

Abstract
The emergence of several fentanyl-related substances in the recreational drug marketplace has resulted in a surge of opioid overdose deaths in the United States. Many of these substances have never been examined in living organisms under controlled conditions. In the present study, seven fentanyl-related substances were tested in adult male Swiss Webster mice for their effects on locomotion and antinociception and compared to those of fentanyl and morphine. In locomotor activity tests, fentanyl (1, 10 mg/kg), morphine (100, 180 mg/kg), isobutyrylfentanyl (10 mg/kg), crotonylfentanyl (10 mg/kg), para-fluorobutyrylfentanyl (10, 100 mg/kg), para-methoxybutyrylfentanyl (10 mg/kg), thiophenefentanyl (100 mg/kg), and benzodioxolefentanyl (0.1 mg/kg) produced significant (p ≤ 0.05) dose-dependent increases in locomotion. Valerylfentanyl, however, was without effects on locomotion up to 100 mg/kg. In warm-water tail-withdrawal tests, all substances produced significant (p ≤ 0.05) dose-dependent increases in antinociception with increasing ED50 values (CI) of isobutyrylfentanyl [0.0768 mg/kg (0.044–0.128)] > fentanyl [0.0800 mg/kg (0.0403–0.164)] > para-methoxybutyrylfentanyl [0.106 mg/kg (0.0516–0.195)] > crotonylfentanyl [0.226 mg/kg (0.176–0.292)] > para-fluorobutyrylfentanyl [0.908 mg/kg (0.459–1.58)] > thiophenefentanyl [4.66 mg/kg (3.65–5.95)] > valerylfentanyl [6.43 mg/kg (3.91–10.5)] > morphine [7.82 mg/kg (5.42–11.0)] > benzodioxolefentanyl [46.3 mg/kg (25.8–83.4)]. Naltrexone (1 mg/kg) increased antinociceptive ED50 values several fold in decreasing magnitudes of isobutyrylfentanyl (233x) > para-methoxybutyrylfentanyl (37.7x) > thiophenefentanyl (34.6x) > valerylfentanyl (11.9x) > para-fluorobutyrylfentanyl (10.9x) > benzodioxolefentanyl (8.42x) > crotonylfentanyl (6.27x) > fentanyl (3.95x) > morphine (1.48x). These findings establish that locomotor and antinociceptive effects of several fentanyl-related substances are similar to those of morphine and fentanyl and are mediated by opioid receptors.
This article is part of the Special Issue entitled ‘New Vistas in Opioid Pharmacology’.
Keywords: Analgesia, Analog, Fentanyl, Locomotion, Mice, Opioid
1. Introduction
It is well-established that fatalities attributed to opioid overdose are rapidly increasing in the United States and that a surge in the supply of illicitly manufactured fentanyl and its analogs is driving this trend (Gladden et al., 2016; Peterson et al., 2016; Rudd et al., 2016; O’Donnell et al., 2017a, 2017b). Fentanyl is a synthetic opioid that is primarily prescribed for acute and chronic pain and is approximately 100 times more potent than morphine as an analgesic. Fentanyl-related substances are structurally similar to fentanyl and therefore often have comparable biological effects while rendering them less detectable to routine screening methods (Maher et al., 2018; Palaty et al., 2018). Many of these substances were originally designed in the early 1960’s for use as combination anesthetic and analgesic agents (Corssen et al., 1964; Stanley, 2014a). However, fentanyl-related substances have emerged in the recreational drug marketplace as heroin adulterants, constituents of counterfeit prescription pills, and even as standalone products designed to circumvent drug control laws (Drug Enforcement Administration, 2016; O’Donnell et al., 2017a; Sutter et al., 2017). Several recent reports of analytically confirmed cases of non-fatal intoxication that involved fentanyl-related substances serve as evidence of this re-emergence (Backberg et al., 2015; Helander et al., 2016, 2017). Furthermore, numerous post-mortem toxicology and medical examiner reports have identified fentanyl-related substances in deceased individuals (Poklis et al., 2015, 2016; Zawilska, 2017).
Fentanyl, along with a number of analogs, were first synthesized in 1960 by Dr. Paul Janssen and first appear in the patent literature beginning in 1964 (Janssen, 1964; Stanley, 2014b). The potential modifications of the fentanyl structure are vast, and illicit drug suppliers have taken advantage of this variety to elude prosecution. In Europe, for example, 38 new opioids have been detected on Europe’s drug market between 2009 and 2017, 28 of which were fentanyl-related substances, and 10 of which were reported for the first time in 2017 (European Monitoring Centre for Drugs and Drug Addiction, 2018). In the United States between January 1, 2015 and December 31, 2016, 8511 reports of fentanyl-related substances were made by state and local forensic laboratories (Drug Enforcement Administration, 2017a). Many of the fentanyl-related substances that are appearing have never been evaluated for their in vivo pharmacological effects under controlled conditions. Today, illicitly manufactured and imported fentanyl and fentanyl-related substances are driving massive increases in opioid overdose deaths in the United States (Drug Enforcement Administration, 2017b). In 2013, 3105 deaths involved synthetic narcotics, and according to provisional estimates, that number rose to 28,466 in 2017, representing an increase of 816% over a period of 4 years (Centers for Disease Control and Prevention, 2018). Synthetic narcotics, including fentanyl and fentanyl-related substances, are now estimated to account for approximately 60% of all opioid overdose deaths in the United States (Centers for Disease Control and Prevention, 2018). As a result, the United States Department of Justice, on February 6, 2018, exercised its emergency scheduling authority under the purview of the Controlled Substances Act by temporarily placing nearly all fentanyl-related substances into a Schedule I status to combat imminent hazards to public safety (United States Department of Justice, 2018). It cannot be assumed, however, that all fentanyl-related substances would have abuse liability similar to fentanyl itself; in fact, some are reported to function as opioid receptor antagonists (Bagley et al., 1989; Wynn et al., 1991).
Several fentanyl-related compounds that have either appeared in recently confiscated drug samples of illicit drug users, or have been discussed in drug user forums on the internet, or in other ways have captured the attention of law enforcement have either been minimally or never studied for their in vivo pharmacological effects (Drug Enforcement Administration, 2018a, b). Increases in locomotor activity and the induction of antinociception in mice are two characteristic effects of abused opioids such as morphine and fentanyl (Wise and Bozarth, 1987; Robinson and Berridge, 1993). Drugs sharing in similar pharmacological effects of abused drugs is one factor that regulatory authorities consider when making predictions of the abuse likelihood of compounds and consequentially for making decisions regarding their level of regulatory control (Beardsley, 2007). In the present study, we examined the effects on locomotion and nociception in mice of seven of these fentanyl-related compounds to determine if their effects were similar in kind to those of morphine and fentanyl as the first step in a more comprehensive pharmacological comparison to these well-known abused opioids. The evaluated drugs included isobutyrylfentanyl, crotonylfentanyl, valerylfentanyl, para-fluorobutyrylfentanyl, para-methoxybutyrylfentanyl, thiophenefentanyl, and benzodioxolefentanyl. Four of these drugs had never been previously examined under controlled laboratory conditions in living organisms, and the rest only minimally so. The results indicated marked differences in potency and in efficacy among the analogs for producing these effects, and their rank orders for doing so were not always predictable by their in vitro receptor binding potency and efficacy relationships.
