

Abstract
Children and young adults with mutant forms of ataxia telangiectasia mutated (ATM), a kinase involved in DNA damage signaling and mitochondrial homeostasis, suffer from recurrent respiratory infections, immune deficiencies, and obstructive airways disease associated with disorganized airway epithelium. We previously showed in mice how Atm was required to mount a protective immune memory response to influenza A virus [IAV; Hong Kong/X31 (HKx31), H3N2]. Here, Atm wildtype (WT) and knockout (Atm-null) mice were used to investigate how Atm is required to regenerate the injured airway epithelium following IAV infection. When compared with WT mice, naive Atm-null mice had increased airway resistance and reduced lung compliance that worsened during infection before returning to naïve levels by 56 days postinfection (dpi). Although Atm-null lungs appeared pathologically normal before infection by histology, they developed an abnormal proximal airway epithelium after infection that contained E-cadherin+, Sox2+, and Cyp2f2+ cells lacking secretoglobin family 1 A member 1 (Scgb1a1) protein expression. Patchy and low expression of Scgb1a1 were eventually observed by 56 dpi. Genetic lineage tracing in HKx31-infected mice revealed club cells require Atm to rapidly and efficiently restore Scgb1a1 expression in proximal airways. Since Scgb1a1 is an immunomodulatory protein that protects the lung against a multitude of respiratory challenges, failure to efficiently restore its expression may contribute to the respiratory diseases seen in individuals with ataxia telangiectasia.
Keywords: airway club cells, ataxia telangiectasia, influenza A virus, stem cells
INTRODUCTION
Ataxia telangiectasia (A-T) is a rare disorder caused by mutations in the ataxia-telangiectasia mutated (ATM) gene that results in an abnormal form of the protein ATM. ATM is a large phosphatidylinositol 3 kinase-related kinase (PIKK) involved in repairing DNA double-stranded breaks (DSBs), regulating oxidative stress, and maintaining mitochondrial homeostasis (1, 2). Because ATM has a wide array of functions, A-T manifests with a variety of symptoms including neurodegeneration, radiosensitivity, susceptibility to cancers, recurrent respiratory infections, immune dysfunction, and lung disease (1, 3, 4). Premature death of individuals with A-T occurs at 25 yr of age with 23%–46% of deaths attributed to respiratory diseases, yet studies investigating pulmonary responses to pathogens are lacking (5–7). Individuals with A-T are susceptible to recurrent respiratory infections that can lead to the development of bronchiectasis. Lung disease is primarily associated with bacterial infection but not opportunistic pathogen infections (8–10). Pneumonia is often preceded by respiratory viral infections, which could account for why viruses have also been seen in morbid individuals with A-T. Individuals with A-T may not be able to control infections because they suffer from immune dysfunction, generally characterized by low immunoglobulin levels and lymphopenia primarily with reduced T cell numbers (11–13). Consistent with Atm playing a critical role in controlling recurrent infections, we previously identified a defect in influenza A virus (IAV)-specific antibody production in mice lacking Atm due to a deletion of exon 4 leading to increased morbidity following viral challenge (14). Failure to produce the full spectrum of antibodies to a primary viral infection helps to explain why individuals with A-T suffer from recurrent respiratory infections.
The frequency, severity, and spectrum of respiratory infections, however, does not fully correlate with the extent of immune defect because individuals with A-T without reported immune defects can still develop lung disease (4, 8), suggesting there is a possible immune cell-extrinsic factor that has a role in the susceptibility of individuals with A-T to recurrent infections. Consistent with this idea, cells from patients with A-T are typically more sensitive to increased oxidative stress and produce more reactive oxygen species (ROS) in response to Streptococcus pneumonia (15, 16). However, increased oxidative stress is not seen in our exon 4 mutant Atm mouse, suggesting oxidative stress may not be required for immune-specific dysfunction (14). Disordered airway epithelium has also been reported in a limited number of individuals with A-T who succumb to respiratory infections (8, 17). Increased airway resistance has been seen in Atm-null mice harboring a mutation in the kinase domain that also exhibits increased airway epithelial injury when exposed to hydrochloric acid (18). How Atm regulates proper airway lung development or regeneration is not known.
We have been infecting mice with heterologous strains of influenza A virus (IAV) because this approach revealed how Atm was required to generate protective antigen-specific antibodies needed to defend against subsequent infections (14). Mice are initially infected with IAV A/Hong Kong/X31 (HKx31, H3N2), a mouse-adapted IAV that is generally considered a low pathogenic virus because it primarily targets airway epithelial cells with minimal morbidity and mortality (19). One airway epithelial cell type that can be infected by HKx31 are club cells. Club cells are best known for their ability to produce secretoglobins particularly secretoglobin family 1 A member 1 [Scgb1a1; also referred to as club cell secretory protein (CCSP), CC10, CC16, or uteroglobin], which is involved in the maintenance of airway epithelial integrity, detoxification of inhaled substances, and blunting inflammation (20–23). These cells are also progenitor cells because they can self-renew and differentiate into airway ciliated and mucus-producing goblet cells (24). Basal cells of the trachea and upper bronchi are capable of differentiating into airway club cells and ciliated cells during development, at steady state, and even after injury (25, 26). Here, we use global Atm-null and airway-specific Atm-null mice to investigate whether dysregulated airway epithelium seen in individuals with A-T is related to a defect in lung development or the ability to effectively restore airway club cells following IAV infection.
MATERIALS AND METHODS
Animals
Mice containing Atm wildtype alleles [AtmWT/WT, referred to as wildtype (WT)], homozygous Atm mutant alleles lacking exon 4 (Atmf/f, Atm-null), or Atm floxed alleles with exon 4 flanked by loxP sites (AtmF/F) have been previously described (27). Scgb1a1-Cre/ERTM (Stock No: 016225; the Jackson Laboratory, Bar Harbor, ME) and Rosa26-mTmG (Stock No. 007676; The Jackson Laboratory) mice were bred with AtmF/F mice to generate club cell-specific GFP+ Atm conditional knockout mice (Scgb1a1-Cre/ERTM+;AtmF/F;Rosa26-mTmG) and club cell-specific GFP+ Atm control mice (Scgb1a1-Cre/ERTM+;AtmWT/F;Rosa26-mTmG). Adult animals were administered 0.1 mg/g tamoxifen (Sigma, St. Louis, MO) in corn oil (Thermo Fisher Scientific, Waltham, MA) intraperitoneally (ip) daily for four consecutive days and infected with IAV 14 days after the last dose of tamoxifen. Pulmonary function tests were performed on mice with a Flexivent small animal ventilator (SCIREQ, Montreal, QC, Canada) as previously described (28). Animal experiments followed Institutional Animal Care and Use Committee guidelines and were approved by the University Committee on Animal Resources at the University of Rochester.
Influenza Infection
Adult mice (8–10-wk old) were infected intranasally (in) with 105 plaque-forming units (PFU) influenza A virus (IAV) HKx31 (H3N2) in 30 µL PBS (Gibco, Grand Island, NY). Virus was obtained from the National Institutes of Health (NIH) Biodefense and Emerging Infections (BEI) Research Resources Repository, NIAID, NIH: Kilbourne F108: A/Aichi/2/68 (HA, NA) x A/Puerto Rico/8/34 (H3N2), Reassortant X-31, NR-3483 (referred to as HKx31 here).
Immunohistochemistry
The right lobes of the lung were inflation fixed, paraffin embedded, sectioned, and stained with hematoxylin and eosin (H + E) or with antibodies against specific proteins (Table 1; 29). TUNEL assay was performed per the manufacturer’s protocol (EMD Millipore Corp., Temecula, CA). Fluorescence was visualized with a Nikon E-800 fluorescence microscope (Nikon Instruments, Melville, NY), and images were captured with a SPOT-RT digital camera (Diagnostic Instruments, Sterling Heights, MI). Brightness was adjusted with Microsoft PowerPoint (Microsoft Corporation, Redmond, WA).
Table 1.
List of antibodies used for immunohistochemical staining
| Antigen | Species | Manufacturer |
|---|---|---|
| Scgb1a1 | Goat | Barry Stripp (Cedars-Sinai, Los Angeles, CA) |
| Anti-epithelial cadherin (E-cadherin) clone 2410 | Rabbit | Cell Signaling Technologies (Danvers, MA) |
| Anti-cytochrome P4502f2 | Mouse | Santa Cruz Biotechnology (Dallas, TX) |
| Anti-acetylated tubulin clone 6-11-B-1 | Mouse | Sigma Chemical Company |
| Anti-FoxJ1 clone 2A5 | Mouse | Invitrogen (Waltham, MA) |
| Anti-mucin 5AC clone 45M1 | Mouse | Thermo Fisher Scientific |
| Anti-Sox2 | Rabbit | 7Hills (Cincinnati, OH) |
| Anti-proSPC | Rabbit | 7Hills (Cincinnati, OH) |
| Anti-podoplanin (T1α) clone 8.1.1 | Hamster | Developmental Studies Hybridoma Bank (Iowa City, IA) |
| Anti-Ki-67 | Rabbit | Abcam (Cambridge, MA) |
| Anti-cytokeratin 5 (K5) clone EP1601Y | Rabbit | Thermo Fisher Scientific |
Scgb1a1, secretoglobin family 1 A member 1.
