

Abstract
Loss of orexin-producing neurons results in narcolepsy with cataplexy and orexin agonists have been shown to increase wakefulness and alleviate narcolepsy symptoms in animal models. Several OX2R agonists have been reported, but with little or no activity at OX1R. We conducted structure-activity relationship (SAR) studies on OX2R agonist YNT-185 (2) and discovered dual agonists such as RTOXA-43 (40) with EC50’s of 24 nM at both OX2R and OX1R. Computational modeling studies based on the agonist bound OX2R cryo-EM structures showed that 40 bound in the same binding pocket and interactions of the pyridylmethyl group of 40 with OX1R may have contributed to its high OX1R potency. Intraperitoneal injection of 40 increased time awake, decreased time asleep and increased sleep/wake consolidation in 12-month old mice. This work provides a promising dual small molecule agonist and supports development of orexin agonists as potential treatments for orexin-deficient disorders such as narcolepsy.
Keywords: orexin, narcolepsy, dual agonist, arylsulfonamide, structure-activity relationship
Graphical Abstract
Introduction
Orexin-A and B (also known as hypocretins 1 and 2) are two hypothalamic neuropeptides and the endogenous ligands for two G protein-coupled receptors (GPCRs), orexin-1 (OX1R) and orexin-2 (OX2R).1, 2 Orexin-expressing neurons are limited in number and located predominantly in a small area of the lateral hypothalamus;2-5 however, the nerve fibers of orexin neurons project throughout the central nervous system (CNS) and their afferents are sent to many brain regions in cortical, limbic and brainstem circuits.3, 6-8 The orexin system has been shown to modulate a variety of important behavioral and physiological processes, including sleep/wakefulness,9, 10 arousal,11, 12 feeding,2 energy homeostasis,2 stress and anxiety,13-16 and learning and memory.17-20
Loss of orexin-producing neurons results in narcolepsy with cataplexy, an incurable chronic neurological disorder characterized with sleep disruptions (excessive daytime sleepiness (EDS), fragmented sleep and intrusions of sleep episodes during the active phase, etc.).21-24 Narcolepsy affects an estimated 200,000 Americans and approximately 3 million worldwide and severely impacts the day-to-day lives of affected individuals. Central orexin-A administration successfully enhances wakefulness and consolidates sleep/wake states.25 Consistent with orexin’s multifaceted role, orexin deficiency has also been linked to abnormalities in energy homeostasis, stress-related behavior, and reward systems,26 and is believed to be associated with pathophysiologies such as obesity and age-related disorders.27 A loss of orexin neurons and/or orexin peptides has been found in Alzheimer’s and Parkinson’s patients,28-32 as well as in aged humans and mice.17-19
Medication development targeting the orexin system has largely focused on antagonists thus far, as over- or abnormal activation of the orexin system leads to insomnia.33, 34 A large number of orexin receptor antagonists, dual or subtype selective, have been developed.35-39 Two dual antagonists, Suvorexant and more recently Lemborexant, have been approved by the FDA for the treatment of insomnia. Orexin antagonism has also shown early promises in the treatment of drug addiction and anxiety disorders.13, 14, 16, 36, 40-42 In contrast, activation of the orexin receptors is primarily accomplished using orexin peptides, particularly the more stable orexin-A (33 AA with two disulfide bridges). Only a limited number of small molecule orexin agonists have been disclosed thus far. Yan7874 (1) was first reported as a small molecule OX2R agonist in a 2010 patent (Figure 1);43 however, it was later confirmed to be a weak agonist of both OX receptors (EC50 > 3.2 μM) and also showed OX receptor independent cytotoxicity.44 YNT-185 (2) was reported in 2015 and displayed good OX2R potency and selectivity over OX1R (EC50 = 28 nM vs. 2750 nM at OX1R).45 Intracerebroventricular (i.c.v., 30–300 nmol) and/or intraperitoneally (i.p., 40 mg/kg) administration of 2 (hydrochloride salt) promoted wakefulness without affecting body temperature in wild-type mice, whereas in orexin knock-out (KO) and orexin neuron-ablated mice, 2 suppressed cataplexy-like episodes.46 In another study, 2 attenuated morphine-induced sedative effects in rats, as assessed by EEG changes and behavioral measures including locomotor activity and startle response latency, without affecting the analgesic effect of morphine.47 More recently, a series of substituted piperidines as OX2R agonists were reported, and Tak-925 (3) is currently in phase 1 clinical trials for the treatment of narcolepsy.48 Another small molecule orexin agonist by Takeda, TAK-994, is in a Phase 2 clinical trial in type 1 and 2 narcoleptic patients (www.clinicaltrials.gov), although its structure is not disclosed. A series of structurally similar OX2R agonists based on a pyrrolidine core (e.g. 4) were reported in 2020 with high potencies at OX2R but no data were reported on OX1R activity.49 Finally, the cryogenic electron microscopy (cryo-EM) structure of the first agonist bound OX2R using agonist 5 was recently reported, providing structural insight for agonist-receptor interactions.50
Figure 1.
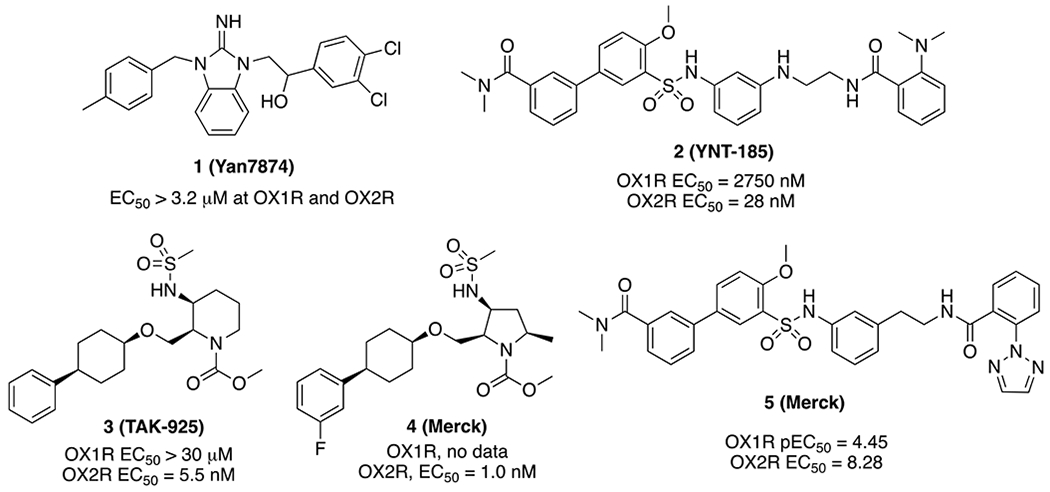
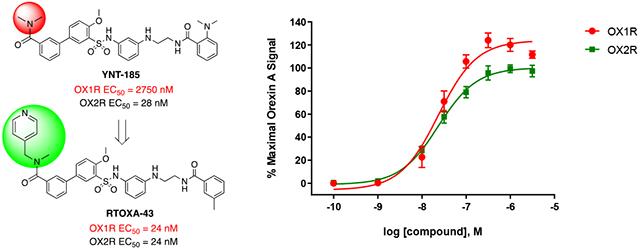
Small molecule orexin agonists reported in the literature
The two orexin receptors (and their mRNAs) have overlapping, but sometimes distinct, patterns of distribution in the brain, 3, 5, 51, 52 suggesting they may play differential physiological roles. For instance, mice lacking OX2R display several abnormalities similar to human narcolepsy;53, 54 however, the behavioral phenotype of OX2R KO mice appears less severe than orexin null mice, and double orexin receptor KO mice displayed sleep/wake disturbances most similar to human narcolepsy.55-57 Similarly, although OX2R antagonists were shown to be efficacious in promoting sleep,58 a number of pre-clinical studies have suggested that antagonizing both orexin receptors was more effective.55, 59, 60 Given the role of the OX1R in many functions and the equal potency of the endogenous peptide orexin-A, agonists that also have OX1R activity are desired. We conducted structure-activity relationship (SAR) studies on 2 with structural modifications at several sites (Figure 2), and developed a series of new orexin agonists (6-44). One of the most potent compounds (RTOXA-43, 40) showed good potencies at both OX1R and OX2R (EC50 = 24 at both OX1R and OX2R) when evaluated in calcium mobilization assays using cells overexpressing these receptors. Excitingly, 40 (40 mg/kg, i.p.) increased time awake, decreased time asleep and increased sleep/wake consolidation when measured using continuous EEG/EMG recordings in 12-month old mice. Hereby we describe the design, synthesis and pharmacological characterization of these orexin receptor agonists.
Figure 2.
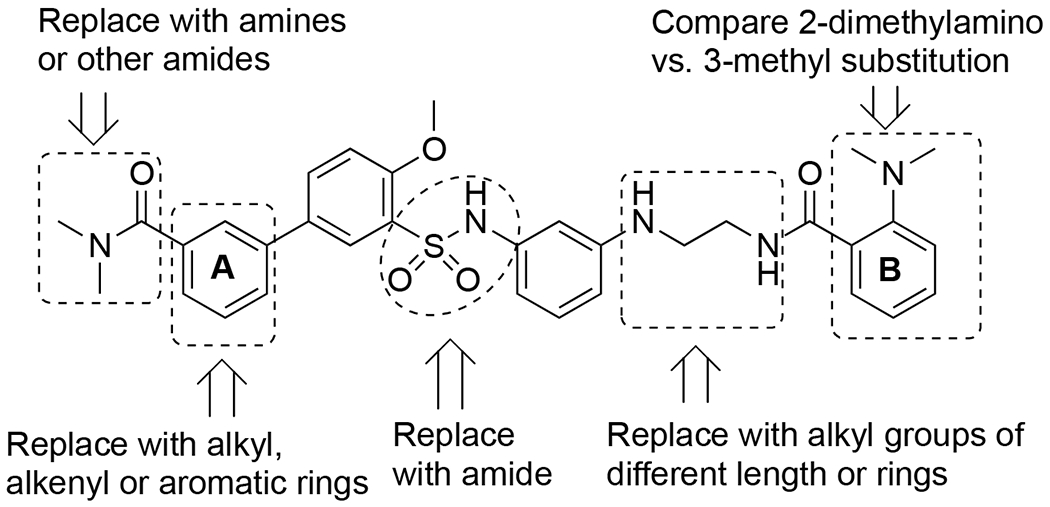
SAR strategies on 2
Results and Discussion
Chemistry.
The syntheses of all target compounds were accomplished following procedures shown in Schemes 1-6. Compounds 2 and 6-16 with modifications on ring-A were synthesized following the procedures described by Nagahara and co-workers.45 Commercially available 1-fluoro-3-nitrobenzene was reacted with excess ethylenediamine at 120°C for 12 hours to afford the substituted aniline, which was immediately reacted with Boc2O to give 45 in 63% over two steps. The aniline group in 45 was then protected by treatment with benzyl bromide in the presence of potassium carbonate in DMF to afford key intermediate 46 in 62% yield. Reduction of the nitro group using iron gave amine 47 almost quantitatively (98%). Reaction of 5-bromo-2-methoxybenzenesulfonyl chloride in THF with 47 in DCM in the presence of pyridine led to intermediate 48 in excellent yield (90%). Suzuki coupling of 48with different boronic acids gave intermediate 49, which was immediately submitted to acidic Boc deprotection and amide coupling to provide 50. Finally, deprotection of the benzyl group of 50 using hydrogenation catalyzed by palladium on carbon afforded target products 2 and 6-16 in 29-45% yield over four steps.
Scheme 1.
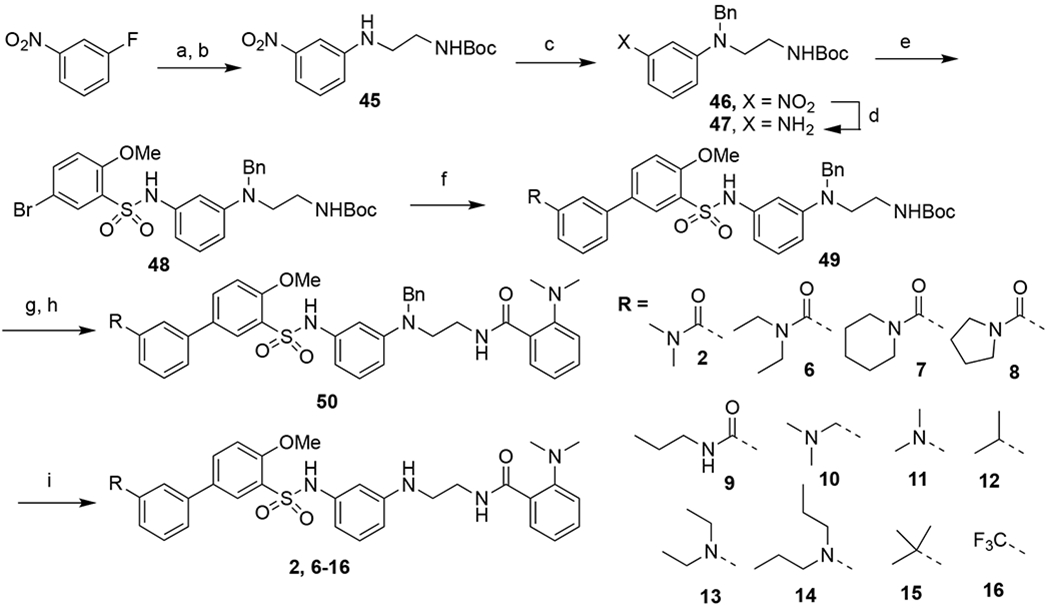
Syntheses of compounds 2 and 6-16
Scheme 6.
