

Abstract
Mitochondria serve numerous critical cellular functions, rapidly responding to extracellular stimuli and cellular demands while dynamically communicating with other organelles. Mitochondrial function in the gastrointestinal epithelium plays a critical role in maintaining intestinal health. Emerging studies implicate the involvement of mitochondrial dysfunction in inflammatory bowel disease (IBD). This review presents mitochondrial metabolism, function, and quality control that converge in intestinal epithelial stemness, differentiation programs, barrier integrity, and innate immunity to influence intestinal inflammation. Intestinal and disease characteristics that set the stage for mitochondrial dysfunction being a key factor in IBD and, in turn, pathogenic mitochondrial mechanisms influencing and potentiating the development of IBD, are discussed. These findings establish the basis for potential mitochondrial-targeted interventions for IBD therapy.
Keywords: intestinal epithelium, ulcerative colitis, Crohn’s disease, inflammation, mitochondria
1. MITOCHONDRIAL STRUCTURE AND FUNCTION
Mitochondria are dynamic organelles that can undergo rapid alteration of structure and intracellular localization in response to cellular need. The double membrane-bound structure of mitochondria is directly related to function (Figure 1). The outer mitochondrial membrane is composed of a phospholipid bilayer separating the mitochondria and its contents from the cytosol. Ions and proteins smaller than 5 kDa diffuse in and out of the mitochondria freely via porins, and therefore, the concentration of small proteins and ions in the mitochondrial intermembrane space matches the cytosol. Proteins larger than 5 kDa possessing a 5′ mitochondrial localization sequence translocate across the outer mitochondrial membrane via binding to translocase of the outer membrane transporters. Proteins transferred into the mitochondria include nuclear DNA–encoded proteins such as those for the electron transport chain and heat-shock proteins (1). The inner mitochondrial membrane phospholipid bilayer houses the five electron transport chain complexes that function to produce adenosine triphosphate (ATP) from acetyl-coenzyme A (CoA) through oxidative phosphorylation (OXPHOS). Transporters called translocases of the inner membrane are required for all ions and proteins to enter the mitochondrial matrix, thereby facilitating the formation of the electrical-chemical gradient in the inner membranous space that drives ATP formation via ATPase. Inward folds of the inner mitochondrial membrane are called cristae, which increase the surface area for OXPHOS to match cellular demand and narrow the space between mitochondrial membranes (2). Enclosed by the inner mitochondrial membrane is the matrix that houses mitochondrial DNA, RNA, ribosomes, and ATP synthase proteins that facilitate the catalysis from adenosine diphosphate (ADP) to ATP (1). Additionally, the matrix contains enzymes involved in the tricarboxylic acid cycle, fatty acid oxidation (FAO), amino acid oxidation, heme synthesis, and iron sulfur cluster formation (3). With numerous specific enzymes, the mitochondrial matrix has a high protein density (4). Importantly, mitochondria are major cellular producers of acetyl-CoA and S-adenosylmethionine, which in turn mediate molecular modifications such as acetylation and methylation, respectively (5). Thus, alterations in mitochondrial biology and metabolism have profound outcomes on the epigenetic state of the genome and post-translational protein modifications. Additionally, mitochondria are also important sites of intracellular calcium storage and the induction of apoptosis during cellular stress predominantly by the release of cytochrome C into the cytosol. Mitochondria are mobile throughout the cell via transportation along microtubules to subcellular locations requiring a local and rapid ATP supply (6).
Figure 1.
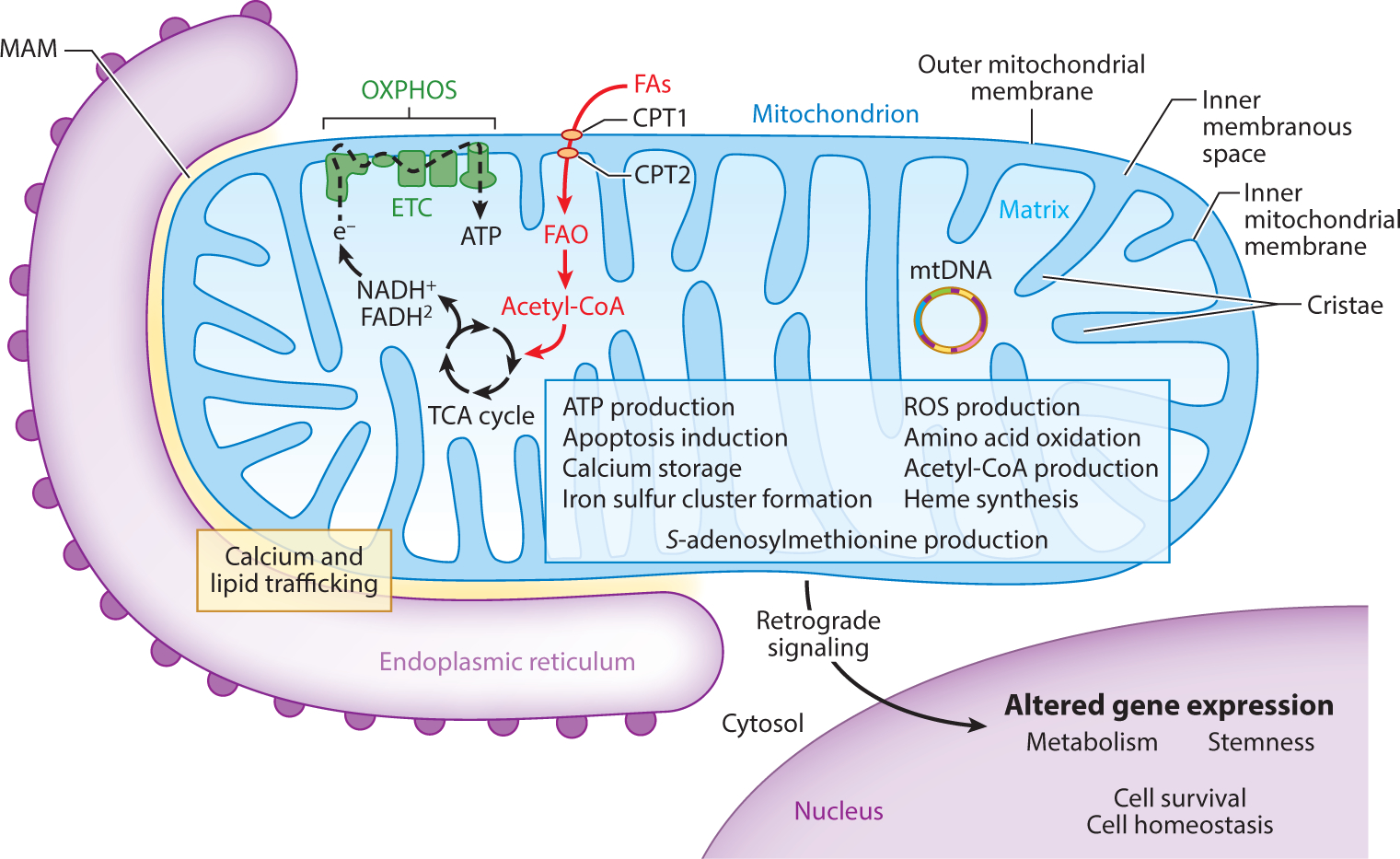
Mitochondrial structure is directly related to function. The structure of mitochondria facilitates numerous functions based on cellular need (blue box) (3–5, 7, 10–12). Arguably, the most important function performed by mitochondria is energy production in the form of ATP through OXPHOS. In Lgr5+ crypt base columnar stem cells, FAO is emerging as an important provider of acetyl-CoA to fuel the TCA cycle and OXPHOS (37–39). Mitochondria also actively communicate with other organelles important for global cell homeostasis. Mitochondria are intimately associated with the endoplasmic reticulum through MAMs, where the exchange of lipids and calcium influences homeostasis of each organelle (11). Retrograde signaling serves as mitochondrial-to-nuclear intraorganelle communication, resulting in altered gene expression based on mitochondrial status (12). Abbreviations: Acetyl-CoA, acetyl-coenzyme A; ATP, adenosine triphosphate; CPT, carnitine palmitoyltransferase; e−, donated electron; ETC, electron transport chain; FAs, fatty acids; FAO, fatty acid oxidation; MAM, mitochondrial-associated membrane; mtDNA, mitochondrial DNA; OXPHOS, oxidative phosphorylation; ROS, reactive oxygen species; TCA, tricarboxylic acid.
OXPHOS:
oxidative phosphorylation, the process of ATP generation via the electron transport chain
Tricarboxylic acid cycle:
chemical reactions occurring in the mitochondrial matrix as a part of aerobic respiration
FAO:
fatty acid oxidation, the mitochondrial process of breaking down fatty acids to acetyl-CoA
Mitochondria are the predominant producers of reactive oxygen species (ROS) as a byproduct of OXPHOS. Approximately 1–2% of oxygen consumed during OXPHOS is converted to superoxide when electrons leak from the electron transport chain and are aberrantly transferred to molecular oxygen (7). Superoxide can be converted spontaneously or by superoxide dismutase (SOD) to hydrogen peroxide, which has a longer half-life, can cross the mitochondrial membranes, and readily induces damage to lipids, proteins, and DNA (7). Hydrogen peroxide can be fully reduced to water by catalase or partially reduced to hydroxyl radical, an extremely strong oxidant. The production of ROS is increased when the electron transport chain function is suboptimal, to which the mitochondrial matrix exhibits antioxidant defense in the form of SOD2 expression. In contrast, catalase is not found in the mitochondria of most tissues (8). Mitochondrial-derived ROS are not only cytotoxic agents but also act as diffusible signaling molecules that modulate numerous intracellular responses, including cell cycle regulation, apoptosis, differentiation programs regulated by Notch, Forkhead box O (FOXO), p53, c-Myc, and peroxisome proliferator-activated receptor-γ coactivator 1-α (PGC1α), longevity, and immune cell effector function (9, 10).
Mitochondrial-derived ROS:
superoxide generated as a byproduct of OXPHOS when electrons leak from the electron transport chain to molecular oxygen
Dynamic intraorganelle communication exists between mitochondria and the endoplasmic reticulum (ER) and the nucleus (Figure 1). Physical interaction between the mitochondria and ER occurs at mitochondrial-associated membranes (MAMs), which are microdomains that facilitate calcium and lipid trafficking between the organelles important for calcium homeostasis and lipid metabolism (11). Mitochondria-to-nuclear cross talk is predominantly facilitated by mitochondrial retrograde signaling. This is achieved by regulation of nuclear transcription factors by cytosolic signaling factors such as calcium, ROS, and NAD/NADH ratio (12). This retrograde signaling influences key aspects of cell physiology such as metabolism, stemness, and survival. The mitochondrial unfolded protein response is activated in response to unfolded or misfolded proteins in the matrix and consists of multiple signaling pathways (AKT, PKR, ATF5) that alter nuclear gene expression to restore mitochondrial homeostasis (10).
MAM:
mitochondrial-associated membrane; region of contact between mitochondria and ER involved in fundamental cellular processes and communication between the two organelles
1.1. Mitochondrial Quality Control
The health of the mitochondrial pool is maintained by various quality control mechanisms (Figure 2). Mitochondrial-derived vesicles are considered a first round of defense for the organelle, constituting a local response to stress not requiring fission or autophagy. Mitochondrial-derived vesicles are 70–150 nm in diameter and contain damaged outer mitochondrial membrane, inner mitochondrial membrane, or matrix proteins that are delivered to and degraded by the lysosomes (13). Mitochondrial fission and fusion not only mediate high plasticity of structure for the organelle but also serve to compartmentalize or pinch off damaged portions (fission) and dilute damaged components with healthy mitochondria (fusion) (14). Mitochondrial fission is crucial for selective autophagy of mitochondria, called mitophagy, by dividing mitochondria into fragments of small enough size to be engulfed by the autophagosome (1). Incorporation of damaged mitochondria into the autophagosome is facilitated by autophagy receptors that localize to the outer mitochondrial membrane and mark damaged mitochondria (15). Autophagy receptors can be grouped based on dependency on Parkin; Parkin, an E3 ubiquitin ligase, is recruited to dysfunctional mitochondria and targets outer mitochondrial membrane proteins for ubiquitination. Polyubiquitinated chains are then recognized by autophagy receptors, thereby incorporating the mitochondrion into the eventual autophagosome via binding to LC3II present in the forming phagosome. Parkin-dependent autophagy receptors that target mitochondria include p62, OPTN, NBR1, and AMBRA1. Other autophagy receptors that target mitochondria and act independently of Parkin include Bnip3, Nix/Bnip3L, Fundc1, and Bcl2l13 (16–18). Parkin-independent autophagy receptors contain transmembrane domains and, upon expression, constitutively localize to the outer mitochondrial membrane where they can directly bind to LC3II.
