

Abstract
The kinase AKT (also known as protein kinase B) is a key regulator of cell proliferation, survival, and metabolism. In addition to being activated by growth factors, AKT is activated in response to DNA damage. Here, we found that the DNA damage response kinase DNA-PK sustains cell survival through a phosphorylation event that leads to increased AKT activity. In various cancer and non-cancer cells in culture, DNA damage caused by ionizing radiation or topoisomerase inhibitors triggered DNA-PK–dependent phosphorylation of the mTOR complex 2 (mTORC2) subunit Sin1, which enabled interaction with the guanine nucleotide exchange factor ECT2. Depleting Sin1 or ECT2 or disrupting the protein interaction or catalytic function of ECT2 attenuated DNA damage-induced AKT activation, thereby enhancing cellular sensitivity to DNA damaging agents. Our findings elucidate a mechanism mediating DNA damage-induced AKT activation and cell survival.
INTRODUCTION
The AKT/PKB is a Ser/Thr kinase involving in virtually all hallmarks of cancer including proliferation, survival, metabolism, invasion and metastasis (1–3). Hyperactivation of AKT promotes cancer progression in multiple mouse models (4–6). Aberrant activation of AKT is also frequently observed in various types of human cancers including lung, breast, ovarian, gastric and pancreatic cancers (7–9), and associated with poor outcome largely due to relapse and resistance to chemo- and radio-therapy (10, 11).
Extensive studies have demonstrated that AKT is a critical regulator of genome stability by modulating DNA damage response and DNA repair (12, 13). For example, direct phosphorylation of BRCA1 by AKT inhibits BRCA1 and RAD51 foci formation at DNA damage sites, leading to deficiency in homologous recombination (14). AKT also suppresses non-homologous end joining pathway by phosphorylating XRCC4-like factor (XLF), leading to the dissociation of XLF from the X-ray repair cross-complementing protein 4/DNA ligase IV (XRCC4/LIG4) complex (15). In addition, AKT phosphorylates additional DNA damage-related factors [including but not limited to checkpoint kinase 1 (CHK1), DNA topoisomerase 2-binding protein 1 (TOPBP1), C-terminal binding protein 1 (CtIP), replication protein A (RPA), and post meiotic segregation increased 2 (PMS2), through which AKT regulates DNA damage sensing, DNA resection, and repair pathway choice (13). Therefore, it is likely that increased AKT activation promotes tumorigenesis and drug resistance both by inducing genome instability that facilitates accumulation of tumor-prone genetic alterations and by antagonizing DNA damage-induced apoptosis to maintain cell survival.
In the classical growth factor-induced activation model, AKT is phosphorylated and activated on plasma membrane by a signaling pathway consisting of phosphoinositide 3-kinase (PI3K), phosphoinositide dependent kinase 1 (PDK1), and mammalian target of rapamycin complex 2 (mTORC2) (1, 16). Specifically, PI3K phosphorylates PtdIns-4,5-P2 (PIP2) to generate PtdIns-3,4,5-P3 (PIP3), which acts as a second messenger binding the pleckstrin homology (PH) domain of AKT for its plasma membrane translocation (17–19). A full AKT activation event requires phosphorylation of AKT at Thr308 (AKT-pThr308) in the activation loop by PDK1 and Ser473 (AKT-pSer473) in the hydrophobic motif (HM) in a manner mediated by mTORC2 (20). Beyond growth signals, other pathophysiological cues also promote AKT activation, such as recently identified MERTK-mediated AKT phosphorylation in responding to apoptotic cells (21). Of note, DNA damage has been associated with its ability to trigger AKT phosphorylation and activation. In response to DNA damage, AKT-pThr308 was found to mainly occur in the cytoplasm in a PDK1-dependent manner (22, 23), whereas AKT-pSer473 was likely co-localized at DNA damage foci in nucleus (22, 24). A handful of kinases—including ataxia telangiectasia mutated protein (ATM), ataxia telangiectasia and Rad3-related protein (ATR), DNA-dependent protein kinase (DNA-PK) and mTORC2—have been reported to be required for DNA damage-induced AKT-pSer473 (23, 25–28). Here, we uncovered a molecular mechanism of DNA-PK-mediated activation of mTORC2/AKT in response to DNA damage.
RESULTS
AKT activation induced by DNA damage requires DNA-PK and mTORC2
Ionizing radiation (IR) and DNA-damaging agents, such as doxorubicin, etoposide and cisplatin, have been widely used to treat various cancers by inducing cell apoptosis (29, 30). However, activation of oncogenic signaling including the mTOR–AKT pathway has been shown to enable cancer cell evasion from lethal DNA damaging conditions (31). Consistent with previous studies (22–24), we found that IR or etoposide treatment induced phosphorylation of AKT at both Thr308 and Ser473 in MCF7 cells (Fig. 1A), and AKT-pSer473 primarily occurred in nucleus (Fig. 1B). Moreover, genetic ablation of PRKDC (the gene encoding DNA-PKcs, the catalytic subunit of DNA-PK) blocked AKT-pSer473 in response to irradiation and etoposide treatment in multiple cell lines including mouse embryonic fibroblasts (MEFs) (fig. S1, A and B), MCF7 (fig. S1C), HeLa (fig. S1D), U2OS (fig. S1E) and HCT116 (fig. S1F). Consistently, inhibition of DNA-PK by NU7026 (32) abolished AKT-pSer473 in irradiated MCF7 cells (fig. S1G). Considering that the mTORC2 complex is the major kinase responsible for AKT-pSer473 by growth factors (20, 33), we investigated whether it is also involved in DNA damage-stimulated AKT-pSer473. Notably, compared to wild-type (WT) counterparts (Sin1+/+ or Rictor+/+), AKT-pSer473 was largely abolished in Sin1−/− or Rictor−/− MEFs (Fig. 1, C and D). Since Sin1 and Rictor are essential components of the mTORC2 complex (34), our data suggest that mTORC2 is indispensable for AKT-pSer473 upon IR treatment. In further support of this notion, depletion of Rictor or Sin1 inhibited IR- or etoposide-induced AKT-pSer473 in several cancer cell lines (fig. S2, A to G). Moreover, comparable amount of Rictor and Sin1 were observed in both cytoplasm and nucleus (fig. S2H), which is a prerequisite for AKT-pSer473 occurring in nucleus under DNA damage conditions. In contrast, phosphorylation of AKT-Thr308 mainly distributed in the cytoplasm, where PDK1 primarily located (fig. S2, I and J). Lastly, a selective mTOR inhibitor PP242 (35) prevented AKT-pSer473 in doxorubicin-treated MCF7 cells (fig. S2K). Together, these results suggest that both DNA-PK and mTORC2 are required for phosphorylation of AKT-Ser473 in response to DNA damage in cells we examined.
Fig. 1. DNA damage-induced AKT-Ser473 phosphorylation is dependent on mTORC2.
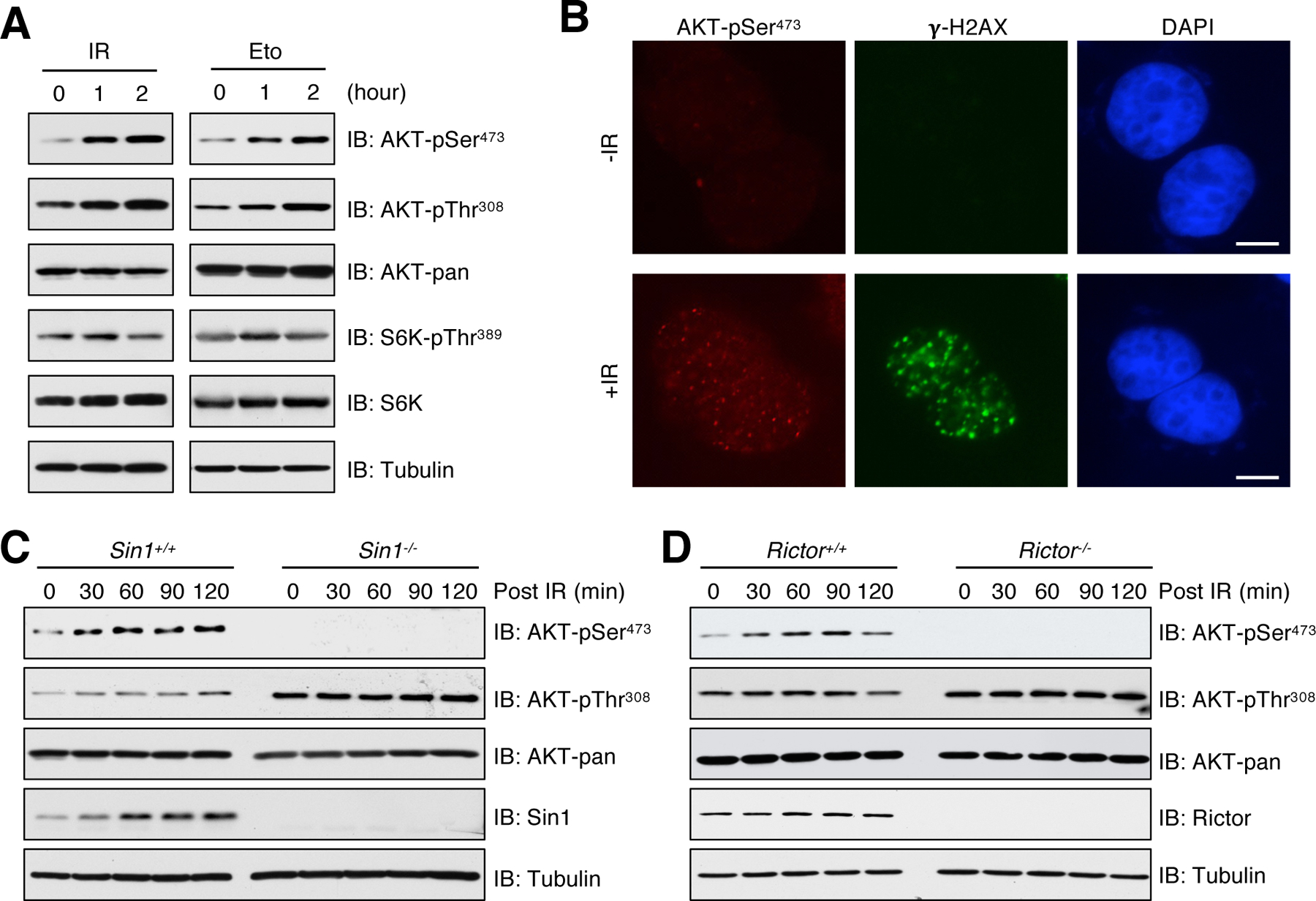
(A) MCF7 cells were irradiated (IR) with 10 Gy or treated with 10 μM etoposide (Eto) for indicated time points before harvesting for immunoblot (IB) analysis. n = 3 independent experiments. (B) Representative immunofluorescent images of AKT-pSer473 and γ-H2AX in MEFs irradiated with 10 Gy and recovered for 60 min. n = 2 independent experiments. Scale bar, 10 μm. (C) Sin1+/+ and Sin1−/− MEFs were irradiated with 10 Gy and then harvested in the indicated time points for IB analysis. n = 2 independent experiments. (D) Rictor+/+ and Rictor−/− MEFs were irradiated with 10 Gy and then harvested in the indicated time points for IB analysis. n = 2 independent experiments.
