

Abstract
While advances in the treatment of pediatric cancers have improved survival to > 80% across all tumor types, drug resistance continues to limit survival for a considerable number of patients. We review the known mechanisms of resistance in pediatric cancers, including processes that impair conventional chemotherapies, newer classes of targeted small molecule antineoplastic drugs, and monoclonal antibodies. We highlight similarities and differences in treatment approach and resistance between pediatric and adult cancers. We also discuss newer areas of research into drug resistance, including extracellular and immune factors.
Keywords: Tumor microenvironment, drug efflux, chemoresistance, inhibition of apoptosis, BCL2, pediatric cancer
Introduction
Pediatric cancer therapies have made significant advances over the last 70 years, with 5-year overall survival rates rising from < 20% in the 1960’s to > 80% today[1]. Those improvements have not been uniform, however, across pediatric cancer histologies, including significant mortality among patients with acute myeloid leukemia, high-risk neuroblastoma, metastatic sarcomas, and specific brain tumors[1,2]. Resistance to cancer therapies, including chemotherapies and radiation therapy, have been an area of study for many decades, identifying some key mechanisms that allow cancer cells to remain viable. The development of new therapeutic approaches, including tyrosine kinase inhibitors, monoclonal antibodies, and immunoncology approaches, have resulted in yet more resistance mechanisms. These various mechanisms include molecular changes at the intracellular, transcellular, and intercellular levels, and include effects at the genetic, epigenetic, transcriptional and translational levels.
Here, we review the various mechanisms of therapeutic resistance against cancer therapies. We begin with a review of common mechanisms of action of traditional antineoplastic therapies, including key pathways that can be exploited by cancer cells. We will then review established mechanisms of resistance to classical treatments, exploratory studies into resistance against modern targeted agents, and new directions of research into cancer cell viability, as they apply to childhood cancers. We specifically highlight similarities and differences in therapy resistance in pediatric and adult cancers, as well as some important approaches being taken by pediatric oncologists and researchers to tackling treatment resistance.
The battlefield of the cancer cell
Traditional chemotherapy and radiation therapy act through diverse mechanisms, including inhibition of cell division kinetics, inhibition of DNA replication, direct DNA damage, and metabolic inhibition. However, there are some cellular activities that must exist for these agents to act against cancer cells. First, the drug must be stably delivered to the cell through the blood supply[3]; some drugs additionally need to be converted from the prodrug to the active agent either at the liver[4,5] or, less commonly, in the target cell[6]. Second, the drug must be able to enter the cell, either through passive diffusion[7,8] or more commonly through exploitation of existing transport mechanisms[9]. Third, the drug must be retained in the cells and reach its target. Fourth, the drug must be able to bind its target. Fifth, the activity of the agent must be able to induce cytotoxicity; this can occur through a number of mechanisms, including intrinsic induction of apoptosis through DNA damage signaling and/or cytochrome C-mediated apoptosis, or through extrinsic activation of caspase-mediated apoptosis. Optionally, the agent must be able to pause cell proliferation by inhibiting cell cycling.
As an example, we can review doxorubicin-mediated cytotoxicity. Doxorubicin is an anthracycline antibiotic that can be derived from Streptomyces species, though it is most commonly generated through recombinant methods today. It is used against a number of childhood cancers including leukemias, lymphomas, and solid tumors[9]. Doxorubicin is a vesicant, so it is administered through central venous catheters directly into the bloodstream, where it is delivered to its targets. It can passively enter the cell but more commonly via the transmembrane channel SLC22A16[10]. Once in the cell, it can activate apoptosis either by DNA intercalation, activating DNA damage pathways, or by generation of reactive oxygen species, causing membrane damage and cytochrome C-mediated apoptosis[11]. Doxorubicin also induces G2 arrest through interference with DNA replication[12]. The activity of doxorubicin identifies key nodes of toxicity that cancer cells can then modify to maintain viability.
Specific considerations of pediatric cancers and pediatric cancer therapy
Just as children are not simply small adults, pediatric cancers and their treatment have significant differences from adult tumors. Whereas adult cancers, particularly carcinomas, harbor high rates of gene mutations and fusion oncogenes, pediatric cancers are comparatively genomically “quiet”[13]. Epigenetic dysregulation, due to various mechanisms including mutations or aberrant imprinting, seems to be of particular import in many types of pediatric cancers (reviewed in[14-20]). Additionally, segmental chromosomal changes, including segmental gains, losses, and translocations, are observed in multiple pediatric cancers and correlate with disease severity[21-30], suggesting that the key genetic changes that lead to malignancy may occur in a more cataclysmic fashion than in adult cancers. These features have had therapeutic implications historically and with regard to new drug development.
Pediatric cancer treatment regimens, like most cancers, were developed since the 1940’s[31] and based on the use of conventional chemotherapies that targeted key processes of DNA replication and cell division. Since then, two hallmarks of treatment have been (1) systemic chemotherapy use, with some augmentation with external beam radiation therapy and/or surgery; and (2) the use of multiagent regimens with dose intensification. The former was based on evidence of hematologic or lymphatic disease dissemination, as exemplified in patients with localized osteosarcoma who had metastatic recurrence despite surgery alone[32]. The latter led to marked improvements in long-term cures across pediatric tumor types (reviewed here[2]), in part by overcoming some of the resistance mechanisms described below. These approaches were possible due to increased hematopoietic recovery in children as compared to adults, improvements in supportive care including blood product transfusions, infection control measures, better baseline organ function in children, and a tolerance of heightened toxicity as a cost of improved survival[33]. This has resulted in long-term morbidity for survivors, including effects on growth, neurocognitive development, endocrine health, predisposition to metabolic syndrome, cardiotoxicity, pulmonary toxicity, nephrotoxicity, fertility issues, and secondary malignancy risk; these have been reviewed more comprehensively elsewhere[34-40]. These outcomes emphasize how the identification of treatment resistance mechanisms and resultant counterstrategies are needed to reduce treatment intensity and toxicity while improving survival, which we now discuss [Figure 1].
Figure 1.
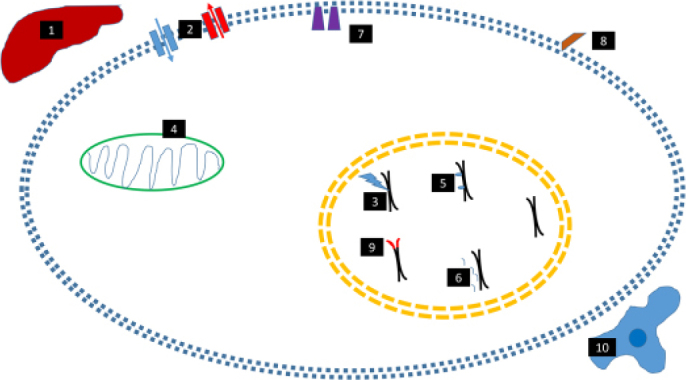
Mechanisms of therapeutic resistance in pediatric cancers. (1) Altered drug metabolism, either at the liver, in the bloodstream, or in the cell; (2) Altered drug influx/effux; (3) Disruption of response to DNA damage; (4) Inhibition of mitochondrial/cytochrome c/ROS-mediated cell death; (5) Epigenetic dysregulation; (6) miRNA dysregulation; (7) Mutations/amplification of tyrosine kinases; (8) Abnormal expression/downregulation of cell surface markers and targets of monoclonal antibodies and other immune therapies; (9) Activation of telomerase/ALT. (10) Tumor microenvironment enabling viability
Cellular mechanisms of therapeutic resistance and counterstrategies
Drug metabolism
A significant number of chemotherapeutic drugs used in pediatric cancers are actually prodrugs that require hepatic or intracellular metabolism for conversion to the active agent. As such, genetic variations in the involved enzymes, usually of the cytochrome P450 (CYP) family, can cause hypometabolism of these agents and diminish their efficacy[41-43]. These mutations or single nucleotide variations (SNVs) can be found at the germline or tumor somatic level. A different set of mutations and SNVs have been well described to be associated with drug hypermetabolism[41,44-46]. In particularly, variations in members of the CYP2 and CYP3A families have been associated with increased drug metabolism, decreased exposure of the tumor to the active agent, and decreased survival. Other involved proteins include glutathione s-transferases, thiopurine methyltransferase (TPMT), methylenetetrahydrofolate reductase, and UDP glucuronosyltransferase 1 family, polypeptide A1. These variations have been identified preclinically and clinically in patients with osteosarcoma[47,48], medulloblastoma[49], neuroblastoma[50], acute lymphoblastic leukemia[51-55]. Many of these studies have been observation and association studies, however, requiring additional work to define the mechanisms of resistance (e.g., does hepatic or tumoral metabolism matter more?) and how to attack these mechanisms without increasing drug toxicity.
One approach taken in pediatric acute lymphoblastic leukemia (ALL) relates to TPMT. Allelic variation and enzymatic activity have been strongly correlated with response to 6-mercaptopurine and 6-thioguanine, with either excessive myelotoxicity (with hypoactive variants) or hypermetabolism and lack of efficacy at standard dosing (with hyperactive variants)[56]. As a result, TPMT testing has now become the standard of care for children with ALL at the start of therapy[57]. While additional pharmacogenomics biomarkers have been studied in pediatric cancers specifically[58], the cost-benefit of testing of these genes continues to be evaluated. Testing of TPMT and other metabolism genes has focused primarily on dose reduction to avoid toxicity, but additional work needs to be done if pharmacogenomics testing could indicate safe dose escalation to overcome disease resistance.
Drug efflux and influx
Drug efflux pumps function in both prokaryotes and eukaryotes, thought to act to clear endogenous or exogenous toxins from normal cells. Of these pumps, the ATP binding cassette (ABC) proteins function “actively”, i.e., by direct energy conversion and conformational change to expel bound molecules out of the cell. The prototypical ABC protein, MDR1, also known as P-glycoprotein or ABCB1, was identified as overexpressed in pediatric solid tumors and leukemias since 1986, with correlation with drug resistance and poor survival[59-67]. Other ABC proteins, including MRP1 and the ABCG proteins, have also been implicated in drug resistance in pediatric cancers[68-79]. Overexpression of these proteins in pediatric tumors is usually induced through other oncogenic pathways, such as MYCN amplification or overexpression in neuroblastoma[60,80,81], CD133 expression in astrocytomas[82], or SORCIN or MDK expression in acute lymphoblastic leukemia[83,84]. There has also been concern that the tumor cells upregulate ABC protein expression in response to therapy, resulting in higher expression during therapy or at recurrence[60,85-87]. Germline or somatic polymorphisms of these proteins have also been associated with increased activity, decreased drug exposure, and worse survival in a number of pediatric cancers[74,88-91]; polymorphisms have also been identified as a risk factor for cancer diagnosis[90,92-94], suggesting there may be other functions of these proteins, as we discuss below. Prior studies generally evaluated expression and polymorphisms of the ABC genes proteins in low- or medium-throughput, however, and there is no clear pattern as to which of the 48 proteins may be expressed in a given tumor. As such, a comprehensive evaluation of these efflux pumps across tumor types is deserved.
In contrast to the large number of studies on drug efflux pumps, there has been very little research on drug influx and alterations that effect outcomes in pediatric cancer. Some work has been studied in the setting of adult cancers, such as platinum resistance due to mutations or downregulation of the CTR1 gene[95] or in regard to drug development generally[96]. As such, investigations into the drug influx mechanisms may be of value for pediatric cancer research.
P-glycoprotein itself has been evaluated as a therapeutic target in cancers generally, but with a single pediatric-specific Phase 1 study of tariquidar[97], a specific inhibitor of this target. Tested in pediatric patients with refractory solid tumors, tariquidar decreased clearance of the taxane docetaxel and the vinca alkaloid vinorelbine, with some disease responses observed. Despite this promising result, it is unclear what the development plan is for this agent in children with cancers.
DNA damage pathways
A number of chemotherapy agents act through DNA damage, which then relies on activation of the kinases ATR or ATM to activate p53, CHEK1, CHEK2, and other DNA damage response pathways. These pathways can then lead to either DNA repair or, more commonly, cell cycle arrest or apoptosis. These pathways are dependent on a number of additional interacting proteins, such as MDM2, which suppresses p53 activation, and multiple downstream proteins such as BRCA1, CDC25A, Rb, p21, CASP2, and CASP3, which are needed to allow either DNA repair, cycle arrest or apoptosis (reviewed in[98]). Thus, there are multiple tumor suppressors which can be targeted in cancer cells to avoid DNA-damage-induced cytotoxicity.
TP53, which encodes the p53 protein, is somatically mutated in < 5% of all pediatric cancers at diagnosis[13], with a notable exceptions in osteosarcoma[99] and adrenocortical carcinomas[100]. Germline mutations, such as in Li Fraumeni Syndrome, are associated with increased cancer risk and increased toxicity from DNA damaging agents[101]. However, somatic TP53 mutations or epigenetic silencing have been associated with increased rates of relapse, with enrichment in these tumors, and with worse prognosis both at diagnosis and relapse, including in glioma[102,103], neuroblastoma[104-107], and leukemia[108,109]. Mutations in other components of the DNA repair and cell cycle pathways in refractory pediatric cancers, including ATM, ATR, PTEN, and CHEK1, have also been associated with poor prognosis, presumably through avoidance of cell death[110-112].
Indirect mechanisms are more commonly found to repress the DNA damage pathways, promoting cell survival and therapeutic resistance. MYCN and MYC basally can upregulate TP53 expression, but when overexpressed, either by amplification, translocation, or epigenetically-driven overexpression, these transcription factors can promote MDM2 expression, which ubiquitinates p53 leading to its degradation. MDM2 is overexpressed in pediatric tumors including leukemias[113-115], neuroblastoma[116], retinoblastoma[117] and associated with poor prognosis and/or treatment resistance[118-121], supporting its role in chemoresistant disease. Additional response pathways, such as MEK/ERK activation and NF-kappaB activation, are being studied in pediatric cancers.
There is also a paradoxical role of DNA repair proteins in chemoresistance. DNA damage can activate p53 to induce apoptosis, if the damage is sufficient, or cell cycle arrest and DNA repair, which would maintain viability. Expression of proteins including CHEK1, CHEK2, ERCC1, ERCC2, PARP and WEE1 predispose cancer cells treated with chemotherapy toward DNA repair and proliferation. As such, these proteins have been identified as either prognostic biomarkers or therapeutic targets to improve chemotherapeutic effects in pediatric tumors[122-132]. The mechanisms that balance the expression and stability of these proteins continue to be areas of active investigation.