Figure 2 :
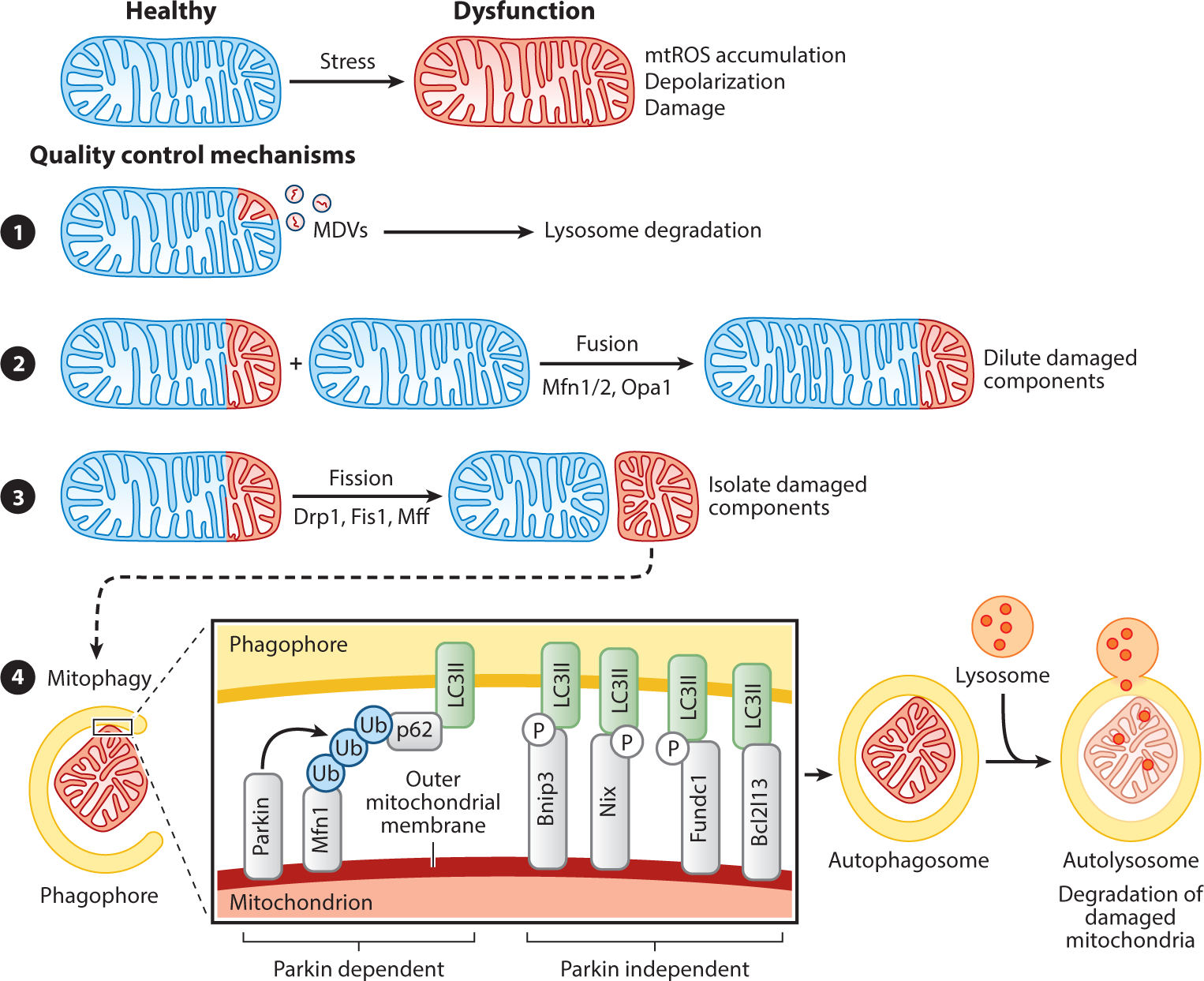
Mitochondrial quality control mechanisms. Mitochondrial dysfunction can be controlled by various mechanisms. (❶) The release of MDVs containing damaged mitochondrial proteins destined for degradation by the lysosomes is considered a first round of defense for the organelle (13). The processes of mitochondrial fission and fusion are mediated by GTPases of the dynamin family. (❷) Mitochondrial fusion, mediated by Mfn1, Mfn2, and Opa1, facilitates the sharing of mtDNA between mitochondria, thereby providing more support for critical functions such as oxidative phosphorylation (14). (❸) Mitochondrial fission, mediated by Drp1, Fis1, and Mff, isolates damaged from healthy components and is crucial for mitophagy by dividing mitochondria into small enough sizes to fit into the autophagosome. (❹) Damaged mitochondria are marked for mitophagy by OMM-localized autophagy receptors, which are either dependent on the E3 ubiquitin ligase Parkin or are Parkin independent. Parkin is recruited to damaged mitochondria by Pink1 and forms polyubiquitinated chains on OMM proteins (such as Mfn1, Mfn2, or VDAC1) that are then recognized by an autophagy receptor (such as p62/sequestosome 1, optineurin, NBR1, or AMBRA1). These autophagy receptors bind to LC3II present in the membrane of the forming phagosome, thereby incorporating the damaged mitochondrion into the autophagy pathway for ultimate degradation in the autolysosome. Parkin-independent autophagy receptors contain transmembrane domains and upon expression constitutively localize to the OMM and interact with autophagy factors such as LC3II (via an LIR domain), ULK1, DFCP1, and WIPI1. Expression of Bnip3 and Nix are controlled by the transcription factors Hif1α, NF-κB, or FOXO3, suggesting that hypoxia, inflammation, and stemness pathways are involved in mitophagy induction (142, 143). Posttranslational modification of Bnip3, Nix, and Fundc1 by phosphorylation within the LIR domain increases their affinity for LC3II binding, suggesting that phosphorylation regulates their pro-mitophagy activity (144). Abbreviations: AMBRA1, autophagy and beclin 1 regulator 1; DFCP1, double FYVE-containing protein 1; Drp1, dynamin-related protein 1; Fis1, fission 1; FOXO3, Forkhead box O 3; GTPases, guanosine triphosphatases; Hif1α, hypoxia-inducible factor 1 alpha; LC3, microtubule-associated protein 1A/1B-light chain 3; LIR, LC3 interacting region; MDV, mitochondrial-derived vesicle; Mff, mitochondria fission factor; Mfn, mitofusin; mtDNA, mitochondrial DNA; mtROS, mitochondrial-derived reactive oxygen species; NBR1, next to BRCA1 gene 1; NF-κB, nuclear factor kappa B; OMM, outer mitochondrial membrane; Opa1, optic atrophy 1; P, phosphorylation; Ub, ubiquitin; ULK1, Unc-51 like autophagy activating kinase 1; VDAC1, voltage-dependent anion channel 1; WIPI1, WD repeat domain phosphoinositide interacting 1.
Mitophagy:
autophagy of mitochondria in which damaged mitochondria are incorporated into the autophagosome as cargo and degraded in the lysosome
2. INTESTINAL EPITHELIAL HOMEOSTASIS AND THE MITOCHONDRIA
2.1. Epithelial Function, Regeneration, and the Mitochondria
The gastrointestinal tract is uniquely equipped to tolerate the luminal microbiota and protect the underlying tissue from potentially harmful pathogens and environmental antigens. The intestinal epithelium, which is composed of a single layer of cells, forms a physical barrier from luminal contents and coordinates mucosal immunity by mediating bacteria and immune cell intercommunication. This barrier is semipermeable, allowing absorption of nutrients and immune cell sensing while blocking harmful intruders. The intestinal epithelium architecture is organized into finger-like protrusions called villi that greatly increase the absorptive surface area in the small intestine (Figure 3). The base of each villus is surrounded by numerous invaginations called crypts. The villi are absent from the colon, forming a flat surface epithelium between crypts. At the crypt base resides the Lgr5+ crypt base columnar (CBC) intestinal stem cells (ISCs), a population of rapidly dividing cells expressing leucine-rich-repeat-containing G protein–coupled receptor 5 (Lgr5) that regenerates the epithelium at a rapid pace of every three to five days (19). CBCs divide into progenitor cells that move upward within the crypt into the transit-amplifying zone where cells differentiate further into a specific intestinal epithelial cell type, including enterocytes, Paneth, goblet, enteroendocrine, tuft, and microfold cells. They continue to migrate toward the villus tip, where senescent intestinal epithelial cells slough off through anoikis, a specific type of programmed cell death for anchorage-dependent cells, and make room for newly formed cells to take their place. Paneth cells are the exception, as these cells migrate to the crypt base in the small intestine and reside between CBCs where they are involved in the innate immune response and express stem cell factors such as epidermal growth factor, Wnt3 and Notch ligands DLL1 and DLL4 that sustain the stem cell niche (20). The major signaling pathways regulating CBC stemness, proliferation, and differentiation are Wnt and Notch (21, 22). CBCs are crucial regulators of tissue homeostasis given the high turnover rate of the intestinal epithelium.
Figure 3 :
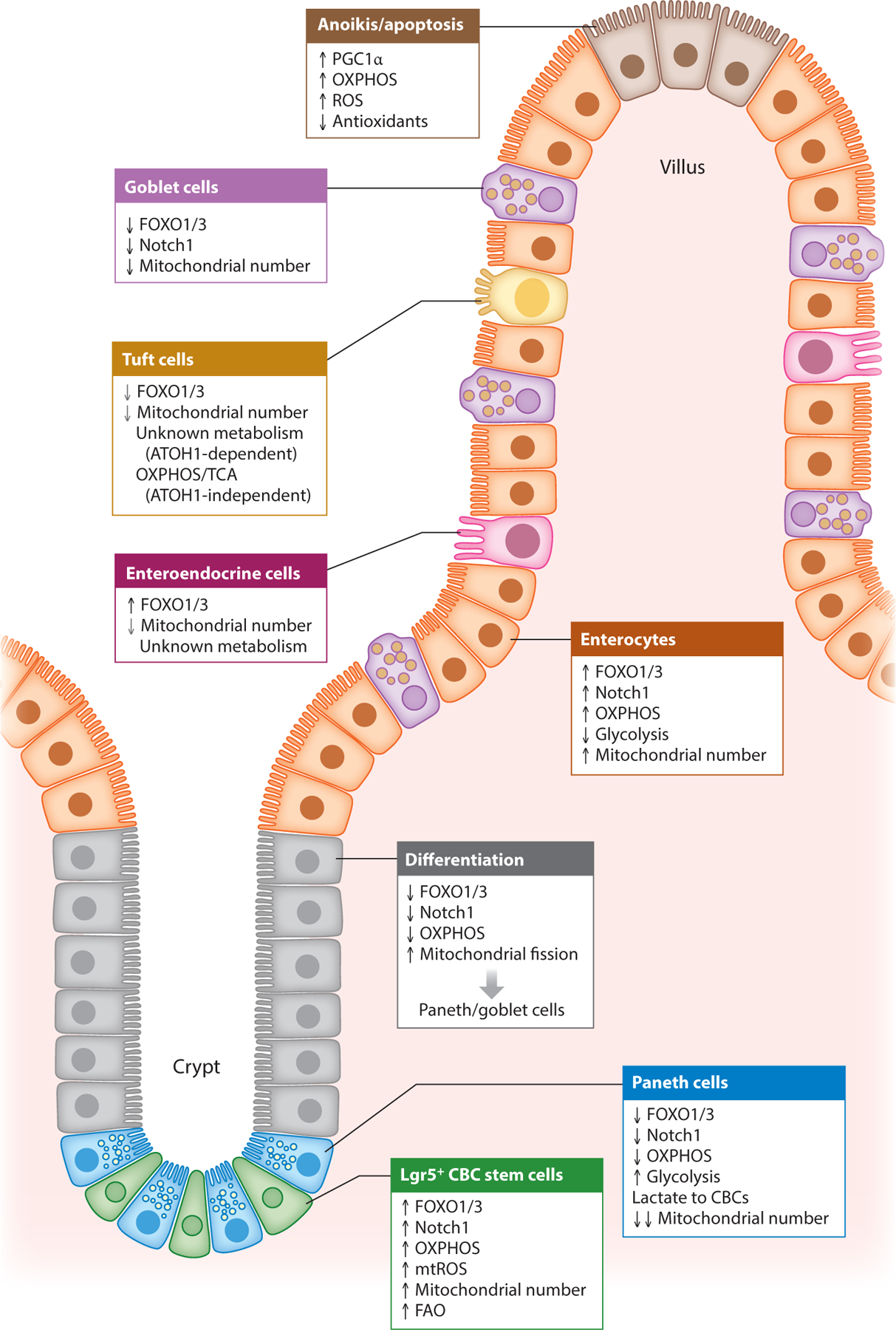
Mitochondrial influence on intestinal epithelial cell fate. Mitochondrial health, metabolism, and numbers play important roles in driving CBC stemness and determining the differentiation of secretory versus absorptive epithelial lineages. CBCs exhibit high mitochondrial respiration and numbers dependent on high expression of FOXO1/3 (27, 28). During differentiation to secretory cells (Paneth cells), a metabolic switch away from OXPHOS is driven by inhibition of FOXO1/3 and enhanced mitochondrial fission (27). During differentiation to absorptive enterocytes, dependency on OXPHOS and expression of FOXO1/3 remains unchanged. Cell death of apical IECs furthest from the crypt base is also influenced by mitochondrial metabolism driven by PGC1α expression (41). Grey arrows indicate that the observed effect is to a lesser extent. Abbreviations: CBC, Lgr5+ crypt base columnar stem cell; IEC, intestinal epithelial cell; FAO, fatty acid oxidation; FOXO, Forkhead box O; mtROS, mitochondrial-derived ROS; OXPHOS, oxidative phosphorylation; PGC1α, peroxisome proliferator-activated receptor-γ coactivator 1-α; ROS, reactive oxygen species; TCA, tricarboxylic acid.
