

ABSTRACT
Glioblastoma (GBM) is a common and aggressive brain cancer that accounts for 60% of adult brain tumors. Anti-angiogenesis therapy is an attractive option due to the high vasculature density of GBM. However, the best-known anti-angiogenic therapeutics, bevacizumab, and aflibercept, have failed to show significant benefits in GBM patients. One of the reasons is the limited brain penetration of antibody-based therapies due to existence of the blood–brain barrier (BBB), which is further strengthened by the blood vessel normalization effects induced by anti-angiogenic therapies. To investigate if increased drug concentration in the brain by transferrin receptor (TfR)-mediated delivery across the BBB can enhance efficacy of anti-angiogenic antibody therapies, we first identified an antibody that binds to the apical domain of the mouse TfR and does not compete with the natural ligand transferrin (Tf) binding to TfR. Then, we engineered two bispecific antibodies fusing a vascular endothelial growth factor (VEGF)-Trap with the TfR-targeting antibody. Characterization of the two bispecific formats using multiple in vitro assays, which include endocytosis, cell surface and whole-cell TfR levels, human umbilical vein endothelial cell growth inhibition, and binding affinity, demonstrated that the VEGF-Trap fused with a monovalent αTfR (VEGF-Trap/moAb4) has desirable endocytosis without the induction of TfR degradation. Peripherally administered VEGF-Trap/moAb4 improved the brain concentration of VEGF-Trap by more than 10-fold in mice. The distribution of VEGF-Trap/moAb4 was validated to be in the brain parenchyma, indicating the molecule was not trapped inside the vasculature. Moreover, improved VEGF-Trap brain distribution significantly inhibited the angiogenesis of U-87 MG GBM tumors in a mouse model.
KEYWORDS: Bispecific antibody, transferrin receptor, BBB, GBM, angiogenesis
Introduction
Diseases in the central nervous system (CNS), including cancer (e.g., glioblastoma (GBM) and glioma), neurodegenerative diseases (e.g., Parkinson’s disease and Alzheimer’s disease), autoimmune diseases (e.g., multiple sclerosis), nervous system disease (e.g., amyotrophic lateral sclerosis), and genetic disorders (e.g., lysosomal storage diseases (LSDs)), are often difficult to treat. Antibody and protein-based drug modalities are promising options in treating CNS diseases, yet a very limited number of approved therapies are in this category.1, 2 One of the major hurdles for developing antibody and protein-based therapeutics for CNS diseases is the low brain entry, which is largely due to existence of the blood-brain barrier (BBB).3 The BBB is formed by the endothelial cell tight and adherent junctions that severely restrict the entry of macromolecules administered peripherally.4–6
In order to facilitate the brain entry of protein-based therapeutics, receptor-mediated transcytosis (RMT) has been exploited to deliver antibodies and proteins across the BBB.7–12 RMT takes advantage of endogenous transportation of biomolecules, such as transferrin (Tf), insulin, and leptin, by engaging receptors expressed on blood vessel endothelial cells. The transferrin receptor (TfR) is one of the most exploited for RMT and has successfully aided the delivery of antibody and protein therapeutics crossing the BBB and demonstrated enhanced therapeutic effects in both mouse and primate models.13–16 While the human TfR and mouse TfR are highly homologous, the anti-mouse and anti-human TfR antibodies reported in the literature are not cross reactive.15,16 In addition, sequences of the anti-mouse TfR antibodies reported previously are not available. There is a need to develop an anti-mouse TfR antibody for testing TfR-mediated brain entry of protein-based therapies in mouse models. Although the TfR-mediated brain entry has been reported in diseases such as Alzheimer’s disease and LSDs, the strategy has not been attempted to improve the brain entry and efficacy of cancer anti-angiogenic therapy.
GBM is the most common malignant brain cancer and has merely a 5% 5-year survival rate.3,17–19 Considering the highly vascularized nature of GBMs, anti-angiogenic therapy has been extensively exploited in clinical trials.20,21 Bevacizumab, an anti-vascular endothelial growth factor (VEGF) antibody, was approved as a second-line treatment for GBM by the US Food and Drug Administration. Although bevacizumab can shrink tumor size, the therapy shows no benefits in the overall survival of GBM patients.22–24 Aflibercept, a VEGFR-Fc fusion protein serves as a “trap” for VEGFA, VEGFB, and placental growth factor, has been studied for GBM due to its substantially higher affinity to VEGFA than bevacizumab.25,26 However, aflibercept was found not to provide significant benefits to GBM patients.25 Anti-angiogenesis therapies have been reported to restore the intactness of the BBB, and therefore further limits the brain access of these therapies and other protein or antibody-based therapeutics.20,27 The self-limiting nature of anti-angiogenesis therapy is known to restrict not only the antibody-based anti-angiogenic therapy, but also delivery of chemotherapy to CNS tumors.20,28,29 Therefore, overcoming the hurdle of the BBB is an attractive strategy to improve the therapeutic efficacy of aflibercept.
In this study, we developed an anti-mouse TfR antibody that does not compete with Tf binding. Using the anti-TfR antibody, we engineered a bispecific construct named VEGF-Trap/moAb4 that improved the delivery of aflibercept across the BBB by more than 10-fold. VEGF-Trap/moAb4 was confirmed to be distributed in the brain parenchyma. More importantly, the VEGF-Trap/moAb4 demonstrated significant improvement in inhibiting U-87 MG xenograft angiogenesis.
Results
Discovery of TfR antibodies
When evaluating therapeutics in mouse models engaging TfR-mediated BBB crossing, the antibodies need to bind mouse TfR (muTfR). To identify antibodies that bind muTfR, we designed a screening strategy from a phage-displayed single-chain variable fragment (scFv) human antibody library.30 Briefly, we first enriched and identified 38 scFv clones that bind to the muTfR extracellular domain (ECD) by phage panning and phage enzyme-linked immunosorbent assay (ELISA) from a total of 400 phage clones picked from the third round of panning (Figure 1(a)). The 38 muTfR-binding scFv clones were then converted into human IgG1 for binding confirmation by the bio-layer interferometry (BLI) assay. We confirmed 6 IgG1 antibodies that bind to muTfR in the BLI assay. Surprisingly, when the 6 IgG1 antibodies were screened for binding the muTfR expressed on cell surface of 293 T cells, only 1 of 6 antibodies (Ab4) bound cell surface-expressed TfR (Figure 1(b)).
Figure 1.
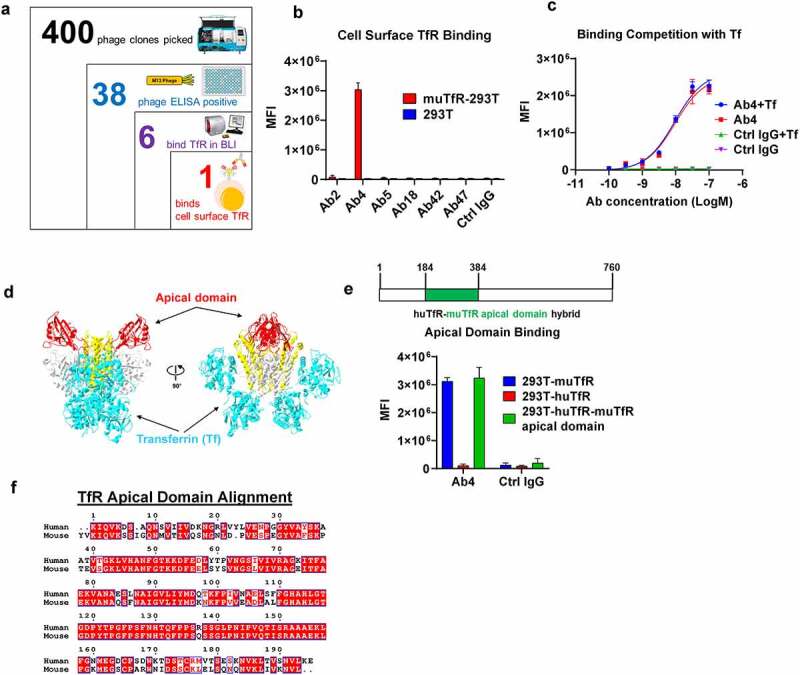
Screening and characterization of anti muTfR mAbs. a. The process of identifying anti-muTfR Ab4. A total of 400 scFv phage colonies were picked using the Qpix instrument from the 3rd round of panning output; and 38 clones were found to be positive in phage ELISA against muTfR-His. After sequencing, 6 unique scFv clones were converted to full IgG1. BLI assay showed 6 antibodies were able to bind to muTfR-His. b. Antibody Ab4 was identified to bind to surface expressed muTfR on HEK293T cells by flow cytometry. MFI is the mean fluorescent intensity of live cells, n = 3 independent repeats. c. Ab4 showed no competition with muTf in muTfR binding. HEK293T-muTfR was incubated with 10 μg/mL Ab4 or Ctrl IgG with or without 10 μM muTf. The amount of antibody binding to the cell surface was presented as the mean fluorescence intensity MFI, n = 3 independent repeats. d. Since muTfR structure is not available, we use the crystal structure of huTfR-huTf complex to demonstrate that the apical domain is distant from where Tf binds TfR.31 Images of the crystal structures were derived from PDB file 1SUV with proteins showing in ribbons. Both TfR and Tf were shown as dimers with their major domains identified: the apical domain of TfR (red), the helical domain of TfR (yellow), the protease-like domain of TfR (white), and Tf (cyan). e. Ab4 binds to muTfR and huTfR-muTfR apical domain hybrid expressed on HEK293T cell surface, but not to huTfR. MFI is the mean fluorescent intensity of live cells, n = 3 independent repeats. The huTfR-muTfR apical domain hybrid was constructed by replacing the apical domain of human TfR (aa 184–384) with the corresponding apical domain from the muTfR. f. Protein sequence alignment of the apical domains of human and muTfRs. Error bars in graphs of b, c, and e represent mean ± SD of three replications.