2. Materials and methods
2.1. Drugs
(1) Morphine, 4R,4aR,7S,7aR,12bS)-3-methyl-2,3,4,4a,7,7a-hexahydro-1H-4,12-methanobenzofuro[3,2-e]isoquinoline-7,9-diol sulfate pentahydrate, was provided by the National Institute on Drug Abuse (Bethesda, MD, USA) Drug Supply Program. (2) Fentanyl, N-(1-phenethylpiperidin-4-yl)–N-phenylpropionamide citrate, was obtained from Sigma-Aldrich (St. Louis, MO, USA). Fentanyl-related substances: (3) isobutyrylfentanyl; N-(1-phenethylpiperidin-4-yl)–N-phenylisobutyramide hydrochloride, (4) crotonylfentanyl; (E)–N-(1-phenethylpiperidin-4-yl)–N-phenylbut-2-enamide, (5) valerylfentanyl; N-(1-phenethylpiperidin-4-yl)–N-phenylpentanamide hydrochloride, (6) para-fluorobutyrylfentanyl; N-(4-fluorophenyl)–N-(1-phenethylpiperidin-4-yl)butyramide hydrochloride, (7) para-methoxybutyrylfentanyl; N-(4-methoxyphenyl)–N-(1-phenethylpiperidin-4-yl)butyramide hydrochloride, (8) thiophenefentanyl; N-(1-phenethylpiperidin-4-yl)–N-phenylthiophene-2-carboxamide hydrochloride, and (9) benzodioxolefentanyl; N-(1-phenethylpiperidin-4-yl)–N-phenylbenzo[d][1,3]dioxole-5-carboxamide were obtained from the Cayman Chemical Company (Ann Arbor, MI, USA) (see Fig. 1 for chemical structures). (10) Naltrexone, (4R,4aS,7aR, 12bS)-3-(cyclopropylmethyl)-4a,9-dihydroxy-2,3,4,4a, 5,6-hexahydro-1H-4,12-methanobenzofuro[3,2-e]isoquinolin-7(7aH)-one hydrochloride, was obtained from Sigma-Aldrich (St. Louis, MO, USA). All drugs were obtained as dry powders and dissolved in either sterile saline (Fisher Scientific, Hampton, NH, USA) or 2% methylcellulose (Sigma-Aldrich, St. Louis, MO, USA) in deionized water and were administered subcutaneously (s.c.) in a volume equivalent to 10 ml/kg body weight.
Fig. 1.
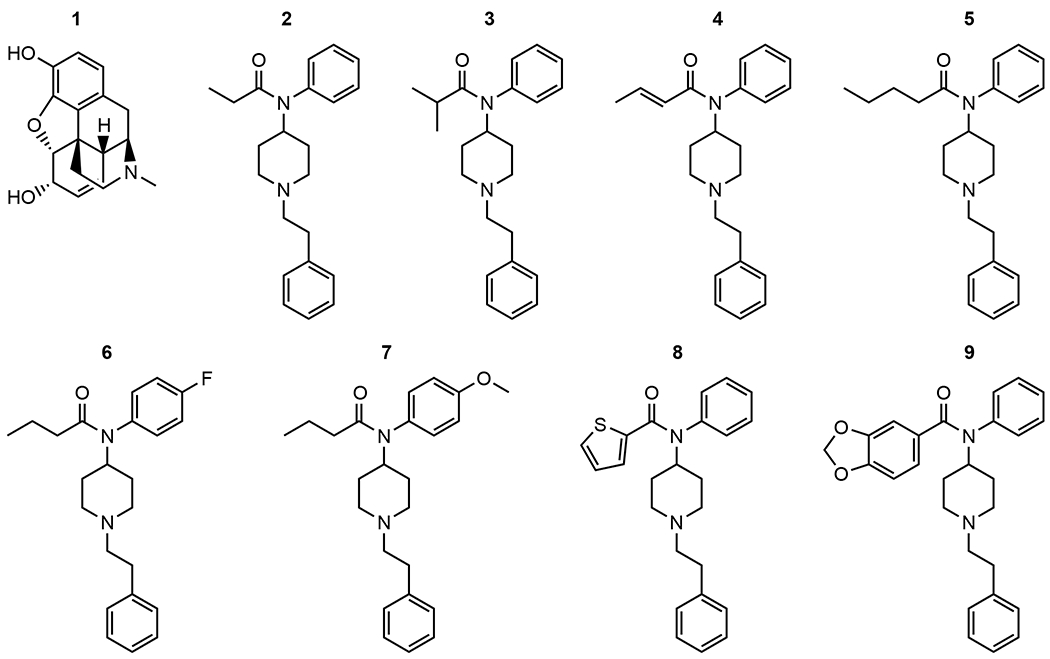
Chemical structures of (1) morphine, (2) fentanyl, (3) isobutyrylfentanyl, (4) crotonylfentanyl, (5) valerylfentanyl, (6) para-fluorobutyrylfentanyl, (7) para-methoxybutyrylfentanyl, (8) thiophenefentanyl, (9) benzodioxolefentanyl.
2.2. Subjects
Adult male CFW Mice [Swiss Webster, Crl:CFW(SW), Charles River Laboratories International, Raleigh, NC, USA] weighing approximately 25–50 g at the time of testing were housed four subjects per cage in Association for Assessment and Accreditation of Laboratory Animal Care-accredited facilities. Mice had ad libitum access to food (Teklad 7012 Rodent Diet; Envigo, Madison, WI, USA) and tap water. Vivaria were maintained at 22 °C ± 2 °C and 45%–50% humidity, with lights set to a 12-h light/dark cycle (lights on at 06:00). All testing occurred during the light cycle. All subjects were acclimated to the vivarium for at least one week before the commencement of studies and were drug-naive before testing. All procedures were carried out in accordance with the Guide for Care and Use of Laboratory Animals as adopted and promulgated by the National Institutes of Health (Committee for the Update of the Guide for the Care and Use of Laboratory Animals, 2011). The experimental protocol was approved by the Institutional Animal Care and Use Committee at Virginia Commonwealth University.
2.3. Locomotor activity
Locomotor activity tests were conducted in eight commercially obtained, automated activity monitoring devices each enclosed in sound- and light-attenuating chambers that recorded distance traveled in cm in 10-min intervals via computer-controlled circuitry (AccuScan Instruments, Columbus, OH, USA). The interior of each device was divided into separate 20 × 20 × 30 cm arenas permitting the independent and simultaneous measurement of two mice. Sixteen photobeam sensors per axis were spaced 2.5 cm apart along the walls of the chamber and were used to detect movement. A fan mounted in each test chamber provided ventilation and masking noise. On a test day, mice were brought to the laboratory where they were allowed to acclimate for approximately 30 min. Mice were injected s.c. with either vehicle or a test substance and immediately placed in the test chambers where their activity was recorded for 120 min in 10-min intervals. A dose range of fentanyl-related substances was tested to include a sufficient number of doses such that: (1) maximal mean effects between at least two doses significantly differed from one another (as defined by non-overlapping SEMs); and (2) at least at one time interval, the effects of at least one dose was significantly different (p ≤ 0.05) from vehicle; or (3) until 100 mg/kg was tested. Based on these criteria, fentanyl-related substances were tested at doses of 0.1, 1, and 10 (and 100 if necessary) mg/kg. Fentanyl (0.1, 1 and 10 mg/kg) and morphine (1, 10, 100 and 180 mg/kg) were tested as comparators. The total distance traveled (cm) within each 10-min bin during the experimental session was recorded for each mouse.