Quantitating Immunohistochemistry
Five fields of view of the lung parenchyma were selected under DAPI fluorescence and imaged. Fields that contained airways and large blood vessels were not used in the analysis. Quantification was performed with ImageJ (National Institutes of Health, Bethesda, MD).
Naphthalene Administration
Adult mice were administered one dose of 275 mg/kg ip naphthalene in corn oil or vehicle as control (30).
Flow Cytometry
Lungs were digested with dispase (5 mg/mL in PBS) as previously described (14). Cells were incubated with anti-CD16/32 (Fc block; BD Biosciences, San Jose, CA), stained with anti-mouse epithelial cell adhesion molecule (EpCAM; CD326), and run through an LSRII flow cytometer (BD Biosciences). The data were collected using FACS Diva software, and FlowJo software (v. 10) was used for final analysis (FlowJo, LLC).
Statistical Analysis
Data were graphed, and statistical analysis was performed in Prism software (GraphPad Software, La Jolla, CA). Pulmonary function data were analyzed for respiratory system resistance (Rrs), Newtonian airway resistance (RN), and respiratory system compliance (Crs), and statistical analyses were performed as previously described (28). Data presented represent means ± standard error (SE). Student’s t tests were performed and P < 0.05 was considered significant between groups.
RESULTS
Airway Histology and Respiratory Mechanics Are Abnormal in Atm-Null Mice
Lung resistance and compliance were analyzed in adult naïve wildtype (WT) and Atm-null mice. Increased respiratory system resistance (Rrs), which includes measurements of both airway and tissue components, was detected in naïve Atm-null mice compared with WT controls (Fig. 1A). This change may reflect greater resistance in large conducting airways as measured by Newtonian resistance (RN; Fig. 1B). Atm-null mice also had a slight but significant decrease in lung compliance (Crs) before infection (Fig. 1C). In addition, naïve Atm-null mice had a downshifted pressure-volume (PV) curve (Fig. 1D). These data reveal naïve Atm-null mice have altered respiratory system mechanics compared with WT mice (Figs. 1 and 2), suggesting Atm may play a role in lung development. In contrast, histologic analysis failed to identify any change in cellularity or structure of the lung. Airways of WT and Atm-null mice were lined by a single layer of well-ordered columnar epithelial cells marked by secretoglobin family 1 A member 1 (Scgb1a1) expression, the main protein expressed by airway club cells (Figs. 1E and 3A; 31). These findings suggest that Atm by itself may not be sufficient to produce the disorganized airway epithelium seen in postmortem tissues of individuals with A-T.
Figure 1.
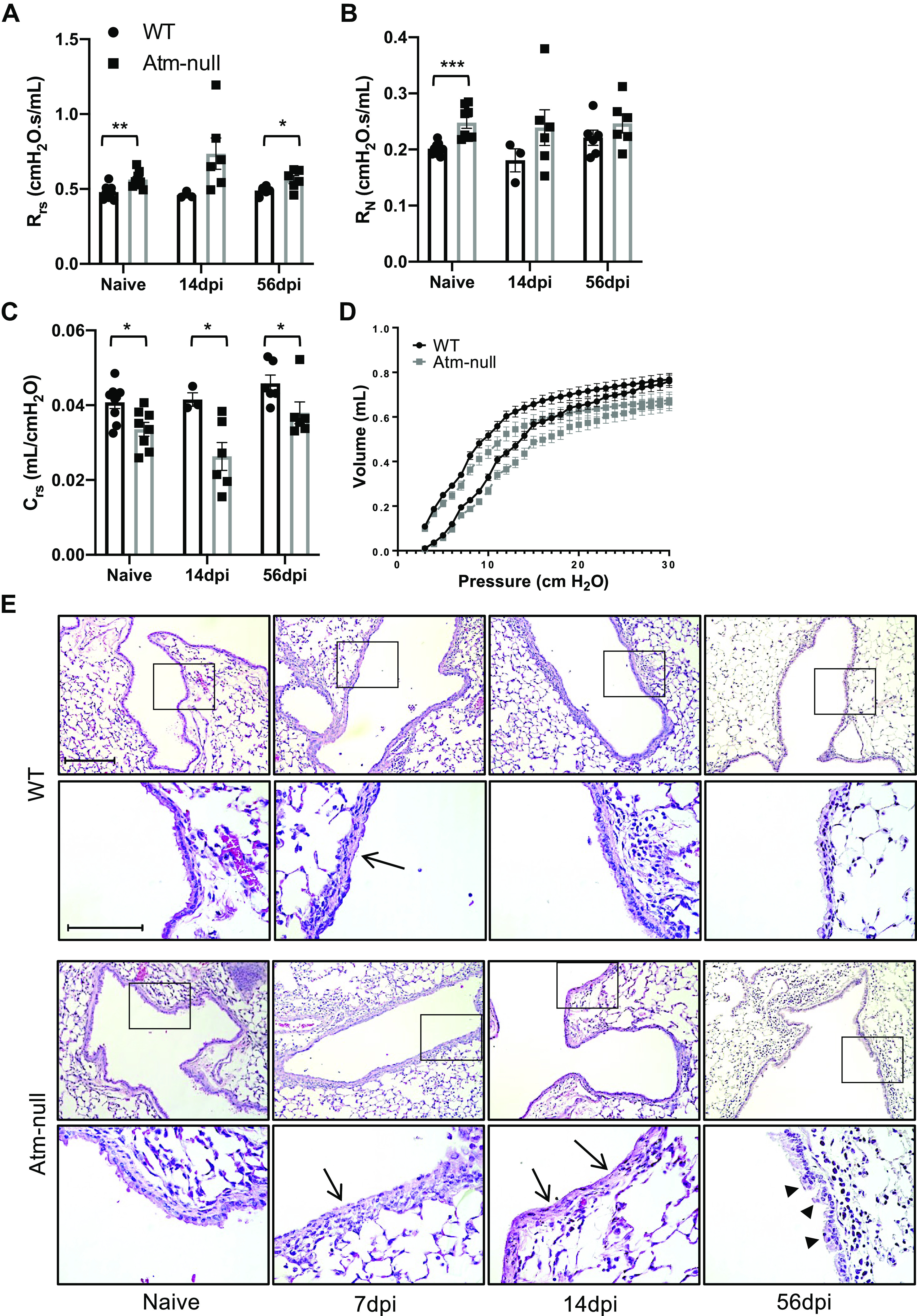
Atm-null mice have abnormal respiratory mechanics in naïve mice. WT and Atm-null mice were infected with 105 plaque-forming units (PFUs) of HKx31. Naïve and HKx31 infected WT (black bars) and Atm-null (gray bars) mice were ventilated on a Flexivent small animal ventilator. Lung function was analyzed on naïve mice and at 14 and 56 dpi. A: total respiratory system resistance (Rrs), Newtonian resistance (RN; B), and compliance (Crs; C) were graphed. D: pressure-volume (PV) curve from naïve WT and Atm-null mice. Values represent means ± standard error with individual values shown as dots. E: lungs were harvested from naïve mice and at 7, 14, and 56 dpi. Representative images of lung sections from WT (top) and Atm-null (bottom) mice stained with hematoxylin and eosin. Inset boxes are enlarged in the lower images. Arrows indicate regions of epithelial thinning. Arrowheads indicate regions of epithelial thickening. Scale bar = 100 µm and inset scale bar = 200 µm. Representative images of n ≥ 3 mice/time point/group are shown. P values were determined by Student’s t test. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001; n ≥ 3/time point/group. dpi, days postinfection; HKx31, Hong Kong/X31; WT, wildtype.
Figure 2.

Naïve Atm-null mice have abnormal respiratory mechanics without lower inspiratory capacity. Naïve and HKx31-infected WT (black bars) and Atm-null (gray bars) mice were ventilated on a Flexivent small animal ventilator. Inspiratory capacity (IC), tissue damping (G), tissue elastance (H), static lung compliance (Cst), tissue hysteresivity (η), elastance (Ers), and area were analyzed on naïve mice and at 14 and 56 dpi. Values represent means ± standard error with individual values shown as dots. P values were determined by Student’s t test. *P ≤ 0.05, **P ≤ 0.01; n ≤ 3/time point/group. dpi, days postinfection; HKx31, Hong Kong/X31; WT, wildtype.
Figure 3.
