na+] calculated 408.0643, found 408.0641.
N-(4-chloro-2,5-dimethoxyphenyl)-4-ethoxy-N-propylbenzenesulfonamide (34).
Compound 34 was synthesized using compound 1 (10 mg, 0.03 mmol) and 1-iodopropane (32b, 2.9 μL, 0.03 mmol) as tan solid (11 mg, yield = 96%). 1H NMR (500 MHz, CHLOROFORM-d) δ 7.59 (d, J = 8.80 Hz, 2H), 6.86 – 6.92 (m, 3H), 6.81 (s, 1H), 4.07 (q, J = 7.09 Hz, 2H), 3.84 (s, 3H), 3.51 (br. s., 2H), 3.36 (s, 3H), 1.44 (sxt, J = 7.30 Hz, 5H), 0.89 (t, J = 7.34 Hz, 3H). 13C NMR (126 MHz, CHLOROFORM-d) δ 162.0, 150.7, 148.6, 131.7, 129.6, 125.7, 122.6, 117.4, 113.9, 113.7, 63.9, 56.8, 55.6, 51.4, 22.2, 14.6, 11.2. HRMS for C19H24ClNO5SNa [M + Na+] calculated 436.0956, found 436.0954.
N-(4-chloro-2,5-dimethoxyphenyl)-4-ethoxy-N-butylbenzenesulfonamide (35).
Compound 35 was synthesized using compound 1 (10 mg, 0.03 mmol) and 1-iodobutane (32c, 2.9 μL, 0.03 mmol) as white solid (11 mg, yield = 96%). 1H NMR (500 MHz, CHLOROFORM-d) δ 7.60 (d, J = 8.80 Hz, 2H), 6.87 – 6.93 (m, 3H), 6.82 (s, 1H), 4.08 (q, J = 6.93 Hz, 2H), 3.85 (s, 3H), 3.48 – 3.61 (m, 2H), 3.37 (s, 3H), 1.45 (t, J = 6.85 Hz, 3H), 1.39 (dd, J = 7.46, 14.79 Hz, 2H), 1.29 – 1.35 (m, 2H), 0.87 (t, J = 7.09 Hz, 3H). 13C NMR (126 MHz, CHLOROFORM-d) δ 162.0, 150.8, 148.6, 131.7, 129.6, 125.6, 122.6, 117.4, 113.9, 113.7, 63.9, 56.8, 55.6, 49.4, 31.0, 19.8, 14.6, 13.7. HRMS for C20H26ClNO5SNa [M + Na+] calculated 450.1112, found 450.1106.
N-(4-chloro-2,5-dimethoxyphenyl)-4-ethoxy-N-pentylbenzenesulfonamide (36).
Compound 36 was synthesized using compound 1 (10 mg, 0.03 mmol) and 1-iodopentane (32d, 3.9 μL, 0.03 mmol) as off-white solid (12 mg, yield = 98%). 1H NMR (500 MHz, CHLOROFORM-d) δ 7.59 (d, J = 8.80 Hz, 2H), 6.87 – 6.92 (m, 3H), 6.82 (s, 1H), 4.08 (q, J = 7.09 Hz, 2H), 3.85 (s, 3H), 3.54 (br. s., 2H), 3.36 (s, 3H), 1.45 (t, J = 6.97 Hz, 3H), 1.37 – 1.42 (m, 2H), 1.26 – 1.30 (m, J = 3.70 Hz, 4H), 0.85 (t, J = 6.85 Hz, 3H). 13C NMR (126 MHz, CHLOROFORM-d) δ 162.0, 150.8, 148.6, 131.7, 129.6, 125.6, 122.6, 117.4, 113.9, 113.7, 63.9, 56.8, 55.5, 49.7, 28.7, 28.5, 22.3, 14.6, 14.0. HRMS for C21H28ClNO5SNa [M + Na+] calculated 464.1269, found 464.1265.
N-(4-chloro-2,5-dimethoxyphenyl)-4-ethoxy-N-hexylbenzenesulfonamide (37).
Compound 37 was synthesized using compound 1 (10 mg, 0.03 mmol) and 1-iodohexane (32e, 4.4 μL, 0.03 mmol) as off-white solid (8 mg, yield = 65%). 1H NMR (500 MHz, CHLOROFORM-d) δ 7.59 (d, J = 8.80 Hz, 2H), 6.86 – 6.91 (m, 3H), 6.81 (s, 1H), 4.07 (q, J = 7.09 Hz, 2H), 3.84 (s, 3H), 3.47 – 3.62 (m, 2H), 3.36 (s, 3H), 1.44 (t, J = 7.10 Hz, 3H), 1.39 (quin, J = 7.60 Hz, 2H), 1.16 – 1.34 (m, 6H), 0.85 (t, J = 6.85 Hz, 3H). 13C NMR (126 MHz, CHLOROFORM-d) δ 162.0, 150.8, 148.6, 131.7, 129.6, 125.6, 122.6, 117.4, 113.9, 113.7, 63.9, 56.8, 55.5, 49.7, 31.4, 28.8, 26.2, 22.6, 14.6, 14.0. HRMS for C22H30ClNO5SNa [M + Na+] calculated 478.1425, found 478.1422.
N-(4-chloro-2,5-dimethoxyphenyl)-4-ethoxy-N-heptylbenzenesulfonamide (38).
Compound 38 was synthesized using compound 1 (10 mg, 0.03 mmol) and 1-iodoheptane (32f, 4.9 μL, 0.03 mmol) as white solid (12 mg, yield = 95%). 1H NMR (500 MHz, CHLOROFORM-d) δ 7.59 (d, J = 8.80 Hz, 2H), 6.87 – 6.91 (m, 3H), 6.81 (s, 1H), 4.07 (q, J = 6.85 Hz, 2H), 3.84 (s, 3H), 3.45 – 3.63 (m, 2H), 3.36 (s, 3H), 1.44 (t, J = 7.10 Hz, 6H), 1.39 (quin, J = 7.40 Hz, 1H), 1.14 – 1.33 (m, 10H), 0.85 (t, J = 6.97 Hz, 3H). 13C NMR (126 MHz, CHLOROFORM-d) δ 161.9, 150.7, 148.5, 131.6, 129.5, 125.6, 122.6, 117.3, 113.9, 113.6, 63.9, 56.7, 55.5, 49.6, 31.7, 28.9, 28.9, 26.5, 22.6, 14.6, 14.1. HRMS for C23H32ClNO5SNa [M + Na+] calculated 492.1582, found 492.1578.
N-(4-chloro-2,5-dimethoxyphenyl)-4-ethoxy-N-dodecylbenzenesulfonamide (39).
Compound 39 was synthesized using compound 1 (10 mg, 0.03 mmol) and 1-bromododecane (32g, 7.1 μL, 0.03 mmol) as white solid (14 mg, yield = 96%). 1H NMR (500 MHz, CHLOROFORM-d) δ 7.59 (d, J = 8.80 Hz, 2H), 6.86 – 6.92 (m, 3H), 6.81 (s, 1H), 4.07 (q, J = 7.09 Hz, 2H), 3.84 (s, 3H), 3.45 – 3.63 (m, 2H), 3.36 (s, 3H), 1.44 (t, J = 6.97 Hz, 3H), 1.38 (quin, J = 7.30 Hz, 2H), 1.23 – 1.30 (m, 10H), 0.88 (t, J = 6.97 Hz, 3H). 13C NMR (126 MHz, CHLOROFORM-d) δ 161.9, 150.7, 148.5, 131.6, 129.6, 125.6, 122.6, 117.3, 113.9, 113.6, 63.9, 56.7, 55.5, 49.7, 31.9, 29.7, 29.6, 29.6, 29.6, 29.4, 29.2, 28.9, 26.6, 22.7, 14.6, 14.1. HRMS for C28H43ClNO5S [M + H+] calculated 540.2545, found 540.2549.
N-(4-chloro-2,5-dimethoxyphenyl)-4-ethoxy-N-isopropylbenzenesulfonamide (40).
Compound 40 was synthesized using compound 1 (10 mg, 0.03 mmol) and 2-iodopropane (32h, 2.96 μL, 0.03 mmol) as white solid (5 mg, yield = 48%). 1H NMR (500 MHz, CHLOROFORM-d) δ 7.76 (d, J = 8.80 Hz, 2H), 6.94 (s, 1H), 6.92 (d, J = 8.80 Hz, 2H), 6.70 (s, 1H), 4.39 (spt, J = 6.70 Hz, 1H), 4.09 (q, J = 7.09 Hz, 2H), 3.81 (s, 3H), 3.61 (s, 3H), 1.46 (t, J = 6.97 Hz, 3H), 1.13 (d, J = 6.60 Hz, 3H), 0.99 (d, J = 6.60 Hz, 3H). 13C NMR (126 MHz, CHLOROFORM-d) δ 161.9, 152.9, 148.3, 132.9, 129.8, 123.3, 122.6, 118.5, 114.0, 113.9, 63.9, 56.8, 55.8, 51.9, 22.2, 20.9, 14.6. HRMS for C19H24ClNO5SNa [M + Na+] calculated 436.0956, found 436.0957.
