

Keywords: chenodeoxycholic, colitis, deoxycholic, lithocholic
Abstract
Bile acids are amphipathic, detergent molecules. The detergent effects of di-α-hydroxy-bile acids are relevant to several colonic diseases. The aims were to review the concentrations of bile acids reaching the human colon in health and disease, the molecular structure of bile acids that determine detergent functions and the relationship to human diseases (neuroendocrine tumors, microscopic colitis, active celiac disease, and ulcerative colitis, Crohn’s disease and ileal resection), the relationship to bacterial uptake into the mucosa, mucin depletion, and epithelial damage, the role of bile acids in mucosal inflammation and microscopic colitis, and the role of sulfation of bile salts in detoxification or prevention of the detergent effects of bile acids. The concentrations of bile acids reaching the human colon range from 2 to 10 mM; di-α-hydroxy bile acids are the only bile acids with detergent effects that include mucin depletion, mucosal damage, bacterial uptake, and microscopic inflammation that may be manifest in diseases associated with no overt inflammation of the mucosa, such as bile acid diarrhea, ileal diseases such as neuroendocrine tumors, ileal resection, and nonalcoholic steatohepatitis. Sulfation inactivates colonic secretion due to primary bile acids, but it may render secondary bile acids proinflammatory in the colon. Other evidence in preclinical models of inflammatory bowel disease (IBD) suggests reduced sulfation causes barrier dysfunction, inflammation, or carcinogenesis. These advances emphasize relevance and opportunities afforded by greater understanding of the chemistry and metabolism of bile acids, which stands to be further enhanced by research into the metabolic interactions of microbiota with bile acids.
INTRODUCTION
Physical Chemistry and Physiology of Bile Acids
Primary bile acids are synthesized from cholesterol by hepatocytes, with the rate-limiting step being the enzyme CYP7A1. In humans, the primary bile acids are cholic acid (CA) and chenodeoxycholic acid (CDCA), which are conjugated to either taurine or glycine. In humans, glycine conjugation predominates, and conjugation lowers the pKa of bile acids and alters their physicochemical properties and biochemical and physiological actions. Thus, conjugated bile acids are fully ionized at the pH of the proximal small intestine, making them impermeable to epithelial cell membranes, and therefore leading to high intraluminal concentrations enabling micellar solubilization of dietary lipids to occur. In the distal ileum, the epithelial cells express the apical sodium-dependent bile acid transporter (ASBT, also known as the ileal bile acid transporter, IBAT), which transports only bile acids and has a greater affinity for conjugated over nonconjugated bile acids (1). Thus, 90%–95% of the bile acids reaching the ileum are reabsorbed through uptake by ASBT, traverse the cytoplasm of the epithelial cells bound to ileal bile acid-binding protein (IBABP), and exit the basolateral domains of the enterocyte via the heterodimeric protein organic solute transporter α and β (OSTα/OSTβ), a facilitated diffusion transporter that is present throughout the small intestine. Once in the interstitium, conjugated bile acids enter villus capillaries to gain access to the portal circulation, probably by passive diffusion. The bile acids thus enter the enterohepatic circulation and are taken up by the hepatocytes through transporters for bile acid in the sinusoidal (basolateral) membrane of hepatocytes, which are Na+-taurocholate cotransporting polypeptide (NTCP) and organic anion transporting polypeptides (2), the latter being Na+-independent [with organic anion transporting polypeptide-C (OATP-C), also called OATP2, being predominant in human liver (3)]. Bile acids also bind to fibroblast growth factor receptor 4 (FGFR-4) to modulate uptake and synthesis by the rate-limiting enzyme of bile acid synthesis, cholesterol 7α-hydroxylase (CYP7A1). After entering the hepatocytes, bile acids are further metabolized by hepatic enzymes and reconjugated to glycine and taurine, after which they are then secreted into the bile and recirculated. The arrival of bile acids in the hepatocytes also provides feedback regulation on the rate of bile acid synthesis.
In the colon, the 5%–10% of primary bile acids not reabsorbed in the ileum are extensively metabolized by the resident microbiota into secondary bile acids, first by deconjugation by bile salt hydrolases (BSH), enzymes that are widely expressed by gram-positive bacteria and some gram-negative bacteria within the gut lumen, and second by 7α-dehydroxylation that converts the bile acids to more hydrophobic molecules, which decreases their ionization and enables bile acid reabsorption by passive diffusion across the colonocyte cell membrane (1).
Detergency and Other Effects of Bile Acids
Di-α-hydroxy bile acids, chenodeoxycholic acid (CDCA, 3α,7α-dihydroxy-5β-cholan-24-oic acid), and deoxycholic acid (DCA, 3α,12α-dihydroxy-5β-cholan-24-oic acid) are detergent molecules that induce mucus denudation from the surface of the colonic mucosa, mucin secretion, and cause mucosal damage, as shown by the appearance of biochemical measurements (e.g., protein-bound hexose, hexosamine, protein, and DNA) or by scanning electron microscopy of the mucosa after perfusion in rat and rabbit colon (4–7). Conversely, 7α-dehydroxylation of CDCA by colonic microbiota, which results in the formation of lithocholic acid (LCA), has anti-inflammatory effects relative to the primary bile acid, CDCA; this anti-inflammatory effect of LCA is mediated, at least in part, by inhibition of epithelial apoptosis and promotion of barrier function (8). However, there is diminution of anti-inflammatory potential by sulfation of LCA, which may also result from effects of colonic microbiota (9). In addition, the hydrophobic bile acid, LCA, has well-documented cytotoxicity in the biliary epithelium (10), and even in diverse cancer cells such as colon cancer cells (11) and prostate cancer cells (12).
The intraluminal concentration of the detergent bile acids is also very relevant to their biological effects. In dose response rabbit colon perfusion studies with CDCA, there were dose-related effects on loss of mucus, mucosal damage, and water and electrolyte secretion (5) with documentation of mucus loss before the accumulation of DNA (marker of mucosal damage) and secretion of water and sodium ions. Pharmacologically, the anticholinergic atropine reduced and the cholinergic agonist carbachol increased mucus secretion, and this was associated with reduction in mucosal damage and fluid secretion (6). These studies suggested that cholinergic modulation of the mucus barrier influenced the resistance to the damaging effects of CDCA in the rabbit colon in vivo.
The effects and efficacious doses of bile acids on the luminal side of the colonic mucosa were confirmed in vitro using T84 colonic epithelial monolayer (13), showing that a concentration of taurodeoxycholate greater than or equal to 1 mM was required for apical addition to result in an electrolyte transport effect, which was associated with an abrupt increase in the permeability of the monolayer. Once tight junction permeability increases, luminal bile salts are able to reach the basolateral membrane of the epithelial cells, increase free cytosolic Ca2+ from extracellular sources, and induce transcellular Cl− secretion (13).
In addition to the detergent effects of bile acids, which are important for solubilizing biliary cholesterol and for micelle formation in the upper gut, and for destructive effects on mucosal membranes, there may be other mechanisms mediating bile acid-induced effects on the epithelium, as reviewed elsewhere (1). Thus, bile acids also affect messenger molecules and interact both with surface receptors [specifically Takeda G protein-coupled receptor (TGR5) or G protein coupled bile acid receptor 1 (GPBAR1)] and nuclear receptors [farnesoid X receptor (FXR) and vitamin D receptor], and they have protean effects on metabolism throughout the body. A recent review assessed the effects of bile acids as receptor agonists and their effects on ion channels (14). Therefore, although this article focuses on detergency, epithelial and other damage, and induction of inflammation, it should not be assumed that the detergent effects are the only mechanisms that could be relevant to the induction of inflammation.