DNA-PK phosphorylates Sin1 at multiple SQ motifs
Having demonstrated the essential role of DNA-PK and mTORC2 in promoting AKT-pSer473, we sought to explore the potential mechanistic connection between these two kinases. DNA-PK prefers to phosphorylate Ser or Thr residue followed by a Gln (termed SQ or TQ), although it has also been shown to phosphorylate non-S/TQ motif in vitro (36–39). Thus, we tested whether mTORC2 specific components could be phosphorylated by DNA-PK. In irradiated cells, Sin1, but not Rictor, was phosphorylated at SQ motifs, which was detected by a specific anti-pSQ/pTQ antibody (Fig. 2A). Moreover, Sin1-pSQ was also detected in cells treated with etoposide and to a lesser extent, doxorubicin, but not insulin (Fig. 2B and fig. S3, A and B), indicating that this phosphorylation event is specifically induced by DNA damage. Furthermore, Sin1-pSQ signals were completely abolished in DNA-PKcs−/− MEFs (Fig. 2C), which prompted us to investigate whether Sin1 is a substrate of DNA-PK. We examined Sin1 protein sequence and identified four SQ motifs that are conserved in both human and mouse (Fig. 2D). Liquid chromatography-tandem mass spectrometry analysis confirmed phosphorylation of Sin1 at Ser186, Ser343, and Ser367 in cells (fig. S3C). Mutating all four Ser residues to Ala (4A), but not each of them singly, blocked DNA-PKcs-mediated phosphorylation of Sin1 in vitro (Fig. 2E). Moreover, IR or etoposide failed to promote Sin1-pSQ signals in Sin1–4A mutant cells (Fig. 2F and fig. S3, D and E). These results suggest that DNA-PK functions as a kinase phosphorylating Sin1-SQ motifs in response to DNA damage.
Fig. 2. Sin1 is phosphorylated at SQ motifs by DNA-PK.
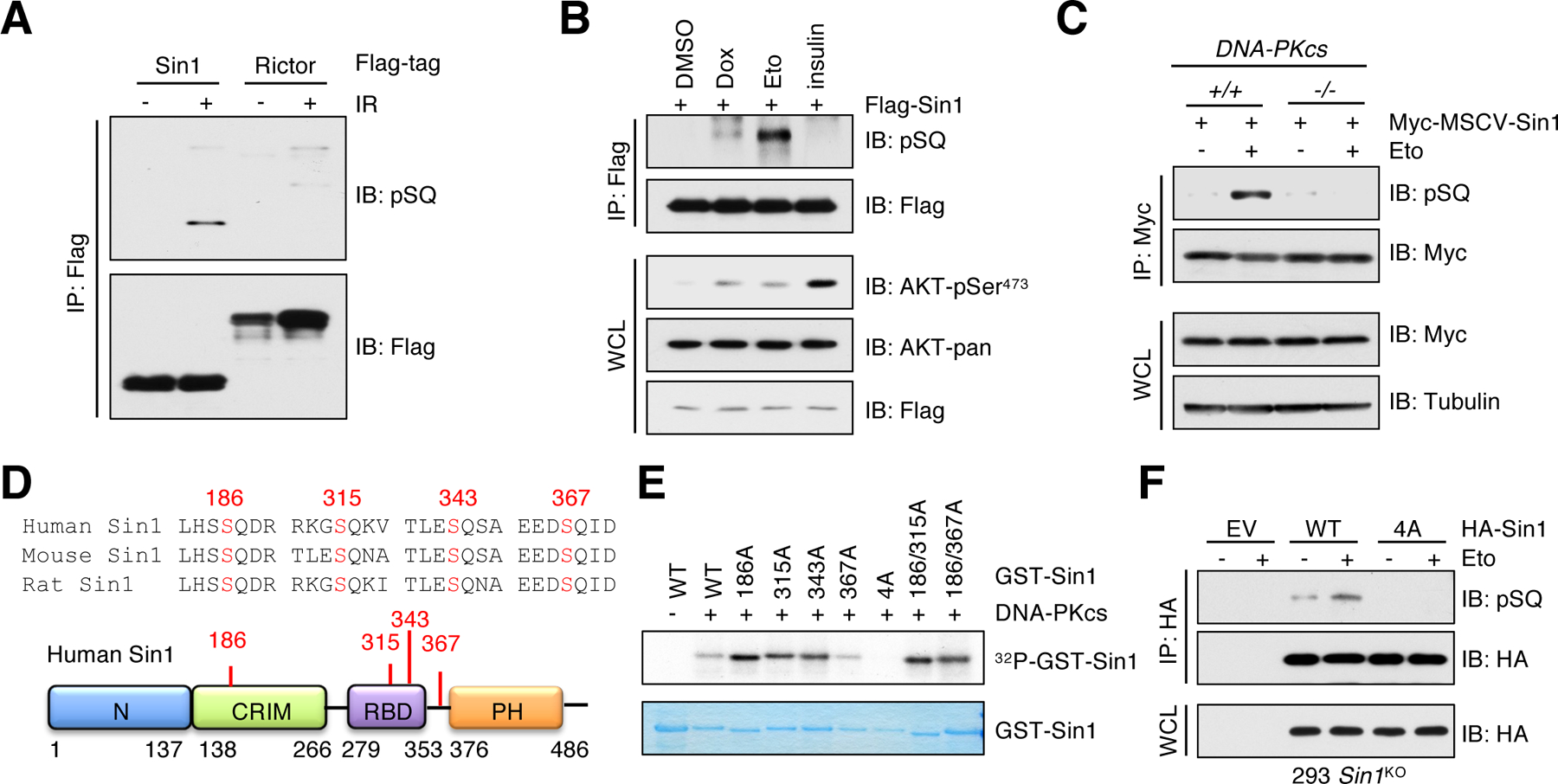
(A) IB analysis of Flag immunoprecipitates (IPs) derived from HEK293 cells transfected with Flag-Sin1 or Flag-Rictor. Cells were irradiated with 10 Gy and harvested at 60 min post irradiation (IR). n = 2 independent experiments. (B) IB analysis of whole cell lysates (WCL) and Flag IPs derived from HEK293 cells transfected with Flag-Sin1. Cells were treated with 1 μM doxorubicin (Dox) or 10 μM etoposide (Eto) for 60 min or serum-starved for 12 hours before 100 nM insulin treatment for 30 min. DMSO was used as a control. n = 2 independent experiments. (C) IB analysis of WCL and Myc IPs derived from DNA-PKcs+/+ and DNA-PKcs−/− MEFs stably expressing Myc-Sin1. Cells were treated with 10 μM etoposide for 60 min before harvesting. n = 2 independent experiments. (D) A schematic presentation of the SQ motifs in Sin1. (E) Recombinant GST-Sin1 proteins were purified from E. coli and the active DNA-PKcs was used as the source of kinase in the in vitro kinase assay. Phosphorus-32 (32P) isotope was used to detect phosphorylated Sin1 species. n = 2 independent experiments. (F) IB analysis of WCL and HA IPs derived from HEK293-Sin1KO cells stably expressing HA-Sin1-WT or Sin1–4A. EV (empty vector) was a negative control. Cells were treated with 10 μM etoposide for 60 min before harvesting. n = 3 independent experiments.
Phosphorylation of Sin1-SQ motifs promotes DNA damage-induced AKT-pSer473
Previous studies demonstrated that genetic ablation of Sin1 abolishes mTORC2 complex formation and AKT-Ser473 phosphorylation in response to growth factors, suggesting an essential role of Sin1 for functional mTORC2 (33, 40). To investigate whether Sin1-pSQ governs mTORC2 activation in response to DNA damage, we reconstituted Sin1-WT or Sin1–4A mutant at comparable levels in Sin1-knockout MCF7 cells (Sin1KO) and Sin1−/− MEFs. Notably, AKT-pSer473 was induced by etoposide in cells expressing Sin1-WT, but not the Sin1–4A mutant (Fig. 3, A and B and fig. S4A). Moreover, we found that compared with Sin1-WT, Sin1–4A immunoprecipitated from etoposide-treated cells was unable to enhance AKT-pSer473 in vitro (Fig. 3C). Given growth factors are another major source stimulating mTORC2 kinase activity (41), we next explored crosstalk between growth factor signaling and DNA damage on mTORC2/AKT activation. Both Sin1-WT and Sin1–4A expressing cells displayed similar levels of AKT-pSer473 in response to insulin stimulation (Fig. 3D), in keeping with our previous finding that Sin1-pSQ was not regulated by insulin (Fig. 2B). Our previous study demonstrated that Sin1-PH domain plays a critical role for its plasma translocation and mTORC2/AKT activation by growth factors (42). However, PH-deleted Sin1 still promoted AKT-pSer473 upon etoposide treatment (fig. S4B), indicating that Sin1-PH domain is dispensable for mTORC2 activation by DNA damage. Moreover, phosphorylation of Sin1 on both Thr86 and Thr398 has been shown to disrupt mTORC2 integrity and consequently inhibit growth factor-induced mTORC2/AKT activation (43). In keeping with this study, Sin1-WT, but not the phosphomimetic mutant Sin1-T86E/T398E, restored AKT-pSer473 in Sin1-knockout cells under insulin stimulation (fig. S4, C to E). Notably, Sin1-T86E/T398E mutant also failed to rescue AKT-pSer473 in Sin1-knockout cells in response to etoposide treatment (fig. S4, F to H). These results suggest that both Sin1-pSQ and mTORC2 integrity are likely required for mTORC2 activation under DNA damage conditions.
Fig. 3. Sin1–4A mutant inhibits AKT-pSer473 in response to DNA damage.