We next studied whether Ab4 competes with Tf, which is the natural ligand of TfR. Tf binds to TfR at high affinity at neutral pH.32 Tf presents in serum at a very high concentration of about 3 mg/mL.33 Therefore, any antibody that competes with Tf will not be able to bind to TfR, rendering the antibody ineffective. Moreover, competing with Tf binding to TfR may impede the normal iron delivery into cells, resulting in potential side effects. Using HEK293T cells overexpressing muTfR, we co-incubated Ab4 with an excessively high concentration of muTf (10 μM) and detected no differences in antibody binding to cell surface muTfR (Figure 1(c)), suggesting that Ab4 binds to muTfR specifically on cell surface without being blocked by Tf.
TfR has three domains: apical domain, protease-like domain, and helical domain.31,34 Tf interactions with TfR mostly involve the helical domain and the protease-like domain. Therefore, we hypothesize that Ab4 most likely binds to the apical domain of TfR (Figure 1(d)). To confirm this hypothesis, we constructed a chimeric huTfR receptor with its apical domain replaced by the corresponding apical domain from muTfR (huTfR-muTfR apical domain), and observed that the muTfR apical domain alone is sufficient to enable Ab4 binding to the chimeric huTfR-muTfR apical domain receptor (Figure 1(e)). It is noted that Ab4 does not cross-react with the human TfR (Figure 1(e)), although the percent of identity between the apical domains of the mouse and human TfRs is 70% (Figure 1(f)). This is consistent with previous reports that antibodies are less likely to cross-react between mouse and human TfRs.15,16
VEGF-Trap bispecific antibody characterization
We next created two bispecific antibodies by incorporating VEGF-Trap and muTfR Ab4 (Figure 2(a)). The VEGF-Trap was designed based on aflibercept, which is the fusion protein of the D2 domain of VEGFR1, D3 domain of VEGFR2, and human antibody crystallizable fragment (Fc)35 (Figure 2(a)). Ab4 was fused to the C terminus of the VEGF-Trap in an antibody-binding fragment (Fab) format. TfR-targeting antibodies with full Fc effector functions have been shown to deplete reticulocytes and cause acute toxicities.36 In order to avoid Fc-mediated effector functions, LALAPG mutations (L234A, L235A, and P329G) were introduced to abolish interactions with Fc receptors in humans and in mice.37,38 For the monovalent TfR bispecific design (VEGF-Trap/moAb4), we used the knobs-into-holes mutations (knob: T366W and S354C; hole: T366S, L368A, Y407V, and Y349C) to promote heterodimerization between the heavy chains.39 The TfR-antibody fusion arm was introduced with the “hole” mutations, while the other arm bears the “knob” mutations (Figure 2(a)). For the bivalent TfR bispecific design (VEGF-Trap/biAb4), a homodimer of VEGF-Trap fusion with Ab4 from the N-terminus to the C-terminus was used, resulting in bivalency for both VEGF-Trap and Ab4.
Figure 2.
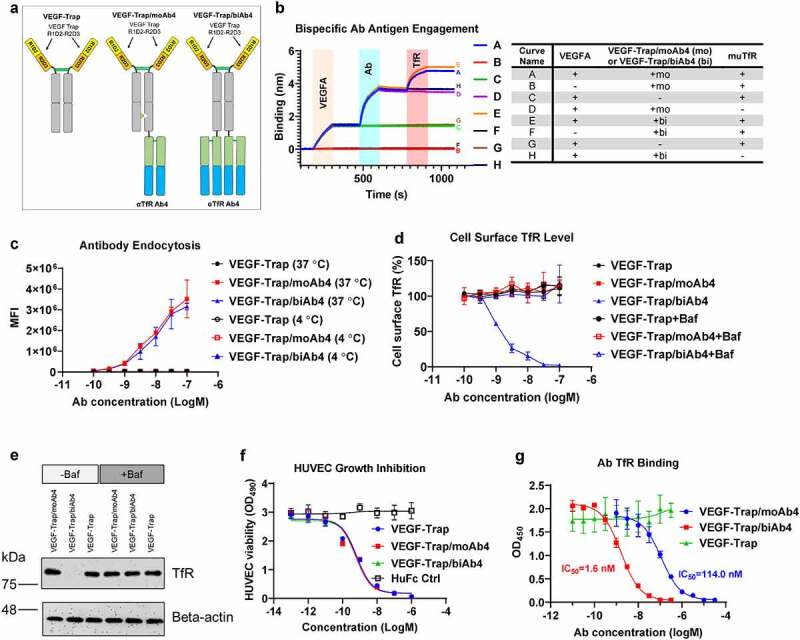
Characterization of VEGF-Trap/αTfR bispecific antibodies. a. Design of VEGF-Trap and VEGF-Trap/αTfR bispecific antibodies. In the VEGF-Trap/moAb4 design, knob-into-hole mutations were introduced to promote heterodimerization. The αTfR Fab was fused to the C-terminus of the CH3. Although not depicted, the human Fc regions contain LALAPG mutations to abolish Fc-mediated immune effector functions. b. Sandwich BLI assay showing the incorporation of both VEGF-Trap and αTfR into the bispecific constructs. The corresponding proteins involved in each curve were labeled in the shaded area and are detailed in the table next to the BLI diagram. c. αTfR bispecific antibodies showed dose-dependent endocytosis in Bend.3 cells. MFI is the mean fluorescent intensity of live cells, n = 3 independent repeats. d. VEGF-Trap/biAb4 showed dose-dependent reduction of cell surface muTfR level. The reduction of TfR surface level by VEGF-Trap/biAb4 was the result of lysosomal degradation. The cell surface level of TfR was measured by flow cytometry using a non-competing antibody against muTfR, n = 3 independent repeats. Baf is bafilomycin A1 at 100 nM. e. Western blotting showing the reduced level of total TfR in Bend.3 cells by VEGF-Trap/biAb4 treatment. The reduction of TfR level by VEGF-Trap/biAb4 was the result lysosomal degradation. Baf is bafilomycin A1 at 100 nM. f. VEGF-Trap/αTfR bispecific antibodies showed similar potency in inhibiting VEGFA-mediated HUVEC proliferation compared to VEGF-Trap, n = 3 independent repeats. g. Titration curves showing dose-dependent binding of TfR by VEGF-Trap/αTfR bispecific antibodies. VEGF-Trap/biAb4 showed significantly stronger binding to TfR than that of VEGF-Trap/moAb4, n = 3 independent repeats. Data points with error bars represent mean ± SD.