2.4. Warm-water tail withdrawal
A commercial warm water bath (Model # JBN5 US; Grant Instruments Ltd., Cambridge, UK) was used to assess nociception. At least 24 h before the start of testing, mice were handled and habituated to a restraint tube crafted from a cotton-lined surgical drape during two 5-min habituation periods. Before starting drug testing, mice were placed in the restraint tube and tested for tail-withdrawal latencies under ambient temperature conditions to ensure they did not perform a tail withdrawal under non-noxious conditions. The distal 3 cm of the tail was immersed into a bath containing 21 ± 0.5 °C water for three trials separated by a 2-min inter-trial interval. Subjects that withdrew their tails before 10s had elapsed in more than one trial were omitted from all subsequent testing. Tail-withdrawal latencies were measured with a digital stopwatch (Model # 14-649-7; Fisher Scientific, Pittsburgh, PA, USA).
All subjects that met inclusion criteria for subsequent antinociception testing were evaluated in a cumulative dosing procedure (see Fig. 2 for the dosing schematic used with fentanyl). Subjects were first tested for their baseline tail-withdrawal latency using 50 °C water, followed immediately thereafter by subcutaneous (s.c.) injections of the test compound’s vehicle and saline (naltrexone’s vehicle). Dosages and inter-injection intervals during cumulative dosing were based upon locomotor activity results, whereby the shortest interval following the injection of the lowest dose that produced a significant difference from vehicle during locomotor activity tests was used as the inter-injection interval, not to exceed 30 min. After the designated pretreatment interval (10 min for fentanyl and all fentanyl-related substances, 20 min for morphine), the tail-withdrawal latency was re-determined in each subject, followed immediately with an injection of the lowest dose of test compound. After the pretreatment interval had elapsed, subjects were assessed again for tail-withdrawal latency and injected with the next dose of the test substance. The process of assessing tail-withdrawal latencies and administering the next cumulative dose proceeded until the highest cumulative dose was tested (see Fig. 2 for dosing schematics using fentanyl as the example). After the highest cumulative dose was administered, a time-course assessment was performed with tail-withdrawal latencies assessed at 10-min intervals for 60 min and then at 30-min intervals up to 120 min. A 10-s cutoff time was imposed across all assessments to minimize potential tissue damage.
Fig. 2.

Schematic of the cumulative dosing testing procedure for the assessment of the antinociceptive effects of fentanyl-related substances using a warm-water tail-withdrawal assay (FEN; fentanyl used as example). SAL and VEH refer to pretreatment of saline (i.e. naltrexone’s vehicle; control for antagonism experiments) and fentanyl’s vehicle (saline), respectively. Time course testing began 10 min after behavioral measurement of the highest cumulative dose. Time course assessment occurred in 10-min intervals for the first hour and 30-min intervals for the second hour.
In a separate experiment, drug-naive mice were tested to evaluate the ability of the opioid receptor antagonist, naltrexone, to attenuate the antinociceptive effects of the test compounds. Naltrexone (1 mg/kg s.c.) was co-administered with the respective agonist vehicle, then testing proceeded as described above except that higher agonist doses (not to exceed 100 mg/kg) were also tested for their ability to surmount naltrexone antagonism, and time course evaluations were not conducted. This procedure is illustrated in Fig. 3 using fentanyl as the example test compound.
Fig. 3.

Schematic of the testing procedure for assessment of 1 mg/kg naltrexone (NTX) vs. vehicle (VEH) and cumulative doses of fentanyl-related substances (FEN; fentanyl used as example) in a warm-water tail-withdrawal assay. “T X” indicates time in minutes following the injection of naltrexone.
2.5. Data analysis
In locomotor activity experiments, distance traveled (in cm) was used as the main dependent variable for statistical analyses and is shown in graphs as mean values (±SEM) for groups of subjects at each drug dose. Statistical significance was assessed by appropriate one-way or two-way analyses of variance (ANOVA). Fisher’s LSD post-hoc tests were used for all pairwise comparisons. Dose-effect curves for locomotion were further analyzed using nonlinear regression ([Agonist] vs. response − Variable slope, Y=Bottom + (X^Hillslope)*(Top-Bottom)/(X^HillSlope + EC50HillSlope)) with the exception of fentanyl citrate that generated an inverted U-shaped curve relating distance traveled to dose and was modeled with bell-shaped function (Y=Dip+((Plateaul-Dip)/(1 + 10^((Log(EC50_1)-X)*nH1)))+((Plateau2-Dip)/(1 + 10^((X-Log(EC50_2))*nH2))). Valerylfentanyl and benzodioxolefentanyl did not produce marked effects on locomotion and therefore were not appropriate for modeling.
In warm-water tail-withdrawal tests, tail-withdrawal latencies were transformed into the percentage of maximum possible effect (%MPE) that was calculated as [100 x (post-treatment latency - baseline latency)/(maximum possible latency - baseline latency)], where the maximum possible latency was 10 s %MPEs are shown in figures as mean values (±SEM) for groups of subjects at each drug dose. Statistical significance for dose-effect analyses was assessed by an appropriate one-way analysis of variance (ANOVA). Fisher’s LSD post-hoc tests were used for all pairwise comparisons. Dose-effect curves for antinociception were further analyzed using nonlinear regression ([Agonist] vs. normalized response, Y = 100*X/(EC50+X); [Agonist] vs. normalized response − Variable slope, Y = 100*(X^HillSlope)/(EC50^HillSlope+(X^HillSlope))) from which ED50 values (interpolated 50% effective dose) with 95% asymmetrical (likelihood) confidence interval (95% CI) values were calculated for each agonist in the presence or absence of naltrexone pretreatment. Time course effects for antinociception were analyzed using nonlinear regression ([Inhibitor] vs. response − Variable slope, Y=Bottom+(Top-Bottom)/(1+(IC50/X)^HillSlope); [Inhibitor] vs. normalized response − Variable slope, Y = 100/(1+(IC50/X)^HillSlope)). For each test compound, the lowest dose that produced complete (arbitrarily defined as ≥80% MPE) or maximal antinociception in saline-pretreated mice was compared to the %MPE value obtained with the identical agonist dose in mice pretreated with 1 mg/kg naltrexone using unpaired t-tests. For all analyses, p < 0.05 was considered statistically significant. Statistical analyses were conducted using computer software (GraphPad Prism 8 for Microsoft Windows 10 × 64; GraphPad Software, San Diego, CA, USA).
