

Abstract
S-Adenosylmethionine (SAM) and S-adenosylhomocysteine (SAH) are important metabolites in the one-carbon cycle that modulates cellular methylation required for proliferation and epigenetic regulation. Their concentrations, synthesis, and turnover are difficult to determine conveniently and reliably. We have developed such a method by coupling a simple and rapid purification scheme that efficiently captures both compounds, with high sensitivity, sample throughput direct infusion nanoelectrospray ultra-high-resolution Fourier transform mass spectrometry (DI-nESI-UHR-FTMS). This method is compatible with Stable Isotope-Resolved Metabolomic (SIRM) analysis of numerous other metabolites. The limits of detection for both SAM and SAH were < 1 nM, and the linearity range was up to 1000 nM. The method was first illustrated for SAM/SAH analysis of mouse livers, and lung adenocarcinoma A549 cells. We then applied the method to track 13C1-CH3-Met incorporation into SAM and 13C6-glucose transformation into SAM and SAH via de novo synthesis. We further used the method to show the distinct effects on A549 and H1299 cells with treatment of anti-cancer methylseleninic acid (MSA), selenite, and selenomethionine, notably SAM depletion and increased SAM to SAH ratio by MSA, which implicates altered epigenetic regulation.
Keywords: S-Adenosylmethionine, S-adenosylhomocysteine, Stable Isotope-Resolved Metabolomics, DI-nESI-UHR-FTMS, methylseleninic acid, selenite
Graphical Abstract
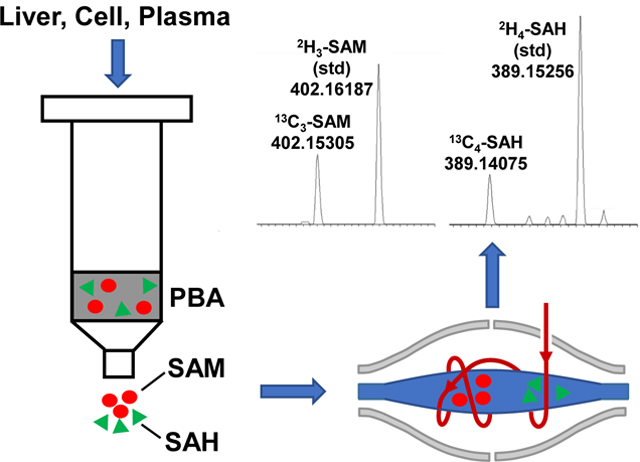
1. Introduction
Methionine is a sulfur-containing essential amino acid participating in the one-carbon metabolism (Figure 1), which continuously interconvert S-adenosylmethionine (SAM) and methionine [1–3]. The cycle begins with the generation of S-adenosylmethionine (SAM) from methionine, utilizing ATP as a co-substrate via methionine adenosyltransferase (MAT) [4–6]. SAM is biologically important in that it is a universal methyl donor in a wide range of methylation reactions and is the second most common cofactor after ATP in various enzymatic reactions [7]. The methyl group on the sulfur atom of SAM can be transferred to various substrates including DNA, RNA, proteins, glycine, polyamines, and phosphatidylethanolamine (PE) through a transmethylation reaction [3, 8]. SAM is thereby irreversibly converted to S-adenosylhomocysteine (SAH). SAH is then hydrolyzed to adenosine plus homocysteine by S-adenosylhomocysteine hydrolase. Homocysteine is either further converted to cysteine via the transsulfuration pathway or is remethylated by methionine synthase using 5-methyl-tetrahydrofolate (5-methyl-THF). This recycling of homocysteine is critical to regenerating methionine and maintaining the transmethylation cycles [9, 10].
Figure 1. Inter-conversion of SAM and SAH in the transmethylation cycle.
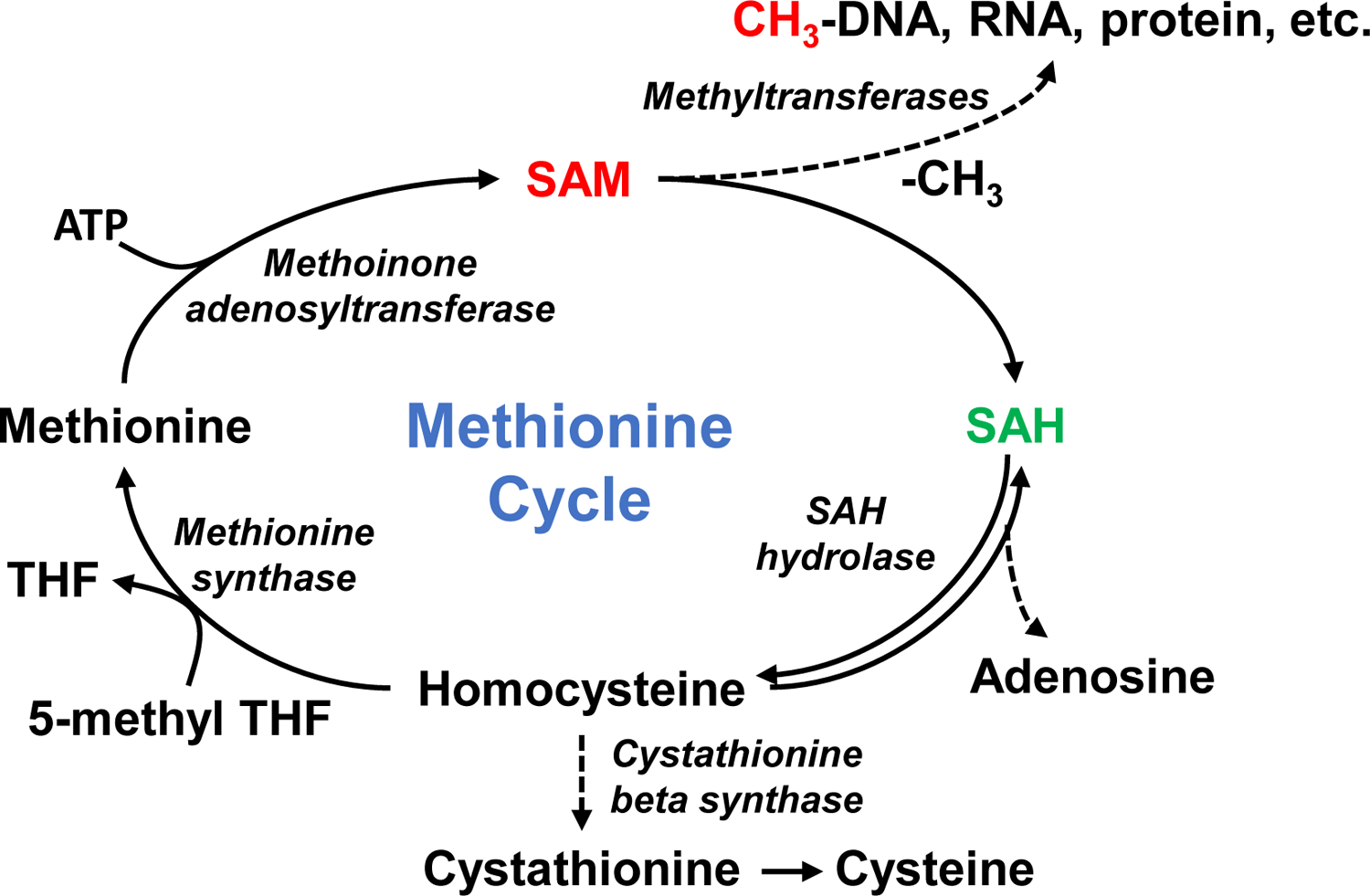
SAM, S-adenosylmethionine; SAH, S-adenosylhomocysteine; THF, tetrahydrofolate; 5-methyl THF, 5-methyltetrahydrofolate.
The ratio of SAM to SAH, which is called “methylation index”, is physiologically important because it controls most biological methylation reactions including epigenetic DNA and histone methylation, which is central to regulating gene expression in both normal and disease states including cancers [11–13]. Increased SAM to SAH ratios have been associated with many diseases such as cardiovascular diseases, renal failure and liver diseases, where protein methylation is altered. The SAM to SAH ratio in human serum has also been reported to be a sensitive indicator of atherosclerosis [10, 11, 14–17]. Therefore, by governing the methylation of various biomolecules, the ratio of SAM to SAH is closely linked to human pathophysiological conditions.
As SAH is typically found at a much lower level than SAM [18], and the ratio changes under pathological conditions, it is therefore important to measure both SAM and SAH accurately. Several methods have been developed for determining this ratio such as the fluorescence polarization immunoassay, colorimetric enzymatic methods [19–21], or liquid chromatography coupled with tandem mass spectrometry (LC-MS/MS) based analysis [22–26]. The former two methods lack selectivity or have poor reproducibility, while the LC-MS/MS method coupled with the use of commercially available stable isotope-enriched standards is preferred due to its high selectivity, sensitivity, and accuracy as well as molecular structural determination, which makes it compatible with SIRM applications. However, the LC-MS/MS method is time-consuming, which makes it less compatible with high throughput applications and data analysis. A fast and robust sample preparation method coupled with direct infusion nanoelectrospray ultra-high-resolution Fourier transform mass spectrometry (DI-nESI-UHR-FTMS) is an attractive alternative method as high-resolution and mass accuracy data are acquired without physical separation for high-speed and robust metabolite analysis [27, 28].