As the roles of DNA repair pathways in pediatric chemoresistance have been elucidated, opportunities to use targeted therapies have arisen. Many of these drugs were developed originally for cancers in adults, but there are ongoing pediatric early phase studies of PARP inhibitors[133] and WEE1 inhibitors[134], alone or in combination with chemotherapy.
Apoptosis via cytochrome C, redox/ROS activation, and caspase activation
Apoptosis can be triggered by a number of different pathways in normal cells. DNA damage can activate p53 to promote expression of pro-apoptotic members of the BCL2 family, NOXA and PUMA. These proteins then translocate to the mitochondria and bind antiapoptotic BCL2 proteins, including BCL2, BCL2L1, and MCL1. This process releases the proteins BID, BIM, BAK and BAX to then release cytochrome C and apoptosis inducing factor (AIF) into the cytosol, which then activates caspase cleavage and apoptosis. Alternatively, the production of reactive oxygen species can cause direct membrane damage of organelles, directly releasing AIF and cytochrome C into the cytosol. New signaling pathways involving NFE2L2[135-137] (aka NRF2) and AKR1C[138] have also been implicated in modulation of and resistance to ROS/redox-mediated cell death. Additionally, the “extrinsic” pathway can be triggered to induce apoptosis; external ligands, such TRAIL, TNFA, or FASL can bind their respective receptors, which then can directly activate CASP8 to trigger apoptosis.
Altered BCL2 family expression occurs in a number of childhood cancers. Overexpression of BCL2, BCL2L1, and MCL2 can stoichiometrically inhibit NOXA and PUMA, effectively squelching apoptosis. This overexpression can be driven by chromosomal translocations, through signal transduction from extracellular signaling[139], alternative splicing[140] or epigenetic dysregulation[141]. Antiapoptotic BCL2 family expression has been seen in numerous pediatric cancers, including neuroblastomas[142-144], leukemias[141,145-147], lymphomas[148,149], brain tumors[150-152], and sarcomas[153-156].
Investigations into caspase expression and processing, in contrast, have variably linked these pathways to therapeutic resistance and poor survival, particularly in leukemias and solid tumors[157-163]. However, the numerous mechanisms that can activate caspase processing and apoptosis have riddled these studies with confounders, suggesting that effects on caspase biology are likely secondary to other oncogenic pathways. Additionally, there are few opportunities to target this pathway therapeutically. As a result, this area of study has largely fallen by the wayside.
From a therapeutic perspective, BCL2 and MCL1 serve as potential targets, alone or in combination with chemotherapy. Venetoclax (ABT-199) and navitoclax (ABT-263) inhibit each protein respectively, and are currently in early phase pediatric clinical trials. Venetoclax specifically has received FDA approval in adult AML and CLL, alone or in combination with one additional drug[164-166]. However, in the pediatric approach, monotherapy is not expected to be effective alone; as such, a novel Phase 1 study has been opened that rapidly allows evaluation of venetoclax in monotherapy and combination chemotherapy[167] This novel approach advocates for the advancement of a targeted therapeutic approach for pediatric cancer patients specifically.
Epigenetic and miRNA-mediated resistance in pediatric cancers
As noted above, pediatric cancers differ from adult cancers in their comparatively quiet genomes; instead, epigenetic dysregulation is of greater impact on disease biology[14,168,169]. Broadly, this includes effects on DNA methylation, histone methylation, and histone acetylation, with differential effects depending on the pathway targeted and proteins involved. Implicated proteins, either due to direct mutation, overexpression, or due to novel interactions with other oncogenes, include KMT2A[170,171] (aka MLL), DOT1L[172], EZH2[173-175], KDM1A[176-178] (aka LSD1), CREBBP[179,180], SWI/SNF, SETD2[181,182], ATRX[183,184], and H3F3A[185-187] (aka Histone 3.3) mutations, to name a few, as well as the HDAC family[188-192]. Most of these proteins have effects across the genome, so elucidation of specific effects is complex. Nonetheless, they have been viewed as key factors that maintain tumor viability in the face of conventional chemotherapies. As such, novel agents targeting these epigenetic modifiers have been developed specifically for pediatric cancers.
In MLL-rearranged ALL, DOT1L was found to be a key driver of disease aggression; the DOT1L inhibitor pinometostat was studied in a pediatric Phase 1 trial, with no objective responses seen in single agent use[193]. There are a number of studies of the HDAC inhibitor vorinostat, alone or in combination, in pediatric cancer patients, though with generally low rates of responses in single use[194-198]. Tazemetostat is an EZH2 inhibitor currently under study in pediatric patients with lymphomas, synovial cell sarcoma, or other relapsed/refractory solid tumors[199]. The most novel agent currently in a pediatric Phase 1 study is the LSD1 inhibitor, seclidemstat (SP-2509), which was specifically identified for its efficacy against Ewing sarcoma[200]. It is likely that these agents will need to be used in combination therapies, as a common challenge for antiepigenetic therapies has been their slow effect, but they still offer novel approaches for the treatment of pediatric cancers.
In parallel to the study of epigenetics of pediatric cancers, the roles of miRNAs are also being evaluated. Specifically, miRNAs have been evaluated as mediators of therapeutic resistance in childhood leukemias[201], neuroblastoma[202], and CNS tumors[203]. However, in contrast to epigenetic modifiers, therapeutic options to inhibit or sponge miRNAs remain in early development, given the challenges of drug targeting and delivery, and no studies yet exist in pediatric oncology. Still, this remains a potential area for study.
New therapies, new mechanisms of resistance
Targeted antineoplastic therapies have been over 20 years, generally falling into two large categories: (1) small molecules targeting specific cellular proteins with inhibitory effects; and (2) monoclonal antibodies targeting cell-surface proteins to inhibit their functions and/or to recruit an anti-tumor immune response. Since their first use in cancers, resistance mechanisms have been identified, including in pediatric cancers. One of the first targeted therapies brought to clinic were inhibitors of MTOR, a key component of the PI3K/AKT oncogenic pathway. These agents, including sirolimus, everolimus, and temsirolimus, were found to be comparatively less toxic than conventional chemotherapies and with some benefits, generally in combination use, against a variety of cancers[204-208]. However, these responses were generally found to be not durable. In some tumors, the MTOR proteins were found to develop a gain-of-function mutation that rendered them immune to inhibition by these first and second-generation inhibitors[209], though third-generation inhibitors are now being developed to sidestep these mutations. More commonly, however, cancers were found to overcome MTOR inhibition by activation of other oncogenic pathways, including overexpression of MYC[210], IDO1[211], other components of the AKT pathway[212], or rebound activation of AKT[213,214], such as through MTORC2 activity (not inhibited by current MTOR inhibitors)[215-217].
A rapidly expanding class of drugs with increasing use in pediatric oncology are kinase inhibitors, including receptor tyrosine kinase inhibitors and other kinase inhibitors targeting intracellular pathways. The development of imatinib, an inhibitor of the BCR-ABL fusion tyrosine kinase, revolutionized the treatment of chronic myelogenous leukemia[218] and other leukemias expressing this fusion protein[219]. While these kinase inhibitors are generally designed for specificity for one kinase, often times they have effects on multiple kinases due to homology among the proteins, with varying toxicity and efficacy[220-223]. With these drugs, resistance often develops with time as well. A common mechanism of resistance is the isolation of a new subpopulation with a mutation resistant to the original drug, such as identified in BCR-ABL in leukemias[224-226] and ALK in neuroblastoma[227,228] (the development of these mutations are described below). In these cases, multiple second and third generation inhibitors have been developed against the same target, allowing for cycling among drugs for durable remission. In other cases, the cause of resistance has been less clear because of the new use of these agents, but extrapolation from adult data suggests resistance could be due to activation of an alternative oncogenic pathway[229-231], increased drug efflux[232], or downregulation of apoptosis[233,234]. As such, sequential or concurrent multiagent therapy may be needed for durable efficacy.
Monoclonal antibodies, included engineered antibodies with or without conjugated drugs, have been developed against a variety of cell surface targets in pediatric cancers[235-241], generally with success but with some notable failures[242-246]. In the latter cases, activation of alternative pathways generally was responsible for lack of efficacy. However, two other important mechanisms of resistance have been identified. In some cases, the cancer became resistant to the monoclonal antibody by losing expression of the target antigen, such as CD20 or CD30 in lymphomas[247,248]. This is incidentally a mechanism of resistance for the first-in-class anti-CD19 chimeric antigen receptor (CAR) T-cells[249]. In other cases, neutralizing antibodies have developed against the therapeutic antibody, with diminished pharmacokinetics and response[250-252]. These neutralizing antibodies are also being identified against other therapeutics, such as asparaginase used against acute lymphoblastic leukemia[253-255], though impact on drug efficacy is variable. Different approaches are being developed against these resistance mechanisms, including combination agent therapies to prevent activation of alternative oncogenic pathways, antibodies capable of multi-antigen binding, and recombinant antibodies and drugs with diminished anti-antibody production.
Future areas of study in pediatric cancer treatment resistance, and key collaborations for advancement
New mechanisms of therapeutic resistance continued to be identified as our understanding of cancer biology deepens. A major question has been the origin of the chemoresistant population, particularly regarding the existence of cells with resistance mutations as a subpopulation from diagnosis or the generation of novel mutations over time. In neuroblastoma, there is evidence to support both mechanisms. Next-generation deep sequencing approaches have allowed for higher resolution identification of tumor subpopulations; sequencing on paired diagnosis-relapse patient samples has demonstrated that mutations identified in relapse samples can also be identified at diagnosis but below the resolution of traditional testing[256,257]. In contrast, a study examining ALK mutations in cell lines derived from a patient with neuroblastoma at original diagnosis and at relapse. In these paired cell lines, different ALK mutations were identified, with differential response to ALK inhibitors[258]. However, it is unclear if these mutations may have occurred ex vivo. These different mechanisms have major implications on diagnostic and therapeutic approach; the presence of cancer subpopulations would support deeper sequencing of tumors and/or empiric use of ALK inhibitors to squelch selection of a resistant population, whereas the generation of de novo mutations would argue against these efforts.
An analogous though more contentious topic has been the role of “cancer stem cells” in therapeutic resistance. These stem cells have been variably defined phenotypically but include the capacity to initiate a new tumor, a slower proliferation rate and/or maintenance of a basal undifferentiated population. These features can inherently induce therapy resistance, as most chemotherapeutic agents rely on damage to the mechanisms of cell proliferation. There is some question about the relevance of “cancer stem cells” in different tumors, but there is also some evidence that markers of these cells can promote therapeutic resistance. For example, CD133, a surface marker associated with stem cells in a number of pediatric solid tumors and leukemias[259-261], can promote therapeutic resistance directly and has been associated with poor survival clinically[82,262-265]. Additional work is needed to clarify the causality of these stem cell features in disease aggression.
Intercellular effects within the tumors also have demonstrated impacts on disease resistance. The tumor microenvironment is a complex landscape of primary neoplastic cells and recruited supporting cells, including immune populations, fibroblasts, and vasculature. Cell signaling among these cells is complex and includes interactions through cytokines[266-269], extracellular vesicles (reviewed in[270]), and even the efflux of chemical signaling via ABC transporters[271,272]. The role of tumor immunosuppressive cells, such as tumor-associated macrophages[273-275], regulatory T cells[276-279], myeloid-derived suppressor cells[280-282], continues to be evaluated in pediatric cancers, and these cells undoubtedly have roles in therapy resistance beyond immunotherapies.
Advances in pediatric cancers have been generally possible only through collaborations between scientists and clinicians around the world, including advances against treatment resistance. The Pediatric Preclinical Testing Program has been an important contributor to this work, with the first comprehensive evaluations of existing and novel therapeutics for childhood cancers[283]. Now the Pediatric Preclinical Testing Consortium, http://www.ncipptc.org/, this group builds on the basic science discoveries of pediatric oncology to elucidate the mechanisms of disease biology in childhood cancers, the efficacy of novel drugs and drug combinations, and the potential roles of these agents in clinical trials and use. This international work is but one of many groups around the world, including the Children’s Oncology Group, The International Society of Pediatric Oncology, and numerous childhood cancer research groups and early phase consortia. Collaboration among these private groups, federal agencies, and pharmaceutical companies will further optimize existing treatments and add in novel therapeutics to overcome treatment resistance in pediatric cancers.
Conclusion
As advances continue to be made in therapeutic approaches for pediatric cancers, known and new mechanisms of therapeutic resistance will need to be considered. Known intracellular mechanisms of resistance can be targeted with new adjunctive therapies to ensure proper drug delivery and retention and optimal induction of apoptosis. New therapeutic approaches will also need to consider the genetic and epigenetic changes that can be selected and/or induced in response to therapy, and adjunctive approaches attacking newly discovered mechanisms of resistance, including effects on the tumor microenvironment, must be designed to further improve therapy. These multidimensional therapeutic approaches will offer the next great leap forward in outcomes for children with pediatric cancers.
Declarations
Authors’ contributions
The author solely contributed to the article.
Availability of data and materials
Not applicable.
Financial support and sponsorship
None.
Conflicts of interest
The author declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2019.