3. Results
3.1. Results from locomotor assays
Fig. 4 (filled circles, right ordinates) shows locomotor activity results (distance traveled in cm) as a function of dose for all drugs. All compounds, with the exception of valerylfentanyl, produced significant increases (p ≤ 0.05) in distance traveled relative to vehicle control at least at one dose tested: morphine (10 and 100 mg/kg); fentanyl (1 and 10 mg/kg); isobutyrylfentanyl (10 mg/kg); crotonylfentanyl (10 mg/kg); para-fluorobutyrylfentanyl (10 and 100 mg/kg); para-methoxybutyrylfentanyl (10 mg/kg); thiophenefentanyl (100 mg/kg); and benzodioxolefentanyl (0.1 mg/kg). Generally, as dose increased, distance traveled increased, except for fentanyl for which an inverted u-shaped dose-response function related distance traveled to increasing dose, and for benzodioxolefentanyl that produced a near-flat dose-effect relationship. Peak distance traveled induced by benzodioxolefentanyl of 1138 (±151) cm, although statistically significant relative to its vehicle, was low and even lower than that induced by valerylfentanyl of 1841 (±142) cm that was not statistically significant.
Fig. 4.

Cumulative dose effects on tail-withdrawal latencies in the warm-water tail-withdrawal assay (open circles, left ordinate) and the acute dose effects on distance traveled (cm) during locomotor activity tests (filled symbols, right ordinate). Symbols above “S+V” and “V” indicate saline + vehicle results in antinociception tests and of results with vehicle in locomotor activity tests, respectively. Symbols for antinociception results are the mean ± SEM for N = 8 mice per group expressed as %MPE. Symbols for locomotor activity results are mean ± SEM for N = 8–16 mice per group, expressed as total distance traveled (cm). Significant differences between a drug’s dose and the respective vehicle condition are indicated by asterisks: *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001 ****p ≤ 0.0001.
Table 1 shows the maximal total distance traveled produced, maximum observed effect as a percentage of fentanyl’s maximal observed effect, and estimated dose (mg/kg) required to produce a level of effect equal to 5000 cm. At the doses tested, fentanyl produced the maximum effect observed of 34905 ± 5355 cm whereas benzodioxolefentanyl was the least efficacious. Based upon the estimated dose required to induce 5000 cm of distanced traveled, the results indicated a rank order potency of: fentanyl > isobutyrylfentanyl > crotonylfentanyl = para-methoxybutyrylfentanyl > morphine > para-fluorobutyrylfentanyl > thiophenefentanyl > > > valerylfentanyl = benzodioxolefentanyl. Fig. 5 shows distance traveled as a function of dose and time for fentanyl, morphine, and valerylfentanyl. Drugs were selected for Fig. 5 to characterize the classical comparators of morphine and fentanyl, whose locomotor activity effects persisted for at least 120 min, with the atypical effects of valerylfentanyl. Time-effect plots for all other substances are provided in the supplemental materials.
Table 1.
Results from locomotor activity tests. For each drug: observed maximum effect (total distance traveled in cm), maximum observed effect as a % of fentanyl’s maximum observed effect, and estimated dose (mg/kg) required to produce a level of effect equal to 5000 cm of travel.
| Drug | Locomotion |
|||
|---|---|---|---|---|
| Maximum observed effect (distance traveled in cm ± SEM) | Maximum observed effect as a % of fentanyl’s maximum effect | Estimated dose (mg/kg) required to elicit 5000 cm of travel | ||
| 1 | Morphine | 22381 ± 3785 | 64.1 ± 10.8 | 4.44 |
| 2 | Fentanyl | 34905 ± 5355 | 100.00 ± 15.3 | 0.167 |
| 3 | Isobutyrylfentanyl | 23870 ± 5811 | 68.4 ± 16.6 | 0.858 |
| 4 | Crotonylfentanyl | 26864 ± 4254 | 77.0 ± 12.2 | 1.14 |
| 5 | Valerylfentanyl | 1841 ± 142 | 5.27 ± 0.405 | N/A * |
| 6 | Para-fluorobutyrylfentanyl | 8594 ± 1462 | 24.6 ± 4.19 | 8.99 |
| 7 | Para-methoxybutyrylfentanyl | 23695 ± 7721 | 67.9 ± 22.1 | 1.14 |
| 8 | Thiophenefentanyl | 7840 ± 2492 | 22.5 ± 7.14 | 59.7 |
| 9 | Benzodioxolefentanyl | 1138 ± 151 | 3.26 ± 0.433 | N/A * |
Impossible to estimate based upon the slope of the dose-effect curve. Data are mean ± SEM for N = 8–16 mice per group.
Fig. 5.

Distance traveled (cm) in 10-min bins following administrations of morphine, fentanyl, and valerylfentanyl. Data are mean ± SEM for N=8–16 mice per group.
3.2. Results from antinociceptive assays
Fig. 4 (empty circles, left ordinate) shows results from antinociception tests (%MPE as a function of dose) for all drugs in the warm-water tail-withdrawal assay. As dose increased, %MPE increased for all drugs. Table 2 shows efficacy (%Emax values) and potency estimates for producing antinociception (ED50 values) in the warm-water tail-withdrawal assay and potency relationships to morphine and fentanyl among the test compounds. The ED50 values for producing antinociception showed a rank order of potency of isobutyrylfentanyl > fentanyl > para-methoxybutyrylfentanyl > crotonylfentanyl > para-fluorobutyrylfentanyl > thiophenefentanyl > valerylfentanyl > morphine > benzodioxolefentanyl. Relative to morphine (ED50=7.82 mg/kg), isobutyrylfentanyl (ED50=0.07 68 mg/kg), para-methoxybutyrylfentanyl (ED50=0.106 mg/kg), crotonylfentanyl (ED50=0.226 mg/kg), para-fluorobutyrylfentanyl (ED50=0.908 mg/kg), and thiophenefentanyl (ED50=4.66 mg/kg) were approximately 102-fold, 74-fold, 35-fold, 9-fold, and 2-fold, respectively, more potent than morphine. Valerylfentanyl (ED50=6.43 mg/kg) was equipotent with morphine, and benzodioxolefentanyl (ED50=46.3 mg/kg) was the least potent compound tested.
Table 2.
Results from antinociceptive assays. Potency estimates in the warm-water tail-withdrawal assay expressed as %Emax (maximum observed efficacy), ED50 (mg/kg) values for drug alone, ED50 values for drug following 1 mg/kg naltrexone pretreatment, fold change between (agonist + antagonist)/(agonist) potency, drug potency ratio to morphine, and drug potency ratio to fentanyl. Data are mean ± SEM or 95% confidence intervals for N = 8 mice per group.