Moreover, the ability of DI-nESI-UHR-FTMS to accurately measure isotopically enriched mass isotopologues makes it particularly suited for stable isotope-resolved metabolomics (SIRM) applications. SIRM is a powerful and rigorous approach for mapping metabolic networks in cells or tissues to uncover metabolic dysregulations induced by disease development or stressor treatments. It has been applied to most biosystems ranging from microbial systems to human subjects [29–31]. As SIRM relies on untargeted MS1 to obtain whole-metabolite isotopic incorporation, it depends on both accurate mass and ultra-high mass resolution to quantify as many metabolite isotopologues as possible. This, in turn, greatly benefits from long (many minutes) data acquisition times afforded by direct infusion than by LC (a few seconds). UHR-FTMS also enables analysis of multiple tracer atoms in the same or different metabolites for multiplexed SIRM applications [22, 32].
There have been a few studies that utilized direct infusion mass spectrometry to measure SAM and SAH [33], but none have been applied to analyzing isotope labeled SAM and SAH resulting from metabolism. The latter applications have a much higher sensitivity requirement due to the distribution of mass signals into various isotopologues. In this study, we describe a simple, rapid, and highly sensitive method for SAM and SAH analysis for cells and tissues by coupling phenylboronic acid (PBA) affinity capture with DI-nESI-UHR-FTMS. The method is fully compatible with comprehensive profiling of metabolites using methods such as ion chromatography-UHR-FTMS. We have applied this method to SAM/SAH analysis of mouse livers and lung adenocarcinoma A549 or H1299 cells. We further used the method to track 13C incorporation into SAM and SAH in A549 cells treated with 13C1-CH3-Methionine or 13C6-glucose, and to probe the distinct effect of anti-cancer selenium treatments in A549 and H1299 cells.
2. Experimental
2.1. Materials and reagents
HPLC grade ammonium acetate, acetonitrile, methanol, formic acid, acetic acid, 28% ammonia solution, and chloroform were purchased from Sigma Aldrich (St. Louis, MO, USA). Bond-Elut SPE phenylboronic acid (PBA) columns were ordered from Agilent Technologies (Santa Clara, CA, USA). Unlabeled SAM and SAH standards were obtained from Fisher Scientific (Pittsburgh, PA, USA). 2H3-SAM was purchased from Medical Isotopes, Inc. (Pelham, NH, USA) and 2H4-SAH was obtained from Cayman Chemical (Ann Arbor, MI, USA).
2.2. Cell culturing, treatment, and extraction
A549 or H1299 human lung adenocarcinoma cells (American Type Culture Collection (Rockville, MD)) were cultured in DMEM (pH 7.4) supplemented with 10% v/v fetal bovine serum, 3.7 g L−1 sodium bicarbonate, 0.2% Glucose, 2 mM Glutamine and 1x Penicillin-Streptomycin (complete DMEM) and maintained at 37 °C in a 5% CO2 humidified atmosphere. Cells were lifted using TripLE (GIBCO, Life technologies, NY) and reseeded at a density of 8.7×105 cells/10 cm plate allowing 60% confluency in 24 hours. For the SIRM experiments, A549 cell media were changed to complete DMEM with glucose replaced by 13C6-glucose (0.2% w/v) or with the addition of 13C1-CH3-methionine (0.2 mM). The cells were also grown in parallel using unlabeled media. For the experiment on treatment with seleno compounds, A549 or H1299 cell media were changed to vehicle (Ctl), 5 μM methylseleninic acid (MSA), 6.25 μM selenite (SeO3), or 0.5 mM selenomethionine (SeM), which are the IC50 concentrations for A549 cells under these conditions ([34, 35] and unpublished data). The MSA medium was renewed after 12 h of treatment due to rapid metabolic degradation. After 24 hours, cells were quenched, polar metabolites extracted as described previously [36]. One eighteenth of the polar extracts lyophilized, dissolved in 0.5 mL 20 mM ammonium acetate (pH 7.4), and processed through PBA columns as described below.
2.3. Mouse liver extraction
Flash-frozen mouse liver tissues were pulverized under liq. N2 and ca. 6–8 mg by wet weight was extracted for polar metabolites, as previously described [37]. One eighth of the polar extracts was lyophilized and dissolved in 0.5 mL 20 mM ammonium acetate for processing through PBA columns as described below.
The procedure to simultaneously quench, lyse, and extract polar metabolites from cells and tissues was as previously reported [38]. Briefly, the metabolism of cells or tissues was quenched and cell lysed with 1 mL of cold CH3CN (stored at −20°C) before 0.75 mL of Nanopure water and 1 mL of cold CHCl3 was added in a final CH3CN:H2O:CHCl3 ratio of 2:1.5:1 (v/v). The polar, non-polar, and insoluble proteinaceous fractions were recovered. The polar fraction was aliquoted, lyophilized, and stored at −80 °C before processing for SAM/SAH analysis. The protein fraction was solubilized in 62.5 mM Tris + 2% SDS + 1 mM dithiothreitol (pH 6.8) for protein determination using the BCA method (Pierce Chemicals).
2.4. Human plasma sample collection and preparation
Blood was collected into a purple top vacutainer containing K2-EDTA from two healthy volunteers (UK094, female and UK168, male) and a patient with (JK016) with intraductal papillary mucinous neoplasms (IPMN) according to approved IRB protocols (IRB #43807 and 48495). Blood samples were placed on ice immediately after collection, centrifuged within 30 min of collection at 3,500×g for 15 min at 4°C as described previously [39]. The plasma was then immediately acidified with 9 μL of 5 M acetic [26] or 4.5 μL formic acid per mL of plasma to stabilize SAM (final pH =4.5–5). Acidified plasmas were frozen in liquid N2 and kept at −80°C. For extracting SAM and SAH, 10 μL of 2H3-SAM (0.115 μM) and 2H4-SAH (0.167 μM) was added to the thawed acidified plasma before neutralization to pH 7–7.5 with 1 M NH4OH and 225–490 μL of which was immediately processed with PBA columns as described below.
2.5. PBA based-solid phase extraction for SAM and SAH
Bond-Elut PBA columns were used to capture SAM and SAH from polar extracts. The column was custom-made to have low sorbent mass (50 mg) in a 1 mL column to reduce the elution volume. Binding and elution were carried out essentially as described previously [24, 26, 40], but with the following modifications. The PBA column was acidified with 0.1 M formic acid (1 mL) before conditioning with 20 mM ammonium acetate (pH 7.4) solution (1 mL, 2 times). Plasma as is or reconstituted polar extract in 0.5 mL 20 mM ammonium acetate was applied to the conditioned column. The column was washed with 1 mL of 20 mM ammonium acetate for cell/tissue extracts or 2×1 mL of ammonium acetate for plasma samples to remove salts. Bound analytes were eluted using 200 μL of 0.1 M formic acid, which was repeated five times using the same eluate to result in a final collected volume of ca 200 μL. 2H4-SAH (16.7 nM) and 2H3-SAM (11.5 nM) were added to final eluates of cell/tissue extracts before dilution 1:1 with methanol containing 0.25 % formic acid (v/v) for DI-nESI-UHR-FTMS analysis. Eluates of plasma samples were lyophilized, resuspended in 15 μL of water and 10 μL of which was diluted to 50 μL with methanol + 0.25 % formic acid for DI-nESI-UHR-FTMS analysis. The final concentration for 2H4-SAH and 2H3-SAM in the spray solution was 22.3 and 15.4 nM, respectively.
2.6. DI-nESI-UHR-FTMS analysis of SAM/SAH and data processing
Samples were introduced using the nanoelectrospray TriVersa NanoMate (Advion Biosciences, Ithaca, NY, USA) with +1.45 kV electrospray voltage and 0.6 psi head pressure. UHR-FTMS data were collected on an Orbitrap Fusion Lumos Tribrid mass spectrometer (Lumos) from Thermo Scientific (San Jose, CA, USA) with 1,000,000 resolving power (at 200 m/z) in positive ion mode. Three different scan modes were used for data collection. MS1 full scans were acquired using 2 microscans per scan in the m/z range of 150 to 1000; SIM scans were obtained to include m/z 399.1445 for SAM (C15H22N6O5S, [M+H]+) and m/z 385.1289 for SAH (C14H20N6O5S, [M+H]+) with 25 m/z isolation windows and 1 microscan per scan. Spectra were acquired over a continuous infusion period of 5–10 minutes, and spectra that exceeded a threshold were averaged. This process increased both signal to noise ratio and the mass accuracy, while removing low intensity scans that could impact quantification using peak area (ion intensity). The absolute quantification was achieved by referencing to the ion intensity of internal deuterated standards.