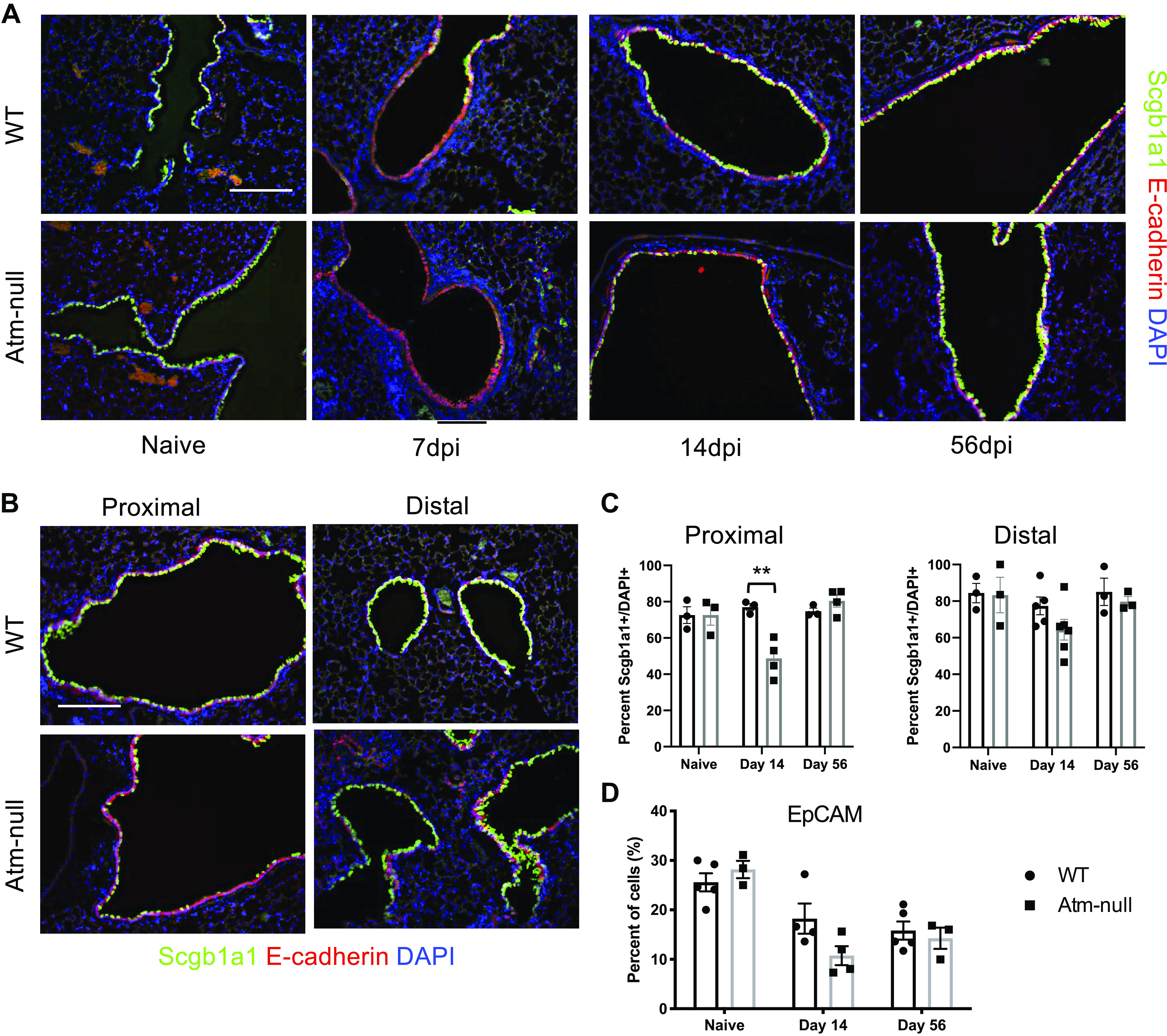
Atm-null mice have aberrant expression of Scgb1a1 following IAV infection. WT and Atm-null mice were infected with 105 PFU of HKx31. Lungs were harvested from naïve mice and at 7, 14, and 56 dpi. A: sections of lung were stained with antibodies against Scgb1a1 (green) and epithelial cadherin (E-cadherin; red), and counterstained with DAPI (blue). B: representative images of proximal and distal airways of WT and Atm-null mice collected at 14 dpi and stained as in A. C: the number of Scgb1a1+ cells in A were counted and graphed as a percentage of the total number of DAPI+ cells per airway. Proximal airways were determined to be larger airways within a section, whereas distal airways are those that contain BADJ. D: whole lung homogenates were stained for EpCAM and run on a flow cytometer. EpCAM+ cells were graphed as a percentage of total cells analyzed. Scale bars = 200 µm. Representative images of n ≥ 3 mice/time point/group are shown. P values were determined by Student’s t test. **P ≤ 0.01; n ≥ 3/time point/group. BADJ, bronchoalveolar duct junction; dpi, days postinfection; EpCAM, epithelial cell adhesion molecule; HKx31, Hong Kong/X31; IAV, influenza A virus; PFU, plaque-forming unit; Scgb1a1, secretoglobin family 1 A member 1; WT, wildtype.
To determine if airway pathology seen in individuals with A-T was related to respiratory infection, adult WT and Atm-null mice were infected with a sublethal dose of influenza A virus (IAV; HKx31, H3N2). We measured lung mechanics and analyzed lung sections at 7, 14, and 56-days postinfection (dpi). At 7 dpi, the airways of both WT and Atm-null mice were composed of a layer of flattened, thin cells (Fig. 1E; arrows). WT airways at 14 and 56 dpi returned to a naïve cuboidal epithelial structure with very few regions of disorganized epithelium. In contrast, Atm-null airways were composed of regions of squamous cells and epithelial thinning at 14 dpi among regions of epithelial thickening (Fig. 1E; arrowheads). Interestingly, the differences seen in airway resistance and lung compliance between WT and Atm-null mice before infection became more pronounced during infection but then returned to naïve differences by 56 dpi (Fig. 1A).
Atm-Null Mice Have an Aberrant Recovery of Scgb1a1 Expression following IAV Infection
We investigated whether Atm influenced the expression of Scgb1a1, the primary secretoglobin produced by airway epithelial club cells. After infection, both WT and Atm-null mice lost expression of Scgb1a1 at 7 dpi. The expression was largely restored in WT mice by 14 dpi and appeared similar to naïve mice by 56 dpi (Fig. 3A; top). In contrast, patchy expression of Scgb1a1 was detected in Atm-null mice at 14 dpi, although Scgb1a1 expression in Atm-null mice was also largely restored by 56 dpi (Fig. 3A; bottom). The delayed recovery of Scgb1a1 expression in Atm-null mice prompted us to take a closer look at the airways of infected mice. We found that WT mice regained Scgb1a1 protein expression by 14 dpi along the entire airway. In contrast, the lack of Scgb1a1 recovery in Atm-null mice was limited to the large proximal airways, although the distal airways typically regained Scgb1a1 expression similar to WT controls (Fig. 3, B and C). Here, we define proximal airways as the larger airways in a lung section and distal airways as those that contain the bronchoalveolar duct junction (BADJ). Despite the lack of Scgb1a1 larger airways contained E-cadherin-positive cells, suggesting that they had replenished epithelial cells and there were no differences in the percentage of epithelial cells as determined by epithelial cell adhesion molecule (EpCAM)+ cells in the total lung at 14 and 56 dpi (Fig. 3, A and D). Taken together, these data suggest that Atm-null mice have a delayed ability to restore Scgb1a1 expression in epithelial cells of the proximal airway.
TUNEL and Ki-67 Staining Are Not Altered in Atm-Null Mouse Airways
We next analyzed apoptosis by TUNEL and proliferation using Ki-67 staining during IAV infection to determine if the abnormal expression of Scgb1a1 in Atm-null mice is due to altered cell death or lack of airway regeneration. IAV-induced cell death occurs early postinfection, whereas there is active virus replication, immune-mediated killing, and damage of infected and bystander cells (32, 33). At 3, 5, and 7 dpi, there was no difference in TUNEL between WT and Atm-null airways (Fig. 4, A and B). As apoptotic airway cells are cleared, new cells must proliferate to regenerate the damaged airway epithelium. There was limited cell proliferation in naive WT and Atm-null mice, however, Ki-67 staining increased by 3 dpi (Fig. 4, C and D). At 7 dpi, ∼25%–30% of cells in the lung were Ki-67+ (Fig. 4D). However, there were no differences in the percent of Ki-67+ cells in the lung between WT and Atm-null mice (Fig. 4D). Atm-deficiency does not alter the extent of apoptosis or cell proliferation in the infected lung, suggesting Atm-null mice do not increased epithelial injury.
Figure 4.