We used a BLI-based sandwich capture assay to characterize the bispecific antibodies. The bispecific antibodies were first captured onto sensors via VEGF165A. After equilibrium in blank buffer, the sensor-captured bispecific antibodies were then incubated with muTfR ECD. The VEGF-Trap/TfR bispecific antibodies were shown simultaneously binding to both VEGF165 and TfR (Figure 2(b), Curves A and E). To further validate the antigen binding, we introduced another three control groups that missed one of the three binding partners (VEGFA, antibody, or TfR). Omitting VEGFA showed a complete flat curve, which confirmed the observed binding signals in curves A and E are dependent on the proteins being captured by VEGFA (Figure 2(b), Curves B and F). Omitting antibody showed a flat curve when the sensors were dipped into TfR solution, which confirmed the observed TfR binding signals in curves A and E are dependent on the existence of antibodies (Figure 2(b), Curves C and G). Finally, the flat curves without TfR confirmed the observed binding signals in curves A and E were antibodies binding to TfR (Figure 2(b), Curves D and H). Taken together, these data showed that both the bivalent and monovalent TfR bispecific antibodies VEGF-Trap/moAb4 and VEGF-Trap/biAb4 can simultaneously engage both VEGF and TfR.
We have demonstrated that Ab4 does not interfere with TfR from binding to its ligand Tf, suggesting that the antibody will not interfere with the natural functions of TfR. We next tested if the bispecific antibodies can trigger cellular endocytosis, which is required for effective transcytosis of the antibodies through TfR. Using the mouse endothelial BEnd.3 cells, we showed concentration-dependent endocytosis of the bispecific antibodies bearing either bivalent or monovalent TfR Ab4 (Figure 2(c)). As a negative control, endocytosis was abolished when the cells and antibodies were incubated at 4°C (Figure 2(c)). Of note, bivalent and monovalent TfR Ab-bearing bispecific antibodies showed similar levels of endocytosis across all concentrations (Figure 2(c)).
We next tested whether antibody-mediated TfR endocytosis has an effect on the level of TfR cell surface expression. Naturally, TfR endocytosis delivers Tf into endosomes, where the Tf releases iron, and the TfR-Tf complex recycles back to the cell surface.33 We measured TfR cell surface levels after incubation with bispecific antibodies. Similar to the VEGF-Trap negative control, monovalent TfR Ab-bearing bispecific antibody VEGF-Trap/moAb4 showed no reduction of cell surface TfR levels (Figure 2(d)). In contrast, the bivalent bispecific antibody (VEGF-Trap/biAb4)-treated BEnd.3 cells demonstrated concentration-dependent reduction of cell surface TfR levels, and at 100 nM, the surface TfR was reduced to undetectable levels (Figure 2(d)). As a control, co-incubation with the lysosomal inhibitor Baf abolished the reduction of surface TfR levels by the bivalent bispecific antibody VEGF-Trap/biAb4 (Figure 2(d)). Collectively, the data suggests that the bivalent TfR antibody VEGF-Trap/biAb4 induces the decrease of cell surface TfR level by promoting its lysosomal degradation. To rule out the possibility that the bivalent antibody-induced TfR re-localization intracellularly, we treated BEnd.3 cells with the two bispecific antibodies and measured total TfR protein levels by Western blotting. The total TfR levels in BEnd.3 cells are similar between VEGF-Trap-treated and VEGF-Trap/moAb4-treated groups (Figure 2(e)). In contrast, the VEGF-Trap/biAb4 treatment significantly diminished the total TfR protein levels (Figure 2(e)). As a control, co-incubation of lysosomal inhibitor Baf was able to prevent the decrease of total TfR levels mediated by VEGF-Trap/biAb4 treatment, confirming that the bivalent antibody induces TfR lysosomal degradation.
Since the human endothelial cell line HUVEC has been the gold standard to evaluate the efficacy of anti-angiogenesis therapeutics,35,40 we studied the inhibition of VEGFA-stimulated HUVEC cell proliferation by the two bispecific antibodies under the conditions of depleted growth factors and cytokines. VEGF-Trap/biAb4 and VEGF-Trap/moAb4 bispecific antibodies showed similar dose-dependent inhibition of VEGFA-mediated HUVEC proliferation as the VEGF-Trap positive control (Figure 2(f)). In comparison, treatment with the same concentration of HuFc Ctrl (human Fc fragment without VEGF Trap or antibody fragments) showed no inhibition in the concentration range tested, suggesting that the inhibition effects are VEGF-Trap-specific.
We next determined the binding affinity of the TfR bispecific antibodies to TfR by a competition ELISA. The ELISA plate was coated with muTfR ECD and a series of concentrations of TfR bispecific antibodies were added in the presence of 1 nM of biotinylated Ab4. The 1 nM Ab4 concentration was predetermined to be sensitive enough in quantifying the amount of unbound TfR, yet the concentration is low enough to not interfere with the binding of bispecific TfR antibodies. Both the VEGF-Trap/moAb4 and VEGF-Trap/biAb4 bispecific antibodies showed dose-dependent binding to muTfR, as indicated by the decreased OD450 signals from the biotinylated Ab4 (Figure 2(g)). In contrast, VEGF-Trap/biAb4 showed a significantly stronger (about 100-fold) occupation (binding) of TfR as compared to the VEGF-Trap/moAb4 (Figure 2(g)). The data suggests that avidity played a significant role in the TfR binding for the bivalent bispecific antibody.
Biodistribution of VEGF-Trap TfR bispecific antibodies in the brain and serum of mice
We next studied the biodistribution of VEGF-Trap/αTfR bispecific antibodies in brain and blood of mice. A day after a single intraperitoneal (IP) injection at 20 mg/kg of the antibodies (designs illustrated in Figure 3(a)), we collected the serum and the brains; the 20 mg/kg dose was chosen based on previous work by others.15,16,27 The brains were collected after thorough phosphate-buffered saline (PBS) perfusion to avoid the interference from residue blood in the vasculature. We first used sandwich ELISA to quantify the concentration of bispecific antibodies inside the brain. In the sandwich ELISA, the bispecific antibody was first captured by plate-coated VEGFA, and then the captured antibody was detected by a secondary antibody. The VEGF-Trap/moAb4 bispecific antibody showed a significantly higher concentration in the brain than that of the VEGF-Trap/biAb4 and the VEGF-Trap (Figure 3(b)). The VEGF-Trap/moAb4 showed a 10-fold increase in brain concentration over the VEGF-Trap/Ctrl and a 5-fold increase over the VEGF-Trap/biAb4 (Figure 3(b)). Even though the VEGF-Trap/biAb4 induces significant TfR lysosomal degradation, it still showed a 2-fold increase in brain concentration over the VEGF-Trap/Ctrl 24 hours after injection (Figure 3(b)). A 7-day time course study showed that the concentrations of VEGF-Trap/moAb4 in the brain decreased as time elapsed (Figure 3(c)). From day 1 to day 5 after injection, the brain concentrations of VEGF-Trap/moAb4 were significantly higher than VEGF-Trap/Ctrl and VEGF-Trap/biAb4. The brain concentration of VEGF-Trap/biAb4 dropped quickly to a level similar to VEGF-Trap/Ctrl on day 3 after injection. The brain concentration of VEGF-Trap/moAb4 also returned to the basal level on day 7.
Figure 3.
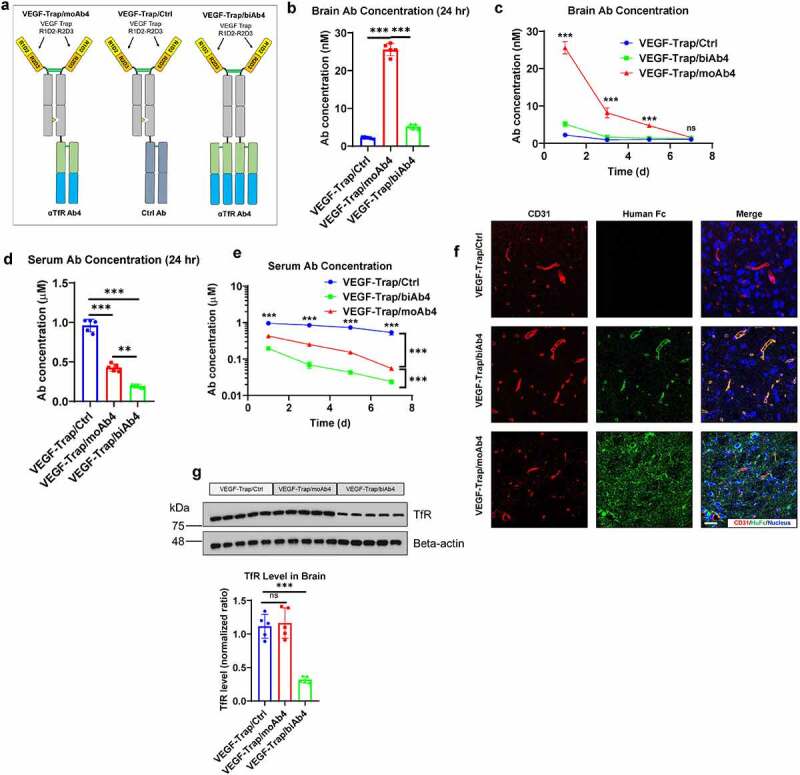
Characterization of VEGF-Trap/αTfR bispecific antibody brain entry. a. Illustration showing the design of bispecific antibodies used in this figure. b. Antibody concentrations in perfused brains 24 hours after the mice were treated with designated antibodies at 20 mg/kg through intraperitoneal injection. c. Antibody concentrations in perfused brains at designated time points after treatment as described in b. d. Antibody concentrations in serum 24 hours after the mice were treated as described in b. e. Antibody concentrations in serum at designated time points after treatment as described in b. f. Immunofluorescent staining of perfused mouse brain tissues 24 hours after treatment as described in b. Scale bar = 20 μm. g. Western blotting showing the level of total TfR in mouse brain lysates 24 hours after treatment as described in b. The Western blotting signals were quantified and shown in a bar graph. For all the animal studies, n = 5 mice per group. Error bars represent mean ± SD. For the statistical analysis, ns = not statistically different, *** P < .001, two-tailed Student t-test.