This review does address the hypothesis that detergency is significant in the cascade of effects that lead to inflammation in view of the likely concentrations of bile acids reaching the colon in different disease states in humans, the alterations in the mucosal barrier, and inflammatory effects of bile acids in the mammalian colon, and the opportunities to reverse those effects through protective mechanisms including bile acid sequestration and sulfation.
CONCENTRATIONS OF BILE ACIDS REACHING THE HUMAN COLON IN HEALTH AND DISEASE
Each day, 7 to 8 L of fluid are secreted into the gastrointestinal tract or taken by mouth. Oral intake and saliva contribute 1.5 L/day, bile and pancreatic secretion each 1 L, and gastric secretion add 1.5 L a day. However, fluxes of fluid during intestinal digestion contribute to the lumen net 3.0 L of fluid, which flows along osmotic gradients through the highly permeable jejunal mucosa and results in the initial loss of fluids from the intravascular space into the lumen. Subsequently, small bowel reabsorption of water and electrolytes recovers much of the secreted fluid, so that only ∼1.2 L of fluid enters the colon each day. Thus, the small bowel reabsorbs 5 L of fluid a day. Colonic reabsorption recovers 1 L of fluid, but the reserve absorptive capacity of the colon might recover up to 3 L/day. Thus, stool volume rarely exceeds 200 mL/day in healthy individuals. The rate of fluid loading to the colon and regional transit rates can determine stool consistency (15, 16).
Conservative estimates of cecal bile acid concentrations in the absence of disease may be calculated based on three assumptions: first, the bile acids are in solution in ∼1.2 L of water reaching the colon daily; second, the 90th percentile of normal fecal bile acid excretion in a large healthy volunteer database is 2,337 µmoles/48 h or 1,200 µmoles/24 h; and third, the human and rabbit colon passively reabsorb 50%–75% of bile acids entering the cecum in perfusion studies (5, 17). Therefore, reasonable estimates of concentrations of bile acids in the cecum in health are 2–3 mM/L.
The highest concentrations of bile acids reaching the colon can be approximated, based on the fecal concentrations observed in patients with ileostomy, ileitis, pouchitis, or ileal resection. Table 1 summarizes data from recently published fecal bile acid excretion measurements in clinical practice at Mayo Clinic (18, 19). Using the medians and 75th percentiles across all groups (2,016 and 3,363 μmol/48 h, respectively) and assuming ∼1.2 L as the average volume of fluid reaching the colon each day and 50%–75% passive absorption of bile acids in the colon, it is estimated that cecal concentrations reach 2–5 mM/L for the third quartile and well above 5 mM/L for the highest quartile. These estimated concentrations are relevant to the detergent, secretory, mucin-depleting, and proinflammatory effects of bile acids reaching the human colon.
Table 1.
Fecal bile acid excretion in patients with diverse disease states presenting with chronic diarrhea
| Data Show Median (IQR) | NET With Ileal Resection (n = 45) |
NET Without Ileal Resection (n = 22) |
Active Celiac Disease (n = 9) |
Collagenous Colitis (n = 34) | Lymphocytic Colitis (n = 32) | Active Ulcer. Colitis (n = 8) | Ileal Pouch(n = 5) | Active Crohn’s Disease (n = 15) | Crohn’s + Ileal Resection (n = 30) | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| n | n | ||||||||||
| Sex (F/M) | 27/18 | 15/7 | 6/3 | 27/7 | 29/3 | 5/3 | 3/2 | 8/7 | 13/17 | ||
| Age | 67 | 64 (56–69) | 22 | 60 (46, 73) | 36 (23–50) | 66 (54–72) | 58 (45–69) | 49 (21–65) | 66 (48–70) | 45 (37–59) | 50 (39–62) |
| Ileum resection | 36 | 32 (15–69) cm | 55 (22–87.5) cm | ||||||||
| Total Fecal BA, μmol/48 h | 52 | 4,448 (3,011–8,483) | 15 | 2,016 (627, 3,363) | 882 (355–2,363) | 727 (450–1,889) | 1,131 (446–2,091) | 859 (371–1,801) | 3,863 (2,471–4,329) | 2,223 (884–4,928) | 6,134 (3,307–12,262) |
| 1° fecal BA, % | 52 | 60 (14–95.7) | 15 | 17.5 (8.9, 52) | 14.5 (1.6–70) | 5.6 (1.1–21.7) | 4.9 (1.2–27) | 7.1 (2.3–46.6) | 95 (78–99) | 73 (4–97) | 20.4 (4.8–81) |
| Fecal fat, g/24 h | 42 | 16.5 (8.7–38) | 12 | 6.5 (5.2, 14.5) | 9.5 (4.3–12.3) | 8 (5–10) | 7 (4–10) | 10 (4–13) | 11 (6–15) | 9 (5–30) | 9 (5.3–30) |
| Fecal wt, g/48 h | 51 | 716 (480–1,089) | 15 | 553 (344, 850) | 577 (458–812) | 645 (409–941) | 546 (447–838) | 602 (398–774) | 1,251 (516–2,103) | 625 (422–1,336) | 1,011 (203–1,348) |
n refers to number of patients in disease group with data available; all data were available where n is not specified. Note the interquartile ranges (IQRs) of total fecal bile acids (BA) per 48 h range from 355 μmol to 12,262 μmol with the median of the medians, and 75th percentiles across all groups being 2,016 and 3,363 μmol/48 h. NET, neuroendocrine tumor.
EFFECT OF MOLECULAR STRUCTURE ON BILE ACID-INDUCED ALTERATIONS IN MUCOSAL ABSORPTIVE FUNCTION, PERMEABILITY, AND MORPHOLOGY
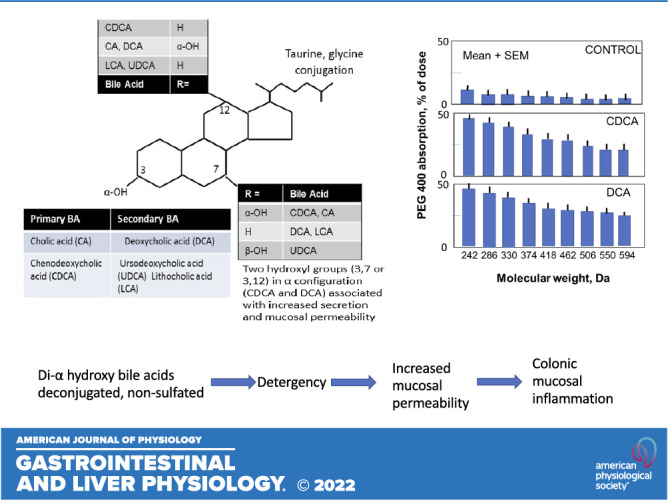
Chadwick et al. (4) performed pioneering studies of the effects of several bile acids perfused at 5 mM concentration in the rabbit colon on absorption/secretion of fluid with permeability measurements using polyethylene glycol 400 (PEG400), which is a polydisperse of nine molecules, spaced by 44 daltons (Da). Using gas chromatography, the individual chemicals were assayed in the perfusate, thereby delineating the spectrum of molecular sizes that permeated through the mucosa (Fig. 1) with the different bile acid moieties. The surface effects of CDCA and DCA were compared with those of ursodeoxycholic acid (UDCA, 3α,7β-dihydroxy-5β-cholan-24-oic acid), cholic acid (CA, 3α,7α,12α-dihydroxy-5β-cholan-24-oic acid), and other chemical-related entities using scanning electron microscopy, confirming the increased permeability and detergent, surface-damaging properties of only CDCA and DCA among the predominant, naturally occurring bile acids in mammals (4). It is important to note that the same studies showed no effect of the 7β-epimer of CDCA, which is ursodeoxycholic acid, that it does not induce secretion or cause altered permeability or mucosal pathology (4), as confirmed in other studies (20, 21). In addition, there is evidence that UDCA exerts antisecretory effects (22).