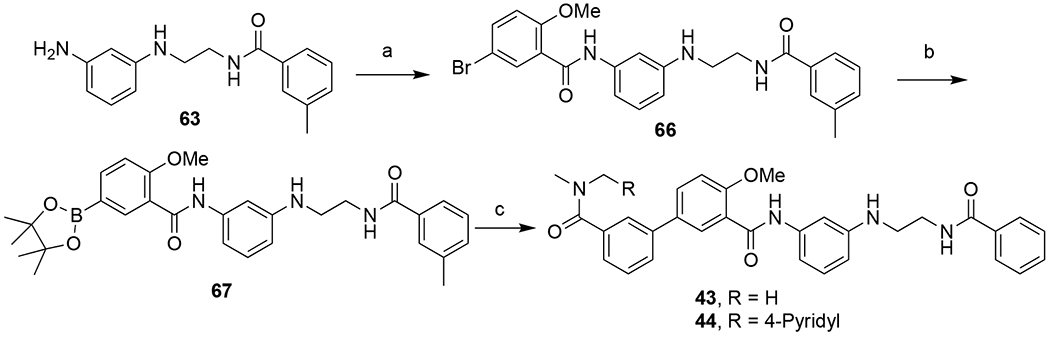
Synthesis of compound 43 and 44
Similarly, compounds 17 to 21 that examine the importance of the ethyl linker were synthesized following a similar sequence as shown in Scheme 2, using different diamines instead of ethylenediamine. 1-Fluoro-3-nitrobenzene underwent fluoro displacement with diamines, Boc protection and benzyl protection to give 52, which was then reduced to give amine 53 in excellent yield (92%). Coupling of 53 with 5-bromo-2-methoxybenzenesulfonyl chloride afforded 54, which was subjected to Suzuki coupling to give intermediate 55. Boc deprotection followed by amide coupling furnished 56, deprotection of which provided final compounds 17-21 in 19-32% yield over four steps.
Scheme 2.
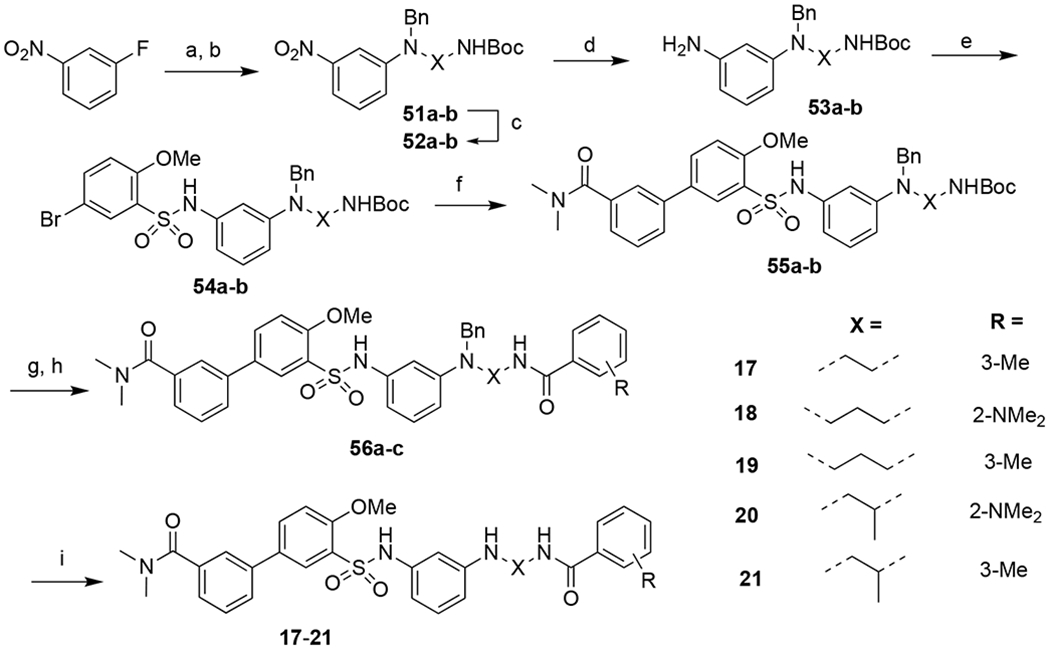
Syntheses of compounds 17-21
For compounds 22 and 23 with a piperidine group, since the protection of nitrogen was not needed, a simplified synthetic route was followed (Scheme 3). 1-Fluoro-3-nitrobenzene was reacted with excess piperazine followed by Boc protection to give 57 in 49% yield. Reduction of the nitro group provided amine 58, which was then reacted with 5-bromo-2-methoxybenzenesulfonyl chloride to afford 59 in 80% yield. Suzuki coupling furnished 60, followed by Boc deprotection and amide coupling to give final compounds 22-23 in 36% and 41% yields, respectively, over three steps.
Scheme 3.
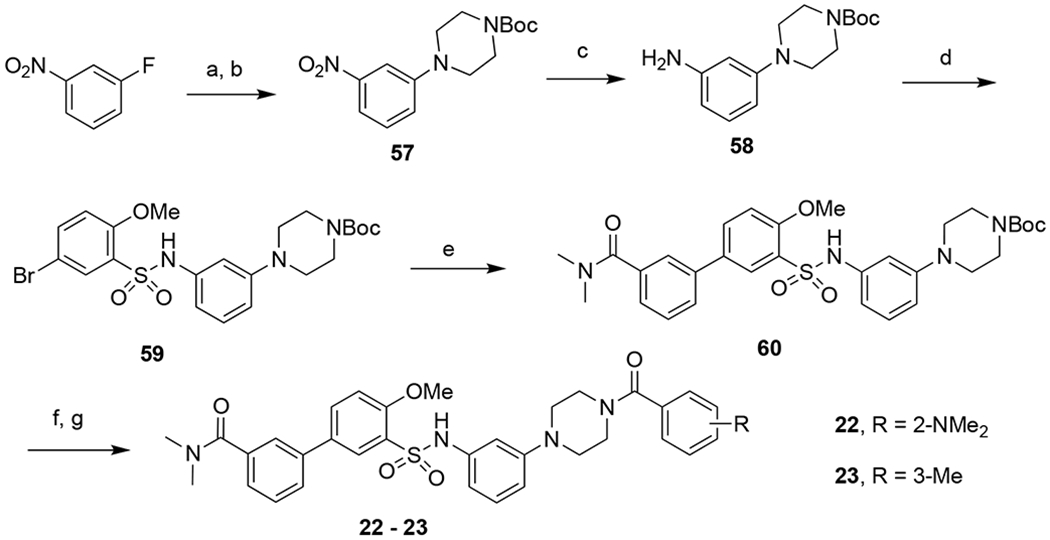
Syntheses of compounds 22 and 23
Compounds 24-32, in which ring-A was replaced with other aromatic or alkyl groups, were synthesized following modified procedures as shown in Scheme 4. In order to facilitate the examination of the SAR on the left-hand side, the amide on the right was installed first. In addition, to simplify the synthetic route, we attempted to not protect the aniline using a benzyl group as previously described but to install the sulfonamide selectively at the more accessible primary amino group. Thus, 45 was treated with 4N HCl in dioxane to afford the amine 61. Amide coupling between compound 61 and 3-methylbenzoic acid led to 62, which was reduced using iron to give 63 in 62% over two steps. Slow addition of a diluted solution of 5-bromo-2-methoxybenzenesulfonyl chloride in THF into a diluted solution of 63 in DCM in the presence of triethylamine and catalytic DMAP at 0°C afforded 64 in 76% yield, with the selective acylation of the less hindered amino groups. This sequence avoided the protection/deprotection steps and afforded the desired product in good yields. The subsequent Miyaura borylation reaction gave boronic acid pinacol ester 65, which was readily reacted with different aromatic bromides or iodides to furnish the final products 24-32 in 63-88% yield.
Scheme 4.
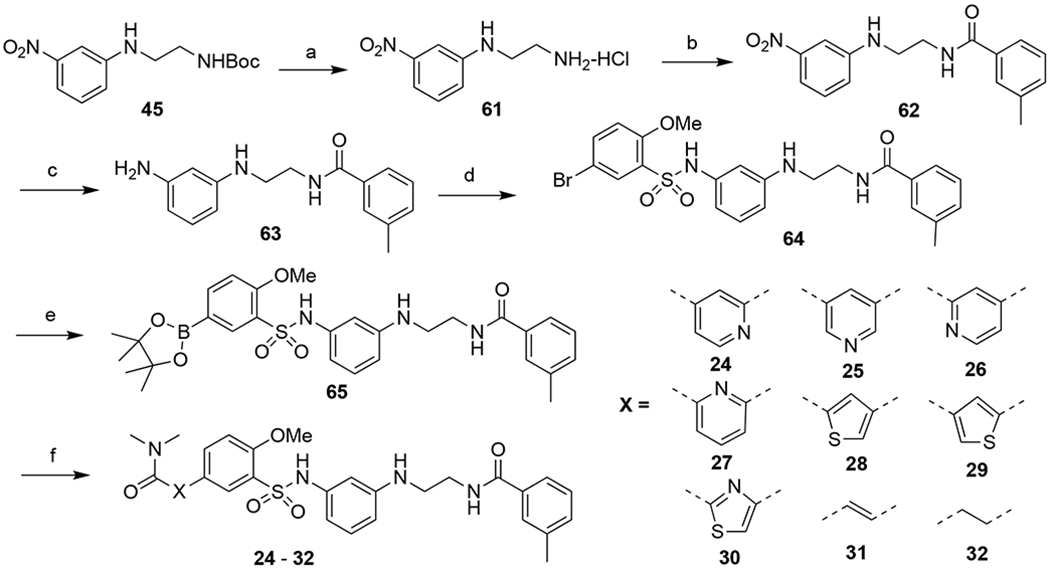
Synthetic route to compounds 24-32
Compounds 33-42 with different amide functionalities on ring-A were prepared in good yields following a Suzuki coupling of intermediate 63 with the appropriate aryl halides under standard conditions in 62-85% yield.
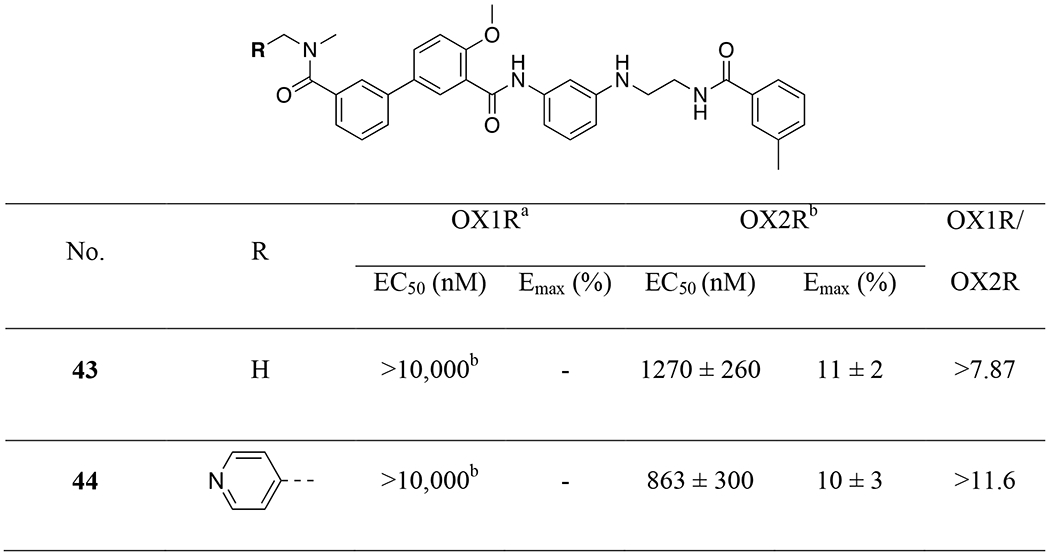
Amide coupling between intermediate 63 and 5-bromo-2-methoxybenzoic acid chloride afforded bromide 66 in 60% yield. Miyaura borylation of 66 using bis(pinacolato)diboron provided 67 quantitatively, which then underwent a Suzuki coupling reaction with the halides to provide compounds 43 and 44 in good yield (78% and 72%, respectively). In these two compounds, the sulfonamide was replaced with an amide functionality.
Potency in OX1R and OX2R calcium mobilization assays.