We quantified serum concentrations of the bispecific antibodies by the same sandwich ELISA as described above. Serum concentrations of VEGF-Trap/moAb4 and VEGF-Trap/biAb4 bispecific antibodies were significantly lower than that of the VEGF-Trap/Ctrl (Figure 3(d)). Of note, the VEGF-Trap/biAb4 concentration was 60% of the VEGF-Trap/moAb4 in the sera (Figure 3(d)). We also tracked the serum antibody concentrations over the time course of a week (Figure 3(e)). The VEGF-Trap/Ctrl showed only a 30% decrease over the time course of a week; in comparison, the VEGF-Trap/moAb4 and VEGF-Trap/biAb4 bispecific antibodies showed significantly faster clearance over the same time period (Figure 3(e)). In comparison to VEGF-Trap/moAb4, VEGF-Trap/biAb4 showed 3–5-fold lower serum concentrations over all the time points, indicating a faster clearance.
We then mapped the location of the antibodies in the brain by immunofluorescence staining. As shown in Figure 3(f), the VEGF-Trap/moAb4 showed prominent brain parenchyma distribution. CD31 was co-stained to discern blood vessels and determine if the antibody is trapped inside the blood vessel. In contrast, the VEGF-Trap/biAb4 showed localization within the blood vessel, and therefore is likely to be trapped in the blood vessel without entering the brain parenchyma (Figure 3(f)). Of note, the VEGF-Trap/Ctrl showed almost no distribution in either the blood vessel or the brain parenchyma (Figure 3(f)).
Since the VEGF-Trap/biAb4 induced quick degradation of TfR in vitro, we tested if the same effects occur in vivo. The VEGF-Trap/biAb4 induced a significant decrease in TfR concentration in the brain as measured by Western blotting (Figure 3(g)). In comparison, VEGF-Trap/Ctrl and the VEGF-Trap/moAb4 showed no differences in the amount of brain TfR (Figure 3(g)). Collectively, these data suggests that the VEGF-Trap/biAb4 bispecific antibody induced degradation of TfR in vivo.
Inhibition of tumor angiogenesis
Aflibercept and other anti-VEGF antibodies have been studied in clinical trials for the treatment of GBM. Although brain tumors have higher blood vessel permeability due to angiogenesis, anti-angiogenesis therapy often reduces the permeability by its blood vessel normalization effect. It has been reported that restored BBB integrity upon anti-angiogenesis therapy limits the brain access to therapeutics.27 We tested if delivering VEGF-Trap via TfR bispecific antibody can improve the anti-angiogenesis effect of VEGF-Trap by overcoming the BBB blockade in the U-87 MG model, which is a human GBM model with known BBB leakage.41
Three antibodies were used in the U-87 MG GBM studies (Figure 4(a)). We first validated the change of BBB permeability after VEGF-Trap treatment. To assess BBB permeability, animals were injected with fluorescently labeled albumin molecules 2 hours before sacrifice. Ctrl/moAb4-treated mice showed significant BBB leakage as indicated by the albumin signals in the tumor (Figure 4(b)). In comparison, the VEGF-Trap/Ctrl treatment showed significantly lower albumin signals in the tumor. The low amount of albumin detected in tumors indicates low BBB permeability. Similar to VEGF-Trap/Ctrl, the amount of detected albumin in VEGF-Trap/moAb4-treated tumors were also low (Figure 4(b)). Collectively, these data suggest that VEGF-Trap treatment restores the intactness of BBB with low permeability to macromolecules. We then tested if the TfR-targeted VEGF-Trap/moAb4 bispecific antibody can enhance the inhibition of angiogenesis by the increased brain access of VEGF-Trap. The angiogenesis in the tumor was characterized by immunostaining of the endothelial cell marker CD31.42 VEGF-Trap/moAb4 bispecific antibody treatment showed significantly reduced CD31 intensity in the tumor in comparison to Ctrl/moAb4 and the VEGF-Trap/Ctrl, and no difference of CD31 intensity between Ctrl/moAb4 and VEGF-Trap/Ctrl was observed (Figure 4(c)). Collectively, the data suggest enhanced VEGF-Trap/moAb4 bispecific brain entry translated into improved anti-angiogenesis efficacy.
Figure 4.
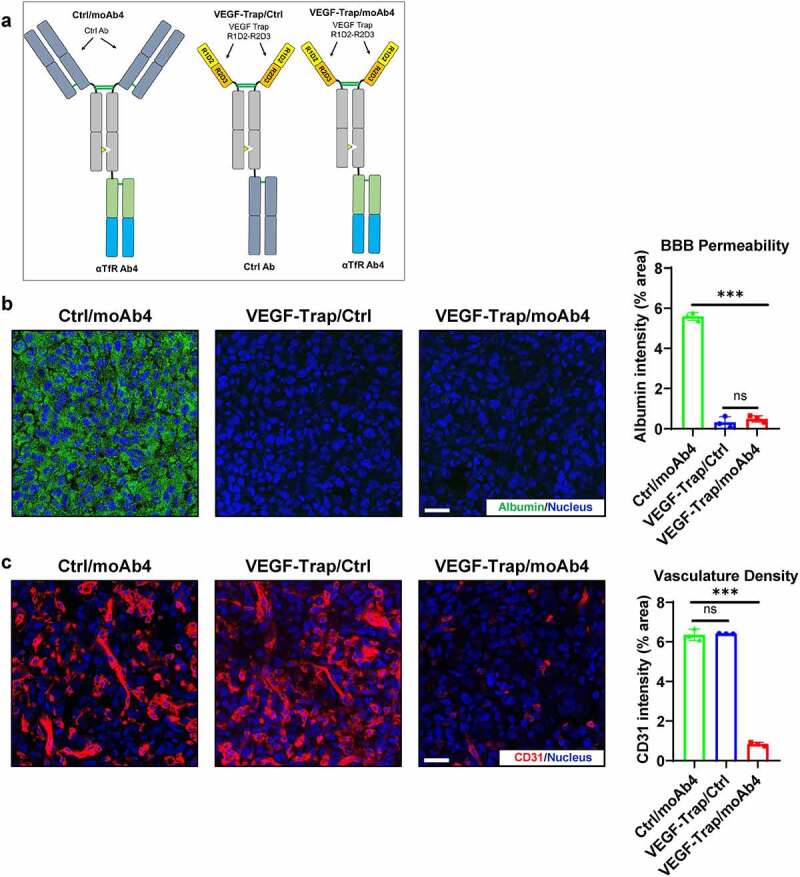
VEGF-Trap/moAb4 bispecific antibody significantly enhanced the anti-angiogenic efficacy of VEGF-Trap. a. Illustration showing the design of bispecific antibodies used in this figure. b. Immunofluorescent staining showing the level of fluorescently labeled albumin in U-87 MG tumors, which serves as an indicator of BBB integrity. Scale bar = 20 μm. The immunofluorescence data was quantified and showed in a bar graph, n = 3 independent mice. c. Immunofluorescent staining showing the level of CD31 in U-87 MG tumors. Scale bar = 20 μm. The immunofluorescence data was quantified and showed in a bar graph, n = 3 independent mice. Bar graphs with error bars represent mean ± SD. For the statistical analysis, ns = not statistically different, *** P < .001, two-tailed Student t-test.