Figure 1.

Absorption of different molecular components of polyethylene glycol 400 in the rabbit colon in response to perfusion with control iso-osmolar electrolyte solution, and iso-osmolar solutions containing 5 mM CDCA or DCA. Note the increased absorption of all molecular species with the di-α-hydroxy bile acids, CDCA and DCA, compared with control, indicating increased colonic mucosal permeability. Data plotted from Ref. (4). CDCA, chenodeoxycholic acid; DCA, deoxycholic acid.
Di-α-hydroxy bile acids also increase mucosal permeability in sigmoid colon biopsies from healthy human participants studied in vitro (23) using concentrations of DCA and CDCA up to 1 mM. Permeability increase was measured using 51CrEDTA (molecular mass 340 Da), and was associated with Escherichia coli uptake into the lamina propria, confirmed by confocal microscopy; these data led to the hypothesis that bile acids may contribute to the development of intestinal inflammation. These in vitro studies with relatively low concentrations of DCA and CDCA can certainly explain the results showing increased permeability to 51CrEDTA [molecular weight 340, with reported molecular diameter of 10.5 Å and estimated molecular diameter 9.6 Å, based on the formula radius = 0.33 × (MM0.46)]. However, the uptake of the bacterium E. coli would not be expected to traverse the epithelial barrier that allows passage of 51CrEDTA, suggesting either the bacterial passage occurs through other mechanism(s) or that the in vitro model does not necessarily replicate in vivo conditions, for example, due to loss of surface mucus or innervation of tight junctions by the submucosal neurons that are absent in vitro.
These effects on colonic permeability have been confirmed by other studies on epithelial integrity in human colonic T84 cells, which showed CDCA and its 7α-dehydroxylated derivative, LCA, have opposite effects on permeability (24), as previously shown in vivo in rabbit colon (4). However, these studies also showed that prolonged exposure of the cell line for 2 h and up to 8 h with 0.5 mM CDCA increased permeability to 10 kDa dextran, suggesting effects of the bile acid on the “leak” pathway of permeability. CDCA was also shown to decrease occludin (but not claudin-2 protein expression) and to decrease occludin localization in the tight junctions (24). The effects of CDCA on permeability [and associated increase in interleukin-8 (IL-8) production] were also enhanced by the proinflammatory cytokines tumor necrosis factor-alpha (TNFα), IL-1β, and IFNγ. These effects on permeability and IL-8 production were all reduced by LCA (24).
Glycodeoxycholate (GDC) and deoxycholate were tested (25) at >2 mM concentrations over 60 min exposure in Caco-2 cells in vitro and at 10 mM ex vivo in isolated rat colonic mucosae mounted in Ussing chambers. Both agents reduced the transepithelial resistance, suggesting that the conjugation with glycine did not reduce the effects on mucosal barrier function. The 10 mM GDC increased the paracellular permeabilities to fluorescein isothiocyanate (FITC)-dextran 4000 (FD4) and the fluorescent peptide, FITC-LKP (leucine-lysine-proline, total molecular mass ∼700 Da), across colonic mucosae and increased paracellular fluxes for hydrophilic molecules including peptides such as octreotide (molar mass 1,019 Da; 25).
Mechanism
The modulation of intestinal permeability by the detergent bile acids is reported to occur via epidermal growth factor (EGF) receptor autophosphorylation, occludin dephosphorylation, and rearrangement at the tight junction level. The effect is mediated by the src family kinases and is abolished by EGF treatment (26). There is also evidence that certain bile acids, such as LCA and UDCA, may reduce apoptosis, which is one of the mechanisms leading to increased mucosal permeability in response to dioctyl sodium sulfosuccinate (DSS; 8). However, it does not appear that programmed cell loss or apoptosis is a direct effect of detergent bile acids, with the exception of LCA when present at relatively high concentrations of 500 µM in permeabilized cell lines such as HT-29 cells and HCT-116 cell (27).
Increased permeability with minimal-to-no intestinal inflammation in disease states.
Two examples are discussed here.
Nonalcoholic steatohepatitis. There is evidence implicating intestinal permeability in the pathogenesis of nonalcoholic steatohepatitis (NASH), but the underlying mechanisms have been unclear. Gupta et al. (28) examined the role of bile acids in Western diet-induced loss of colonic epithelial barrier function in junctional adhesion molecule A knockout mice, which have impaired intestinal epithelial barrier function. Knockout mice fed a Western diet developed severe NASH, which was associated with selectively increased unconjugated primary bile acid concentration in the cecum and increased abundance of microbial taxa linked to bile acid metabolism. In vitro assays revealed that CDCA (elevated in the cecum of the mice) increased paracellular permeability, whereas sevelamer hydrochloride, a bile acid-binding resin, protected against CDCA-induced loss of barrier function in vitro. In vivo, sevelamer attenuated colonic mucosal inflammation, improved barrier function [measured by blood levels of FITC-conjugated dextran (4 kDa) 3 h after oral gavage], reduced hepatic inflammation and fibrosis, and improved metabolic derangements associated with NASH. The authors concluded that increased primary bile acids in the colon impair the intestinal barrier in NASH (28).
Bile acid diarrhea. Intestinal and colonic permeability were compared in vivo using oral sugar probes, [13C]mannitol and lactulose, in 44 patients with bile acid diarrhea [(BAD) elevated fasting serum 7αC4, with lower fasting serum fibroblast growth factor 19 (FGF-19) and higher fecal primary bile acids in random stool sample], in 161 patients with diarrhea-predominant irritable bowel syndrome (IBS-D) with no evidence of bile acid diarrhea, and in 60 healthy controls (29). The patients with BAD had significantly higher [13C]mannitol urinary excretion, corresponding to both small bowel (2–8 h excretion) and colon (8–24 and 2–24 h excretion), compared with the patients with IBS-D whose permeability was not significantly different than the healthy controls.
DI-α-HYDROXY BILE ACIDS AND BACTERIAL UPTAKE IN HUMAN COLONIC AND DUODENAL BIOPSIES IN VITRO
Colonic Biopsies
As indicated earlier, the permeability of 51CrEDTA in human colonic biopsies after adding 100, 500, or 1,000 µmol/L of CDCA and DCA for 120 min was increased, when measured as percentage passage of mucosal concentration. This increased permeability was associated with greater uptake of E. coli during exposure to CDCA and DCA (23).