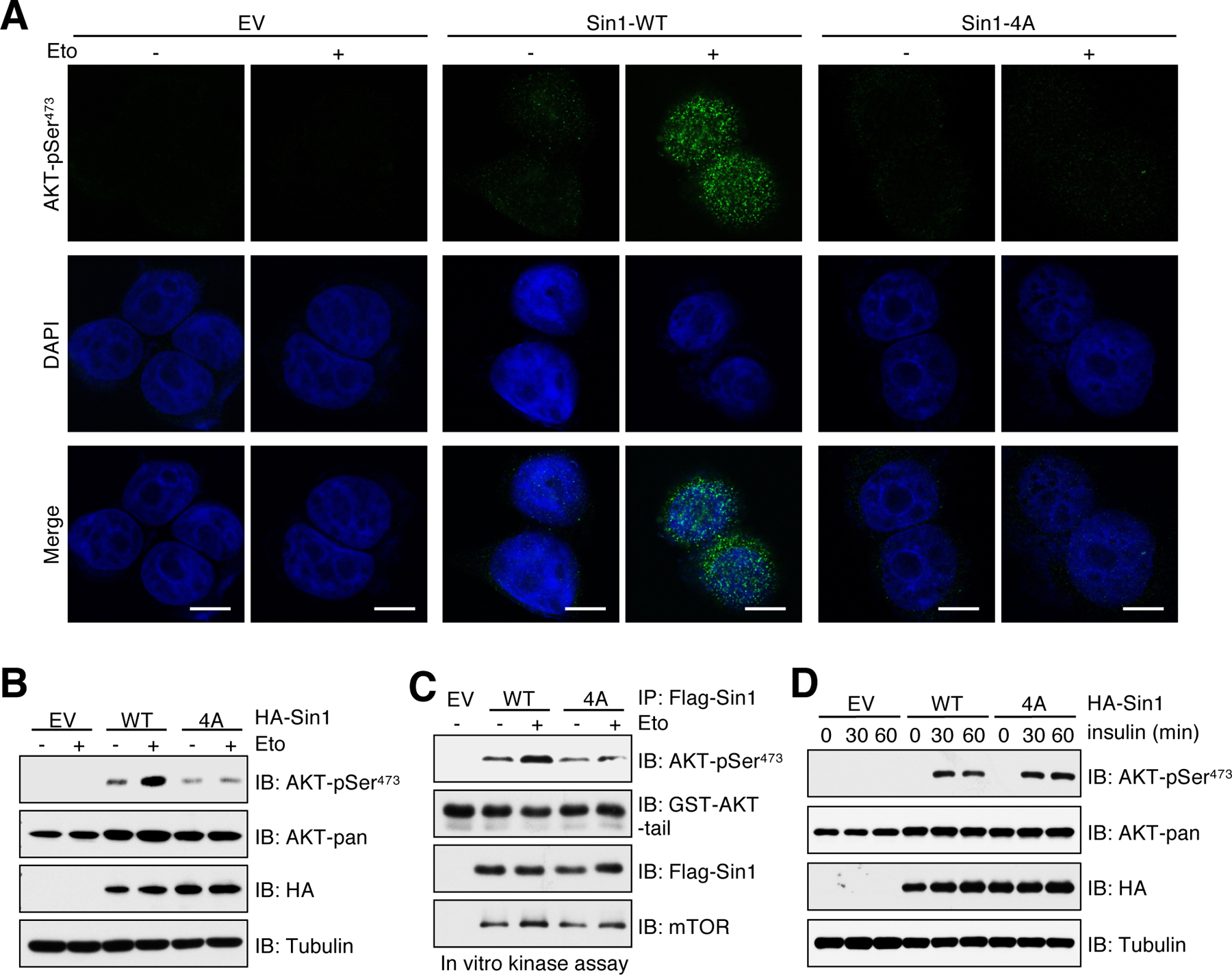
(A) MCF7-Sin1KO cells were reconstituted with Sin1-WT or Sin1–4A. EV was a negative control. Cells were treated with 10 μM etoposide (Eto) for 30 min before performing immunofluorescent staining. Representative images of AKT-pSer473 and DAPI are shown; n = 3 independent experiments. Scale bar, 10 μm. (B) IB analysis of WCL derived from MCF7-Sin1KO cells stably expressing HA-Sin1-WT or Sin1–4A. EV was a negative control. Cells were treated with 10 μM etoposide for 60 min before harvesting. n = 3 independent experiments. (C) In vitro kinase assays analysis of mTORC2 kinase activity. Flag-Sin1 IP was derived from MCF7-Sin1KO cells reconstituted with Flag-Sin1-WT or Sin1–4A. EV was a negative control. Cells were treated with 10 μM etoposide for 60 min before harvesting. n = 3 biological replicates. (D) MCF7-Sin1KO cells reconstituted with Flag-Sin1-WT or Sin1–4A were serum-starved for 12 hours and then stimulated with 100 nM insulin for 30 and 60 min before harvesting for IB analysis. n = 3 independent experiments.
Sin1-SQ phosphorylation mediates its interaction with ECT2 through the BRCT domain
To elucidate the molecular mechanisms by which DNA-PK-mediated phosphorylation of Sin1-SQ promotes mTORC2 kinase activity, we first examined if it regulates mTORC2 complex formation. Compared to Sin1-WT, Sin1–4A showed comparable interactions with mTOR, Rictor and GβL (also termed mLST8) in response to DNA damaging agents (fig. S5, A and B), suggesting that Sin1-pSQ unlikely affects the mTORC2 complex integrity.
It has been well characterized that tandem BRCT domains are the reader of pS/pTQ (44, 45). A number of BRCT domain-containing proteins play important roles on formation of protein complexes in DNA damage signaling cascades (46). Therefore, we hypothesized that Sin1-pSQ might be recognized by BRCT domain-containing protein(s), which in turn promotes mTORC2 activation (Fig. 4A). To this end, we performed immunoprecipitation assays and found that Sin1 specifically interacted with ECT2, but not BRCA1, p53-binding protein 1 (53BP1) and DNA ligase 4 (LIG4) (Fig. 4B), all of which contain BRCT domains (46) (fig. S5C). Moreover, the association between ECT2 and Sin1 was enhanced by IR or etoposide in a Sin1-pSQ dependent manner (Fig. 4, B and C and fig. S5D). Reciprocally, ECT2 specifically bound to Sin1, but not other subunits of the mTORC2 complex (Fig. 4D). We further validated the enhanced interaction between BRCT domains of ECT2 and Sin1-WT, but not Sin1–4A, under DNA damage conditions (Fig. 4E and fig. S5E). However, mutating all these Ser residues to Asp (4D) or Gln (4E), which is considered as phosphomimetic mutant, was incapable of enhancing its interaction with ECT2 and AKT-pSer473 (fig. S5, F and G). These results suggest that these 4D and 4E mutants may not faithfully mimic the protein confirmation changes as Sin1-SQ phosphorylation. Previous structural studies revealed that Thr153 and Lys195 residues in the first BRCT domain of ECT2 are essential for its BRCT domain binding to pSQ/pTQ motif (47). To further define the pivotal role of BRCT domains in mediating ECT2 binding to Sin1, we generated an ECT2-T153A/K195M mutant (termed ECT2-TK hereafter) and found that it impaired interaction with Sin1 (Fig. 4F and fig. S5, H to J). These data collectively demonstrate that DNA-PK-mediated phosphorylation of Sin1 at SQ motifs is specifically recognized by the BRCT domains of ECT2, leading to formation of the ECT2/Sin1 complex.
Fig. 4. ECT2 binds Sin1 through BRCT domain-mediated recognition of Sin1-pSQ.
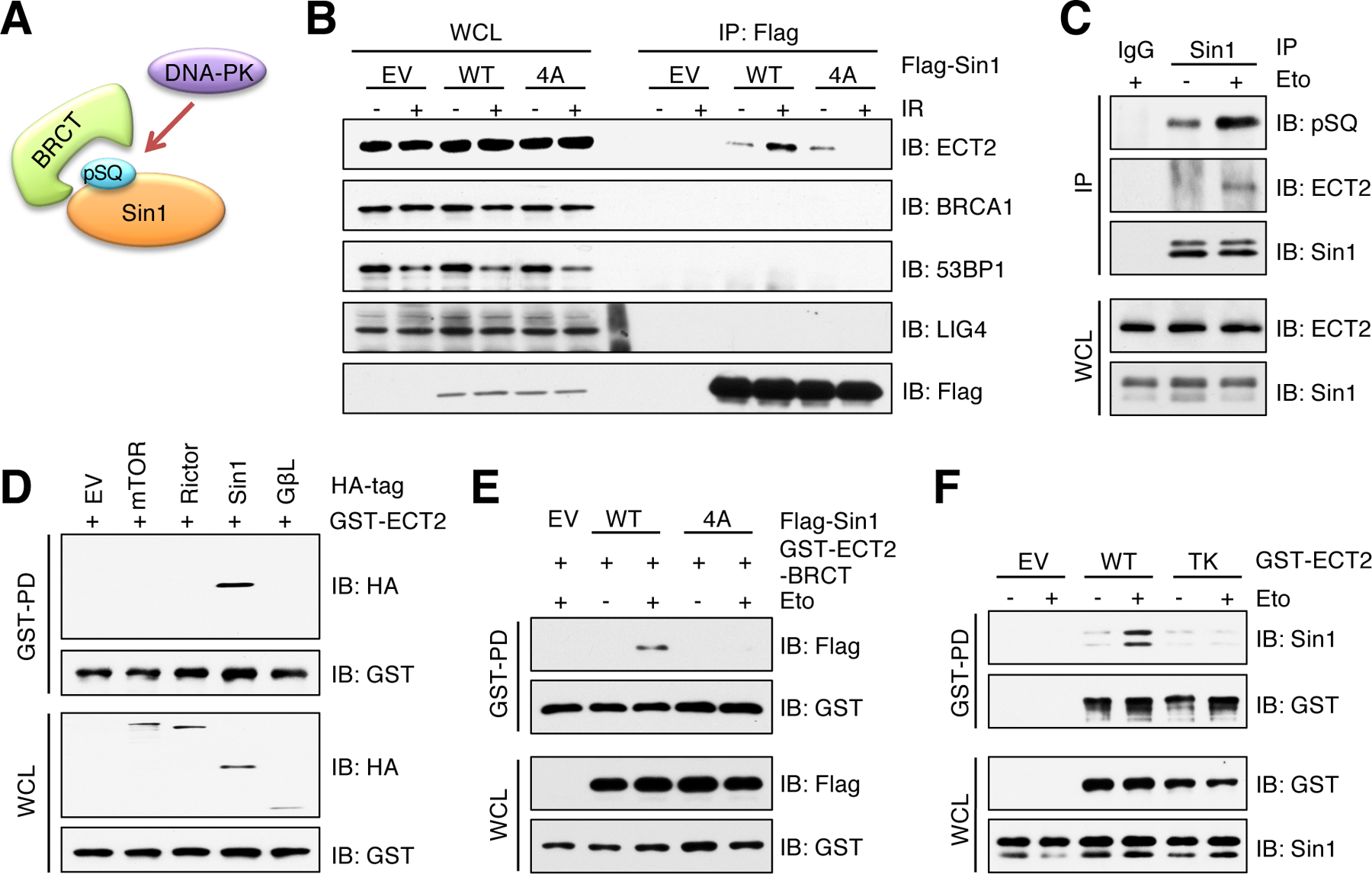
(A) A schematic presentation of hypothesis that BRCT domain interacts with DNA-PK mediated phosphorylation of Sin1-SQ. (B) IB analysis of WCL and Flag IPs derived from HEK293 cells transfected with Flag-Sin1-WT or Sin1–4A. EV was a negative control. Cells were irradiated (IR) with 10 Gy followed by 60 min incubation before harvesting. n = 2 independent experiments. (C) IB analysis of WCL and Sin1 IPs derived from HEK293 cells. IgG was a negative control. Cells were treated with 10 μM etoposide (Eto) for 60 min before harvesting. n = 3 independent experiments. (D) IB analysis of WCL and GST pulldown products (GST-PD) derived from HEK293 cells transfected with indicated HA-tagged constructs and CMV-GST-ECT2. EV was a negative control. n = 2 independent experiments. (E) IB analysis of WCL and GST-PD products derived from MCF7 cells transfected with ECT2-BRCT domains and Sin1-WT or Sin1–4A. EV was a negative control. Cells were treated with 10 μM etoposide for 60 min before harvesting. n = 2 independent experiments. (F) IB analysis of WCL and GST-PD products derived from HEK293 cells transfected with ECT2-WT or ECT2-TK. EV was a negative control. Cells were treated with 10 μM etoposide for 60 min before harvesting. n = 3 independent experiments.