Discussion
Although TfR has been exploited in facilitating the delivery of antibody and protein therapeutics into the brain for neurodegenerative diseases such as Alzheimer’s disease,43 a similar strategy has not been used in improving the delivery of anti-angiogenic protein therapies for brain cancer. In this study, we validated the reduced BBB permeability after VEGF-Trap treatment, which limits the delivery of proteins through the BBB. We, therefore, engineered a bispecific antibody strategy to enable VEGF-Trap brain access through the BBB. To obtain a suitable αTfR antibody for the bispecific constructs, we panned our phage-displayed human scFv library. During the screening process, we have noticed that antibody clones showing positive binding in ELISA or BLI assays against TfR ECD have a higher rate of failure in binding cell surface TfR. Therefore, we included cell surface receptor binding as a key parameter in the screening protocol to avoid false positives.
Since TfR exists as a homodimer, we did not use BLI to measure the antibody affinity to avoid complications from the avidity effect. Therefore, we choose to detect the amount of available TfR using a low concentration of biotinylated Ab4. The concentration is too low to pose significant competition to the antibodies. A similar approach has been used in previous studies that characterize the binding affinity of antibodies against TfR and CD98.16,44 The avidity effects from bivalent αTfR resulted in a significantly stronger occupation of TfR than the monovalent αTfR. The stronger binding to TfR by the bivalent antibody may explain the fact that the bivalent TfR VEGF-Trap antibody was trapped inside blood vessels instead of crossing the BBB into the brain parenchyma. In addition, the lack of dissociation between bivalent anti-TfR and TfR may lead to more lysosomal degradation of the TfR/antibody complex. Future work is warranted to validate this hypothesis to provide insights on the impact of antibody valency and dissociation on TfR degradation.
After a single IP injection in mice, we observed that brain concentration of the monovalent αTfR bispecific molecule (VEGF-Trap/moAb4) was 10-fold higher that of the control (VEGF-Trap/Ctrl). Immunofluorescent imaging validated that VEGF-Trap/moAb4 indeed crossed the BBB and showed a broad distribution inside the brain parenchyma. Although the bivalent αTfR (VEGF-Trap/biAb4) also showed increased brain concentration over the control as determined by ELISA, immunofluorescence imaging revealed that VEGF-Trap/biAb4 was trapped inside the vasculature instead of entering the brain parenchyma. The increased concentration of bivalent αTfR is the result of antibody binding to the vasculature, which is consistent with previous reports.14,16 Our finding is also consistent with a previous study that showed monovalent αTfR crossed the vasculature while the bivalent αTfRs were trapped inside the vasculature.14
The preference of TfR-based transcytosis toward monovalent αTfR over bivalent αTfR could be also explained by the fact that bivalent αTfR induces lysosomal degradation of TfR, reducing both the cell surface and overall cellular TfR levels. The reduced TfR level in turn limits bivalent αTfR crossing the BBB. In addition, bivalent αTfR binds 100-fold stronger to TfR than monovalent αTfR due to avidity effects and the stronger binding limits the efficient dissociation of bivalent αTfR after being transported through the luminal side of endothelial cells. Although both formats bind equally well in BLI assay and showed similar levels of endocytosis, the combination of slower dissociation from TfR and induction of TfR degradation results in the suboptimal brain distribution of the bivalent αTfR. It has been reported previously that affinity and avidity are the key criteria in developing TfR antibody-based brain delivery platforms.14,16 The ideal TfR antibody should not have too high affinity, which will limit the release of transported antibodies, and monovalent TfR antibody is preferred over bivalent.
Bivalent TfR binding is expected to have a faster internalization rate. In this study, we quantified the antibody endocytosis 2 hours after adding the antibody to cells, and no differences were observed for VEGF-Trap/moAb4 and VEGF-Trap-biAb4, which suggests that an endocytosis plateau has reached at 2 hours. The reason that we did not measure the antibody endocytosis at shorter time intervals is that the in vivo exposure time was beyond 2 hours. In addition, endocytosis may be affected by the cell surface availability of TfR available for uptake. The fact that bivalent TfR binding induces TfR lysosomal degradation also complicates the comparison of endocytosis rate.
We showed that the VEGF-Trap/αTfR has a significantly quicker clearance than Ctrl IgG in the sera, which may be explained by the broad distribution of TfR in the BBB and accumulation of the VEGF-Trap/αTfR in the brain. Our findings are consistent to other bispecific antibodies targeting the TfR, which exhibit a quicker serum clearance than the antibody without αTfR.15,16 It was reported that bevacizumab treatment restored BBB integrity in the U-87 MG GBM mouse model.27 We observed a similar phenomenon that VEGF-Trap treatment alone significantly restored the integrity of BBB in the U-87 MG GBM mouse model. The reduced BBB permeability could further restrict the amount of VEGF-Trap from entering the brain, and therefore limiting the therapeutic effect.
We found that the VEGF-Trap showed minimal differences in tumor blood vessel density while the VEGF-Trap delivered by the monovalent αTfR showed significant inhibition of angiogenesis, which is likely due to enhanced brain entry of the monovalent VEGF-Trap/αTfR bispecific antibody. Although with significant inhibition of tumor angiogenesis, we did not observe any benefits in the survival rate or the tumor growth inhibition. It has been previously reported that the anti-angiogenic therapy bevacizumab demonstrated only marginal tumor growth inhibition in a GBM rat model.45 Although bevacizumab showed inhibition of angiogenesis, it induced a more hypoxic tumor microenvironment as indicated the upregulation of the HIF1α, PI3K, and Wnt pathways.45 The shifted tumor metabolism to glycolysis in the hypoxic tumor microenvironment underlines the need to combine anti-angiogenic therapies with therapeutics that can overcome the metabolic adaptions, which warrants future studies.
Materials and methods
Cell lines
HEK293T, U-87 MG, and BEnd.3 cell lines were acquired from the American Type Culture Collection (ATCC) and cultured in DMEM+10% fetal bovine serum (FBS). HUVEC was also acquired from the ATCC and maintained in F-12 K medium supplemented with 0.1 mg/mL heparin, 10% FBS, 30 μg/mL Endothelial Cell Growth Supplement.
Panning of phage-displayed antibody library
A phage-displayed scFv antibody library was prepared previously.30 Panning of the library for muTfR specific antibodies was carried out as described previously with modifications.30 Briefly, MaxiSorp Nunc-Immuno tubes (Thermo Fisher Scientific) were coated with 20 μg/mL muTfR-His (Sino Biological 50741-M07H) in DPBS overnight at 4°C. Unbound antigen was removed after washing with DPBS. After blocking the surface with 5% milk in DPBS, the phage library was incubated with the coated-muTfR for 2 hours at room temperature in 5% milk. After washing with PBS+0.05% tween-20 to remove unbound phage, captured phage was eluted by incubating with 100 mM triethylamine for 20 min. Eluted phage-infected log-phase growing E. coli TG1 were amplified on 2× YTAG agar 500 cm2 square plate (Corning) at 30°C overnight. The amplified phage-infected TG1 was used to prepare the phage for the next round of panning using the M13 KO7 helper phage. The enrichment process was done in three rounds using the output from the previous round as the input for the next round.
After three rounds of panning, the output titer was measured and single colonies were used to prepare phage for ELISA. High-binding ELISA plates (Corning) were coated with muTfR-His (Sino Biological 50741-M07H) at 2 μg/mL overnight at 4°C. After blocking with 5% milk in PBS, phage prepared from single TG1 colonies in 5% milk PBS was incubated with coated muTfR for 1 hour at room temperature. After washing with PBS+0.05% Tween-20, anti-M13-HRP (Santa Cruz Biotechnology sc-53004) was added at 1:2000 concentration and incubated for 1 hour at room temperature. After washing with PBS+0.05% Tween-20, TMB substrate (Thermo Fisher Scientific) was added and incubated for 5 min before stopping by 1 N H2SO4. OD values were read at 450 nm. Top 20% high-binding clones were selected. Phagemids were extracted using Qiagen BioRobot Universal System in 96-well format. After DNA sequencing, sequences were analyzed using the IMGT V-quest service to identify antibody sequences with unique CDR3 regions.