CBC:
Lgr5+ crypt base columnar stem cell that gives rise to all intestinal epithelial cell types
Human mitochondrial DNA is a ~16.6-kb circular, multicopy genome found within the mitochondrial matrix that encodes 13 essential subunits of the OXPHOS system together with 22 transfer RNAs (tRNAs) and 2 ribosomal RNAs (rRNAs) to support synthesis of mitochondrial DNA-encoded proteins within the organelle. The mitochondria have a rudimentary DNA repair mechanism compared to the nuclear genome, which makes mitochondrial DNA more prone to oxidative damage and secondary mutations. A unique feature of the gut is seen in the ability of CBC ISCs to pass on or propagate their mitochondrial DNA to new daughter cells. Greaves et al. (23) showed that the progeny of a morphologically normal human colonic CBC ISC that contains a mitochondrial DNA mutation can expand to occupy a whole crypt. Thus, conditions that promote mitochondrial DNA mutations associated with a deleterious change in mitochondrial function in a CBC ISC (e.g., during aging or chronic inflammation) can influence the overall epithelial phenotype if born from a mutated CBC ISC. It has been shown that mitochondrial DNA mutations increase exponentially with age and that these mutations can lead to a reduction or complete loss of expression of OXPHOS complexes in intestinal tissue (24). Whether mitochondrial DNA mutations affect CBC ISC function or their ability to differentiate, particularly in IBD, has not yet been studied in detail.
2.2. Metabolism in the Intestinal Epithelium Influences Cell Fate
Each of the differentiated intestinal epithelial cells carries out unique functions. Enterocytes (called colonocytes in the colon) are absorptive cells responsible for nutrient and water absorption and are the most abundant intestinal epithelial cell type. Secretory cells include goblet cells that secrete mucins that comprise the mucus barrier, enteroendocrine cells that secrete hormones such as glucagon-like peptide 1 and peptide YY, and Paneth cells that secrete factors crucial for stem cell homeostasis and antimicrobial peptides that shape the gut microbiota composition (20). Chemosensory and secretory tuft cells provide defense against luminal pathosymbionts such as helminths (25). Microfold cells are crucial to the immune surveillance due to their sampling, uptake, and presentation of luminal antigens (26). These intestinal epithelial cells are found throughout the small intestine and colon, with the exception of Paneth cells and microfold cells, which are located only in the small intestine during homeostasis. Additionally, turnover time of these cells varies with enterocytes/colonocytes and goblet cells at 3–5 days, enteroendocrine cells at 3–10 days or longer, tufts cells at 7–14 days, and the long-lived Paneth cells at 30–60 days (20).
Bioenergetic need is dynamically matched to cellular function, life span, and stress response. It is not surprising given the distinct functions and longevity of intestinal epithelial cells that they also have distinct cellular metabolisms (Figure 3). Recent studies have demonstrated the importance of mitochondria and metabolic transition in determining CBC stem cell fate (27, 28), which has also been shown in other stem cell systems (29). CBCs exhibit high OXPHOS activity, high production of mitochondrial ROS, and a relatively high number of mitochondria compared to neighboring Paneth cells (28). This maintains CBC self-renewal and the proliferative capacity necessary to match intestinal epithelial cell turnover. Furthermore, optimal mitochondrial function is crucial in maintaining CBC stemness. This was demonstrated in mouse models exhibiting intestinal epithelial cell mitochondrial stress driven by genetic deletion of heat-shock protein 60 (Hsp60) (30). HSP60 is a chaperone protein essential for protein maturation in the mitochondrial matrix (10). Loss of Hsp60 activated the mitochondrial unfolded protein response, which comprises distinct signaling pathways tasked to restore the mitochondrial proteome, induce mitophagy, regulate mitochondrial respiration, and induce proliferation (reviewed in 10). Additionally, loss of Hsp60 induced mitochondrial dysfunction, caused a loss in CBC stemness and a deficiency in CBC numbers, and inhibited intestinal epithelial cell proliferation, thereby demonstrating the reliance of ISC homeostasis on mitochondrial health (30).
Compared to CBCs, Paneth cells exhibit a lower number of mitochondria and high glycolytic activity, producing lactate that is secreted to adjacent CBCs to fuel their high OXPHOS activity (28). During differentiation to Paneth cells or goblet cells, CBCs undergo a metabolic switch driven by inhibition of FOXO1/3 transcription factors to downregulate dependency on mitochondrial activity (27). Specifically, FOXO1/3 downregulation led to inhibition of Notch signaling [a determinant of secretory lineage differentiation (31)] and induction of mitochondrial fission necessary for differentiation into Paneth and goblet cells (27). During differentiation to enterocytes, Foxo1/3 and Notch1 expression remained unchanged and mitochondrial abundance was maintained with reliance on mitochondrial OXPHOS activity (27, 32). Accordingly, in Drosophila, ISCs with impaired OXPHOS failed to differentiate into enterocytes (33), and activation of OXPHOS enhanced ISC function (34). Progression to enteroendocrine differentiation was similar, with unchanged Foxo1/3 and Notch1 expression but a modest decrease in mitochondrial numbers compared to the decrease noted in Paneth cells and goblet cells (27). Tuft cell differentiation results in heterogeneous populations during homeostasis that, in the small intestine, can be driven by atonal homolog 1 (ATOH1)-dependent and -independent signaling (35, 36). A recent study revealed that progression of ATOH1-independent tuft cell differentiation was accompanied by an increase in genes related to the tricarboxylic acid cycle and OXPHOS, which was driven by the metabolite succinate from the intestinal microbiome (35). These studies demonstrate the importance of mitochondria and metabolism in determining CBC function and differentiation routes. Further studies are needed to characterize the metabolic phenotype of goblet cells, ATOH1-dependent tuft cells, and enteroendocrine cells.
Recent studies suggest that FAO is an important metabolic process that fuels mitochondrial respiration in CBCs. Mice fed a high-fat diet exhibited increased numbers of CBCs (37). This was corroborated in vitro with palmitic acid treatment of intestinal organoids, giving rise to more CBCs with enhanced stemness and less dependency on Paneth cells (37), presumably due to less need for Paneth cell–derived lactate in the presence of palmitic acid to drive OXPHOS. Deletion of Cpt1a, the gene encoding the rate-limiting step in FAO, decreased CBC numbers and function (38). In addition, the transcription factor PRDM16, which promotes FAO, was shown to be necessary for growth and differentiation of upper intestinal progenitor cells (39). Recently, it was demonstrated that the transcription factors hepatocyte nuclear factor (HNF)4α and HNF4γ regulate the expression of genes required for FAO in the intestine (40). Mice deficient in HNF4α/γ exhibited decreased FAO and loss of CBCs through an inability to self-renew. The same study showed that restoration of acetyl-CoA with acetate (a precursor of acetyl-CoA) or dichloroacetate rescued stem cell function and renewal capacity during deletion of Hnf4α/γ (40).
Mitochondrial metabolism is also crucial at the villus tip in determining epithelial cell fate (Figure 3). PGC1α is a transcriptional coactivator driving mitochondrial biogenesis and a metabolic shift toward mitochondrial respiration. PGC1α was shown to be highly expressed in apical intestinal epithelial cells farthest from the crypt base that exhibit high mitochondrial activity and ROS production and lacking a balanced increase in antioxidant enzyme production (41). This accumulation of ROS fosters a proapoptotic fate, thereby preserving tissue homeostasis by facilitating balanced intestinal epithelial cell turnover (41).
3. MITOCHONDRIAL HEALTH AND INFLAMMATORY BOWEL DISEASE
3.1. Inflammatory Bowel Disease
Inflammatory bowel disease (IBD) is complex, multifactorial, chronic, and relapsing immune-mediated inflammatory disease of the gastrointestinal tract. The two most common forms are Crohn’s disease (CD) and ulcerative colitis (UC), which are separate entities but share many clinical and pathological features (Table 1). CD can affect the entire gastrointestinal tract with discontinuous, transmural inflammation, including characteristic histological granulomas. UC presents with inflammation limited to the mucosa and involving the rectum that can extend proximally in the colon in a continuous fashion. IBD is considered a global health problem, with accelerating incidence in newly industrialized countries and stabilizing incidence but increasing prevalence in countries with a westernized lifestyle (Europe and North America) (42). The peak age of onset occurs between 15 and 30 years, with ~10% of patients diagnosed before adulthood. IBD carries lifelong morbidity. Current therapeutics include biologics, immunomodulators, aminosalicylates, and corticosteroids, but due to the heterogeneous nature of the diseases across patients [disease location, disease progression, related but distinct mechanisms (43)], therapeutic outcomes are variable and present a challenge for treatment strategies. For instance, although antitumor necrosis factor alpha (anti-TNFα) biologics provide a major breakthrough in IBD therapy, 30% of patients are primary nonresponders and 50% are secondary nonresponders to anti-TNFα therapies. In fact, anti-TNFα therapy exhibits a limited efficacy of ~45% mucosal healing, a key clinical end point, at 12 and 24 months (44).
Table 1.
Summary of clinical features of Crohn’s disease and ulcerative colitis
| Crohn’s disease | Ulcerative colitis | |
|---|---|---|
| Sex | Higher incidence in females | Equal incidence in males and females |
| Clinical presentation | Chronic diarrhea, abdominal pain, fever, malnourishment, fatigue, and weight loss | Most commonly bloody diarrhea or rectal bleeding |
| Affected areas | Can affect any part of gastrointestinal tract; terminal ileum is often involved | Affects the colon only |
| Pattern of inflammation | Patchy with aphthous ulcers with granuloma formation | Continuous confluent is mainly subepithelial inflammation |
| Environmental factors | Cigarette smoking worsens disease | Cigarette smoking is protective |
| Genetics | NOD2, ATG16L1, 1L-23 | Human leukocyte antigen |
| Serological markers | Antibodies to microbiota, including anti-Saccharomyces cerevisiae antibodies (ASCAs) | Anti-neutrophil cytoplasmic antibodies (ANCAs) |
Although the etiology of IBD is unknown, it is regarded to be a multifactorial condition dependent on the interplay of various genetic, environmental, immunological, and microbial factors. With the exception of over 50 genes identified to cause monogenic and rare very early-onset IBD that develops in children <6 years of age (45), IBD is a polygenic disorder driven by multiple common genetic polymorphisms. To date, genome-wide association studies (GWAS) have identified 242 loci that confer IBD susceptibility (46). Several risk loci (68%) are shared between CD and UC, whereas others are unique to each disease, suggesting related mechanisms and distinct pathologies (recently reviewed in 47). Additionally, ~70% of IBD risk loci are shared with other complex immune-mediated diseases (48). Through GWAS, multiple pathways involved in IBD have been identified and include microbial defense, innate immune regulation, adaptive immune regulation, autophagy, epithelial barrier function and restitution, and ER stress. However, GWAS only explain part of IBD heritability, limited by the inability to identify rarer genetic variants.