References
- 1. Howlader N, Krapcho M, Miller D, Brest A, Yu M, et al. SEER Cancer Statistics Review, 1975-2016, National Cancer Institute. Bethesda MD, based on November 2018 SEER data submission, posted to the SEER web site, April 2019. Avaliable from: https://seer.cancer.gov/csr/1975_2016/ [Last accessed on 26 June 2019]
- 2.Adamson PC. Improving the outcome for children with cancer: development of targeted new agents. CA Cancer J Clin. 2015;65:212–20. doi: 10.3322/caac.21273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vaupel PW. The influence of tumor blood flow and microenvironmental factors on the efficacy of radiation, drugs and localized hyperthermia. Klin Padiatr. 1997;209:243–9. doi: 10.1055/s-2008-1043957. [DOI] [PubMed] [Google Scholar]
- 4.Emadi A, Jones RJ, Brodsky RA. Cyclophosphamide and cancer: golden anniversary. Nat Rev Clin Oncol. 2009;6:638–47. doi: 10.1038/nrclinonc.2009.146. [DOI] [PubMed] [Google Scholar]
- 5.Stewart CF, Zamboni WC, Crom R, Gajjar A, Heideman RL, et al. Topoisomerase I interactive drugs in children with cancer. Invest New Drugs. 1996;14:37–47. doi: 10.1007/BF00173681. [DOI] [PubMed] [Google Scholar]
- 6.Masetti R, Biagi C, Zama D, Vendemini F, Martoni A, et al. Retinoids in pediatric onco-hematology: the model of acute promyelocytic leukemia and neuroblastoma. Adv Ther. 2012;29:747–62. doi: 10.1007/s12325-012-0047-3. [DOI] [PubMed] [Google Scholar]
- 7.Isakoff MS, Bielack SS, Meltzer P, Gorlick R. Osteosarcoma: current treatment and a collaborative pathway to success. J Clin Oncol. 2015;33:3029–35. doi: 10.1200/JCO.2014.59.4895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jakacki RI, Cohen KJ, Buxton A, Krailo, Burger PC, et al. Phase 2 study of concurrent radiotherapy and temozolomide followed by temozolomide and lomustine in the treatment of children with high-grade glioma: a report of the Children’s Oncology Group ACNS0423 study. Neuro Oncol. 2016;18:1442–50. doi: 10.1093/neuonc/now038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lipshultz SE, Cochran TR, Franco VI, Miller TL. Treatment-related cardiotoxicity in survivors of childhood cancer. Nat Rev Clin Oncol. 2013;10:697–710. doi: 10.1038/nrclinonc.2013.195. [DOI] [PubMed] [Google Scholar]
- 10.Okabe M, Unno M, Harigae H, Kaku M, Okitsu Y, et al. Characterization of the organic cation transporter SLC22A16: a doxorubicin importer. Biochem Biophys Res Commun. 2005;333:754–62. doi: 10.1016/j.bbrc.2005.05.174. [DOI] [PubMed] [Google Scholar]
- 11.Zhao H, Shi P, Deng M, Jiang Z, Li Y, et al. Low dose triptolide reverses chemoresistance in adult acute lymphoblastic leukemia cells via reactive oxygen species generation and DNA damage response disruption. Oncotarget. 2016;7:85515–28. doi: 10.18632/oncotarget.13454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hayes FA, Green AA, Mauer AM. Correlation of cell kinetic and clinical response to chemotherapy in disseminated neuroblastoma. Cancer Res. 1977;37:3766–70. [PubMed] [Google Scholar]
- 13.Gröbner SN, Worst BC, Weischenfeldt J, Buchhalter I, Kleinheinz K, et al. The landscape of genomic alterations across childhood cancers. Nature. 2018;555:321–7. doi: 10.1038/nature25480. [DOI] [PubMed] [Google Scholar]
- 14.Fruhwald MC, Witt O. The epigenetics of cancer in children. Klin Padiatr. 2008;220:333–41. doi: 10.1055/s-0028-1086026. [DOI] [PubMed] [Google Scholar]
- 15.Moore SW. Developmental genes and cancer in children. Pediatr Blood Cancer. 2009;52:755–60. doi: 10.1002/pbc.21831. [DOI] [PubMed] [Google Scholar]
- 16.Lawlor ER, Thiele CJ. Epigenetic changes in pediatric solid tumors: promising new targets. Clin Cancer Res. 2012;18:2768–79. doi: 10.1158/1078-0432.CCR-11-1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yiu TT, Li W. Pediatric cancer epigenome and the influence of folate. Epigenomics. 2015;7:961–73. doi: 10.2217/epi.15.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.La Madrid AM, Kieran MW. Epigenetics in clinical management of children and adolescents with brain tumors. Curr Cancer Drug Targets. 2018;18:57–64. doi: 10.2174/1568009617666170203164456. [DOI] [PubMed] [Google Scholar]
- 19.Nordlund J, Syvanen AC. Epigenetics in pediatric acute lymphoblastic leukemia. Semin Cancer Biol. 2018;51:129–38. doi: 10.1016/j.semcancer.2017.09.001. [DOI] [PubMed] [Google Scholar]
- 20.Filbin M, Monje M. Developmental origins and emerging therapeutic opportunities for childhood cancer. Nat Med. 2019;25:367–76. doi: 10.1038/s41591-019-0383-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schleiermacher G, Mosseri V, London WB, Maris JM, Brodeur GM, et al. Segmental chromosomal alterations have prognostic impact in neuroblastoma: a report from the INRG project. Br J Cancer. 2012;107:1418–22. doi: 10.1038/bjc.2012.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chicard M, Boyault S, Colmet Daage L, Richer W, Gentien D, et al. Genomic copy number profiling using circulating free tumor DNA highlights heterogeneity in neuroblastoma. Clin Cancer Res. 2016;22:5564–73. doi: 10.1158/1078-0432.CCR-16-0500. [DOI] [PubMed] [Google Scholar]
- 23.Defferrari R, Mazzocco K, Ambros IM, Ambros PF, Bedwell C, et al. Influence of segmental chromosome abnormalities on survival in children over the age of 12 months with unresectable localised peripheral neuroblastic tumours without MYCN amplification. Br J Cancer. 2015;112:290–5. doi: 10.1038/bjc.2014.557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Squire JA, Pei J, Marrano P, Beheshti B, Bayani J, et al. High-resolution mapping of amplifications and deletions in pediatric osteosarcoma by use of CGH analysis of cDNA microarrays. Genes Chromosomes Cancer. 2003;38:215–25. doi: 10.1002/gcc.10273. [DOI] [PubMed] [Google Scholar]
- 25.Smida J, Xu H, Zhang Y, Baumhoer D, Ribi S, et al. Genome-wide analysis of somatic copy number alterations and chromosomal breakages in osteosarcoma. Int J Cancer. 2017;141:816–28. doi: 10.1002/ijc.30778. [DOI] [PubMed] [Google Scholar]
- 26.Behjati S, Tarpey PS, Haase K, Ye H, Young MD, et al. Recurrent mutation of IGF signalling genes and distinct patterns of genomic rearrangement in osteosarcoma. Nat Commun. 2017;8:15936. doi: 10.1038/ncomms15936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ratnaparkhe M, Wong JKL, Wei PC, Hlevnjak M, Kolb T, et al. Defective DNA damage repair leads to frequent catastrophic genomic events in murine and human tumors. Nat Commun. 2018;9:4760. doi: 10.1038/s41467-018-06925-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rausch T, Jones DT, Zapatka M, Stutz AM, Zichner T, et al. Genome sequencing of pediatric medulloblastoma links catastrophic DNA rearrangements with TP53 mutations. Cell. 2012;148:59–71. doi: 10.1016/j.cell.2011.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gratias EJ, Dome JS, Jennings LJ, Chi YY, Tian J, et al. Association of Chromosome 1q Gain With Inferior Survival in Favorable-Histology Wilms Tumor: a report from the children’s oncology group. J Clin Oncol. 2016;34:3189–94. doi: 10.1200/JCO.2015.66.1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Radtke I, Mullighan CG, Ishii M, Su X, Cheng J, et al. Genomic analysis reveals few genetic alterations in pediatric acute myeloid leukemia. Proc Natl Acad Sci U S A. 2009;106:12944–9. doi: 10.1073/pnas.0903142106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Farber S. Some observations on the effect of folic acid antagonists on acute leukemia and other forms of incurable cancer. Blood. 1949;4:160–7. [PubMed] [Google Scholar]
- 32.Price CH, Zhuber K, Salzer-Kuntschik M, Salzer M, Willert HG, et al. Osteosarcoma in children. A study of 125 cases. J Bone Joint Surg Br. 1975;57:341–5. [PubMed] [Google Scholar]
- 33.Smith M, Abrams J, Trimble EL, Ungerleider RS. Dose Intensity of Chemotherapy for Childhood Cancers. Oncologist. 1996;1:293–304. [PubMed] [Google Scholar]
- 34.Neil EC, Hanmantgad S, Khakoo Y. Neurological Complications of Pediatric Cancer. J Child Neurol. 2016;31:1412–20. doi: 10.1177/0883073815620673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Weyl-Ben-Arush M. The price of the successful treatment of pediatric malignancies. Curr Pediatr Rev. 2017;13:4–7. doi: 10.2174/1573396312666161213103558. [DOI] [PubMed] [Google Scholar]
- 36.Pluimakers VG, van Waas M, Neggers S, van den Heuvel-Eibrink MM. Metabolic syndrome as cardiovascular risk factor in childhood cancer survivors. Crit Rev Oncol Hematol. 2019;133:129–41. doi: 10.1016/j.critrevonc.2018.10.010. [DOI] [PubMed] [Google Scholar]
- 37.Roeca C, Dovey S, Polotsky AJ. Recommendations for assessing ovarian health and fertility potential in survivors of childhood cancer. Maturitas. 2019;122:57–9. doi: 10.1016/j.maturitas.2019.01.009. [DOI] [PubMed] [Google Scholar]
- 38.Sklar CA, Antal Z, Chemaitilly W, Cohen LE, Follin C, et al. Hypothalamic-pituitary and growth disorders in survivors of childhood cancer: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. 2018;103:2761–84. doi: 10.1210/jc.2018-01175. [DOI] [PubMed] [Google Scholar]
- 39.Dixon SB, Bjornard KL, Alberts NM, Armstrong GT, Brinkman TM, et al. Factors influencing risk-based care of the childhood cancer survivor in the 21st century. CA Cancer J Clin. 2018;68:133–52. doi: 10.3322/caac.21445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ehrhardt MJ, Hochberg J, Bjornard KL, Brinkman TM. Long-term survivors of childhood, adolescent and young adult non-Hodgkin lymphoma. Br J Haematol. 2019;185:1099–110. doi: 10.1111/bjh.15775. [DOI] [PubMed] [Google Scholar]
- 41.Ekhart C, Doodeman VD, Rodenhuis S, Smits PH, Beijnen JH, et al. Influence of p olymorphisms of drug metabolizing enzymes (CYP2B6, CYP2C9, CYP2C19, CYP3A4, CYP3A5, GSTA1, GSTP1, ALDH1A1 and ALDH3A1) on the pharmacokinetics of cyclophosphamide and 4-hydroxycyclophosphamide. Pharmacogenet Genomics. 2008;18:515–23. doi: 10.1097/FPC.0b013e3282fc9766. [DOI] [PubMed] [Google Scholar]
- 42.Algeciras-Schimnich A, O’Kane DJ, Snozek CL. Pharmacogenomics of tamoxifen and irinotecan therapies. Clin Lab Med. 2008;28:553–67. doi: 10.1016/j.cll.2008.05.004. [DOI] [PubMed] [Google Scholar]
- 43.Queckenberg C, Erlinghagen V, Baken BC, Van Os SH, Wargenau M, et al. Pharmacokinetics and pharmacogenetics of capecitabine and its metabolites following replicate administration of two 500 mg tablet formulations. Cancer Chemother Pharmacol. 2015;76:1081–91. doi: 10.1007/s00280-015-2840-6. [DOI] [PubMed] [Google Scholar]
- 44.de Man FM, Goey AKL, van Schaik RHN, Mathijssen RHJ, Bins S. Individualization of Irinotecan Treatment: a review of pharmacokinetics, pharmacodynamics, and pharmacogenetics. Clin Pharmacokinet. 2018;57:1229–54. doi: 10.1007/s40262-018-0644-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mittal B, Tulsyan S, Kumar S, Mittal RD, Agarwal G. Cytochrome P450 in Cancer Susceptibility and Treatment. Adv Clin Chem. 2015;71:77–139. doi: 10.1016/bs.acc.2015.06.003. [DOI] [PubMed] [Google Scholar]
- 46.Sim SC, Kacevska M, Ingelman-Sundberg M. Pharmacogenomics of drug-metabolizing enzymes: a recent update on clinical implications and endogenous effects. Pharmacogenomics J. 2013;13:1–11. doi: 10.1038/tpj.2012.45. [DOI] [PubMed] [Google Scholar]
- 47.Alini TP, Francine TG, Antonio SP, Maria TSA, Reynaldo JGF, et al. CYP genes in osteosarcoma: Their role in tumorigenesis, pulmonary metastatic microenvironment and treatment response. Oncotarget. 2017;8:38530–40. doi: 10.18632/oncotarget.15869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Melanie MH, Marieke JHC, Hans G, Remco RM, Hanneke IV, et al. A First Step toward Personalized Medicine in Osteosarcoma: Pharmacogenetics as Predictive Marker of Outcome after Chemotherapy-Based Treatment. Clin Cancer Res. 2015;21:3436–41. doi: 10.1158/1078-0432.CCR-14-2638. [DOI] [PubMed] [Google Scholar]