Duodenal Biopsies
Duodenal biopsies from patients with functional dyspepsia and healthy volunteers were investigated using Ussing chamber technique (30). The transepithelial electrical resistance (TEER) of duodenal biopsies from the patients was lower than with healthy volunteers, consistent with increased mucosal permeability. Conjugated bile acids in duodenal aspirates from the patients showed the ratio of cholic acid (CA) and CDCA to UDCA correlated positively with transepithelial electrical resistance in the patients. However, E. coli passage after 120 min was surprisingly lower in biopsies from the patients than in biopsies from the healthy controls, with no correlation observed between mucosal resistance and bacterial passage.
Overall, these data show increased mucosal permeability in association with bile acid exposure at both sites, but they only support a potential for bacterial passage into colonic rather than small bowel mucosa. These data may suggest this mechanism is more relevant in the colon, given the higher permeability of the small bowel compared with the colon and the higher microbial concentrations in the colon.
MUCUS DEPLETION AND EPITHELIAL DAMAGE
Mucin depletion is often the first protection to the colonic epithelium that is damaged by the detergent effects of bile acids. This was demonstrated by the timing of mucin depletion, which preceded biochemical evidence of mucosal damage and fluid secretion in perfusion studies in the rabbit colon (5, 6).
These in vivo observations were confirmed in vitro in a cell culture system. Thus, following removal of the mucus layer by incubation with the mucolytic agent, N-acetyl-l-cysteine, there was increased absorption of hydrophilic markers (mannitol, polyethylene glycols 900 and 4000) of permeability, consistent with greater paracellular permeability (31).
MICROSCOPIC INFLAMMATION INDUCED BY DETERGENT BILE ACIDS
Terminal ileal biopsies from 15 patients with irritable bowel syndrome (IBS) showed increased farnesoid X receptor (FXR) mRNA expression compared with those from 15 healthy controls (32). FXR is a nuclear receptor in the ileal enterocytes, and it is stimulated by bile acids absorbed by ileal enterocytes. It has also been demonstrated (33) that FXR has anti-inflammatory effects (inhibition of proinflammatory cytokine production), preserves intestinal barrier function (measured by FITC-dextran 4 kDa absorption in blood at 4 h after oral gavage), and reduces goblet cell loss in inflammatory bowel disease (IBD) models [dextran sulfate sodium (DSS)- and trinitrobenzene sulfonic acid (TNBS)-induced colitis] in mice.
In addition, in vitro studies performed using Caco-2 cells demonstrated that CDCA significantly decreased transepithelial electrical resistance (TEER), increased permeability, and increased interleukin-8 (IL-8) release. The increased IL-8 release was inhibited by preincubation of the Caco-2 cells with the FXR antagonist, guggulsterone, but the increased permeability was not reversed (22). The potential for FXR agonists to reduce inflammation, enhance barrier function, and regulate immunity in the gut has been reviewed elsewhere (34).
Relevance of Bile Acid-Induced Inflammation to Microscopic Colitis
There is evidence of bile acid malabsorption (BAM) in microscopic colitis. Based on the 75SeHCAT retention test, 43% of patients with microscopic colitis [lymphocytic (60%) and collagenous (27%) colitis] had evidence of BAM, and 86% of those patients responded to cholestyramine (35). Evidence of BAM in microscopic colitis in other studies has not been consistent (36–38), though recent study showed 9/14 (64%) patients with microscopic colitis had BAM based on serum 7αC4 > 48.3 ng/mL and FGF-19 < 60 pg/mL (39). Nevertheless, the mechanistic basis for BAM is supported by evidence of villous atrophy, inflammation, and collagen deposition in the ileum reported in patients with microscopic colitis (40, 41). Indeed, suppression if inflammation with budesonide is associated with increased ability to absorb bile acids based on 75SeHCAT retention (42). Nevertheless, there is still no published evidence that bile acid sequestration alone results in reversal of the colonic inflammation in microscopic colitis.
ROLE OF SULFATION OF BILE SALTS IN DETOXIFICATION OR PREVENTION OF DETERGENT EFFECTS
Sulfotransferase-mediated sulfation is an essential conjugation reaction in mammalian physiology. The substrates of sulfation include endogenous and exogenous chemicals, as well as protein peptides (43). Sulfotransferases catalyze the transfer of a sulfonate group from the universal sulfate donor, 3′-phosphoadenosine-5′-phosphosulfate (PAPS), to a nucleophilic group of their substrates to generate hydrophilic products. In mammals, PAPS is generated from adenosine trisphosphate and inorganic sulfate (SO42–), in which the 3′-phosphoadenosine-5′-phosphosulfate synthase 2 (PAPSS2) is the key enzyme to catalyze the formation of PAPS (44, 45). Decreased intestinal sulfation has been observed in patients with gastrointestinal diseases, including inflammatory bowel disease (46–48).
Sulfation of bile salt was first documented by Palmer (49), who identified partially sulfated lithocholate in human bile. Subsequently, sulfate esters of cholate and chenodeoxycholate were also identified in serum and urine of patients with cholestasis. Sulfation increases the solubility of bile salts in water and changes their metabolism, excretion, and toxicity. Specifically, sulfated bile salts are more rapidly excreted in the urine, and sulfated lithocholate is less efficiently reabsorbed in the intestine than nonsulfated lithocholate. Hydrophobic bile acids, such as LCA, cause bile duct injury and destructive cholangitis with periductal fibrosis resembling sclerosing cholangitis (50). Sulfated bile salts seem less toxic than nonsulfated molecules. Sulfation of lithocholate is an important metabolic pathway and reduces lithocholate hepatotoxicity by intrahepatic sulfation in healthy participants (51). In contrast, sulfation of cholate, deoxycholate, and chenodeoxycholate only becomes quantitatively important in patients with cholestasis (52).
Sulfotransferases are also identified in the human intestine, particularly the small intestinal mucosa (53, 54). There are potential beneficial or detoxifying effects of bile acid sulfation, reversing their detergency, including secretory and inflammatory effects.
Sulfation Inhibits Bile Acid-Induced Secretion
Breuer et al. (7) used a single-pass perfusion system in conscious restrained rats to measure changes in water and electrolyte transport, and mucosal damage after perfusing 5–15 mM solutions of sulfated or nonsulfated bile acids through the colon. In contrast to 5 mM, nonsulfated DCA or CDCA perfusion with 5 and 15 mM sulfated DCA or with 5 mM sulfated CDCA (7) had minimal secretion and caused only modest changes in protein and DNA output (markers of mucosal damage). These results suggest that sulfation prevents secretion caused by the di-α-hydroxy bile acids in the colon (7).
GYY4137, a novel and synthetic (H2S) hydrogen sulfide donor, reduced injury of intestinal barrier induced by sodium deoxycholate both in vivo and in vitro (55). In Caco-2 monolayers, pretreatment with GYY4137 markedly reduced barrier dysfunction and improved distribution of tight junctions and phosphorylation level of myosin light chain kinase. In vivo in mice, gavaged GYY4137 reduced the sodium deoxycholate-induced injury of the intestinal barrier including histological score, and improved expression level of tight junctions (55).