| Drug | Antinociception |
|||||||
|---|---|---|---|---|---|---|---|---|
| %Emax ± SEM | Agonist ED50 mg/kg (95% CI) | Agonist + Antagonist ED50 mg/kg (95% CI) | Fold Change (agonist + antagonist/agonist) | Potency ratio to morphine | Potency ratio to fentanyl | |||
| 1 | Morphine | 94.0 ± 5.98 | 7.82 (5.42–11.0) | 11.6 (5.98–23.8) | 1.48 | 1.00 | 0.010 | |
| 2 | Fentanyl | 89.2 ± 10.8 | 0.0800 (0.0403–0.164) | 0.316 (0.154–0.615) | 3.95 | 97.7 | 1.00 | |
| 3 | Isobutyrylfentanyl | 100.0 | 0.0768 (0.044–0.128) | 17.9 (6.33–75.2) | 233 | 102 | 1.04 | |
| 4 | Crotonylfentanyl | 100.0 | 0.226 (0 .176–0.292) | 1.42 (0.599–3.44) | 6.27 | 34.5 | 0.354 | |
| 5 | Valerylfentanyl | 91.9 ± 5.54 | 6.43 (3.91–10.5) | 76.5 (43.6–140) | 11.9 | 1.22 | 0.0125 | |
| 6 | Para-fluorobutyrylfentanyl | 96.1 ± 3.87 | 0.908 (0.459–1.58) | 9.87 (5.46–20.5) | 10.9 | 8.61 | 0.0882 | |
| 7 | Para-methoxybutyrylfentanyl | 96.7 ± 3.31 | 0.106 (0.0516–0.195) | 3.99 (0.918–81.8) | 37.7 | 74.0 | 0.758 | |
| 8 | Thiophenefentanyl | 88.6 ± 4.89 | 4.66 (3.65–5.95) | 162 (84.2–369) | 34.6 | 1.68 | 0.0172 | |
| 9 | Benzodioxolefentanyl | 49.1 ± 7.05 | 46.3 (25.8–83.4) | 390 (195–1420) | 8.42 | 0.169 | 0.00173 | |
Fig. 6 shows %MPE of the highest tested dose of each drug as a function of time as determined by nonlinear regression obtained during the warm-water tail-withdrawal tests. The time to peak effect for all compounds occurred within 10 min. Degeneration of antinociceptive effects occurred most rapidly for para-methoxybutyrylfentanyl, thiophenefentanyl, para-fluorobutyrylfentanyl, valerylfentanyl, benzodioxolefentanyl, and isobutyrylfentanyl. Antinociceptive effects by crotonylfentanyl, fentanyl, and morphine persisted longer than the other drugs and were still above 50% MPE levels 120 min following drug administration. Benzodioxolefentanyl was the least efficacious fentanyl-related substance tested with an observed maximum possible effect of approximately 50% at a dose of 100 mg/kg and had only transient effects in warm-water tail-withdrawal time-course assessments.
Fig. 6.

Antinociceptive effects of the highest cumulative dose of each drug tested as a function of time in the warm-water tail-withdrawal test, i.e. (1) 32 mg/kg morphine, (2) 1 mg/kg fentanyl, (3) 0.1 mg/kg isobutyrylfentanyl, (4) 10 mg/kg crotonylfentanyl, (5) 32 mg/kg valerylfentanyl, (6) 10 mg/kg para-fluorobutyrylfentanyl, (7) 1 mg/kg para-methoxybutyrylfentanyl, (8) 32 mg/kg thiophenefentanyl, (9) 100 mg/kg benzodioxolefentanyl. Data are represented as curves determined by nonlinear regression with N = 8 mice per drug.
The effects of 1 mg/kg naltrexone pretreatment on the antinociceptive effects of the test compounds are shown in Fig. 7 and Table 2 (second column). Pretreatment with naltrexone attenuated the antinociceptive effects of all drugs. The relative antagonism of antinociception varied across drugs. Naltrexone pretreatment increased antinociceptive ED50 values several fold in decreasing magnitudes of isobutyrylfentanyl (233x) > para-methoxybutyrylfentanyl (37.7x) > thiophenefentanyl (34.6x) > valerylfentanyl (11.9x) > para-fluorobutyrylfentanyl (10.9x) > benzodioxolefentanyl (8.42x) > crotonylfentanyl (6.27x) > fentanyl (3.95x) > morphine (1.48x).
Fig. 7.

Antinociceptive effects in the warm-water tail-withdrawal assay of (1) 32 mg/kg morphine, (2) 1 mg/kg fentanyl, (3) 0.1 mg/kg isobutyrylfentanyl (4) 10 mg/kg crotonylfentanyl, (5) 32 mg/kg valerylfentanyl, (6) 10 mg/kg para-fluorobutyrylfentanyl, (7) 1 mg/kg para-methoxybutyrylfentanyl, (8) 32 mg/kg thiophenefentanyl, (9) 100 mg/kg benzodioxolefentanyl accompanied by either by 1 mg/kg naltrexone [NTX; filled bars] pretreatment or by naltrexone’s vehicle, saline [VEH; open bars]. Each bar represents the mean %MPE (±SEM) of N = 8 mice per group. Significant differences between agonist + saline (open bars) vs agonist + 1 mg/kg naltrexone (filled bars) condition are indicated by asterisks: **p ≤ 0.01, ***p ≤ 0.001 ****p ≤ 0.0001.
4. Discussion
Results from the present locomotor activity and warm-water tail-withdrawal tests, to our knowledge, represent the first known assessments of para-fluorobutyrylfentanyl, para-methoxybutyrylfentanyl, thiophenefentanyl, and benzodioxolefentanyl in living organisms under controlled-laboratory conditions. In locomotor activity tests, all drugs, except for valerylfentanyl, produced significant dose-dependent hyperlocomotion with fentanyl eliciting the greatest increases in distance traveled within the doses examined. In warm-water tail-withdrawal tests, all drugs produced significant dose-dependent antinociception with the apparent potency to do so of isobutyrylfentanyl > fentanyl > para-methoxybutyrylfentanyl > crotonylfentanyl > para-fluorobutyrylfentanyl > thiophenefentanyl > valerylfentanyl > morphine > benzodioxolefentanyl. To further explore the role of opioid receptors in mediating the in vivo effects of fentanyl-related substances, we examined the antinociceptive effects of the drugs in warm-water tail-withdrawal tests with naltrexone pretreatment. These tests demonstrated a significant blockade of antinociception induced by all drugs indicating that their antinociceptive effects are, indeed, opioid-receptor mediated.
Several groups have reported the effects of three of the fentanyl-related substances tested in the present study including isobutyrylfentanyl, crotonylfentanyl, and valerylfentanyl. Higashikawa and Suzuki (2008) reported the ED50 of orally administered isobutyrylfentanyl to be 0.261 mg/kg in a mouse acetic acid writhing assay with a relative potency to morphine and fentanyl of 1.3 and 0.02, respectively (Higashikawa and Suzuki, 2008). By contrast, we found that subcutaneously administered isobutyrylfentanyl had an ED50 of 0.0768 (0.044–0.128) mg/kg with a relative potency to morphine and fentanyl of 102 and 1.04, respectively. Differences between the two studies, such as the assays (warm-water tail withdrawal vs acetic acid writhing) and routes of administration used, likely account for the observed differences in ED50 values. For instance, fentanyl analogs have poor oral bioavailability due to their first-pass metabolism (Prekupec et al., 2017), and consequentially, potency estimates from Higashikawa and Suzuki (2008) would likely be lower (i.e., higher ED50 values) than in the present study that administered drugs subcutaneously. Nonetheless, isobutyrylfentanyl was more potent than morphine in both studies. However, Higashikawa and Suzuki (2008) reported that isobutyrylfentanyl was about 50x less potent than fentanyl, whereas we found the two drugs to be about equipotent. Woods and colleagues reported isobutyrylfentanyl to be a mu opioid receptor agonist in the mouse vas deferens preparation and that its potency in the binding assay was similar to that of morphine (Woods et al., 1987). Woods’ (1987) in vitro findings are consistent with binding affinity determinations derived from competition radioligand assays made by Janowsky et al. (A. J. Janowsky, personal communication, October 3, 2018) which demonstrated approximately equal binding affinities for isobutyrylfentanyl and morphine. In addition, Baumann and colleagues recently found isobutyrylfentanyl to be about 100 times more selective for mu-opioid receptors than delta or kappa (Baumann et al., 2018a). Moreover, Aceto et al. (1987) reported that isobutyrylfentanyl prevented the emergence of withdrawal effects in morphine-dependent monkeys when substituted for morphine at a dose of 0.1 mg/kg, s.c. (with partial substitution at 0.025 mg/kg, s.c.) and estimated that it was 30 times more potent than morphine (Aceto et al., 1987). The relative magnitude of greater potency of isobutyrylfentanyl to that of morphine approached that observed in the present study (102x) using the mouse warm-water tail-withdrawal assay. Overall, isobutyrylfentanyl appears to mediate its antinociceptive and locomotor stimulating effects, like fentanyl and morphine, via the mu opioid receptor.