The SIM m/z windows were chosen to accommodate all possible 13C isotopologues. The MS2 scans were collected via quadrupole isolation using 0.5 m/z isolation window and HCD (High Energy Collision Dissociation) set at 30 % collision energy with 100,000 or 500,000 resolving power to confirm standards and isotope labeled SAM and SAH. The AGC target was 105 with a maximal 1000 ms injection time for both MS1 and MS2 scans. The data collection time for MS1 and MS2 scans was 10 and 3 minutes, respectively.
The linearity of SAM/SAH analysis was performed by diluting a 10 μM standard mixture to concentrations ranging from 25 to 1000 nM using 50% methanol containing 0.1% formic acid. The UHR-FTMS analysis for each standard sample was repeated three times to check for precision.
Limits of detection were assessed by the apparent signal to noise ratio at the accurate mass with 1 ppm mass error for SAM and SAH, using a value of 5X above background noise (for detection) and 10X for the limit of quantification.
For data processing, the exact (accurate mass) m/z values and peak intensities were exported as an Excel file. Then, isotopologues of SAM and SAH were assigned using an in-house software PREMISE (PRecalculated Exact Mass Isotopologue Search Engine) [41] by matching theoretical m/z values against detected m/z values. The raw, observed m/z value was compared to the theoretical m/z values of SAM and SAH within a 0.002 m/z window for assignment; when internal standards were used, the resulting m/z correction resulted in a 0.001 m/z window- matching criterion. The peak intensity of all assigned isotopologues was corrected for natural abundance [42] to determine isotopic enrichment.
2.7. Ion chromatography-UHR-FTMS
Polar fractions of A549 cells were concentrated by nitrogen flushing using Evaporex® microplate evaporators (Apricot Designs, Covina, CA, USA). Typically 10% of total polar extracts (ca. 150 μL) were placed in the wells of 96-well plates and evaporated by continuous nitrogen flow (45 L min−1) at 40 °C for 20 minutes, which concentrated the analytes by 3–4 fold. The concentrates were the analyzed as previously described [36] by IC-UHR-FTMS using a Dionex ICS-5000+ ion chromatograph connected to an Orbitrap Fusion Tribrid mass spectrometer (ThermoFisher) operated in negative ion mode. A Dionex IonPac AG11-HC-4 μm reagent-free ion chromatography and high pressure ion chromatography guard column (2 × 50 mm) was connected prior to anionic Dionex IonPac AC11-HC 4 μm RFIC&HPIC (2 × 250 mm) column. The mass resolution was set to 500,000 (FWHM at m/z 200). Peak areas and 13C isotopologues of metabolites were determined using TraceFinder 3.3 (Thermo) and were corrected for natural abundance distribution for each isotopologue [36].
3. Results & Discussion
3.1. DI-nESI-UHR-FTMS analysis of SAM and SAH standards
Figure 2a shows UHR mass spectra of standard SAH, SAM and the deuterium-labeled standards recorded with 10 min of signal averaging at a mass resolution of 1 million (at 200.0 m/z). The theoretical m/z values of proton adducts of SAH and SAM are 385.12887 and 399.14451 and in this case the observed m/z values were 385.12867 and 399.14427 respectively. The mass error of both SAM and SAH ions was 0.6 ppm, which was the same mass error for the 2H3-SAM and 2H4-SAH standards (Table 1). These deuterated external standards were well-resolved from the 13C isotopologues of SAM and SAH in A549 cell extracts that had the same number of 13C as that of 2H atoms (Fig. 3a and b), which make them suitable for use in quantification for SIRM studies. In addition, as the resolution of FTMS was increased from 120,000 to 106 (Fig. S1), the natural abundance M+1 13C isotope of SAM (m/z=400.14691) was progressively separated from other unknown peaks. These data showed that it is crucially important to use UHR-FTMS to quantify labeled metabolites from crude extracts, including sample introduction via direct infusion. To confirm the structures and define the fragmentation patterns that are important for isotopomer analysis in SIRM experiments (see below), MS2 spectra of each standard were analyzed at 120,000 resolution with 30% HCD and shown in Fig. 2b and c for SAH (385.1287 m/z) and 2H4-SAH (389.1537 m/z), respectively. Several major fragment ions showed 4 Da differences between unlabeled and labeled SAH but the 136.06 m/z fragment ion did not, which corresponds to the adenine group released by cleavage at the glycosyl bond. Fig. S2a and b shows the MS2 spectra of SAM (399.1443 m/z) and its 2H3 standard (402.1631 m/z). Unlike the fragmentation patterns for SAH, only one minor fragment ion showed 3 m/z differences between SAM (298.10) and 2H3-SAM (301.11) as the major fragment ions did not contain the C2H3 group.
Figure 2. Ultrahigh resolution mass spectra of SAM and SAH.
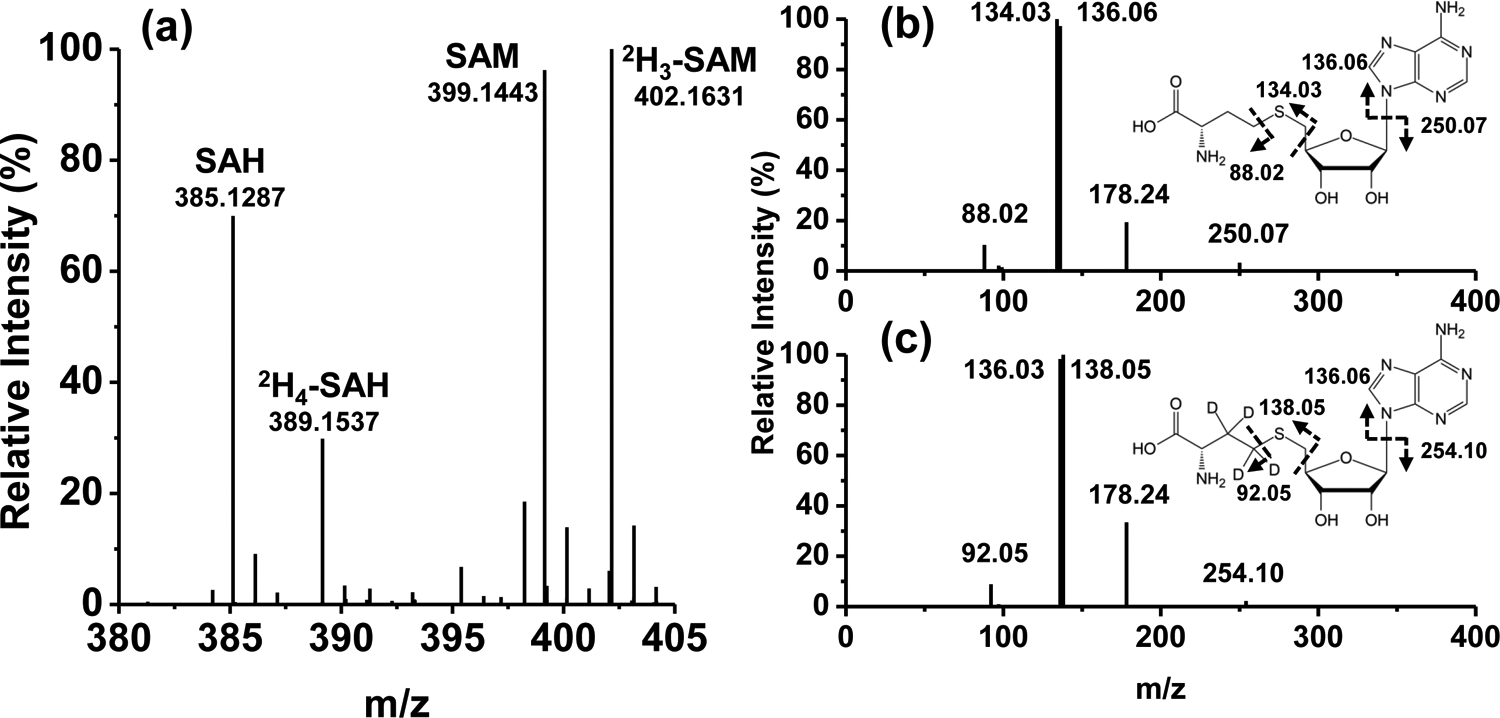
Standards and deuterated versions were prepared and introduced into the Orbitrap as described in Experimental. a) MS spectrum of S-adenosylmethionine (SAM, 399.1443 m/z), S-adenosylhomocysteine (SAH, 385.1287 m/z), 2H3-labeled SAM (402.1631 m/z), and 2H4-labeled SAH (389.1537 m/z) standards; b) MS2 spectrum and structure of SAH with bond cleavages and resulting mass fragments; c) MS2 spectrum of 2H4-labeled SAH with bond cleavages and the resulting mass fragments.
Table 1.