- 49.Valencia-Cervantes J, Huerta-Yepez S, Aquino-Jarquín G, Rodríguez-Enríquez S, Martínez-Fong D, et al. Hypoxia increases chemoresistance in human medulloblastoma DAOY cells via hypoxiainducible factor 1alphamediated downregulation of the CYP2B6, CYP3A4 and CYP3A5 enzymes and inhibition of cell proliferation. Oncol Rep. 2019;41:178–90. doi: 10.3892/or.2018.6790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Darwish MH, Farah RA, Farhat GN, Torbey PH, Ghandour FA, et al. Association of CYP3A4/5 genotypes and expression with the survival of patients with neuroblastoma. Mol Med Rep. 2015;11:1462–68. doi: 10.3892/mmr.2014.2835. [DOI] [PubMed] [Google Scholar]
- 51.Egbelakin A, Ferguson MJ, MacGill EA, Lehmann AS, Topletz AR, et al. Increased risk of vincristine neurotoxicity associated with low CYP3A5 expression genotype in children with acute lymphoblastic leukemia. Pediatr Blood Cancer. 2011;56:361–7. doi: 10.1002/pbc.22845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Araoz HV, D’Aloi K, Foncuberta ME, Sanchez La Rosa CG, Alonso CN, et al. Pharmacogenetic studies in children with acute lymphoblastic leukemia in Argentina. Leuk Lymphoma. 2015;56:1370–78. doi: 10.3109/10428194.2014.951844. [DOI] [PubMed] [Google Scholar]
- 53.Borst L, Wallerek S, Dalhoff K, Rasmussen KK, Wesenberg F, et al. The impact of CYP3A5*3 on risk and prognosis in childhood acute lymphoblastic leukemia. Eur J Haematol. 2011;86:477–83. doi: 10.1111/j.1600-0609.2011.01608.x. [DOI] [PubMed] [Google Scholar]
- 54.Gezsi A, Lautner-Csorba O, Erdelyi DJ, Hullam G, Antal P, et al. In interaction with gender a common CYP3A4 polymorphism may influence the survival rate of chemotherapy for childhood acute lymphoblastic leukemia. Pharmacogenomics J. 2015;15:241–47. doi: 10.1038/tpj.2014.60. [DOI] [PubMed] [Google Scholar]
- 55.Sims RP. The effect of race on the CYP3A-mediated metabolism of vincristine in pediatric patients with acute lymphoblastic leukemia. J Oncol Pharm Pract. 2016;22:76–81. doi: 10.1177/1078155214553143. [DOI] [PubMed] [Google Scholar]
- 56.Lennard L, Lilleyman JS, Van Loon J, Weinshilboum RM. Genetic variation in response to 6-mercaptopurine for childhood acute lymphoblastic leukaemia. Lancet. 1990;336:225–9. doi: 10.1016/0140-6736(90)91745-v. [DOI] [PubMed] [Google Scholar]
- 57.Lennard L. Implementation of TPMT testing. Br J Clin Pharmacol. 2014;77:704–14. doi: 10.1111/bcp.12226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mlakar V, Huezo-Diaz Curtis P, Satyanarayana Uppugunduri CR, Krajinovic M, Ansari M. Pharmacogenomics in pediatric oncology: review of gene-drug associations for clinical use. Int J Mol Sci. 2016;17:E1502. doi: 10.3390/ijms17091502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Roninson IB, Chin JE, Choi KG, Gros P, Housman DE, et al. Isolation of human mdr DNA sequences amplified in multidrug-resistant KB carcinoma cells. Proc Natl Acad Sci U S A. 1986;83:4538–42. doi: 10.1073/pnas.83.12.4538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Goldstein LJ, Fojo AT, Ueda K, Crist W, Green A, et al. Expression of the multidrug resistance, MDR1, gene in neuroblastomas. J Clin Oncol. 1990;8:128–36. doi: 10.1200/JCO.1990.8.1.128. [DOI] [PubMed] [Google Scholar]
- 61.Nooter K, Sonneveld P, Oostrum R, Herweijer H, Hagenbeek T, et al. Overexpression of the mdr1 gene in blast cells from patients with acute myelocytic leukemia is associated with decreased anthracycline accumulation that can be restored by cyclosporin-A. Int J Cancer. 1990;45:263–8. doi: 10.1002/ijc.2910450210. [DOI] [PubMed] [Google Scholar]
- 62.Benard J, Bourhis J, de Vathaire F, Ferrandis E, Terrier-Lacombe MJ, et al. Prognostic value of MDR1 gene expression in neuroblastoma: results of a multivariate analysis. Prog Clin Biol Res. 1994;385:111–6. [PubMed] [Google Scholar]
- 63.Gsur A, Zochbauer S, Gotzl M, Kyrle P A, Lechner K, et al. MDR1 RNA expression as a prognostic factor in acute myeloid leukemia: an update. Leuk Lymphoma. 1993;12:91–4. doi: 10.3109/10428199309059575. [DOI] [PubMed] [Google Scholar]
- 64.Wunder JS, Bell RS, Wold L, Andrulis IL. Expression of the multidrug resistance gene in osteosarcoma: a pilot study. J Orthop Res. 1993;11:396–403. doi: 10.1002/jor.1100110311. [DOI] [PubMed] [Google Scholar]
- 65.Pirker R, Wallner J, Geissler K, Linkesch W, Haas OA, et al. MDR1 gene expression and treatment outcome in acute myeloid leukemia. J Natl Cancer Inst. 1991;83:708–12. doi: 10.1093/jnci/83.10.708. [DOI] [PubMed] [Google Scholar]
- 66.Molina-Ortiz D, Torres-Zarate C, Cardenas-Cardos R, Palacios-Acosta JM, Hernandez-Arrazola D, et al. MDR1 not CYP3A4 gene expression is the predominant mechanism of innate drug resistance in pediatric soft tissue sarcoma patients. Cancer Biomark. 2018;22:317–24. doi: 10.3233/CBM-171027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chan HS, Haddad G, Thorner PS, DeBoer G, Lin YP, et al. P-glycoprotein expression as a predictor of the outcome of therapy for neuroblastoma. N Engl J Med. 1991;325:1608–14. doi: 10.1056/NEJM199112053252304. [DOI] [PubMed] [Google Scholar]
- 68.Hodorova I, Rybarova S, Vecanova J, Solar P, Plank L, et al. Relation between expression pattern of wild-type p53 and multidrug resistance proteins in human nephroblastomas. Acta Histochem. 2013;115:273–8. doi: 10.1016/j.acthis.2012.08.001. [DOI] [PubMed] [Google Scholar]
- 69.Fazlina N, Maha A, Zarina AL, Hamidah A, Zulkifli SZ, et al. Assessment of P-gp and MRP1 activities using MultiDrugQuant Assay Kit: a preliminary study of correlation between protein expressions and its functional activities in newly diagnosed acute leukaemia patients. Malays J Pathol. 2008;30:87–93. [PubMed] [Google Scholar]
- 70.Kourti M, Vavatsi N, Gombakis N, Sidi V, Tzimagiorgis G, et al. Expression of multidrug resistance 1 (MDR1), multidrug resistance-related protein 1 (MRP1), lung resistance protein (LRP), and breast cancer resistance protein (BCRP) genes and clinical outcome in childhood acute lymphoblastic leukemia. Int J Hematol. 2007;86:166–73. doi: 10.1532/IJH97.E0624. [DOI] [PubMed] [Google Scholar]
- 71.Haber M, Smith J, Bordow SB, Flemming C, Cohn SL, et al. Association of high-level MRP1 expression with poor clinical outcome in a large prospective study of primary neuroblastoma. J Clin Oncol. 2006;24:1546–53. doi: 10.1200/JCO.2005.01.6196. [DOI] [PubMed] [Google Scholar]
- 72.Li Y, Zhao L, Li N, Miao Y, Zhou H, et al. miR-9 regulates the multidrug resistance of chronic myelogenous leukemia by targeting ABCB1. Oncol Rep. 2017;37:2193–200. doi: 10.3892/or.2017.5464. [DOI] [PubMed] [Google Scholar]
- 73.Hontecillas-Prieto L, Garcia-Dominguez DJ, Vaca DP, Garcia-Mejias R, Marcilla D, et al. Multidrug resistance transporter profile reveals MDR3 as a marker for stratification of blastemal Wilms tumour patients. Oncotarget. 2017;8:11173–86. doi: 10.18632/oncotarget.14491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ruiz-Pinto S, Pita G, Patino-Garcia A, Garcia-Miguel P, Alonso J, et al. Identification of genetic variants in pharmacokinetic genes associated with Ewing Sarcoma treatment outcome. Ann Oncol. 2016;27:1788–93. doi: 10.1093/annonc/mdw234. [DOI] [PubMed] [Google Scholar]
- 75.Kuss BJ, Deeley RG, Cole SP, Willman CL, Kopecky KJ, et al. The biological significance of the multidrug resistance gene MRP in inversion 16 leukemias. Leuk Lymphoma. 1996;20:357–64. doi: 10.3109/10428199609052416. [DOI] [PubMed] [Google Scholar]
- 76.Oda Y, Rose I, Radig K, Wagemann W, Mittler U, et al. Expression of MDR1/p-glycoprotein and multidrug resistance-associated protein in childhood solid tumours. Virchows Arch. 1997;430:99–105. doi: 10.1007/BF01008030. [DOI] [PubMed] [Google Scholar]
- 77.El-Sharnouby JA, Abou El-Enein AM, El Ghannam DM, El-Shanshory MR, Hagag AA, et al. Expression of lung resistance protein and multidrug resistance-related protein (MRP1) in pediatric acute lymphoblastic leukemia. J Oncol Pharm Pract. 2010;16:179–88. doi: 10.1177/1078155209351329. [DOI] [PubMed] [Google Scholar]
- 78.Steinbach D, Sell W, Voigt A, Hermann J, Zintl F, et al. BCRP gene expression is associated with a poor response to remission induction therapy in childhood acute myeloid leukemia. Leukemia. 2002;16:1443–47. doi: 10.1038/sj.leu.2402541. [DOI] [PubMed] [Google Scholar]
- 79.Rodrigo MAM, Buchtelova H, Jimenez AMJ, Adam P, Babula P, et al. Transcriptomic Landscape of Cisplatin-Resistant Neuroblastoma Cells. Cells. 2019;8:E235. doi: 10.3390/cells8030235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ferrandis E, Da Silva J, Riou G, Benard I. Coactivation of the MDR1 and MYCN genes in human neuroblastoma cells during the metastatic process in the nude mouse. Cancer Res. 1994;54:2256–61. [PubMed] [Google Scholar]
- 81.Haber M, Bordow SB, Haber PS, Marshall GM, Stewart BW, et al. The prognostic value of MDR1 gene expression in primary untreated neuroblastoma. Eur J Cancer. 1997;33:2031–36. doi: 10.1016/s0959-8049(97)00229-3. [DOI] [PubMed] [Google Scholar]
- 82.Xi G, Li YD, Grahovac G, Rajaram V, Wadhwani N, et al. Targeting CD133 improves chemotherapeutic efficacy of recurrent pediatric pilocytic astrocytoma following prolonged chemotherapy. Mol Cancer. 2017;16 doi: 10.1186/s12943-017-0593-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Dabaghi M, Rahgozar S, Moshtaghian J, Moafi A, Abedi M, et al. Overexpression of SORCIN is a Prognostic Biomarker for Multidrug-Resistant Pediatric Acute Lymphoblastic Leukemia and Correlates with Upregulated MDR1/P-gp. Genet Test Mol Biomarkers. 2016;20:516–21. doi: 10.1089/gtmb.2016.0031. [DOI] [PubMed] [Google Scholar]
- 84.Hu R, Yan Y, Li Q, Lin Y, Jin W, et al. Increased drug efflux along with midkine gene high expression in childhood B-lineage acute lymphoblastic leukemia cells. Int J Hematol. 2010;92:105–10. doi: 10.1007/s12185-010-0613-x. [DOI] [PubMed] [Google Scholar]
- 85.Shi CJ, Wang F, Ren MF, Mi YJ, Yan YY, et al. Up-regulation of ABCB1/P-glycoprotein by escaping promoter hypermethylation indicates poor prognosis in hematologic malignancy patients with and without bone marrow transplantation. Leuk Res. 2011;35:73–9. doi: 10.1016/j.leukres.2010.04.019. [DOI] [PubMed] [Google Scholar]
- 86.Warmann S, Hunger M, Teichmann B, Flemming P, Gratz KF, et al. The role of the MDR1 gene in the development of multidrug resistance in human hepatoblastoma: clinical course and in vivo model. Cancer. 2002;95:1795–801. doi: 10.1002/cncr.10858. [DOI] [PubMed] [Google Scholar]
- 87.Janiszewska H, Styczynski J, Kolodziej B, Wysocki M, Haus O. Changes in the MDR1 gene expression after short-term ex vivo therapy with prednisolone have prognostic impact in childhood acute lymphoblastic leukemia. Ann Hematol. 2009;88:1193–98. doi: 10.1007/s00277-009-0739-1. [DOI] [PubMed] [Google Scholar]
- 88.Pongstaporn W, Pakakasama S, Chaksangchaichote P, Pongtheerat T, Hongeng, et al. MDR1 C3435T and C1236T polymorphisms: association with high-risk childhood acute lymphoblastic leukemia. Asian Pac J Cancer Prev. 2015;16:2839–43. doi: 10.7314/apjcp.2015.16.7.2839. [DOI] [PubMed] [Google Scholar]
- 89.Schaich M, Kestel L, Pfirrmann M, Robel K, Illmer T, et al. A MDR1 (ABCB1) gene single nucleotide polymorphism predicts outcome of temozolomide treatment in glioblastoma patients. Ann Oncol. 2009;20:175–81. doi: 10.1093/annonc/mdn548. [DOI] [PubMed] [Google Scholar]