ECT2 is required for DNA damage induced AKT-pSer473
Next, we sought to investigate the role of ECT2 in modulating mTORC2/AKT activation by DNA damage agents. Of note, endogenous ECT2 predominantly localized in nucleus (fig. S6, A and B), where AKT-pSer473 is observed upon DNA damage. Depletion of ECT2 prevented the induction of AKT-pSer473 in irradiated or etoposide-treated cells, including MCF7 (Fig. 5, A and B and fig. S6C), HeLa (fig. S6D) and MEFs (fig. S6E), accompanied by minor changes of AKT-pThr308. In contrast, insulin effectively induced AKT-pThr308 and pSer473 in both Ect2+/+ and Ect2−/− MEFs (fig. S6F). ECT2 depletion also attenuated protein kinase C (PKC) phosphorylation, another characterized mTORC2 substrate (48), in MCF7 cells treated with etoposide (fig. S6G). Moreover, Sin1 immunoprecipitated from etoposide-treated control cells, but not ECT2-depleted cells, phosphorylated AKT at Ser473 in vitro (Fig. 5C). Notably, ECT2-depleted cells exhibited similar reduction of AKT-pSer473 as cells expressing Sin1–4A, and depletion of ECT2 in Sin1–4A expressing cells had no additive effects in AKT-pSer473 reduction (Fig. 5D). Lastly, neither Sin1-pSQ nor mTORC2 integrity was affected by ECT2 depletion (fig. S6, H and I). These data suggest that ECT2 largely promotes mTORC2/AKT activation in responding to DNA damage through Sin1-pSQ.
Fig. 5. Functional ECT2 promotes AKT-pSer473 in response to DNA damage.
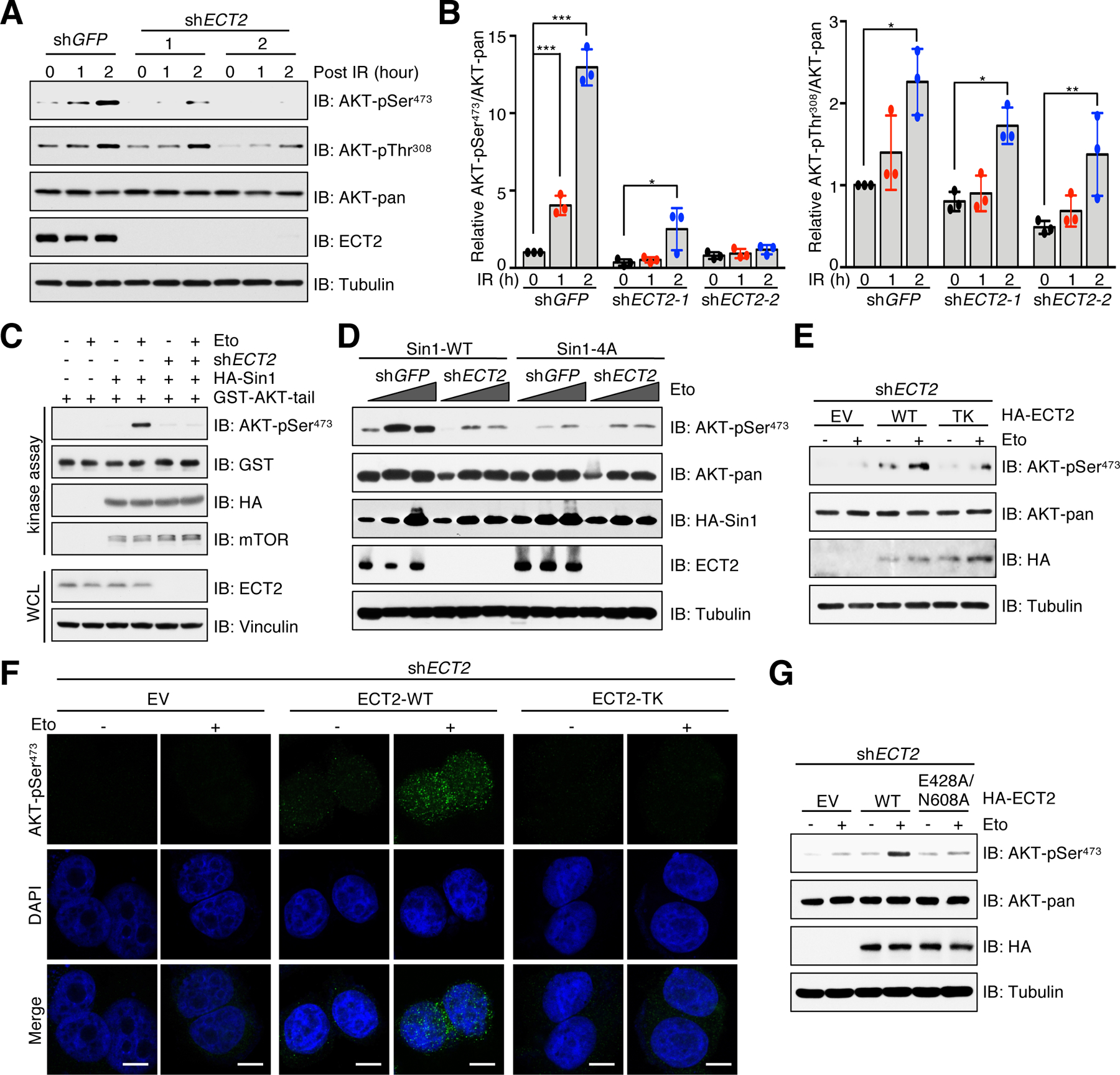
(A) IB analysis of WCL derived from ECT2-depleted MCF7 cells. shGFP was a negative control. Cells were irradiated (IR) with 10 Gy and incubated for indicated time periods before harvesting. n = 3 independent experiments. (B) Quantification of relative AKT phosphorylation by normalizing phospho-signal to total AKT. Data are means ± SD from three independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001 by one-way ANOVA and Tukey post hoc test. (C) In vitro kinase assays analysis of mTORC2 kinase activity. HA-Sin1 was purified from MCF7-Sin1KO cells that were reconstituted with HA-Sin1 and depleted of ECT2. Cells were treated with 10 μM etoposide (Eto) for 60 min before harvesting. n = 3 independent experiments. (D) MCF7-Sin1KO cells were reconstituted Sin1-WT or Sin1–4A and then infected with ECT2 shRNA. shGFP was a negative control. Cells were treated with 5 or 10 μM etoposide for 60 min before harvesting for IB analysis. n = 3 independent experiments. (E) ECT2-depleted MCF7 cells were reconstituted with EV, ECT-WT or ECT2-TK. Cells were treated with 10 μM etoposide for 60 min before harvesting for IB analysis. n = 3 independent experiments. (F) Cells generated in (E) were treated with 10 μM etoposide for 30 min before performing immunofluorescent staining. Representative images of AKT-pSer473 and DAPI are shown. n = 3 independent experiments. Scale bar, 10 μm. (G) ECT2-depleted MCF7 cells were reconstituted with EV, ECT-WT or ECT2-E428A/N608A. Cells were treated with 10 μM etoposide for 60 min before harvesting for IB analysis. n = 3 independent experiments.
To obtain more experimental evidence supporting the pivotal role of ECT2 in promoting AKT-pSer473, we reintroduced ECT2-WT or the ECT2-TK mutant into ECT2-depleted cells. Consistent with a loss of interaction between Sin1 and the ECT2-TK mutant, etoposide treatment failed to enhance AKT-pSer473 in cells expressing ECT2-TK compared to cells expressing ECT2-WT (Fig. 5, E and F). ECT2 not only contains a tandem of BRCT domains in its N-terminus, but it also has a Dbl homology (DH) domain next to the C-terminus (fig. S5C), which confers ECT2 guanine nucleotide exchange factor (GEF) activity for Rho family GTPases (49). To test whether the GEF activity of ECT2 is required for AKT-pSer473, we ectopically expressed a GEF-deficient mutant ECT2-E428A/N608A (50, 51) in ECT2-depleted cells. Although ECT2-WT and ECT2-E428A/N608A mutant comparably interacted with Sin1 (fig. S7A), AKT-pSer473 was only induced by etoposide in cells expressing ECT2-WT, but not the ECT2-E428A/N608A mutant (Fig. 5G and fig. S7B). It is well established that ECT2 expression fluctuates during the cell cycle and its GEF activity is required for cytokinesis (52–54). Consistent with these studies, cells expressing ECT2-TK or ECT2-E428A/N608A were defective in cytokinesis, leading to an increase of multinucleated cells (fig. S7C). However, AKT-pSer473 was induced upon DNA damage in both mononucleated and multinucleated cells expressing ECT2-WT (fig. S7D), but not ECT2-TK (fig. S7E) or ECT2-E428A/N608A (fig. S7F). These results suggest that ECT2 is likely capable of directly promoting mTORC2/AKT activation in a BRCT domain and GEF activity-dependent manner under DNA damage conditions, although the exact molecular mechanism underlying how ECT2 promotes mTORC2 kinase activity is unclear, which warrants more in-depth studies.
Deficiency in Sin1/ECT2 signaling sensitizes cells to DNA damaging agents
One important function of AKT is antagonizing apoptosis to enable cell survival under cytotoxic conditions (55). Having revealed the crucial role of the Sin1/ECT2 axis in mTORC2/AKT activation in response to DNA damage, we next examined whether abrogation of this axis affects cellular sensitivity to DNA damaging agents. Notably, reconstitution of Sin1-WT, but not Sin1–4A in MCF7-Sin1KO cells, significantly increased cell viability and colony formation upon etoposide treatment (Fig. 6A and fig. S8, A and B), which is in part due to reduction of cell apoptosis (Fig. 6B). Similarly, compared to control (shGFP), ECT2 depletion decreased colony formation ability upon IR (fig. S8, C and D). Moreover, compared to cells re-expressing ECT2-WT, cells expressing the ECT2-TK or ECT2-E428A/N608A mutant were more sensitive to etoposide in part due to enhanced cell apoptosis (Fig. 6, C to F). These results together indicate that the Sin1/ECT2 axis promotes cell survival in part by activating mTORC2/AKT pathway to prevent cell from apoptosis upon DNA damage.