Conversion of phage scFv to IgG
Unique scFv clones were converted into human IgG1 using mixed universal primers with degeneracy as reported previously.30 Individual heavy and light variable chains were amplified using PrimeStar GXL polymerase (Takara Bio). Gel-purified variable chain fragments were cloned into digested vectors using In-Fusion HD cloning enzyme mix (Takara Bio). After the converted plasmid was sequenced, sequences of verified IgG plasmids were transfected into Expi293 cells at the 2-mL scale. After culturing for 5 days, cells were removed and antibody-containing supernatant was collected for screening assay.
For milligram-scale antibody purification, Expi293-produced antibodies were purified using CaptivA Protein A affinity resin (Repligen) and eluted with 0.1 M glycine (pH = 2.5) and then neutralized with 1/20 volume 1 M Tris-HCl (pH = 9). Buffer exchange to DPBS was done using Amicon Ultra-15 ultrafiltration units (Mw cutoff = 30k) (MilliporeSigma).
TfR-expressing 293 T generation
HEK293T expressing full-length mouse and human TfR or the chimeric receptor were generated using lentivirus. Briefly, the receptor genes were cloned into the pCDH-CMV-MCS-EF1α-Puro vector downstream of the CMV promoter. The 293 T cell lines were generated by transducing with packaged lentivirus (generated using the transfer plasmid, pCMV-VSV-G (Addgene 8454), pCMV delta R8.2 (Addgene 12263)). Cells expressing the transgene were selected by 1 μg/mL puromycin until a sufficient number of cells with transgene emerged.
Bispecific antibody validation by BLI
tStreptavidin sensors (Fortebio) were used to capture biotinylated VEGFA proteins (Sino Biological 11066-H27H-B). During all incubation steps, samples were kept at room temperature with 1000 rpm shaking. In the VEGFA loading step, 100 nM biotinylated VEGFA proteins were incubated with the sensors for the designated time. In the bispecific antibody interaction steps, 200 nM antibodies were used. In the muTfR incubation step, 100 nM muTfR-His (Sino Biological 50741-M07H) were used. Between incubations, the sensors were dipped into blank kinetic buffers to allow the free dissociation of proteins.
Antibody endocytosis
A total of 5 × 104 BEnd.3 cells were incubated with antibodies at designated concentrations and temperature for 2 hours. The antibodies were pre-labeled with Alexa Fluor 488 NHS (Thermo Fisher). After incubation, unbound antibodies were removed by centrifugation at 500 g for 5 min. Trypan blue solution (0.2%) was incubated with cells for 5 min to quench the cell surface-bound antibody fluorescence. Cells were then transferred into a V-bottom 96-well plate and washed twice by 350 g 5 min centrifugation. The endocytosis was quantified using the iQue3 high throughput flow cytometer (Sartorius) with at least 10,000 live cells collected.
Immunoblotting
Cell lysate or brain lysates were obtained by lysing cells or brain tissues using NP-40 lysis buffer (1% NP40, 50 mM Tris-HCl, pH = 8, 150 mM NaCl) with Halt™ Protease and Phosphatase Inhibitor Cocktail (100X) (Thermo Fisher) for 1 hour with shaking. After removing debris by centrifugation, the total protein amount was normalized by Pierce BCA Protein Assay Kit (Thermo Fisher). Protein samples were resolved by 10% sodium dodecyl sulfate-polyacrylamide gels (Biorad) and later transferred onto Immun-Blot PVDF membranes (Biorad). Proteins were probed with specific primary antibodies and secondary antibodies diluted in 5% bovine serum albumin (BSA) TBST.46–48 Antibodies used were TfR (Santa Cruz, 1:1000, sc-59112) and actin-beta (Santa Cruz, 1:1000, sc-8432). The immunoreactive bands were visualized with the West Pico PLUS Chemiluminescent Substrate (Thermo Fisher). The immunoreactive bands were quantified using ImageJ. Three independent treatment replicates were conducted with the representative immunoblot shown.
HUVEC cell growth assay
HUVEC cells maintained in the full growth medium were seeded 1d before the assay into 96-well plates at the density of 1 × 104 per well in assay medium (F12K+ 2% FBS) with 50 ng/μL human VEGFA (Sino Biological 11066-HNAH) but without the growth factor supplement. To start the assay, the medium was replaced with assay medium with designated antibodies and cultured for another 2 days. The cell viability was quantified using MTS assay (Promega) according to the manufacturer’s protocol.
TfR occupation assay
High-binding ELISA plates (Corning) were coated with muTfR-His (Sino Biological 50741-M07H) at 2 μg/mL overnight at 4°C. After blocking with 1% BSA PBS, individual antibodies (at designated concentrations) and 1 nM biotinylated TfR Ab4 in 1% BSA PBS were incubated with coated muTfR for 2 hours at room temperature. After washing with PBS+0.05% Tween-20, streptavidin-HRP (Jackson ImmunoResearch 016–030-084) was added at 1:5000 concentration and incubated for 1 hour at room temperature. After washing with PBS+0.05% Tween-20, TMB substrate (Thermo Fisher Scientific) was added and incubated for 5 min before being stopped by 1 N H2SO4. OD values were read at 450 nm.
Antibody brain distribution study
The animal experiments were conducted according to the institutional guidelines with approved protocol AWC-19-0051. BALB/C mice (female, 8-week-old, Jackson Laboratory) were randomly grouped into five mice per group. Mice received IP injections of antibodies (20 mg/kg) in 0.1 mL DPBS. At the designated time points, blood was collected via tail vein and mice then received transcardial perfusion at 2 mL/min by DPBS for 10 min. Brains were collected with half flash-frozen in liquid nitrogen and another half prepared for cryo-sectioning. For immunofluorescence, the half mouse brains were dipped into 4% paraformaldehyde for 1d, then 30% sucrose for 2d before being embedded into OCT medium (Sakura) and sectioned using Leica Cryostat CM1950 into 40 μm floating sections. The floating sections were stored at 4°C in PBS with 0.01% sodium azide until use.
Measurement of antibody concentration in brain and serum
High-binding ELISA plates (Corning) were coated with human VEGFA (Sino Biological 11066-HNAH) at 2 μg/mL overnight at 4°C. After blocking with 1% BSA PBS, individual brain lysates were incubated with coated VEGFA for 2 hours at room temperature. After washing with PBS+0.05% Tween-20, anti-human Fc-HRP (Jackson ImmunoResearch, 109–035-088) was added at 1:5000 concentration and incubated for 1 hour at room temperature. After washing with PBS+0.05% Tween-20, TMB substrate (Thermo Fisher Scientific) was added and incubated for 5 min before being stopped by 1 N H2SO4. OD values were read at 450 nm. Standard curves were established using purified corresponding bispecific antibodies following the same method described above.
Immunofluorescence staining of mouse brains
Floating sections were first blocked in 1% BSA PBS with 0.3% Triton X-100 for 2 hours, then stained with corresponding antibodies: CD31 (1:500, R&D system AF3628), human Fc (1:1000, Jackson ImmunoResearch 109–035-190), or streptavidin-Alexa Fluor 488 (1:500, Jackson ImmunoResearch 016–540-084) in 1% BSA PBS with 0.3% Triton X-100 for overnight at 4°C with gentle rocking. After washing in PBS 0.3% Triton X-100, corresponding secondary antibodies with fluorescent labeling were incubated with brain slices for 2 hours at 4°C with gentle rocking. The nucleus was stained with TO-PRO-3 (2 µM) in DPBS for 30 min. Brains slices were imaged using a Leica confocal microscope.
U-87 MG xenograft model
NSG mice (female, 8-week-old, Jackson Laboratory) were randomly grouped into three mice per group. The mice were implanted with 5 × 105 U-87 MG cells in the caudate nucleus using a stereotaxic injection frame. Five days after tumor implantation, mice received an IP injection with designated antibodies at 20 mg/kg in 0.2 mL sterile PBS. Four days after injection, all mice were sacrificed and the brains were preserved and cryo-sectioned as described above. For observing mouse survival, the body weight was recorded daily. Any mouse that reaches a 20% bodyweight decrease is considered reaching the experiment endpoint and was euthanized.
Protein sequence analysis
Protein sequence alignment was performed using the T-Coffee multiple sequence alignment server and the alignment figures were generated in ESPript – http://espript.ibcp.fr.49 The crystal structure was visualized using DeepView-Swiss-PdbViewer, ver 4.1.
Statistical analysis
GraphPad Prism (v8, GraphPad Software) was used to generate plots and perform statistical analysis. Statistical differences were determined to be significant at p < .05 using a two-tailed Student t-test. Data are presented as mean ± SD.