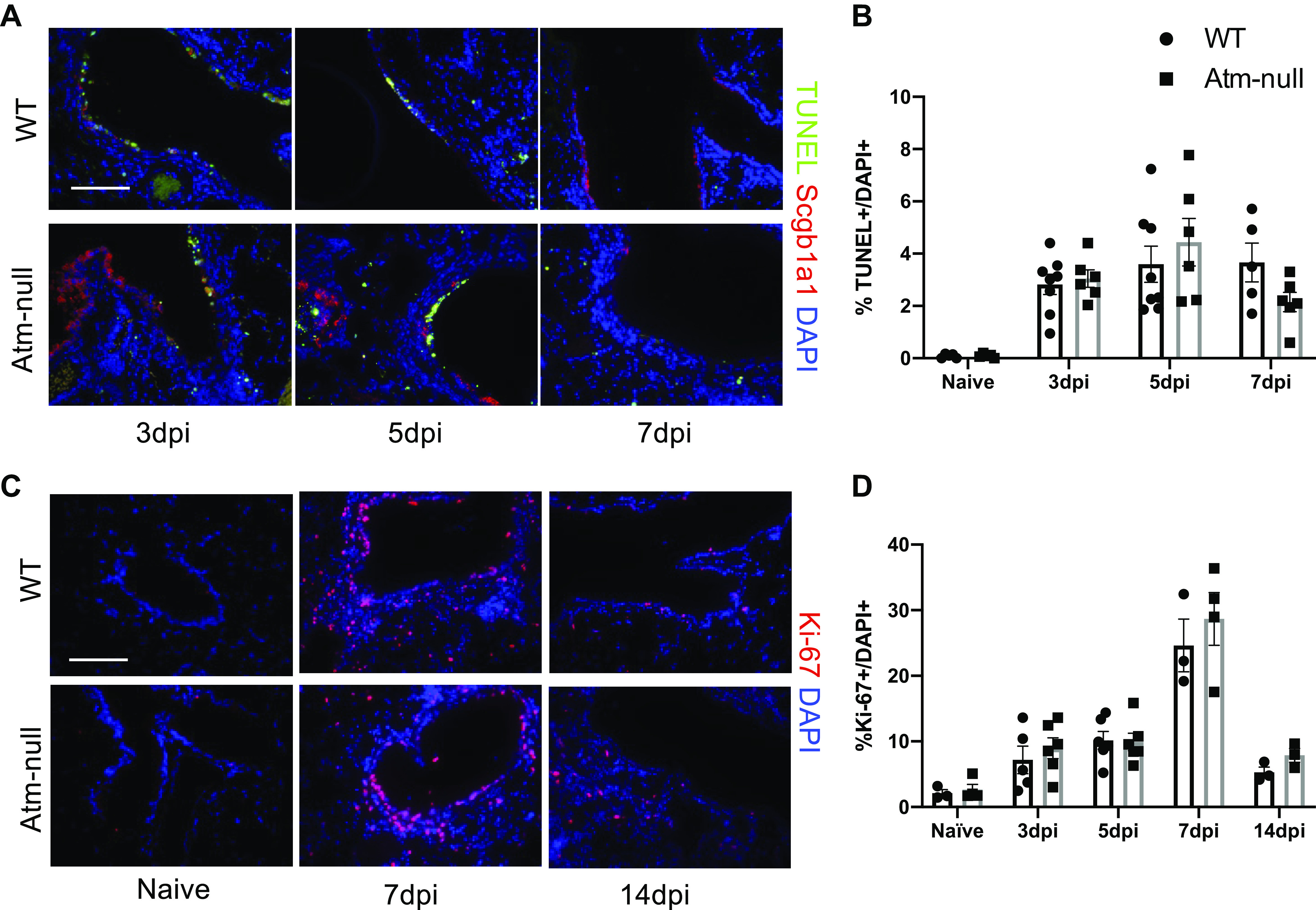
Atm is not required for apoptosis or proliferation of airway epithelial cells following IAV infection. WT and Atm-null mice were infected with 105 PFU of HKx31. A: lungs were harvested at 3, 5, and 7 dpi and were stained with terminal deoxynucleotidyl transferase (TdT) dUTP nick end labeling (TUNEL; green), an antibody recognizing Scgb1a1 (red), and counterstained with DAPI (blue). Scale bar = 100 µm. B: the number of TUNEL+ and DAPI+ cells at 3, 5, and 7 dpi were counted on n ≥ 5 images/time point and graphed as a percent of TUNEL+ to total DAPI+ values. C: lungs harvested from naïve mice at 7 and 14 dpi and were stained for Ki-67 (red) and counterstained with DAPI (blue). Scale bar = 100 µm. D: the number of Ki-67+ and DAPI+ cells in naïve mice and at 3, 5, 7, and 14 dpi were counted on n ≥ 4 images/time point and graphed as a percent of Ki-67+ of total DAPI+. Representative images of n ≥ 3 mice/time point/group are shown. dpi, days postinfection; HKx31, Hong Kong/X31; IAV, influenza A virus; PFU, plaque-forming unit; Scgb1a1, secretoglobin family 1 A member 1; WT, wildtype.
Loss of Scgb1a1 is Not Associated with an Increase in Alternative Airway Cell Phenotypes
Since Atm-null mice fail to efficiently restore Scgb1a1 expression but have no differences in apoptosis or proliferation following IAV infection, we investigated whether they also had a delayed ability to restore cytochrome P450 2f2 (Cyp2f2) expression, another protein expressed by club cells. Cytochrome P450 2f2 (Cyp2f2) is expressed by all club cells except variant club cells located at branch points and bronchoalveolar stem cells (BASCs) located at BADJ (34, 35). Before infection, Cyp2f2 staining overlapped with Scgb1a1 in the airways of both WT and Atm-null mice (Fig. 5A). Cyp2f2 staining was greatly reduced in WT and Atm-null mice at 7 dpi and was restored by 14 dpi in both groups of mice. However, we identified cells that expressed both Scgb1a1 and Cyp2f2, which are likely canonical club cells (Fig. 5A; yellow arrows), and cells that only expressed Cyp2f2 (Fig. 5A; red arrow). Importantly, we did not identify cells that expressed Scgb1a1 but not Cyp2f2 at 14 dpi, suggesting that Atm affects Scgb1a1 but not Cyp2f2 expression in club cells.
Figure 5.
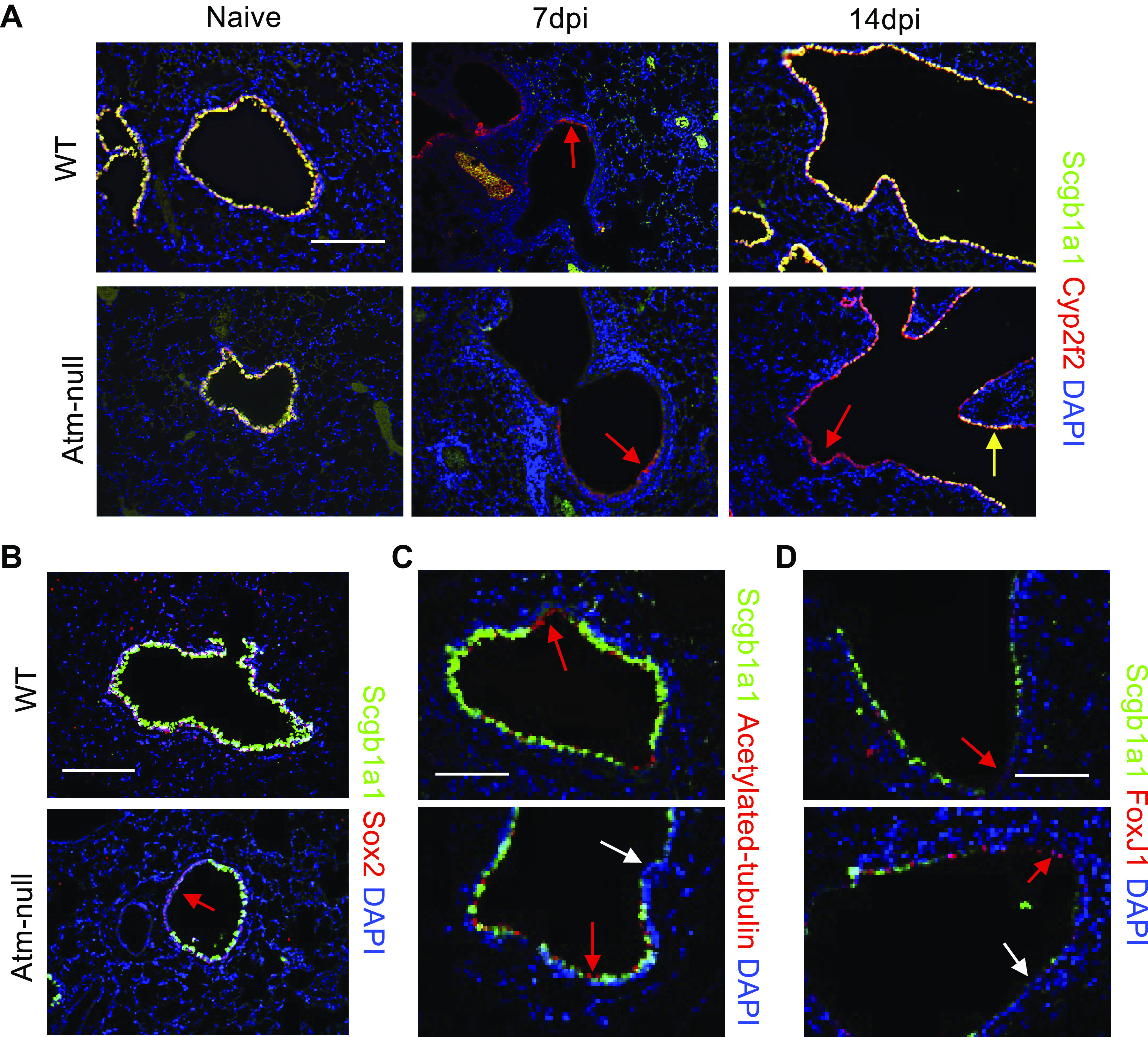
Scgb1a1− cells in proximal airways following IAV in Atm-null mice do not express markers of other airway cells. WT and Atm-null mice were infected with 105 PFU of HKx31. Lungs were harvested from naïve mice and at 7 and 14 dpi. A: lungs were stained for Scgb1a1 (green), Cyp2f2 (red), and counterstained with DAPI (blue). Red arrows indicate Cyp2f2+ cells, yellow arrows indicate Scgb1a1+; Cyp2f2+ cells, and lungs harvested at 14 dpi were stained for Scgb1a1 (green) and Sox2 (red; B), acetylated tubulin (red; C), or FoxJ1 (red; D). White arrows indicate DAPI+ cells that do not stain positive for the indicated marker. Sections were then counterstained with DAPI (blue). A and B: scale bar = 200 µm. C and D: scale bar = 100 µm. Representative images of n ≥ 3 mice/time point/group are shown. Cyp2f2, cytochrome P450 2f2; dpi, days postinfection; HKx31, Hong Kong/X31; IAV, influenza A virus; PFU, plaque-forming unit; Scgb1a1, secretoglobin family 1 A member 1; WT, wildtype.