- 90.Jamroziak K, Mlynarski W, Balcerczak E, Mistygacz M, Trelinska J, et al. Functional C3435T polymorphism of MDR1 gene: an impact on genetic susceptibility and clinical outcome of childhood acute lymphoblastic leukemia. Eur J Haematol. 2004;72:314–21. doi: 10.1111/j.1600-0609.2004.00228.x. [DOI] [PubMed] [Google Scholar]
- 91.Chung ES, Bok E, Sohn S, Lee YD, Baik HH, et al. GT1b-induced neurotoxicity is mediated by the Akt/GSK-3/tau signaling pathway but not caspase-3 in mesencephalic dopaminergic neurons. BMC Neurosci. 2010;11:74. doi: 10.1186/1471-2202-11-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Mhaidat NM, Alshogran OY, Khabour OF, Alzoubi KH, Matalka II, et al. Multi-drug resistance 1 genetic polymorphism and prediction of chemotherapy response in Hodgkin’s Lymphoma. J Exp Clin Cancer Res. 2011;30:68. doi: 10.1186/1756-9966-30-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Jamroziak K, Robak T. Do polymorphisms in ABC transporter genes influence risk of childhood acute lymphoblastic leukemia? Leuk Res. 2008;32:1173–75. doi: 10.1016/j.leukres.2008.01.009. [DOI] [PubMed] [Google Scholar]
- 94.Yue Q, Xiong B, Chen L, Chen Y, Bu F, et al. MDR1 C3435T polymorphism and childhood acute lymphoblastic leukemia susceptibility: an updated meta-analysis. Biomed Pharmacother. 2015;69:76–81. doi: 10.1016/j.biopha.2014.10.015. [DOI] [PubMed] [Google Scholar]
- 95.Howell SB, Safaei R, Larson CA, Sailor MJ. Copper transporters and the cellular pharmacology of the platinum-containing cancer drugs. Mol Pharmacol. 2010;77:887–94. doi: 10.1124/mol.109.063172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.International Transporter C. Giacomini KM, Huang SM, Tweedie DJ, Benet LZ, et al. Membrane transporters in drug development. Nat Rev Drug Discov. 2010;9:215–36. doi: 10.1038/nrd3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Fox E, Widemann BC, Pastakia D, Chen CC, Yang SX, et al. Pharmacokinetic and pharmacodynamic study of tariquidar (XR9576), a P-glycoprotein inhibitor, in combination with doxorubicin, vinorelbine, or docetaxel in children and adolescents with refractory solid tumors. Cancer Chemother Pharmacol. 2015;76:1273–83. doi: 10.1007/s00280-015-2845-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Harper JW, Elledge SJ. The DNA damage response: ten years after. Mol Cell. 2007;28:739–45. doi: 10.1016/j.molcel.2007.11.015. [DOI] [PubMed] [Google Scholar]
- 99.Chen X, Bahrami A, Pappo A, Easton J, Dalton J, et al. Recurrent somatic structural variations contribute to tumorigenesis in pediatric osteosarcoma. Cell Rep. 2014;7:104–12. doi: 10.1016/j.celrep.2014.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Wasserman JD, Novokmet A, Eichler-Jonsson C, Ribeiro RC, Rodriguez-Galindo C, et al. Prevalence and functional consequence of TP53 mutations in pediatric adrenocortical carcinoma: a children’s oncology group study. J Clin Oncol. 2015;33:602–9. doi: 10.1200/JCO.2013.52.6863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Malkin D. Li-fraumeni syndrome. Genes Cancer. 2011;2:475–84. doi: 10.1177/1947601911413466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Izycka-Swieszewska E, Bien E, Stefanowicz J, Szurowska E, Szutowicz-Zielinska E, et al. Malignant Gliomas as Second Neoplasms in Pediatric Cancer Survivors: Neuropathological Study. Biomed Res Int. 2018;2018:4596812. doi: 10.1155/2018/4596812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Pollack IF, Finkelstein SD, Woods J, Burnham J, Holmes EJ, et al. Expression of p53 and prognosis in children with malignant gliomas. N Engl J Med. 2002;346:420–7. doi: 10.1056/NEJMoa012224. [DOI] [PubMed] [Google Scholar]
- 104.Ackermann S, Cartolano M, Hero B, Welte A, Kahlert Y, et al. A mechanistic classification of clinical phenotypes in neuroblastoma. Science. 2018;362:1165–70. doi: 10.1126/science.aat6768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Cattelani S, Ferrari-Amorotti G, Galavotti S, Defferrari R, Tanno B, et al. The p53 codon 72 Pro/Pro genotype identifies poor-prognosis neuroblastoma patients: correlation with reduced apoptosis and enhanced senescence by the p53-72P isoform. Neoplasia. 2012;14:634–43. doi: 10.1593/neo.12594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Layfield LJ, Thompson JK, Dodge RK, Kerns BJ. Prognostic indicators for neuroblastoma: stage, grade, DNA ploidy, MIB-1-proliferation index, p53, HER-2/neu and EGFr--a survival study. J Surg Oncol. 1995;59:21–27. doi: 10.1002/jso.2930590107. [DOI] [PubMed] [Google Scholar]
- 107.Veschi V, Liu Z, Voss TC, Ozbun L, Gryder B, et al. Epigenetic siRNA and Chemical Screens Identify SETD8 Inhibition as a Therapeutic Strategy for p53 Activation in High-Risk Neuroblastoma. Cancer Cell. 2017;31:50–63. doi: 10.1016/j.ccell.2016.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Felix CA, Hosler MR, Provisor D, Salhany K, Sexsmith EA, et al. The p53 gene in pediatric therapy-related leukemia and myelodysplasia. Blood. 1996;87:4376–81. [PubMed] [Google Scholar]
- 109.Yoshizato T, Nannya Y, Atsuta Y, Shiozawa Y, Iijima-Yamashita Y, et al. Genetic abnormalities in myelodysplasia and secondary acute myeloid leukemia: impact on outcome of stem cell transplantation. Blood. 2017;129:2347–58. doi: 10.1182/blood-2016-12-754796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Takagi M, Yoshida M, Nemoto Y, Tamaichi H, Tsuchida R, et al. Loss of DNA Damage Response in Neuroblastoma and Utility of a PARP Inhibitor. J Natl Cancer Inst. 2017;109 doi: 10.1093/jnci/djx062. [DOI] [PubMed] [Google Scholar]
- 111.Ballinger ML, Goode DL, Ray-Coquard I, James PA, Mitchell G, et al. Monogenic and polygenic determinants of sarcoma risk: an international genetic study. Lancet Oncol. 2016;17:1261–71. doi: 10.1016/S1470-2045(16)30147-4. [DOI] [PubMed] [Google Scholar]
- 112.Chen C, Bartenhagen C, Gombert M, Okpanyi V, Binder V, et al. Next-generation-sequencing of recurrent childhood high hyperdiploid acute lymphoblastic leukemia reveals mutations typically associated with high risk patients. Leuk Res. 2015;39:990–1001. doi: 10.1016/j.leukres.2015.06.005. [DOI] [PubMed] [Google Scholar]
- 113.Richmond J, Carol H, Evans K, High L, Mendomo A, et al. Effective targeting of the P53-MDM2 axis in preclinical models of infant MLL-rearranged acute lymphoblastic leukemia. Clin Cancer Res. 2015;21:1395–405. doi: 10.1158/1078-0432.CCR-14-2300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Gu L, Zhu N, Findley HW, Zhou M. MDM2 antagonist nutlin-3 is a potent inducer of apoptosis in pediatric acute lymphoblastic leukemia cells with wild-type p53 and overexpression of MDM2. Leukemia. 2008;22:730–9. doi: 10.1038/leu.2008.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Zhou M, Gu L, Findley HW, Jiang R, Woods WG. PTEN reverses MDM2-mediated chemotherapy resistance by interacting with p53 in acute lymphoblastic leukemia cells. Cancer Res. 2003;63:6357–62. [PubMed] [Google Scholar]
- 116.Inomistova MV, Svergun NM, Khranovska NM, Skachkova OV, Gorbach OI, et al. Prognostic significance of MDM2 gene expression in childhood neuroblastoma. Exp Oncol. 2015;37:111–5. [PubMed] [Google Scholar]
- 117.McEvoy JD, Dyer MA. Genetic and Epigenetic Discoveries in Human Retinoblastoma. Crit Rev Oncog. 2015;20:217–25. doi: 10.1615/critrevoncog.2015013711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Kang MH, Reynolds CP, Kolb EA, Gorlick R, Carol H, et al. Initial Testing (Stage 1) of MK-8242-A Novel MDM2 Inhibitor-by the Pediatric Preclinical Testing Program. Pediatr Blood Cancer. 2016;63:1744–52. doi: 10.1002/pbc.26064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Tisato V, Voltan R, Gonelli A, Secchiero P, Zauli G. MDM2/X inhibitors under clinical evaluation: perspectives for the management of hematological malignancies and pediatric cancer. J Hematol Oncol. 2017;10:133. doi: 10.1186/s13045-017-0500-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Zhou M, Gu L, Abshire TC, Homans A, Billett AL, et al. Incidence and prognostic significance of MDM2 oncoprotein overexpression in relapsed childhood acute lymphoblastic leukemia. Leukemia. 2000;14:61–7. doi: 10.1038/sj.leu.2401619. [DOI] [PubMed] [Google Scholar]
- 121.Gamble LD, Kees UR, Tweddle DA, Lunec J. MYCN sensitizes neuroblastoma to the MDM2-p53 antagonists Nutlin-3 and MI-63. Oncogene. 2012;31:752–63. doi: 10.1038/onc.2011.270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Cole KA, Huggins J, Laquaglia M, Hulderman CE, Russell MR, et al. RNAi screen of the protein kinome identifies checkpoint kinase 1 (CHK1) as a therapeutic target in neuroblastoma. Proc Natl Acad Sci U S A. 2011;108:3336–41. doi: 10.1073/pnas.1012351108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Hoglund A, Nilsson LM, Muralidharan SV, Hasvold LA, Merta P, et al. Therapeutic implications for the induced levels of Chk1 in Myc-expressing cancer cells. Clin Cancer Res. 2011;17:7067–79. doi: 10.1158/1078-0432.CCR-11-1198. [DOI] [PubMed] [Google Scholar]
- 124.Hattinger CM, Michelacci F, Sella F, Magagnoli G, Benini S, et al. Excision repair cross-complementation group 1 protein expression predicts survival in patients with high-grade, non-metastatic osteosarcoma treated with neoadjuvant chemotherapy. Histopathology. 2015;67:338–47. doi: 10.1111/his.12653. [DOI] [PubMed] [Google Scholar]
- 125.Lowery CD, VanWye AB, Dowless M, Blosser W, Falcon BL, et al. The Checkpoint Kinase 1 Inhibitor Prexasertib Induces Regression of Preclinical Models of Human Neuroblastoma. Clin Cancer Res. 2017;23:4354–63. doi: 10.1158/1078-0432.CCR-16-2876. [DOI] [PubMed] [Google Scholar]
- 126.Zhang Q, Lv LY, Li BJ, Zhang J, Wei F. Investigation of ERCC1 and ERCC2 gene polymorphisms and response to chemotherapy and overall survival in osteosarcoma. Genet Mol Res. 2015;14:11235–41. doi: 10.4238/2015.September.22.17. [DOI] [PubMed] [Google Scholar]
- 127.Chen Z, Wang L, Yao D, Yang T, Cao WM, et al. Wip1 inhibitor GSK2830371 inhibits neuroblastoma growth by inducing Chk2/p53-mediated apoptosis. Sci Rep. 2016;6:38011. doi: 10.1038/srep38011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Sarmento LM, Povoa V, Nascimento R, Real G, Antunes I, et al. CHK1 overexpression in T-cell acute lymphoblastic leukemia is essential for proliferation and survival by preventing excessive replication stress. Oncogene. 2015;34:2978–90. doi: 10.1038/onc.2014.248. [DOI] [PubMed] [Google Scholar]
- 129.Duan Y, Dong X, Nie J, Li P, Lu F, et al. Wee1 kinase inhibitor MK-1775 induces apoptosis of acute lymphoblastic leukemia cells and enhances the efficacy of doxorubicin involving downregulation of Notch pathway. Oncol Lett. 2018;16:5473–81. doi: 10.3892/ol.2018.9291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Matheson CJ, Venkataraman S, Amani V, Harris PS, Backos DS, et al. A WEE1 Inhibitor Analog of AZD1775 Maintains Synergy with Cisplatin and Demonstrates Reduced Single-Agent Cytotoxicity in Medulloblastoma Cells. ACS Chem Biol. 2016;11:921–30. doi: 10.1021/acschembio.5b00725. [DOI] [PubMed] [Google Scholar]
- 131.Mueller S, Hashizume R, Yang X, Kolkowitz I, Olow AK, et al. Targeting Wee1 for the treatment of pediatric high-grade gliomas. Neuro Oncol. 2014;16:352–60. doi: 10.1093/neuonc/not220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Sanmartin E, Munoz L, Piqueras M, Sirerol JA, Berlanga P, et al. Deletion of 11q in Neuroblastomas Drives Sensitivity to PARP Inhibition. Clin Cancer Res. 2017;23:6875–87. doi: 10.1158/1078-0432.CCR-17-0593. [DOI] [PubMed] [Google Scholar]
- 133. https://clinicaltrials.gov/ct2/results?term=PARP&Search=Apply&age_v=&age=0&gndr=&type=&rslt=
- 134. https://clinicaltrials.gov/ct2/results?cond=&term=wee1&cntry=&state=&city=&dist=&Search=Search&age=0 .