Chemical formula, theoretical and observed m/z ([M+H]+) values, and mass error of SAM, SAH and their isotopically enriched standard in Fig. 2a.
| Formula | Theoretical mass (m/z), [M+H]+ | Observed mass (m/z), [M+H]+ | Mass error (ppm) | |
|---|---|---|---|---|
| SAM | C15H22N6O5S | 399.14451 | 399.14427 | −0.6 |
| D3-SAM | C15H19D3N6O5S | 402.16335 | 402.16309 | −0.6 |
| SAH | C14H20N6O5S | 385.12887 | 385.12862 | −0.6 |
| D4-SAH | C14H16D4N6O5S | 389.15397 | 389.15372 | −0.6 |
Figure 3. MS spectra of 2H standards of SAH and SAM added to A549 cell extracts containing 13C-labeled SAH and SAM.

Sample preparation, experimental condirions and analyses were as as described in Experimental.
We also determined the linear range of the MS response for SAM and SAH standards for optimizing of the SIRM analysis. As shown in Table S1 and Fig. S3, both SAM and SAH showed a good linear and reproducible response over the range of 25 to 1000 nM. The detection limit was somewhat better than published LC/MS methods [22, 43], illustrating the high sensitivity of UHR-FTMS along with its higher sample throughput. Together with the low coefficient of variation, this new method enabled rapid and reliable quantification of SAM and SAH.
3.2. Optimization of extraction conditions for A549 cells
It is crucially important to minimize the salt content in polar biological extracts to maintain stable electrospray ionization using direct infusion MS [28]. We took two simple measures to reduce the salt content in A549 cell extracts for SAM and SAH analysis. One is to perform a quick Nanopure water rinse of the cell culture before extraction. The other is to capture SAM and SAH by a PBA column via interaction of SAM/SAH’s vicinal cis-diol group with PBA at neutral pH, followed by wash with ammonium acetate, elution with volatile formic acid, and lyophilization to remove formic acid. Similar PBA cleanup methods have been used by other researchers for SAM and SAH analysis [24, 26, 40]. We found that the electrospray ionization stability was significantly improved with this desalting approach (data not shown).
As SAM has been reported to be unstable at pH above 6.0 [44], the effect of pH on cell lysis and extraction was also evaluated. Simultaneous cell lysis and extraction was performed in aqueous 60% CH3CN containing 18 MOhm deionized water, 0.01 M formic acid, and 0.1 M formic acid. The pH of the final extracts were 6.0, 5.0, and 3.5 respectively. Fig. S4a to c show the MS1 spectra of SAM from these polar extracts of A549 cells, which showed the lowest MS intensity for the extract at pH 3.5. We have also performed lysis and extraction at pH 1.5 by adding 0.05 M HCl, but the MS intensity of SAM was no higher than that at pH 5 (data not shown). Although the SAM stability is known to be greater in acidic conditions than at neutral pH, we found that moderately acidic conditions (pH 5.0 to 6.0) for recovering SAM from A549 cell were better suited for DI-nESI-UHR-FTMS analysis. SAH also displayed the lowest MS intensity at pH 3.5, followed by pH 5.0 and 6.0 (Fig. S4). There appeared to be some advantage to extraction at pH 5.0 for SAM analysis but the number of metabolites analyzable by IC-UHR-FTMS was lower at pH 5 than at pH 6 (see below). Using the external standards, we determined recoveries of 93± 4% for SAH and 91± 2.5% for SAM at pH 6.
Our goal is to have a lysis/extraction method that not only reliably recovers SAM and SAH but also many other metabolites in SIRM experiments. We analyzed the A549 extracts at the three different pH’s by IC-UHR-FTMS to determine the number of assignable metabolites. Table S2 lists the peak areas of metabolites detected in the negative ion mode. The number of negatively charged metabolites was highest for pH 6 (123), followed by pH 5 (117) and pH 3.5 (78) extractions, due at least in part to the instability of some metabolites (e.g. ATP, fructose-1,6-bisphosphate) at lower pH values. Thus, 60% CH3CN in Nanopure water (pH 6) was chosen for recovering SAM/SAH from biological systems (except for plasma) for DI-nESI-UHR-FTMS analysis. It should be noted that this lysis/extraction method has been used by us in most of our SIRM studies [28, 45, 46].
In addition to SAM and SAH, other metabolites that contain vicinal hydroxyls are expected to be retained on the PBA column. Table S2 shows a total of 35 such quantifiable analytes in the pH 6 eluate of the PBA column including nucleotides (e.g. AMP) and several 2-hydroxy acids (e.g. 2-hydroxyglutarate).
3.3. Quantification of SAM and SAH in mouse liver tissue and human plasma
We applied the above optimized method to analyze SAM and SAH in mouse liver tissue extracts. Fig. 4a shows a MS1 spectrum acquired over 4 minutes of infusion of a 1/8 fraction of polar extract from about 8 mg wet weight of mouse liver tissue. The MS intensity of SAM (3.1.5e3) and SAH (5.5e3) was higher than that of the spiked external standards and well-resolved from other unknown peaks in full scan mode (384–403 m/z shown). Note the extremely small deviation from theoretical m/z for uncorrected spectra, in each case < 0.0004 m/z. Repeated analyses showed the concentrations of SAM and SAH in the polar extract of this mouse liver tissue were 0.254 μM and 0.117 μM, respectively, which corresponded to 0.052 and 0.023 μmole g−1 protein and a SAM/SAH ratio of 2.3. The recoveries determined by spiking of external standards were 89±1.5% for SAH and 89±6% for SAM.
Figure 4. MS1 spectra of SAM and SAH from mouse liver extract and human plasma, with internal standards SAM-2H3 and SAH-2H4.
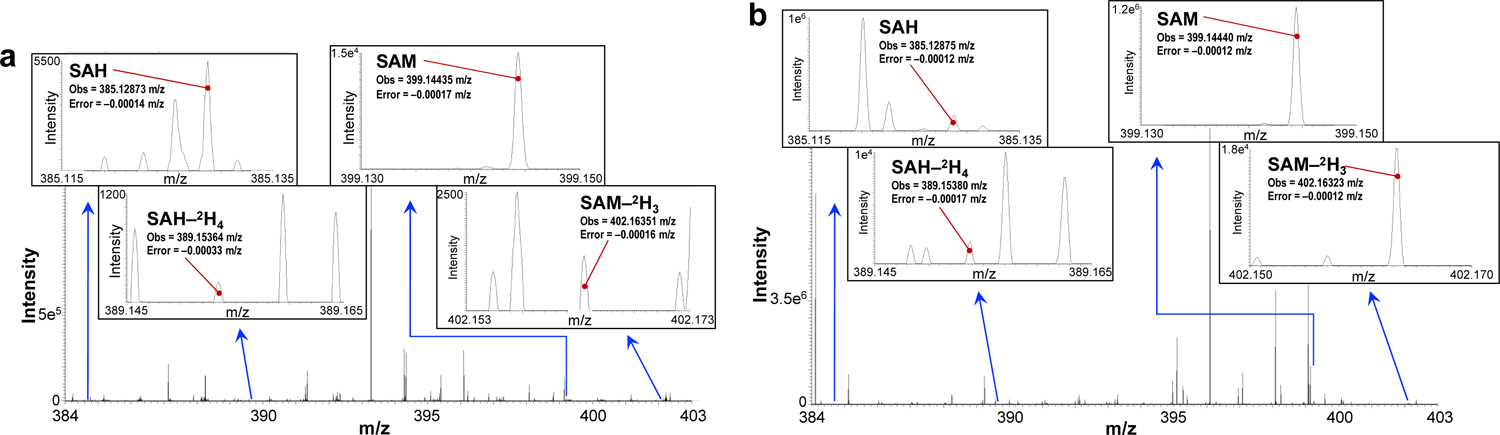
Samples were prepared as described in the Experimental. a) Mouse liver extract spectrum comprising 69 FT spectra from over approx. 4 min acquisition under full scan MS1 100–1000 m/z at 1M resolution setting and divided by the number of summed scans to provide an average. The averaged peak intensity or area is normalized to that of the internal deuterated standard to determine the concentration. The region displayed is 384 – 403 m/z and the insets show 0.02 m/z windows for greater detail. Errors stated are accurate mass errors from theoretical m/z values. b) Human plasma spectrum of a pancreatic cancer patient. The spectrum comprises 82 spectra averaged over approx. 9 min acquisition under full scan MS1 150–1000 m/z at 1 M resolution setting. The averaged intensity is normalized to that of the internal deuterated standard to determine the concentration. The region displayed is 384 – 403 m/z and the insets show 0.02 m/z windows for greater detail. Errors stated are accurate mass deviation from theoretical m/z values.