To determine whether these undefined cells were still airway epithelial cells, we stained sections for Sox2, an airway epithelial-specific Sox protein. In addition, we identified ciliated cells using acetylated tubulin and FoxJ1. At 14 dpi, proximal airways of WT and Atm-null mice expressed E-cadherin and Sox2 (Figs. 3A and 5B), suggesting they are epithelial cells that retained an airway-specific lineage marker. Scgb1a1− proximal airway cells seen in Atm-null mice were not acetylated tubulin+ or FoxJ1+, suggesting they were not ciliated cells (Fig. 5, C and D). Together, these findings suggest that the proximal airways of Atm-null mice contain partially differentiated club cells that are Cyp2f2+, Sox2+, and E-cadherin+ but Scgb1a1−.
Basal cells (cytokeratin 5+; K5+) are stem cells residing in the trachea and mainstem bronchi that are able to differentiate into club cells (25). Importantly, these cells were rarely found in the distal airways of naïve mice (Fig. 6). However, at 7 dpi, the proximal airways of WT and Atm-null mice had K5+ cells underlying the airways, suggesting these cells were recruited into the lung during injury (Fig. 6). At 14 dpi, the K5+ cells had severely diminished in both WT and Atm-null mice and there were only a few K5+ cells present underlying the airways. By 56 dpi, very rare K5+ cells were detected in the airways of WT and Atm-null mice. These findings reveal K5+ progenitor cells are recruited into both WT and Atm-null lungs, but they do not appear to accumulate as partially differentiated club cells in the Atm-null airway.
Figure 6.
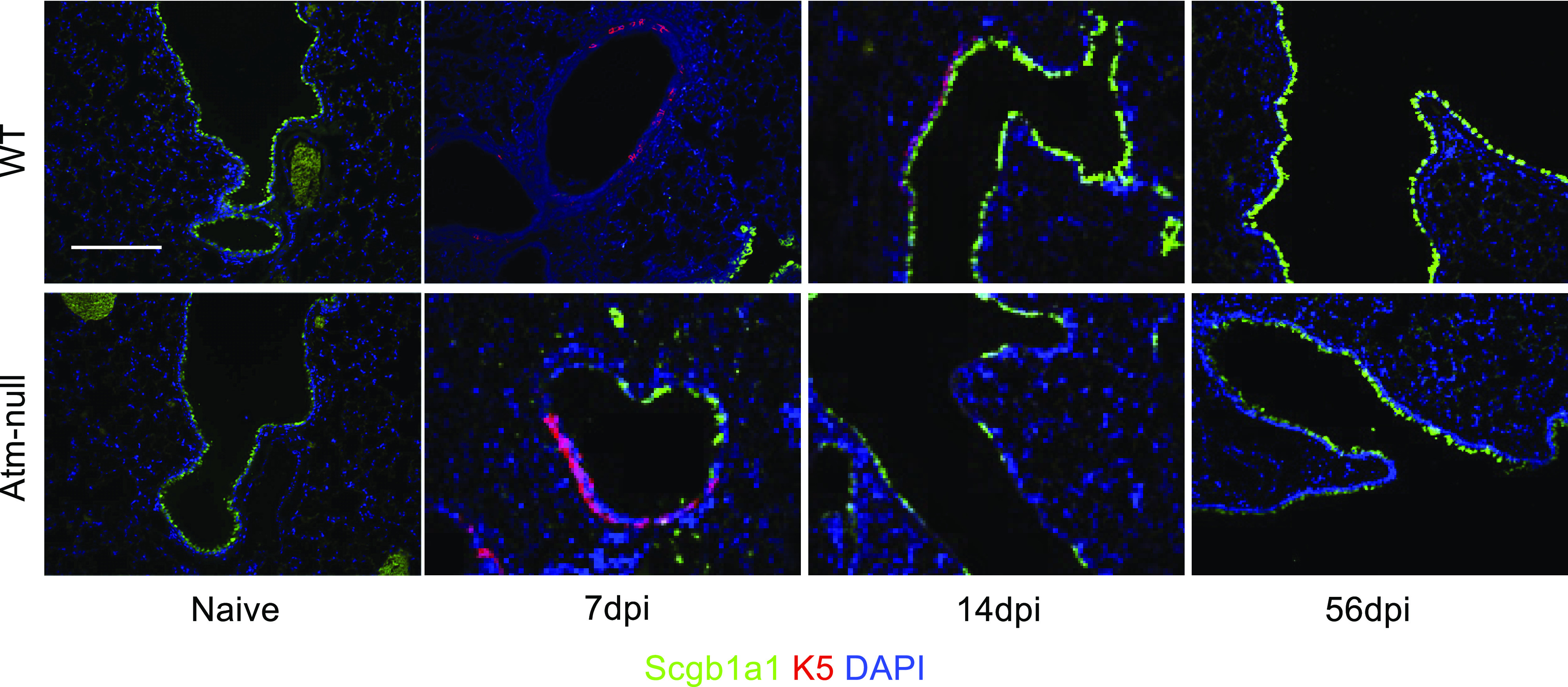
Atm is not required for K5+ cells to expand into the distal airways following IAV infection. Lungs from naïve and HKx31 infected mice at 7, 14, and 56 dpi were stained with Scgb1a1 (green), cytokeratin 5 (K5; red) and counterstained with DAPI (blue). Images are representative data from n ≥ 3 mice/time point/group. Scale bar = 200 µm. Representative images of n ≥ 3 mice/time point/group are shown. dpi, days postinfection; HKx31, Hong Kong/X31; IAV, influenza A virus; Scgb1a1, secretoglobin family 1 A member 1.
Loss of Atm Does Not Alter Variant Club Cell and Bronchoalveolar Stem Cell Regeneration
Naphthalene is converted into a toxic byproduct by Cyp2f2 causing cell death of club cells (36). However, variant club cells and BASCs survive naphthalene exposure because they lack Cyp2f2. These Scgb1a1+; Cyp2f2− cells then proliferate following naphthalene exposure and fully differentiate into club cells expressing Cyp2f2 (34, 35). To determine if Atm was required in variant club cells and BASCs to regenerate the airway epithelium, WT and Atm-null mice were administered naphthalene and recovered for 14 days. Club cells in proximal and distal airways were depleted by 3 days postexposure in control mice as indicated by the loss of Scgb1a1 staining (Fig. 7). At 14 days postexposure, Scgb1a1+ cells were detected in the large airways and at the bronchoalveolar duct junction of both WT and Atm-null mice (Fig. 7). These data suggest that Atm was not required by Scgb1a1+; Cyp2f2− variant club cells or BASCs (Scgb1a1+; Sftpc+) to regenerate the airway epithelium following naphthalene-dependent depletion of club cells.
Figure 7.

Atm is not required to regenerate the airway following naphthalene. WT and Atm-null mice were administered 275 mg/kg ip naphthalene in corn oil at 8 wk of age. Lungs were harvested at 3 or 14 days post naphthalene and stained for Scgb1a1 (green) and counterstained with DAPI (blue). Scale bar = 200 µm. Representative images of n ≥ 3 mice/time point/group are shown. Scgb1a1, secretoglobin family 1 A member 1; WT, wildtype.
Atm is Required within Club Cells for Proximal Airway Regeneration
To determine the role of Atm specifically in club cells, we sought to delete Atm specifically within Scgb1a1+ cells and lineage trace these cells following IAV infection with an enhanced green fluorescent protein (EGFP) reporter. Adult Scgb1a1-CreERTM;AtmWT/F;mTmG (control) mice and Scgb1a1-CreERTM;AtmF/F;mTmG mice were administered tamoxifen at 8 wk of age, and lung mechanics were measured 2 wk following tamoxifen administration. Unlike global Atm-null mice that displayed changes in lung mechanics before infection, lung mechanics were not different between naïve Scgb1a1-CreERTM;AtmF/F;mTmG and control mice where Atm was selectively deleted in the adult airway club cells (Fig. 8A).
Figure 8.