- 135.Tsai WC, Hueng DY, Lin CR, Yang TC, Gao HW. Nrf2 Expressions Correlate with WHO Grades in Gliomas and Meningiomas. Int J Mol Sci. 2016;17:E722. doi: 10.3390/ijms17050722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Park JY, Kim YW, Park YK. Nrf2 expression is associated with poor outcome in osteosarcoma. Pathology. 2012;44:617–21. doi: 10.1097/PAT.0b013e328359d54b. [DOI] [PubMed] [Google Scholar]
- 137.Zhang J, Wang X, Wu W, Dang H, Wang B. Expression of the Nrf2 and Keap1 proteins and their clinical significance in osteosarcoma. Biochem Biophys Res Commun. 2016;473:42–6. doi: 10.1016/j.bbrc.2016.03.047. [DOI] [PubMed] [Google Scholar]
- 138.Bortolozzi R, Bresolin S, Rampazzo E, Paganin M, Maule F, et al. AKR1C enzymes sustain therapy resistance in paediatric T-ALL. Br J Cancer. 2018;118:985–94. doi: 10.1038/s41416-018-0014-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Klein S, Abraham M, Bulvik B, Dery E, Weiss ID, et al. CXCR4 Promotes Neuroblastoma Growth and Therapeutic Resistance through miR-15a/16-1-Mediated ERK and BCL2/Cyclin D1 Pathways. Cancer Res. 2018;78:1471–83. doi: 10.1158/0008-5472.CAN-17-0454. [DOI] [PubMed] [Google Scholar]
- 140.Laetsch TW, Liu X, Vu A, Sliozberg M, Vido M, et al. Multiple components of the spliceosome regulate Mcl1 activity in neuroblastoma. Cell Death Dis. 2014;5:e1072. doi: 10.1038/cddis.2014.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Aries IM, Bodaar K, Karim SA, Chonghaile TN, Hinze L, et al. PRC2 loss induces chemoresistance by repressing apoptosis in T cell acute lymphoblastic leukemia. J Exp Med. 2018;215:3094–114. doi: 10.1084/jem.20180570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Ham J, Costa C, Sano R, Lochmann TL, Sennott EM, et al. Exploitation of the Apoptosis-Primed State of MYCN-Amplified Neuroblastoma to Develop a Potent and Specific Targeted Therapy Combination. Cancer Cell. 2016;29:159–72. doi: 10.1016/j.ccell.2016.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Jubierre L, Soriano A, Planells-Ferrer L, Paris-Coderch L, Tenbaum SP, et al. BRG1/SMARCA4 is essential for neuroblastoma cell viability through modulation of cell death and survival pathways. Oncogene. 2016;35:5179–90. doi: 10.1038/onc.2016.50. [DOI] [PubMed] [Google Scholar]
- 144.Lestini BJ, Goldsmith KC, Fluchel MN, Liu X, Chen NL, et al. Mcl1 downregulation sensitizes neuroblastoma to cytotoxic chemotherapy and small molecule Bcl2-family antagonists. Cancer Biol Ther. 2009;8:1587–95. doi: 10.4161/cbt.8.16.8964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Avgeris M, Stamati L, Kontos CK, Piatopoulou D, Marmarinos A, et al. BCL2L12 improves risk stratification and prediction of BFM-chemotherapy response in childhood acute lymphoblastic leukemia. Clin Chem Lab Med. 2018;56:2104–18. doi: 10.1515/cclm-2018-0507. [DOI] [PubMed] [Google Scholar]
- 146.Jing D, Bhadr VA, Beck D, Thoms JA, Yakob NA, et al. Opposing regulation of BIM and BCL2 controls glucocorticoid-induced apoptosis of pediatric acute lymphoblastic leukemia cells. Blood. 2015;125:273–83. doi: 10.1182/blood-2014-05-576470. [DOI] [PubMed] [Google Scholar]
- 147.Tahir IM, Iqbal T, Jamil A, Saqib M. Association of BCL-2 with oxidative stress and total antioxidant status in pediatric acute lymphoblastic leukemia. J Biol Regul Homeost Agents. 2017;31:1023–27. [PubMed] [Google Scholar]
- 148.Pedersen MO, Gang AO, Brown P, Pedersen M, Knudsen H, et al. Real world data on young patients with high-risk diffuse large B-cell lymphoma treated with R-CHOP or R-CHOEP - MYC, BCL2 and BCL6 as prognostic biomarkers. PLoS One. 2017;12:e0186983. doi: 10.1371/journal.pone.0186983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Prutsch N, Gurnhofer E, Suske T, Liang HC, Schlederer M, et al. Dependency on the TYK2/STAT1/MCL1 axis in anaplastic large cell lymphoma. Leukemia. 2019;33:696–709. doi: 10.1038/s41375-018-0239-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Karpel-Massler G, Ishida CT, Bianchetti E, Zhang Y, Shu C, et al. Induction of synthetic lethality in IDH1-mutated gliomas through inhibition of Bcl-xL. Nat Commun. 2017;8:1067. doi: 10.1038/s41467-017-00984-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Das P, Puri T, Suri V, Sharma MC, Sharma BS, et al. Medulloblastomas: a correlative study of MIB-1 proliferation index along with expression of c-Myc, ERBB2, and anti-apoptotic proteins along with histological typing and clinical outcome. Childs Nerv Syst. 2009;25:825–35. doi: 10.1007/s00381-009-0884-9. [DOI] [PubMed] [Google Scholar]
- 152.McLendon RE, Friedman HS, Fuchs HE, Kun LE, Bigner SH. Diagnostic markers in paediatric medulloblastoma: a Paediatric Oncology Group Study. Histopathology. 1999;34:154–62. doi: 10.1046/j.1365-2559.1999.00577.x. [DOI] [PubMed] [Google Scholar]
- 153.Thallinger C, Wolschek MF, Maierhofer H, Skvara H, Pehamberger H, et al. Mcl-1 is a novel therapeutic target for human sarcoma: synergistic inhibition of human sarcoma xenotransplants by a combination of mcl-1 antisense oligonucleotides with low-dose cyclophosphamide. Clin Cancer Res. 2004;10:4185–91. doi: 10.1158/1078-0432.CCR-03-0774. [DOI] [PubMed] [Google Scholar]
- 154.Krskova L, Kalinova M, Brizova H, Mrhalova M, Sumerauer D, et al. Molecular and immunohistochemical analyses of BCL2, KI-67, and cyclin D1 expression in synovial sarcoma. Cancer Genet Cytogenet. 2009;193:1–8. doi: 10.1016/j.cancergencyto.2009.03.008. [DOI] [PubMed] [Google Scholar]
- 155.Haydn T, Metzger E, Schuele R, Fulda S. Concomitant epigenetic targeting of LSD1 and HDAC synergistically induces mitochondrial apoptosis in rhabdomyosarcoma cells. Cell Death Dis. 2017;8:e2879. doi: 10.1038/cddis.2017.239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Javaheri T, Kazemi Z, Pencik J, Pham HT, Kauer M, et al. Increased survival and cell cycle progression pathways are required for EWS/FLI1-induced malignant transformation. Cell Death Dis. 2016;7:e2419. doi: 10.1038/cddis.2016.268. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 157.Sanchez R, St-Cyr J, Lalonde ME, Healy J, Richer C, et al. Impact of promoter polymorphisms in key regulators of the intrinsic apoptosis pathway on the outcome of childhood acute lymphoblastic leukemia. Haematologica. 2014;99:314–21. doi: 10.3324/haematol.2013.085340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Marston E, Weston V, Jesson J, Maina E, McConville C, et al. Stratification of pediatric ALL by in vitro cellular responses to DNA double-strand breaks provides insight into the molecular mechanisms underlying clinical response. Blood. 2009;113:117–26. doi: 10.1182/blood-2008-03-142950. [DOI] [PubMed] [Google Scholar]
- 159.Holleman A, den Boer ML, Kazemier KM, Beverloo HB, von Bergh AR, et al. Decreased PARP and procaspase-2 protein levels are associated with cellular drug resistance in childhood acute lymphoblastic leukemia. Blood. 2005;106:1817–23. doi: 10.1182/blood-2004-11-4296. [DOI] [PubMed] [Google Scholar]
- 160.Grotzer MA, Eggert A, Zuzak TJ, Janss AJ, Marwaha S, et al. Resistance to TRAIL-induced apoptosis in primitive neuroectodermal brain tumor cells correlates with a loss of caspase-8 expression. Oncogene. 2000;19:4604–10. doi: 10.1038/sj.onc.1203816. [DOI] [PubMed] [Google Scholar]
- 161.Teitz T, Lahti JM, Kidd VJ. Aggressive childhood neuroblastomas do not express caspase-8: an important component of programmed cell death. J Mol Med (Berl) 2001;79:428–36. doi: 10.1007/s001090100233. [DOI] [PubMed] [Google Scholar]
- 162.Dukers DF, Meijer CJ, ten Berge RL, Vos W, Ossenkoppele GJ, et al. High numbers of active caspase 3-positive Reed-Sternberg cells in pretreatment biopsy specimens of patients with Hodgkin disease predict favorable clinical outcome. Blood. 2002;100:36–42. doi: 10.1182/blood.v100.1.36. [DOI] [PubMed] [Google Scholar]
- 163.Pingoud-Meier C, Lang D, Janss AJ, Rorke LB, Phillips PC, et al. Loss of caspase-8 protein expression correlates with unfavorable survival outcome in childhood medulloblastoma. Clin Cancer Res. 2003;9:6401–09. [PubMed] [Google Scholar]
- 164.Konopleva M, Pollyea DA, Potluri J, Chyla B, Hogdal L, et al. Efficacy and Biological Correlates of Response in a Phase II Study of Venetoclax Monotherapy in Patients with Acute Myelogenous Leukemia. Cancer Discov. 2016;6:1106–17. doi: 10.1158/2159-8290.CD-16-0313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Jain N, Keating M, Thompson P, Ferrajoli A, Burger J, et al. Ibrutinib and Venetoclax for First-Line Treatment of CLL. N Engl J Med. 2019;380:2095–103. doi: 10.1056/NEJMoa1900574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Coutre S, Choi M, Furman RR, Eradat H, Heffner L, et al. Venetoclax for patients with chronic lymphocytic leukemia who progressed during or after idelalisib therapy. Blood. 2018;131:1704–11. doi: 10.1182/blood-2017-06-788133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Place AE, Goldsmith K, Bourquin JP, Loh ML, Gore L, et al. Accelerating drug development in pediatric cancer: a novel Phase I study design of venetoclax in relapsed/refractory malignancies. Future Oncol. 2018;14:2115–29. doi: 10.2217/fon-2018-0121. [DOI] [PubMed] [Google Scholar]
- 168.Huether R, Dong L, Chen X, Wu G, Parker M, et al. The landscape of somatic mutations in epigenetic regulators across 1,000 paediatric cancer genomes. Nat Commun. 2014;5:3630. doi: 10.1038/ncomms4630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.McKenna ES, Roberts CW. Epigenetics and cancer without genomic instability. Cell Cycle. 2009;8:23–6. doi: 10.4161/cc.8.1.7290. [DOI] [PubMed] [Google Scholar]
- 170.Thirman MJ, Gill HJ, Burnett RC, Mbangkollo D, McCabe NR, et al. Rearrangement of the MLL gene in acute lymphoblastic and acute myeloid leukemias with 11q23 chromosomal translocations. N Engl J Med. 1993;329:909–14. doi: 10.1056/NEJM199309233291302. [DOI] [PubMed] [Google Scholar]
- 171.Peterson JF, Baughn LB, Pearce KE, Williamson CM, Benevides Demasi JC, et al. KMT2A (MLL) rearrangements observed in pediatric/young adult T-lymphoblastic leukemia/lymphoma: A 10-year review from a single cytogenetic laboratory. Genes Chromosomes Cancer. 2018;57:541–6. doi: 10.1002/gcc.22666. [DOI] [PubMed] [Google Scholar]
- 172.Bernt KM, Zhu N, Sinha AU, Vempati S, Faber J, et al. MLL-rearranged leukemia is dependent on aberrant H3K79 methylation by DOT1L. Cancer Cell. 2011;20:66–78. doi: 10.1016/j.ccr.2011.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Li Z, Takenobu H, Setyawati AN, Akita N, Haruta M, et al. EZH2 regulates neuroblastoma cell differentiation via NTRK1 promoter epigenetic modifications. Oncogene. 2018;37:2714–27. doi: 10.1038/s41388-018-0133-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Chen L, Alexe G, Dharia NV, Ross L, Iniguez AB, et al. CRISPR-Cas9 screen reveals a MYCN-amplified neuroblastoma dependency on EZH2. J Clin Invest. 2018;128:446–62. doi: 10.1172/JCI90793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Wang C, Liu Z, Woo CW, Li Z, Wang L, et al. EZH2 Mediates epigenetic silencing of neuroblastoma suppressor genes CASZ1, CLU, RUNX3, and NGFR. Cancer Res. 2012;72:315–24. doi: 10.1158/0008-5472.CAN-11-0961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Feng Z, Yao Y, Zhou C, Chen F, Wu F, et al. Pharmacological inhibition of LSD1 for the treatment of MLL-rearranged leukemia. J Hematol Oncol. 2016;9:24. doi: 10.1186/s13045-016-0252-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Bennani-Baiti IM, Machado I, Llombart-Bosch A, Kovar H. Lysine-specific demethylase 1 (LSD1/KDM1A/AOF2/BHC110) is expressed and is an epigenetic drug target in chondrosarcoma, Ewing’s sarcoma, osteosarcoma, and rhabdomyosarcoma. Hum Pathol. 2012;43:1300–7. doi: 10.1016/j.humpath.2011.10.010. [DOI] [PubMed] [Google Scholar]
- 178.Sankar S, Theisen ER, Bearss J, Mulvihill T, Hoffman LM, et al. Reversible LSD1 inhibition interferes with global EWS/ETS transcriptional activity and impedes Ewing sarcoma tumor growth. Clin Cancer Res. 2014;20:4584–97. doi: 10.1158/1078-0432.CCR-14-0072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Gao C, Zhang RD, Liu SG, Zhao XX, Cui L, et al. Low CREBBP expression is associated with adverse long-term outcomes in paediatric acute lymphoblastic leukaemia. Eur J Haematol. 2017;99:150–9. doi: 10.1111/ejh.12897. [DOI] [PubMed] [Google Scholar]
- 180.Robinson G, Parker M, Kranenburg TA, Lu C, Chen X, et al. Novel mutations target distinct subgroups of medulloblastoma. Nature. 2012;488:43–8. doi: 10.1038/nature11213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Viaene AN, Santi M, Rosenbaum J, Li MM, Surrey LF, et al. SETD2 mutations in primary central nervous system tumors. Acta Neuropathol Commun. 2018;6:123. doi: 10.1186/s40478-018-0623-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Zhu X, He F, Zeng H, Ling S, Chen A, et al. Identification of functional cooperative mutations of SETD2 in human acute leukemia. Nat Genet. 2014;46:287–93. doi: 10.1038/ng.2894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Olar A, Tran D, Mehta VP, Reinhardt A, Manekia JH, et al. ATRX protein loss and deregulation of PI3K/AKT pathway is frequent in pilocytic astrocytoma with anaplastic features. Clin Neuropathol. 2019;38:59–73. doi: 10.5414/NP301105. [DOI] [PubMed] [Google Scholar]
- 184.Cheung NK, Zhang J, Lu C, Parker M, Bahrami A, et al. Association of age at diagnosis and genetic mutations in patients with neuroblastoma. JAMA. 2012;307:1062–71. doi: 10.1001/jama.2012.228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Pages M, Beccaria K, Boddaert N, Saffroy R, Besnard A, et al. Co-occurrence of histone H3 K27M and BRAF V600E mutations in paediatric midline grade I ganglioglioma. Brain Pathol. 2018;28:103–11. doi: 10.1111/bpa.12473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Gielen GH, Gessi M, Hammes J, Kramm CM, Waha A, et al. H3F3A K27M mutation in pediatric CNS tumors: a marker for diffuse high-grade astrocytomas. Am J Clin Pathol. 2013;139:345–9. doi: 10.1309/AJCPABOHBC33FVMO. [DOI] [PubMed] [Google Scholar]
- 187.Schwartzentruber J, Korshunov A, Liu XY, Jones DT, Pfaff E, et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature. 2012;482:226–31. doi: 10.1038/nature10833. [DOI] [PubMed] [Google Scholar]
- 188.Lucio-Eterovic AK, Cortez MA, Valera ET, Motta FJ, Queiroz RG, et al. Differential expression of 12 histone deacetylase (HDAC) genes in astrocytomas and normal brain tissue: class II and IV are hypoexpressed in glioblastomas. BMC Cancer. 2008;8:243. doi: 10.1186/1471-2407-8-243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Oehme I, Deubzer HE, Wegener D, Pickert D, Linke JP, et al. Histone deacetylase 8 in neuroblastoma tumorigenesis. Clin Cancer Res. 2009;15:91–9. doi: 10.1158/1078-0432.CCR-08-0684. [DOI] [PubMed] [Google Scholar]
- 190.Wittenburg LA, Ptitsyn AA, Thamm D. A systems biology approach to identify molecular pathways altered by HDAC inhibition in osteosarcoma. J Cell Biochem. 2012;113:773–83. doi: 10.1002/jcb.23403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Warrell RP, Jr, He LZ, Richon V, Calleja E, Pandolfi P. Therapeutic targeting of transcription in acute promyelocytic leukemia by use of an inhibitor of histone deacetylase. J Natl Cancer Inst. 1998;90:1621–5. doi: 10.1093/jnci/90.21.1621. [DOI] [PubMed] [Google Scholar]
- 192.Bhatla T, Wang J, Morrison DJ, Raetz EA, Burke MJ, et al. Epigenetic reprogramming reverses the relapse-specific gene expression signature and restores chemosensitivity in childhood B-lymphoblastic leukemia. Blood. 2012;119:5201–10. doi: 10.1182/blood-2012-01-401687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Shukla N, Wetmore C, O’Brien MM, Silverman LB, Brown P, et al. Final Report of Phase 1 Study of the DOT1L Inhibitor, Pinometostat (EPZ-5676), in Children with Relapsed or Refractory MLL-r Acute Leukemia. Blood. 2016;128:2780. [Google Scholar]
- 194.Fouladi M, Park JR, Stewart CF, Gilbertson RJ, Schaiquevich P, et al. Pediatric phase I trial and pharmacokinetic study of vorinostat: a Children’s Oncology Group phase I consortium report. J Clin Oncol. 2010;28:3623–9. doi: 10.1200/JCO.2009.25.9119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Witt O, Milde T, Deubzer HE, Oehme I, Witt R, et al. Phase I/II intra-patient dose escalation study of vorinostat in children with relapsed solid tumor, lymphoma or leukemia. Klin Padiatr. 2012;224:398–403. doi: 10.1055/s-0032-1323692. [DOI] [PubMed] [Google Scholar]
- 196.Muscal JA, Thompson PA, Horton TM, Ingle AM, Ahern CH, et al. A phase I trial of vorinostat and bortezomib in children with refractory or recurrent solid tumors: a Children’s Oncology Group phase I consortium study (ADVL0916). Pediatr Blood Cancer. 2013;60:390–5. doi: 10.1002/pbc.24271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Hummel TR, Wagner L, Ahern C, Fouladi M, Reid JM, et al. A pediatric phase 1 trial of vorinostat and temozolomide in relapsed or refractory primary brain or spinal cord tumors: a Children’s Oncology Group phase 1 consortium study. Pediatr Blood Cancer. 2013;60:1452–7. doi: 10.1002/pbc.24541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.DuBois SG, Groshen S, Park JR, Haas-Kogan DA, Yang X, et al. Phase I Study of Vorinostat as a Radiation Sensitizer with 131I-Metaiodobenzylguanidine (131I-MIBG) for Patients with Relapsed or Refractory Neuroblastoma. Clin Cancer Res. 2015;21:2715–21. doi: 10.1158/1078-0432.CCR-14-3240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. https://clinicaltrials.gov/ct2/results?term=ezh2&age_v=&age=0&gndr=&type=&rslt=&Search=Apply .