For the human plasma samples (Figure 4b), both SAM and SAH were readily observed in the full scan mode spectrum. Here again, note the extremely small deviation from theoretical m/z for the uncorrected spectra, in each case < 0.0002 m/z. Table S3 lists the plasma concentrations of SAM and SAH for two healthy and one IPMN patient volunteers. Previous studies have reported the concentration of SAM and SAH in healthy human plasma to be in the nanomolar range (50 to 155 nM for SAM) using LC/MS or ELISA methods [26, 43, 47]. Our analysis showed that the SAM concentration of the healthy subjects was at the low end of the literature range while that of one IPMN patient was much above the range. Our analysis also showed that SAH was below detectable limits in the two healthy plasma samples and the SAM/SAH ratio for the IPMN patient was 71, which was much higher than the literature range for healthy adults (2.7–12.4) [26]. As SAH is an indicator of not only SAM metabolism but also a principal degradation product of SAM due to sample handling and processing, this lack of detectable SAH (i.e. below the sub nM detection limit) in our healthy plasma samples indicates very low SAM breakdown resulting from sample handling. Whether the abnormally high SAM concentration in the IPMN patient is representative of this disease awaits further investigation.
3.4. SAM/SAH synthesis from 13C6 glucose and 13CH3-methionine in A549 cells.
To demonstrate the method for tracking newly synthesized SAM and SAH in cell cultures, we incubated A549 cells for 24 hours in media containing 13C6 glucose, 13CH3-methionine, or the unlabeled counterparts. The average total abundances of SAM and SAH were determined to be about 1.5 and 0.35–0.5 μmol g−1 protein, respectively, with a SAM to SAH ratio of 2.9–4.5 (Table S4).
We also determined the fractional enrichment of different isotopologues of SAM, SAH, and ATP as well as their PPP precursors (analyzed by IC-UHR-FT-MS) with each tracer treatment and corrected for natural abundance [42] as shown in Fig. 5. With 13C6-glucose as tracer (Fig. 5A), the most abundant 13C isotopologues for the PPP metabolites were the uniformly 13C labeled species (e.g. 5 or 13C5-R5P in c and 7 or 13C7-S7P in d), as expected from the atom-resolved pathway tracing. Also consistent with the pathway tracing was the significant presence of species such as 13C2-/13C5-S7P and 13C4-/13C5-F6P (f). 13C5-R5P was then converted to 13C5-PRPP (g) before incorporation into ATP (h), SAM (i), and SAH (j). Thus, the predominant 5 isotopologue in ATP/SAM/SAH is consistent with the high extent of 13C5-ribose incorporation, which we have shown for ATP previously [48]. Less abundant isotopologues of the three metabolites but present significantly were the 13C6- (6) and 13C7- (7) species, which represents 13C1- and 13C2-adenine (generated via one-carbon metabolism) plus 13C5-ribose. This 13C labeling pattern of ATP in 13C6-glucose-grown A549 cells recapitulated that in our previous study [28]. In SAM, the higher ratio for the fractional enrichment of the 13C6- (6) over 13C7- (7) isotopologue than that in ATP (Fig. 5B) could be accounted for by the contribution of the 13CH3 + 13C5-ribose (in addition to the 13C1-adenine + 13C5-ribose) species to the 6 isotopologue in SAM.
Figure 5. 13C Isotopologue distributions of SAM, SAH and their precursors in A549 cells grown on 13C6-glucose or 13CH3-methionine.
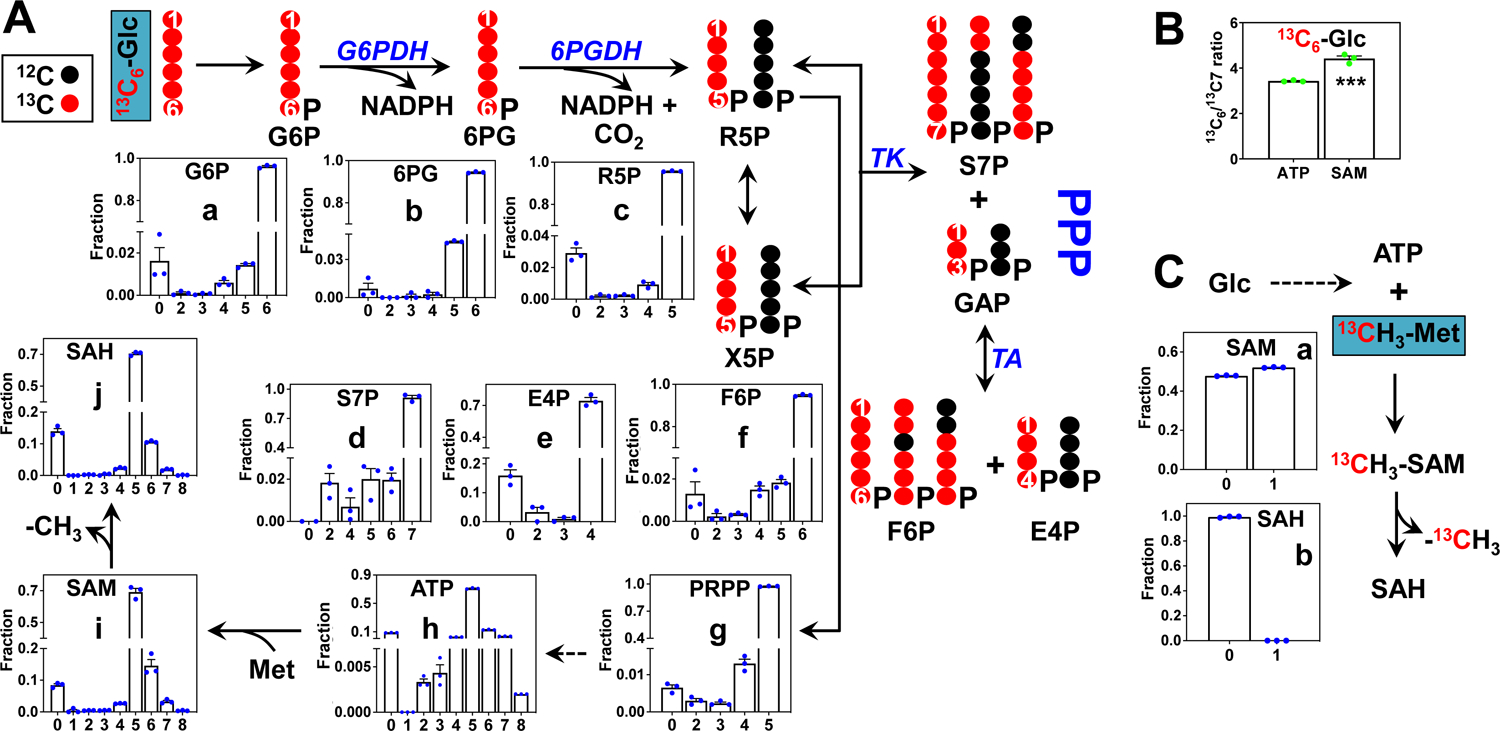
- Fractional distribution of metabolite isotopologues in the pentose phosphate pathway (PPP) and those of the downstream products phosphoribosyl pyrophosphate (PRPP), ATP, SAM, and SAH in 13C6-glucose grown samples. The atom tracing scheme depicts the more abundant isotopologue species resulting from the transformation of 13C6-Glc via the PPP. G6P: glucose-6-phosphate; 6PG: 6-phosphogluconate; R5P: ribose/ribulose-5-phosphate; X5P: xylulose-5-phosphate; S7P: sedoheptulose-7-phosphate; E4P: erythrose-4-phosphate; F6P: fructose-6-phosphate; G6PDH: glucose-6-phosphate dehydrogenase; 6PGDH: 6-phosphogluconate dehydrogenase; TK: transketolase; TA: transaldolase.
- Ratio of the 13C6- to 13C7-isotopologues of ATP and SAM derived from 13C6-Glc. A higher ratio in SAM than that in ATP reflect 13C labeling in the methyl group of SAM. ***: p-value 0.0012
- Fractional distribution of isotopologues of SAM and SAH in 13CH3-Met grown samples.
In comparison, with 13CH3-methionine as tracer, SAM (a) but not SAH (b) showed significant 13C1 labeling, i.e. ca. 50% in fractional enrichment (Fig. 5C), which was comparable to that of methionine supplied in the medium. This indicates an efficient conversion of exogenous methionine to SAM presumably via the action of methionine adenosyltransferase (MAT) and loss of the 13CH3 group in SAH via the action of methyltransferases in A549 cells.
To confirm the position of incorporated 13C in SAM and SAH, we acquired and analyzed the MS/MS patterns of m0, m1, and m5 ions of SAM. Fig. 6a shows the tandem mass spectrum of the m0 ion. The three most abundant ions at m/z 250.09, 136.06, and 102.06 corresponded respectively to (i) the product of cleavage at the S-methionine to C5-ribose bond to release adenosine, (ii) that at the N9-adenine to C1-ribose bond to release adenine and (iii) that at the S to C4 bond of methionine to release 2-aminobutanoate. The other product ions at m/z 150.05, 264.09, and 298.10, have relatively lower intensities. In the tandem mass spectrum of m1 SAM (Fig. 6b), the m/z values of fragment ions containing the methyl-group of SAM were +1 m/z shifted (298.10->299.10, 150.05 ->151.05, and 264.09->265.09), which confirmed 13C labeling at the methyl position of SAM with 13C1-methionine as tracer. With 13C6-glucose as tracer, the m5 ion of SAM in Fig. 6c yielded ribose-containing fragment ions that were +5 Da shifted (298.10 -> 303.10, 250.09 -> 255.11, and 264.09 -> 269.09). These data confirmed the m5 ion to arise from 13C5-ribose of the adenosyl subunit of SAM. Thus, SAM and SAH analysis using the new method demonstrated efficient SAM synthesis from methionine and de novo synthesis of SAM/SAH from glucose in A549 cells.