N-(4-chloro-2,5-dimethoxyphenyl)-4-ethoxy-N-isobutylbenzenesulfonamide (41).
Compound 41 was synthesized using compound 1 (10 mg, 0.03 mmol) and 1-iodo-2-methylpropane (32i, 3.4 μL, 0.03 mmol) as white solid (9 mg, yield = 77%). 1H NMR (500 MHz, CHLOROFORM-d) δ 7.56 (d, J = 8.80 Hz, 2H), 6.92 (s, 1H), 6.89 (d, J = 8.80 Hz, 2H), 6.80 (s, 1H), 4.07 (q, J = 7.09 Hz, 2H), 3.85 (s, 3H), 3.29 – 3.51 (m, 2H), 3.33 (s, 3H), 1.59 (spt, J = 7.00 Hz, 2H), 1.44 (t, J = 6.97 Hz, 3H), 0.91 (br. s., 6H). 13C NMR (126 MHz, CHLOROFORM-d) δ 161.9, 150.6, 148.5, 131.5, 129.6, 126.0, 122.5, 117.2, 113.9, 113.7, 63.9, 57.1, 56.8, 55.5, 27.6, 20.1, 14.6. HRMS for C20H26ClNO5SNa [M + Na+] calculated 450.1112, found 450.1109.
N-(4-chloro-2,5-dimethoxyphenyl)-4-ethoxy-N-(prop-2-yn-1-yl)benzenesulfonamide (42).
Compound 42 was synthesized using compound 1 (10 mg, 0.03 mmol) and propargyl bromide solution in toluene (32j, 2.8 μL, 0.03 mmol) as white solid (9 mg, yield = 81%). 1H NMR (500 MHz, CHLOROFORM-d) δ 7.62 (d, J = 8.80 Hz, 2H), 6.96 (s, 1H), 6.90 (d, J = 8.80 Hz, 2H), 6.85 (s, 1H), 4.44 (br. s., 2H), 4.08 (q, J = 7.09 Hz, 2H), 3.82 (s, 3H), 3.44 (s, 3H), 2.12 – 2.23 (m, 1H), 1.45 (t, J = 6.85 Hz, 3H). 13C NMR (126 MHz, CHLOROFORM-d) δ 162.3, 150.5, 148.6, 131.1, 129.8, 125.0, 123.1, 117.2, 114.1, 113.7, 78.4, 73.2, 64.0, 56.7, 55.8, 39.6, 14.6. HRMS for C19H20ClNO5SNa [M + Na+] calculated 432.0643, found 432.0639.
N-(but-3-yn-1-yl)-N-(4-chloro-2,5-dimethoxyphenyl)-4-ethoxybenzenesulfonamide (43).
Compound 43 was synthesized using compound 1 (40 mg, 0.11 mmol) and 4-bromo-1-butyne (32k, 11.1 μL, 0.11 mmol) as white solid (3 mg, yield = 7%). 1H NMR (500 MHz, CHLOROFORM-d) δ 7.59 (d, J = 8.80 Hz, 2H), 6.98 (s, 1H), 6.90 (d, J = 8.80 Hz, 2H), 6.81 (s, 1H), 4.08 (q, J = 7.09 Hz, 2H), 3.85 (s, 3H), 3.72 (br. s., 2H), 3.35 (s, 3H), 2.42 (dt, J = 2.57, 7.40 Hz, 2H), 1.95 (t, J = 2.57 Hz, 1H), 1.45 (t, J = 6.97 Hz, 3H). 13C NMR (126 MHz, CHLOROFORM-d) δ 162.1, 150.3, 148.6, 131.4, 129.6, 125.2, 123.0, 117.6, 114.0, 113.6, 81.0, 70.0, 63.9, 56.7, 55.5, 48.6, 19.7, 14.6. HRMS for C20H22ClNO5SNa [M + Na+] calculated 446.0799, found 446.0798.
N-(4-chloro-2,5-dimethoxyphenyl)-4-ethoxy-N-(pent-4-yn-1-yl)benzenesulfonamide (44).
Compound 44 was synthesized using compound 1 (40 mg, 0.11 mmol) and 5-iodopent-1-yne (32l, 13.5 μL, 0.12 mmol) as off-white solid (40 mg, yield = 84%). 1H NMR (500 MHz, CHLOROFORM-d) δ 7.59 (d, J = 8.80 Hz, 2H), 6.90 (d, J = 8.80 Hz, 2H), 6.87 (s, 1H), 6.83 (s, 1H), 4.08 (q, J = 7.09 Hz, 2H), 3.84 (s, 3H), 3.64 (br. s., 2H), 3.38 (s, 3H), 2.26 (dt, J = 2.45, 7.21 Hz, 2H), 1.91 (t, J = 2.57 Hz, 1H), 1.67 (quin, J = 7.09 Hz, 2H), 1.44 (t, J = 6.97 Hz, 3H). 13C NMR (126 MHz, CHLOROFORM-d) δ 162.1, 150.7, 148.6, 131.3, 129.6, 125.5, 122.8, 117.0, 114.0, 113.7, 83.4, 68.7, 63.9, 56.8, 55.6, 48.9, 27.8, 15.8, 14.6. HRMS for C21H24ClNO5SNa [M + Na+] calculated 460.0956, found 460.0954.
N-(4-chloro-2,5-dimethoxyphenyl)-4-ethoxy-N-(2-(2-(2-(prop-2-yn-1yloxy)ethoxy)ethoxy)ethyl)benzenesulfonamide (45).
Compound 45 was synthesized using compound 1 (40 mg, 0.11 mmol) and propargyl-PEG3-bromide (32m, 29.7 μL, 0.12 mmol) as clear oil (48 mg, yield = 82%). 1H NMR (500 MHz, METHANOL-d4) δ 7.59 (d, J = 8.80 Hz, 2H), 7.02 (d, J = 8.80 Hz, 2H), 6.97 (s, 1H), 6.96 (s, 1H), 4.16 (d, J = 2.20 Hz, 2H), 4.11 (q, J = 7.09 Hz, 2H), 3.72 – 3.85 (m, 5H), 3.61 – 3.65 (m, 2H), 3.57 – 3.60 (m, 2H), 3.48 – 3.54 (m, 6H), 3.38 (s, 3H), 2.85 (t, J = 2.32 Hz, 1H), 1.41 (t, J = 6.97 Hz, 3H). 13C NMR (126 MHz, METHANOL-d4) δ 164.0, 152.4, 150.1, 132.9, 131.0, 127.1, 124.3, 119.1, 115.5, 115.0, 80.7, 76.1, 71.7, 71.5, 71.3, 70.5, 70.2, 65.3, 59.2, 57.4, 56.4, 50.4, 15.1. HRMS for C25H32ClNO8SNa [M + Na+] calculated 564.1429, found 564.1431.
N-benzyl-N-(4-chloro-2,5-dimethoxyphenyl)-4-ethoxybenzenesulfonamide (46).
Compound 46 was synthesized using compound 1 (10 mg, 0.03 mmol) and benzyl chloride (32n, 3.4 μL, 0.03 mmol) as white solid (12 mg, yield = 94%). 1H NMR (500 MHz, CHLOROFORM-d) δ 7.65 (d, J = 8.80 Hz, 2H), 7.10 – 7.25 (m, 5H), 6.92 (d, J = 8.80 Hz, 2H), 6.75 (s, 1H), 6.63 (s, 1H), 4.74 (br. s., 2H), 4.09 (q, J = 6.85 Hz, 2H), 3.67 (s, 3H), 3.35 (s, 3H), 1.46 (t, J = 6.85 Hz, 3H). 13C NMR (126 MHz, CHLOROFORM-d) δ 162.1, 150.5, 148.4, 136.5, 131.7, 129.6, 128.8, 128.2, 127.6, 125.1, 122.6, 117.9, 114.0, 113.5, 63.9, 56.6, 55.5, 53.4, 14.6. HRMS for C23H24ClNO5SNa [M + Na+] calculated 484.0956, found 484.0952.
N-(4-chloro-2,5-dimethoxyphenyl)-4-ethoxy-N-phenethylbenzenesulfonamide (47).