Fig. 6. The Sin1/ECT2 complex promotes cell viability in response to DNA damaging agents.
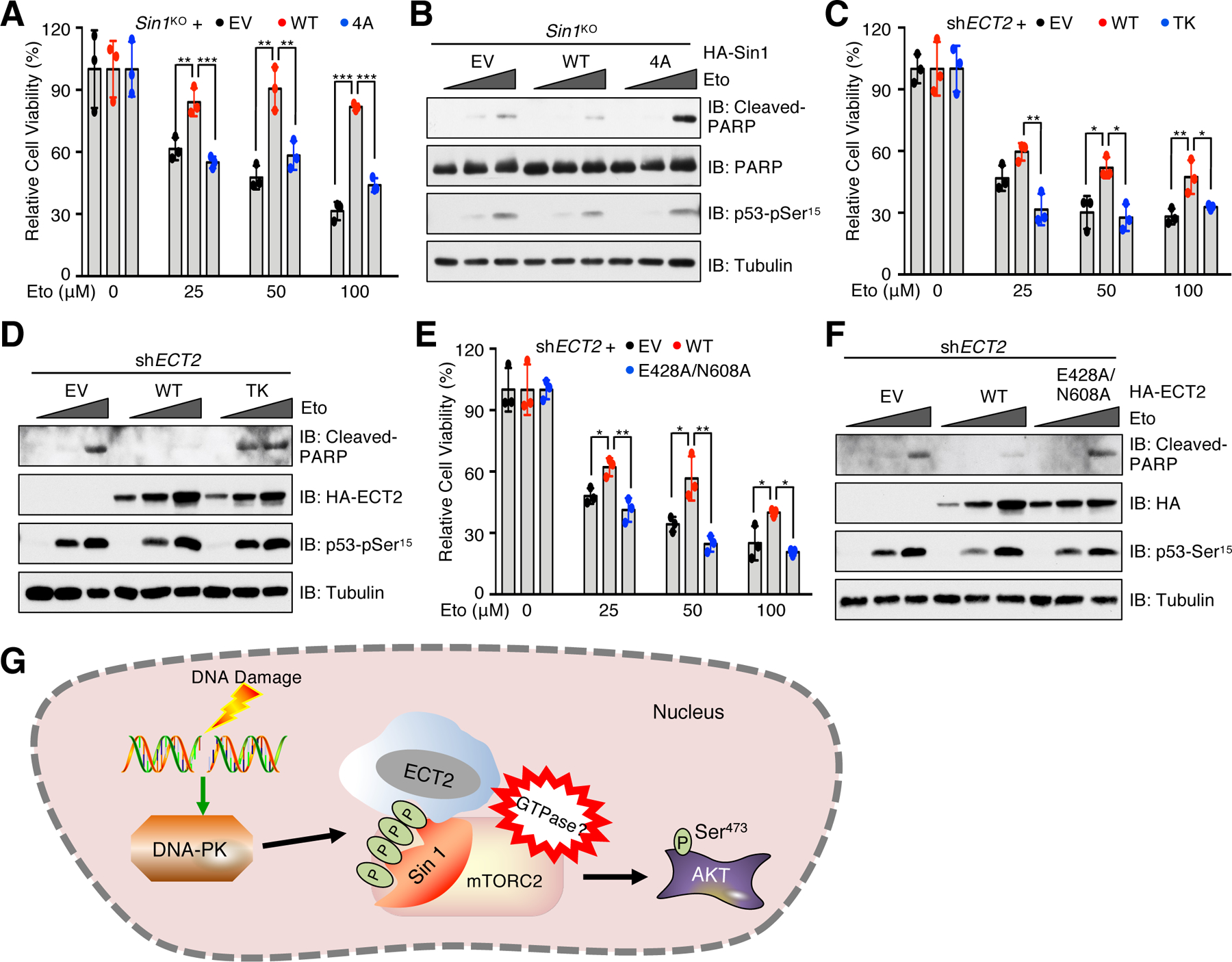
(A) MCF7-Sin1KO cells reconstituted with EV, Sin1-WT or Sin1–4A were treated with etoposide (Eto) at the indicated doses for 48 hours before examining cell viability. Data are means ± SD from three independent experiments. (B) IB analysis of WCL derived from cells generated in (A). The cells were treated with 0, 50 or 100 μM etoposide for 48 hours before harvesting. n = 3 independent experiments. (C) ECT2-depleted MCF7 cells reconstituted with EV, ECT2-WT or ECT2-TK were treated with etoposide at indicated doses for 48 hours before examining cell viability. Data are means ± SD from three independent experiments. (D) IB analysis of WCL derived from cells generated in (C). The cells were treated with 0, 100 or 300 μM etoposide for 48 hours before harvesting. n = 3 independent experiments. (E) ECT2-depleted MCF7 cells reconstituted with EV, ECT2-WT or ECT2-E428A/N608A were treated with etoposide at indicated doses for 48 hours before examining cell viability. Data are means ± SD from three independent experiments. In (A, C and E), *P < 0.05, **P < 0.01, ***P < 0.001 by one-way ANOVA and Tukey post hoc test. (F) IB analysis of WCL derived from cells generated in (E). The cells were treated with 0, 100 or 300 μM etoposide for 48 hours before harvesting. Blots represent 3 independent experiments. (G) A proposed model illustrating how the DNA-PK/Sin1/ECT2 signaling axis promotes mTORC2 activation to phosphorylate AKT-Ser473 in response to DNA damage.
DISCUSSION
Compared to the well-characterized molecular mechanisms of growth factor-induced PI3K-dependent AKT activation (2, 16, 56), mechanisms of DNA damage-stimulated AKT activation remain elusive. Previous studies showed that AKT-pSer473, but not AKT-pThr308, is induced in nucleus and colocalizes with γH2AX and ATM-pSer1981 at the DNA damage sites (22, 24), indicating that AKT-pSer473 is spatially separated from AKT-pThr308. Consistent with these studies, our study showed that AKT-pSer473, Rictor, Sin1 and ECT2 localize in nucleus, whereas AKT-pThr308 and PDK1 predominately locate in cytoplasm. Although previous study demonstrates that both DNA-PK and PDK1 are required for AKT-pThr308 in irradiated cells, it is unknown whether there is a connection between DNA-PK and PDK1. Consistently, our data showed that ECT2, but not DNA-PK, is dispensable for AKT-pThr308 in response to DNA damage, which might be due to the different cellular localization of ECT2 and PDK1/AKT-pThr308. However, it warrants a follow-up study to investigate how DNA-PK/PDK1 and/or other proteins, but not ECT2, promote AKT-pThr308 under DNA damage conditions.
Notably, several DNA damage response proteins including ATM, ATR, DNA-PK, meiotic recombination 11 (MRE11) and ring finger protein 168 (RNF168) have been reported to participate in DNA damage induced AKT-pSer473 in a PI3K-dependent or independent manner (12). Bozulic et al. found that both DNA-PK inhibitor and PI3K inhibitor suppress irradiation-induced AKT-pSer473 in HUVEC cells (23), whereas Fraser et al. revealed that in U2OS and DU145 cancer cells, nuclear AKT-pSer473 induced by irradiation relies on MRE11/ATM, but not DNA-PK, ATR nor PI3K (24). It is also controversial regarding whether mTORC2 or DNA-PK is responsible for AKT-pSer473 during DNA damage response. A study showed that depletion of DNA-PKcs, but not Rictor, blocks platinum-mediated AKT-pSer473 in platinum-resistant SKOV3 cells (27). In Rictor−/− MEFs, AKT-pSer473 induced by doxorubicin was partially inhibited (23). In contrast, knockdown of Rictor or administration of mTOR inhibitors markedly suppressed irradiation-stimulated AKT-pSer473 in breast cancer cells (28). Our study provided additional lines of evidence demonstrating the indispensable role of the DNA-PK/ECT2/Sin1 axis in phosphorylating AKT-Ser473 in nucleus in response to DNA damage (Fig. 6G). Collectively, these studies suggest a context-dependent phosphorylation of AKT-Ser473 by mTORC2 or DNA-PK upon DNA damage.
Posttranslational modifications (PTMs), particular phosphorylation and ubiquitination, play central roles in the initiation and execution of DNA damage response signaling cascade (57). Kinases (including DNA-PK, ATM and ATR) are the “writers” of protein phosphorylation, whereas three major types of domains [including BRCT domains, forkhead-associated (FHA) domains and 14–3-3 proteins] are the “readers” of phosphor-Ser/Thr (46). Of note, Sin1 has been shown to undergo phosphorylation and ubiquitination. Ribosomal S6 kinase (S6K)- or AKT-mediated phosphorylation of Sin1 on Thr86/Thr398 impaired its interaction with Rictor and mTOR, leading to mTORC2 inactivation in response to growth factor stimulation (43). In contrast, AKT-mediated phosphorylation of Sin1 on Thr86 alone led to activation of mTORC2 (58). The Sin1 protein is ubiquitinated by the CUL5-SOCS6 E3 complex, leading to its degradation (59). Here, we identified that activated DNA-PK directly phosphorylates Sin1 at four SQ motifs, which is subsequently recognized by the BRCT domain-containing protein ECT2, leading to mTORC2 activation and AKT-Ser473 phosphorylation. Thus, it will be interesting to identify additional Sin1 PTMs, which will help to elucidate the regulatory mechanisms of mTORC2 activation.
mTOR forms two distinct kinase complexes, termed mTORC1 and mTORC2. In the past twenty-five years, the knowledge of mTORC1 biology has been exponentially expanded. A number of components were identified to execute amino acid- and growth factor-induced activation of mTORC1 signaling (60, 61). A landmark finding is the discovery of tuberous sclerosis complex (TSC) that functions as a GTPase-activating protein (GAP) to inhibit RAS homolog enriched in brain (Rheb) activity and mTORC1 activation induced by growth factors (62). Another milestone in the mTORC1 research field is the identification of the RAS-related GTPases (RagA-D) that dictate mTORC1 core complex translocation to the lysosome for amino acid-induced mTORC1 activation (63, 64). Based on these findings, subsequent studies identified the Ragulator complex and GAP activity toward Rags 1 (GATOR1) complex as the GEF and GAP for RagA/B GTPases, respectively, and the folliculin (FLCN) complex as the GAP for RagC/D GTPases (60, 61). Therefore, the Rags and Rheb GTPases together with their upstream GEFs/GAPs form the foundation of mTORC1 signaling. Similar to the mTORC1 pathway, studies have shed light on the activation mTORC2 by small GTPases. Rac family small GTPase 1 (Rac1) directly binds mTOR to promote both mTORC1 and mTORC2 activation independent of its nucleotide loading activity and of the PI3K/AKT pathway (65). Moreover, RAS directly associates with both mTOR and Sin1 to enable mTORC2 activation at the plasma membrane (66). Of note, RAS may also promotes mTORC2 kinase activity through the PI3K pathway (67). RIT, another member of the RAS family GTPases, interacts with Sin1 to promote oxidative stress-induced mTORC2 activation (68). Moreover, GDP-bound RacE associates with Tor (mTOR homologue) and PiaA (Rictor homologue) to promote mTORC2/AKT signaling in Dictyostelium cells, but unlikely in mammalian cells (69). Our study found that ECT2, a GEF of the Rho family GTPases (70), interacts with Sin1 through BRCT domain-mediated recognition of Sin1-pSQ in the context of DNA damage, leading to activation of mTORC2/AKT signaling pathway. However, it remains unknown whether the Rho family GTPases regulated by ECT2 play a role in promoting mTORC2 kinase activity during DNA damage response, which is an important question and warrants a follow-up study.