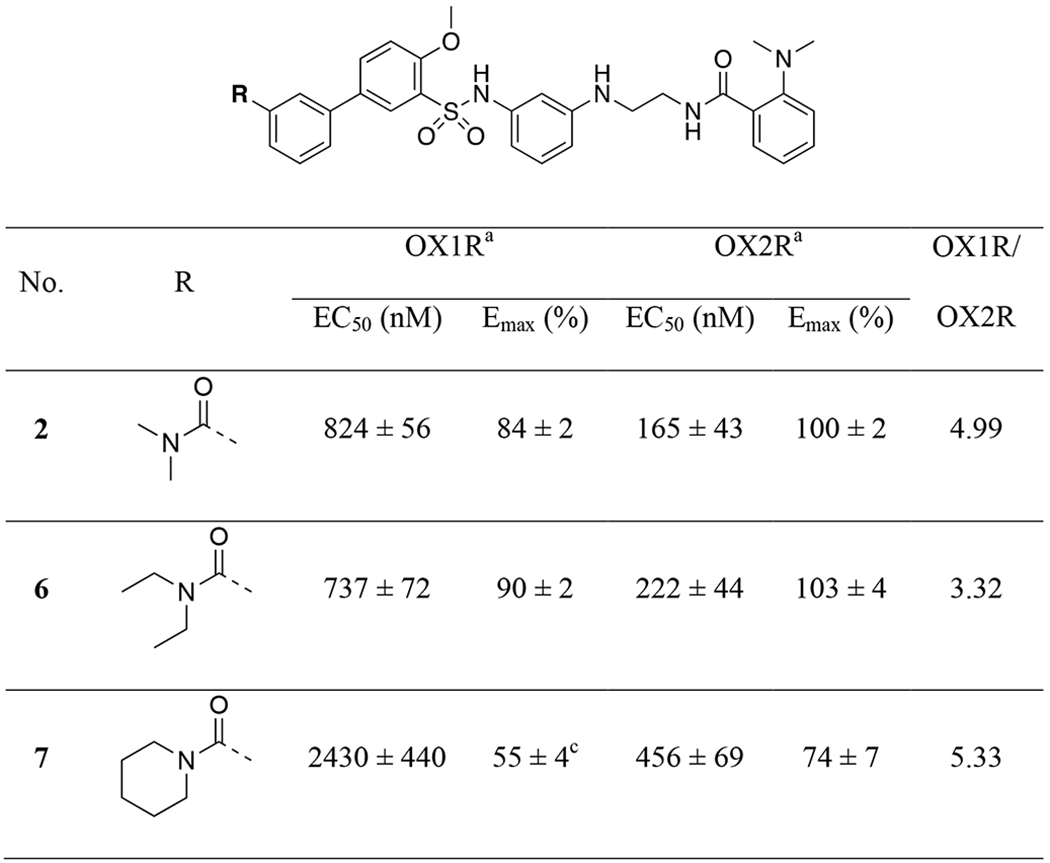
All target compounds were evaluated for their agonist potencies in calcium mobilization assays using CHO RD-HGA16 cells (Molecular Devices) engineered to stably express either the human OX2R or OX1R as previously described.61 In this assay platform, receptor activation is measured by an increase in fluorescence, which is directly proportional to an increase in internal calcium. The EC50 values are listed in Tables 1-5. YNT-185 (2) was previously reported to have EC50 values of 28 nM at OX2R and 2750 nM at OX1R.45 However, in our orexin calcium mobilization assays, 2 displayed potencies of 165 nM and 824 nM at OX2R and OX1R, respectively. The potency differences may have resulted from the differently engineered cell lines. While both groups use calcium mobilization assays to measure receptor activation, Nagahara and colleagues used CHO cells co-expressing the orexin receptors and a luciferase reporter,45 and our group used Gα16-expressing CHO cells. In their assays, orexin-A had EC50’s = 1.0 nM and 1.5 nM at OX2R and OX1R, respectively, whereas the EC50 values are 0.6 nM and 0.3 nM in our assays, respectively.
Table 1.
|
|
EC50 and Emax values (% of orexin-A control) are the means ± S.E.M. of at least three independent experiments conducted in duplicate.
Values are from two independent experiments conducted in duplicate.
Concentration-response curve does not have a top plateau.
Table 5:
|
EC50 and Emax values (% of orexin-A control) are the means ± S.E.M. of at least three independent experiments conducted in duplicate.
Values are from two independent experiments conducted in duplicate.
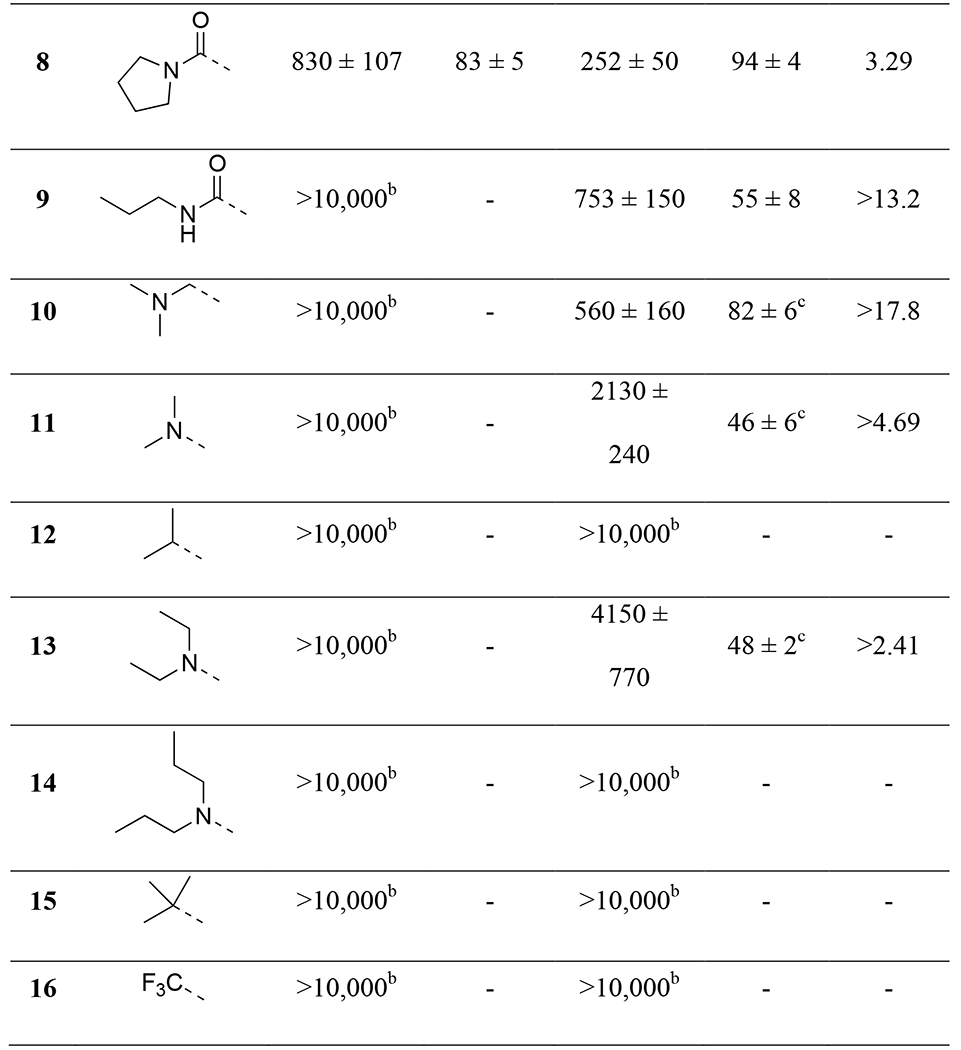
In our structure-activity relationship (SAR) studies, we first examined substitutions on aromatic ring-A on the left-hand side of the structure (Table 1). The dimethylamino amide on 2 was replaced with several differently substituted alkyl amides including diethylamino (6), cyclic (7, 8) or secondary amides (9). However, most of the compounds showed similar or even reduced potencies at OX2R and/or OX1R, with 9 showing no activity at OX1R. We then attempted to remove the carbonyl functionality, which resulted in a decrease in potency at OX2R (10, EC50 = 560 nM), whereas no activity was observed at OX1R. None of the other amino or alkyl groups (11-16) showed activity at either receptor. Together, these results confirmed the importance of the carbonyl functionality for orexin receptor activation.
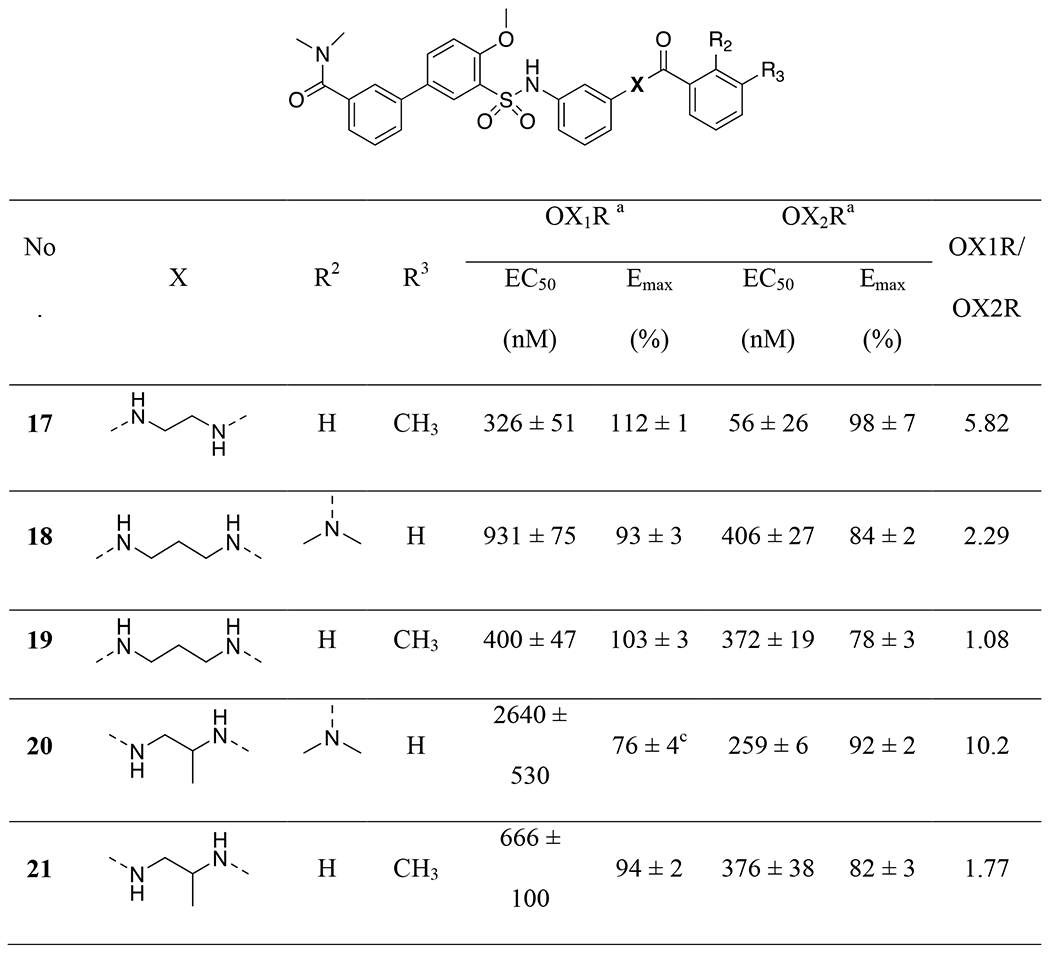
We next examined substitutions on aromatic ring-B on the right-hand side as well as the ethyl linker (Table 2). Consistent with results reported by Nagahara etc.,45 replacement of the 2-dimethylamino with a 3-methyl group (17) slightly improved potencies at both OX2R and OX1R (56 vs. 165 nM at OX2R; 326 vs. 826 nM at OX1R). Elongation of the ethyl group to the 3-carbon propyl group (18-19) or addition of a methyl group (20-21) resulted in little change in potency. However, when the ethyl group was converted to a rigid piperazine group (22-23), all activities were lost. In general, in this series, the 3-methyl analogs appeared to provide better potencies at OX1R than the corresponding 2-dimethylamino analogs (2, 18, 20 vs. 17, 19, 21), and we therefore used 3-methylamino in the subsequent SAR studies.
Table 2.
|

|
EC50 and Emax values (% of orexin-A control) are the means ± S.E.M. of at least three independent experiments conducted in duplicate.
Values are from two independent experiments conducted in duplicate.
Concentration-response curve does not have a top plateau.