Figure 6. MS/MS spectra of SAM from A549 cells exposed to 13C6-glucose or 13C1-CH3- methionine.
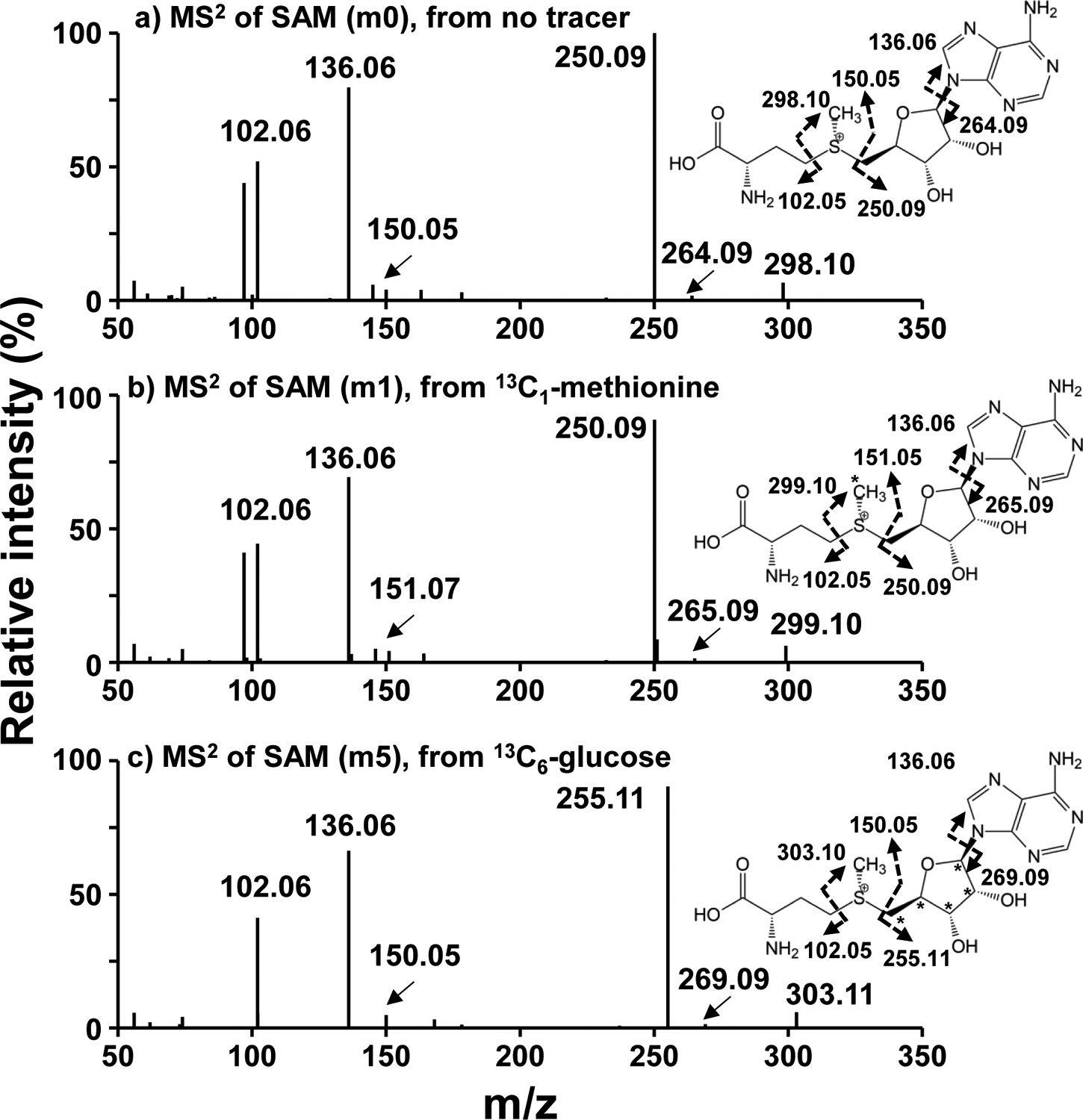
- MS/MS of m0 isotopologue of SAM (399.1443 m/z) from an unlabeled sample
- MS/MS of m1 isotopologue of SAM (400.1479 m/z) from a 13C1-methionine labeled A549 cell extract,
- MS/MS of m5 isotopologue of SAM (404.1613 m/z) from a 13C6-glucose labeled A549 cell extract.
Data were acquired at a setting of 500 K mass resolution with 10 μscan/scan. Asterisks(*) represent 13C.
Dashed arrows represent the fragmentation positions giving rise to the observed m/z.
3.5. Cell line-dependent perturbations of SAM/SAH levels and ratios by MSA, selenite, and SeM treatments
We further demonstrated the new method by analyzing the effect of three different anti-cancer selenium agents (MSA, SeO3, and SeM) on SAM and SAH status in two lung adenocarcinoma (A549 and H1299) cell lines. Figure 7a shows the depleting effect of the three agents on SAM and SAH levels in A549 cells, which was similarly observed in H1299 cells except for the SeO3 treatment (Fig. 7c). This effect could result from decreased synthesis and/or increased utilization of SAM and SAH. However, SAM depletion without the buildup of the product SAH is more consistent with a block in SAM synthesis, which is corroborated by the attenuating effect of SeO3 [34] and MSA [49] on the synthesis of SAM’s precursor, ATP in A549 cells. In contrast, the SAM to SAH ratio was unaltered in all but one case, i.e. it more than doubled in MSA- versus in control-treated A549 cells (Fig. 7b, d). This increase in SAM to SAH ratio despite the depletion of SAM levels in A549 cells points to an inhibition of the methyltransferases that catalyze DNA/RNA/protein methylation (cf. Fig. 1), besides the block in SAM synthesis. In addition, the buildup of SAM with the lack of change in SAM to SAH ratio in SeO3-treated H1299 cells (Fig. 7c, d) suggests activation of SAM synthesis. Pending further confirmation, the SAM/SAH analysis using our new method revealed cell line- and Se form-dependence in perturbing one-carbon metabolism in lung cancer cells.
Figure 7. Cell line-dependent perturbations of SAM/SAH levels and ratios by MSA, selenite, and SeM treatments.
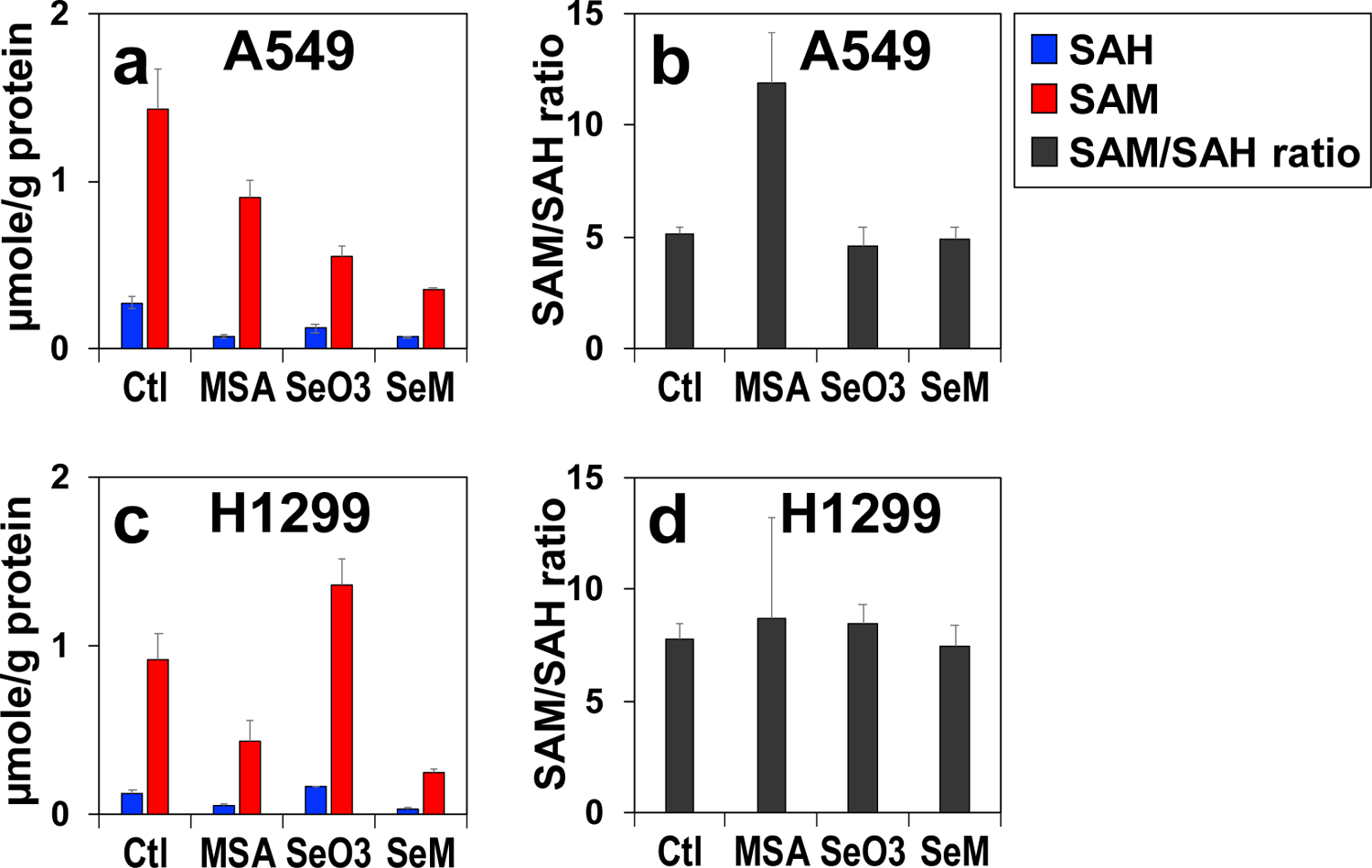
A549 (a,b) and H1299 (a,b) cells were treated with 5 μM MSA, 6.25 μM selenite, or 0.5 mM SeM for 24 h (n = 3), extracted for polar metabolites, and analyzed for SAM/SAH as described in Experimental. ■: SAH; ■: SAM; ■: SAM/SAH ratio.