Relevance of Bile Acid Sulfation in Disease States
Functional constipation in children.
Hofmann et al. (56) investigated fecal bile acid composition in 103 children with functional constipation and in 104 nonconstipated control children (56). Whereas the proportions of DCA did not differ in the two groups, monosulfated dihydroxy bile acids were greater in the children with functional constipation, and the difference was attributable to six constipated children whose major fecal bile acid was the 3-sulfate of CDCA. Sulfation of CDCA is known to abolish its secretory activity and may contribute to constipation.
There is also other evidence of colonic bile acid deficiency in association with constipation; thus, 15% of patients with irritable bowel syndrome (IBS)-constipation have reduced total bile acids and level of DCA in fecal samples collected over 48 h on a 100 g fat diet. In these patients, lower levels of excretion of bile acids into feces correlated with slower colonic transit (57). Although the state of sulfation of the bile acids was not measured, these data suggest that the absence of the physiological laxative effects of the bile acids resulted in retardation of colonic transit and presentation with constipation. Moreover, the IBAT inhibitor, elobixibat, is an approved treatment for chronic constipation in Japan (58).
Sulfation of Secondary Bile Acids May Be Proinflammatory
Inflammatory bowel disease.
In a study of patients with colonic inflammatory bowel disease (IBD; n = 42) and healthy subjects (n = 29), fecal-conjugated and 3-OH-sulfated bile acids were significantly higher in active IBD, whereas secondary bile acids rates were significantly lower. In addition, the deconjugation, transformation, and de-sulfation activities of the microbiota were impaired in the patients with IBD (9).
The impact of bile acids on the inflammatory response was investigated in vitro using Caco-2 cells stimulated by IL-1β. These studies showed that secondary bile acids exerted anti-inflammatory effects, but sulfation of secondary bile acids abolished their anti-inflammatory properties. These data suggest that impaired microbiota enzymatic activity in IBD-associated dysbiosis leads to modifications in the luminal bile acid pool composition that may be a factor leading to chronic inflammation in IBD (9).
Sulfation Prevents Inflammation and Carcinogenesis
A more recent study came to a different conclusion based on studies of PAPSS2, which is the key enzyme to generate PAPS, the universal sulfonate donor for all sulfation reactions. PAPSS2 mRNA expression was downregulated in IBD colon biopsies from patients with actively inflamed IBD (both ulcerative colitis and Crohn’s disease) compared with non-IBD colon biopsies (59). The decreased expression of PAPSS2 was associated with decreased expression of tight junction proteins including occludin and zonula occludens-1, and increased expression of inflammatory marker genes such as interleukin-6 (IL-6), IL-1b, and tumor necrosis factor-α (TNFα).
Intestinal-specific PAPSS2 knockout (PAPSS2ΔIE) mice showed heightened sensitivity to colitis and colon cancer from damaging the intestinal mucosal barrier, increasing intestinal permeability and bacteria infiltration, and worsening the intestinal tumor microenvironment. These mice also exhibited reduced intestinal sulfomucin content; sulfomucin content is essential for gut barrier function, and the effect on barrier function was demonstrated in the knockout mice using serum levels of FITC-dextran in both untreated and dioctyl sodium sulfosuccinate (DSS)-treated mice. Metabolomic analyses revealed the accumulation of bile acids and deficiency in the formation of bile acid sulfates in the colon (59). Finally, the expression of PAPSS2 is decreased in the colon cancers of mice and humans. The lower expression of PAPSS2 in patients with colon cancer is correlated with worse survival.
Overall, these studies show that PAPSS2 protects against colitis and associated colonic carcinogenesis through intestinal sulfomucin and promotion of bile acid homeostasis via the PAPSS2-PAPS-sulfation axis. The authors also suggested that intestinal sulfation may represent a potential diagnostic marker and that PAPSS2 may serve as a potential therapeutic target for inflammatory bowel disease and colon cancer (59).
The discrepancies in the results and conclusions of the two studies may reflect the differences in the cells studied, which ranged from CacO2 cells to biopsies from patients with active inflammatory bowel disease. Thus, further research is necessary to investigate the effect of sulfation of bile acids on inflammation.
POTENTIAL ROLE OF URSODEOXYCHOLIC ACID AS AN ANTI-INFLAMMATORY AGENT
There is increasing evidence that UDCA has anti-inflammatory effects, which include inhibition of TNFα-induced release of IL-8 from monocytes (60), regulation of human beta defensin-1 and 2 secretion by colonic epithelial cells (61), attenuation of the release of proinflammatory cytokines from colonic epithelial cells in vitro, and protection against the development of colonic inflammation in vivo (62), inhibition of epithelial apoptosis (8), and regulation of colonic epithelial wound healing (63). These and other data are summarized in a prior review (64). The potential clinical impact of these effects was documented by the observation that UDCA significantly decreased the risk for developing colorectal dysplasia or cancer in patients with ulcerative colitis and primary sclerosing cholangitis (65).
A potential role of the microbiome in the beneficial effects of UDCA is supported by the evidence that UDCA was able to reverse inflammation and infection due to recurrent Clostridioides difficile in a patient with pouchitis, and the ability of UDCA to inhibit the germination and the growth of C. difficile in vitro (66).
CONCLUSIONS
The detergent effects of di-α-hydroxy bile acids are relevant to several colonic diseases, and the studies of sulfation as a metabolic pathway for toxic bile acids suggest there is a significant impact that could be expected with further research of the biochemical as well as metabolic effects of nonsulfated and sulfated bile acids. Such studies could impact bile acid diarrhea, functional constipation, inflammatory bowel disease, and even colon carcinogenesis. These insights also emphasize the relevance and opportunities afforded by greater understanding of the chemistry and metabolism of bile acids. Further research into the metabolic interactions of microbiota and bile acids has additional potential.
GRANTS
This study was supported by National Institutes of Health Grant R01-DK115950 (to M. Camilleri).
DISCLAIMERS
Its contents are solely the responsibility of the authors and do not necessarily represent the official view of NIH.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
M.C. prepared figures; drafted manuscript; edited and revised manuscript; approved final version of manuscript.
ACKNOWLEDGMENTS
The author thanks Cindy Stanislav for secretarial support.