Phenyl ring-A was then replaced with other aromatic rings or a rigid alkenyl or an ethyl group (Table 3). Out of the four pyridine groups (24-27), the 2,6-substituted pyridine (27) afforded the best potencies at both OX1R and OX2R (EC50’s = 74 nM at OX2R and 226 nM at OX1R), with EC50 values lower than 2, and appeared to be a full agonist at both OX1R and OX2R. We then investigated several 5-membered heteroaromatic rings (28-30), including thiophenes and a thiazole, and they were similar or less potent than 2 at OX2R, with little activity at OX1R. Interestingly, these 5-membered analogs (28-30) all acted as partial agonists at both OX1R and OX2R. The ethylenyl (31) and the ethyl (32) analogs, which were designed to provide structural flexibility, were both inactive at both receptors. These results suggest that an aromatic group is preferred at this position.
Table 3:
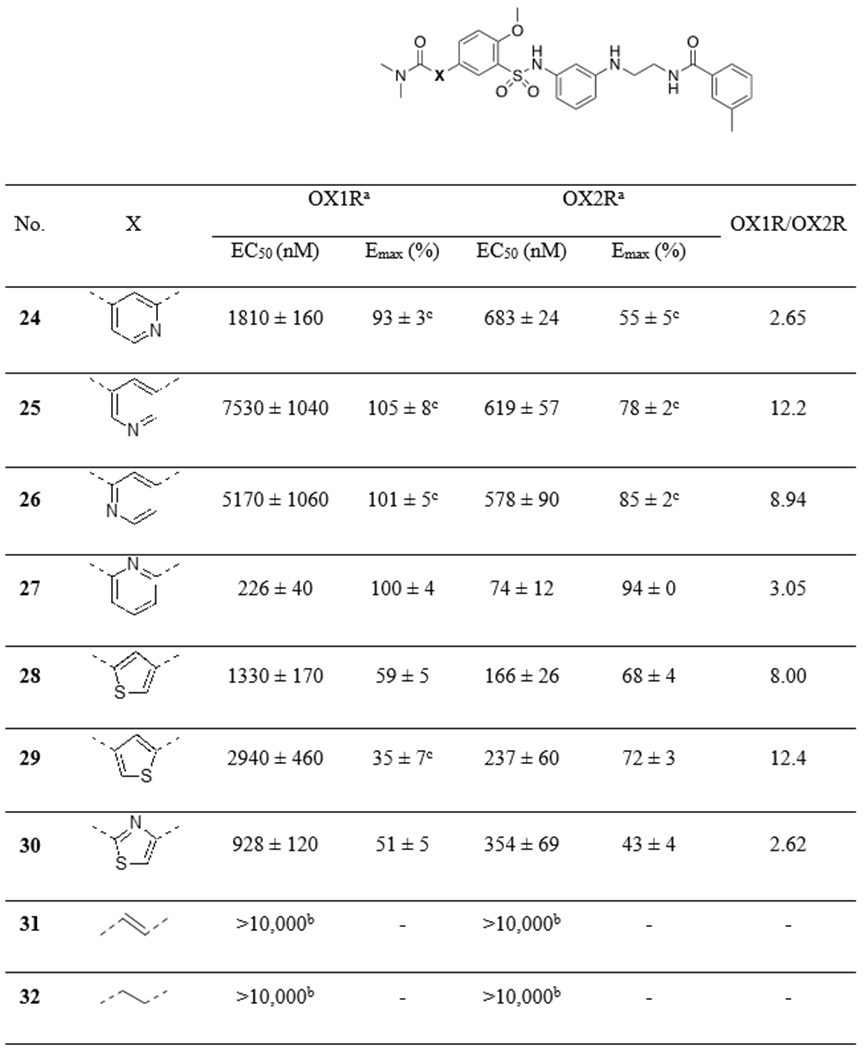
|
EC50 and Emax values (% of orexin-A control) are the means ± S.E.M. of at least three independent experiments conducted in duplicate.
Values are from two independent experiments conducted in duplicate.
Concentration-response curve does not have a top plateau.
Given the importance of the carbonyl group of the amide functionality on ring-A, we retained the amide and explored further substitutions at this position (Table 4). First, we replaced one of the methyl groups on the amide with a dimethylaminoethyl group (33), which offers a site for salt formation to improve solubility. However, a slight reduction in potency was observed (EC50 = 293 nM at OX2R and 638 nM at OX1R). Notably, removal of the other methyl on the nitrogen resulted in a sharp decrease of potency at OX2R (33 vs 34) and complete loss of potency at OX1R. This is consistent with earlier observations that primary amides were not favored (9). Introduction of a pyridyl (35) group in place of a methyl on the dimethylamino group led to a significant decrease in potency at both receptors. Interestingly, while a benzyl (36) showed lowered potency at OX2R and no activity at OX1R, 2-pyridylmethyl group (37) led to better potencies than 2 at both OX receptors (EC50 = 107 nM at OX2R and 235 nM at OX1R). This suggests that hydrogen-bonding or polar-polar interaction between the pyridyl group and the OX receptors may be present. Similarly, removal of the other methyl on the dimethylamino group again led to complete loss of the OX1R potency (38). Considering the potency enhancement by the pyridyl group, we began to finely tune the substitution and examine different pyridylmethyl groups. While 3-pydidylmethyl (39) showed a slight drop in potency (EC50 = 134 nM at OX2R; 460 nM at OX1R), excitingly, 40 with a 4-pyridylmethyl group showed high and equal potencies at both OX2R and OX1R (EC50 = 24 nM), significantly more potent than 2. Compound 40 was a full agonist at both OX2R and OX1R, with Emax of ~100% orexin-A (Figure 3). Replacement of the pyridylmethyl with longer pyridylethyl groups resulted in decrease on potency at both receptors (41 and 42).
Table 4:
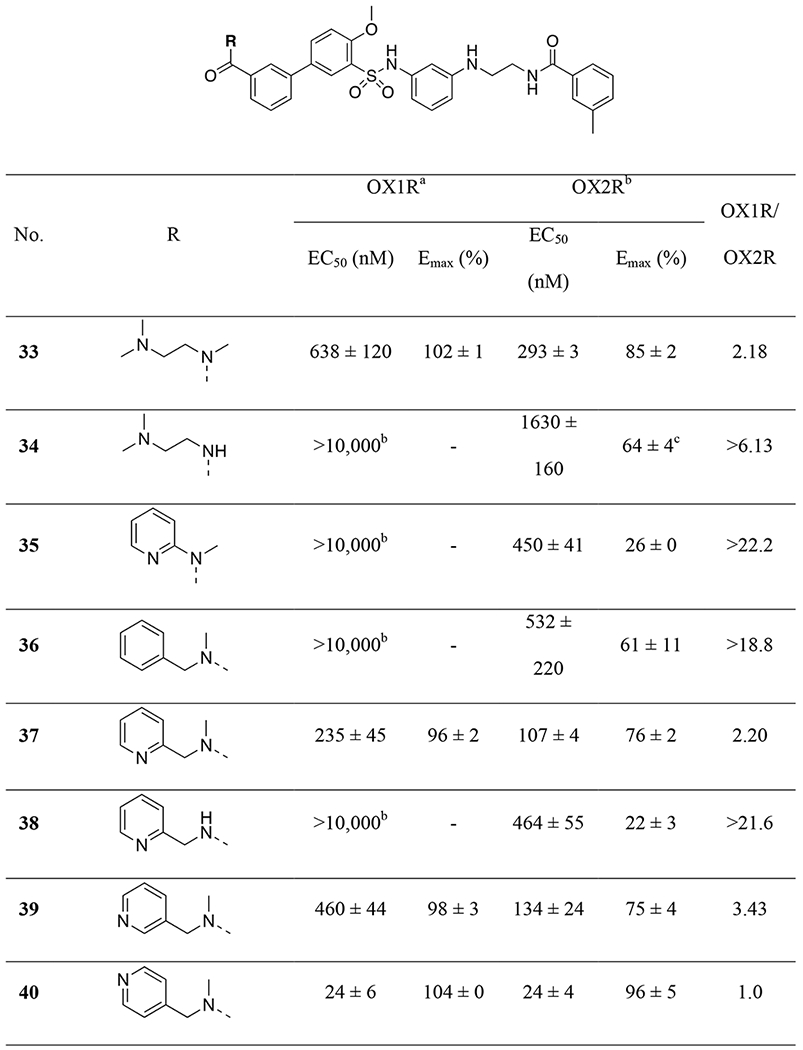
|

|
EC50 and Emax values (% of orexin-A control) are the means ± S.E.M. of at least three independent experiments conducted in duplicate.
Values are from two independent experiments conducted in duplicate.
Concentration-response curve does not have a top plateau.
Figure 3.
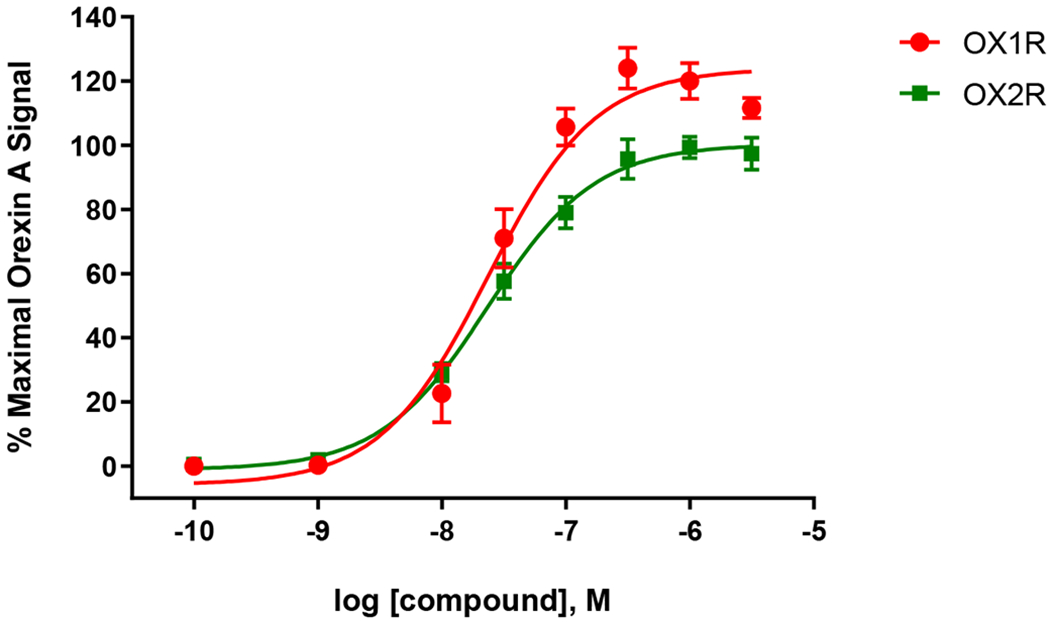
Activity of 40 in the OX1R and OX2R calcium mobilization assays. Each data point is the mean ± S.E.M. of three independent experiments conducted in duplicate
Finally, we examined whether the sulfonamide could be replaced with an amide (43-44, Table 5). Unfortunately, this structural modification resulted in significant reduction in potency at OX2R and total loss of activity at OX1R, similar to previous report.45 This clearly indicates the importance of the sulfonamide for activity at the orexin receptors.
Computational modeling studies.
We built a full-length OX2R model based on the cryo-electron microscopy (cryo-EM) structures of agonist 5 (PDBID: 7L1V) and orexin-B bound OX2R (PDBID: 7L1U) recently reported by Hong and colleagues,50 and a homology OX1R model based on the backbone and fold templates of the human OX2R structure, which shares 64% sequence identity with human OX1R.2 The initial models were processed through a series of refinement steps employing MODELLER (simulated annealing with topological constraints),62 AMBER18 low-mode exploration,63-65 energy minimization and dynamical equilibration in a lipid/KCl/water system employing the LIPID14 and ff14SB forcefields.66, 67 We then explored agonists 5, 2 and 40 with OX2R and/or OX1R models via GLIDE-XP or SP docking,68 distilling 120 minimized docked poses to the best 5, followed by induced fit modeling.69 Molecular dynamics (MD) simulations were conducted with 40 in both OX2R and OX1R to identify key receptor–ligand interactions and probe their stability. Consistent with the cryo-EM OX2R structure, all compounds (5, 2 and 40) formed a “Z” shape in our OX2R model, kinked around the sulfonamide functionality with the two adjacent aromatic rings perpendicular to each other. The right end of these molecules inserted into the hydrophobic bottom of the pocket with the left end extending toward extracellular space (Figure 4A-C). This is consistent with the orexin-B bound cryo-EM structure showing the largely hydrophobic C-terminus residues of orexin-B, G24-I25-L26-T27-M28, bound at the hydrophobic bottom of the binding pocket.50
Figure 4.