In a short communication, Essawi (1999) reported that crotonylfentanyl was “clearly less potent than fentanyl”, however, it also showed an extended duration of analgesia that was transiently reversed by naloxone (Essawi, 1999). This result is consistent with our findings which showed an ED50 of 0.08 mg/kg for fentanyl and 0.226 mg/kg for crotonyl fentanyl, making it approximately two-thirds less potent than fentanyl in antinociceptive tests. Furthermore, these findings are consistent with the extended duration of antinociception observed in the warm-water tail-withdrawal time course evaluation (Fig. 5). Moreover, we also noted a reversal of the antinociceptive effects of crotonylfentanyl when tested with a naltrexone pretreatment.
Aceto et al. (1987) reported that 5 mg/kg, s.c. valerylfentanyl completely substituted for 3 mg/kg, s.c. morphine, with partial substitution at 2.5 mg/kg s.c., 15 h following the last administration of morphine in the monkey single dose suppression test, an indicator of cross-dependency (Aceto et al., 1987). Moreover, Aceto reported that valerylfentanyl did not antagonize the antinociceptive effects of morphine in the mouse tail-flick assay at a dose of 1 mg/kg. Woods reported that the ED50 of valerylfentanyl was 7.5 (5.4–10.3) mg/kg in a mouse hot plate antinociception assay and that it was inactive upon the isolated, electrically stimulated mouse vas deferens preparation in concentrations that ranged from 3×10−9M to 10−8M (Woods et al., 1988). Our in vivo assessments in the warm-water tail-withdrawal test in which we determined the ED50 of valerylfentanyl to be 6.43 (3.91–10.5) mg/kg are generally consistent with Woods’s antinociceptive findings in the mouse hot plate assay. Woods concluded that valerylfentanyl did not show opioid activity in the mouse vas deferens assay; however, it did show displacement at the etorphine specific binding site. The findings by Woods and colleagues for valerylfentanyl in the mouse vas deferens preparation are unexpected given their report of its displacement at the etorphine specific binding site, as well as with recent findings of its binding affinity and partial receptor functional activation of mu- and kappa-opioid receptors (A. J. Janowsky, personal communication, October 3, 2018). In addition, binding affinity in rat brain membranes indicated that valerlyfentanyl is more selective for mu-opioid receptors than delta (65x) or kappa (37x) (Baumann et al., 2018a). In contrast to fentanyl and morphine, valerylfentanyl did not elicit hyperlocomotion in our locomotor activity tests, which may suggest that it does not possess abuse-related effects. However, Walentiny et al., (this issue) report that valerylfentanyl completely substitutes for oxycodone in mice in an assay of drug discrimination (Walentiny et al., 2019). It is possible that inability of valerylfentanyl to produce locomotor activation in mice, even when tested up to 100 mg/kg, may be explained by its low efficacy at the mu opioid receptor as well as significant kappa opioid receptor activation at high doses (Kuzmin et al., 2000).
In summary, fentanyl, morphine, and seven fentanyl-related substances were efficacious opioid receptor agonists as determined by in vivo locomotor and antinociceptive tests in the present studies. To our knowledge, the in vivo effects of para-fluorobutyrylfentanyl, para-methoxybutyrylfentanyl, thiophenefentanyl and benzodioxolefentanyl had never before been reported in the published literature. It was previously reported that the potency ratios of para-fluorobutyrylfentanyl and para-methoxybutyrylfentanyl to fentanyl and morphine were unknown (Suzuki and El-Haddad, 2017). Here we report that the both para-fluorobutyrylfentanyl and para-methoxybutyrylfentanyl produce dose-dependent antinociception and locomotor activation. We also found that both thiophenefentanyl and benzodioxolefentanyl elicit antinociceptive activity in a dose-dependent manner and produce minimal locomotor activation relative to morphine and fentanyl. In general, the present findings support older preclinical literature describing the pharmacology of isobutyrylfentanyl, crotonylfentanyl, and valerylfentanyl in rodent models. Consistent with findings for other synthetic opioids (Baumann et al., 2018b), we found that in vitro receptor binding affinity and receptor functional activation were poorly predictive of the relative in vivo antinociceptive potency or efficacy. Other laboratories have also demonstrated that binding affinity alone, for various opioids, cannot always be used to compare the relative safety and efficacy of drugs and only provides a limited indication of clinical potency and risk (Volpe et al., 2011). Overall, most of the fentanyl-related substances tested exhibit lower receptor affinity and activation than morphine (A. J. Janowsky, personal communication, October 3, 2018; Brandt, 2018a, b), but were more potent than morphine in the warm-water tail-withdrawal test. One possible explanation for the imperfect ability of results from in vitro receptor affinity and activation studies to predict relative in vivo potencies may be attributed to enhanced blood-brain barrier penetration conferred by lipophilic chemical structures. For instance, while we found that morphine and valerylfentanyl were equipotent in vivo, predicted physiochemical properties of valerylfentanyl (cLogP=4.68), an in vitro partial mu- and kappa-opioid receptor agonist with a maximum observed effect at the mu opioid receptor less than half of that observed with morphine (A. J. Janowsky, personal communication, October 3, 2018), indicate that it is significantly more hydrophobic than is morphine (cLogP=0.572), which may account for its morphine-like potency. Others have argued that physiochemical properties of the synthetic opioids could account for the imperfect relationship between in vitro properties (affinity and efficacy) and in vivo activity (Baumann et al., 2018b). In contrast to other compounds, benzodioxolefentanyl, which was found to be a partial kappa opioid receptor agonist and completely devoid of activity at both mu- and delta-opioid receptors in vitro (A. J. Janowsky, personal communication, October 3, 2018; Brandt, 2018a, b), produced significant, dose-dependent, and naltrexone-sensitive antinociception though the maximal observed effect of approximately 50% MPE at a dose of 100 mg/kg was less than that of all other fentanyl-related substances tested.