REFERENCES
- 1.Hegyi P, Maléth J, Walters JR, Hofmann AF, Keely SJ. Guts and gall: bile acids in regulation of intestinal epithelial function in health and disease. Physiol Rev 98: 1983–2023, 2018. doi: 10.1152/physrev.00054.2017. [DOI] [PubMed] [Google Scholar]
- 2.Slijepcevic D, Roscam Abbing RLP, Katafuchi T, Blank A, Donkers JM, van Hoppe S, de Waart DR, Tolenaars D, van der Meer JHM, Wildenberg M, Beuers U, Oude Elferink RPJ, Schinkel AH, van de Graaf SFJ. Hepatic uptake of conjugated bile acids is mediated by both sodium taurocholate cotransporting polypeptide and organic anion transporting polypeptides and modulated by intestinal sensing of plasma bile acid levels in mice. Hepatology 66: 1631–1643, 2017. doi: 10.1002/hep.29251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Takikawa H. Hepatobiliary transport of bile acids and organic anions. J Hepatobiliary Pancreat Surg 9: 443–447, 2002. doi: 10.1007/s005340200055. [DOI] [PubMed] [Google Scholar]
- 4.Chadwick VS, Gaginella TS, Carlson GL, Debongnie JC, Phillips SF, Hofmann AF. Effect of molecular structure on bile acid-induced alterations in absorptive function, permeability, and morphology in the perfused rabbit colon. J Lab Clin Med 94: 661–674, 1979. [PubMed] [Google Scholar]
- 5.Camilleri M, Murphy R, Chadwick VS. Dose-related effects of chenodeoxycholic acid in the rabbit colon. Dig Dis Sci 25: 433–438, 1980. doi: 10.1007/BF01395507. [DOI] [PubMed] [Google Scholar]
- 6.Camilleri M, Murphy R, Chadwick VS. Pharmacological inhibition of chenodeoxycholate-induced fluid and mucus secretion and mucosal injury in the rabbit colon. Dig Dis Sci 27: 865–869, 1982. doi: 10.1007/BF01316567. [DOI] [PubMed] [Google Scholar]
- 7.Breuer NF, Rampton DS, Tammar A, Murphy GM, Dowling RH. Effect of colonic perfusion with sulfated and nonsulfated bile acids on mucosal structure and function in the rat. Gastroenterology 84: 969–977, 1983. doi: 10.1016/0016-5085(83)90199-3. [DOI] [PubMed] [Google Scholar]
- 8.Lajczak-McGinley NK, Porru E, Fallon CM, Smyth J, Curley C, McCarron PA, Tambuwala MM, Roda A, Keely SJ. The secondary bile acids, ursodeoxycholic acid and lithocholic acid, protect against intestinal inflammation by inhibition of epithelial apoptosis. Physiol Rep 8: e14456, 2020. doi: 10.14814/phy2.14456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Duboc H, Rajca S, Rainteau D, Benarous D, Maubert M-A, Quervain E, Thomas G, Barbu V, Humbert L, Despras G, Bridonneau C, Dumetz F, Grill J-P, Masliah J, Beaugerie L, Cosnes J, Chazouillères O, Poupon R, Wolf C, Mallet J-M, Langella P, Trugnan G, Sokol H, Seksik P. Connecting dysbiosis, bile-acid dysmetabolism and gut inflammation in inflammatory bowel diseases. Gut 62: 531–539, 2013. doi: 10.1136/gutjnl-2012-302578. [DOI] [PubMed] [Google Scholar]
- 10.Baiocchi L, Zhou T, Liangpunsakul S, Lenci I, Santopaolo F, Meng F, Kennedy L, Glaser S, Francis H, Alpini G. Dual role of bile acids on the biliary epithelium: friend or foe? Int J Mol Sci 20: 1869, 2019. doi: 10.3390/ijms20081869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Singh M, Singh A, Kundu S, Bansal S, Bajaj A. Deciphering the role of charge, hydration, and hydrophobicity for cytotoxic activities and membrane interactions of bile acid based facial amphiphiles. Biochim Biophys Acta 1828: 1926–1937, 2013. doi: 10.1016/j.bbamem.2013.04.003. [DOI] [PubMed] [Google Scholar]
- 12.Gafar AA, Draz HM, Goldberg AA, Bashandy MA, Bakry S, Khalifa MA, AbuShair W, Titorenko VI, Sanderson JT. Lithocholic acid induces endoplasmic reticulum stress, autophagy and mitochondrial dysfunction in human prostate cancer cells. PeerJ 4: e2445, 2016. doi: 10.7717/peerj.2445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dharmsathaphorn K, Huott PA, Vongkovit P, Beuerlein G, Pandol SJ, Ammon HV. Cl-secretion induced by bile salts. A study of the mechanism of action based on a cultured colonic epithelial cell line. J Clin Invest 84: 945–953, 1989. doi: 10.1172/JCI114257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Keely SJ, Urso A, Ilyaskin AV, Korbmacher C, Bunnett NW, Poole DP, Carbone SE. Contributions of bile acids to gastrointestinal physiology as receptor agonists and modifiers of ion channels. Am J Physiol Gastrointest Liver Physiol 322: G201–G222, 2022. doi: 10.1152/ajpgi.00125.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Debongnie JC, Phillips SF. Capacity of the human colon to absorb fluid. Gastroenterology 74: 698–703, 1978. [PubMed] [Google Scholar]
- 16.Hammer J, Phillips SF. Fluid loading of the human colon: effects on segmental transit and stool composition. Gastroenterology 105: 988–998, 1993. doi: 10.1016/0016-5085(93)90941-5. [DOI] [PubMed] [Google Scholar]
- 17.Mekhjian HS, Phillips SF, Hofmann AF. Colonic absorption of unconjugated bile acids: perfusion studies in man. Dig Dis Sci 24: 545–550, 1979. doi: 10.1007/BF01489324. [DOI] [PubMed] [Google Scholar]
- 18.Khanna L, Halfdanarson TR, Sonbol MB, Eiring R, Prond T, Camilleri M. Bile acid malabsorption in patients with neuroendocrine tumors. Dig Dis Sci, 2021. doi: 10.1007/s10620-021-07189-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vijayvargiya P, Gonzalez Izundegui D, Calderon G, Tawfic S, Batbold S, Saifuddin H, Duggan P, Melo V, Thomas T, Heeney M, Beyde A, Miller J Jr, Valles K, Oyemade K, Brant JF, Atieh J, Donato LJ, Camilleri M. Increased fecal bile acid excretion in a significant subset of patients with other inflammatory diarrheal diseases. Dig Dis Sci, 2021. doi: 10.1007/s10620-021-06993-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gordon SJ, Kinsey MD, Magen JS, Joseph RE, Kowlessar OD. Structure of bile acids associated with secretion in the rat cecum. Gastroenterology 77: 38–44, 1979. [PubMed] [Google Scholar]
- 21.Igimi H, Carey MC. pH-Solubility relations of chenodeoxycholic and ursodeoxycholic acids: physical-chemical basis for dissimilar solution and membrane phenomena. J Lipid Res 21: 72–90, 1980. [PubMed] [Google Scholar]