4. Conclusions
We have developed a rapid, sensitive, and robust method for SAM and SAH analysis in biological extracts by coupling PBA affinity capture with DI-nESI-UHR-FTMS, that supports metabolic tracing using 13C (SIRM). As the lysis/extraction solvent system (CH3CN in mildly acidic Nanopore water) used to efficiently recovered SAM/SAH was the same as that for extracting polar metabolites in general, this method is compatible with comprehensive metabolite profiling studies including SIRM applications. This is an added advantage over the existing methods that call for extensive sample acidification, which leads to degradation of acid-labile metabolites. In addition, harnessing UHR-FTMS to resolve the same number of 13C from 2H labels in SAM and SAH enabled the use of 2H-labeled external standards for reliable quantification of SAM/SAH in complex biological matrices including those containing 13C labeled metabolites. We first demonstrated the utility of the method on SAM/SAH quantification in mouse liver, A549 cells, and human plasma samples. Then, we applied the method to track 13C incorporation from 13C1-methionine and 13C6-glucose into SAM and SAH in A549 cells via the MAT activity and the PPP-purine nucleotide synthesis pathway, respectively. Moreover, we used the method to discover that MSA may block both SAM synthesis and its use in methylation reactions. Thus, the new method is applicable to both high throughput and tracer-based metabolic pathway analysis for SAM and SAH, thereby facilitating the understanding of epigenetic metabolism and regulation.
Supplementary Material
Acknowledgments.
We thank Ms. T. Cassel and Ms. H. Yu for expert technical assistance.
Funding
This work was supported in part by NIH grants 1U24DK097215-01A1, P01CA163223-01A1, 5P20GM121327, 1P30ES026529-01A1, and Shared Resource(s) of the University of Kentucky Markey Cancer Center P30CA177558.
Abbreviations:
- DI-nESI-UHR-FTMS
direct infusion nanoelectrospray ultra-high-resolution Fourier transform mass spectrometry
- DMEM
Dulbecco’s modified Eagle’s medium
- ESI
electrospray ionization
- MSA
methylseleninic acid
- PBA
phenylboronic acid
- SeO3
selenite
- SAH
S-adenosylhomocysteine
- SAM
S-adenosylmethionine
- SeM
selenomethionine
- SIRM
stable isotope-resolved metabolomics
- THF
tetrahydrofolate
Footnotes
Competing Interests: None
References
- [1].Sanderson SM, Gao X, Dai Z, Locasale JW, Methionine metabolism in health and cancer: a nexus of diet and precision medicine, Nat Rev Cancer, 19 (2019) 625–637. [DOI] [PubMed] [Google Scholar]
- [2].Locasale JW, Serine, glycine and one-carbon units: cancer metabolism in full circle, Nat Rev Cancer, 13 (2013) 572–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Finkelstein JD, Methionine metabolism in mammals, J Nutr Biochem, 1 (1990) 228–237. [DOI] [PubMed] [Google Scholar]
- [4].Newman AC, Maddocks ODK, One-carbon metabolism in cancer, Br J Cancer, 116 (2017) 1499–1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Ducker GS, Rabinowitz JD, One-Carbon Metabolism in Health and Disease, Cell Metab, 25 (2017) 27–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Lu SC, Mato JM, S-adenosylmethionine in liver health, injury, and cancer, Physiol Rev, 92 (2012) 1515–1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Zhang J, Zheng YG, SAM/SAH Analogs as Versatile Tools for SAM-Dependent Methyltransferases, ACS Chem Biol, 11 (2016) 583–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Mudd SH, Brosnan JT, Brosnan ME, Jacobs RL, Stabler SP, Allen RH, Vance DE, Wagner C, Methyl balance and transmethylation fluxes in humans, Am J Clin Nutr, 85 (2007) 19–25. [DOI] [PubMed] [Google Scholar]
- [9].Green TJ, Skeaff CM, McMahon JA, Venn BJ, Williams SM, Devlin AM, Innis SM, Homocysteine-lowering vitamins do not lower plasma S-adenosylhomocysteine in older people with elevated homocysteine concentrations, Br J Nutr, 103 (2010) 1629–1634. [DOI] [PubMed] [Google Scholar]
- [10].Zhang H, Liu Z, Ma S, Zhang H, Kong F, He Y, Yang X, Wang Y, Xu H, Yang A, Tian J, Zhang M, Cao J, Jiang Y, Guo X, Ratio of S-adenosylmethionine to S-adenosylhomocysteine as a sensitive indicator of atherosclerosis, Mol Med Rep, 14 (2016) 289–300. [DOI] [PubMed] [Google Scholar]
- [11].Mato JM, Corrales FJ, Lu SC, Avila MA, S-Adenosylmethionine: a control switch that regulates liver function, FASEB J, 16 (2002) 15–26. [DOI] [PubMed] [Google Scholar]
- [12].Elshorbagy AK, Jerneren F, Samocha-Bonet D, Refsum H, Heilbronn LK, Serum S-adenosylmethionine, but not methionine, increases in response to overfeeding in humans, Nutr Diabetes, 6 (2016) e192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Mentch SJ, Mehrmohamadi M, Huang L, Liu X, Gupta D, Mattocks D, Gomez Padilla P, Ables G, Bamman MM, Thalacker-Mercer AE, Nichenametla SN, Locasale JW, Histone Methylation Dynamics and Gene Regulation Occur through the Sensing of One-Carbon Metabolism, Cell Metab, 22 (2015) 861–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Pizzolo F, Blom HJ, Choi SW, Girelli D, Guarini P, Martinelli N, Stanzial AM, Corrocher R, Olivieri O, Friso S, Folic acid effects on s-adenosylmethionine, s-adenosylhomocysteine, and DNA methylation in patients with intermediate hyperhomocysteinemia, J Am Coll Nutr, 30 (2011) 11–18. [DOI] [PubMed] [Google Scholar]
- [15].Tran TQ, Lowman XH, Kong M, Molecular Pathways: Metabolic Control of Histone Methylation and Gene Expression in Cancer, Clin Cancer Res, 23 (2017) 4004–4009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Mato JM, Martinez-Chantar ML, Lu SC, S-adenosylmethionine metabolism and liver disease, Ann Hepatol, 12 (2013) 183–189. [PMC free article] [PubMed] [Google Scholar]
- [17].Kruglova MP, Grachev SV, Bulgakova PO, Ivanov AV, Virus ED, Nikiforova KA, Fedoseev AN, Savina GD, Kubatiev AA, Low S-adenosylmethionine/ S-adenosylhomocysteine Ratio in Urine is Associated with Chronic Kidney Disease, Lab Med, 51 (2020) 80–85. [DOI] [PubMed] [Google Scholar]
- [18].Mato JM, Alvarez L, Ortiz P, Pajares MA, S-adenosylmethionine synthesis: molecular mechanisms and clinical implications, Pharmacol Ther, 73 (1997) 265–280. [DOI] [PubMed] [Google Scholar]
- [19].Hanson NQ, Eckfeldt JH, Schwichtenberg K, Aras O, Tsai MY, Interlaboratory variation of plasma total homocysteine measurements: results of three successive homocysteine proficiency testing surveys, Clin Chem, 48 (2002) 1539–1545. [PubMed] [Google Scholar]
- [20].O’Broin SD, Kelleher BP, McPartlin J, O’Gorman P, Browne M, White B, Smith OP, Optimization of routine plasma homocysteine monitoring, Blood Coagul Fibrinolysis, 11 (2000) 367–369. [DOI] [PubMed] [Google Scholar]
- [21].Melnyk S, Pogribna M, Pogribny IP, Yi P, James SJ, Measurement of plasma and intracellular S-adenosylmethionine and S-adenosylhomocysteine utilizing coulometric electrochemical detection: alterations with plasma homocysteine and pyridoxal 5’-phosphate concentrations, Clinical chemistry, 46 (2000) 265–272. [PubMed] [Google Scholar]
- [22].Guiraud SP, Montoliu I, Da Silva L, Dayon L, Galindo AN, Corthesy J, Kussmann M, Martin FP, High-throughput and simultaneous quantitative analysis of homocysteine-methionine cycle metabolites and co-factors in blood plasma and cerebrospinal fluid by isotope dilution LC-MS/MS, Anal Bioanal Chem, 409 (2017) 295–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Struys EA, Jansen EE, de Meer K, Jakobs C, Determination of S-adenosylmethionine and S-adenosylhomocysteine in plasma and cerebrospinal fluid by stable-isotope dilution tandem mass spectrometry, Clin Chem, 46 (2000) 1650–1656. [PubMed] [Google Scholar]