Compound 47 was synthesized using compound 1 (10 mg, 0.03 mmol) and (2-iodoethyl)benzene (32o, 3.4 μL, 0.03 mmol) as off-white solid (12 mg, yield = 95%). 1H NMR (500 MHz, CHLOROFORM-d) δ 7.45 – 7.52 (m, 2H), 7.18 (d, J = 7.60 Hz, 2H), 7.12 (t, J = 7.30 Hz, 1H), 7.06 (d, J = 7.60 Hz, 2H), 6.76 – 6.83 (m, J = 8.80 Hz, 2H), 6.72 (s, 1H), 6.66 (s, 1H), 3.99 (q, J = 6.85 Hz, 2H), 3.61 – 3.85 (m, 5H), 3.25 (s, 3H), 2.74 (t, J = 7.70 Hz, 2H), 1.37 (t, J = 6.97 Hz, 3H). 13C NMR (126 MHz, CHLOROFORM-d) δ 162.0, 150.4, 148.5, 138.4, 131.4, 129.5, 128.9, 128.3, 126.4, 125.6, 122.6, 117.5, 113.9, 113.4, 63.9, 56.6, 55.5, 51.2, 35.8, 14.6. HRMS for C24H26ClNO5SNa [M + Na+] calculated 498.1112, found 498.111.
N-(4-chloro-2,5-dimethoxyphenyl)-N-((4-ethoxyphenyl)sulfonyl)acetamide (48).
To a solution of compound 1 (10 mg, 0.03 mmol) in anhydrous CH2Cl2 were added, acetyl chloride (32p, 3.8 μL, 0.03 mmol) and triethylamine (15 μL, 0.12 mmol) and the reaction was heated at 45 °C with stirring for 20h upon which more acetyl chloride (3.8 μL, 0.03 mmol) was added to drive the reaction to completion. The solvent was then removed, and the residue was dissolved in EtOAc, washed with water and brine, dried over MgSO4 and concentrated under vacuum to obtain the residue which was purified by silica gel column chromatography (30% EtOAc/hexanes) to obtain compound 48 as white solid (5 mg, yield = 45%). 1H NMR (500 MHz, CHLOROFORM-d) δ 7.98 (d, J = 8.80 Hz, 2H), 7.05 (s, 1H), 6.85 – 7.01 (m, 3H), 4.12 (q, J = 7.10 Hz, 2H), 3.91 (s, 3H), 3.67 – 3.76 (m, 3H), 1.86 (s, 3H), 1.46 (t, J = 6.97 Hz, 3H). 13C NMR (126 MHz, CHLOROFORM-d) δ 170.1, 163.2, 149.8, 149.3, 131.8, 130.0, 124.9, 123.9, 115.7, 114.1, 113.8, 64.0, 56.9, 56.0, 24.0, 14.6. HRMS for C18H20ClNO6SNa [M + Na+] calculated 436.0592, found 436.0592.
Ethyl N-(4-chloro-2,5-dimethoxyphenyl)-N-((4-ethoxyphenyl)sulfonyl)glycinate (49).
Compound 49 was synthesized using compound 1 (10 mg, 0.03 mmol) and ethylbromoacetate (32q, 3.3 μL, 0.03 mmol) as off-white solid (12 mg, yield = 97%). 1H NMR (500 MHz, CHLOROFORM-d) δ 7.60 (d, J = 8.80 Hz, 2H), 7.18 (s, 1H), 6.89 (d, J = 8.80 Hz, 2H), 6.80 (s, 1H), 4.38 (s, 2H), 4.16 (d, J = 7.09 Hz, 2H), 4.07 (d, J = 7.09 Hz, 2H), 3.83 (s, 3H), 3.39 (s, 3H), 1.44 (t, J = 6.97 Hz, 3H), 1.25 (t, J = 7.09 Hz, 3H). 13C NMR (126 MHz, CHLOROFORM-d) δ 169.4, 162.3, 149.8, 148.6, 131.3, 129.7, 125.6, 123.0, 117.8, 114.0, 113.5, 63.9, 61.3, 56.7, 55.7, 51.0, 14.6, 14.2. HRMS for C20H25ClNO7S [M + H+] calculated 458.1035, found 458.1035.
Ethyl 4-((N-(4-chloro-2,5-dimethoxyphenyl)-4-ethoxyphenyl)sulfonamido)butanoate (50).
Compound 50 was synthesized using compound 1 (40 mg, 0.11 mmol) and ethyl-4-bromobutyrate (32r, 16.9 μL, 0.12 mmol) as white solid (47 mg, yield = 91%). 1H NMR (500 MHz, CHLOROFORM-d) δ 7.58 (d, J = 8.80 Hz, 2H), 6.89 (d, J = 8.80 Hz, 2H), 6.87 (s, 1H), 6.82 (s, 1H), 4.09 (q, J = 7.20 Hz, 2H), 4.07 (q, J = 7.00 Hz, 2H), 3.84 (s, 3H), 3.60 (br. s., 2H), 3.37 (s, 3H), 2.41 (t, J = 7.46 Hz, 2H), 1.74 (quin, J = 7.09 Hz, 2H), 1.44 (t, J = 6.97 Hz, 3H), 1.23 (t, J = 7.21 Hz, 3H). 13C NMR (126 MHz, CHLOROFORM-d) δ 173.1, 162.1, 150.7, 148.6, 131.3, 129.6, 125.3, 122.9, 117.0, 114.0, 113.7, 63.9, 60.4, 56.8, 55.6, 49.0, 31.1, 24.1, 14.6, 14.2. HRMS for C22H28ClNO7SNa [M + Na+] calculated 508.1167, found 508.117.
N-(4-chloro-2,5-dimethoxyphenyl)-4-ethoxy-N-(3-hydroxypropyl)benzenesulfonamide (51).
Compound 51 was synthesized using compound 1 (10 mg, 0.03 mmol) and 3-bromo-1-propanol (32s, 2.7 μL, 0.03 mmol) as off-white solid (5 mg, yield = 42%). 1H NMR (500 MHz, DMSO-d6) δ 7.55 (d, J = 8.80 Hz, 2H), 7.14 (s, 1H), 7.08 (d, J = 8.80 Hz, 2H), 6.76 (s, 1H), 4.43 (t, J = 4.89 Hz, 1H), 4.11 (q, J = 6.85 Hz, 2H), 3.72 (s, 3H), 3.50 (br. s., 2H), 3.42 (s, 3H), 3.33 – 3.36 (m, 2H), 1.46 (quin, J = 6.80 Hz, 2H), 1.34 (t, J = 6.85 Hz, 3H). 13C NMR (126 MHz, DMSO-d6) δ 161.8, 151.1, 147.9, 130.8, 129.5, 125.9, 121.5, 116.4, 114.5, 114.3, 63.8, 58.1, 56.5, 56.1, 47.0, 31.7, 14.5. HRMS for C19H24ClNO6SNa [M + Na+] calculated 452.0905, found 452.0907.
tert-Butyl (3-((N-(4-chloro-2,5-dimethoxyphenyl)-4-ethoxyphenyl)sulfonamido)propyl)carbamate (52).
Compound 52 was synthesized using compound 1 (50 mg, 0.13 mmol) and tert-butyl (3-bromopropyl)carbamate (32t, 35.2 μL, 0.15 mmol) as off-white solid (60 mg, yield = 84%). 1H NMR (500 MHz, CHLOROFORM-d) δ 7.59 (d, J = 8.80 Hz, 2H), 6.91 (d, J = 8.80 Hz, 2H), 6.84 (s, 1H), 6.84 (s, 1H), 5.00 (br. s., 1H), 4.08 (q, J = 6.85 Hz, 2H), 3.84 (s, 3H), 3.61 (br. s., 2H), 3.38 (s, 3H), 3.27 (br. s., 2H), 1.56 (quin, J = 6.40 Hz, 2H), 1.40 – 1.50 (m, 12H). 13C NMR (126 MHz, CHLOROFORM-d) δ 162.1, 156.0, 150.7, 148.7, 131.2, 129.6, 125.2, 123.0, 117.0, 114.0, 113.8, 79.1, 63.9, 56.8, 55.6, 47.0, 37.1, 28.8, 28.4, 14.6. HRMS for C24H33ClN2O7SNa [M + Na+] calculated 551.1589, found 551.1587.
4-((N-(4-chloro-2,5-dimethoxyphenyl)-4-ethoxyphenyl)sulfonamido)butanoic acid (53).