MATERIALS AND METHODS
Cell culture and reagents
HEK293, HEK293T, HeLa, MCF7, U2OS and HCT116 cells were obtained from American Type Culture Collection (ATCC). Sin1+/+ and Sin1−/− MEFs were provided by Dr. Bing Su (Shanghai JiaoTong University, China). Rictor+/+ and Rictor−/− MEFs were obtained from Dr. Mark Magnuson (Vanderbilt University School of Medicine, USA). DNA-PKcs+/+ and DNA-PKcs−/− MEFs were provided by Dr. Cyrus Vaziri (University of North Carolina at Chapel Hill). Ect2fl/fl MEFs were obtained from Dr. Channing Der (The University of North Carolina at Chapel Hill). All cell lines were maintained in Dulbecco’s Modified Eagle’s Medium (DMEM) containing 10% fetal bovine serum (FBS), 100 units/ml penicillin and 100 μg/ml streptomycin. All the cell lines were tested to confirm no mycoplasma contamination. Etoposide (E1383), Doxorubicin hydrochloride (D1515) and PP242 (475988) were purchased from Sigma Aldrich. KU-55933 (S1092), VE-821 (S8007) and NU7026 (S2893) were purchased from Selleckchem. Insulin (41400045) was purchased from Thermo Fisher.
Transfection and Viral Infection
For protein expression in cells, transfection was performed using Lipofectamine 3000 and Plus reagents following manufacturer’s instructions. For lentiviral virus production, 293FT cells were transfected with target plasmid and packaging plasmids (pMD2G and psPAX2) with Polyethylenimine (PEI). Twenty-four hours post transfection, fresh medium was replaced. Virus containing supernatants were harvested at forty-eight hours post transfection and filtered with 0.45 μm PES filter. Targeted cells were infected with virus and selected with hygromycin (200 μg/ml), puromycin (1 to 2 μg/ml) or blasticidin (10 μg/ml) for four days to eliminate the non-infected cells.
Plasmid construction
pLJM1-hygro-HA-Sin1 and pLJM1-hygro-HA-ECT2 were generated by cloning the corresponding cDNA into the pLJM1-HA vector. CMV-GST-ECT2 and CMV-GST-ECT2-BRCT were generated by cloning the corresponding cDNA into the CMV-GST vector. Flag-Rictor was generated by cloning the corresponding cDNA into the super-Flag vector. Flag-Sin1, HA-mTOR, HA-Sin1, HA-Rictor, HA-GβL and GST-AKT-tail were described previously (43, 71). All mutants were generated using the QuikChange XL site-directed mutagenesis kit (Agilent) according to the manufacturer’s instructions.
shRNAs and sgRNAs
All shRNAs were purchased from Sigma Aldrich. shRictor: TRCN0000074291 and TRCN0000074290. shSin1: TRCN0000010762 and TRCN0000003151. shECT2: N0000299290 and TRCN0000299362. DNA-PKcs-sg1: TAAGTTCACCCAGAAAAGCG and DNA-PKcs-sg2: AAATGTGGATTCGAACAACA. sgRNA (GGACACCGATTTCCCCCCGC) was used to generated Sin1 knockout cells.
Antibodies
All antibodies were used at a dilution of 1:1,000–3,000 in TBST buffer with 5% non-fat milk for Western blotting. Antibodies to mTOR (2932), GβL (3274), Sin1 (12860), phospho-AKT Ser473 (4060), phospho-AKT Thr308 (13038), AKT (pan; 4691), Rictor (2114), phospho-p70 S6 kinase Thr389 (9234), p70 S6 kinase (9202), PDK1 (13037), histone H3 (4499), phospho-PKC (pan; 9371), PKCζ (9368), cleaved PARP (5625), DNA-PKcs (4602), phospho-ATM/ATR substrate (2851), phospho-p53 Ser15 (9286), GST (2622), rabbit antibody to HA (3724) and Myc-tag (2278), and mouse antibody to Myc-tag (2276) were purchased from Cell Signaling Technology. Mouse monoclonal antibody to HA (901503) was purchased from BioLegend. Antibodies to mouse IgG (A4416), rabbit IgG (A4914), rabbit-anti-Flag (F7425) and mouse-anti-Flag (F3165) were purchased from Sigma Aldrich. Antibodies to α-tubulin (66031–1-Ig) and PARP1 (13371–1-AP) were purchased from Proteintech. Antibodies to ECT2 (sc-1005, discontinued) and LIG4 (sc-28232, discontinued) were purchased from Santa Cruz Biotechnology. Antibody to BRCA1 (MABC 199) was purchased from EMD Millipore. Antibody to 53BP1 (612523) was purchased from BD Biosciences.
Immunoblot (IB) and immunoprecipitation (IP) analyses
Cells were lysed in EBC buffer (50 mM Tris pH 7.5, 120 mM NaCl and 0.5% NP-40) or Triton buffer (40 mM HEPES pH 7.4, 150 mM NaCl, 2.5 mM MgCl2, 1 mM EDTA and 1% Triton X-100) supplemented with protease inhibitors (Thermo Fisher, A32953) and phosphatase inhibitors (phosphatase inhibitor cocktail Set I and II, Calbiochem). The cell lysates were clarified by centrifugation at 13,200 r.p.m. at 4 °C for 10 min. The protein concentrations of lysates were measured using Nanodrop (Thermo Fisher) with Bio-Rad protein assay reagent. Equal amounts of whole cell lysates were resolved by SDS-PAGE and immunoblotted with indicated antibodies. For immunoprecipitation, 1000–3000 μg lysates were incubated with 50% slurry of agarose conjugated antibody for 3 to 5 hours at 4 °C or with the indicated antibody (3 to 5 μg) for 3 to 4 hours followed by one-hour incubation with Protein A Sepharose beads (GE Healthcare). Immunoprecipitants were washed three times with NETN buffer (20 mM Tris, pH 8.0, 150 mM NaCl, 1 mM EDTA and 0.5% NP-40) or Triton buffer before being resolved by SDS-PAGE.
Immunofluorescence staining
Cells were grown on glass coverslips for 24 hours before irradiated with 10 Gy or treated with 10 μM etoposide for 60 min. Cells were fixed with 3.7% formaldehyde in PBS for 15 min at room temperature and permeabilized with 0.1% Triton X-100 in PBS for 5 min. Cells were then rinsed three times with PBS, followed incubation with 5% BSA for 30 min. Cells were incubated with primary antibodies for 60 min, washed with PBST three times, and then incubated with Alexa-488 conjugated anti-Rabbit secondary antibody or Alexa-594 conjugated anti-mouse secondary antibody for 60 min. Nuclei were stained with 4,6-diamidino-2-phenylindole (DAPI) for 10 min. Coverslips were rinsed with PBS for two times and mounted onto slides using VECTASHIELD Antifade Mounting Media (Vector Laboratories). At least 100 cells were imaged for each sample.
In vitro kinase assays
mTORC2 in vitro kinase assays were performed as described previously (42). Briefly, HEK293 cells transfected with Flag-Sin1 were treated with 10 μM etoposide for 60 min before harvesting for immunoprecipitation (IP). Flag-Sin1 IPs were washed extensively in CHAPS buffer and used as the kinase sources. 10 μL of Flag-Sin1 IPs were incubated with 2 μg of GST-Akt1-tail (aa 409–480) and 200 μM cold ATP in the kinase assay buffer (50 μM HEPES pH 7.5, 10 μM MnCl2, 10 μM MgCl2, 2 μM DTT and 0.5 μM EGTA) at 30˚C for 60 min. The reactions were stopped by addition of SDS containing lysis buffer and resolved by SDS-PAGE. Phosphorylation of GST-Akt1-tail was detected using the AKT-pSer473 antibody.
DNA-PK in vitro kinase assays were performed as described previously (72). Briefly, various GST-Sin1 mutant proteins were purified from Escherichia coli (E. coli) and incubated with active DNA-PKcs and 10 μCi of [γ−32P]ATP in Z′ buffer (25 mM HEPES-KOH at pH 7.9, 50 mM KCl, 10 mM MgCl2, 20% glycerol, 0.1% NP-40, 1 mM DTT and 200 μM ATP) for at 30 °C for 30 min. The reaction was stop by adding SDS containing lysis buffer and subjected to SDS-PAGE, followed by autoradiography.
Analysis of Sin1-SQ phosphorylation in cells
HEK293 cells transfected with Flag-Sin1 were treated with 10 μM etoposide for 60 min before harvesting with EBC buffer for immunoprecipitation. The Flag immunoprecipitates were then resolved on SDS-PAGE and visualized by GelCode Blue Stain (Thermo Scientific, 24590). The band containing Flag-Sin1 was excised and digested with trypsin, followed analysis by C18 microcapillary liquid chromatography tandem mass spectrometry.
Cell viability assays
Cells were seeded in 96-well plates (2000 cells/well) for 24 hours and treated with indicated doses of etoposide for 48 hours. Assays were performed with the Cell Titer-Glo luminescent cell viability assay kit according to the manufacturer’s instructions (Promega).
Clonogenic survival assays
Cells were seeded in 6-well plates (500 cells/well) for 24 hours and irradiated with dose as indicated or treated with etoposide for 2 hours. Etoposide containing medium was then removed and replaced with fresh media. Cells were grown for 10 to 15 days until formation of visible colonies. Colonies were washed with PBS and fixed with 10% acetic acid/10% methanol for 20 min, then stained with 0.4% crystal violet for 20 min. After staining, the plates were washed with distilled water and air-dried.
Statistical analysis
Most experiments were repeated at least three times; N are noted in each legend. All quantitative data were presented as mean ± SD Results were analyzed by one-way ANOVA with Tukey post hoc test. *P< 0.05 was considered significant.