One approach to respond more effectively to the health hazards of fentanyl-related substances is to identify those with narrow separation between their abuse-related and respiratory depressant potencies. The rapid pace with which fentanyl-related substances are appearing in the recreational drug marketplace warrants additional preclinical testing to evaluate their in vivo antinociceptive, respiratory depressant, and abuse-related effects. An investigation of fentanyl-related substances may illuminate a range of compounds from those with high abuse liability and respiratory toxicity to those with low abuse liability, toxicity and potential therapeutic efficacy as analgesics. Separating the analgesic effects of opioids from their unwanted effects has long been an unmet goal of opioid research, and unexplored synthetic opioids may offer improved side-effect profiles relative to current analgesics. Drugs exhibiting a separation in dose ranges eliciting their effects (i.e., producing complete antinociception and minimal locomotor activation) may be ideal candidates for further investigation such as with valerylfentanyl, para-fluorobutyrylfentanyl, and thiophenefentanyl. To date, the evaluation of many emerging fentanyl-related substances concerning their antinociceptive, respiratory, and abuse-related effects has not yet been performed (Prekupec et al., 2017; Baumann and Pasternak, 2018). The findings from such studies will have vast implications for drug discovery and medication development and immense public health relevance.
Supplementary Material
HIGHLIGHTS.
Morphine, fentanyl, and most tested fentanyl analogs elicit hyperlocomotion.
Fentanyl-related substances produce antinociception in the tail-withdrawal assay.
Valerylfentanyl elicits antinociception but not hyperlocomotion up to 100 mg/kg.
Benzodioxolefentanyl shows incomplete antinociception (~50% levels) up to 100 mg/kg.
Naltrexone (1 mg/kg) attenuates antinociceptive effects of all tested substances.
Acknowledgements
This work was supported by the United States Drug Enforcement Administration (DJD-17-HQ-P-0641). N. B. Varshneya was supported by the National Institute on Drug Abuse (T32DA007027). The authors wish to thank Molly Creighton for exceptional technical assistance.
Footnotes
Declaration of interest
Authors T. D. Walker and L. R. Akinfiresoye were employees of the United States Drug Enforcement Administration at time of publication. There were no other interests to declare.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.neuropharm.2019.03.023.
References
- Aceto M, Bowman E, Harris L, May E, 1987. Dependence studies of new compounds in the rhesus monkey, rat and mouse (1987). Nat. Inst. Drug Abuse Res. Monogr. Ser. Problem. Drug Depend 1987. [Google Scholar]
- Backberg M, Beck O, Jonsson KH, Helander A, 2015. Opioid intoxications involving butyrfentanyl, 4-fluorobutyrfentanyl, and fentanyl from the Swedish STRIDA project. Clin. Toxicol 53, 609–617. [DOI] [PubMed] [Google Scholar]
- Bagley JR, Wynn RL, Rudo FG, Doorley BM, Spencer HK, Spaulding T, 1989. New 4-(heteroanilido)piperidines, structurally related to the pure opioid agonist fentanyl, with agonist and/or antagonist properties. J. Med. Chem 32, 663–671. [DOI] [PubMed] [Google Scholar]
- Baumann MH, Kopajtic TA, Madras BK, 2018a. Pharmacological research as a key component in mitigating the opioid overdose crisis. Trends Pharmacol. Sci 39, 995–998. [DOI] [PubMed] [Google Scholar]
- Baumann MH, Majumdar S, Le Rouzic V, Hunkele A, Uprety R, Huang XP, Xu J, Roth BL, Pan YX, Pasternak GW, 2018b. Pharmacological characterization of novel synthetic opioids (NSO) found in the recreational drug marketplace. Neuropharmacology 134, 101–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumann MH, Pasternak GW, 2018. Novel synthetic opioids and overdose deaths: tip of the iceberg? Neuropsychopharmacology 43, 216–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beardsley PM, 2007. Assessing dependency and abuse potential. In: Sietsema Schwen (Eds.), Nonclinical Drug Safety Assessment: Practical Considerations for Successful Registration. FDAnews, pp. 441–463. [Google Scholar]
- Centers for Disease Control and Prevention, 2018. Multiple cause of death 1999-2017 on CDC WONDER online Database, released December, 2018. Data are from the Multiple cause of death Files, 1999-2017 as compiled from data provided by the 57 vital statistics jurisdictions through the vital Statistics cooperative Program. National Center for Health Statistics. [Google Scholar]
- Committee for the Update of the Guide for the Care and Use of Laboratory Animals, 2011. Guide for the Care and Use of Laboratory Animals. Institute for Laboratory Animal Research; Division on Earth and Life Studies; National Research Council of the National Academies. [Google Scholar]
- Corssen G, Domino EF, Sweet RB, 1964. Neuroleptanalgesia and anesthesia. Anesth. Analg 43, 748–763. [PubMed] [Google Scholar]
- Drug Enforcement Administration, 2016. Counterfeit Prescription Pills Containing Fentanyls: A Global Threat. DEA Intelligence Brief. [Google Scholar]
- Drug Enforcement Administration, 2017a. NFLIS Brief: Fentanyl and Fentanyl-Related Substances Reported in NFLIS, 2015–2016. National Forensic Laboratory Information System. [Google Scholar]
- Drug Enforcement Administration, 2017b. NFLIS Brief: Fentanyl, 2001–2015. NFLIS Brief: Fentanyl, pp. 2001–2015. [Google Scholar]
- Drug Enforcement Administration, 2018a. Emerging Threat Report. Special Testing and Research Laboratory. [Google Scholar]
- Drug Enforcement Administration, 2018b. Fentanyl Remains the Most Significant Synthetic Opioid Threat and Poses the Greatest Threat to the Opioid User Market in the United States. DEA Intelligence Brief. [Google Scholar]
- Essawi MYH, 1999. Fentanyl analogues with a modified propanamido group as potential affinity labels: synthesis and in vivo activity. Pharmazie 54, 307–308. [PubMed] [Google Scholar]
- European Monitoring Centre for Drugs and Drug Addiction, 2018. European Drug Report 2018: Trends and Developments. Publications Office of the European Union, Luxembourg. [Google Scholar]
- Gladden RM, Martinez P, Seth P, 2016. Fentanyl Law Enforcement Submissions and Increases in Synthetic Opioid–Involved Overdose Deaths — 27 States, 2013–2014. Morbidity and Mortality Weekly Report, vol. 65. [DOI] [PubMed] [Google Scholar]
- Helander A, Backberg M, Beck O, 2016. Intoxications involving the fentanyl analogs acetylfentanyl, 4-methoxybutyrfentanyl and furanylfentanyl: results from the Swedish STRIDA project. Clin. Toxicol 54, 324–332. [DOI] [PubMed] [Google Scholar]