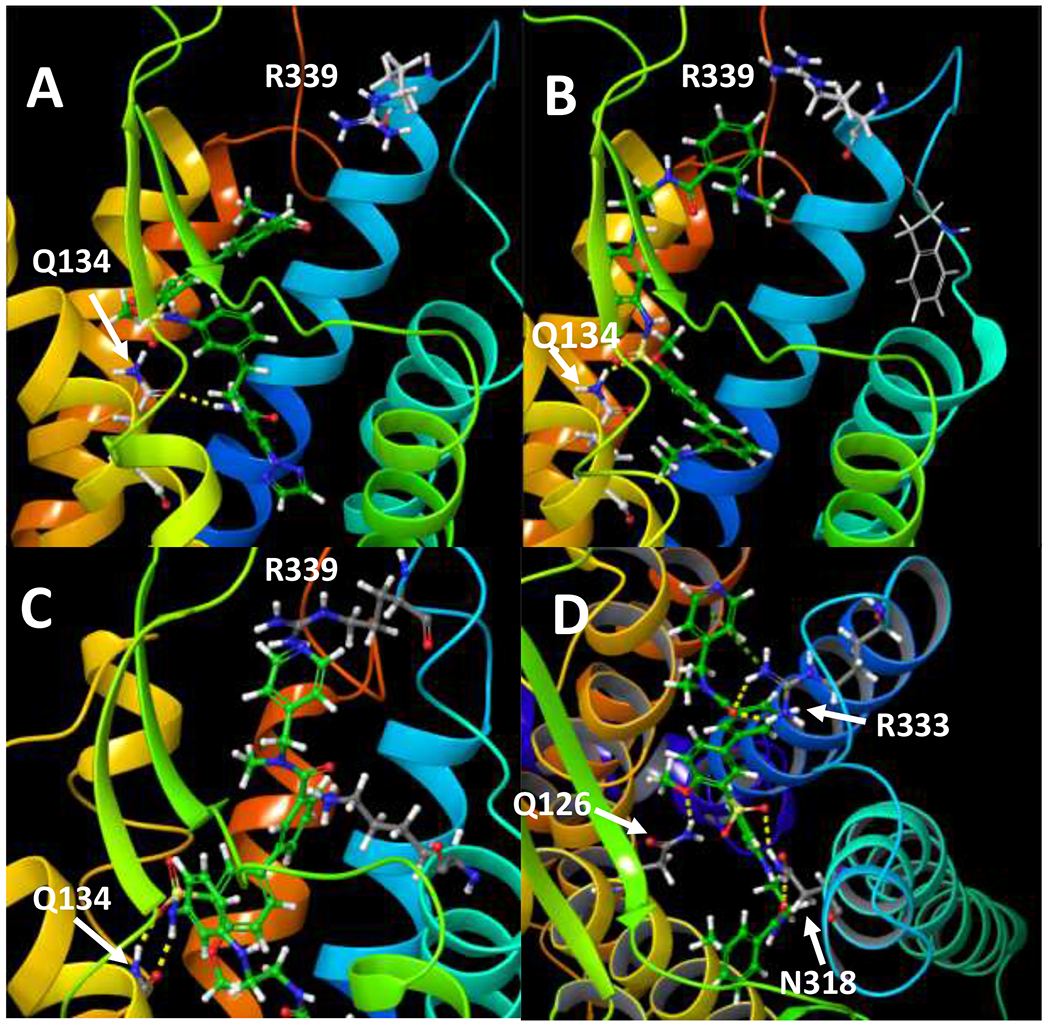
Induced fit top Emodel poses for A) 5 in OX2R, B) 2 in OX2R, C) 40 in OX2R, D) 40 in OX1R full-length models based on the agonist bound cryo-EM OX2R structures (7L1U and 7L1V). Ligands are in green and residues in gray.
Q1343.32 (Ballesteros-Weinstein numbering in superscript) was identified as the residue essential for binding and receptor activation in the agonist 5 and orexin-B bound OX2R cryo-EM structures.50 In both agonist bound structures, the sidechain of Q1343.32 was extended and projected upward toward the extracellular side, as opposed to pointing more downward toward the cytoplasm in the antagonist bound OX2R structures.50, 70, 71 We observed the agonist-like orientation of the Q1343.32 side chain in our OX2R model and such a conformation was largely maintained during 180 nanosecond (ns) MD simulations of 40 in OX2R. Q1343.32 was in close proximity of the sulfonamide group, as observed by Hong etc. in the 5 bound OX2R cryo-EM structure,50 but was also close in space to the methoxy group on the neighboring aromatic ring in the initial induced fit docked poses in these agonists (Figure 4A-B). When MD simulations were performed on 40, intimate and persistent hydrogen bond interactions of the Q1343.32 amide group with the sulfonamide, sometimes bidentate as reported by Hong etc., were observed within 15 ns of the 180 ns MD simulation equilibration at 300K, with additional hydrogen bond interactions, although slightly more distant, with the methoxy on the neighboring phenyl group (Figure 4C). These results clearly demonstrated the importance of the sulfonamide and is consistent with the SAR results where replacement of the sulfonamide with the corresponding amide (43, 44) resulted in significant drop on potency at OX2R. In addition, it should be noted that all the active orexin agonists reported thus far (e.g. 2-5, Figure 1) have a sulfonamide functionality.
In the OX1R homology model, 40 formed a similar “Z” shape, although slightly rotated (Fig. 4D). Similar to the OX2R model, Q1263.32 of OX1R, the equivalent residue of Q1343.32 in OX2R, formed hydrogen bonding with the sulfonamide group and/or the neighboring methoxy group during the 180 ns molecular dynamics simulations. In addition, hydrogen bonding between the sulfonamide group and N3186.55 was also observed (Figure 4D). Markedly, as opposed to only transient interactions with R3397.28 at the top of helix 7 in OX2R, the pyridylmethyl group on the left-hand side of 40 formed persistent hydrogen bonds or π-cation interactions with R3337.29 in OX1R during the 180 ns MD simulations. Although additional studies are clearly needed, these receptor-ligand interactions, particularly via the pyridylmethyl group, may have contributed to the high potency of 40 in OX1R, in contrast to OX2R agonists 2 and 5, in which the pyridylmethyl motif is absent.
Sleep modulation studies.
It has been previously shown that upon i.p. administration at 40 or 60 mg/kg, 2 increased wake time, as expected with OX2R agonists and decreased direct transitions from wakefulness to REM sleep, a measure that is used to assess cataplexy, for 3 hours.45, 46 To test whether our OX agonists influence time spent awake and the transitions between sleep/wake states, we injected 40 into 12-month old female mice (40 mg/kg, i.p. during the quiet, lights-on phase) and measured sleep/wake parameters using continuous EEG/EMG recordings for 4 h post-injection. Compound 40 significantly increased active wakefulness and reduced NREM and REM sleep relative to that after the vehicle injection (Figure 4A-C). There was a main effect of treatment on active wakefulness (Figure 4A, P<0.01), NREM sleep (Figure 4B, P < 0.05) and REM sleep (Figure 4C, P < 0.001) during the 0–4 h post-injection time period.
To test whether 40 influences sleep patterns and quality, the total number of transitions between states, number and the mean duration of episodes of each vigilance state were determined. Our data demonstrated that 40 stabilized behavior and reduced sleep/wake fragmentation as indicated by reduced transitions between states (Figure 5D). In addition, 40-induced enhancement of wake time was associated with an increase in average duration of wake episodes (Figure 5E). Whereas the agonist reduced number of wake episodes (Figure 5F), the increased duration of wake episodes resulted in overall enhancement of wake time following treatment. Similar to wake episodes, 40 increased the duration of NREM sleep episodes (Figure 5E). Reduced number of NREM sleep episodes (Figure 5F) following agonist administration resulted in overall reduction in time spent in NREM sleep. Surprisingly, 40 did not affect the duration of REM sleep episodes (Figure 5E), but fewer episodes of REM sleep (Figures 5F) resulted in overall reduction in REM sleep time following agonist administration. Together, these data demonstrate that 40 enhances wake, suppresses sleep and reduces sleep/wake fragmentation. In particular, 40 enhances wake by increasing episode duration, and decreases NREM and REM sleep by reducing the number, but not the duration, of episodes of NREM and REM sleep.
Figure 5.
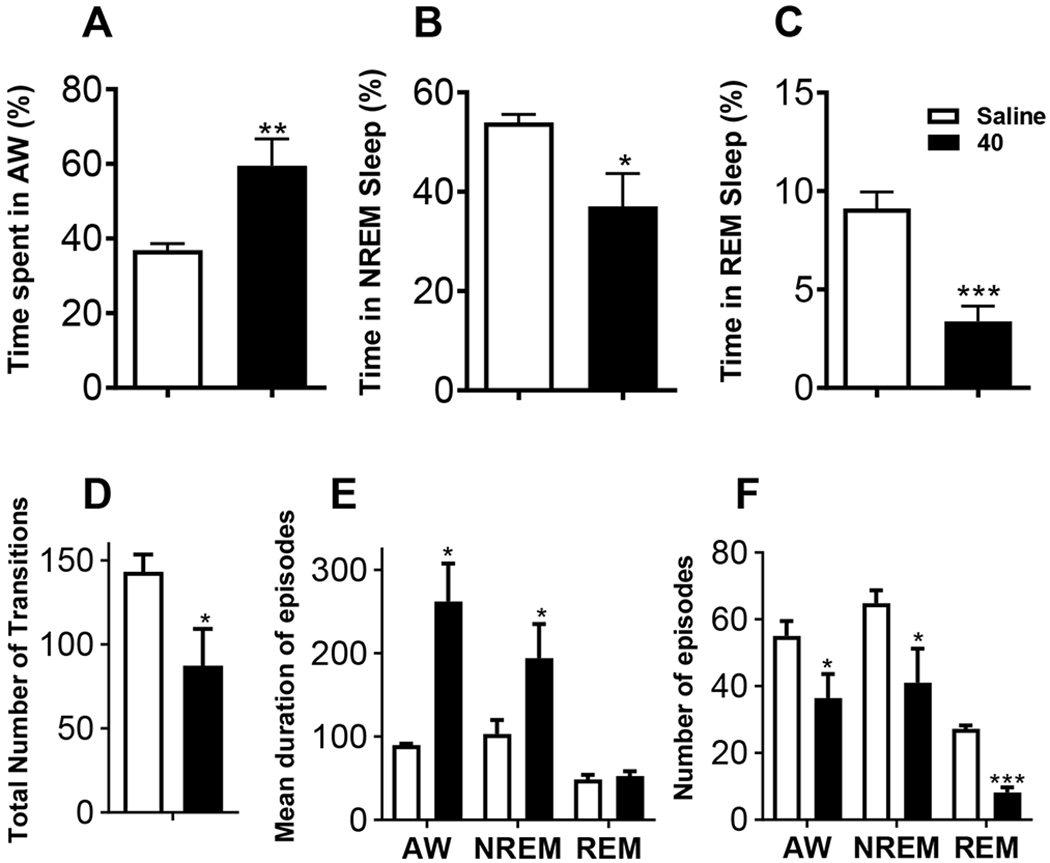
Intraperitoneal injection of 40 (40 mg/kg) during the quiet phase (lights-on) increased time spent in wakefulness (A), reduced time spent in NREM sleep (B) and REM sleep (C) 4-h post-injection. Agonist 40 also reduced the number of state transitions compared to saline injections (D), increased the duration of wake and NREM sleep episodes, without effecting the duration of REM sleep episodes (E), and reduced the number of episodes of wake, NREM and REM sleep (F) 4-h post-injection. n = 5. Data represented as mean ± SEM. *P < 0.05, **P < 0.01 and ***P < 0.001. Note different scaling on y-axes.
The wake-promoting effects observed with 40 were similar to those of orexin-A and the OX2R agonist 2 reported earlier. We have shown increased wakefulness and reduced NREM and REM sleep following ventrolateral preoptic area injection of orexin-A in rats, which was associated with decreased NREM sleep and wake episodes 1 h post-injection.25 Intracerebroventricular administration of orexin-A (3 nmol) enhanced wakefulness and suppressed both NREM and REM sleep in mice up to 3 h following treatment.54 Similarly, i.c.v. administered 2 (30, 100, 300 nmol doses) during the light phase decreased percent time in NREM sleep and increased time in wakefulness up to 3 h post injection. In addition, i.p. injected 2 (40 and 60 mg/kg, 6 h into the light phase) increased percent time in wakefulness and decreased percent time in REM sleep for up to 3 h following injections and the 40 mg/kg (i.p) dose also reduced the percent time in NREM sleep in mice.46 Notably, both the 40 and 60 mg/kg doses had identical wake-promoting effects, whereas the 20 mg/kg dose (i.p.) did not have any effect on the sleep/wake parameters. The relatively high doses required to affect sleep/wake cycles suggest that while 2 and 40 are able to reach the OX2R in the brain to modulate sleep, their brain penetration may be limited.
At 40 mg/kg 40 and 2 produced similar wake promoting effects upon i.p. administration, despite the significantly higher OX1R potency of 40. This may reflect that OX2R plays a pivotal role in the modulation of sleep/wakefulness. It has been reported that OX2R knockout (KO) mice exhibited a narcoleptic phenotype, whereas OX1R knockout mice showed only a mild fragmentation of sleep and awake states.72 In addition, while orexin-A effects on wake promotion and NREM sleep suppression were attenuated in both OX2R and OX1R KO mice, substantially greater reductions in OX2R KO mice (relative to OX1R KO mice) were observed.54 Together, these studies indicate that sleep appears to be primarily regulated by OX2R and to a lesser extent by OX1R; however, the role of the OX1R in sleep/wakefulness regulation may be best addressed when selective OX1R agonists become available.