To a solution of compound 50 (38 mg, 0.08 mmol) in MeOH (0.5 mL) was added a solution of lithium hydroxide monohydrate (16.4 mg, 0.39 mmol) in water (0.5 mL) and the reaction was stirred overnight. The solvent was then removed and the residue was dissolved in acidified water (3N aq. HCl) and extracted with EtOAc. The organic layer was dried over MgSO4 and concentrated under vacuum to obtain the residue which was purified by silica gel column chromatography (8% MeOH/CH2Cl2) to obtain compounds 53 as white solid (26 mg, yield = 73%). 1H NMR (500 MHz, DMSO-d6) δ 11.74 – 12.24 (m, 1H), 7.54 (d, J = 9.05 Hz, 2H), 7.14 (s, 1H), 7.07 (d, J = 9.05 Hz, 2H), 6.75 (s, 1H), 4.11 (q, J = 7.09 Hz, 2H), 3.70 (s, 3H), 3.45 – 3.47 (m, 2H), 3.41 (s, 3H), 2.26 (t, J = 7.34 Hz, 2H), 1.50 (quin, J = 7.03 Hz, 2H), 1.34 (t, J = 6.85 Hz, 3H). 13C NMR (126 MHz, DMSO-d6) δ 174.1, 161.8, 151.1, 148.0, 130.7, 129.5, 125.8, 121.6, 116.2, 114.5, 114.3, 63.8, 56.5, 56.1, 48.9, 30.4, 23.5, 14.5. HRMS for C20H23ClNO7S [M − H]− calculated 456.0889, found 456.0889.
4-((N-(4-chloro-2,5-dimethoxyphenyl)-4-ethoxyphenyl)sulfonamido)-N-ethylbutanamide (54).
To a solution of compound 53 (7 mg, 0.02 mmol) in anhydrous DMF were added, HATU (6.4 mg, 0.02), 2M ethyl amine solution in THF (8.36 μL, 0.02 mmol), and triethylamine (4.3 μL, 0.03 mmol) and the reaction was stirred until completion. The solvent was then removed under vacuum and the residue was purified by silica gel column chromatography (5% MeOH/CH2Cl2) to obtain compound 54 as white solid (6 mg, yield = 81%). 1H NMR (500 MHz, CHLOROFORM-d) δ 7.56 (d, J = 8.80 Hz, 2H), 6.91 (d, J = 8.80 Hz, 2H), 6.85 (s, 1H), 6.79 (s, 1H), 5.90 (br. s., 1H), 4.08 (q, J = 7.09 Hz, 2H), 3.83 (s, 3H), 3.58 (br. s., 2H), 3.40 (s, 3H), 3.32 (quin, J = 6.85 Hz, 2H), 2.33 (t, J = 6.85 Hz, 2H), 1.73 (quin, J = 6.48 Hz, 2H), 1.45 (t, J = 6.97 Hz, 3H), 1.18 (t, J = 7.34 Hz, 3H). 13C NMR (126 MHz, CHLOROFORM-d) δ 172.3, 162.2, 150.7, 148.7, 131.1, 129.6, 125.2, 123.1, 116.8, 114.1, 114.0, 63.9, 56.8, 55.7, 48.9, 34.4, 33.3, 24.6, 14.8, 14.6. HRMS for C22H30ClN2O6S [M + H+] calculated 485.1508, found 485.1511.
N-(3-aminopropyl)-N-(4-chloro-2,5-dimethoxyphenyl)-4-ethoxybenzenesulfonamide hydrochloride (55).
Compound 52 (48 mg, 0.09 mmol) was dissolved in 4N HCl solution in dioxane (1 mL) and stirred at room temperature for 20h. Solvent was then removed under vacuum, reside was suspended in CH2Cl2 followed by removal of the solvent under vacuum to obtain compound 55 as white solid (37.1 mg, yield = 95%). 1H NMR (500 MHz, DMSO-d6) δ 7.85 (br. s., 3H), 7.56 (d, J = 8.80 Hz, 2H), 7.17 (s, 1H), 7.09 (d, J = 8.80 Hz, 2H), 6.78 (s, 1H), 4.11 (q, J = 6.85 Hz, 2H), 3.71 – 3.75 (m, 3H), 3.53 (br. s., 2H), 3.42 (s, 3H), 2.83 (t, J = 8.10 Hz, 2H), 1.61 (td, J = 7.00, 14.61 Hz, 2H), 1.34 (t, J = 6.97 Hz, 3H). 13C NMR (126 MHz, DMSO-d6) δ 161.9, 151.0, 148.0, 130.5, 129.5, 125.5, 121.8, 116.3, 114.6, 114.4, 63.8, 56.6, 56.2, 47.1, 36.6, 26.4, 14.5. HRMS for C19H26ClN2O5S [M + H+] calculated 429.1245, found 429.1246.
N-(6-((3-((N-(4-chloro-2,5-dimethoxyphenyl)-4-ethoxyphenyl)sulfonamido)propyl)amino)-6-oxohexyl)-3’,6’-dihydroxy-3-oxo-3H-spiro[isobenzofuran-1,9’-xanthene]-5-carboxamide (56).
To a solution of compound 55 (4 mg, 0.01 mmol) in anhydrous DMF were added, triethylamine (3.6 μL, 0.03 mmol) and fluorescein-5(6)-carboxamidocaproic acid N-succinimidyl ester (5(6)-SFX SE, Chemodex, #F0044) (5 mg, 0.01 mmol). After completion of the reaction, solvent was removed, and the residue was purified using silica gel column chromatography (10% MeOH/CH2Cl2) to obtain compound 56 as bright yellow solid (5.4 mg, yield = 70%). 1H NMR (500 MHz, METHANOL-d4) δ 8.12 (d, J = 8.07 Hz, 1H), 8.06 (d, J = 8.10 Hz, 1H), 7.60 (s, 1H), 7.55 (d, J = 8.80 Hz, 2H), 6.99 – 7.03 (m, 2H), 6.97 (s, 1H), 6.87 (s, 1H), 6.65 – 6.73 (m, 2H), 6.60 (br. s., 2H), 6.54 (s, 2H), 4.10 (q, J = 7.09 Hz, 2H), 3.78 (s, 3H), 3.54 – 3.64 (m, 2H), 3.35 – 3.37 (m, 3H), 3.12 – 3.28 (m, 4H), 2.12 (s, 2H), 1.48 – 1.69 (m, 8H), 1.40 (t, J = 6.97 Hz, 3H). HRMS for C46H47ClN3O12S [M + H+] calculated 900.2563, found 900.2563.
N-(9-(2-carboxy-4-((3-((N-(4-chloro-2,5-dimethoxyphenyl)-4-ethoxyphenyl)sulfonamido)propyl)carbamoyl)phenyl)-6-(diethylamino)-3H-xanthen-3-ylidene)-N-ethylethanaminium (57).
To a solution of compound 55 (5 mg, 0.01 mmol) in anhydrous DMF were added, triethylamine (4.5 μL, 0.03 mmol) and 5(6)-carboxy-tetramethylrhodamine succinimidyl ester (NHS-Rhodamine, 6 mg, 0.01 mmol). After completion of the reaction, solvent was removed, and the residue was purified using silica gel column chromatography (20% MeOH/CH2Cl2) to obtain compound 57 as dark purple solid (8.5 mg, yield = 94%). 1H NMR (500 MHz, acetone) δ 8.38 (s, 1H), 7.99 – 8.27 (m, 2H), 7.58 – 7.73 (m, 2H), 7.55 (d, J = 8.80 Hz, 1H), 7.33 (d, J = 7.83 Hz, 1H), 6.93 – 7.11 (m, 4H), 6.52 – 6.69 (m, 5H), 4.11 – 4.19 (m, 2H), 3.86 (s, 2H), 3.68 – 3.83 (m, 3H), 3.59 (s, 2H), 3.47 (s, 2H), 3.37 (s, 1H), 2.97 – 3.12 (m, 12H), 1.78 (quin, J = 6.80 Hz, 1H), 1.59 – 1.68 (m, J = 6.85, 6.85, 6.85, 6.85 Hz, 1H), 1.37 – 1.42 (m, 3H). HRMS for C44H46ClN4O9S [M+] calculated 841.2669, found 841.2659.
N-(3-((N-(4-chloro-2,5-dimethoxyphenyl)-4-ethoxyphenyl)sulfonamido)propyl)-5-((3aR,4R,6aS)-2-oxohexahydro-1H-thieno[3,4-d]imidazol-4-yl)pentanamide (58).
To a solution of compound 55 (10 mg, 0.02 mmol) in anhydrous DMF were added, HATU (14.9 mg, 0.04 mmol), biotin (5.8 mg, 0.02 mmol) and triethylamine (11 μL, 0.08 mmol). After stirring overnight, the solvent was removed and the residue was purified using silica gel column chromatography (10% MeOH/CH2Cl2) to obtain compound 58 as off-white solid (14 mg, quantitative yield). 1H NMR (500 MHz, METHANOL-d4) δ 7.57 (d, J = 8.80 Hz, 2H), 7.03 (d, J = 8.80 Hz, 2H), 6.99 (s, 1H), 6.89 (s, 1H), 4.48 (dd, J = 4.89, 7.83 Hz, 1H), 4.30 (dd, J = 4.40, 7.83 Hz, 1H), 4.12 (q, J = 7.09 Hz, 2H), 3.80 (s, 3H), 3.63 (br. s., 2H), 3.39 (s, 3H), 3.17 – 3.29 (m, 3H), 2.92 (dd, J = 4.89, 12.72 Hz, 1H), 2.70 (d, J = 12.72 Hz, 1H), 2.17 (t, J = 7.34 Hz, 2H), 1.57 – 1.75 (m, 6H), 1.36 – 1.48 (m, 5H). 13C NMR (126 MHz, METHANOL-d4) δ 176.2, 166.3, 164.1, 152.6, 150.2, 132.4, 131.0, 126.8, 124.4, 118.5, 115.5, 115.2, 65.3, 63.5, 61.8, 57.3, 57.1, 56.5, 41.2, 37.8, 37.0, 29.9, 29.8, 29.6, 27.1, 15.1. HRMS for C29H40ClN4O7S2 [M + H+] calculated 655.2021, found 655.2025.