- Helander A, Backberg M, Signell P, Beck O, 2017. Intoxications involving acrylfentanyl and other novel designer fentanyls - results from the Swedish STRIDA project. Clin. Toxicol 55, 589–599. [DOI] [PubMed] [Google Scholar]
- Higashikawa Y, Suzuki S, 2008. Studies on 1-(2-phenethyl)-4-(N-propionylanilino)piperidine (fentanyl) and its related compounds. VI. Structure-analgesic activity relationship for fentanyl, methyl-substituted fentanyls and other analogues. Forensic Toxicol. 26, 1–5. [Google Scholar]
- Janowsky A, 2018. 4-Methoxy-butyryl fentanyl. N-(4-Methoxyphenyl)-N-(1-phenethylpiperidin-4-yl)butyramide, monohydrochloride. Binding and functional activity at delta, kappa and mu opioid receptors. DEA-VA interagency agreement title: “in vitro receptor and transporter assays for abuse liability testing for the DEA by the VA”. March 2016. In: Brandt SD (Ed.), Critical Review Report: P-Methoxy-Butyrylfentanyl. Expert Committee on Drug Dependence Forty-first Meeting, Geneva, Switzerland. [Google Scholar]
- Janssen PAJ, 1964. Compose a activite pharmacologique a base de 1-arylalcoyl-4-(N-arylalcanamido)-piperidines. N. V. Research Laboratorium, Dr. C. Janssen, FR. [Google Scholar]
- Kuzmin A, Sandin J, Terenius L, Ogren S, 2000. Dose- and time-dependent Bimodal effects of k-opioid agonists on locomotor activity in mice. J. Pharmacol. Exp. Ther 295. [PubMed] [Google Scholar]
- Maher S, Elliott SP, George S, 2018. The analytical challenges of cyclopropylfentanyl and crotonylfentanyl: an approach for toxicological analysis. Drug Test. Anal 10, 1483–1487. [DOI] [PubMed] [Google Scholar]
- O’Donnell JK, Gladden RM, Seth P, 2017a. Trends in Deaths Involving Heroin and Synthetic Opioids Excluding Methadone, and Law Enforcement Drug Product Reports, by Census Region — United States, 2006–2015. Morbidity and Mortality Weekly Report 66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Donnell JK, Halpin J, Mattson CL, Goldberger BA, Gladden RM, 2017b. Deaths Involving Fentanyl, Fentanyl Analogs, and U-47700 — 10 States, July–December 2016. Morbidity and Mortality Weekly Report 66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palaty J, Konforte D, Karakosta T, Wong E, Stefan C, 2018. Rapid identification of cyclopropyl fentanyl/crotonyl fentanyl in clinical urine specimens: a case study of clinical laboratory collaboration in Canada. Clin. Biochem 53, 164–167. [DOI] [PubMed] [Google Scholar]
- para-Fluorobutyryl Fentanyl (p-FBF), 2018. N-(4-Fluorophenyl)-N-[1-(2-phenylethyl)-4-piperidinyl]-butanamide, monohydrochloride. Binding and functional activity at delta, kappa and mu opioid receptors. DEA-VA interagency agreement title: “In vitro receptor and transporter assays for abuse liability testing for the DEA by the VA”. March 2016, as cited in: Brandt SD In: Critical Review Report: P-Fluoro-Butyrylfentanyl, Expert Committee on Drug Dependence Forty-First Meeting. World Health Organization, Geneva, Switzerland. [Google Scholar]
- Peterson AB, Gladden RM, Delcher C, Spies E, Garcia-Williams A, Wang Y, Halpin J, Zibbell J, McCarty CL, DeFiore-Hyrmer J, DiOrio M, Goldberger BA, 2016. Increases in Fentanyl-Related Overdose Deaths — Florida and Ohio, 2013–2015. Morbidity and Mortality Weekly Report 65. [DOI] [PubMed] [Google Scholar]
- Poklis J, Poklis A, Wolf C, Hathaway C, Arbefeville E, Chrostowski L, Devers K, Hair L, Mainland M, Merves M, Pearson J, 2016. Two fatal intoxications involving Butyryl fentanyl. J. Anal. Toxicol 40, 703–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poklis J, Poklis A, Wolf C, Mainland M, Hair L, Devers K, Chrostowski L, Arbefeville E, Merves M, Pearson J, 2015. Postmortem tissue distribution of acetyl fentanyl, fentanyl and their respective nor-metabolites analyzed by ultrahigh performance liquid chromatography with tandem mass spectrometry. Forensic Sci. Int 257, 435–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prekupec MP, Mansky PA, Baumann MH, 2017. Misuse of novel synthetic opioids: a deadly new trend. J. Addict. Med 11, 256–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson TE, Berridge KC, 1993. The neural basis of drug craving: an incentive-sensitization theory of addiction. Brain Res. Brain Res. Rev 18, 247–291. [DOI] [PubMed] [Google Scholar]
- Rudd RA, Seth P, David F, Scholl L, 2016. Increases in Drug and Opioid-Involved Overdose Deaths — United States, 2010–2015. Morbidity and Mortality Weekly Report, vol. 65. [DOI] [PubMed] [Google Scholar]
- Stanley TH, 2014a. The fentanyl story. J. Pain 15, 1215–1226. [DOI] [PubMed] [Google Scholar]
- Stanley TH, 2014b. The History of Opioid Use in Anesthetic Delivery. The Wondrous Story of Anesthesia, pp. 641–659. [Google Scholar]
- Sutter ME, Gerona RR, Davis MT, Roche BM, Colby DK, Chenoweth JA, Adams AJ, Owen KP, Ford JB, Black HB, Albertson TE, 2017. Fatal fentanyl: one pill can kill. Acad. Emerg. Med 24, 106–113. [DOI] [PubMed] [Google Scholar]
- Suzuki J, El-Haddad S, 2017. A review: fentanyl and non-pharmaceutical fentanyls. Drug Alcohol Depend. 171, 107–116. [DOI] [PubMed] [Google Scholar]
- United States Department of Justice, 2018. Schedules of controlled substances: temporary placement of fentanyl related substances in Schedule I. Fed. Regist 83, 21 CFR Part 1308. [PubMed] [Google Scholar]
- Volpe DA, McMahon Tobin GA, Mellon RD, Katki AG, Parker RJ, Colatsky T, Kropp TJ, Verbois SL, 2011. Uniform assessment and ranking of opioid mu receptor binding constants for selected opioid drugs. Regul. Toxicol. Pharmacol 59, 385–390. [DOI] [PubMed] [Google Scholar]
- Walentiny DM, Moisa LT, Beardsley PM, 2019. Oxycodone-like Discriminative Stimulus Effects of Fentanyl-Related Emerging Drugs of Abuse in Mice, This Issue. [DOI] [PubMed] [Google Scholar]
- Wise RA, Bozarth MA, 1987. A psychomotor stimulant theory of addiction. Psychol. Rev 94, 469–492. [PubMed] [Google Scholar]
- Woods JH, Medzihradsky F, Smith C, Winger G, France C, 1988. Evaluation of new compounds for opioid activity: 1988 annual report. Nat. Inst. Drug Abuse Res. Monogr. Ser. Problem. Drug Depend 1988. [Google Scholar]
- Woods JH, Medzihradsky F, Smith C, Winger G, Gmerek D, 1987. Evaluation of new compounds for opioid activity: 1987 annual report. Nat. Inst. Drug Abuse Res. Monogr. Ser. Problem. Drug Depend 1987. [PubMed] [Google Scholar]
- Wynn RL, Bagley JR, Spencer HK, Spaulding TC, 1991. Evaluation of the morphine reversal actions and antinociceptive activity of a new class of opiate antagonists structurally related to fentanyl. Drug Dev. Res 22, 189–195. [Google Scholar]
- Zawilska JB, 2017. An expanding world of novel psychoactive substances: opioids. Front. Psychiatry 8, 110. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.