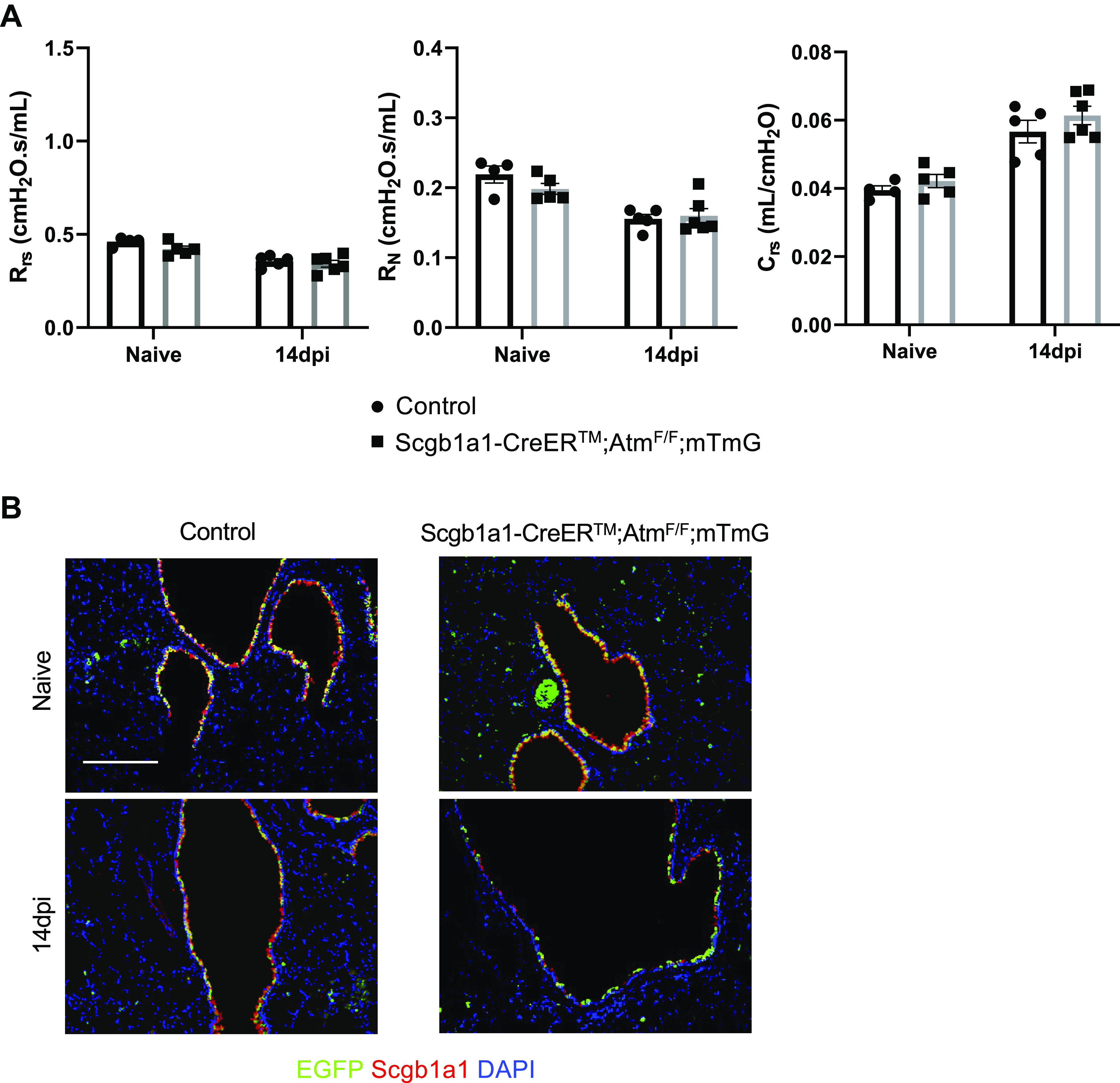
Atm is required within Scgb1a1+ cells for regeneration of the airway epithelium at day 14 postinfection. Control and Scgb1a1-CreERTM;AtmF/F;mTmG mice were administered 0.1 mg/g tamoxifen in corn oil for 4 days. Two weeks after tamoxifen, control, and Scgb1a1−CreERTM;AtmF/F;mTmG mice were infected with 105 PFU of HKx31. A: lung function was analyzed on naïve (tamoxifen induced) mice at 14 dpi. The total respiratory system resistance (Rrs; left), Newtonian resistance (RN; middle), and compliance (Crs; right) were graphed. Values represent means ± standard error with individual values shown as dots. B: lung sections were stained from naïve mice and at 14 dpi for EGFP (green), Scgb1a1 (red), and counterstained with DAPI (blue). Scale bar = 200 µm. Representative images of n ≥ 3 mice/time point/group are shown. dpi, days postinfection; EGFP, enhanced green fluorescent protein; HKx31, Hong Kong/X31; Scgb1a1, secretoglobin family 1 A member 1.
We next wanted to determine if Atm was required specifically in club cells for their regeneration following infection. Following tamoxifen administration, control and Scgb1a1-CreERTM;AtmF/F;mTmG mice were infected with HKx31. Before infection, control and Scgb1a1-CreERTM;AtmF/F;mTmG lungs contained EGFP+/Scgb1a1+ airways (Fig. 8B; top). At 14 dpi, control mice restored their proximal airways with EGFP and Scgb1a1 single positive and double-positive cells (Fig. 8B; bottom). These data suggest that non-EGFP labeled cells and EGFP+ cells are recruited to regenerate the proximal airways and express Scgb1a1 in WT mice in response to HKx31 infection. In contrast, Scgb1a1-CreERTM;AtmF/F;mTmG mouse proximal airways contained few EGFP+ and Scgb1a1+ cells, similar to Atm-null mice (Fig. 8). These data suggest that Atm is required in Scgb1a1+ cells to properly restore the proximal airway epithelium at 14 dpi.
DISCUSSION
Pulmonary epithelial cells are constantly being exposed and damaged by inhaled pathogens and other environmental pollutants (37). IAV is a seasonal respiratory pathogen that infects and kills airway and alveolar epithelial cells (38). Airway club cells that survive infection mobilize to restore normal lung function through their ability to self-renew and differentiate into ciliated or goblet cells. Failure to effectively regenerate the infected lung can lead to airway hyperreactivity, obstructive airways disease, and other types of chronic lung disease. Lung disease is responsible for 23%–46% of deaths in individuals with A-T, and autopsy results demonstrate abnormal airway epithelial cell architecture (5–7). Patients with A-T are also susceptible to recurrent respiratory infections (3, 8). Whether the airway pathology seen in these individuals is related to infection or an underlying defect in lung development is not clear, prompting us to determine if Atm is required for effective repair of the lung following IAV infection.
The data presented here confirm previous reports using a different Atm-mutant mouse showing that the naïve Atm-null lung is mechanically abnormal compared with the WT lung (18). However, like that study, analysis of lung histology failed to detect an obvious explanation for why global loss of Atm affects adult lung mechanics. ATM-null mice had baseline increased respiratory system and airway resistance, decreased lung compliance, and downshifted PV curves compared with WT controls, indicating Atm is required for normal lung development. These changes persisted despite similar inspiratory capacity (a surrogate for total lung capacity) and body weight between naïve mice, indicating the differences are unlikely attributable solely to differences of animal or lung size. Interestingly, airway mechanics were normal in Scgb1a1-CreERTM;AtmF/F;mTmG, where Atm was conditionally deleted in adult airway club cells. Together, this suggests that Atm is required during development in club cells to build a mechanically normal lung or that it is required in other lung cells such as smooth muscle or fibroblasts. Following HKx31 infection, changes in airway resistance and alveolar compliance worsened before returning to the naïve levels by 56 dpi. The large spread in the data during infection suggest that some Atm-null mice had more severe functional impairments than other Atm-null and WT mice. Whether this variability is associated with an overall diverse pathological presentation as is seen in individuals with A-T needs to be determined. The patchy nature of viral infection in a susceptible model like A-T makes it a challenge to discern how loss of Atm versus failure to effectively restore Scgb1a1 contributes to increased airway resistance after infection compared with baseline measurements before infection.
Despite our inability to discern how the loss of Atm alters lung mechanics, we found that it was required for rapid and efficient regeneration of proximal airway club cells as defined by delayed expression of Scgb1a1. Although WT mice expressed Scgb1a1 by 14 dpi, Atm-null mice failed to express Scgb1a1. These airway cells did not express markers of basal cells (K5) or ciliated cells (acetylated tubulin, FoxJ1). Importantly, they did express markers of airway epithelial cells in addition to Cyp2f2, including Sox2 and E-cadherin, suggesting they were immature or partially differentiated club cells. Interestingly, expression of Scgb1a1 was seen by 56 dpi indicating that Atm was required for rapid or efficient expression of Scgb1a1 early after IAV infection. Failure to rapidly restore expression of Scgb1a1 may contribute to the respiratory disease seen in individuals with A-T because defects in Scgb1a1 expression are associated with various lung diseases, including asthma, chronic obstructive pulmonary disease (COPD), and bronchopulmonary dysplasia (BPD; 39–41). Scgb1a1 knockout mice are viable but have some structural abnormalities and fail to modulate immune responses to viruses (21, 42). Thus, reduced or delayed expression of Scgb1a1 could render Atm-null mice, and theoretically, individuals with A-T more susceptible to viral infections if they were exposed in the window of time in which club cells do not express Scgb1a1. Recurrent infections of airway club cells deficient in Scgb1a1 could also lead to continual abnormal repair following each infection, propagating development of lung disease. Interestingly, Scgb1a1 replacement therapy has been proposed as a treatment for asthma and BPD either through the direct administration of recombinant Scgb1a1 or the augmentation Scgb1a1 expression with the use of retinoids or stimulation upstream transcription factors (39, 43, 44). Thus, Scgb1a1 replacement therapy could be a promising treatment for AT individuals with respiratory viral infections.