N-(4-azido-2,5-dimethoxyphenyl)-4-ethoxybenzenesulfonamide (59).
To a solution of compound 10 (84 mg, 0.24 mmol) in anhydrous acetonitrile were added, tert-butyl nitrite (37 mg, 0.36 mmol) and azidotrimethylsilane (33 mg, 0.29 mmol) and the reaction was heated at 45 °C for 1h. The solvent was then removed, and the residue was purified using silica gel column chromatography (20% EtOAc/hexanes) under low-light conditions to obtain compound 59 as white solid (52 mg, yield = 57%). 1H NMR (500 MHz, CHLOROFORM-d) δ 7.64 (d, J = 8.80 Hz, 2H), 7.19 (s, 1H), 6.81 – 6.89 (m, 3H), 6.36 (s, 1H), 4.04 (q, J = 6.85 Hz, 2H), 3.86 (s, 3H), 3.56 (s, 3H), 1.42 (t, J = 6.97 Hz, 3H). 13C NMR (126 MHz, CHLOROFORM-d) δ 162.5, 146.3, 144.2, 130.1, 129.3, 124.7, 122.9, 114.3, 107.3, 103.8, 63.9, 56.7, 56.3, 14.6. HRMS for C16H18N4O5SNa [M + Na+] calculated 401.089, found 401.0894.
3-Amino-N-(4-chloro-2,5-dimethoxyphenyl)-4-methoxybenzenesulfonamide (60).
Combine 1,2-dimethylethylenediamine (7.2 μL, 0.069 mmol), CuI (9.2 mg, 0.057 mmol) and sodium ascorbate (8.7 mg, 0.057 mmol) in a microwave reaction vial. Seal and evacuate the vial and add H2O (300 μL). Separately combine compound 25 (50 mg, 0.11 mmol) and NaN3 (41.6 mg, 0.23 mmol) in EtOH (350 μL) and DMF (350 μL) and add to the reaction vial. Fill vial with argon gas and irradiate reaction using microwave at 100 °C for 1h. Water was poured into the reaction mixture and extracted with EtOAc. The organic layer was collected, solvent was removed to obtain the residue which was purified using silica gel column chromatography (30% EtOAc/hexanes) to obtain compound 60 as a tan solid (28 mg, yield = 63%). 1H NMR (500 MHz, METHANOL-d4) δ 7.16 (s, 1H), 7.02 – 7.10 (m, 2H), 6.89 (s, 1H), 6.84 (d, J = 8.56 Hz, 1H), 3.86 (s, 3H), 3.80 (s, 3H), 3.55 (s, 3H). 13C NMR (126 MHz, METHANOL-d4) δ 152.1, 150.5, 147.1, 138.9, 132.7, 127.1, 119.6, 119.0, 114.7, 113.7, 110.4, 109.8, 57.2, 57.2, 56.4. HRMS for C15H18ClN2O5S [M + H+] calculated 373.0619, found 373.062.
3-Azido-N-(4-chloro-2,5-dimethoxyphenyl)-4-methoxybenzenesulfonamide (61).
To a solution of compound 60 (23 mg, 0.06 mmol) in anhydrous acetonitrile were added, tert-butyl nitrite (10 mg, 0.09 mmol) and azidotrimethylsilane (9 mg, 0.07 mmol) and the reaction was heated at 45 °C for 1h. The solvent was then removed, and the residue was purified using silica gel column chromatography (25% EtOAc/hexanes) under low-light conditions to obtain compound 61 as light yellow solid (22 mg, yield = 88%). 1H NMR (500 MHz, CHLOROFORM-d) δ 7.49 (dd, J = 1.96, 8.56 Hz, 1H), 7.38 (d, J = 1.71 Hz, 1H), 7.24 (s, 1H), 6.95 (s, 1H), 6.85 (d, J = 8.56 Hz, 1H), 6.80 (s, 1H), 3.91 (s, 3H), 3.88 (s, 3H), 3.66 (s, 3H). 13C NMR (126 MHz, CHLOROFORM-d) δ 155.4, 149.3, 143.5, 131.2, 129.2, 125.4, 124.8, 119.2, 118.2, 113.1, 111.2, 106.2, 56.8, 56.4, 56.3. HRMS for C15H15ClN4O5SNa [M + Na+] calculated 421.0344, found 421.0348.
N-(4-azido-2,5-dimethoxyphenyl)-4-ethoxy-N-(prop-2-yn-1-yl)benzenesulfonamide (62).
To a solution of compound 61 (10 mg, 0.03 mmol) in anhydrous DMF were added, potassium carbonate (7.0 mg, 0.06 mmol) and propargyl bromide solution in toluene (32j, 3.23 μL, 0.03 mmol). The reaction was heated at 45 °C for 2h, followed by removal of the solvent. The residue was then dissolved in EtOAc and washed by water and brine, dried over sodium sulfate, concentrated under vacuum to obtain the residue which was purified by silica gel column chromatography (28% EtOAc/hexanes) to obtain compound 62 as off-white solid (10 mg, yield = 89%). 1H NMR (500 MHz, CHLOROFORM-d) δ 7.62 (d, J = 8.80 Hz, 2H), 6.81 – 6.97 (m, 3H), 6.41 (s, 1H), 4.43 (br. s., 2H), 4.08 (q, J = 7.10 Hz, 2H), 3.80 (s, 3H), 3.42 (s, 3H), 2.17 (t, J = 2.40 Hz, 1H), 1.44 (t, J = 6.97 Hz, 3H). 13C NMR (126 MHz, CHLOROFORM-d) δ 162.3, 151.0, 145.6, 131.2, 129.8, 129.2, 122.6, 117.4, 114.1, 104.2, 78.6, 73.1, 63.9, 56.6, 55.7, 39.7, 14.6. HRMS for C19H20N4O5SNa [M + Na+] calculated 439.1047, found 439.105.
N-(3-aminopropyl)-N-(4-azido-2,5-dimethoxyphenyl)-4-ethoxybenzenesulfonamide (63).
To a solution of compound 61 (27 mg, 0.07 mmol) in anhydrous DMF were added, potassium carbonate (19.5 mg, 0.14 mmol) and tert-butyl (3-bromopropyl)carbamate (32t, 22 mg, 0.09 mmol). The reaction was heated at 45 °C for 2h, followed by removal of the solvent. The residue was then dissolved in EtOAc and washed by water and brine, dried over sodium sulfate, concentrated under vacuum to obtain the residue which was purified by silica gel column chromatography (35% EtOAc/hexanes) to obtain N-Boc protected intermediate (14 mg) which was stirred in a solution of 4N HCl in dioxane for 1h. The solvent was then removed to obtain the compound 63 as tan solid (12 mg, yield = 36%). 1H NMR (500 MHz, METHANOL-d4) δ 7.58 (d, J = 8.80 Hz, 2H), 7.04 (d, J = 8.80 Hz, 2H), 6.79 (s, 1H), 6.56 (s, 1H), 4.12 (q, J = 7.09 Hz, 2H), 3.79 (s, 3H), 3.67 – 3.72 (m, 2H), 3.40 (s, 3H), 3.13 (t, J = 7.58 Hz, 2H), 1.77 (quin, J = 7.00 Hz, 2H), 1.42 (t, J = 6.97 Hz, 3H). 13C NMR (126 MHz, METHANOL-d4) δ 164.3, 153.1, 147.8, 132.1, 131.2, 131.0, 124.2, 118.3, 115.6, 106.5, 65.3, 57.5, 56.4, 38.6, 28.0, 15.1. HRMS for C19H26N5O5S [M + H+] calculated 436.1649, found 436.1652.
N-(3-((N-(4-azido-2,5-dimethoxyphenyl)-4-ethoxyphenyl)sulfonamido)propyl)-5((3aR,4R,6aS)-2-oxohexahydro-1H-thieno[3,4-d]imidazol-4-yl)pentanamide (64).