Acknowledgments
We thank Dr. Zhengmei Mao at UTHealth for the using the confocal microscope.
Funding Statement
This study was funded in part by The Cancer Prevention and Research Institute of Texas (CPRIT) Grants RP150551 and RP190561 and Welch Foundation grant no. AU-0042-20030616.
Abbreviations
Ab,Antibody
ALS,Amyotrophic lateral sclerosis
ATCC,American Type Culture Collection
BBB,Blood–brain barrier
BLI,Bio-layer interferometry
CNS,Central nervous system
ECD,Extracellular domain
ELISA,Enzyme-linked immunosorbent assay
FBS,Fetal bovine serum
Fc,Fragment crystallizable
GBM,Glioblastoma
IP,intraperitoneal
LSD,Lysosomal storage disease
PBS,phosphate-buffered saline
PGF,Placental growth factor
RMT,Receptor-mediated transcytosis
scFv,Single-chain variable fragment
Tf,Transferrin
TfR,Transferrin receptor
VEGF,Vascular Endothelial Growth Factor
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding authors on reasonable request.
Authors’ contributions
Conceptualization, PZ, NZ, ZA; Methodology, PZ, XF, LL, YA, PG; Resources, NZ, ZA, KT; writing—original draft preparation, PZ, ZA; writing—review and editing, NZ, ZA, PZ; Supervision, NZ, ZA; Project administration, NZ, ZA; Funding acquisition, NZ, ZA. All authors read and approved the final manuscript.
Disclosure statement
The University of Texas System has filed a patent application on the TfR targeting antibodies and PZ, NZ, and ZA are named inventors of the patent application.
References
- 1.Freskgård PO, Urich E.. Antibody therapies in CNS diseases. Neuropharmacology. 2017;120:38–12. doi: 10.1016/j.neuropharm.2016.03.014. [DOI] [PubMed] [Google Scholar]
- 2.Wahl A-S, Correa D, Imobersteg S, Maurer MA, Kaiser J, Augath MA, Schwab ME, Imobersteg S, Maurer MA, Kaiser J, et al. Targeting therapeutic antibodies to the CNS: a comparative study of intrathecal, intravenous, and subcutaneous Anti-Nogo a antibody treatment after stroke in rats. Neurotherapeutics. 2020;17(3):1153–59. doi: 10.1007/s13311-020-00864-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Taylor OG, Brzozowski JS, Skelding KA. Glioblastoma multiforme: an overview of emerging therapeutic targets. Front Oncol. 2019;9:963. doi: 10.3389/fonc.2019.00963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Daneman R, Prat A. The blood-brain barrier. Cold Spring Harb Perspect Biol. 2015;7(1):a020412–a020412. doi: 10.1101/cshperspect.a020412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sharif Y, Jumah F, Coplan L, Krosser A, Sharif K, Tubbs RS. Blood brain barrier: a review of its anatomy and physiology in health and disease. Clinical Anatomy. 2018;31(6):812–23. doi: 10.1002/ca.23083. [DOI] [PubMed] [Google Scholar]
- 6.Chow BW, Gu C. The molecular constituents of the blood–brain barrier. Trends Neurosci. 2015;38(10):598–608. doi: 10.1016/j.tins.2015.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Banks WA. From blood-brain barrier to blood-brain interface: new opportunities for CNS drug delivery. Nat Rev Drug Discov. 2016;15(4):275–92. doi: 10.1038/nrd.2015.21. [DOI] [PubMed] [Google Scholar]
- 8.Barar J, Rafi MA, Pourseif MM, Omidi Y. Blood-brain barrier transport machineries and targeted therapy of brain diseases. Bioimpacts. 2016;6(4):225–48. doi: 10.15171/bi.2016.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.de Boer AG, van der Sandt IC, Gaillard PJ. The role of drug transporters at the blood-brain barrier. Annu Rev Pharmacol Toxicol. 2003;43(1):629–56. doi: 10.1146/annurev.pharmtox.43.100901.140204. [DOI] [PubMed] [Google Scholar]
- 10.Jones AR, Shusta EV. Blood-brain barrier transport of therapeutics via receptor-mediation. Pharm Res. 2007;24(9):1759–71. doi: 10.1007/s11095-007-9379-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pardridge WM. Drug transport across the blood–brain barrier. Journal of Cerebral Blood Flow & Metabolism. 2012;32(11):1959–72. doi: 10.1038/jcbfm.2012.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Razpotnik R, Novak N, Čurin Šerbec V, Rajcevic U. Targeting malignant brain tumors with antibodies. Front Immunol. 2017;8:1181–1181. doi: 10.3389/fimmu.2017.01181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kariolis MS, Wells RC, Getz JA, Kwan W, Mahon CS, Tong R, Kim DJ, Srivastava A, Bedard C, Henne KR, et al. Brain delivery of therapeutic proteins using an Fc fragment blood-brain barrier transport vehicle in mice and monkeys. Sci Transl Med. 2020;12(545). doi: 10.1126/scitranslmed.aay1359. [DOI] [PubMed] [Google Scholar]
- 14.Niewoehner J, Bohrmann B, Collin L, Urich E, Sade H, Maier P, Rueger P, Stracke JO, Lau W, Tissot AC, et al. Increased brain penetration and potency of a therapeutic antibody using a monovalent molecular shuttle. Neuron. 2014;81(1):49–60. doi: 10.1016/j.neuron.2013.10.061. [DOI] [PubMed] [Google Scholar]
- 15.Yu YJ, Atwal JK, Zhang Y, Tong RK, Wildsmith KR, Tan C, Bien-Ly N, Hersom M, Maloney JA, Meilandt WJ, et al. Therapeutic bispecific antibodies cross the blood-brain barrier in nonhuman primates. Sci Transl Med. 2014;6(261):261ra154. doi: 10.1126/scitranslmed.3009835. [DOI] [PubMed] [Google Scholar]
- 16.Yu YJ, Zhang Y, Kenrick M, Hoyte K, Luk W, Lu Y, Atwal J, Elliott JM, Prabhu S, Watts RJ, et al. Boosting brain uptake of a therapeutic antibody by reducing its affinity for a transcytosis target. Sci Transl Med. 2011;3(84):84ra44. doi: 10.1126/scitranslmed.3002230. [DOI] [PubMed] [Google Scholar]
- 17.Wen PY, Weller M, Lee EQ, Alexander BM, Barnholtz-Sloan JS, Barthel FP, Batchelor TT, Bindra RS, Chang SM, Chiocca EA, et al. Glioblastoma in adults: a Society for Neuro-Oncology (SNO) and European Society of Neuro-Oncology (EANO) consensus review on current management and future directions. Neuro-Oncology. 2020;22(8):1073–113. doi: 10.1093/neuonc/noaa106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tan AC, Ashley DM, López GY, Malinzak M, Friedman HS, Khasraw M. Management of glioblastoma: state of the art and future directions. CA Cancer J Clin. 2020;70(4):299–312. doi: 10.3322/caac.21613. [DOI] [PubMed] [Google Scholar]
- 19.Omuro A, DeAngelis LM. Glioblastoma and other malignant gliomas: a clinical review. JAMA. 2013;310(17):1842–50. doi: 10.1001/jama.2013.280319. [DOI] [PubMed] [Google Scholar]
- 20.Thompson EM, Frenkel EP, Neuwelt EA. The paradoxical effect of bevacizumab in the therapy of malignant gliomas. Neurology. 2011;76(1):87–93. doi: 10.1212/WNL.0b013e318204a3af. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Das S, Marsden PA, Phimister EG. Angiogenesis in glioblastoma. N Engl J Med. 2013;369(16):1561–63. doi: 10.1056/NEJMcibr1309402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Friedman HS, Prados MD, Wen PY, Mikkelsen T, Schiff D, Abrey LE, Yung WK, Paleologos N, Nicholas MK, Jensen R, et al. Bevacizumab alone and in combination with irinotecan in recurrent glioblastoma. J Clin Oncol. 2009;27(28):4733–40. doi: 10.1200/JCO.2008.19.8721. [DOI] [PubMed] [Google Scholar]
- 23.Kreisl TN, Kim L, Moore K, Duic P, Royce C, Stroud I, Garren N, Mackey M, Butman JA, Camphausen K, et al. Phase II trial of single-agent bevacizumab followed by bevacizumab plus irinotecan at tumor progression in recurrent glioblastoma. J Clin Oncol. 2009;27(5):740–45. doi: 10.1200/JCO.2008.16.3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wong ET, Hess KR, Gleason MJ, Jaeckle KA, Kyritsis AP, Prados MD, Levin VA, Yung WK. Outcomes and prognostic factors in recurrent glioma patients enrolled onto phase II clinical trials. J Clin Oncol. 1999;17(8):2572–78. doi: 10.1200/JCO.1999.17.8.2572. [DOI] [PubMed] [Google Scholar]