Supplementary Material
Acknowledgments:
We thank the members of Liu, Wei and Gan laboratories for critical reading and discussion of the manuscript and the visiting PhD students Lichong Yan and Ziqi Wang in Dr. Wei’s laboratory for the technical support. We also thank Dr. Bing Su (Shanghai JiaoTong University, China) for providing Sin+/+ and Sin1−/− MEFs, Dr. Mark Magnuson (Vanderbilt University School of Medicine, USA) for sharing Rictor+/+ and Rictor−/− MEFs, Dr. Cyrus Vaziri (University of North Carolina at Chapel Hill) for sharing the DNA-PKcs+/+ and DNA-PKcs−/− MEFs and Dr. Channing Der (The University of North Carolina at Chapel Hill) for sharing the Ect2 fl/fl MEFs and the ECT2 construct.
Funding:
This work was supported by the NIH grant R00CA207867 (W.G.), NIH grant R35CA253027 (W.W.) and NIH grant R01CA177910 (W.W.). This work was also supported in part by the Biostatistics Shared Resource, Hollings Cancer Center, Medical University of South Carolina (P30 CA138313). S.Y. was supported by Hollings Cancer Center Abney Postdoctoral Fellowship.
Footnotes
Competing interests: W.W. is a co-founder and consultant for the ReKindle Therapeutics. Other authors declare no conflicts of interest.
Data and materials availability: All data are present in the main text or the supplementary materials.
REFERENCES AND NOTES:
- 1.Hemmings BA, Restuccia DF, PI3K-PKB/Akt pathway. Cold Spring Harb Perspect Biol 4, a011189 (2012). doi: 10.1101/cshperspect.a011189; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Manning BD, Toker A, AKT/PKB Signaling: Navigating the Network. Cell 169, 381–405 (2017). doi: 10.1016/j.cell.2017.04.001; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hanahan D, Weinberg RA, Hallmarks of cancer: the next generation. Cell 144, 646–674 (2011). doi: 10.1016/j.cell.2011.02.013; [DOI] [PubMed] [Google Scholar]
- 4.Ackler S, Ahmad S, Tobias C, Johnson MD, Glazer RI, Delayed mammary gland involution in MMTV-AKT1 transgenic mice. Oncogene 21, 198–206 (2002). doi: 10.1038/sj.onc.1205052; [DOI] [PubMed] [Google Scholar]
- 5.Hutchinson J, Jin J, Cardiff RD, Woodgett JR, Muller WJ, Activation of Akt (protein kinase B) in mammary epithelium provides a critical cell survival signal required for tumor progression. Mol Cell Biol 21, 2203–2212 (2001). doi: 10.1128/MCB.21.6.2203-2212.2001; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Blanco-Aparicio C, Canamero M, Cecilia Y, Pequeno B, Renner O, Ferrer I, Carnero A, Exploring the gain of function contribution of AKT to mammary tumorigenesis in mouse models. PLoS One 5, e9305 (2010). doi: 10.1371/journal.pone.0009305; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bellacosa A, Kumar CC, Di Cristofano A, Testa JR, Activation of AKT kinases in cancer: implications for therapeutic targeting. Adv Cancer Res 94, 29–86 (2005). doi: 10.1016/S0065-230X(05)94002-5; [DOI] [PubMed] [Google Scholar]
- 8.Hers I, Vincent EE, Tavare JM, Akt signalling in health and disease. Cell Signal 23, 1515–1527 (2011). doi: 10.1016/j.cellsig.2011.05.004; [DOI] [PubMed] [Google Scholar]
- 9.Altomare DA, Testa JR, Perturbations of the AKT signaling pathway in human cancer. Oncogene 24, 7455–7464 (2005). doi: 10.1038/sj.onc.1209085; [DOI] [PubMed] [Google Scholar]
- 10.Avan A, Narayan R, Giovannetti E, Peters GJ, Role of Akt signaling in resistance to DNA-targeted therapy. World J Clin Oncol 7, 352–369 (2016). doi: 10.5306/wjco.v7.i5.352; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jabbarzadeh Kaboli P, Salimian F, Aghapour S, Xiang S, Zhao Q, Li M, Wu X, Du F, Zhao Y, Shen J, Cho CH, Xiao Z, Akt-targeted therapy as a promising strategy to overcome drug resistance in breast cancer - A comprehensive review from chemotherapy to immunotherapy. Pharmacol Res 156, 104806 (2020). doi: 10.1016/j.phrs.2020.104806; [DOI] [PubMed] [Google Scholar]
- 12.Liu Q, Turner KM, Alfred Yung WK, Chen K, Zhang W, Role of AKT signaling in DNA repair and clinical response to cancer therapy. Neuro Oncol 16, 1313–1323 (2014). doi: 10.1093/neuonc/nou058; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xu N, Lao Y, Zhang Y, Gillespie DA, Akt: a double-edged sword in cell proliferation and genome stability. J Oncol 2012, 951724 (2012). doi: 10.1155/2012/951724; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Plo I, Laulier C, Gauthier L, Lebrun F, Calvo F, Lopez BS, AKT1 inhibits homologous recombination by inducing cytoplasmic retention of BRCA1 and RAD51. Cancer Res 68, 9404–9412 (2008). doi: 10.1158/0008-5472.CAN-08-0861; [DOI] [PubMed] [Google Scholar]
- 15.Liu P, Gan W, Guo C, Xie A, Gao D, Guo J, Zhang J, Willis N, Su A, Asara JM, Scully R, Wei W, Akt-mediated phosphorylation of XLF impairs non-homologous end-joining DNA repair. Mol Cell 57, 648–661 (2015). doi: 10.1016/j.molcel.2015.01.005; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fruman DA, Chiu H, Hopkins BD, Bagrodia S, Cantley LC, Abraham RT, The PI3K Pathway in Human Disease. Cell 170, 605–635 (2017). doi: 10.1016/j.cell.2017.07.029; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Franke TF, Kaplan DR, Cantley LC, Toker A, Direct regulation of the Akt proto-oncogene product by phosphatidylinositol-3,4-bisphosphate. Science 275, 665–668 (1997). doi: 10.1126/science.275.5300.665; [DOI] [PubMed] [Google Scholar]
- 18.Stephens L, Anderson K, Stokoe D, Erdjument-Bromage H, Painter GF, Holmes AB, Gaffney PR, Reese CB, McCormick F, Tempst P, Coadwell J, Hawkins PT, Protein kinase B kinases that mediate phosphatidylinositol 3,4,5-trisphosphate-dependent activation of protein kinase B. Science 279, 710–714 (1998). doi: 10.1126/science.279.5351.710; [DOI] [PubMed] [Google Scholar]
- 19.Alessi DR, James SR, Downes CP, Holmes AB, Gaffney PR, Reese CB, Cohen P, Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Balpha. Curr Biol 7, 261–269 (1997). doi: 10.1016/s0960-9822(06)00122-9; [DOI] [PubMed] [Google Scholar]
- 20.Sarbassov DD, Guertin DA, Ali SM, Sabatini DM, Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 307, 1098–1101 (2005). doi: 10.1126/science.1106148; [DOI] [PubMed] [Google Scholar]
- 21.Jiang Y, Zhang Y, Leung JY, Fan C, Popov KI, Su S, Qian J, Wang X, Holtzhausen A, Ubil E, Xiang Y, Davis I, Dokholyan NV, Wu G, Perou CM, Kim WY, Earp HS, Liu P, MERTK mediated novel site Akt phosphorylation alleviates SAV1 suppression. Nat Commun 10, 1515 (2019). doi: 10.1038/s41467-019-09233-7; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Boehme KA, Kulikov R, Blattner C, p53 stabilization in response to DNA damage requires Akt/PKB and DNA-PK. Proc Natl Acad Sci U S A 105, 7785–7790 (2008). doi: 10.1073/pnas.0703423105; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bozulic L, Surucu B, Hynx D, Hemmings BA, PKBalpha/Akt1 acts downstream of DNA-PK in the DNA double-strand break response and promotes survival. Mol Cell 30, 203–213 (2008). doi: 10.1016/j.molcel.2008.02.024; [DOI] [PubMed] [Google Scholar]
- 24.Fraser M, Harding SM, Zhao H, Coackley C, Durocher D, Bristow RG, MRE11 promotes AKT phosphorylation in direct response to DNA double-strand breaks. Cell Cycle 10, 2218–2232 (2011). doi: 10.4161/cc.10.13.16305; [DOI] [PubMed] [Google Scholar]
- 25.Viniegra JG, Martinez N, Modirassari P, Hernandez Losa J, Parada Cobo C, Sanchez-Arevalo Lobo VJ, Aceves Luquero CI, Alvarez-Vallina L, Ramon y Cajal S, Rojas JM, Sanchez-Prieto R, Full activation of PKB/Akt in response to insulin or ionizing radiation is mediated through ATM. J Biol Chem 280, 4029–4036 (2005). doi: 10.1074/jbc.M410344200; [DOI] [PubMed] [Google Scholar]
- 26.Caporali S, Levati L, Starace G, Ragone G, Bonmassar E, Alvino E, D’Atri S, AKT is activated in an ataxia-telangiectasia and Rad3-related-dependent manner in response to temozolomide and confers protection against drug-induced cell growth inhibition. Mol Pharmacol 74, 173–183 (2008). doi: 10.1124/mol.107.044743; [DOI] [PubMed] [Google Scholar]
- 27.Stronach EA, Chen M, Maginn EN, Agarwal R, Mills GB, Wasan H, Gabra H, DNA-PK mediates AKT activation and apoptosis inhibition in clinically acquired platinum resistance. Neoplasia 13, 1069–1080 (2011). doi: 10.1593/neo.111032; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Silvera D, Ernlund A, Arju R, Connolly E, Volta V, Wang J, Schneider RJ, mTORC1 and −2 Coordinate Transcriptional and Translational Reprogramming in Resistance to DNA Damage and Replicative Stress in Breast Cancer Cells. Mol Cell Biol 37, (2017). doi: 10.1128/MCB.00577-16; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Baskar R, Lee KA, Yeo R, Yeoh KW, Cancer and radiation therapy: current advances and future directions. Int J Med Sci 9, 193–199 (2012). doi: 10.7150/ijms.3635; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cheung-Ong K, Giaever G, Nislow C, DNA-damaging agents in cancer chemotherapy: serendipity and chemical biology. Chem Biol 20, 648–659 (2013). doi: 10.1016/j.chembiol.2013.04.007; [DOI] [PubMed] [Google Scholar]
- 31.Delou JMA, Souza ASO, Souza LCM, Borges HL, Highlights in Resistance Mechanism Pathways for Combination Therapy. Cells 8, (2019). doi: 10.3390/cells8091013; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Veuger SJ, Curtin NJ, Richardson CJ, Smith GC, Durkacz BW, Radiosensitization and DNA repair inhibition by the combined use of novel inhibitors of DNA-dependent protein kinase and poly(ADP-ribose) polymerase-1. Cancer Res 63, 6008–6015 (2003). doi: [PubMed] [Google Scholar]
- 33.Jacinto E, Facchinetti V, Liu D, Soto N, Wei S, Jung SY, Huang Q, Qin J, Su B, SIN1/MIP1 maintains rictor-mTOR complex integrity and regulates Akt phosphorylation and substrate specificity. Cell 127, 125–137 (2006). doi: 10.1016/j.cell.2006.08.033; [DOI] [PubMed] [Google Scholar]