To a solution of compound 63 (9 mg, 0.02 mmol) in anhydrous DMF were added, HATU (8.6 mg, 0.02 mmol), biotin (4.3 mg, 0.02 mmol), and triethylamine (6.6 μL, 0.05 mmol). Th reaction was stirred overnight, followed by removal of the solvent to obtain the residue which was purified using silica gel column chromatography (10% MeOH/CH2Cl2) to obtain compound 64 as off-white solid (8.4 mg, 67%). 1H NMR (500 MHz, METHANOL-d4) δ 7.91 (br. s., 1H), 7.57 (d, J = 8.80 Hz, 2H), 7.02 (d, J = 8.80 Hz, 2H), 6.84 (s, 1H), 6.51 (s, 1H), 4.48 (dd, J = 5.01, 7.70 Hz, 1H), 4.30 (dd, J = 4.40, 7.83 Hz, 1H), 4.12 (q, J = 6.85 Hz, 2H), 3.80 (s, 3H), 3.61 (br. s., 2H), 3.36 (s, 3H), 3.16 – 3.29 (m, 3H), 2.92 (dd, J = 5.14, 12.72 Hz, 1H), 2.70 (d, J = 12.72 Hz, 1H), 2.17 (t, J = 7.34 Hz, 2H), 1.51 – 1.80 (m, 6H), 1.36 – 1.49 (m, 5H). 13C NMR (126 MHz, METHANOL-d4) δ 176.3, 176.2, 166.3, 164.1, 153.0, 147.6, 132.5, 131.0, 130.7, 124.3, 118.8, 115.5, 106.2, 65.3, 63.5, 61.8, 57.5, 57.1, 56.3, 41.2, 37.8, 37.0, 29.9, 29.7, 29.6, 27.1, 15.1. HRMS for C29H39N7O7S2Na [M + Na+] calculated 684.2245, found 684.2241.
Biology: Cell lines and reagents
The THP1-Blue™ NF-κB cell line was purchased from Invivogen (San Diego, CA) which contains a stably integrated NF-κB-inducible secreted embryonic alkaline phosphatase (SEAP). ISRE-bla THP-1 cell line was generated by us as described earlier.42 QuantiBlue was purchased from Invivogen, MTT (3-[4,5-dimethylthiazol-2-yl]-2,5-dipheyl tetrazolium bromide) was purchased from Acros Organics, LPS (lps-eb) from Invivogen, and IFN-α from R&D Systems (#11200–2).
Measurement of NF-κB activation using THP1-Blue™ NF-κB cells
THP1-Blue™ NF-κB cells were plated in 96-well plates at 105 cells/well in 100 μl RPMI supplemented with 10% fetal bovine serum (FBS, Omega Scientific, Inc., Tarzana, CA), 100 U/mL penicillin, 100 μg/ml streptomycin (Thermo Fisher Scientific) and Normocin (Invivogen). LPS was prepared in assay medium at a concentration of 20 μg/mL. Tested compounds were dissolved in DMSO at 1 mM as a stock solution and were further diluted in the LPS solution to a final concentration of 10 μM. 100 μL of this solution was then transferred to the plated cells to obtain a final concentration of LPS at 10 μg/mL and compound at 5 μM (0.05% DMSO). The culture supernatants were harvested after a 20h incubation period. SEAP activity in the culture supernatants was determined by a colorimetric assay using QuantiBlue (Invivogen). Plate absorbance was read at 630 nm using a Tecan Infinite M200 plate reader (Männedorf, Switzerland). The SEAP concentration was directly proportional to NF-κB activity, which was 2-point normalized to yield activity of compound 1 + LPS as 200% and activity for LPS as 100%.
Measurement of ISRE activity in ISRE-bla THP-1 cells
ISRE-bla THP-1 cells were plated in 96-well plates at 5×104 cells/well in 50 μl RPMI supplemented with 10% dialyzed FBS (Atlanta Biologicals, Inc., GA), 0.1 mM non-essential amino acids, 1 mM sodium pyruvate, 100 U/mL penicillin and 100 μg/ml streptomycin. Type I IFN-α (R&D Systems, #11200–2) solution was prepared in assay medium at a concentration of 200 U/mL Tested compounds were dissolved in DMSO at 1 mM and were further diluted in the IFN-α solution to a final concentration of 10 μM. 50 μL of this solution was then transferred to the plated cells to obtain a final concentration of IFN-α at 100 U/mL and compound at 5 μM (0.05% DMSO). The cells were incubated for 16h, after which 20 μL of 6xLiveBLAzer™ FRET B/G Substrate (CCF4-AM) mixture (prepared according to the manufacturer’s instructions) was added to each well. Plates were incubated at room temperature in the dark for 3h. Fluorescence was measured on a Tecan Infinite M200 plate reader at an excitation wavelength of 405 nm, and emission wavelengths of 465 nm and 535 nm. Background values (cell free wells at the same fluorescence wavelength) were subtracted from the raw fluorescence intensity values and the emission ratios were calculated as the ratio of background subtracted fluorescence intensities at 465 nm to background subtracted fluorescence intensities at 535 nm. The ISRE activity values for these compounds were 2-point normalized to yield activity of compound 1 + IFN-α as 200% and activity for IFN-α as 100%.
Cell viability assay
THP-1 cells were plated in 96-well plates (105 cells/well) in 100 μL RPMI supplemented with 10% FBS, 100 U/mL penicillin and 100 μg/ml streptomycin. Compounds were dissolved in DMSO at 1 mM stock solution and were further diluted to 10 μM in the assay medium. 100 μL of this solution was added to the cells to obtain a final compound concentration of 5 μM (0.05% DMSO). After 18h incubation, a solution of MTT in assay media (0.5 mg/mL) was added to each well and further incubated for 4 to 6 h, followed by addition of cell lysis buffer (15% w/v SDS and 0.12% v/v 12N HCl aqueous solution), incubated overnight, and then absorbance measured at 570 nm using 650 nm as reference using Tecan Infinite M200 plate reader.
Animals
Seven to nine-week-old C57BL/6 (wild-type, WT) mice were purchased from The Jackson Laboratories (Bar Harbor, MA). All animal experiments received prior approval from the UCSD Institutional Animal Care and Use Committee.
In vivo adjuvant activity study
WT mice (n=8 per group) were immunized in the gastronemius muscle with ovalbumin (20 μg/animal) mixed with MPLA (10 ng/animal) and compound 1 or 12 or 33 (50 nmol/animal) on days 0 and 21. On day 28, immunized mice were bled and OVA-specific IgG titers were measured by ELISA as previously described.60
Statistical analysis
Data are represented as mean ± standard error of the mean (SEM). Origin 7 (Origin Lab, Northampton, MA) graphing software was used for figure preparation while Prism 4 (GraphPad, San Diego, CA) software was used for statistical calculations.
Supplementary Material
Acknowledgements
We are grateful for the assistance provided by Dr. Yongxuan Su of the Department of Chemistry at University of California, San Diego for high resolution mass spectrometry.
Funding Sources
We acknowledge the NIH Adjuvant Discovery Program for funding (HHSN272200900034C and HHSN272201400051C, Principal Investigator-DAC). The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
ABBREVIATIONS
- APC
antigen presenting cells
- AS04
Adjuvant System 04
- DCs
dendritic cells
- DMF
N,N-dimethylformamide
- DMSO
dimethyl sulfoxide
- ELISA
enzyme-linked immunosorbent assay
- FDA
Food and Drug Administration
- FRET
Förster resonance energy transfer
- HRMS
high resolution mass spectrometry
- HTS
high-throughput screening
- IFN
interferon
- Ig
immunoglobulin
- IL
interleukin
- IRAK-M
interleukin-1 receptor-associated kinase-M
- ISRE
interferon stimulating response element
- LPS
lipopolysaccharide
- MPLA
monophosphoryl Lipid A
- MTT
(3-[4,5-dimethylthiazol-2-yl]-2,5-dipheyl tetrazolium bromide)
- NF-κB
nuclear factor kappa B
- NLR
nucleotide-binding oligomerization domain-like receptors (NLRs)
- OD
optical density
- OVA
ovalbumin
- PRR
pattern recognition receptors
- RLR
RIG-I-like receptors
- RR
response ratio
- SAR
structure-activity relationship
- SEAP
secreted embryonic alkaline phosphatase
- THF
tetrahydrofuran
- Th
T helper
- TLC
thin layer chromatography
- TLR
Toll-like receptor
Footnotes
Supporting Information. The following files are available free of charge. Supporting Information, Figure S1, Scheme S1, chemistry experimental section with compound names, reaction yields for syntheses of reagents 3a-f, 1H, 13C NMR spectra, HRMS characterization data, and LC-MS spectra for all the final compounds (PDF). Molecular formula strings for all the final compounds is also available (CSV).
Conflict of interest
All authors declare that there is no conflict of interest regarding the publication of this manuscript.