- 200. https://clinicaltrials.gov/ct2/show/NCT03600649?term=lsd1&age=0&rank=2 .
- 201.Gabra MM, Salmena L. microRNAs and Acute Myeloid Leukemia Chemoresistance: A Mechanistic Overview. Front Oncol. 2017;7:255. doi: 10.3389/fonc.2017.00255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Galardi A, Colletti M, Businaro P, Quintarelli C, Locatelli F, et al. MicroRNAs in Neuroblastoma: Biomarkers with Therapeutic Potential. Curr Med Chem. 2018;25:584–600. doi: 10.2174/0929867324666171003120335. [DOI] [PubMed] [Google Scholar]
- 203.Kumar V, Kumar V, McGuire T, Coulter DW, Sharp JG, et al. Challenges and Recent Advances in Medulloblastoma Therapy. Trends Pharmacol Sci. 2017;38:1061–84. doi: 10.1016/j.tips.2017.09.002. [DOI] [PubMed] [Google Scholar]
- 204.Tarek N, Hayes-Jordan A, Salvador L, McAleer MF, Herzog CE, et al. Recurrent desmoplastic small round cell tumor responding to an mTOR inhibitor containing regimen. Pediatr Blood Cancer. 2018;65:1. doi: 10.1002/pbc.26768. [DOI] [PubMed] [Google Scholar]
- 205.Becher OJ, Gilheeney SW, Khakoo Y, Lyden DC, Haque S, et al. A phase I study of perifosine with temsirolimus for recurrent pediatric solid tumors. Pediatr Blood Cancer. 2017;64:7. doi: 10.1002/pbc.26409. [DOI] [PubMed] [Google Scholar]
- 206.Vo KT, Karski EE, Nasholm NM, Allen S, Hollinger F, et al. Phase 1 study of sirolimus in combination with oral cyclophosphamide and topotecan in children and young adults with relapsed and refractory solid tumors. Oncotarget. 2017;8:23851–61. doi: 10.18632/oncotarget.12904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Krueger DA, Care MM, Holland K, Agricola K, Tudor C, et al. Everolimus for subependymal giant-cell astrocytomas in tuberous sclerosis. N Engl J Med. 2010;363:1801–11. doi: 10.1056/NEJMoa1001671. [DOI] [PubMed] [Google Scholar]
- 208.Bagatell R, Norris R, Ingle AM, Ahern C, Voss S, et al. Phase 1 trial of temsirolimus in combination with irinotecan and temozolomide in children, adolescents and young adults with relapsed or refractory solid tumors: a Children’s Oncology Group Study. Pediatr Blood Cancer. 2014;61:833–9. doi: 10.1002/pbc.24874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Rodrik-Outmezguine VS, Okaniwa M, Yao Z, Novotny CJ, McWhirter C, et al. Overcoming mTOR resistance mutations with a new-generation mTOR inhibitor. Nature. 2016;534:272–6. doi: 10.1038/nature17963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Hosoi H, Dilling MB, Liu LN, Danks MK, Shikata T, et al. Studies on the mechanism of resistance to rapamycin in human cancer cells. Mol Pharmacol. 1998;54:815–24. doi: 10.1124/mol.54.5.815. [DOI] [PubMed] [Google Scholar]
- 211.Folgiero V, Miele E, Carai A, Ferretti E, Alfano V, et al. IDO1 involvement in mTOR pathway: a molecular mechanism of resistance to mTOR targeting in medulloblastoma. Oncotarget. 2016;7:52900–11. doi: 10.18632/oncotarget.9284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Pelloski CE, Lin E, Zhang L, Yung WK, Colman H, et al. Prognostic associations of activated mitogen-activated protein kinase and Akt pathways in glioblastoma. Clin Cancer Res. 2006;12:3935–41. doi: 10.1158/1078-0432.CCR-05-2202. [DOI] [PubMed] [Google Scholar]
- 213.Alvarenga AW, Machado LE, Rodrigues BR, Lupinacci FC, Sanemastu P, et al. Evaluation of Akt and RICTOR Expression Levels in Astrocytomas of All Grades. J Histochem Cytochem. 2017;65:93–103. doi: 10.1369/0022155416675850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.King D, Yeomanson D, Bryant H. PI3King the lock: targeting the PI3K/Akt/mTOR pathway as a novel therapeutic strategy in neuroblastoma. J Pediatr Hematol Oncol. 2015;37:245–51. doi: 10.1097/MPH.0000000000000329. [DOI] [PubMed] [Google Scholar]
- 215.Miyahara H, Yadavilli S, Natsumeda M, Rubens JA, Rodgers L, et al. The dual mTOR kinase inhibitor TAK228 inhibits tumorigenicity and enhances radiosensitization in diffuse intrinsic pontine glioma. Cancer Lett. 2017;400:110–6. doi: 10.1016/j.canlet.2017.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Flannery PC, DeSisto JA, Amani V, Venkataraman S, Lemma RT, et al. Preclinical analysis of MTOR complex 1/2 inhibition in diffuse intrinsic pontine glioma. Oncol Rep. 2018;39:455–64. doi: 10.3892/or.2017.6122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Akgul S, Li Y, Zheng S, Kool M, Treisman DM, et al. Opposing Tumor-Promoting and -Suppressive Functions of Rictor/mTORC2 Signaling in Adult Glioma and Pediatric SHH Medulloblastoma. Cell Rep. 2018;24:463–78. doi: 10.1016/j.celrep.2018.06.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Talpaz M, Silver RT, Druker BJ, Goldman JM, Gambacorti-Passerini C, et al. Imatinib induces durable hematologic and cytogenetic responses in patients with accelerated phase chronic myeloid leukemia: results of a phase 2 study. Blood. 2002;99:1928–37. doi: 10.1182/blood.v99.6.1928. [DOI] [PubMed] [Google Scholar]
- 219.Schultz KR, Carroll A, Heerema NA, Bowman WP, Aledo A, et al. Long-term follow-up of imatinib in pediatric Philadelphia chromosome-positive acute lymphoblastic leukemia: Children’s Oncology Group study AALL0031. Leukemia. 2014;28:1467–71. doi: 10.1038/leu.2014.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Rutkowski P, Magnan H, Chou AJ, Benson C. Treatment of gastrointestinal stromal tumours in paediatric and young adult patients with sunitinib: a multicentre case series. BMC Cancer. 2017;17:717. doi: 10.1186/s12885-017-3727-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Wedekind MF, Ranalli M, Shah N. Clinical efficacy of cabozantinib in two pediatric patients with recurrent renal cell carcinoma. Pediatr Blood Cancer. 2017;64:11. doi: 10.1002/pbc.26586. [DOI] [PubMed] [Google Scholar]
- 222.Truffaux N, Philippe C, Paulsson J, Andreiuolo F, Guerrini-Rousseau L, et al. Preclinical evaluation of dasatinib alone and in combination with cabozantinib for the treatment of diffuse intrinsic pontine glioma. Neuro Oncol. 2015;17:953–64. doi: 10.1093/neuonc/nou330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Glade Bender JL, Lee A, Reid JM, Baruchel S, Roberts T, et al. Phase I pharmacokinetic and pharmacodynamic study of pazopanib in children with soft tissue sarcoma and other refractory solid tumors: a children’s oncology group phase I consortium report. J Clin Oncol. 2013;31:3034–43. doi: 10.1200/JCO.2012.47.0914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Yang K, Fu L. Mechanisms of resistance to BCR-ABL TKIs and the therapeutic strategies: A review. Crit Rev Oncol Hematol. 2015;93:277–92. doi: 10.1016/j.critrevonc.2014.11.001. [DOI] [PubMed] [Google Scholar]
- 225.Aoe M, Shimada A, Muraoka M, Washio K, Nakamura Y, et al. ABL kinase mutation and relapse in 4 pediatric Philadelphia chromosome-positive acute lymphoblastic leukemia cases. Int J Hematol. 2014;99:609–15. doi: 10.1007/s12185-014-1565-3. [DOI] [PubMed] [Google Scholar]
- 226.Zwaan CM, Rizzari C, Mechinaud F, Lancaster DL, Lehrnbecher T, et al. Dasatinib in children and adolescents with relapsed or refractory leukemia: results of the CA180-018 phase I dose-escalation study of the Innovative Therapies for Children with Cancer Consortium. J Clin Oncol. 2013;31:2460–8. doi: 10.1200/JCO.2012.46.8280. [DOI] [PubMed] [Google Scholar]
- 227.Bresler SC, Weiser DA, Huwe PJ, Park JH, Krytska K, et al. ALK mutations confer differential oncogenic activation and sensitivity to ALK inhibition therapy in neuroblastoma. Cancer Cell. 2014;26:682–94. doi: 10.1016/j.ccell.2014.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Tucker ER, Danielson LS, Innocenti P, Chesler L. Tackling Crizotinib Resistance: The Pathway from Drug Discovery to the Pediatric Clinic. Cancer Res. 2015;75:2770–4. doi: 10.1158/0008-5472.CAN-14-3817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Sabnis AJ, Bivona T. Principles of Resistance to Targeted Cancer Therapy: Lessons from Basic and Translational Cancer Biology. Trends Mol Med. 2019;25:185–97. doi: 10.1016/j.molmed.2018.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Cooper J, Giancotti F. Integrin Signaling in Cancer: Mechanotransduction, Stemness, Epithelial Plasticity, and Therapeutic Resistance. Cancer Cell. 2019;35:347–67. doi: 10.1016/j.ccell.2019.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Marona P, Gorka J, Kotlinowski J, Majka M, Jura J, et al. C-Met as a Key Factor Responsible for Sustaining Undifferentiated Phenotype and Therapy Resistance in Renal Carcinomas. Cells. 2019;8:E272. doi: 10.3390/cells8030272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Harivenkatesh N, Kumar L, Bakhshi S, Sharma A, Kabra M, et al. Influence of MDR1 and CYP3A5 genetic polymorphisms on trough levels and therapeutic response of imatinib in newly diagnosed patients with chronic myeloid leukemia. Pharmacol Res. 2017;120:138–45. doi: 10.1016/j.phrs.2017.03.011. [DOI] [PubMed] [Google Scholar]
- 233.Brown LM, Hanna DT, Khaw SL, Ekert P. Dysregulation of BCL-2 family proteins by leukemia fusion genes. J Biol Chem. 2017;292:14325–33. doi: 10.1074/jbc.R117.799056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Morgillo F, Della Corte CM, Fasano M, Ciardiello F. Mechanisms of resistance to EGFR-targeted drugs: lung cancer. ESMO Open. 2016;1:e000060. doi: 10.1136/esmoopen-2016-000060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Yu AL, Gilman AL, Ozkaynak MF, London WB, Kreissman SG, et al. Anti-GD2 antibody with GM-CSF, interleukin-2, and isotretinoin for neuroblastoma. N Engl J Med. 2010;363:1324–34. doi: 10.1056/NEJMoa0911123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Shusterman S, London WB, Gillies SD, Hank JA, Voss SD, et al. 18-IL2 in patients with relapsed/refractory neuroblastoma: a Children’s Oncology Group (COG) phase II study. J Clin Oncol. 2010;28:4969–75. doi: 10.1200/JCO.2009.27.8861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Cheung NK, Cheung IY, Kushner BH, Ostrovnaya I, Chamberlain E, et al. Murine anti-GD2 monoclonal antibody 3F8 combined with granulocyte-macrophage colony-stimulating factor and 13-cis-retinoic acid in high-risk patients with stage 4 neuroblastoma in first remission. J Clin Oncol. 2012;30:3264–70. doi: 10.1200/JCO.2011.41.3807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Mody R, Naranjo A, Van Ryn C, Yu AL, London WB, et al. Irinotecan-temozolomide with temsirolimus or dinutuximab in children with refractory or relapsed neuroblastoma (COG ANBL1221): an open-label, randomised, phase 2 trial. Lancet Oncol. 17;18:946–57. doi: 10.1016/S1470-2045(17)30355-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Goldman S, Smith L, Galardy P, Perkins SL, Frazer JK, et al. Rituximab with chemotherapy in children and adolescents with central nervous system and/or bone marrow-positive Burkitt lymphoma/leukaemia: a Children’s Oncology Group Report. Br J Haematol. 2014;167:394–401. doi: 10.1111/bjh.13040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Meinhardt A, Burkhardt B, Zimmermann M, Borkhardt A, Kontny U, et al. Phase II window study on rituximab in newly diagnosed pediatric mature B-cell non-Hodgkin’s lymphoma and Burkitt leukemia. J Clin Oncol. 2010;28:3115–21. doi: 10.1200/JCO.2009.26.6791. [DOI] [PubMed] [Google Scholar]
- 241.Locatelli F, Mauz-Koerholz C, Neville K, Llort A, Beishuizen A, et al. Brentuximab vedotin for paediatric relapsed or refractory Hodgkin’s lymphoma and anaplastic large-cell lymphoma: a multicentre, open-label, phase 1/2 study. Lancet Haematol. 18;5:450–61. doi: 10.1016/S2352-3026(18)30153-4. [DOI] [PubMed] [Google Scholar]