- 25.de Groot JF, Lamborn KR, Chang SM, Gilbert MR, Cloughesy TF, Aldape K, Yao J, Jackson EF, Lieberman F, Robins HI, et al. Phase II study of aflibercept in recurrent malignant glioma: a North American Brain Tumor Consortium study. J Clin Oncol. 2011;29(19):2689–95. doi: 10.1200/JCO.2010.34.1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gaya A, Tse V. A preclinical and clinical review of aflibercept for the management of cancer. Cancer Treat Rev. 2012;38(5):484–93. doi: 10.1016/j.ctrv.2011.12.008. [DOI] [PubMed] [Google Scholar]
- 27.Stegmayr C, Oliveira D, Niemietz N, Willuweit A, Lohmann P, Galldiks N, Shah NJ, Ermert J, Langen KJ. Influence of bevacizumab on blood-brain barrier permeability and O-(2-(18)F-Fluoroethyl)-l-tyrosine uptake in rat gliomas. J Nucl Med. 2017;58(5):700–05. doi: 10.2967/jnumed.116.187047. [DOI] [PubMed] [Google Scholar]
- 28.Claes A, Wesseling P, Jeuken J, Maass C, Heerschap A, Leenders WP. Antiangiogenic compounds interfere with chemotherapy of brain tumors due to vessel normalization. Mol Cancer Ther. 2008;7(1):71–78. doi: 10.1158/1535-7163.MCT-07-0552. [DOI] [PubMed] [Google Scholar]
- 29.Bergers G, Hanahan D. Modes of resistance to anti-angiogenic therapy. Nat Rev Cancer. 2008;8(8):592–603. doi: 10.1038/nrc2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhao S, Zhu Y, Schultz RD, Li N, He Z, Zhang Z, Caron A, Zhu Q, Sun K, Xiong W, et al. Partial leptin reduction as an insulin sensitization and weight loss strategy. Cell Metab. 2019;30(4):706–719.e6. doi: 10.1016/j.cmet.2019.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cheng Y, Zak O, Aisen P, Harrison SC, Walz T. Structure of the human transferrin receptor-transferrin complex. Cell. 2004;116(4):565–76. doi: 10.1016/S0092-8674(04)00130-8. [DOI] [PubMed] [Google Scholar]
- 32.Giannetti AM, Snow PM, Zak O, Björkman PJ, Thornton J. Mechanism for multiple ligand recognition by the human transferrin receptor. PLoS Biol. 2003;1(3):E51. doi: 10.1371/journal.pbio.0000051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wessling-Resnick M. Crossing the iron gate: why and how transferrin receptors mediate viral entry. Annu Rev Nutr. 2018;38(1):431–58. doi: 10.1146/annurev-nutr-082117-051749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lawrence CM, Ray S, Babyonyshev M, Galluser R, Borhani DW, Harrison SC. Crystal structure of the ectodomain of human transferrin receptor. Science. 1999;286(5440):779–82. doi: 10.1126/science.286.5440.779. [DOI] [PubMed] [Google Scholar]
- 35.Holash J, Davis S, Papadopoulos N, Croll SD, Ho L, Russell M, Boland P, Leidich R, Hylton D, Burova E, et al. VEGF-Trap: a VEGF blocker with potent antitumor effects. Proc Natl Acad Sci U S A. 2002;99(17):11393–98. doi: 10.1073/pnas.172398299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Couch JA, Yu YJ, Zhang Y, Tarrant JM, Fuji RN, Meilandt WJ, Solanoy H, Tong RK, Hoyte K, Luk W, et al. Addressing safety liabilities of TfR bispecific antibodies that cross the blood-brain barrier. Sci Transl Med. 2013;5(183):183ra57, 1–12. doi: 10.1126/scitranslmed.3005338. [DOI] [PubMed] [Google Scholar]
- 37.Wang X, Mathieu M, Brezski RJ. IgG Fc engineering to modulate antibody effector functions. Protein Cell. 2018;9(1):63–73. doi: 10.1007/s13238-017-0473-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schlothauer T, Herter S, Koller CF, Grau-Richards S, Steinhart V, Spick C, Kubbies M, Klein C, Umaña P, Mössner E. Novel human IgG1 and IgG4 Fc-engineered antibodies with completely abolished immune effector functions. Protein Eng Des Sel. 2016;29(10):457–66. doi: 10.1093/protein/gzw040. [DOI] [PubMed] [Google Scholar]
- 39.Schaefer W, Regula JT, Bähner M, Schanzer J, Croasdale R, Dürr H, Gassner C, Georges G, Kettenberger H, Imhof-Jung S, et al. Immunoglobulin domain crossover as a generic approach for the production of bispecific IgG antibodies. Proc Natl Acad Sci U S A. 2011;108(27):11187–92. doi: 10.1073/pnas.1019002108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Robinson CJ, Das RG, Stammers R, Rafferty B. The World Health Organization reference reagent for vascular endothelial growth factor, VEGF165. Growth Factors. 2006;24(4):285–90. doi: 10.1080/01674820601051034. [DOI] [PubMed] [Google Scholar]
- 41.Brighi C, Reid L, Genovesi LA, Kojic M, Millar A, Bruce Z, White AL, Day BW, Rose S, Whittaker AK, et al. Comparative study of preclinical mouse models of high-grade glioma for nanomedicine research: the importance of reproducing blood-brain barrier heterogeneity. Theranostics. 2020;10(14):6361–71. doi: 10.7150/thno.46468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Scholz A, Harter PN, Cremer S, Yalcin BH, Gurnik S, Yamaji M, Di Tacchio M, Sommer K, Baumgarten P, Bähr O, et al. Endothelial cell-derived angiopoietin-2 is a therapeutic target in treatment-naive and bevacizumab-resistant glioblastoma. EMBO Mol Med. 2016;8(1):39–57. doi: 10.15252/emmm.201505505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yu YJ, Watts RJ. Developing therapeutic antibodies for neurodegenerative disease. Neurotherapeutics: The Journal of the American Society for Experimental NeuroTherapeutics. 2013;10(3):459–72. doi: 10.1007/s13311-013-0187-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zuchero YJ, Chen X, Bien-Ly N, Bumbaca D, Tong RK, Gao X, Zhang S, Hoyte K, Luk W, Huntley MA, et al. Discovery of novel blood-brain barrier targets to enhance brain uptake of therapeutic antibodies. Neuron. 2016;89(1):70–82. doi: 10.1016/j.neuron.2015.11.024. [DOI] [PubMed] [Google Scholar]
- 45.Keunen O, Johansson M, Oudin A, Sanzey M, Rahim SA, Fack F, Thorsen F, Taxt T, Bartos M, Jirik R, et al. Anti-VEGF treatment reduces blood supply and increases tumor cell invasion in glioblastoma. Proc Natl Acad Sci U S A. 2011;108(9):3749–54. doi: 10.1073/pnas.1014480108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhong L, Wang Z, Wang D, Wang Z, Martens YA, Wu L, Xu Y, Wang K, Li J, Huang R, et al. Amyloid-beta modulates microglial responses by binding to the triggering receptor expressed on myeloid cells 2 (TREM2). Mol Neurodegener. 2018;13(1):15. doi: 10.1186/s13024-018-0247-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhao Y, Wu X, Li X, Jiang LL, Gui X, Liu Y, Sun Y, Zhu B, Piña-Crespo JC, Zhang M, et al. TREM2 is a receptor for β-amyloid that mediates microglial function. Neuron. 2018;97(5):1023–1031.e7. doi: 10.1016/j.neuron.2018.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen H-M, van der Touw W, Wang YS, Kang K, Mai S, Zhang J, Alsina-Beauchamp D, Duty JA, Mungamuri SK, Zhang B, et al. Blocking immunoinhibitory receptor LILRB2 reprograms tumor-associated myeloid cells and promotes antitumor immunity. J Clin Invest. 2018;128(12):5647–62. doi: 10.1172/JCI97570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Robert X, Gouet P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 2014. Web Server issue;42(W1):W320–4. doi: 10.1093/nar/gku316. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets used and/or analyzed during the current study are available from the corresponding authors on reasonable request.