The cells present in Atm-null proximal airways were Cyp2f2+ but lacked the canonical club cell marker, Scgb1a1, suggesting they could be immature club cells. Club cells are stem cells that can self-renew and differentiate into ciliated and goblet cells (24, 45). Most club cells are sensitive to naphthalene due to their expression of Cyp2f2 that converts it to a cytotoxic product. Rare variant club cells that lack Cyp2f2 and are resistant to naphthalene exist at airway branchpoints and at the BADJs. It does not appear that these cells require Atm to regenerate the injured lung because club cell regeneration was detected in Atm-null mice administered naphthalene. In addition, basal cells expressing K5 are able to differentiate into club cells (25), however, we did not detect differences in staining for K5+ cells at 7 or 14 dpi between WT and Atm-null mice. It has been previously reported that Sox2+ epithelial cells migrate into the alveoli following severe injury with H1N1 (46). These cells express K5 and form “pods” when they migrate to the alveoli and do not express canonical AT2 or AT1 cell markers. Interestingly, the Sox2+ cells do not form canonical airway cell lineages such as club cells or basal cells, despite their K5 expression (46). Thus, it is possible that injury in Atm-null mice recapitulated this type of regeneration, in which Scgb1a1-lineage negative cells regenerated the airway following HKx31 injury and did not fully differentiate into club cells. But the most conservative conclusion comes from our Scgb1a1-CreERTTM;AtmF/F;mTmG mice lineage mapping studies showing that Atm is required to regenerate EGFP+ and EGFP− airway club cells. In other words, it appears Scgb1a1-lineage positive and Scgb1a1-lineage negative progenitor cells require Atm to efficiently restore Scgb1a1 expression in proximal airways. How Atm regulates Scgb1a1 expression during club cell differentiation remains to be determined. Atm is an extremely large kinase that regulates the phosphorylation status of thousands of proteins involved in DNA damage signaling and mitochondrial oxidative stress. A single cell phospho-proteome study may be required to discern how Atm regulates the Scgb1a1 expression during club cell differentiation.
In summary, we found that Atm is required for normal airway and alveolar function. It is also required for rapid and efficient proximal airway epithelial regeneration following influenza A virus infection. Recurrent infections and obstructive lung disease seen in individuals with A-T are widely believed to be related to failure to generate sufficient memory immune responses needed to reduce lung injury during secondary infection. The findings reported here suggest that it may also be related to base changes in lung function and failure to effectively regenerate injured airway epithelium.
DATA AVAILABILITY
The data that support this study are available upon request from the corresponding author.
GRANTS
This work was funded in part by National Institutes of Health (NIH) Grants R01 HL091968 (to M.A.O.), R03 NS061339 (to M.M.-P.), and R56 NS111927 (to M.M.-P.) and Action for A-T (to M.M.-P.; M.A.O.). R.W. was supported by NIH T32 training Grants (GM068411 and AI118689). NIH Center Grant P30 ES001247 supported the tissue processing core. L.M.-S. is partially funded by the New York Influenza Center of Excellence (NYICE, NIH 272201400005C), a member of the National Institute of Allergy and Infectious Diseases (NIAID), National Institutes of Health (NIH), Department of Health and Human Services, Centers of Excellence for Influenza Research and Surveillance (CEIRS) contract No. HHSN272201400005C (NYICE), by the NIH/NIAID 1R01AI145332, and by the Department of Defense (DoD) Peer Reviewed Medical Research Program (PRMRP) Grant W81XWH-18-1-0460.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
R.W., A.M.D., W.D., M.Y., M.M.-P., L.M.-S., and M.A.O. conceived and designed research; R.W., A.M.D., M.B., W.D., and M.Y. performed experiments; R.W., A.M.D., M.B., W.D., M.Y., M.M.-P., and M.A.O. analyzed data; R.W., A.M.D., M.B., W.D., M.Y., M.M.-P., L.M.-S., and M.A.O. interpreted results of experiments; R.W. and A.M.D. prepared figures; R.W. and A.M.D. drafted manuscript; R.W., A.M.D., M.B., W.D., M.Y., M.M.-P., L.M.-S., and M.A.O. edited and revised manuscript; R.W., A.M.D., M.B., W.D., M.Y., M.M.-P., L.M.-S., and M.A.O. approved final version of manuscript.
REFERENCES
- 1.McKinnon PJ. ATM and ataxia telangiectasia. EMBO Rep 5: 772–776, 2004. doi: 10.1038/sj.embor.7400210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McKinnon PJ. ATM and the molecular pathogenesis of ataxia telangiectasia. Annu Rev Pathol 7: 303–321, 2012. doi: 10.1146/annurev-pathol-011811-132509. [DOI] [PubMed] [Google Scholar]
- 3.Rothblum-Oviatt C, Wright J, Lefton-Greif MA, McGrath-Morrow SA, Crawford TO, Lederman HM. Ataxia telangiectasia: a review. Orphanet J Rare Dis 11: 159, 2016. doi: 10.1186/s13023-016-0543-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bott L, Lebreton J, Thumerelle C, Cuvellier J, Deschildre A, Sardet A. Lung disease in ataxia-telangiectasia. Acta Paediatr 96: 1021–1024, 2007. doi: 10.1111/j.1651-2227.2007.00338.x. [DOI] [PubMed] [Google Scholar]
- 5.Crawford TO, Skolasky RL, Fernandez R, Rosquist KJ, Lederman HM. Survival probability in ataxia telangiectasia. Arch Dis Child 91: 610–611, 2006. doi: 10.1136/adc.2006.094268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pump KK, Dunn HG, Meuwissen H. A Study of the bronchial and vascular structures of a lung: from a case of ataxia-telangiectasia. Dis Chest 47: 473–486, 1965. doi: 10.1378/chest.47.5.473. [DOI] [PubMed] [Google Scholar]
- 7.Swift M. Genetics and epidemiology of ataxia-telangiectasia. Kroc Found Ser 19: 133–146, 1985. [PubMed] [Google Scholar]
- 8.Schroeder SA, Zielen S. Infections of the respiratory system in patients with ataxia-telangiectasia. Pediatr Pulmonol 49: 389–399, 2014. doi: 10.1002/ppul.22817. [DOI] [PubMed] [Google Scholar]
- 9.McGrath-Morrow SA, Gower WA, Rothblum-Oviatt C, Brody AS, Langston C, Fan LL, Lefton-Greif MA, Crawford TO, Troche M, Sandlund JT, Auwaerter PG, Easley B, Loughlin GM, Carroll JL, Lederman HM. Evaluation and management of pulmonary disease in ataxia-telangiectasia. Pediatr Pulmonol 45: 847–859, 2010. doi: 10.1002/ppul.21277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McGrath-Morrow SA, Lederman HM, Aherrera AD, Lefton-Greif MA, Crawford TO, Ryan T, Wright J, Collaco JM. Pulmonary function in children and young adults with ataxia telangiectasia. Pediatr Pulmonol 49: 84–90, 2014. doi: 10.1002/ppul.22760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Aucouturier P, Bremard-Oury C, Griscelli C, Berthier M, Preud’homme JL. Serum IgG subclass deficiency in ataxia-telangiectasia. Clin Exp Immunol 68: 392–396, 1987. [PMC free article] [PubMed] [Google Scholar]
- 12.Schubert R, Reichenbach J, Zielen S. Deficiencies in CD4+ and CD8+ T cell subsets in ataxia telangiectasia. Clin Exp Immunol 129: 125–132, 2002. doi: 10.1046/j.1365-2249.2002.01830.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nowak-Wegrzyn A, Crawford TO, Winkelstein JA, Carson KA, Lederman HM. Immunodeficiency and infections in ataxia-telangiectasia. J Pediatr 144: 505–511, 2004. doi: 10.1016/j.jpeds.2003.12.046. [DOI] [PubMed] [Google Scholar]
- 14.Warren R, Domm W, Yee M, Campbell A, Malone J, Wright T, Mayer-Pröschel M, O’Reilly MA. Ataxia-telangiectasia mutated is required for the development of protective immune memory after influenza A virus infection. Am J Physiol Lung Cell Mol Physiol 317: L591–L601, 2019. doi: 10.1152/ajplung.00031.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yeo AJ, Fantino E, Czovek D, Wainwright CE, Sly PD, Lavin MF. Loss of ATM in airway epithelial cells is associated with susceptibility to oxidative stress. Am J Respir Crit Care Med 196: 391–393, 2017. doi: 10.1164/rccm.201611-2210LE. [DOI] [PubMed] [Google Scholar]
- 16.Yeo AJ, Henningham A, Fantino E, Galbraith S, Krause L, Wainwright CE, Sly PD, Lavin MF. Increased susceptibility of airway epithelial cells from ataxia-telangiectasia to S. pneumoniae infection due to oxidative damage and impaired innate immunity. Sci Rep 9: 2627, 2019. [Erratum in Sci Rep 10: 12742, 2020]. doi: 10.1038/s41598-019-38901-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schroeder SA, Swift M, Sandoval C, Langston C. Interstitial lung disease in patients with ataxia-telangiectasia. Pediatr Pulmonol 39: 537–543, 2005. doi: 10.1002/ppul.20209. [DOI] [PubMed] [Google Scholar]