- 242.Anderson PM, Bielack SS, Gorlick RG, Skubitz K, Daw NC, et al. A phase II study of clinical activity of SCH 717454 (robatumumab) in patients with relapsed osteosarcoma and Ewing sarcoma. Pediatr Blood Cancer. 2016;63:1761–70. doi: 10.1002/pbc.26087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Pappo AS, Vassal G, Crowley JJ, Bolejack V, Hogendoorn PC, et al. A phase 2 trial of R1507, a monoclonal antibody to the insulin-like growth factor-1 receptor (IGF-1R), in patients with recurrent or refractory rhabdomyosarcoma, osteosarcoma, synovial sarcoma, and other soft tissue sarcomas: results of a Sarcoma Alliance for Research Through Collaboration study. Cancer. 2014;120:2448–56. doi: 10.1002/cncr.28728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Peters KB, Lipp ES, Miller E, Herndon JE, 2nd, McSherry F, et al. Phase I/II trial of vorinostat, bevacizumab, and daily temozolomide for recurrent malignant gliomas. J Neurooncol. 2018;137:349–56. doi: 10.1007/s11060-017-2724-1. [DOI] [PubMed] [Google Scholar]
- 245.Navid F, Santana VM, Neel M, McCarville MB, Shulkin BL, et al. A phase II trial evaluating the feasibility of adding bevacizumab to standard osteosarcoma therapy. Int J Cancer. 2017;141:1469–77. doi: 10.1002/ijc.30841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.DeWire M, Fouladi M, Turner DC, Wetmore C, Hawkins C, et al. An open-label, two-stage, phase II study of bevacizumab and lapatinib in children with recurrent or refractory ependymoma: a collaborative ependymoma research network study (CERN). J Neurooncol. 2015;123:85–91. doi: 10.1007/s11060-015-1764-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Hiraga J, Tomita A, Sugimoto T, Shimada K, Ito M, et al. Down-regulation of CD20 expression in B-cell lymphoma cells after treatment with rituximab-containing combination chemotherapies: its prevalence and clinical significance. Blood. 2009;113:4885–93. doi: 10.1182/blood-2008-08-175208. [DOI] [PubMed] [Google Scholar]
- 248.Al-Rohil RN, Torres-Cabala CA, Patel A, Tetzlaff MT, Ivan D, et al. Loss of CD30 expression after treatment with brentuximab vedotin in a patient with anaplastic large cell lymphoma: a novel finding. J Cutan Pathol. 2016;43:1161–6. doi: 10.1111/cup.12797. [DOI] [PubMed] [Google Scholar]
- 249.Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med. 2013;368:1509–18. doi: 10.1056/NEJMoa1215134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Ozkaynak MF, Sondel PM, Krailo MD, Gan J, Javorsky B, et al. 18) with granulocyte-macrophage colony-stimulating factor in children with neuroblastoma immediately after hematopoietic stem-cell transplantation: a Children’s Cancer Group Study. J Clin Oncol. 2000;18:4077–85. doi: 10.1200/JCO.2000.18.24.4077. [DOI] [PubMed] [Google Scholar]
- 251.Desai AV, Fox E, Smith LM, Lim AP, Maris JM, et al. Pharmacokinetics of the chimeric anti-GD2 antibody, ch14. Cancer Chemother Pharmacol. 2014;74:1047–55. doi: 10.1007/s00280-014-2575-9. [DOI] [PubMed] [Google Scholar]
- 252.Cheung IY, Kushner BH, Modak S, Basu EM, Roberts SS, et al. Phase I trial of anti-GD2 monoclonal antibody hu3F8 plus GM-CSF: Impact of body weight, immunogenicity and anti-GD2 response on pharmacokinetics and survival. Oncoimmunology. 2017;6:1358331. doi: 10.1080/2162402X.2017.1358331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.van der Sluis IM, de Groot-Kruseman H, Te Loo M, Tissing WJE, van den Bos C, et al. coli asparaginase in children with previously untreated acute lymphoblastic leukemia: A randomized multicenter study of the Dutch Childhood Oncology Group. Pediatr Blood Cancer. 2018;65:27083. doi: 10.1002/pbc.27083. [DOI] [PubMed] [Google Scholar]
- 254.Paillassa J, Leguay T, Thomas X, Huguet F, Audrain M, et al. Monitoring of asparagine depletion and anti-L-asparaginase antibodies in adult acute lymphoblastic leukemia treated in the pediatric-inspired GRAALL-2005 trial. Blood Cancer J. 2018;8:45. doi: 10.1038/s41408-018-0084-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Tram Henriksen L, Gottschalk Hojfeldt S, Schmiegelow K, Frandsen TL, Skov Wehner P, et al. Prolonged first-line PEG-asparaginase treatment in pediatric acute lymphoblastic leukemia in the NOPHO ALL2008 protocol-Pharmacokinetics and antibody formation. Pediatr Blood Cancer. 2017;64:12. doi: 10.1002/pbc.26686. [DOI] [PubMed] [Google Scholar]
- 256.Schleiermacher G, Javanmardi N, Bernard V, Leroy Q, Cappo J, et al. Emergence of new ALK mutations at relapse of neuroblastoma. J Clin Oncol. 2014;32:2727–34. doi: 10.1200/JCO.2013.54.0674. [DOI] [PubMed] [Google Scholar]
- 257.Bellini A, Bernard V, Leroy Q, Rio Frio T, Pierron G, et al. Deep Sequencing Reveals Occurrence of Subclonal ALK Mutations in Neuroblastoma at Diagnosis. Clin Cancer Res. 2015;21:4913–21. doi: 10.1158/1078-0432.CCR-15-0423. [DOI] [PubMed] [Google Scholar]
- 258.Chen L, Humphreys A, Turnbull L, Bellini A, Schleiermacher G, et al. Identification of different ALK mutations in a pair of neuroblastoma cell lines established at diagnosis and relapse. Oncotarget. 2016;7:87301–11. doi: 10.18632/oncotarget.13541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Mehrazma M, Madjd Z, Kalantari E, Panahi M, Hendi A, et al. Expression of stem cell markers, CD133 and CD44, in pediatric solid tumors: a study using tissue microarray. Fetal Pediatr Pathol. 2013;32:192–204. doi: 10.3109/15513815.2012.701266. [DOI] [PubMed] [Google Scholar]
- 260.Choi SA, Wang KC, Phi JH, Lee JY, Park CK, et al. A distinct subpopulation within CD133 positive brain tumor cells shares characteristics with endothelial progenitor cells. Cancer Lett. 2012;324:221–30. doi: 10.1016/j.canlet.2012.05.026. [DOI] [PubMed] [Google Scholar]
- 261.Toren A, Bielorai B, Jacob-Hirsch J, Fisher T, Kreiser D, et al. CD133-positive hematopoietic stem cell “stemness” genes contain many genes mutated or abnormally expressed in leukemia. Stem Cells. 2005;23:1142–53. doi: 10.1634/stemcells.2004-0317. [DOI] [PubMed] [Google Scholar]
- 262.Sartelet H, Imbriglio T, Nyalendo C, Haddad E, Annabi B, et al. CD133 expression is associated with poor outcome in neuroblastoma via chemoresistance mediated by the AKT pathway. Histopathology. 2012;60:1144–55. doi: 10.1111/j.1365-2559.2012.04191.x. [DOI] [PubMed] [Google Scholar]
- 263.Bahnassy AA, Fawzy M, El-Wakil M, Zekri AR, Abdel-Sayed A, et al. Aberrant expression of cancer stem cell markers (CD44, CD90, and CD133) contributes to disease progression and reduced survival in hepatoblastoma patients: 4-year survival data. Transl Res. 2015;165:396–406. doi: 10.1016/j.trsl.2014.07.009. [DOI] [PubMed] [Google Scholar]
- 264.Friedman GK, Moore BP, Nan L, Kelly VM, Etminan T, et al. Pediatric medulloblastoma xenografts including molecular subgroup 3 and CD133+ and CD15+ cells are sensitive to killing by oncolytic herpes simplex viruses. Neuro Oncol. 2016;18:227–35. doi: 10.1016/j.omto.2017.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Zambo I, Hermanova M, Zapletalova D, Skoda J, Mudry P, et al. Expression of nestin, CD133 and ABCG2 in relation to the clinical outcome in pediatric sarcomas. Cancer Biomark. 2016;17:107–16. doi: 10.3233/CBM-160623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.O’Hagan-Wong K, Nadeau S, Carrier-Leclerc A, Apablaza F, Hamdy R, et al. Increased IL-6 secretion by aged human mesenchymal stromal cells disrupts hematopoietic stem and progenitor cells’ homeostasis. Oncotarget. 2016;7:13285–96. doi: 10.18632/oncotarget.7690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Zhang LJ, Liu W, Gao YM, Qin YJ, Wu R. The expression of IL-6 and STAT3 might predict progression and unfavorable prognosis in Wilms’ tumor. Biochem Biophys Res Commun. 2013;435:408–13. doi: 10.1016/j.bbrc.2013.04.102. [DOI] [PubMed] [Google Scholar]
- 268.Bid HK, Roberts RD, Cam M, Audino A, Kurmasheva RT, et al. DeltaNp63 promotes pediatric neuroblastoma and osteosarcoma by regulating tumor angiogenesis. Cancer Res. 2014;74:320–9. doi: 10.1158/0008-5472.CAN-13-0894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Gross AC, Cam H, Phelps DA, Saraf AJ, Bid HK, et al. IL-6 and CXCL8 mediate osteosarcoma-lung interactions critical to metastasis. JCI Insight. 2018;3:99791. doi: 10.1172/jci.insight.99791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Samuel P, Fabbri M, Carter D. Mechanisms of Drug Resistance in Cancer: The Role of Extracellular Vesicles. Proteomics. 2017;17:23–4. doi: 10.1002/pmic.201600375. [DOI] [PubMed] [Google Scholar]
- 271.Mittapalli RK, Chung AH, Parrish KE, Crabtree D, Halvorson KG, et al. ABCG2 and ABCB1 Limit the Efficacy of Dasatinib in a PDGF-B-Driven Brainstem Glioma Model. Mol Cancer Ther. 2016;15:819–29. doi: 10.1158/1535-7163.MCT-15-0093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Ingram WJ, Crowther LM, Little EB, Freeman R, Harliwong I, et al. ABC transporter activity linked to radiation resistance and molecular subtype in pediatric medulloblastoma. Exp Hematol Oncol. 2013;2:26. doi: 10.1186/2162-3619-2-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Asgharzadeh S, Salo JA, Ji L, Oberthuer A, Fischer M, et al. Clinical significance of tumor-associated inflammatory cells in metastatic neuroblastoma. J Clin Oncol. 2012;30:3525–32. doi: 10.1200/JCO.2011.40.9169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Majzner RG, Simon JS, Grosso JF, Martinez D, Pawel BR, et al. Assessment of programmed death-ligand 1 expression and tumor-associated immune cells in pediatric cancer tissues. Cancer. 2017;123:3807–15. doi: 10.1002/cncr.30724. [DOI] [PubMed] [Google Scholar]
- 275.Fujiwara T, Fukushi J, Yamamoto S, Matsumoto Y, Setsu N, et al. Macrophage infiltration predicts a poor prognosis for human ewing sarcoma. Am J Pathol. 2011;179:1157–70. doi: 10.1016/j.ajpath.2011.05.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Alanee S, Shah S, Vijai J, Schrader K, Hamilton R, et al. Prevalence of HOXB13 mutation in a population of Ashkenazi Jewish men treated for prostate cancer. Fam Cancer. 2013;12:597–600. doi: 10.1007/s10689-013-9618-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Li X, Li D, Huang X, Zhou P, Shi Q, et al. Helios expression in regulatory T cells promotes immunosuppression, angiogenesis and the growth of leukemia cells in pediatric acute lymphoblastic leukemia. Leuk Res. 2018;67:60–6. doi: 10.1016/j.leukres.2018.02.007. [DOI] [PubMed] [Google Scholar]
- 278.Rigo V, Emionite L, Daga A, Astigiano S, Corrias MV, et al. Combined immunotherapy with anti-PDL-1/PD-1 and anti-CD4 antibodies cures syngeneic disseminated neuroblastoma. Sci Rep. 2017;7:14049. doi: 10.1038/s41598-017-14417-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Parsons E, Otieno JA, Ong’echa JM, Nixon CE, Vulule J, et al. Regulatory T Cells in Endemic Burkitt Lymphoma Patients Are Associated with Poor Outcomes: A Prospective, Longitudinal Study. PLoS One. 2016;11:e0167841. doi: 10.1371/journal.pone.0167841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Mao Y, Eissler N, Blanc KL, Johnsen JI, Kogner P, et al. Targeting Suppressive Myeloid Cells Potentiates Checkpoint Inhibitors to Control Spontaneous Neuroblastoma. Clin Cancer Res. 2016;22:3849–59. doi: 10.1158/1078-0432.CCR-15-1912. [DOI] [PubMed] [Google Scholar]
- 281.Salem ML, El-Shanshory MR, Abdou SH, Attia MS, Sobhy SM, et al. Chemotherapy alters the increased numbers of myeloid-derived suppressor and regulatory T cells in children with acute lymphoblastic leukemia. Immunopharmacol Immunotoxicol. 2018;40:158–67. doi: 10.1080/08923973.2018.1424897. [DOI] [PubMed] [Google Scholar]
- 282.Long AH, Highfill SL, Cui Y, Smith JP, Walker AJ, et al. Reduction of MDSCs with All-trans Retinoic Acid Improves CAR Therapy Efficacy for Sarcomas. Cancer Immunol Res. 2016;4:869–80. doi: 10.1158/2326-6066.CIR-15-0230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Houghton PJ, Morton CL, Tucker C, Payne D, Favours E, et al. The pediatric preclinical testing program: description of models and early testing results. Pediatr Blood Cancer. 2007;49:928–40. doi: 10.1002/pbc.21078. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.