- 34.Oh WJ, Jacinto E, mTOR complex 2 signaling and functions. Cell Cycle 10, 2305–2316 (2011). doi: 10.4161/cc.10.14.16586; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Apsel B, Blair JA, Gonzalez B, Nazif TM, Feldman ME, Aizenstein B, Hoffman R, Williams RL, Shokat KM, Knight ZA, Targeted polypharmacology: discovery of dual inhibitors of tyrosine and phosphoinositide kinases. Nat Chem Biol 4, 691–699 (2008). doi: 10.1038/nchembio.117; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bannister AJ, Gottlieb TM, Kouzarides T, Jackson SP, c-Jun is phosphorylated by the DNA-dependent protein kinase in vitro; definition of the minimal kinase recognition motif. Nucleic Acids Res 21, 1289–1295 (1993). doi: 10.1093/nar/21.5.1289; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kim ST, Lim DS, Canman CE, Kastan MB, Substrate specificities and identification of putative substrates of ATM kinase family members. J Biol Chem 274, 37538–37543 (1999). doi: 10.1074/jbc.274.53.37538; [DOI] [PubMed] [Google Scholar]
- 38.Blackford AN, Jackson SP, ATM ATR, and DNA-PK: The Trinity at the Heart of the DNA Damage Response. Mol Cell 66, 801–817 (2017). doi: 10.1016/j.molcel.2017.05.015; [DOI] [PubMed] [Google Scholar]
- 39.Jette N, Lees-Miller SP, The DNA-dependent protein kinase: A multifunctional protein kinase with roles in DNA double strand break repair and mitosis. Prog Biophys Mol Biol 117, 194–205 (2015). doi: 10.1016/j.pbiomolbio.2014.12.003; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yang Q, Inoki K, Ikenoue T, Guan KL, Identification of Sin1 as an essential TORC2 component required for complex formation and kinase activity. Genes Dev 20, 2820–2832 (2006). doi: 10.1101/gad.1461206; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fu W, Hall MN, Regulation of mTORC2 Signaling. Genes (Basel) 11, (2020). doi: 10.3390/genes11091045; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu P, Gan W, Chin YR, Ogura K, Guo J, Zhang J, Wang B, Blenis J, Cantley LC, Toker A, Su B, Wei W, PtdIns(3,4,5)P3-Dependent Activation of the mTORC2 Kinase Complex. Cancer Discov 5, 1194–1209 (2015). doi: 10.1158/2159-8290.CD-15-0460; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu P, Gan W, Inuzuka H, Lazorchak AS, Gao D, Arojo O, Liu D, Wan L, Zhai B, Yu Y, Yuan M, Kim BM, Shaik S, Menon S, Gygi SP, Lee TH, Asara JM, Manning BD, Blenis J, Su B, Wei W, Sin1 phosphorylation impairs mTORC2 complex integrity and inhibits downstream Akt signalling to suppress tumorigenesis. Nat Cell Biol 15, 1340–1350 (2013). doi: 10.1038/ncb2860; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yu X, Chini CC, He M, Mer G, Chen J, The BRCT domain is a phospho-protein binding domain. Science 302, 639–642 (2003). doi: 10.1126/science.1088753; [DOI] [PubMed] [Google Scholar]
- 45.Manke IA, Lowery DM, Nguyen A, Yaffe MB, BRCT repeats as phosphopeptide-binding modules involved in protein targeting. Science 302, 636–639 (2003). doi: 10.1126/science.1088877; [DOI] [PubMed] [Google Scholar]
- 46.Mohammad DH, Yaffe MB, 14-3-3 proteins, FHA domains and BRCT domains in the DNA damage response. DNA Repair (Amst) 8, 1009–1017 (2009). doi: 10.1016/j.dnarep.2009.04.004; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zou Y, Shao Z, Peng J, Li F, Gong D, Wang C, Zuo X, Zhang Z, Wu J, Shi Y, Gong Q, Crystal structure of triple-BRCT-domain of ECT2 and insights into the binding characteristics to CYK-4. FEBS Lett 588, 2911–2920 (2014). doi: 10.1016/j.febslet.2014.07.019; [DOI] [PubMed] [Google Scholar]
- 48.Ikenoue T, Inoki K, Yang Q, Zhou X, Guan KL, Essential function of TORC2 in PKC and Akt turn motif phosphorylation, maturation and signalling. EMBO J 27, 1919–1931 (2008). doi: 10.1038/emboj.2008.119; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tatsumoto T, Xie X, Blumenthal R, Okamoto I, Miki T, Human ECT2 is an exchange factor for Rho GTPases, phosphorylated in G2/M phases, and involved in cytokinesis. J Cell Biol 147, 921–928 (1999). doi: 10.1083/jcb.147.5.921; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Justilien V, Ali SA, Jamieson L, Yin N, Cox AD, Der CJ, Murray NR, Fields AP, Ect2-Dependent rRNA Synthesis Is Required for KRAS-TRP53-Driven Lung Adenocarcinoma. Cancer Cell 31, 256–269 (2017). doi: 10.1016/j.ccell.2016.12.010; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Huff LP, Decristo MJ, Trembath D, Kuan PF, Yim M, Liu J, Cook DR, Miller CR, Der CJ, Cox AD, The Role of Ect2 Nuclear RhoGEF Activity in Ovarian Cancer Cell Transformation. Genes Cancer 4, 460–475 (2013). doi: 10.1177/1947601913514851; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhao WM, Fang G, MgcRacGAP controls the assembly of the contractile ring and the initiation of cytokinesis. Proc Natl Acad Sci U S A 102, 13158–13163 (2005). doi: 10.1073/pnas.0504145102; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Liot C, Seguin L, Siret A, Crouin C, Schmidt S, Bertoglio J, APC(cdh1) mediates degradation of the oncogenic Rho-GEF Ect2 after mitosis. PLoS One 6, e23676 (2011). doi: 10.1371/journal.pone.0023676; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kim JE, Billadeau DD, Chen J, The tandem BRCT domains of Ect2 are required for both negative and positive regulation of Ect2 in cytokinesis. J Biol Chem 280, 5733–5739 (2005). doi: 10.1074/jbc.M409298200; [DOI] [PubMed] [Google Scholar]
- 55.Franke TF, Hornik CP, Segev L, Shostak GA, Sugimoto C, PI3K/Akt and apoptosis: size matters. Oncogene 22, 8983–8998 (2003). doi: 10.1038/sj.onc.1207115; [DOI] [PubMed] [Google Scholar]
- 56.Vivanco I, Sawyers CL, The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat Rev Cancer 2, 489–501 (2002). doi: 10.1038/nrc839; [DOI] [PubMed] [Google Scholar]
- 57.Polo SE, Jackson SP, Dynamics of DNA damage response proteins at DNA breaks: a focus on protein modifications. Genes Dev 25, 409–433 (2011). doi: 10.1101/gad.2021311; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Humphrey SJ, Yang G, Yang P, Fazakerley DJ, Stockli J, Yang JY, James DE, Dynamic adipocyte phosphoproteome reveals that Akt directly regulates mTORC2. Cell Metab 17, 1009–1020 (2013). doi: 10.1016/j.cmet.2013.04.010; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cui B, Gong L, Chen M, Zhang Y, Yuan H, Qin J, Gao D, CUL5-SOCS6 complex regulates mTORC2 function by targeting Sin1 for degradation. Cell Discov 5, 52 (2019). doi: 10.1038/s41421-019-0118-6; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Saxton RA, Sabatini DM, mTOR Signaling in Growth, Metabolism, and Disease. Cell 168, 960–976 (2017). doi: 10.1016/j.cell.2017.02.004; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kim J, Guan KL, mTOR as a central hub of nutrient signalling and cell growth. Nat Cell Biol 21, 63–71 (2019). doi: 10.1038/s41556-018-0205-1; [DOI] [PubMed] [Google Scholar]
- 62.Huang J, Manning BD, The TSC1-TSC2 complex: a molecular switchboard controlling cell growth. Biochem J 412, 179–190 (2008). doi: 10.1042/BJ20080281; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sancak Y, Peterson TR, Shaul YD, Lindquist RA, Thoreen CC, Bar-Peled L, Sabatini DM, The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science 320, 1496–1501 (2008). doi: 10.1126/science.1157535; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kim E, Goraksha-Hicks P, Li L, Neufeld TP, Guan KL, Regulation of TORC1 by Rag GTPases in nutrient response. Nat Cell Biol 10, 935–945 (2008). doi: 10.1038/ncb1753; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Saci A, Cantley LC, Carpenter CL, Rac1 regulates the activity of mTORC1 and mTORC2 and controls cellular size. Mol Cell 42, 50–61 (2011). doi: 10.1016/j.molcel.2011.03.017; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kovalski JR, Bhaduri A, Zehnder AM, Neela PH, Che Y, Wozniak GG, Khavari PA, The Functional Proximal Proteome of Oncogenic Ras Includes mTORC2. Mol Cell 73, 830–844 e812 (2019). doi: 10.1016/j.molcel.2018.12.001; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Castellano E, Downward J, RAS Interaction with PI3K: More Than Just Another Effector Pathway. Genes Cancer 2, 261–274 (2011). doi: 10.1177/1947601911408079; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cai W, Andres DA, mTORC2 is required for rit-mediated oxidative stress resistance. PLoS One 9, e115602 (2014). doi: 10.1371/journal.pone.0115602; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Senoo H, Kamimura Y, Kimura R, Nakajima A, Sawai S, Sesaki H, Iijima M, Phosphorylated Rho-GDP directly activates mTORC2 kinase towards AKT through dimerization with Ras-GTP to regulate cell migration. Nat Cell Biol 21, 867–878 (2019). doi: 10.1038/s41556-019-0348-8; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Fields AP, Justilien V, The guanine nucleotide exchange factor (GEF) Ect2 is an oncogene in human cancer. Adv Enzyme Regul 50, 190–200 (2010). doi: 10.1016/j.advenzreg.2009.10.010; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhang J, Xu K, Liu P, Geng Y, Wang B, Gan W, Guo J, Wu F, Chin YR, Berrios C, Lien EC, Toker A, DeCaprio JA, Sicinski P, Wei W, Inhibition of Rb Phosphorylation Leads to mTORC2-Mediated Activation of Akt. Mol Cell 62, 929–942 (2016). doi: 10.1016/j.molcel.2016.04.023; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yavuzer U, Smith GC, Bliss T, Werner D, Jackson SP, DNA end-independent activation of DNA-PK mediated via association with the DNA-binding protein C1D. Genes Dev 12, 2188–2199 (1998). doi: 10.1101/gad.12.14.2188; [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.