Epidemiological studies suggest a key role of environmental factors in the pathogenesis and natural history of IBD (42). Environmental factors include those that influence disease risk at an individual level and those that influence the entire population of a region. To date, the best-characterized environmental factors influencing the risk of IBD include cigarette smoking, diet, antibiotic use, nonsteroidal anti-inflammatory drug (NSAID) use, breastfeeding, urban living, and air pollution. Emerging studies suggest that the influence of environmental factors on IBD risk can vary depending on geographical location. For example, breastfeeding confers protection against IBD in all regions studied, whereas smoking increases the risk of developing CD in Europe and North America, but this association is lacking in Asia (42). Further studies are needed to better understand whether certain environmental risk factors are regional and how they drive IBD in different parts of the world. The proposed mechanisms of action for many of these environmental factors are, at least in part, through alterations of the gut microbiome (associated with cigarette smoking, diet, antibiotic use, and breastfeeding) or disruption of the epithelial barrier (associated with cigarette smoking, diet, and NSAID use) (49). It is well established that patients with IBD exhibit altered composition of the gut microbiota with the expansion of potential pathogenic bacteria and decreased overall diversity (reviewed in 50). Whether dysbiosis of the microbiota causes disease via immune activation or is a consequence of chronic inflammation is unknown. Importantly, profiling gut microbiota composition in intestinal tissue and fecal samples, coupled with genome-wide molecular phenotyping, has revealed molecular signatures that define IBD subtypes that associate with clinical phenotypes (47). This includes clinical phenotypes such as fibrosis, stricture, deep ulcers, penetrating disease, treatment escalation, and anti-TNF response. This is an important step toward personalized disease management to overcome the heterogeneous and complex nature of IBD.
3.2. Pathogenic Mechanisms of Mitochondrial Dysfunction in Inflammatory Bowel Disease
Mitochondrial dysfunction plays an important role in a wide array of chronic inflammatory diseases, ranging from cancer to neurodegenerative disorders (see the sidebar titled Mitochondrial Dysfunction in the Gut–Brain Axis). Pertinent to our review, two fundamental questions arise: First, why is mitochondrial dysfunction a key factor in IBD, and second, how does it contribute to the specific development of IBD (Supplemental Figure 1)? The gut contains high concentrations of bacteria with their metabolic products, immune-active ligands, damage-associated molecular patterns (DAMPs), xenobiotics, and environmental toxins that can damage the mitochondria. In a proteomic analysis across 14 different mouse tissues, Pagliarini et al. (51) demonstrated that intestinal epithelial mitochondria have distinct proteomic profiles and, notably, they have a higher expression of ATP-binding cassette transporters, likely reflecting tissue-specific demands to adapt to the gut environment. Furthermore, it is also prescient that the gut (unlike other tissues and organs) relies substantially on the gut microbiota as its main source of energy and to maintain the overall health of the enterocytes that house the mitochondria (52). Mitochondrial dynamics (fission/fusion) in cultured gut epithelial lines become compromised when in contact with pathobionts such as adherent-invasive E. coli LF82 (53). Such close vicinity and interdependence with the microbiota place a significant demand on the mechanisms that regulate mitochondria function. Animal knockout models in genes with such epithelial protective roles (e.g., Mdr1a−/−, Irgm1−/−, and Sod2−/− transgenic murine models) show an accumulation of damaged mitochondria within gut enterocytes and are more prone to induced experimental colitis (54, 55).
Somewhat less appreciated, 5% of the IBD genetic susceptibility factors identified from human GWAS are involved in mitochondrial homeostasis. The top three positional candidate gene associations are SLC25A28 encoding mitoferrin 2, VARS encoding valine-tRNA ligase, and RNF5 encoding E3 ubiquitin-protein ligase RNF5. These genes encode proteins involved in mitochondria iron transport, tRNA transport, and ubiquitination, respectively (3, 56, 57). Notable also are IBD associations with HSPA1-A, -B, and -L genes that encode heat-shock protein 70, which is integrally involved in the mitochondrial unfolded protein response (58). Specific mitophagy genes such as PARK7 and LRKK2 are associated with UC and CD, respectively (54, 59, 60), and there are notably many other autophagy genes implicated in IBD discussed in Sections 3.7 and 3.8 below. Recently, a CD variant in C13orf31 loci, previously encoding an unknown protein, has been shown to have major immunometabolic function, now called the FAMIN gene (61). Thus, IBD patients may have inherent susceptibility to mitochondrial dysfunction that can be unmasked by the gut environment. Intriguing association with genes such as mitoferrin 2 suggest potentially very specific pathogenic defects, which at the moment are not fully elucidated.
Certain in-built specialized intestinal epithelial cell features, for example, high rates of enterocyte replenishment driven by ISC fission, the maintenance of longer-lived Paneth cells, and the requirement of metabolic fuel from the gut microbiota, are precariously balanced on optimal mitochondrial fitness. Perturbations of such a finely tuned biological system in the face of a very dynamic gut luminal environment can have several potential pathogenic consequences. First, mitochondrial damage can lead to energy-deficient states that compromise epithelial barrier function by increasing susceptibility to cell death, reducing secreted barrier function (Paneth and goblet cells in particular), and the ability to regenerate in response to damaging stimuli (54, 62, 63). In conplastic murine lines with the same nuclear genome but different mitochondrial genomes, naturally occurring mitochondrial DNA polymorphisms have been linked to the level of mitochondrial activity (64). Those conplastic murine lines carrying mitochondrial DNA polymorphisms that were associated with higher intestinal ATP production from more efficient OXPHOS activity were protected from dextran sodium sulfate–induced colitis (64). Second, mitochondrial damage can influence the immune cell phenotype and function via metabolic programming (65). In proinflammatory cells, such as activated monocytes and activated T and B cells, cellular energy is generated by increasing glycolysis, while in T regulatory cells and anti-inflammatory M2 macrophages, energy is generated by increasing mitochondrial-dependent mechanisms and FAO (66). Thus, situations of mitochondrial damage can augment and perpetuate the proinflammatory state with the absence of anti-inflammatory immune cell activation. Third, mitochondrial DAMPs such as mitochondrial DNA and N-formylated peptides can act as direct inflammatory signals and contribute to the inflammatory milieu that sustains the abnormal innate and adaptive immune response, which in turn further potentiates mitochondrial dysfunction in the gut (67–69).
It is noteworthy that in rare maternally transmitted mitochondrial genetic disorders, cardiac and neurological manifestations are more common than intestinal phenotypes (70). From a gut perspective, mitochondrial genetic disorders can result in chronic intestinal pseudo-obstruction that presents with a significant dilatation of the small and large bowel in the absence of mechanical cause. In this regard, a myopathic failure of the intestinal smooth muscle resulting in the loss of gut motility is seen rather than an inflammatory phenotype (71). Hence, a complex picture is evident even in inherited mitochondrial DNA conditions where clinical features manifest in tissue- and organ-dependent manners depending on the influence of local conditions and/or of the functional change driven by the specific mitochondrial DNA mutation. Such complex outcomes driven by mitochondrial alterations are also likely in UC and CD.
3.3. Mitochondria and Ulcerative Colitis
As early as in the 1980s, UC was postulated as a colonic energy deficient disease by Roediger (72). Recently, in a bulk RNA-seq study involving 206 newly diagnosed pediatric UC participants (PROTECT cohort), Haberman et al. (73) showed a striking downregulation of both mitochondrial- and nuclear-encoded genes involved in mitochondrial function in UC patients. Notably, all 13 mitochondrial-encoded genes regulating ATP production (complex I, III, IV, and V) and nuclear PPARGC1A that encodes PGC1α, integral for mitochondrial biogenesis, were decreased in UC patients. An earlier adult IBD microarray data set also showed that genes involved in mitochondrial function were the most differentially expressed genes in the distal colon where UC commonly affects (74). This extends from the observations of mitochondrial morphological abnormalities (even preceding overt gut inflammation) (75–77) and functional electron transport chain deficits (up to 60%) of complexes II, III, and IV in UC patients (78–80). In a proteomic analysis of UC gut mucosa, a clear group of downregulated proteins that are involved in energy generation, mitochondrial stress response, and antioxidant defense are notable, including heat-shock protein 90 (HSPA9B), heat-shock protein 60 (HSP60), H+-transporting two-sector ATPase (ATP5B), prohibitin 1 (PHB1), mitochondrial malate dehydrogenase 2 (MDH2), voltage-dependent anion channel 1 (VDAC1), thioredoxin peroxidase 1 (PRDX1), and thiol-specific antioxidant 2 (PRDX2) (76). These lines of data suggest that local gene expression changes in the UC gut mucosa are important in mediating mitochondrial dysfunction.
The colonic epithelium derives 70% of its energy from the short-chain fatty acids (SCFAs) butyrate, propionate, and acetate, which are products of anaerobic bacterial fermentation of undigested fiber (81). High levels of colonic bacteria Clostridium, Eubacterium, and Butyrivibrio ferment dietary fiber into butyrate at very high concentrations (82, 83). SCFAs are transported into colonocytes and further into mitochondria where they drive ATP production. In UC, ATP levels in the colon are significantly decreased and are linked to SCFA deficiency (84). Two studies suggest an association of mitochondrial DNA mutation MT-ND4 11719 A/G polymorphism with UC (85, 86). This gene encodes for a subunit of the complex I of the electron transport chain. Microbial shifts in UC promote metabolic products such as nitric oxide and hydrogen sulfide that can damage the mitochondria and inhibit cytochrome C and SCFA oxidation (87). Furthermore, Donohoe et al. (52) demonstrated that in gut energy-deficient states, colonocytes turn to autophagy as an alternate energy source. Together with IBD-associated autophagy defects (discussed in Section 3.8), this is likely to foment mitochondrial dysfunction as a key component of UC inflammation. In this context, we discuss two pertinent downstream effects in detail: mitochondrial ROS and the release of mitochondrial DAMPs.
3.4. Mitochondrial ROS and the Inflammatory Response in Ulcerative Colitis
Mitochondrial OXPHOS is a major cellular source of ROS (88). Data have shown that mitochondrial ROS are an important part of host defense and immunity, particularly in antibacterial (direct killing) and antiviral (mediating interferon) responses (89). Under pathologic conditions, however, mitochondria can produce high levels of superoxide and hydrogen peroxide that can combine with iron to generate free radicals that damage macromolecules within mitochondria and oxidize mitochondrial DNA (90). Mitochondrial DNA is vulnerable to oxidative damage by mitochondrial ROS owing to both its close proximity to the electron transport chain, which is the source of mitochondrial ROS, and to the lack of protective histones in mitochondrial DNA structure. In this context, mitochondrial ROS are a driver of the inflammatory response. For example, oxidized mitochondrial DNA is the danger signal that activates the NLRP3 inflammasome (91). Mitochondrial ROS promote the cytosolic release of mitochondrial DNA that is necessary for NLRP3 engagement (92). During failure of mitophagy, ROS-producing mitochondria can accumulate to further amplify the inflammatory response (92). Mitochondrial ROS are also important in metabolic reprograming of macrophages to a proinflammatory phenotype (65).
The deleterious effects of mitochondrial ROS are likely to be highly relevant in UC colonic mucosa. The balance of mitochondrial ROS has been shown to be tonically required to maintain intestinal epithelial homeostasis (93). As discussed earlier, electron transport chain deficits/damage are observed in UC colon that result in higher mitochondrial ROS production (76, 78–80). Furthermore, mitochondria are exposed to significant levels of general oxidative stress associated with active UC (94). SOD2, which is required to detoxify mitochondrial ROS, is highly expressed in UC mucosa, underlying the importance of this specific detoxification mechanism to protect the mitochondria in the gut (54, 95, 96). In mouse models, pharmacologic induction of mitochondrial ROS can trigger colitis, while conversely, inhibition of mitochondrial ROS can lessen disease severity. Ho et al. (54) showed that colonic administration of rotenone (a complex I inhibitor that increases mitochondrial ROS levels) can accelerate the development of chronic colitis in susceptible Mdr1a-deficient mice and in acute dextran sodium sulfate–induced colitis. In the same study, the use of MitoQ (a mitochondrial antioxidant) can mitigate colitis in these two models. MitoQ is a derivative of coenzyme Q10. Owing to its structure and charge, MitoQ is efficient at migrating specifically into the mitochondria. The primary component of MitoQ (mitoquinol, a variant of ubiquinol) migrates inside the inner mitochondrial membrane and donates a hydrogen atom to combat the radical superoxide species, thereby decreasing oxidative stress (97). Similarly in other murine models, genetic-driven knockouts of proteins that are crucial in the regulation of mitochondrial ROS, such as SOD2 (54), uncoupling protein 2 (UCP2) (98), and immediate early response gene X-1 (IEX-1) (99), all showed increased susceptibility to colitis. Of relevance to the human condition, UCP2 genetic variants are associated with UC and CD patients (100).