Conclusions
The orexin system is implicated in many physiological processes and loss or decline has been associated with narcolepsy and other neurological diseases. Orexin agonists suitable for systemic administration have been suggested as the most promising strategy for the treatment of orexin deficiency-associated conditions among all orexin replacement therapies.73, 74 Compound 2 was one of the first small molecule orexin agonists reported thus far, although it mainly activates OX2R with limited agonist activity at OX1R. Our SAR studies at multiple sites suggested that an amide functionality is required on ring-A at the left-hand side. Excitingly, introduction of a pyridylmethyl group at this amide increased potency at both OX1R and OX2R. Computational modeling studies based on the recently reported cryo-EM structure of OX2R bound with agonist 5 showed that 40 formed hydrogen bonding or π-cation interactions via the pyridylmethyl group with R3337.29 in OX1R, which may have contributed to the observed high OX1R potency of 40. Through this effort, we have identified dual OX agonists, including 40 (RTOXA-43), which acted as a full agonist with EC50 values of 24 nM at both OX receptors. These are the first and only small molecule dual orexin agonists discovered thus far. When measured using continuous EEG/EMG recordings in 12-month old mice, 40 (40 mg/kg, i.p.) increased time awake by increasing episode duration, decreased NREM and REM sleep and improved sleep/wake consolidation by reducing the number, but not the duration, of episodes of NREM and REM sleep. The current results provide a promising lead for the discovery of small molecule agonists with OX1R activities and support development of orexin agonists as potential treatments for orexin-deficient disorders such as narcolepsy.
EXPERIMENTAL SECTION
Chemistry.
All solvents and chemicals were reagent grade. Unless otherwise mentioned, all reagents and solvents were purchased from commercial vendors and used as received. Flash column chromatography was carried out on a Teledyne ISCO CombiFlash Rf system using prepacked columns. Solvents used include hexane, ethyl acetate (EtOAc), dichloromethane, methanol, and Chloroform/methanol/ammonium hydroxide (80:18:2) (CMA-80). Purity and characterization of compounds were established by a combination of HPLC, TLC, mass spectrometry, and NMR analyses. Melting point was recorded by the Mel-Temp II instrument (Laboratory Devices Inc. , U.S.). 1H and 13C NMR spectra were recorded on a Bruker Avance DPX-300 (300 MHz) spectrometer and were determined in Chloroform-d, DMSO-d6, or methanol-d4 with tetramethylsilane (TMS) (0.00 ppm) or solvent peaks as the internal reference. Chemical shifts are reported in ppm relative to the reference signal, and coupling constant (J) values are reported in hertz (Hz). Thin layer chromatography (TLC) was performed on EMD precoated silica gel 60 F254 plates, and spots were visualized with UV light or iodine staining. Low resolution mass spectra were obtained using a Waters Alliance HT/Micromass ZQ system (ESI). All test compounds were greater than 95% pure as determined by HPLC on an Agilent 1100 system using an Agilent Zorbax SB-Phenyl, 2.1 mm × 150 mm, 5 μm column using a 15 minute gradient elution of 5-95% solvent B at 1 mL/min followed by 10 minutes at 95% solvent B (solvent A, water with 0.1% TFA; solvent B, acetonitrile with 0.1% TFA and 5% water; absorbance monitored at 220 and 280 nm).
tert-butyl (2-((3-nitrophenyl)amino)ethyl)carbamate (45).
3-nitrofluorobenzene (5.0 mmol, 35.4 mmol) and ethylenediamine (11.8 mL, 177.2 mmol) were mixed in a sealed tube and the reaction was heated at 120°C overnight. After cooling down, the volatile was evaporated under reduced pressure at 60°C. The residue was then redissolved in a mixture of THF (30 mL) and water (30 mL) followed by the addition of potassium carbonate (14.7 g, 106.2 mmol) and di-tert-butyl dicarbonate (19.3 g, 88.5 mmol). The reaction was then stirred overnight and diluted by brine (150 mL). Ethyl acetate (150 mL) was then added and the organic layer was separated and dried. The solvent was removed under reduced pressure and the residue was purified by ISCO to afford pure desired product. 6.28 g brown oil, yield: 63%. 1H NMR (300 MHz, Chloroform-d) δ 7.51 (dd, J = 1.60, 8.01 Hz, 1H), 7.37 (t, J = 2.26 Hz, 1H), 7.22 - 7.31 (m, 1H), 6.87 (dd, J = 2.07, 8.10 Hz, 1H), 4.78 - 4.94 (m, 1H), 4.56 - 4.73 (m, 1H), 3.36 - 3.50 (m, 2H), 3.21 - 3.34 (m, 2H), 1.38 - 1.50 (m, 9H). MS (ESI) m/z: 282.3. [M+H]+; LCMS: >95% purity.
tert-Butyl (2-(benzyl(3-nitrophenyl)amino)ethyl)carbamate (46).
Compound 45 (6.28 g, 22.30 mmol) was dissolved in DMF (110 mL), followed by the addition of potassium carbonate (6.17 g, 44.65 mmol) and benzyl bromide (3.2 mL, 26.79 mmol). The reaction was then heated at 60°C overnight. Water (500 mL) and ethyl acetate (200 mL) were added and the organic layer was separated and dried. The solvent was removed under reduced pressure and the residue was purified by ISCO to afford pure desired product. 5.11 g orange syrup, yield: 62%. 1H NMR (300 MHz, Chloroform-d) δ 7.46 - 7.59 (m, 2H), 7.34 - 7.40 (m, 1H), 7.26 - 7.34 (m, 3H), 7.17 (d, J = 7.16 Hz, 2H), 7.02 (d, J = 6.22 Hz, 1H), 4.58 - 4.76 (m, 3H), 3.57 - 3.72 (m, 2H), 3.37 (q, J = 6.47 Hz, 2H), 1.35 - 1.48 (m, 9H). MS (ESI) m/z: 372.4. [M+H]+.
tert-Butyl (2-((3-aminophenyl)(benzyl)amino)ethyl)carbamate (47).
Compound 46 (5.11 g, 13.76 mmol) was dissolved in a mixture of ethanol and water (55 mL/22 mL), followed by the addition of ammonium chloride (7.36 g, 137.6 mmol) and iron powder (5.38 g, 96.3 mmol). The reaction was then heated at reflux for 3 hours. After cooling down, DCM (100 mL) was added and the mixture was filtered through celite. The organic layer was then separated and dried. The solvent was then removed under reduced pressure and the residue was purified by ISCO to afford the desired product. 4.61 g brown oil, yield: 98%. 1H NMR (300 MHz, Chloroform-d) δ 7.08 - 7.46 (m, 6H), 6.98 (t, J = 8.19 Hz, 1H), 6.02 - 6.26 (m, 2H), 4.62 - 4.77 (m, 1H), 4.40 - 4.60 (m, 2H), 3.41 - 3.58 (m, 2H), 3.22 - 3.39 (m, 2H), 1.55 - 2.23 (m, 2H), 1.32 - 1.53 (m, 9H). MS (ESI) m/z: 342.2. [M+H]+.
tert-Butyl (2-(benzyl(3-((5-bromo-2-methoxyphenyl)sulfonamido)phenyl)amino)ethyl)carb-amate (48).
Under the protection of nitrogen, compound 47 (3.52 g, 10.31 mmol) was dissolved in anhydrous DCM (50 mL) at 0°C. Pyridine (1 mL, 12.37 mmol) was added to the reaction, followed by the addition of 2-methoxy-5-bromobenzenesulfonyl chloride (3.24 g, 11.34 mmol). The reaction was warmed up to room temperature and stirred overnight. The reaction was quenched by saturated NaHCO3 (30 mL) and DCM (100 mL) was added. The organic layer was separated and dried. The solvent was removed under reduced pressure and the residue was purified by ISCO to afford the desired product. 5.46 g off-white solid, yield: 90%. 1H NMR (300 MHz, Chloroform-d) δ 7.87 (d, J = 2.45 Hz, 1H), 7.55 (dd, J = 2.45, 8.85 Hz, 1H), 7.18 - 7.35 (m, 4H), 7.10 (d, J = 6.59 Hz, 2H), 7.00 (t, J = 8.38 Hz, 1H), 6.90 (br. s., 1H), 6.78 (d, J = 8.85 Hz, 1H), 6.49 (d, J = 8.48 Hz, 1H), 6.39 (d, J = 4.71 Hz, 2H), 4.59 - 4.73 (m, 1H), 4.48 (s, 2H), 3.84 (s, 3H), 3.41 - 3.53 (m, 2H), 3.19 - 3.34 (m, 2H), 1.36 - 1.47 (m, 9H). MS (ESI) m/z: 592.2. [M+H]+.
General procedure for the synthesis of compounds 2 and 6-16:
Compound 48 (1.0 eq.), boronic acid (1.2 eq.), Pd(PPh3)4 (0.1 eq.) and potassium carbonate (2.0 eq.) were placed in a round-bottomed flask with and condenser. The system was flushed with nitrogen and a mixture of 1,4-dioxane/water (4/1, 0.1 M) was then added. The reaction was refluxed for 2 hours. After cooling down, DCM (50 ml) was added and the organic layer was separated and dried. The solvent was removed to give crude compound 49 that was then dissolved in 4 N HCl in 1,4-dioxane (10 eq.). The reaction was stirred for 2 hours at room temperature and the solvent was then removed under reduced pressure. The residue was then dissolved in DMF (0.1 M) followed by the addition of 2-dimethylamino benzoic acid (1.1 eq.), HATU (1.2 eq) and DIPEA (1.5 eq.). The reaction was stirred overnight at room temperature and quenched by saturated NaHCO3. DCM (50 mL) was added and the organic layer was separated and dried. The solvent was removed under reduced pressure to afford the crude product (compound 50), which was then mixed with Pd/C (0.1 eq) in MeOH (0.1 M) under the atmosphere of hydrogen (40 psi) for 12 hours. The reaction mixture was filtered and the solvent of the filtrate was removed under reduced pressure. The residue was purified by ISCO to afford the desired final products (2, 6-16).
3’-(N-(3-((2-(2-(dimethylamino)benzamido)ethyl)amino)phenyl)sulfamoyl)-4’-methoxy-N,-N-dimethyl-[1,1’-biphenyl]-3-carboxamide (2).
Yield: 45% over four steps. 1H NMR (300 MHz, Chloroform-d) δ 9.89 (br. s., 1H), 8.00 - 8.17 (m, 2H), 7.64 (dd, J = 1.88, 8.67 Hz, 1H), 7.48 - 7.58 (m, 2H), 7.25 - 7.46 (m, 3H), 7.10 - 7.23 (m, 3H), 7.01 (d, J = 8.67 Hz, 1H), 6.94 (t, J = 8.01 Hz, 1H), 6.45 (br. s., 1H), 6.34 (dd, J = 8.01, 14.79 Hz, 2H), 4.33 (br. s., 1H), 3.94 - 4.09 (m, 3H), 3.54 - 3.73 (m, 2H), 3.28 (br. s., 2H), 2.86 - 3.20 (m, 6H), 2.42 - 2.66 (m, 6H). MS (ESI) m/z: 616.3. [M+H]+.
3’-(N-(3-((2-(2-(dimethylamino)benzamido)ethyl)amino)phenyl)sulfamoyl)-N, N-diethyl-4’-methoxy-[1,1’-biphenyl]-3-carboxamide (6).
Yield: 29% over four steps. 1H NMR (300 MHz, Chloroform-d) δ 9.90 (br. s., 1H), 8.07 - 8.16 (m, 1H), 7.97 - 8.06 (m, 2H), 7.65 (dd, J = 2.26, 8.67 Hz, 1H), 7.46 - 7.54 (m, 2H), 7.41 (t, J = 7.54 Hz, 2H), 7.30 (d, J = 7.35 Hz, 1H), 7.12 - 7.22 (m, 2H), 7.03 (d, J = 8.67 Hz, 1H), 6.95 (t, J = 8.01 Hz, 1H), 6.44 (s, 1H), 6.34 (d, J = 5.09 Hz, 2H), 4.31 (t, J = 5.27 Hz, 1H), 4.07 (s, 3H), 3.65 (q, J = 5.84 Hz, 2H), 3.55 (br. s., 2H), 3.17 - 3.36 (m, 4H), 2.75 - 2.99 (m, 6H), 1.26 (br. s., 3H), 1.11 (br. s., 3H). MS (ESI) m/z: 644.2. [M+H]+.
2-(Dimethylamino)-N-(2-((3-((4-methoxy-3’-(piperidine-1-carbonyl)-[1,1’-biphenyl])-3-sulf-onamido)phenyl)amino)ethyl)benzamide (7)
Yield: 36% over four steps. 1H NMR (300 MHz, Chloroform-d) δ 9.67 - 9.90 (m, 1H), 8.03 (d, J = 2.07 Hz, 2H), 7.65 - 7.74 (m, 1H), 7.56 - 7.65 (m, 1H), 7.36 - 7.56 (m, 6H), 7.31 (s, 2H), 7.07 (s, 2H), 6.92 (br. s., 2H), 6.79 - 6.87 (m, 1H), 4.04 (s, 3H), 3.79 (br. s., 4H), 3.44 (br. s., 4H), 3.14 (s, 6H), 1.69 (br. s., 4H), 1.44 - 1.59 (m, 2H). MS (ESI) m/z: 656.3. [M+H]+.