- 18.Eickmeier O, Kim SY, Herrmann E, Döring C, Duecker R, Voss S, Wehner S, Hölscher C, Pietzner J, Zielen S, Schubert R. Altered mucosal immune response after acute lung injury in a murine model of ataxia telangiectasia. BMC Pulm Med 14: 93, 2014. doi: 10.1186/1471-2466-14-93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tate MD, Schilter HC, Brooks AG, Reading PC. Responses of mouse airway epithelial cells and alveolar macrophages to virulent and avirulent strains of influenza A virus. Viral Immunol 24: 77–88, 2011. doi: 10.1089/vim.2010.0118. [DOI] [PubMed] [Google Scholar]
- 20.Johnston CJ, Mango GW, Finkelstein JN, Stripp BR. Altered pulmonary response to hyperoxia in Clara cell secretory protein deficient mice. Am J Respir Cell Mol Biol 17: 147–155, 1997. doi: 10.1165/ajrcmb.17.2.2676. [DOI] [PubMed] [Google Scholar]
- 21.Harrod KS, Mounday AD, Stripp BR, Whitsett JA. Clara cell secretory protein decreases lung inflammation after acute virus infection. Am J Physiol Lung Cell Mol Physiol 275: L924–L930, 1998. doi: 10.1152/ajplung.1998.275.5.L924. [DOI] [PubMed] [Google Scholar]
- 22.Mango GW, Johnston CJ, Reynolds SD, Finkelstein JN, Plopper CG, Stripp BR. Clara cell secretory protein deficiency increases oxidant stress response in conducting airways. Am J Physiol Lung Cell Mol Physiol 275: L348–L356, 1998. doi: 10.1152/ajplung.1998.275.2.L348. [DOI] [PubMed] [Google Scholar]
- 23.Watson TM, Reynolds SD, Mango GW, Boe IM, Lund J, Stripp BR. Altered lung gene expression in CCSP-null mice suggests immunoregulatory roles for Clara cells. Am J Physiol Lung Cell Mol Physiol 281: L1523–L1530, 2001. doi: 10.1152/ajplung.2001.281.6.L1523. [DOI] [PubMed] [Google Scholar]
- 24.Rawlins EL, Okubo T, Xue Y, Brass DM, Auten RL, Hasegawa H, Wang F, Hogan BL. The role of Scgb1a1+ Clara cells in the long-term maintenance and repair of lung airway, but not alveolar, epithelium. Cell Stem Cell 4: 525–534, 2009. doi: 10.1016/j.stem.2009.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rock JR, Onaitis MW, Rawlins EL, Lu Y, Clark CP, Xue Y, Randell SH, Hogan BL. Basal cells as stem cells of the mouse trachea and human airway epithelium. Proc Natl Acad Sci USA 106: 12771–12775, 2009. doi: 10.1073/pnas.0906850106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hong KU, Reynolds SD, Watkins S, Fuchs E, Stripp BR. Basal cells are a multipotent progenitor capable of renewing the bronchial epithelium. Am J Pathol 164: 577–588, 2004. doi: 10.1016/S0002-9440(10)63147-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Campbell A, Krupp B, Bushman J, Noble M, Pröschel C, Mayer-Pröschel M. A novel mouse model for ataxia-telangiectasia with a N-terminal mutation displays a behavioral defect and a low incidence of lymphoma but no increased oxidative burden. Hum Mol Genet 24: 6331–6349, 2015. doi: 10.1093/hmg/ddv342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dylag AM, Haak J, Yee M, O’Reilly MA. Pulmonary mechanics and structural lung development after neonatal hyperoxia in mice. Pediatr Res 87: 1201–1210, 2020. doi: 10.1038/s41390-019-0723-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yee M, Vitiello PF, Roper JM, Staversky RJ, Wright TW, McGrath-Morrow SA, Maniscalco WM, Finkelstein JN, O’Reilly MA. Type II epithelial cells are critical target for hyperoxia-mediated impairment of postnatal lung development. Am J Physiol Lung Cell Mol Physiol 291: L1101–L1111, 2006. doi: 10.1152/ajplung.00126.2006. [DOI] [PubMed] [Google Scholar]
- 30.Stripp BR, Maxson K, Mera R, Singh G. Plasticity of airway cell proliferation and gene expression after acute naphthalene injury. Am J Physiol Lung Cell Mol Physiol 269: L791–L799, 1995. doi: 10.1152/ajplung.1995.269.6.L791. [DOI] [PubMed] [Google Scholar]
- 31.Reynolds SD, Reynolds PR, Pryhuber GS, Finder JD, Stripp BR. Secretoglobins SCGB3A1 and SCGB3A2 define secretory cell subsets in mouse and human airways. Am J Respir Crit Care Med 166: 1498–1509, 2002. doi: 10.1164/rccm.200204-285OC. [DOI] [PubMed] [Google Scholar]
- 32.Li N, Parrish M, Chan TK, Yin L, Rai P, Yoshiyuki Y, Abolhassani N, Tan KB, Kiraly O, Chow VT, Engelward BP. Influenza infection induces host DNA damage and dynamic DNA damage responses during tissue regeneration. Cell Mol Life Sci 72: 2973–2988, 2015. doi: 10.1007/s00018-015-1879-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Khanna M, Ray A, Rawall S, Chandna S, Kumar B, Vijayan VK. Detection of influenza virus induced ultrastructural changes and DNA damage. Indian J Virol 21: 50–55, 2010. doi: 10.1007/s13337-010-0004-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Giangreco A, Reynolds SD, Stripp BR. Terminal bronchioles harbor a unique airway stem cell population that localizes to the bronchoalveolar duct junction. Am J Pathol 161: 173–182, 2002. doi: 10.1016/S0002-9440(10)64169-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Reynolds SD, Giangreco A, Power JH, Stripp BR. Neuroepithelial bodies of pulmonary airways serve as a reservoir of progenitor cells capable of epithelial regeneration. Am J Pathol 156: 269–278, 2000. doi: 10.1016/S0002-9440(10)64727-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Buckpitt A, Chang AM, Weir A, Van Winkle L, Duan X, Philpot R, Plopper C. Relationship of cytochrome P450 activity to Clara cell cytotoxicity. IV. Metabolism of naphthalene and naphthalene oxide in microdissected airways from mice, rats, and hamsters. Mol Pharmacol 47: 74–81, 1995. [PubMed] [Google Scholar]
- 37.Zanin M, Baviskar P, Webster R, Webby R. The Interaction between respiratory pathogens and mucus. Cell Host Microbe 19: 159–168, 2016. doi: 10.1016/j.chom.2016.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tripathi S, Batra J, Cao W, Sharma K, Patel JR, Ranjan P, Kumar A, Katz JM, Cox NJ, Lal RB, Sambhara S, Lal SK. Influenza A virus nucleoprotein induces apoptosis in human airway epithelial cells: implications of a novel interaction between nucleoprotein and host protein Clusterin. Cell Death Dis 4: e562, 2013. doi: 10.1038/cddis.2013.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhu L, An L, Ran D, Lizarraga R, Bondy C, Zhou X, Harper RW, Liao SY, Chen Y. The Club cell marker SCGB1A1 downstream of FOXA2 is reduced in asthma. Am J Respir Cell Mol Biol 60: 695–704, 2019. doi: 10.1165/rcmb.2018-0199OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Laucho-Contreras ME, Polverino F, Gupta K, Taylor KL, Kelly E, Pinto-Plata V, Divo M, Ashfaq N, Petersen H, Stripp B, Pilon AL, Tesfaigzi Y, Celli BR, Owen CA. Protective role for club cell secretory protein-16 (CC16) in the development of COPD. Eur Respir J 45: 1544–1556, 2015. doi: 10.1183/09031936.00134214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ramsay PL, DeMayo FJ, Hegemier SE, Wearden ME, Smith CV, Welty SE. Clara cell secretory protein oxidation and expression in premature infants who develop bronchopulmonary dysplasia. Am J Respir Crit Care Med 164: 155–161, 2001. doi: 10.1164/ajrccm.164.1.2008022. [DOI] [PubMed] [Google Scholar]
- 42.Stripp BR, Reynolds SD, Boe IM, Lund J, Power JH, Coppens JT, Wong V, Reynolds PR, Plopper CG. Clara cell secretory protein deficiency alters clara cell secretory apparatus and the protein composition of airway lining fluid. Am J Respir Cell Mol Biol 27: 170–178, 2002. doi: 10.1165/ajrcmb.27.2.200200270c. [DOI] [PubMed] [Google Scholar]
- 43.Chen Y, Vasquez MM, Zhu L, Lizarraga RE, Krutzsch M, Einspahr J, Alberts DS, Di PYP, Martinez FD, Guerra S. Effects of retinoids on augmentation of club cell secretory protein. Am J Respir Crit Care Med 196: 928–931, 2017. doi: 10.1164/rccm.201608-1611LE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Levine CR, Gewolb IH, Allen K, Welch RW, Melby JM, Pollack S, Shaffer T, Pilon AL, Davis JM. The safety, pharmacokinetics, and anti-inflammatory effects of intratracheal recombinant human Clara cell protein in premature infants with respiratory distress syndrome. Pediatr Res 58: 15–21, 2005. doi: 10.1203/01.PDR.0000156371.89952.35. [DOI] [PubMed] [Google Scholar]
- 45.Tsao PN, Wei SC, Wu MF, Huang MT, Lin HY, Lee MC, Lin KM, Wang IJ, Kaartinen V, Yang LT, Cardoso WV. Notch signaling prevents mucous metaplasia in mouse conducting airways during postnatal development. Development 138: 3533–3543, 2011. doi: 10.1242/dev.063727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ray S, Chiba N, Yao C, Guan X, McConnell AM, Brockway B, Que L, McQualter JL, Stripp BR. Rare SOX2+ airway progenitor cells generate KRT5+ cells that repopulate damaged alveolar parenchyma following influenza virus infection. Stem Cell Reports 7: 817–825, 2016. doi: 10.1016/j.stemcr.2016.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support this study are available upon request from the corresponding author.