3.5. Mitochondrial DAMPs in Ulcerative Colitis
The mitochondria share several features with bacteria, including a double membrane–bound structure, a circular genome that replicates independently of nuclear DNA, and the synthesis of N-formylated proteins. As the innate immune system recognizes conserved bacterial molecules, mitochondrial constituents are similarly immunogenic, acting as DAMPs when released into the cytosol and extracellular environment (101). Mitochondrial DNA contains a high frequency of unmethylated CpG dinucleotide repeats that can elicit an immune response similar to that seen against bacterial DNA. Boyapati et al. (67) first showed higher mitochondrial DNA in circulation and stools in a prospective cohort of IBD consisting mainly of active severe UC patients. Interestingly, there was normalization of mitochondrial DNA levels in a longitudinal analysis of a smaller subset of patients before and after surgical removal of the colon (67). Here, it is proposed that mitochondrial DNA acts as a circulating DAMP that is released from the active UC mucosa. Using tissue-specific transgenic approaches in mice, Oka et al. (102) showed that local mitochondrial DNA release was beyond simply a biomarker of tissue damage and could directly result in organ inflammatory pathology (in this study focusing on the heart). The seminal study by Hauser’s group (69) showed that when released extracellularly into the circulation, mitochondrial DNA functionally contributes to the inflammatory process by triggering IL-8 production in peripheral circulating neutrophils. In systemic lupus erythematosus, neutrophils with impaired mitophagy have the propensity to release oxidized mitochondria DNA in neutrophil extracellular traps that drive the disease process (103). Although there is no direct evidence yet to link neutrophils as the main source of mitochondrial DAMP release, UC is notable for uncontrolled neutrophil death that may also contribute to mitochondrial DNA release during active disease (104). Taken together, both mitochondrial ROS and DNA (together with the processes that lead to their increase) represent attractive therapeutic targets for intervention in UC.
3.6. Mitochondrial Dysfunction and Crohn’s Disease
Much like in UC, emerging studies suggest the central involvement of mitochondrial health in CD. Polymorphisms in genes involved in mitochondrial homeostasis are significantly associated with CD susceptibility and/or clinical course, including SLC22A5 (encoding OCTN2) (105), IRGM (106), and UCP2 (100). The pediatric RISK stratification study using RNA sequencing analysis on CD mucosal biopsies demonstrated that patients predicted to be at high risk of stricturing who remained complication free through follow-up at 36 months expressed an enhanced mitochondrial function gene signature (107). In a proteome study of pediatric CD patients, impaired mitochondrial function, specifically mitochondrial proteins involved in H2S detoxification, were downregulated and correlated with increased disease severity (108). Furthermore, ultrastructural studies of epithelial cell mitochondria demonstrated dissolved/irregular mitochondrial cristae indicative of impaired function in CD patients prior to other early events during the progression of inflammation, such as alterations to the tight junctions controlling barrier function (109). This suggests that intestinal epithelial cell mitochondrial alterations may serve as early molecular events during the progression of CD inflammation.
Animal models have provided further insight into the role of mitochondrial dysfunction in the pathogenesis of ileitis. The TNFΔARE mouse model exhibits enhanced TNFα mRNA stability, resulting in elevated TNFα levels and the development of spontaneous ileitis due to a genetic deletion within the adenylate/uridylate-rich elements (AREs) of the TNF mRNA 3′ untranslated region. Ileitis in this model was associated with impaired mitochondrial function in ileal crypts, including decreased ATP levels, enhanced mitochondrial unfolded protein response, ER stress, and degenerative mitochondria in Paneth cells (110). Additionally, ileitis in TNFΔARE mice and CD patients was associated with a deficiency of tuft cells that could be restored by microbiota-derived succinate-induced expression of genes that regulate the tricarboxylic acid cycle and OXPHOS, thereby suppressing inflammation (35). This study suggests that microbial metabolites influence intestinal epithelial cell metabolism and differentiation to modulate ileitis. Mice with intestinal epithelial cell–specific double knockout of VIL1 (encoding villin1) and GSN (encoding gelsolin) developed CD-like ileitis associated with intestinal epithelial cell mitochondrial stress and overexpression of IRGM1 (111). These results were corroborated using distal ileum specimens showing decreased expression of VIL1 and GSN and increased levels of IRGM in CD patients (111). In other spontaneous mouse models of CD-like ileitis, including SAMP/YitFc (112), Xbp1ΔIEC (113), or Ship1−/− (114), the status of mitochondrial health was not reported.
3.7. Mitochondria and Paneth Cell Function in Crohn’s Disease
As implicated by the TNFΔARE mouse model, additional recent evidence suggests an important role of impaired mitochondria, specifically in the health and function of Paneth cells associated with ileitis. A recent study utilized an inducible mouse model of intestinal epithelial cell mitochondrial dysfunction via deletion of prohibitin 1 (Phb1iΔIEC), an abundant inner mitochondrial membrane protein that serves numerous roles in mitochondrial function, including stabilizing mitochondrial DNA-encoded proteins, regulating mitochondrial fusion, and maintaining optimal activity of the electron transport chain (115, 116). Importantly, expression of PHB1 is decreased in mucosal biopsies from CD and UC patients (76, 117). Phb1iΔIEC mice developed spontaneous ileitis that is preceded by mitochondrial dysfunction in all epithelial cells, crypt cell death, and early defects in Paneth cells (62). Phb1iΔIEC mice administered a mitochondrial-targeted antioxidant (SOD2 mimetic), Mito-Tempo, were protected from mitochondrial dysfunction, Paneth cell abnormalities, and ileitis, suggesting that mitochondrial ROS contribute to disease progression in this model of intestinal epithelial cell mitochondrial dysfunction (62). Furthermore, Phb1 deficiency induced loss of viability of the ISC niche and Paneth cell defects in cultured ileal enteroids that were ameliorated by Mito-Tempo. In the same study, Paneth cell–specific deletion of Phb1 driven by Defα6-Cre or Mist1-CreERT2 was sufficient to drive spontaneous ileitis, suggesting a causative role of mitochondrial dysfunction in ileitis that initiates from Paneth cell dysfunction (62).
Additional recent studies demonstrated that induction of mitochondrial stress in CBCs via Hsp60 deletion causes the development of abnormal Paneth cells and loss of ISC niche function in mouse models (110). Ileal Paneth cells of CD patients have been reported to exhibit mitochondrial swelling and necrotic cell death (118). Together these results identify Paneth cells as highly susceptible to mitochondrial dysfunction as compared to other intestinal epithelial cell types. Mitochondrial health may be especially important in Paneth cells owing to their physiology, as these cells are terminally differentiated and long lived (30–60 days), providing a cellular environment in which damaged mitochondria are not diluted by cell replication (20). Additionally, Paneth cells have extensive ER to support their secretory functions, and through intimate communication with the mitochondria via MAMs, mitochondrial dysfunction could elicit further cellular stress through induction of ER stress. In the same manner, ER stress could impact the mitochondrial compartment. Further studies are necessary to elucidate the influence of organellar communication on Paneth cell health.
Several CD susceptibility genes integrate into cellular processes that converge on Paneth cell homeostasis, including autophagy (ATG16L1 T300A, LRRK2, IRGM), ER stress (XBP1), and bacterial sensing (NOD2) (119). In work pioneered by Stappenbeck and colleagues, ileal CD patients can be stratified based on Paneth cell phenotypes defined by lysozyme granule staining characteristics (normal, disordered, diminished, diffuse, and excluded) (120–122). They showed that 20–50% of Crohn’s disease patients have ≥20% abnormal Paneth cells (called the type I phenotype). This phenotype was independent of active inflammation and remained stable over time (up to >20 years) (120, 121). The type I phenotype was associated with an immune activation signature, altered gut microbiota composition, and early postoperative recurrence after resection. Interestingly, this phenotype also correlated with the decreased expression of genes that regulate OXPHOS (121), suggesting that patients with the type I phenotype may have altered mucosal mitochondrial function. In CD patients, Paneth cell defects seem to be at the intersection of susceptibility genes (ATG16L1, NOD2, LRRK2) with environmental triggers such as smoking and the gut microbiome (122–124). For instance, tobacco smoke was shown to drive Paneth cell defects in CD subjects carrying the ATG16L1 T300A CD susceptibility allele (122). Mice with deficiency of Irgm1 or Atg16L1 that resulted in Paneth cell defects and decreased antimicrobial peptide production are dependent on specific environmental factors such as interaction with the gut microbiota or infection with mouse norovirus, respectively (125, 126). Paneth cell defects were evident before severe inflammation in TNFΔARE mice and in uninflamed tissue of CD patients that could predict early endoscopic recurrence, representing a molecular indicator of early inflammation prior to macroscopic lesions (110). It has been proposed that Paneth cell phenotyping is one method to stratify CD patients by disease mechanism to overcome the heterogeneity of disease and to enhance treatment outcomes (124). As a next step, careful characterization of Paneth cell mitochondria in type I CD patients will indicate whether mitochondrial-targeted therapeutics may have translational utility in this particular subtype of CD.
3.8. Autophagy/Mitophagy Impairment and Inflammatory Bowel Disease
Numerous genes related to autophagy are strongly associated with IBD: ATG16L1, LRRK2, and ULK1 are linked to CD, whereas IRGM and PTPN2 are linked to both CD and UC (127). Mouse models deficient in autophagy-related genes (Atg16L1, Atg7, Atg5, Irgm1, Lrrk2, Atg7, Optn, VDR, or Tfeb) demonstrate the importance of autophagy in myeloid and Paneth cells. In myeloid cells, deficient autophagy results in the exaggerated production of inflammatory cytokines, reduced microbial killing, and increased susceptibility to colitis (128). In Paneth cells, the role of autophagy is multifaceted and includes secreting antimicrobial peptides (secretory autophagy), degrading invasive microbes (xenophagy), and resolving ER stress (ERphagy). Deficiency in Atg16L1, Atg7, Atg5, Irgm1, Tfeb, or VDR causes Paneth cell defects (128, 129). Given the known role of autophagy in mitochondrial quality control, it is not surprising that damaged mitochondria in Paneth cells are associated with deficient autophagy (122, 130). A limitation of these autophagy-deficient models is their inability to discern the specific impact of mitophagy in the resulting pathology. A recent study overcomes this limitation by demonstrating that mice with deletion of the mitophagy receptor Nix/Bnip3l (Nix−/−) were more susceptible to dextran sodium sulfate–induced colitis and exhibited a higher number of damaged mitochondria in the colonic epithelium during colitis. Mito-Tempo, a mitochondrial-targeted ROS scavenger, could ameliorate dextran sodium sulfate–induced colitis and subsequent increased NIX expression (131). Furthermore, NIX was increased in the intestinal epithelial cells of UC patients and during murine colitis in a manner dependent on mitochondrial ROS and hypoxia-inducible factor 1 alpha (HIF1α) stabilization (131). These findings support an important role of mitophagy as a protective response induced by mitochondrial-derived ROS during intestinal inflammation.
4. THERAPEUTIC OPTIONS
Although many preclinical studies in mouse models demonstrate a beneficial effect in targeting mitochondrial dysfunction in inflammation, direct evidence in human clinical intervention is lacking until now. An upcoming phase 2b randomized controlled trial, MARVEL (ClinicalTrials.gov Identifier: NCT04276740), will investigate the use of mitochondrial antioxidant therapy, MitoQ, as a drug repurposing study in adults and children with UC (132). Analogous to MitoQ, the related mitochondrial superoxide scavenger, Mito-Tempo, as discussed earlier, has a protective role for Paneth cells and the development of ileitis (62). This suggests that a human clinical trial should be similarly contemplated for CD (62). Two phase 2 human studies were conducted with MitoQ in the mid-2000s on hepatitis C (133) and Parkinson’s disease (134). Both conditions have prior data to implicate mitochondrial oxidative stress, but notably, they are not archetypal inflammatory diseases like IBD (135, 136).