- [24].Kirsch SH, Knapp JP, Geisel J, Herrmann W, Obeid R, Simultaneous quantification of S-adenosyl methionine and S-adenosyl homocysteine in human plasma by stable-isotope dilution ultra performance liquid chromatography tandem mass spectrometry, J Chromatogr B Analyt Technol Biomed Life Sci, 877 (2009) 3865–3870. [DOI] [PubMed] [Google Scholar]
- [25].Jiang Z, Liang Q, Luo G, Hu P, Li P, Wang Y, HPLC-electrospray tandem mass spectrometry for simultaneous quantitation of eight plasma aminothiols: application to studies of diabetic nephropathy, Talanta, 77 (2009) 1279–1284. [DOI] [PubMed] [Google Scholar]
- [26].Gellekink H, van Oppenraaij-Emmerzaal D, van Rooij A, Struys EA, den Heijer M, Blom HJ, Stable-isotope dilution liquid chromatography-electrospray injection tandem mass spectrometry method for fast, selective measurement of S-adenosylmethionine and S-adenosylhomocysteine in plasma, Clinical chemistry, 51 (2005) 1487–1492. [DOI] [PubMed] [Google Scholar]
- [27].Lane AN, Fan TW, Higashi RM, Tan J, Bousamra M, Miller DM, Prospects for clinical cancer metabolomics using stable isotope tracers, Exp Mol Pathol, 86 (2009) 165–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Lorkiewicz P, Higashi RM, Lane AN, Fan TW, High information throughput analysis of nucleotides and their isotopically enriched isotopologues by direct-infusion FTICR-MS, Metabolomics, 8 (2012) 930–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Bruntz RC, Lane AN, Higashi RM, Fan TW, Exploring cancer metabolism using stable isotope-resolved metabolomics (SIRM), The Journal of biological chemistry, 292 (2017) 11601–11609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Fan TW, Lorkiewicz PK, Sellers K, Moseley HN, Higashi RM, Lane AN, Stable isotope-resolved metabolomics and applications for drug development, Pharmacol Ther, 133 (2012) 366–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Lane AN, Fan TW, Bousamra M 2nd, Higashi RM, Yan J, Miller DM, Stable isotope-resolved metabolomics (SIRM) in cancer research with clinical application to nonsmall cell lung cancer, OMICS, 15 (2011) 173–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Yang Y, Fan TW, Lane AN, Higashi RM, Quantification of Isotopologues of Amino Acids by Multiplexed Stable Isotope-Resolved Metabolomics Using Ultrahigh-Resolution Mass Spectrometry Coupled with Direct Infusion, in: Alterman M (Ed.) Amino Acid analysis, Humana, New York, NY, 2019, pp. 57–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Maegley KA, Krivacic C, Bingham P, Liu W, Brooun A, Comparison of a High-Throughput Mass Spectrometry Method and Radioactive Filter Binding to Assay the Protein Methyltransferase PRMT5, Assay Drug Dev Technol, 13 (2015) 235–240. [DOI] [PubMed] [Google Scholar]
- [34].Fan T, Bandura L, Higashi R, Lane A, Metabolomics-edited transcriptomics analysis of Se anticancer action in human lung cancer cells, Metabolomics, 1 (2005) 325–339 [Google Scholar]
- [35].Fan TW-M, Tan JL, McKinney MM, Lane AN, Stable Isotope Resolved Metabolomics Analysis of Ribonucleotide and RNA Metabolism in Human Lung Cancer Cells., Metabolomics, 8 (2012) 517–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Sun Q, Fan TW-M, Lane AN, Higashi RM, Ion Chromatography-Ultra High-resolution MS1/MS2 Method for Stable Isotope-Resolved Metabolomics (SIRM) Reconstruction of Metabolic Networks., Anal. Chem, 93 (2021) 2749–2757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Sun RC, Fan TWM, Deng P, Higashi RM, Lane AN, Le A-T, Scott TL, Sun Q, Warmoes MO, Yang Y, Noninvasive liquid diet delivery of stable isotopes into mouse models for deep metabolic network tracing, Nature communications, 8 (2017) 1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Fan TW-M, Sample preparation for metabolomics investigation, in: Fan TW-M, Lane AN, Higashi RM (Eds.) The Handbook of Metabolomics: Pathway and Flux Analysis, Methods in Pharmacology and Toxicology, Springer Science, New York, 2012, pp. 7–27. [Google Scholar]
- [39].Fan TWM, Zhang X, Wang C, Yang Y, Kang W-Y, Arnold S, Higashi RM, Liu J, Lane AN, Exosomal lipids for classifying early and late stage non-small cell lung cancer, Analytica chimica acta, 1037 (2018) 256–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Iglesias Gonzalez T, Cinti M, Montes-Bayon M, Fernandez de la Campa MR, Blanco-Gonzalez E, Reversed phase and cation exchange liquid chromatography with spectrophotometric and elemental/molecular mass spectrometric detection for S-adenosyl methionine/S-adenosyl homocysteine ratios as methylation index in cell cultures of ovarian cancer, J Chromatogr A, 1393 (2015) 89–95. [DOI] [PubMed] [Google Scholar]
- [41].Lane AN, Fan TW, Xie Z, Moseley HN, Higashi RM, Isotopomer analysis of lipid biosynthesis by high resolution mass spectrometry and NMR, Anal Chim Acta, 651 (2009) 201–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Moseley HN, Correcting for the effects of natural abundance in stable isotope resolved metabolomics experiments involving ultra-high resolution mass spectrometry, BMC Bioinformatics, 11 (2010) 139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Krijt J, Duta A, Kozich V, Determination of S-Adenosylmethionine and S-Adenosylhomocysteine by LC-MS/MS and evaluation of their stability in mice tissues, J Chromatogr B Analyt Technol Biomed Life Sci, 877 (2009) 2061–2066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Hoffman JL, Chromatographic analysis of the chiral and covalent instability of S-adenosyl-L-methionine, Biochemistry, 25 (1986) 4444–4449. [DOI] [PubMed] [Google Scholar]
- [45].Sengupta D, Cassel T, Teng KY, Aljuhani M, Chowdhary VK, Hu P, Zhang X, Fan TW, Ghoshal K, Regulation of hepatic glutamine metabolism by miR-122, Molecular metabolism, 34 (2020) 174–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Sun Q, Fan TWM, Lane AN, Higashi RM, Applications of chromatography-ultra high-resolution MS for stable isotope-resolved metabolomics (SIRM) reconstruction of metabolic networks, TrAC Trends in Analytical Chemistry, 123 (2020) 115676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Caudill MA, Wang JC, Melnyk S, Pogribny IP, Jernigan S, Collins MD, Santos-Guzman J, Swendseid ME, Cogger EA, James SJ, Intracellular S-adenosylhomocysteine concentrations predict global DNA hypomethylation in tissues of methyl-deficient cystathionine beta-synthase heterozygous mice, J Nutr, 131 (2001) 2811–2818. [DOI] [PubMed] [Google Scholar]
- [48].Sun Q, Fan TWM, Lane AN, Higashi RM, An Ion Chromatography–Ultrahigh-Resolution-MS1/Data-Independent High-Resolution MS2 Method for Stable Isotope-Resolved Metabolomics Reconstruction of Central Metabolic Networks, Analytical Chemistry, 93 (2021) 2749–2757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Tarrado-Castellarnau M, Cortés R, Zanuy M, Tarragó-Celada J, Polat IH, Hill R, Fan TWM, Link W, Cascante M, Methylseleninic acid promotes antitumour effects via nuclear FOXO3a translocation through Akt inhibition, Pharmacological Research, 102 (2015) 218–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.