2-(Dimethylamino)-N-(2-((3-((4-methoxy-3’-(pyrrolidine-1-carbonyl)-[1,1’-biphenyl])-3-sul-fonamido)phenyl)amino)ethyl)benzamide (8).
Yield: 39% over four steps. 1H NMR (300 MHz, Chloroform-d) δ 9.90 (br. s., 1H), 7.99 - 8.15 (m, 2H), 7.59 - 7.71 (m, 2H), 7.49 - 7.55 (m, 1H), 7.35 - 7.47 (m, 3H), 7.10 - 7.22 (m, 2H), 6.86 - 7.07 (m, 3H), 6.44 (s, 1H), 6.34 (dd, J = 4.71, 7.54 Hz, 2H), 4.23 - 4.39 (m, 1H), 4.06 (s, 3H), 3.56 - 3.72 (m, 8H), 3.42 (t, J = 6.50 Hz, 2H), 3.29 (t, J = 5.65 Hz, 2H), 2.82 (d, J = 8.10 Hz, 2H), 2.56 (s, 6H), 1.94 - 2.03 (m, 2H), 1.85 - 1.91 (m, 2H). MS (ESI) m/z: 642.2. [M+H]+.
3’-(N-(3-((2-(2-(dimethylamino)benzamido)ethyl)amino)phenyl)sulfamoyl)-4’-methoxy-N-p-ropyl-[1,1’-biphenyl]-3-carboxamide (9).
Yield: 32% over four steps. 1H NMR (300 MHz, Chloroform-d) δ 9.87 (br. s., 1H), 7.97 - 8.17 (m, 3H), 7.90 (s, 1H), 7.63 - 7.75 (m, 2H), 7.57 (d, J = 7.72 Hz, 1H), 7.33 - 7.49 (m, 2H), 7.10 - 7.23 (m, 2H), 7.02 (d, J = 4.33 Hz, 1H), 6.88 - 6.98 (m, 1H), 6.51 (br. s., 1H), 6.44 (s, 1H), 6.32 (t, J = 6.59 Hz, 2H), 4.30 (br. s., 1H), 4.05 (s, 3H), 3.60 (q, J = 5.78 Hz, 2H), 3.43 (q, J = 6.47 Hz, 2H), 3.26 (br. s., 2H), 2.76 - 2.83 (m, 6H), 1.60 - 1.71 (m, 2H), 0.98 (t, J = 7.44 Hz, 3H). MS (ESI) m/z: 630.2. [M+H]+.
2-(Dimethylamino)-N-(2-((3-((3’-((dimethylamino)methyl)-4-methoxy-[1,1’-biphenyl])-3-sulfonamido)phenyl)amino)ethyl)benzamide (10).
Yield: 26% over 4 steps. 1H NMR (300 MHz, CHLOROFORM-d) δ 9.89 (br. s., 1H), 8.01 - 8.16 (m, 2H), 7.67 (dd, J = 2.35, 8.57 Hz, 1H), 7.22 - 7.51 (m, 6H), 7.06 - 7.22 (m, 3H), 6.81 - 7.04 (m, 2H), 6.46 (d, J = 1.88 Hz, 1H), 6.25 - 6.42 (m, 2H), 4.14 - 4.41 (m, 1H), 3.98 - 4.11 (m, 3H), 3.63 (q, J = 5.78 Hz, 2H), 3.47 (s, 2H), 3.28 (t, J = 5.65 Hz, 2H), 2.47 - 2.62 (m, 6H), 2.26 (s, 6H). MS (ESI) m/z: 602.4. [M+H]+.
2-(Dimethylamino)-N-(2-((3-((3’-(dimethylamino)-4-methoxy-[1,1’-biphenyl])-3-sulfonamid-o)phenyl)amino)ethyl)benzamide (11).
Yield: 35% over four steps. 1H NMR (300 MHz, CDCl3) δ 9.92 (br. s., 1H), 8.11 (d, J = 7.72 Hz, 1H), 8.05 (d, J = 2.26 Hz, 1H), 7.66 (dd, J = 2.26, 8.48 Hz, 1H), 7.31 - 7.51 (m, 1H), 7.06 - 7.24 (m, 3H), 6.84 - 7.05 (m, 3H), 6.73 - 6.84 (m, 2H), 6.70 (d, J = 10.17 Hz, 1H), 6.45 (s, 1H), 6.31 (d, J = 7.91 Hz, 2H), 3.97 - 4.09 (m, 3H), 3.53 - 3.69 (m, 2H), 3.29 (t, J = 5.75 Hz, 2H), 2.29 - 3.06 (m, 12H). MS (ESI) m/z: 588.2. [M+H]+.
2-(Dimethylamino)-N-(2-((3-((3’-isopropyl-4-methoxy-[1,1’-biphenyl])-3-sulfonamido)phen-yl)amino)ethyl)benzamide (12).
Yield: 41% over four steps. 1H NMR (300 MHz, Chloroform-d) δ 9.91 (br. s., 1H), 8.11 (dd, J = 1.70, 7.91 Hz, 1H), 8.05 (d, J = 2.26 Hz, 1H), 7.65 (dd, J = 2.35, 8.57 Hz, 1H), 7.37 - 7.45 (m, 1H), 7.28 - 7.36 (m, 3H), 7.20 (d, J = 7.35 Hz, 2H), 7.08 - 7.16 (m, 1H), 7.02 (d, J = 8.67 Hz, 1H), 6.94 (t, J = 8.10 Hz, 1H), 6.89 (s, 1H), 6.45 (t, J = 2.07 Hz, 1H), 6.33 (td, J = 2.28, 8.05 Hz, 2H), 4.17 - 4.54 (m, 1H), 4.05 - 4.09 (m, 3H), 3.64 (q, J = 5.97 Hz, 2H), 3.29 (t, J = 5.75 Hz, 2H), 2.87 - 2.99 (m, 1H), 2.54 (s, 6H), 1.27 (d, J = 6.97 Hz, 6H). MS (ESI) m/z: 587.2. [M+H]+.
N-(2-((3-((3’-(diethylamino)-4-methoxy-[1,1’-biphenyl])-3-sulfonamido)phenyl)amino)ethyl-)-2-(dimethylamino)benzamide (13).
Yield: 32% over four steps. 1H NMR (300 MHz, Chloroform-d) δ 9.93 (br. s., 1H), 8.11 (dd, J = 1.70, 7.91 Hz, 1H), 8.04 (d, J = 2.26 Hz, 1H), 7.64 (dd, J = 2.26, 8.67 Hz, 1H), 7.36 - 7.46 (m, 1H), 7.07 - 7.24 (m, 3H), 7.00 (d, J = 8.67 Hz, 1H), 6.87 - 6.96 (m, 2H), 6.69 - 6.77 (m, 2H), 6.65 (d, J = 8.10 Hz, 1H), 6.45 (d, J = 2.07 Hz, 1H), 6.27 - 6.38 (m, 2H), 4.05 (s, 3H), 3.64 (q, J = 5.97 Hz, 2H), 3.38 (q, J = 7.16 Hz, 3H), 3.29 (t, J = 5.75 Hz, 2H), 2.43 - 2.60 (m, 6H), 1.17 (t, J = 7.06 Hz, 6H). MS (ESI) m/z: 616.2. [M+H]+.
2-(Dimethylamino)-N-(2-((3-((3’-(dipropylamino)-4-methoxy-[1,1’-biphenyl])-3-sulfonamid-o)phenyl)amino)ethyl)benzamide (14).
Yield: 33% over four steps. 1H NMR (300 MHz, Chloroform-d) δ 9.93 (br. s., 1H), 8.11 (dd, J = 1.70, 7.72 Hz, 1H), 8.03 (d, J = 2.45 Hz, 1H), 7.63 (dd, J = 2.35, 8.57 Hz, 1H), 7.40 (dt, J = 1.70, 7.72 Hz, 1H), 7.10 - 7.24 (m, 3H), 7.01 (d, J = 8.67 Hz, 1H), 6.93 (t, J = 8.01 Hz, 1H), 6.88 (s, 1H), 6.65 - 6.73 (m, 2H), 6.61 (dd, J = 2.26, 8.29 Hz, 1H), 6.44 (t, J = 2.07 Hz, 1H), 6.27 - 6.36 (m, 2H), 4.05 (s, 3H), 3.64 (q, J = 5.84 Hz, 1H), 3.16 - 3.33 (m, 4H), 2.49 - 2.58 (m, 6H), 1.57 - 1.64 (m, 4H), 0.93 (t, J = 7.44 Hz, 6H). MS (ESI) m/z: 644.2. [M+H]+.
N-(2-((3-((3’-(tert-butyl)-4-methoxy-[1,1’-biphenyl])-3-sulfonamido)phenyl)amino)ethyl)-2-(dimethylamino)benzamide (15).
Yield: 39% over four steps. 1H NMR (300 MHz, Chloroform-d) δ 9.77 - 10.00 (m, 1H), 8.11 (dd, J = 1.60, 7.82 Hz, 1H), 8.04 (d, J = 2.26 Hz, 1H), 7.65 (dd, J = 2.35, 8.57 Hz, 1H), 7.47 (s, 1H), 7.39 (dd, J = 1.70, 7.54 Hz, 1H), 7.32 - 7.37 (m, 1H), 7.28 - 7.31 (m, 1H), 7.26 (s, 1H), 7.15 - 7.22 (m, 1H), 7.12 (d, J = 8.10 Hz, 1H), 7.03 (d, J = 8.67 Hz, 1H), 6.88 - 6.99 (m, 2H), 6.43 - 6.49 (m, 1H), 6.27 - 6.38 (m, 2H), 4.06 (s, 3H), 3.57 - 3.70 (m, 2H), 3.29 (t, J = 5.84 Hz, 2H), 2.42 - 2.60 (m, 6H), 1.29 - 1.38 (m, 9H). MS (ESI) m/z: 601.2. [M+H]+.
2-(Dimethylamino)-N-(2-((3-((4-methoxy-3’-(trifluoromethyl)-[1,1’-biphenyl])-3-sulfonami-do)phenyl)amino)ethyl)benzamide (16).
Yield: 31% over four steps. 1H NMR (300 MHz, Chloroform-d) δ 9.91 (br. s., 1H), 8.11 (dd, J = 1.70, 7.91 Hz, 1H), 8.04 (d, J = 2.45 Hz, 1H), 7.70 (s, 1H), 7.61 - 7.68 (m, 2H), 7.55 - 7.60 (m, 1H), 7.52 (d, J = 7.54 Hz, 1H), 7.42 (dt, J = 1.79, 7.68 Hz, 1H), 7.20 (d, J = 7.72 Hz, 1H), 7.14 (d, J = 8.10 Hz, 1H), 7.06 (d, J = 8.67 Hz, 1H), 6.89 - 6.99 (m, 2H), 6.39 - 6.49 (m, 1H), 6.34 (d, J = 8.10 Hz, 2H), 4.21 - 4.57 (m, 1H), 4.06 - 4.13 (m, 3H), 3.65 (q, J = 5.97 Hz, 2H), 3.29 (t, J = 5.75 Hz, 2H), 2.48 - 2.62 (m, 6H). MS (ESI) m/z: 613.2. [M+H]+.
General procedure for the synthesis of compound 51a-b:
3-Nitrofluorobenzene (1 eq.) and the appropriate diamine (5 eq.) were mixed in a sealed tube and the reaction was heated at 120°C overnight. After cooling down, the volatile was evaporated under reduced pressure at 60°C. The residue was then redissolved in a mixture of THF (30 mL) and water (30 mL) followed by the addition of potassium carbonate (3.0 eq.) and di-tert-butyl dicarbonate (2.5 eq.). The reaction was then stirred overnight and diluted by brine (150 mL). Ethyl acetate (150 mL) was then added and the organic layer was separated and dried. The solvent was removed under reduced pressure and the residue was purified by ISCO to afford pure desired product.
tert-Butyl (3-((3-nitrophenyl)amino)propyl)carbamate (51a).
Yield: 72%. 1H NMR (300 MHz, Chloroform-d) δ 7.50 (dd, J = 1.60, 8.01 Hz, 1H), 7.39 (t, J = 2.26 Hz, 1H), 7.25 - 7.30 (m, 1H), 6.88 (dd, J = 2.07, 8.10 Hz, 1H), 4.56 - 4.69 (m, 1H), 3.25 (dq, J = 3.58, 6.34 Hz, 4H), 2.72 (d, J = 7.16 Hz, 1H), 1.79 (t, J = 6.50 Hz, 2H), 1.45 (s, 9H). MS (ESI) m/z: 296.2. [M+H]+.
tert-Butyl (1-((3-nitrophenyl)amino)propan-2-yl)carbamate (51b).