It is noteworthy that these therapeutic opportunities present a very different approach to the current treatments of IBD with potential mechanisms such as enhancing mitochondrial biogenesis (via PGC1α) (137) and dynamics (138). PGC1α is a transcriptional factor that regulates mitochondrial biogenesis and several stress-response programs, hence integrating mitochondrial function and cellular stress adaption. In the latter, Mancini et al. (138) showed that in the mouse dextran sodium sulfate–induced colitis model, there are high levels of mitochondrial fragmentation where the mitochondrial antifission drug P110 can improve colitis. Pharmacologic modulation of mitophagy is actively explored in conditions such as Parkinson’s disease, which is directly relevant to IBD with its shared genetic susceptibility with PARK7 and LRRK2 (139–141) (see the sidebar titled Mitochondrial Dysfunction in the Gut–Brain Axis). Our work and that by others suggest that mitochondrial dysfunction may be dominant in certain groups of patients, for example, in those with early-onset UC (73), with high mitochondrial DNA levels (102), or manifesting Paneth cell defects (62). Therefore, better ways to identify and stratify patients for potential future mitochondrial-specific therapies are needed. An ongoing study (MUSIC; mitochondrial DAMPs as mechanistic biomarkers in IBD) is currently evaluating the role of mitochondrial DAMPs for this purpose (ClinicalTrials.gov Identifier: NCT04760964).
5. CONCLUDING REMARKS
Our understanding of the importance of mitochondria in the gastrointestinal system is growing, with fascinating insights into the active roles that these ancient organelles play in this dynamic host-environment interphase. Considering the influence mitochondria play on cell homeostasis, and in turn, tissue homeostasis, mitochondrial dysfunction provides a potential therapeutic avenue for multiple chronic diseases, including IBD. Future clinical trials testing new mitochondrial-targeted treatments that may emerge from this would ideally combine clinical outcome data, genetic mutation status, and analyses of intestinal molecular biology to better understand IBD subtypes that will best respond to this type of therapy and to further address the heterogeneous and complex nature of IBD.
Supplementary Material
MITOCHONDRIAL DYSFUNCTION IN THE GUT–BRAIN AXIS.
Much of our understanding of mitochondrial dysfunction as an integral player in disease originates from studies in Parkinson’s disease that began over 40 years ago (145). This includes the discovery of PARKIN and the causative role of impaired mitophagy in autosomal recessive Parkinson’s disease, as demonstrated by mutations in genes such as PARKIN and PINK1 (146, 147). These seminal discoveries have shaped research in many other chronic inflammatory diseases to investigate the involvement of mitochondrial health. Interestingly, IBD patients are at a higher risk of developing Parkinson’s disease later in life. Recent studies suggest involvement of the gut–brain axis with the potential of shared disease mechanisms underlying these disorders, including mitochondrial dysfunction (148).
SUMMARY POINTS.
Mitochondria are dynamic organelles that play a crucial role in cell homeostasis.
The intestinal epithelium is comprised of a single layer of various cell types, including enterocytes Paneth, goblet, enteroendocrine, tuft, and microfold cells that exhibit distinct functions and mitochondrial metabolisms.
Mitochondrial respiration and metabolic transition are important drivers of leucine-rich-repeat-containing G protein–coupled receptor 5 (Lgr5+) crypt base columnar stem cell fate.
Mitochondrial ROS and the release of mitochondrial damage-associated molecular patterns contribute to ulcerative colitis (UC) inflammation.
Paneth cells are highly susceptible to mitochondrial dysfunction related to Crohn’s disease.
The health of the mitochondrial pool is maintained by various quality control mechanisms, of which mitophagy has been shown to protect against colitis.
Treatment of underlying mitochondrial dysfunction provides a novel therapeutic strategy for inflammatory bowel disease (IBD).
FUTURE ISSUES.
Are mitochondrial alterations distinct across particular subtypes of IBD patients?
Does mitochondrial dysfunction in IBD remain stable over the course of disease, including during cycles of flare-ups/remission and increased duration of disease?
What proportion of pathology in deficient autophagy models is driven by deficient mitophagy?
What will be the effect of the mitochondrial antioxidant, MitoQ, in the new phase 2b clinical trial MARVEL in adults and children with UC?
ACKNOWLEDGMENTS
This work was supported by National Institutes of Health grant R01-DK117001 and Litwin IBD Pioneers-391869 (to A.L.T.) and The Leona M. and Harry B. Helmsley Charitable Trust grant G-1911-03343, Jon Moulton Charitable Foundation grant R46040, and Crohn’s and Colitis UK grant M2018-2 (to G.T.H.).
Footnotes
DISCLOSURE STATEMENT
The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
LITERATURE CITED
- 1.Ni HM, Williams JA, Ding WX. 2015. Mitochondrial dynamics and mitochondrial quality control. Redox Biol. 4:6–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Frey TG, Mannella CA. 2000. The internal structure of mitochondria. Trends Biochem. Sci. 25:319–24 [DOI] [PubMed] [Google Scholar]
- 3.Ryan MT, Hoogenraad NJ. 2007. Mitochondrial-nuclear communications. Annu. Rev. Biochem. 76:701–22 [DOI] [PubMed] [Google Scholar]
- 4.Kuhlbrandt W 2015. Structure and function of mitochondrial membrane protein complexes. BMC Biol. 13:89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Minocherhomji S, Tollefsbol TO, Singh KK. 2012. Mitochondrial regulation of epigenetics and its role in human diseases. Epigenetics 7:326–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shen J, Zhang JH, Xiao H, Wu JM, He KM, et al. 2018. Mitochondria are transported along microtubules in membrane nanotubes to rescue distressed cardiomyocytes from apoptosis. Cell Death Dis. 9:81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Turrens JF. 2003. Mitochondrial formation of reactive oxygen species. J. Physiol. 552:335–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Phung CD, Ezieme JA, Turrens JF. 1994. Hydrogen peroxide metabolism in skeletal muscle mitochondria. Arch. Biochem. Biophys. 315:479–82 [DOI] [PubMed] [Google Scholar]
- 9.D’Autreaux B, Toledano MB. 2007. ROS as signalling molecules: mechanisms that generate specificity in ROS homeostasis. Nat. Rev. Mol. Cell Biol. 8:813–24 [DOI] [PubMed] [Google Scholar]
- 10.Rath E, Moschetta A, Haller D. 2018. Mitochondrial function—gatekeeper of intestinal epithelial cell homeostasis. Nat. Rev. Gastroenterol. Hepatol. 15:497–516 [DOI] [PubMed] [Google Scholar]
- 11.Szymański J, Janikiewicz J, Michalska B, Patalas-Krawczyk P, Perrone M, et al. 2017. Interaction of mitochondria with the endoplasmic reticulum and plasma membrane in calcium homeostasis, lipid trafficking and mitochondrial structure. Int. J. Mol. Sci. 18:1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chandel NS. 2015. Evolution of mitochondria as signaling organelles. Cell Metab. 22:204–6 [DOI] [PubMed] [Google Scholar]
- 13.Sugiura A, McLelland GL, Fon EA, McBride HM. 2014. A new pathway for mitochondrial quality control: mitochondrial-derived vesicles. EMBO J. 33:2142–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Youle RJ, van der Bliek AM. 2012. Mitochondrial fission, fusion, and stress. Science 337:1062–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.El-Hattab AW, Suleiman J, Almannai M, Scaglia F. 2018. Mitochondrial dynamics: biological roles, molecular machinery, and related diseases. Mol. Genet. Metab. 125:315–21 [DOI] [PubMed] [Google Scholar]
- 16.Li M, Jia J, Zhang X, Dai H. 2020. Selective binding of mitophagy receptor protein Bcl-rambo to LC3/GABARAP family proteins. Biochem. Biophys. Res. Commun. 530:292–300 [DOI] [PubMed] [Google Scholar]
- 17.Lv M, Wang C, Li F, Peng J, Wen B, et al. 2017. Structural insights into the recognition of phosphorylated FUNDC1 by LC3B in mitophagy. Protein Cell 8:25–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhu Y, Massen S, Terenzio M, Lang V, Chen-Lindner S, et al. 2013. Modulation of serines 17 and 24 in the LC3-interacting region of Bnip3 determines pro-survival mitophagy versus apoptosis. J. Biol. Chem. 288:1099–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Barker N, van Oudenaarden A, Clevers H. 2012. Identifying the stem cell of the intestinal crypt: strategies and pitfalls. Cell Stem Cell 11:452–60 [DOI] [PubMed] [Google Scholar]
- 20.Clevers HC, Bevins CL. 2013. Paneth cells: maestros of the small intestinal crypts. Annu. Rev. Physiol. 75:289–311 [DOI] [PubMed] [Google Scholar]
- 21.Horvay K, Abud HE. 2013. Regulation of intestinal stem cells by Wnt and Notch signalling. Adv. Exp. Med. Biol. 786:175–86 [DOI] [PubMed] [Google Scholar]
- 22.VanDussen KL, Carulli AJ, Keeley TM, Patel SR, Puthoff BJ, et al. 2012. Notch signaling modulates proliferation and differentiation of intestinal crypt base columnar stem cells. Development 139:488–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Greaves LC, Preston SL, Tadrous PJ, Taylor RW, Barron MJ, et al. 2006. Mitochondrial DNA mutations are established in human colonic stem cells, and mutated clones expand by crypt fission. PNAS 103:714–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nooteboom M, Johnson R, Taylor RW, Wright NA, Lightowlers RN, et al. 2010. Age-associated mitochondrial DNA mutations lead to small but significant changes in cell proliferation and apoptosis in human colonic crypts. Aging Cell 9:96–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schneider C, O’Leary CE, Locksley RM. 2019. Regulation of immune responses by tuft cells. Nat. Rev. Immunol. 19:584–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dillon A, Lo DD. 2019. M cells: intelligent engineering of mucosal immune surveillance. Front. Immunol. 10:1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ludikhuize MC, Meerlo M, Gallego MP, Xanthakis D, Burgaya Julia M, et al. 2020. Mitochondria define intestinal stem cell differentiation downstream of a FOXO/Notch axis. Cell Metab. 32:889–900.e7 [DOI] [PubMed] [Google Scholar]
- 28.Rodriguez-Colman MJ, Schewe M, Meerlo M, Stigter E, Gerrits J, et al. 2017. Interplay between metabolic identities in the intestinal crypt supports stem cell function. Nature 543:424–27 [DOI] [PubMed] [Google Scholar]
- 29.Ludikhuize MC, Rodríguez Colman MJ. 2020. Metabolic regulation of stem cells and differentiation: a Forkhead box O transcription factor perspective. Antioxid. Redox Signal. 34:1004–24 [DOI] [PubMed] [Google Scholar]
- 30.Berger E, Rath E, Yuan D, Waldschmitt N, Khaloian S, et al. 2016. Mitochondrial function controls intestinal epithelial stemness and proliferation. Nat. Commun. 7:13171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liang SJ, Li XG, Wang XQ. 2019. Notch signaling in mammalian intestinal stem cells: determining cell fate and maintaining homeostasis. Curr. Stem Cell. Res. Ther. 14:583–90 [DOI] [PubMed] [Google Scholar]