References
- 1.Shukla NM; Chan M; Hayashi T; Carson DA; Cottam HB Recent advances and perspectives in small-molecule TLR ligands and their modulators. ACS Med Chem Lett 2018, 9, 1156–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Reed SG; Orr MT; Fox CB Key roles of adjuvants in modern vaccines. Nat Med 2013, 19, 1597–1608. [DOI] [PubMed] [Google Scholar]
- 3.Shukla NM; Salunke DB; Balakrishna R; Mutz CA; Malladi SS; David SA Potent adjuvanticity of a pure TLR7-agonistic imidazoquinoline dendrimer. PLoS One 2012, 7, e43612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vasilakos JP; Tomai MA The use of toll-like receptor 7/8 agonists as vaccine adjuvants. Expert Rev Vaccines 2013, 12, 809–819. [DOI] [PubMed] [Google Scholar]
- 5.Maisonneuve C; Bertholet S; Philpott DJ; De Gregorio E Unleashing the potential of NOD- and toll-like agonists as vaccine adjuvants. Proc Natl Acad Sci U S A 2014, 111, 12294–12299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Basto AP; Leitao A Targeting TLR2 for vaccine development. J Immunol Res 2014, 2014, 619410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wheeler CM; Skinner SR; Del Rosario-Raymundo MR; Garland SM; Chatterjee A; Lazcano-Ponce E; Salmeron J; McNeil S; Stapleton JT; Bouchard C; Martens MG; Money DM; Quek SC; Romanowski B; Vallejos CS; Ter Harmsel B; Prilepskaya V; Fong KL; Kitchener H; Minkina G; Lim YK; Stoney T; Chakhtoura N; Cruickshank ME; Savicheva A; da Silva DP; Ferguson M; Molijn AC; Quint WG; Hardt K; Descamps D; Suryakiran PV; Karkada N; Geeraerts B; Dubin G; Struyf F Efficacy, safety, and immunogenicity of the human papillomavirus 16/18 AS04-adjuvanted vaccine in women older than 25 years: 7-year follow-up of the phase 3, double-blind, randomised controlled viviane study. Lancet Infect Dis 2016, 16, 1154–1168. [DOI] [PubMed] [Google Scholar]
- 8.Ho NI; Huis In ‘t Veld, L. G. M.; Raaijmakers, T. K.; Adema, G. J. Adjuvants enhancing cross-presentation by dendritic cells: The key to more effective vaccines? Front Immunol 2018, 9, 2874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Proietti E; Bracci L; Puzelli S; Di Pucchio T; Sestili P; De Vincenzi E; Venditti M; Capone I; Seif I; De Maeyer E; Tough D; Donatelli I; Belardelli F Type I IFN as a natural adjuvant for a protective immune response: lessons from the influenza vaccine model. J Immunol 2002, 169, 375–383. [DOI] [PubMed] [Google Scholar]
- 10.Pavot V; Climent N; Rochereau N; Garcia F; Genin C; Tiraby G; Vernejoul F; Perouzel E; Lioux T; Verrier B; Paul S Directing vaccine immune responses to mucosa by nanosized particulate carriers encapsulating NOD ligands. Biomaterials 2016, 75, 327–339. [DOI] [PubMed] [Google Scholar]
- 11.Probst P; Grigg JB; Wang M; Munoz E; Loo YM; Ireton RC; Gale M Jr.; Iadonato SP; Bedard KM A small-molecule IRF3 agonist functions as an influenza vaccine adjuvant by modulating the antiviral immune response. Vaccine 2017, 35, 1964–1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tovey MG; Lallemand C Adjuvant activity of cytokines. Methods Mol Biol 2010, 626, 287–309. [DOI] [PubMed] [Google Scholar]
- 13.Chan M; Hayashi T; Mathewson RD; Nour A; Hayashi Y; Yao S; Tawatao RI; Crain B; Tsigelny IF; Kouznetsova VL; Messer K; Pu M; Corr M; Carson DA; Cottam HB Identification of substituted pyrimido[5,4-b]indoles as selective toll-like receptor 4 ligands. J Med Chem 2013, 56, 4206–4223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Alving CR; Peachman KK; Rao M; Reed SG Adjuvants for human vaccines. Curr Opin Immunol 2012, 24, 310–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hyer RN; Janssen R 2287. Recently approved HEPLISAV-B(R) [hepatitis b vaccine (recombinant), adjuvanted] shows a higher proportion of subjects achieving seroprotection with a more consistent immune response compared with ENGERIX-B(R) [hepatitis b vaccine (recombinant)] in three comparative trials. Open Forum Infect Dis 2018, 5, S677–678. [Google Scholar]
- 16.Fraser CK; Diener KR; Brown MP; Hayball JD Improving vaccines by incorporating immunological coadjuvants. Expert Rev Vaccines 2007, 6, 559–578. [DOI] [PubMed] [Google Scholar]
- 17.Brady RC; Treanor JJ; Atmar RL; Keitel WA; Edelman R; Chen WH; Winokur P; Belshe R; Graham IL; Noah DL; Guo K; Hill H Safety and immunogenicity of a subvirion inactivated influenza A/H5N1 vaccine with or without aluminum hydroxide among healthy elderly adults. Vaccine 2009, 27, 5091–5095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mori A; Oleszycka E; Sharp FA; Coleman M; Ozasa Y; Singh M; O’Hagan DT; Tajber L; Corrigan OI; McNeela EA; Lavelle EC The vaccine adjuvant alum inhibits IL-12 by promoting PI3 kinase signaling while chitosan does not inhibit IL-12 and enhances th1 and th17 responses. Eur J Immunol 2012, 42, 2709–2719. [DOI] [PubMed] [Google Scholar]
- 19.Campbell JD Development of the CpG adjuvant 1018: A case study. Methods Mol Biol 2017, 1494, 15–27. [DOI] [PubMed] [Google Scholar]
- 20.Gürser M; Gregoriadis G Interleukin-2 as a co-adjuvant for liposomal tetanus toxoid. In Vaccines: New generation immunological adjuvants, Gregoriadis G; McCormack B; Allison AC, Eds. Springer US: Boston, MA, 1995; pp 45–50. [Google Scholar]
- 21.Beran J Safety and immunogenicity of a new hepatitis B vaccine for the protection of patients with renal insufficiency including pre-haemodialysis and haemodialysis patients. Expert Opin Biol Ther 2008, 8, 235–247. [DOI] [PubMed] [Google Scholar]
- 22.Fabrizi F; Cerutti R; Nardelli L; Tripodi F; Messa P HBV vaccination with fendrix is effective and safe in pre-dialysis CKD population. Clin Res Hepatol Gastroenterol 2019. [DOI] [PubMed] [Google Scholar]
- 23.Lin L; Parra MM; Sierra VY; Cespedes AS; Granados MA; Luque A; Damaso S; Castrejon Alba MM; Romano-Mazzotti L; Struyf F Safety and immunogenicity of the HPV-16/18 AS04-adjuvanted vaccine in 4–6-year-old girls: Results to month 12 from a randomized trial. Pediatr Infect Dis J 2018, 37, e93–e102. [DOI] [PubMed] [Google Scholar]
- 24.Schwarz T; Spaczynski M; Kaufmann A; Wysocki J; Galaj A; Schulze K; Suryakiran P; Thomas F; Descamps D Persistence of immune responses to the HPV-16/18 AS04-adjuvanted vaccine in women aged 15–55 years and first-time modelling of antibody responses in mature women: Results from an open-label 6-year follow-up study. Bjog 2015, 122, 107–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mutwiri G; Gerdts V; van Drunen Littel-van den Hurk S; Auray G; Eng N; Garlapati S; Babiuk LA; Potter A Combination adjuvants: The next generation of adjuvants? Expert Rev Vaccines 2011, 10, 95–107. [DOI] [PubMed] [Google Scholar]
- 26.Ebensen T; Delandre S; Prochnow B; Guzman CA; Schulze K The combination vaccine adjuvant system alum/c-di-AMP results in quantitative and qualitative enhanced immune responses post immunization. Front Cell Infect Microbiol 2019, 9, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ignacio BJ; Albin TJ; Esser-Kahn AP; Verdoes M Toll-like receptor agonist conjugation: A chemical perspective. Bioconjug Chem 2018, 29, 587–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Levast B; Awate S; Babiuk L; Mutwiri G; Gerdts V; van Drunen Littel-van den Hurk, S. Vaccine potentiation by combination adjuvants. Vaccines (Basel) 2014, 2, 297–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Forster R; Braun A; Worbs T Lymph node homing of T cells and dendritic cells via afferent lymphatics. Trends Immunol 2012, 33, 271–280. [DOI] [PubMed] [Google Scholar]
- 30.Qian C; Cao X Regulation of toll-like receptor signaling pathways in innate immune responses. Ann N Y Acad Sci 2013, 1283, 67–74. [DOI] [PubMed] [Google Scholar]