- 22.Kelly OB, Mroz MS, Ward JB, Colliva C, Scharl M, Pellicciari R, Gilmer JF, Fallon PG, Hofmann AF, Roda A, Murray FE, Keely SJ. Ursodeoxycholic acid attenuates colonic epithelial secretory function. J Physiol 591: 2307–2318, 2013. doi: 10.1113/jphysiol.2013.252544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Munch A, Ström M, Söderholm JD. Dihydroxy bile acids increase mucosal permeability and bacterial uptake in human colon biopsies. Scand J Gastroenterol 42: 1167–1174, 2007. doi: 10.1080/00365520701320463. [DOI] [PubMed] [Google Scholar]
- 24.Sarathy J, Detloff SJ, Ao M, Khan N, French S, Sirajuddin H, Nair T, Rao MC. The Yin and Yang of bile acid action on tight junctions in a model colonic epithelium. Physiol Rep 5: e13294, 2017. doi: 10.14814/phy2.13294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brayden DJ, Stuettgen V. Sodium glycodeoxycholate and sodium deoxycholate as epithelial permeation enhancers: in vitro and ex vivo intestinal and buccal bioassays. Eur J Pharm Sci 159: 105737, 2021. doi: 10.1016/j.ejps.2021.105737. [DOI] [PubMed] [Google Scholar]
- 26.Raimondi F, Santoro P, Barone MV, Pappacoda S, Barretta ML, Nanayakkara M, Apicella C, Capasso L, Paludetto R. Bile acids modulate tight junction structure and barrier function of Caco-2 monolayers via EGFR activation. Am J Physiol Gastrointest Liver Physiol 294: G906–G913, 2008. doi: 10.1152/ajpgi.00043.2007. [DOI] [PubMed] [Google Scholar]
- 27.Katona BW, Anant S, Covey DF, Stenson WF. Characterization of enantiomeric bile acid-induced apoptosis in colon cancer cell lines. J Biol Chem 284: 3354–3364, 2009. doi: 10.1074/jbc.M805804200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gupta B, Liu Y, Chopyk DM, Rai RP, Desai C, Kumar P, Farris AB, Nusrat A, Parkos CA, Anania FA, Raeman R. Western diet-induced increase in colonic bile acids compromises epithelial barrier in nonalcoholic steatohepatitis. FASEB J 34: 7089–7102, 2020. doi: 10.1096/fj.201902687R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Magnus Y, BouSaba J, Sannaa W, McKinzie S, Busciglio I, Camilleri M. Bile acid diarrhea is associated with increased intestinal permeability compared with irritable bowel syndrome-diarrhea. Gastroenterology, 2021. doi: 10.1053/j.gastro.2021.12.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Beeckmans D, Farré R, Riethorst D, Keita ÅV, Augustijns P, Söderholm JD, Vanuytsel T, Vanheel H, Tack J. Relationship between bile salts, bacterial translocation, and duodenal mucosal integrity in functional dyspepsia. Neurogastroenterol Motil 32: e13788, 2020. doi: 10.1111/nmo.13788. [DOI] [PubMed] [Google Scholar]
- 31.Meaney C, O’Driscoll C. Mucus as a barrier to the permeability of hydrophilic and lipophilic compounds in the absence and presence of sodium taurocholate micellar systems using cell culture models. Eur J Pharm Sci 8: 167–175, 1999. doi: 10.1016/S0928-0987(99)00007-X. [DOI] [PubMed] [Google Scholar]
- 32.Horikawa T, Oshima T, Li M, Kitayama Y, Eda H, Nakamura K, Tamura A, Ogawa T, Yamasaki T, Okugawa T, Kondo T, Kono T, Tozawa K, Tomita T, Fukui H, Watari J, Miwa H. Chenodeoxycholic acid releases proinflammatory cytokines from small intestinal epithelial cells through the farnesoid X receptor. Digestion 100: 286–294, 2019. doi: 10.1159/000496687. [DOI] [PubMed] [Google Scholar]
- 33.Gadaleta RM, van Erpecum KJ, Oldenburg B, Willemsen ECL, Renooij W, Murzilli S, Klomp LWJ, Siersema PD, Schipper MEI, Danese S, Penna G, Laverny G, Adorini L, Moschetta A, van Mil SWC. Farnesoid X receptor activation inhibits inflammation and preserves the intestinal barrier in inflammatory bowel disease. Gut 60: 463–472, 2011. doi: 10.1136/gut.2010.212159. [DOI] [PubMed] [Google Scholar]
- 34.Anderson KM, Gayer CP. The pathophysiology of farnesoid X receptor (FXR) in the GI tract: inflammation, barrier function and innate immunity. Cells 10: 3206, 2021. doi: 10.3390/cells10113206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fernandez-Banares F, Esteve M, Salas A, Forne TM, Espinos JC, Martin-Comin J, Viver JM. Bile acid malabsorption in microscopic colitis and in previously unexplained functional chronic diarrhea. Dig Dis Sci 46: 2231–2238, 2001. doi: 10.1023/a:1011927302076. [DOI] [PubMed] [Google Scholar]
- 36.Giardiello FM, Bayless TM, Jessurun J, Hamilton SR, Yardley JH. Collagenous colitis: Physiologic and histopathologic studies in seven patients. Ann Intern Med 106: 46–49, 1987. doi: 10.7326/0003-4819-106-1-46. [DOI] [PubMed] [Google Scholar]
- 37.Kingham JG, Levison DA, Ball JA, Dawson AM. Microscopic colitis-a cause of chronic watery diarrhoea. Br Med J (Clin Res Ed) 285: 1601–1604, 1982. doi: 10.1136/bmj.285.6355.1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Eusufzai S, Löfberg R, Veress B, Einarsson K, Angelin B. Studies on bile acid metabolism in collagenous colitis: no evidence of bile acid malabsorption as determined by the SeHCAT test. Eur J Gastroenterol Hepatol 4: 317–321, 1992. [Google Scholar]
- 39.Lyutakov I, Lozanov V, Sugareva P, Valkov H, Penchev P. Serum 7-alfa-hydroxy-4-cholesten-3-one and fibroblast growth factor-19 as biomarkers diagnosing bile acid malabsorption in microscopic colitis and inflammatory bowel disease. Eur J Gastroenterol Hepatol 33: 380–387, 2021. doi: 10.1097/MEG.0000000000001925. [DOI] [PubMed] [Google Scholar]
- 40.Einarsson K, Eusufzai S, Johansson U, Lofberg R, Theodorsson E, Veress B. Villous atrophy of distal ileum and lymphocytic colitis in a woman with bile acid malabsorption. Eur J Gastroenterol Hepatol 4: 585–590, 1992. [Google Scholar]
- 41.Marteau P, Lavergne-Slove A, Lemann M, Bouhnik Y, Bertheau P, Becheur H, Galian A, Rambaud JC. Primary ileal villous atrophy is often associated with microscopic colitis. Gut 41: 561–564, 1997. doi: 10.1136/gut.41.4.561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bajor A, Kilander A, Gälman C, Rudling M, Ung KA. Budesonide treatment is associated with increased bile acid absorption in collagenous colitis. Aliment Pharmacol Ther 24: 1643–1649, 2006. doi: 10.1111/j.1365-2036.2006.03168.x. [DOI] [PubMed] [Google Scholar]
- 43.Kauffman FC. Sulfonation in pharmacology and toxicology. Drug Metab Rev 36: 823–843, 2004. doi: 10.1081/dmr-200033496. [DOI] [PubMed] [Google Scholar]