- 32.Stringari C, Edwards RA, Pate KT, Waterman ML, Donovan PJ, Gratton E. 2012. Metabolic trajectory of cellular differentiation in small intestine by Phasor Fluorescence Lifetime Microscopy of NADH. Sci. Rep. 2:568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang F, Pirooznia M, Xu H. 2020. Mitochondria regulate intestinal stem cell proliferation and epithelial homeostasis through FOXO. Mol. Biol. Cell 31:1538–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hochmuth CE, Biteau B, Bohmann D, Jasper H. 2011. Redox regulation by Keap1 and Nrf2 controls intestinal stem cell proliferation in Drosophila. Cell Stem Cell 8:188–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Banerjee A, Herring CA, Chen B, Kim H, Simmons AJ, et al. 2020. Succinate produced by intestinal microbes promotes specification of tuft cells to suppress ileal inflammation. Gastroenterology 159:2101–15.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Herring CA, Banerjee A, McKinley ET, Simmons AJ, Ping J, et al. 2018. Unsupervised trajectory analysis of single-cell RNA-seq and imaging data reveals alternative tuft cell origins in the gut. Cell Syst. 6:37–51.e9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Beyaz S, Mana MD, Roper J, Kedrin D, Saadatpour A, et al. 2016. High-fat diet enhances stemness and tumorigenicity of intestinal progenitors. Nature 531:53–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mihaylova MM, Cheng CW, Cao AQ, Tripathi S, Mana MD, et al. 2018. Fasting activates fatty acid oxidation to enhance intestinal stem cell function during homeostasis and aging. Cell Stem Cell 22:769–78.e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stine RR, Sakers AP, TeSlaa T, Kissig M, Stine ZE, et al. 2019. PRDM16 maintains homeostasis of the intestinal epithelium by controlling region-specific metabolism. Cell Stem Cell 25:830–45.e8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen L, Vasoya RP, Toke NH, Parthasarathy A, Luo S, et al. 2020. HNF4 regulates fatty acid oxidation and is required for renewal of intestinal stem cells in mice. Gastroenterology 158:985–99.e9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.D’Errico I, Salvatore L, Murzilli S, Lo Sasso G, Latorre D, et al. 2011. Peroxisome proliferator-activated receptor-γ coactivator 1-α (PGC1α) is a metabolic regulator of intestinal epithelial cell fate. PNAS 108:6603–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Windsor JW, Kaplan GG. 2019. Evolving epidemiology of IBD. Curr. Gastroenterol. Rep. 21:40. [DOI] [PubMed] [Google Scholar]
- 43.Gerich ME, McGovern DP. 2014. Towards personalized care in IBD. Nat. Rev. Gastroenterol. Hepatol. 11:287–99 [DOI] [PubMed] [Google Scholar]
- 44.Colombel JF, Panaccione R, Bossuyt P, Lukas M, Baert F, et al. 2018. Effect of tight control management on Crohn’s disease (CALM): a multicentre, randomised, controlled phase 3 trial. Lancet 390:2779–89 [DOI] [PubMed] [Google Scholar]
- 45.Uhlig HH, Schwerd T, Koletzko S, Shah N, Kammermeier J, et al. 2014. The diagnostic approach to monogenic very early onset inflammatory bowel disease. Gastroenterology 147:990–1007.e3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mirkov MU, Verstockt B, Cleynen I. 2017. Genetics of inflammatory bowel disease: beyond NOD2. Lancet Gastroenterol. Hepatol. 2:224–34 [DOI] [PubMed] [Google Scholar]
- 47.Furey TS, Sethupathy P, Sheikh SZ. 2019. Redefining the IBDs using genome-scale molecular phenotyping. Nat. Rev. Gastroenterol. Hepatol. 16:296–311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jostins L, Ripke S, Weersma RK, Duerr RH, McGovern DP, et al. 2012. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 491:119–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vedamurthy A, Ananthakrishnan AN. 2019. Influence of environmental factors in the development and outcomes of inflammatory bowel disease. Gastroenterol. Hepatol. 15:72–82 [PMC free article] [PubMed] [Google Scholar]
- 50.Ni J, Wu GD, Albenberg L, Tomov VT. 2017. Gut microbiota and IBD: Causation or correlation? Nat. Rev. Gastroenterol. Hepatol. 14:573–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pagliarini DJ, Calvo SE, Chang B, Sheth SA, Vafai SB, et al. 2008. A mitochondrial protein compendium elucidates complex I disease biology. Cell 134:112–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Donohoe DR, Garge N, Zhang X, Sun W, O’Connell TM, et al. 2011. The microbiome and butyrate regulate energy metabolism and autophagy in the mammalian colon. Cell Metab. 13:517–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mancini NL, Rajeev S, Jayme TS, Wang A, Keita AV, et al. 2021. Crohn’s disease pathobiont adherent-invasive E coli disrupts epithelial mitochondrial networks with implications for gut permeability. Cell. Mol. Gastroenterol. Hepatol. 11:551–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ho GT, Aird RE, Liu B, Boyapati RK, Kennedy NA, et al. 2017. MDR1 deficiency impairs mitochondrial homeostasis and promotes intestinal inflammation. Mucosal Immunol. 11:120–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liu B, Gulati AS, Cantillana V, Henry SC, Schmidt EA, et al. 2013. Irgm1-deficient mice exhibit Paneth cell abnormalities and increased susceptibility to acute intestinal inflammation. Am. J. Physiol. Gastrointest. Liver Physiol. 305:G573–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lawrence M, Huber W, Pages H, Aboyoun P, Carlson M, et al. 2013. Software for computing and annotating genomic ranges. PLOS Comput. Biol. 9:e1003118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhong B, Zhang L, Lei C, Li Y, Mao AP, et al. 2009. The ubiquitin ligase RNF5 regulates antiviral responses by mediating degradation of the adaptor protein MITA. Immunity 30:397–407 [DOI] [PubMed] [Google Scholar]
- 58.Rath E, Haller D. 2012. Mitochondria at the interface between danger signaling and metabolism: role of unfolded protein responses in chronic inflammation. Inflamm. Bowel Dis. 18:1364–77 [DOI] [PubMed] [Google Scholar]
- 59.Anderson CA, Boucher G, Lees CW, Franke A, D’Amato M, et al. 2011. Meta-analysis identifies 29 additional ulcerative colitis risk loci, increasing the number of confirmed associations to 47. Nat. Genet. 43:246–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Franke A, McGovern DP, Barrett JC, Wang K, Radford-Smith GL, et al. 2010. Genome-wide meta-analysis increases to 71 the number of confirmed Crohn’s disease susceptibility loci. Nat. Gen. 42:1118–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cader MZ, Boroviak K, Zhang Q, Assadi G, Kempster SL, et al. 2016. C13orf31 (FAMIN) is a central regulator of immunometabolic function. Nat. Immunol. 17:1046–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jackson DN, Panopoulos M, Neumann WL, Turner K, Cantarel BL, et al. 2020. Mitochondrial dysfunction during loss of prohibitin 1 triggers Paneth cell defects and ileitis. Gut 69:1928–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sunderhauf A, Hicken M, Schlichting H, Skibbe K, Ragab M, et al. 2021. Loss of mucosal p32/gC1qR/HABP1 triggers energy deficiency and impairs goblet cell differentiation in ulcerative colitis. Cell. Mol. Gastroenterol. Hepatol. 12:229–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bar F, Bochmann W, Widok A, von Medem K, Pagel R, et al. 2013. Mitochondrial gene polymorphisms that protect mice from colitis. Gastroenterology 145:1055–63.e3 [DOI] [PubMed] [Google Scholar]
- 65.Mills EL, Kelly B, Logan A, Costa ASH, Varma M, et al. 2016. Succinate dehydrogenase supports metabolic repurposing of mitochondria to drive inflammatory macrophages. Cell 167:457–70.e13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Faas MM, de Vos P. 2020. Mitochondrial function in immune cells in health and disease. Biochim. Biophys. Acta. Mol. Basis Dis. 1866:165845. [DOI] [PubMed] [Google Scholar]
- 67.Boyapati RK, Dorward DA, Tamborska A, Kalla R, Ventham NT, et al. 2018. Mitochondrial DNA is a pro-inflammatory damage-associated molecular pattern released during active IBD. Inflamm. Bowel Dis. 24:2113–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.West AP, Shadel GS. 2017. Mitochondrial DNA in innate immune responses and inflammatory pathology. Nat. Rev. Immunol. 17:363–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, et al. 2010. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 464:104–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Valle DL, Antonarakis S, Ballabio A, Beaudet AL, Mitchell GA. 2019. The Online Metabolic and Molecular Bases of Inherited Disease. New York: McGraw-Hill Education [Google Scholar]
- 71.Sekino Y, Inamori M, Yamada E, Ohkubo H, Sakai E, et al. 2012. Characteristics of intestinal pseudo-obstruction in patients with mitochondrial diseases. World J. Gastroenterol. 18:4557–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Roediger WEW. 1980. The colonic epithelium in ulcerative colitis: An energy-deficiency disease? Lancet 2:712–15 [DOI] [PubMed] [Google Scholar]
- 73.Haberman Y, Karns R, Dexheimer PJ, Schirmer M, Somekh J, et al. 2019. Ulcerative colitis mucosal transcriptomes reveal mitochondriopathy and personalized mechanisms underlying disease severity and treatment response. Nat. Commun. 10:38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Noble CL, Abbas AR, Cornelius J, Lees CW, Ho GT, et al. 2008. Regional variation in gene expression in the healthy colon is dysregulated in ulcerative colitis. Gut 57:1398–405 [DOI] [PubMed] [Google Scholar]
- 75.O’Morain C, Smethurst P, Levi J, Peters TJ. 1985. Subcellular fractionation of rectal biopsy homogenates from patients with inflammatory bowel disease. Scand. J. Gastroenterol. 20:209–14 [DOI] [PubMed] [Google Scholar]
- 76.Hsieh SY, Shih TC, Yeh CY, Lin CJ, Chou YY, Lee YS. 2006. Comparative proteomic studies on the pathogenesis of human ulcerative colitis. Proteomics 6:5322–31 [DOI] [PubMed] [Google Scholar]
- 77.Delpre G, Avidor I, Steinherz R, Kadish U, Ben-Bassat M. 1989. Ultrastructural abnormalities in endoscopically and histologically normal and involved colon in ulcerative colitis. Am. J. Gastroenterol. 84:1038–46 [PubMed] [Google Scholar]
- 78.Sifroni KG, Damiani CR, Stoffel C, Cardoso MR, Ferreira GK, et al. 2010. Mitochondrial respiratory chain in the colonic mucosal of patients with ulcerative colitis. Mol. Cell. Biochem. 342:111–15 [DOI] [PubMed] [Google Scholar]
- 79.Santhanam S, Rajamanickam S, Motamarry A, Ramakrishna BS, Amirtharaj JG, et al. 2012. Mitochondrial electron transport chain complex dysfunction in the colonic mucosa in ulcerative colitis. Inflamm. Bowel Dis. 18:2158–68 [DOI] [PubMed] [Google Scholar]
- 80.Santhanam S, Venkatraman A, Ramakrishna BS. 2007. Impairment of mitochondrial acetoacetyl CoA thiolase activity in the colonic mucosa of patients with ulcerative colitis. Gut 56:1543–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Parada Venegas D, De la Fuente MK, Landskron G, Gonzalez MJ, Quera R, et al. 2019. Short chain fatty acids (SCFAs)-mediated gut epithelial and immune regulation and its relevance for inflammatory bowel diseases. Front. Immunol. 10:277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Barcenilla A, Pryde SE, Martin JC, Duncan SH, Stewart CS, et al. 2000. Phylogenetic relationships of butyrate-producing bacteria from the human gut. Appl. Environ. Microbiol. 66:1654–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Pryde SE, Duncan SH, Hold GL, Stewart CS, Flint HJ. 2002. The microbiology of butyrate formation in the human colon. FEMS Microbiol. Lett. 217:133–39 [DOI] [PubMed] [Google Scholar]
- 84.Kameyama J, Narui H, Inui M, Sato T. 1984. Energy level in large intestinal mucosa in patients with ulcerative colitis. Tohoku J. Exp. Med. 143:253–54 [DOI] [PubMed] [Google Scholar]
- 85.Rosa A, Abrantes P, Sousa I, Francisco V, Santos P, et al. 2016. Ulcerative colitis is under dual (mitochondrial and nuclear) genetic control. Inflamm. Bowel Dis. 22:774–81 [DOI] [PubMed] [Google Scholar]
- 86.Dankowski T, Schroder T, Moller S, Yu X, Ellinghaus D, et al. 2016. Male-specific association between MT-ND4 11719 A/G polymorphism and ulcerative colitis: a mitochondria-wide genetic association study. BMC Gastroenterol. 16:118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Szabo C 2007. Hydrogen sulphide and its therapeutic potential. Nat. Rev. Drug Dis. 6:917–35 [DOI] [PubMed] [Google Scholar]
- 88.Orrenius S, Gogvadze V, Zhivotovsky B. 2007. Mitochondrial oxidative stress: implications for cell death. Annu. Rev. Pharmacol. Toxicol. 47:143–83 [DOI] [PubMed] [Google Scholar]
- 89.West AP, Brodsky IE, Rahner C, Woo DK, Erdjument-Bromage H, et al. 2011. TLR signalling augments macrophage bactericidal activity through mitochondrial ROS. Nature 472:476–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Van Houten B, Woshner V, Santos JH. 2006. Role of mitochondrial DNA in toxic responses to oxidative stress. DNA Repair 5:145–52 [DOI] [PubMed] [Google Scholar]
- 91.Shimada K, Crother TR, Karlin J, Dagvadorj J, Chiba N, et al. 2012. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 36:401–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Nakahira K, Haspel JA, Rathinam VA, Lee SJ, Dolinay T, et al. 2011. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 12:222–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Formentini L, Santacatterina F, Nunez de Arenas C, Stamatakis K, Lopez-Martinez D, et al. 2017. Mitochondrial ROS production protects the intestine from inflammation through functional M2 macrophage polarization. Cell Rep. 19:1202–13 [DOI] [PubMed] [Google Scholar]