- 31.Turnis ME; Song XT; Bear A; Foster AE; Gottschalk S; Brenner MK; Chen SY; Rooney CM IRAK-M removal counteracts dendritic cell vaccine deficits in migration and longevity. J Immunol 2010, 185, 4223–4232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yuk JM; Shin DM; Lee HM; Kim JJ; Kim SW; Jin HS; Yang CS; Park KA; Chanda D; Kim DK; Huang SM; Lee SK; Lee CH; Kim JM; Song CH; Lee SY; Hur GM; Moore DD; Choi HS; Jo EK The orphan nuclear receptor SHP acts as a negative regulator in inflammatory signaling triggered by toll-like receptors. Nat Immunol 2011, 12, 742–751. [DOI] [PubMed] [Google Scholar]
- 33.Ho PC; Tsui YC; Feng X; Greaves DR; Wei LN NF-kB-mediated degradation of the coactivator RIP140 regulates inflammatory responses and contributes to endotoxin tolerance. Nat Immunol 2012, 13, 379–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu X; Zhan Z; Xu L; Ma F; Li D; Guo Z; Li N; Cao X MicroRNA 48/152 impair innate response and antigen presentation of TLR-triggered dendritic cells by targeting CaMkIIalpha. J Immunol 2010, 185, 7244–7251. [DOI] [PubMed] [Google Scholar]
- 35.Ma F; Liu X; Li D; Wang P; Li N; Lu L; Cao X MicroRNA-466l upregulates IL-10 expression in TLR-triggered macrophages by antagonizing RNA-binding protein tristetraprolin-mediated IL-10 mRMA degradation. J Immunol 2010, 184, 6053–6059. [DOI] [PubMed] [Google Scholar]
- 36.An H; Zhao W; Hou J; Zhang Y; Xie Y; Zheng Y; Xu H; Qian C; Zhou J; Yu Y; Liu S; Feng G; Cao X SHP-2 phosphatase negatively regulates the TRIF adaptor protein-dependent type I interferon and proinflammatory cytokine production. Immunity 2006, 25, 919–928. [DOI] [PubMed] [Google Scholar]
- 37.Kondo T; Kawai T; Akira S Dissecting negative regulation of toll-like receptor signaling. Trends Immunol 2012, 33, 449–458. [DOI] [PubMed] [Google Scholar]
- 38.Martin-Fontecha A; Lanzavecchia A; Sallusto F Dendritic cell migration to peripheral lymph nodes. Handb Exp Pharmacol 2009, 31–49. [DOI] [PubMed] [Google Scholar]
- 39.Kobayashi K; Hernandez LD; Galan JE; Janeway CA Jr.; Medzhitov R; Flavell RA IRAK-M is a negative regulator of toll-like receptor signaling. Cell 2002, 110, 191–202. [DOI] [PubMed] [Google Scholar]
- 40.Kim KI; Malakhova OA; Hoebe K; Yan M; Beutler B; Zhang DE Enhanced antibacterial potential in UBP43-deficient mice against salmonella typhimurium infection by up-regulating type I IFN signaling. J Immunol 2005, 175, 847–854. [DOI] [PubMed] [Google Scholar]
- 41.Chan M; Ahmadi A; Yao S; Sato-Kaneko F; Messer K; Pu M; Nguyen B; Hayashi T; Corr M; Carson DA; Cottam HB; Shukla NM Identification of biologically active pyrimido[5,4-b]indoles that prolong NF-kB activation without intrinsic activity. ACS Comb Sci 2017, 19, 533–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shukla NM; Arimoto KI; Yao S; Fan JB; Zhang Y; Sato-Kaneko F; Lao FS; Hosoya T; Messer K; Pu M; Cottam HB; Carson DA; Hayashi T; Zhang DE; Corr M Identification of compounds that prolong type I interferon signaling as potential vaccine adjuvants. SLAS Discov 2018, 23, 960–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schenone M; Dancik V; Wagner BK; Clemons PA Target identification and mechanism of action in chemical biology and drug discovery. Nat Chem Biol 2013, 9, 232–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pan S; Zhang H; Wang C; Yao SC; Yao SQ Target identification of natural products and bioactive compounds using affinity-based probes. Nat Prod Rep 2016, 33, 612–620. [DOI] [PubMed] [Google Scholar]
- 45.Smith E; Collins I Photoaffinity labeling in target- and binding-site identification. Future Med Chem 2015, 7, 159–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sumranjit J; Chung SJ Recent advances in target characterization and identification by photoaffinity probes. Molecules 2013, 18, 10425–10451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kan T; Kita Y; Morohashi Y; Tominari Y; Hosoda S; Tomita T; Natsugari H; Iwatsubo T; Fukuyama T Convenient synthesis of photoaffinity probes and evaluation of their labeling abilities. Org Lett 2007, 9, 2055–2058. [DOI] [PubMed] [Google Scholar]
- 48.Ban HS; Naik R; Kim HM; Kim BK; Lee H; Kim I; Ahn H; Jang Y; Jang K; Eo Y; Song KB; Lee K; Won M Identification of targets of the HIF-1 inhibitor IDF-11774 using alkyne-conjugated photoaffinity probes. Bioconjug Chem 2016, 27, 1911–1920. [DOI] [PubMed] [Google Scholar]
- 49.Kawada K; Dolence EK; Morita H; Kometani T; Watt DS; Balapure A; Fitz TA; Orlicky DJ; Gerschenson LE Prostaglandin photoaffinity probes: Synthesis and biological activity of azide-substituted 16-phenoxy- and 17-phenyl-PGF2 alpha prostaglandins. J Med Chem 1989, 32, 256–264. [DOI] [PubMed] [Google Scholar]
- 50.Shukla NM; Mutz CA; Ukani R; Warshakoon HJ; Moore DS; David SA Syntheses of fluorescent imidazoquinoline conjugates as probes of toll-like receptor 7. Bioorg Med Chem Lett 2010, 20, 6384–6386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shukla NM; Lewis TC; Day TP; Mutz CA; Ukani R; Hamilton CD; Balakrishna R; David SA Toward self-adjuvanting subunit vaccines: Model peptide and protein antigens incorporating covalently bound toll-like receptor-7 agonistic imidazoquinolines. Bioorg Med Chem Lett 2011, 21, 3232–3236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fagan V; Hussein WM; Su M; Giddam AK; Batzloff MR; Good MF; Toth I; Simerska P Synthesis, characterization and immunological evaluation of self-adjuvanting group a streptococcal vaccine candidates bearing various lipidic adjuvanting moieties. ChemBioChem 2017, 18, 545–553. [DOI] [PubMed] [Google Scholar]
- 53.Gential GPP; Hogervorst TP; Tondini E; van de Graaff MJ; Overkleeft HS; Codee JDC; van der Marel GA; Ossendorp F; Filippov DV Peptides conjugated to 2-alkoxy-8-oxo-adenine as potential synthetic vaccines triggering TLR7. Bioorg Med Chem Lett 2019, 29, 1340–1344. [DOI] [PubMed] [Google Scholar]
- 54.Li Q; Guo Z Recent advances in toll like receptor-targeting glycoconjugate vaccines. Molecules 2018, 23, E1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liao J; Pan B; Liao G; Zhao Q; Gao Y; Chai X; Zhuo X; Wu Q; Jiao B; Pan W; Guo Z Synthesis and immunological studies of beta-1,2-mannan-peptide conjugates as antifungal vaccines. Eur J Med Chem 2019, 173, 250–260. [DOI] [PubMed] [Google Scholar]
- 56.Nevagi RJ; Dai W; Khalil ZG; Hussein WM; Capon RJ; Skwarczynski M; Toth I Structure-activity relationship of group a streptococcus lipopeptide vaccine candidates in trimethyl chitosan-based self-adjuvanting delivery system. Eur J Med Chem 2019, 179, 100–108. [DOI] [PubMed] [Google Scholar]
- 57.Ramesh UV; Look GC; Singh R; Issakani SD Preparation of biaryl ether sulfonamides and related derivatives as ubiquitin ligase inhibitors. US20050009871A1, 2005.
- 58.Cooks T; Pateras IS; Tarcic O; Solomon H; Schetter AJ; Wilder S; Lozano G; Pikarsky E; Forshew T; Rosenfeld N; Harpaz N; Itzkowitz S; Harris CC; Rotter V; Gorgoulis VG; Oren M Mutant p53 prolongs NF-kB activation and promotes chronic inflammation and inflammation-associated colorectal cancer. Cancer Cell 2013, 23, 634–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Perez JM; Chirieleison SM; Abbott DW An IkB kinase-regulated feedforward circuit prolongs inflammation. Cell Rep 2015, 12, 537–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chan M; Hayashi T; Kuy CS; Gray CS; Wu CCN; Corr M; Wrasidlo W; Cottam HB; Carson DA Synthesis and immunological characterization of toll-like receptor 7 agonistic conjugates. Bioconjugate Chem 2009, 20, 1194–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.