- 44.Leung AW, Backstrom I, Bally MB. Sulfonation, an underexploited area: from skeletal development to infectious diseases and cancer. Oncotarget 7: 55811–55827, 2016. doi: 10.18632/oncotarget.10046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mueller JW, Idkowiak J, Gesteira TF, Vallet C, Hardman R, van den Boom J, Dhir V, Knauer SK, Rosta E, Arlt W. Human DHEA sulfation requires direct interaction between PAPS synthase 2 and DHEA sulfotransferase SULT2A1. J Biol Chem 293: 9724–9735, 2018. doi: 10.1074/jbc.RA118.002248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dawson PA, Huxley S, Gardiner B, Tran T, McAuley JL, Grimmond S, McGuckin MA, Markovich D. Reduced mucin sulfonation and impaired intestinal barrier function in the hyposulfataemic NaS1 null mouse. Gut 58: 910–919, 2009. doi: 10.1136/gut.2007.147595. [DOI] [PubMed] [Google Scholar]
- 47.Corfield AP, Myerscough N, Bradfield N, Corfield Cdo A, Gough M, Clamp JR, Durdey P, Warren BF, Bartolo DC, King KR, Williams JM. Colonic mucins in ulcerative colitis: evidence for loss of sulfation. Glycoconj J 13: 809–822, 1996. doi: 10.1007/BF00702345. [DOI] [PubMed] [Google Scholar]
- 48.Raouf AH, Tsai HH, Parker N, Hoffman J, Walker RJ, Rhodes JM. Sulphation of colonic and rectal mucin in inflammatory bowel disease: reduced sulphation of rectal mucus in ulcerative colitis. Clin Sci (Lond) 83: 623–626, 1992. doi: 10.1042/cs0830623. [DOI] [PubMed] [Google Scholar]
- 49.Palmer RH. The formation of bile acid sulfates: a new pathway of bile acid metabolism in humans. Proc Natl Acad Sci USA 58: 1047–1050, 1967. doi: 10.1073/pnas.58.3.1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Trauner M, Fickert P, Baghdasaryan A, Claudel T, Halilbasic E, Moustafa T, Wagner M, Zollner G. New insights into autoimmune cholangitis through animal models. Dig Dis 28: 99–104, 2010. doi: 10.1159/000282072. [DOI] [PubMed] [Google Scholar]
- 51.Hofmann AF. Detoxification of lithocholic acid, a toxic bile acid: relevance to drug hepatotoxicity. Drug Metab Rev 36: 703–722, 2004. doi: 10.1081/dmr-200033475. [DOI] [PubMed] [Google Scholar]
- 52.Stiehl A. Sulfation of bile salts: a new metabolic pathway. Digestion 11: 406–413, 1974. doi: 10.1159/000197609. [DOI] [PubMed] [Google Scholar]
- 53.Dew MJ, Hawker PC, Nutter S, Allan RN. Human intestinal sulfation of lithocholate: a new site for bile acid metabolism. Life Sci 27: 317–323, 1980. doi: 10.1016/0024-3205(80)90199-X. [DOI] [PubMed] [Google Scholar]
- 54.Chen G, Zhang D, Jing N, Yin S, Falany CN, Radominska-Pandya A. Human gastrointestinal sulfotransferases: identification and distribution. Toxicol Appl Pharmacol 187: 186–197, 2003. doi: 10.1016/s0041-008x(02)00073-x. [DOI] [PubMed] [Google Scholar]
- 55.Chen Z, Tang J, Wang P, Zhu J, Liu Y. GYY4137 attenuates sodium deoxycholate-induced intestinal barrier injury both in vitro and in vivo. Biomed Res Int 2019: 5752323, 2019. doi: 10.1155/2019/5752323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hofmann AF, Loening-Baucke V, Lavine JE, Hagey LR, Steinbach JH, Packard CA, Griffin TL, Chatfield DA. Altered bile acid metabolism in childhood functional constipation: inactivation of secretory bile acids by sulfation in a subset of patients. J Pediatr Gastroenterol Nutr 47: 598–606, 2008. doi: 10.1097/MPG.0b013e31816920a6. [DOI] [PubMed] [Google Scholar]
- 57.Vijayvargiya P, Busciglio I, Burton D, Donato L, Lueke A, Camilleri M. Bile acid deficiency in a subgroup of patients with irritable bowel syndrome with constipation based on biomarkers in serum and fecal samples. Clin Gastroenterol Hepatol 16: 522–527, 2018. doi: 10.1016/j.cgh.2017.06.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Khanna L, Camilleri M. Elobixibat: a novel treatment for chronic constipation. Aliment Pharmacol Ther 53: 234–242, 2021. doi: 10.1111/apt.16143. [DOI] [PubMed] [Google Scholar]
- 59.Xu P, Xi Y, Zhu J, Zhang M, Luka Z, Stolz DB, Cai X, Xie Y, Xu M, Ren S, Huang Z, Yang D, York JD, Ma X, Xie W. Intestinal sulfation is essential to protect against colitis and colonic carcinogenesis. Gastroenterology 161: 271–286, 2021. doi: 10.1053/j.gastro.2021.03.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.O’Dwyer AM, Lajczak NK, Keyes JA, Ward JB, Greene CM, Keely SJ. Ursodeoxycholic acid inhibits TNFα-induced IL-8 release from monocytes. Am J Physiol Gastrointest Liver Physiol 311: G334–G341, 2016. doi: 10.1152/ajpgi.00406.2015. [DOI] [PubMed] [Google Scholar]
- 61.Lajczak NK, Saint-Criq V, O’Dwyer AM, Perino A, Adorini L, Schoonjans K, Keely SJ. Bile acids deoxycholic acid and ursodeoxycholic acid differentially regulate human β-defensin-1 and -2 secretion by colonic epithelial cells. FASEB J 31: 3848–3857, 2017. doi: 10.1096/fj.201601365R. [DOI] [PubMed] [Google Scholar]
- 62.Ward JBJ, Lajczak NK, Kelly OB, O’Dwyer AM, Giddam AK, Ní Gabhann J, Franco P, Tambuwala MM, Jefferies CA, Keely S, Roda A, Keely SJ. Ursodeoxycholic acid and lithocholic acid exert anti-inflammatory actions in the colon. Am J Physiol Gastrointest Liver Physiol 312: G550–G558, 2017. doi: 10.1152/ajpgi.00256.2016. [DOI] [PubMed] [Google Scholar]
- 63.Mroz MS, Lajczak NK, Goggins BJ, Keely S, Keely SJ. The bile acids, deoxycholic acid and ursodeoxycholic acid, regulate colonic epithelial wound healing. Am J Physiol Gastrointest Liver Physiol 314: G378–G387, 2018. doi: 10.1152/ajpgi.00435.2016. [DOI] [PubMed] [Google Scholar]
- 64.Keely SJ, Steer CJ, Lajczak-McGinley NK. Ursodeoxycholic acid: a promising therapeutic target for inflammatory bowel diseases? Am J Physiol Gastrointest Liver Physiol 317: G872–G881, 2019. doi: 10.1152/ajpgi.00163.2019. [DOI] [PubMed] [Google Scholar]
- 65.Pardi DS, Loftus EV Jr, Kremers WK, Keach J, Lindor KD. Ursodeoxycholic acid as a chemopreventive agent in patients with ulcerative colitis and primary sclerosing cholangitis. Gastroenterology 124: 889–893, 2003. doi: 10.1053/gast.2003.50156. [DOI] [PubMed] [Google Scholar]
- 66.Weingarden AR, Chen C, Zhang N, Graiziger CT, Dosa PI, Steer CJ, Shaughnessy MK, Johnson JR, Sadowsky MJ, Khoruts A. Ursodeoxycholic acid inhibits Clostridium difficile spore germination and vegetative growth, and prevents the recurrence of ileal pouchitis associated with the infection. J Clin Gastroenterol 50: 624–630, 2016. doi: 10.1097/MCG.0000000000000427. [DOI] [PMC free article] [PubMed] [Google Scholar]