- 94.Campbell EL, Colgan SP. 2019. Control and dysregulation of redox signalling in the gastrointestinal tract. Nat. Rev. Gastroenterol. Hepatol. 16:106–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Camarillo GF, Goyon EI, Zuniga RB, Salas LAS, Escarcega AEP, Yamamoto-Furusho JK. 2020. Gene expression profiling of mediators associated with the inflammatory pathways in the intestinal tissue from patients with ulcerative colitis. Mediat. Inflamm. 2020:9238970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ikumoto T, Hayashi S, Tomita S, Miwa S, Mitomi H, et al. 2014. Manganese superoxide dismutase plays an important role in the inflammatory process and predicts disease severity and activity in patients with ulcerative colitis. APMIS 122:512–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.James AM, Smith RA, Murphy MP. 2004. Antioxidant and prooxidant properties of mitochondrial Coenzyme Q. Arch. Biochem. Biophys. 423:47–56 [DOI] [PubMed] [Google Scholar]
- 98.Zhang H, Kuai XY, Yu P, Lin L, Shi R. 2012. Protective role of uncoupling protein-2 against dextran sodium sulfate-induced colitis. J. Gastroenterol. Hepatol. 27:603–8 [DOI] [PubMed] [Google Scholar]
- 99.Shen L, Zhi L, Hu W, Wu MX. 2009. IEX-1 targets mitochondrial F1Fo-ATPase inhibitor for degradation. Cell Death Differ. 16:603–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Yu X, Wieczorek S, Franke A, Yin H, Pierer M, et al. 2009. Association of UCP2 −866 G/A polymorphism with chronic inflammatory diseases. Genes. Immun. 10:601–5 [DOI] [PubMed] [Google Scholar]
- 101.Boyapati RK, Rossi AG, Satsangi J, Ho GT. 2016. Gut mucosal DAMPs in IBD: from mechanisms to therapeutic implications. Mucosal Immunol. 9:567–82 [DOI] [PubMed] [Google Scholar]
- 102.Oka T, Hikoso S, Yamaguchi O, Taneike M, Takeda T, et al. 2012. Mitochondrial DNA that escapes from autophagy causes inflammation and heart failure. Nature 485:251–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Lood C, Blanco LP, Purmalek MM, Carmona-Rivera C, De Ravin SS, et al. 2016. Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nat. Med. 22:146–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Drury B, Hardisty G, Gray RD, Ho GT. 2021. Neutrophil extracellular traps in inflammatory bowel disease: pathogenic mechanisms and clinical translation. Cell. Mol. Gastroenterol. Hepatol. 12:321–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Peltekova VD, Wintle RF, Rubin LA, Amos CI, Huang Q, et al. 2004. Functional variants of OCTN cation transporter genes are associated with Crohn disease. Nat. Genet. 36:471–75 [DOI] [PubMed] [Google Scholar]
- 106.Parkes M, Barrett JC, Prescott NJ, Tremelling M, Anderson CA, et al. 2007. Sequence variants in the autophagy gene IRGM and multiple other replicating loci contribute to Crohn’s disease susceptibility. Nat. Genet. 39:830–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Kugathasan S, Denson LA, Walters TD, Kim MO, Marigorta UM, et al. 2017. Prediction of complicated disease course for children newly diagnosed with Crohn’s disease: a multicentre inception cohort study. Lancet 389:1710–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Mottawea W, Chiang CK, Muhlbauer M, Starr AE, Butcher J, et al. 2016. Altered intestinal microbiotahost mitochondria crosstalk in new onset Crohn’s disease. Nat. Commun. 7:13419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Söderholm JD, Olaison G, Peterson KH, Franzén LE, Lindmark T, et al. 2002. Augmented increase in tight junction permeability by luminal stimuli in the non-inflamed ileum of Crohn’s disease. Gut 50:307–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Khaloian S, Rath E, Hammoudi N, Gleisinger E, Blutke A, et al. 2020. Mitochondrial impairment drives intestinal stem cell transition into dysfunctional Paneth cells predicting Crohn’s disease recurrence. Gut 69:1939–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Roy S, Esmaeilniakooshkghazi A, Patnaik S, Wang Y, George SP, et al. 2018. Villin-1 and gelsolin regulate changes in actin dynamics that affect cell survival signaling pathways and intestinal inflammation. Gastroenterology 154:1405–20.e2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Vidrich A, Buzan JM, Barnes S, Reuter BK, Skaar K, et al. 2005. Altered epithelial cell lineage allocation and global expansion of the crypt epithelial stem cell population are associated with ileitis in SAMP1/YitFc mice. Am. J. Pathol. 166:1055–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Adolph TE, Tomczak MF, Niederreiter L, Ko HJ, Bock J, et al. 2013. Paneth cells as a site of origin for intestinal inflammation. Nature 503:272–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Kerr WG, Park MY, Maubert M, Engelman RW. 2011. SHIP deficiency causes Crohn’s disease-like ileitis. Gut 60:177–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Merkwirth C, Dargazanli S, Tatsuta T, Geimer S, Lower B, et al. 2008. Prohibitins control cell proliferation and apoptosis by regulating OPA1-dependent cristae morphogenesis in mitochondria. Genes Dev. 22:476–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Nijtmans LG, de Jong L, Artal Sanz M, Coates PJ, Berden JA, et al. 2000. Prohibitins act as a membrane-bound chaperone for the stabilization of mitochondrial proteins. EMBO J. 19:2444–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Theiss AL, Idell RD, Srinivasan S, Klapproth JM, Jones DP, et al. 2007. Prohibitin protects against oxidative stress in intestinal epithelial cells. FASEB J. 21:197–206 [DOI] [PubMed] [Google Scholar]
- 118.Gunther C, Martini E, Wittkopf N, Amann K, Weigmann B, et al. 2011. Caspase-8 regulates TNF-α-induced epithelial necroptosis and terminal ileitis. Nature 477:335–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Yang E, Shen J. 2020. The roles and functions of Paneth cells in Crohn’s disease: a critical review. Cell Prolif. 54:e12958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Liu TC, Gao F, McGovern DP, Stappenbeck TS. 2014. Spatial and temporal stability of Paneth cell phenotypes in Crohn’s disease: implications for prognostic cellular biomarker development. Inflamm. Bowel Dis. 20:646–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Liu TC, Gurram B, Baldridge MT, Head R, Lam V, et al. 2016. Paneth cell defects in Crohn’s disease patients promote dysbiosis. JCI Insight 1:e86907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Liu TC, Kern JT, VanDussen KL, Xiong S, Kaiko GE, et al. 2018. Interaction between smoking and ATG16L1T300A triggers Paneth cell defects in Crohn’s disease. J. Clin. Investig. 128:5110–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Liu TC, Naito T, Liu Z, VanDussen KL, Haritunians T, et al. 2017. LRRK2 but not ATG16L1 is associated with Paneth cell defect in Japanese Crohn’s disease patients. JCI Insight 2:e91917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.VanDussen KL, Liu TC, Li D, Towfic F, Modiano N, et al. 2014. Genetic variants synthesize to produce Paneth cell phenotypes that define subtypes of Crohn’s disease. Gastroenterology 146:200–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Cadwell K, Patel KK, Maloney NS, Liu TC, Ng AC, et al. 2010. Virus-plus-susceptibility gene interaction determines Crohn’s disease gene Atg16L1 phenotypes in intestine. Cell 141:1135–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Rogala AR, Schoenborn AA, Fee BE, Cantillana VA, Joyce MJ, et al. 2018. Environmental factors regulate Paneth cell phenotype and host susceptibility to intestinal inflammation in Irgm1-deficient mice. Dis. Model Mech. 11:dmm031070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Nguyen HT, Lapaquette P, Bringer MA, Darfeuille-Michaud A. 2013. Autophagy and Crohn’s disease. J. Innate Immun. 5:434–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Kim S, Eun HS, Jo EK. 2019. Roles of autophagy-related genes in the pathogenesis of inflammatory bowel disease. Cells 8:77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Lu R, Zhang Y, Xia Y, Zhang J, Kaser A, et al. 2021. Paneth cell alertness to pathogens maintained by vitamin D receptors. Gastroenterology 160:1269–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Cadwell K, Patel KK, Komatsu M, Virgin HW IV, Stappenbeck TS. 2009. A common role for Atg16L1, Atg5 and Atg7 in small intestinal Paneth cells and Crohn disease. Autophagy 5:250–2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Vincent G, Novak EA, Siow VS, Cunningham KE, Griffith BD, et al. 2020. Nix-mediated mitophagy modulates mitochondrial damage during intestinal inflammation. Antioxid. Redox Signal. 33:1–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Natl. Inst. Health. 2020. MARVEL: Mitochondrial Anti-oxidant Therapy to Resolve Inflammation in Ulcerative Colitis (MARVEL). Natl. Inst. Health, Washington, DC. https://clinicaltrials.gov/ct2/show/NCT04276740 [Google Scholar]
- 133.Gane EJ, Weilert F, Orr DW, Keogh GF, Gibson M, et al. 2010. The mitochondria-targeted anti-oxidant mitoquinone decreases liver damage in a phase II study of hepatitis C patients. Liver Int. 30:1019–26 [DOI] [PubMed] [Google Scholar]
- 134.Snow BJ, Rolfe FL, Lockhart MM, Frampton CM, O’Sullivan JD, et al. 2010. A double-blind, placebo-controlled study to assess the mitochondria-targeted antioxidant MitoQ as a disease-modifying therapy in Parkinson’s disease. Mov. Disord. 25:1670–74 [DOI] [PubMed] [Google Scholar]
- 135.Nicoletti V, Palermo G, Del Prete E, Mancuso M, Ceravolo R. 2021. Understanding the multiple role of mitochondria in Parkinson’s disease and related disorders: lesson from genetics and protein-interaction network. Front. Cell Dev. Biol. 9:636506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Okuda M, Li K, Beard MR, Showalter LA, Scholle F, et al. 2002. Mitochondrial injury, oxidative stress, and antioxidant gene expression are induced by hepatitis C virus core protein. Gastroenterology 122:366–75 [DOI] [PubMed] [Google Scholar]
- 137.Hofer A, Noe N, Tischner C, Kladt N, Lellek V, et al. 2014. Defining the action spectrum of potential PGC-1α activators on a mitochondrial and cellular level in vivo. Hum. Mol. Genet. 23:2400–15 [DOI] [PubMed] [Google Scholar]
- 138.Mancini NL, Goudie L, Xu W, Sabouny R, Rajeev S, et al. 2020. Perturbed mitochondrial dynamics is a novel feature of colitis that can be targeted to lessen disease. Cell. Mol. Gastroenterol. Hepatol. 10:287–307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Moskal N, Riccio V, Bashkurov M, Taddese R, Datti A, et al. 2020. ROCK inhibitors upregulate the neuroprotective Parkin-mediated mitophagy pathway. Nat. Commun. 11:88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Tolosa E, Vila M, Klein C, Rascol O. 2020. LRRK2 in Parkinson disease: challenges of clinical trials. Nat. Rev. Neurol. 16:97–107 [DOI] [PubMed] [Google Scholar]
- 141.Repici M, Giorgini F. 2019. DJ-1 in Parkinson’s disease: clinical insights and therapeutic perspectives. J. Clin. Med. 8:1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Bellot G, Garcia-Medina R, Gounon P, Chiche J, Roux D, et al. 2009. Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol. Cell. Biol. 29:2570–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Chaanine AH, Kohlbrenner E, Gamb SI, Guenzel AJ, Klaus K, et al. 2016. FOXO3a regulates BNIP3 and modulates mitochondrial calcium, dynamics, and function in cardiac stress. Am. J. Physiol. Heart Circ. Physiol. 311:H1540–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Rogov VV, Suzuki H, Marinkovic M, Lang V, Kato R, et al. 2017. Phosphorylation of the mitochondrial autophagy receptor Nix enhances its interaction with LC3 proteins. Sci. Rep. 7:1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Langston JW, Ballard P, Tetrud JW, Irwin I. 1983. Chronic Parkinsonism in humans due to a product of meperidine-analog synthesis. Science 219:979–80 [DOI] [PubMed] [Google Scholar]
- 146.Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, et al. 1998. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392:605–8 [DOI] [PubMed] [Google Scholar]
- 147.Valente EM, Abou-Sleiman PM, Caputo V, Muqit MM, Harvey K, et al. 2004. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science 304:1158–60 [DOI] [PubMed] [Google Scholar]
- 148.Lee HS, Lobbestael E, Vermeire S, Sabino J, Cleynen I. 2021. Inflammatory bowel disease and Parkinson’s disease: common pathophysiological links. Gut 70:408–17 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.