Yield: 30%. 1H NMR (300 MHz, Chloroform-d) δ 7.47 - 7.52 (m, 2H), 7.35 (t, J = 2.26 Hz, 2H), 7.24 - 7.29 (m, 1H), 6.87 (dd, J = 1.88, 8.10 Hz, 1H), 4.78 - 4.87 (m, 1H), 4.46 - 4.59 (m, 1H), 3.92 - 4.03 (m, 1H), 3.21 (s, 2H), 3.06 - 3.15 (m, 1H), 2.74 - 2.92 (m, 1H), 1.40 - 1.47 (m, 9H), 1.24 - 1.28 (m, 3H). MS (ESI) m/z: 296.2. [M+H]+.
General procedure for the synthesis of compound 52a-b:
Compound 51a or 51b (1 eq.) was dissolved in DMF (0.2 M) followed by the addition of potassium carbonate (2 eq.) and benzyl bromide (1.2 eq.). The reaction was then heated at 60°C overnight. Water and ethyl acetate were added and the organic layer was separated and dried. The solvent was removed under reduced pressure and the residue was purified by ISCO to afford the desired product 52a-b.
tert-Butyl (3-(benzyl(3-nitrophenyl)amino)propyl)carbamate (52a)
Yield: 84%. 1H NMR (300 MHz, Chloroform-d) δ 7.46 - 7.54 (m, 2H), 7.28 - 7.39 (m, 3H), 7.25 - 7.27 (m, 1H), 7.18 (d, J = 6.78 Hz, 2H), 6.86 - 6.96 (m, 1H), 4.60 (s, 2H), 3.45 - 3.58 (m, 1H), 3.21 (d, J = 6.40 Hz, 1H), 1.82 - 1.94 (m, 2H), 1.44 (s, 9H). MS (ESI) m/z: 386.2. [M+H]+.
tert-Butyl (1-(benzyl(3-nitrophenyl)amino)propan-2-yl)carbamate (52b).
Yield: 81%. 1H NMR (300 MHz, Chloroform-d) δ 7.58 (br. s., 1H), 7.48 (dd, J = 1.51, 7.91 Hz, 1H), 7.37 (d, J = 4.33 Hz, 1H), 7.25 - 7.32 (m, 4H), 7.17 (s, 1H), 6.97 - 7.12 (m, 1H), 4.59 - 4.77 (m, 3H), 4.27 - 4.50 (m, 1H), 4.09 (d, J = 6.97 Hz, 1H), 3.21 - 3.82 (m, 2H), 1.31 - 1.46 (m, 9H). MS (ESI) m/z: 386.1. [M+H]+.
General procedure for the synthesis of compound 53a-b:
Compound 52a or 52b (1 eq.) was dissolved in the mixture of ethanol and water (5 : 2, 0.2 M) followed by the addition of ammonium chloride (10 eq.) and iron powder (7 eq.). The reaction was then heated at reflux for 3 hours. After cooling down, DCM (100 mL) was added and the mixture was filtered through celite. The organic layer was then separated and dried. The solvent was then removed under reduced pressure and the residue was purified by ISCO to afford the desired products 53a-b.
tert-Butyl N-{3-[(3-aminophenyl)(benzyl)amino]propyl}carbamate (53a).
Yield: 92%. 1H NMR (300 MHz, Chloroform-d) δ 7.27 - 7.40 (m, 2H), 7.18 - 7.25 (m, 3H), 6.97 (t, J = 8.01 Hz, 1H), 5.98 - 6.21 (m, 3H), 4.49 (m, 3H), 3.29 - 3.45 (m, 2H), 3.15 (d, J = 6.22 Hz, 2H), 1.74 - 1.88 (m, 2H), 1.43 (s, 9H). MS (ESI) m/z: 356.2. [M+H]+.
tert-Butyl N-{1-[(3-aminophenyl)(benzyl)amino]propan-2-yl}carbamate (53b).
Yield: 92%. 1H NMR (300 MHz, Chloroform-d) δ 7.37 (d, J = 4.33 Hz, 1H), 7.26 - 7.32 (m, 2H), 7.18 (t, J = 6.69 Hz, 2H), 6.96 (t, J = 8.10 Hz, 1H), 6.23 (dd, J = 2.45, 8.29 Hz, 1H), 6.12 - 6.19 (m, 1H), 6.06 (dd, J = 1.51, 7.72 Hz, 1H), 4.49 - 4.73 (m, 2H), 4.35 - 4.48 (m, 1H), 3.95 - 4.09 (m, 1H), 3.45 - 3.69 (m, 2H), 3.08 - 3.22 (m, 1H), 1.29 - 1.48 (m, 9H), 1.18 (d, J = 6.78 Hz, 3H). MS (ESI) m/z: 356.2. [M+H]+.
General procedure for the synthesis of compound 54a-b:
Under the protection of nitrogen, compound 53a or 53b (1 eq.) was dissolved in anhydrous DCM (0.2 eq.) at 0°C, pyridine (1.2 eq.) was added followed by the addition of 2-methoxy-5-bromobenzenesulfonyl chloride (1.1 eq.). The reaction was warmed up to room temperature and stirred overnight. The reaction was quenched with saturated NaHCO3 (10 mL) and DCM (30 mL) was added. The organic layer was separated and dried. The solvent was removed under reduced pressure and the residue was purified by ISCO to afford the desired products 54a-b.
tert-Butyl (3-(benzyl(3-((5-bromo-2-methoxyphenyl)sulfonamido)phenyl)amino)propyl)car-bamate (54a).
Yield: 85%. 1H NMR (300 MHz, Chloroform-d) δ 7.88 (d, J = 2.64 Hz, 1H), 7.55 (dd, J = 2.64, 8.85 Hz, 1H), 7.28 (d, J = 7.54 Hz, 2H), 7.23 (s, 1H), 7.11 (d, J = 6.59 Hz, 2H), 6.97 (d, J = 8.10 Hz, 1H), 6.92 (s, 1H), 6.79 (d, J = 8.85 Hz, 1H), 6.37 - 6.46 (m, 2H), 6.33 (d, J = 8.67 Hz, 1H), 4.54 - 4.64 (m, 1H), 4.44 (s, 2H), 3.85 (s, 3H), 3.30 - 3.42 (m, 2H), 3.15 (d, J = 6.22 Hz, 2H), 1.70 - 1.83 (m, 2H), 1.44 (s, 9H). MS (ESI) m/z: 606.2. [M+H]+.
tert-Butyl (1-(benzyl(3-((5-bromo-2-methoxyphenyl)sulfonamido)phenyl)amino)propan-2-yl)carbamate (54b).
Yield: 83%. 1H NMR (300 MHz, Chloroform-d) δ 7.86 (d, J = 2.64 Hz, 1H), 7.54 (dd, J = 2.45, 8.85 Hz, 1H), 7.25 - 7.30 (m, 3H), 7.07 (d, J = 6.59 Hz, 2H), 6.99 (t, J = 8.10 Hz, 1H), 6.90 (br. s., 1H), 6.70 - 6.83 (m, 1H), 6.52 (d, J = 8.48 Hz, 1H), 6.34 - 6.46 (m, 2H), 4.42 - 4.64 (m, 2H), 4.33 (d, J = 12.06 Hz, 1H), 3.90 - 4.05 (m, 1H), 3.79 - 3.88 (m, 3H), 3.58 (br. s., 1H), 3.18 (br. s., 1H), 1.37 (s, 9H), 1.16 (d, J = 6.59 Hz, 2H). MS (ESI) m/z: 606.2. [M+H]+.
General procedure for the synthesis of compounds 17 - 21:
Compound 48, 54a or 54b (1.0 eq.), boronic acid (1.2 eq.), Pd(PPh3)4 (0.1 eq.) and potassium carbonate (2.0 eq.) were placed in round-bottomed flask with an efficient condenser. The system was then flushed with nitrogen and a mixture of 1,4-dioxane/water (4/1, 0.1 M) was added. The reaction was refluxed for 2 hours. After cooling down, DCM (50 ml) was added and the organic layer was separated and dried. The solvent was then removed and the residue (intermediate compound 53) was dissolved in 4 N HCl in 1,4-dioxane (10 eq.). The reaction was stirred for 2 hours at room temperature and the solvent was then removed under reduced pressure to give a brown solid that was then dissolved in DMF (0.1 M), followed by the addition of the corresponding benzoic acid (1.1 eq.), HATU (1.2 eq) and DIPEA (1.5 eq.). The reaction was stirred overnight at room temperature and quenched by saturated NaHCO3. DCM (50 mL) was added and the organic layer was separated and dried. The solvent was removed under reduced pressure to afford the crude product (compound 54 a-c), which was then mixed with Pd/C (0.1 eq) in MeOH (0.1 M) under the atmosphere of hydrogen (40 psi) for 12 hours. The reaction mixture was filtered and the solvent of the filtrate was removed under reduced pressure. The residue was purified by ISCO to afford the desired final products 17-21.
4’-Methoxy-N,N-dimethyl-3’-(N-(3-((2-(3-methylbenzamido)ethyl)amino)phenyl)sulfamoyl-)-[1,1’-biphenyl]-3-carboxamide (17).
1H NMR (300 MHz, Chloroform-d) δ 9.89 (br. s., 1H), 8.00 - 8.17 (m, 2H), 7.64 (dd, J = 1.88, 8.67 Hz, 1H), 7.48 - 7.58 (m, 2H), 7.25 - 7.46 (m, 3H), 7.10 - 7.23 (m, 3H), 7.01 (d, J = 8.67 Hz, 1H), 6.94 (t, J = 8.01 Hz, 1H), 6.45 (br. s., 1H), 6.34 (dd, J = 8.01, 14.79 Hz, 2H), 4.33 (br. s., 1H), 3.94 - 4.09 (m, 3H), 3.54 - 3.73 (m, 2H), 3.28 (br. s., 2H), 2.86 - 3.20 (m, 6H), 2.42 - 2.66 (m, 6H). MS (ESI) m/z: 587.2. [M+H]+.
3’-(N-(3-((3-(2-(dimethylamino)benzamido)propyl)amino)phenyl)sulfamoyl)-4’-methoxy-N, N-dimethyl-[1,1’-biphenyl]-3-carboxamide (18).
Yield: 29% over four steps. 1H NMR (300 MHz, Chloroform-d) δ 9.71 (br. s., 1H), 7.97 - 8.15 (m, 2H), 7.62 - 7.80 (m, 2H), 7.50 - 7.57 (m, 2H), 7.30 - 7.48 (m, 4H), 7.14 - 7.22 (m, 2H), 7.02 - 7.10 (m, 1H), 6.88 - 6.97 (m, 2H), 6.39 - 6.53 (m, 1H), 6.20 - 6.37 (m, 2H), 4.03 - 4.10 (m, 3H), 3.31 - 3.54 (m, 2H), 2.78 - 3.21 (m, 12H), 2.38 - 2.64 (m, 2H), 1.74 - 1.90 (m, 2H). MS (ESI) m/z: 630.2. [M+H]+.
4’-Methoxy-N,N-dimethyl-3’-(N-(3-((3-(3-methylbenzamido)propyl)amino)phenyl)sulfamo-yl)-[1,1’-biphenyl]-3-carboxamide (19).
Yield: 32% over four steps. 1H NMR (300 MHz, Chloroform-d) δ 8.08 (d, J = 2.45 Hz, 1H), 7.71 - 8.04 (m, 1H), 7.67 (dd, J = 2.35, 8.57 Hz, 1H), 7.57 (s, 1H), 7.52 (td, J = 1.53, 3.53 Hz, 3H), 7.29 - 7.45 (m, 3H), 7.04 (d, J = 8.85 Hz, 2H), 6.85 - 6.98 (m, 2H), 6.48 (d, J = 1.88 Hz, 1H), 6.25 - 6.35 (m, 2H), 4.13 (s, 1H), 4.02 - 4.08 (m, 4H), 3.36 (q, J = 6.22 Hz, 2H), 2.76 - 3.23 (m, 14H), 2.35 (s, 3H), 1.69 - 1.74 (m, 3H). MS (ESI) m/z: 601.2. [M+H]+.
3’-(N-(3-((2-(2-(dimethylamino)benzamido)propyl)amino)phenyl)sulfamoyl)-4’-methoxy-N, N-dimethyl-[1,1’-biphenyl]-3-carboxamide (20).
Yield: 22% over four steps. 1H NMR (300 MHz, Chloroform-d) δ 9.70 - 9.79 (m, 1H), 8.01 - 8.11 (m, 2H), 7.64 (dd, J = 2.45, 8.67 Hz, 1H), 7.49 - 7.56 (m, 2H), 7.38 - 7.47 (m, 2H), 7.30 - 7.37 (m, 1H), 7.11 - 7.23 (m, 2H), 6.97 - 7.06 (m, 2H), 6.93 (t, J = 8.01 Hz, 1H), 6.42 (t, J = 1.98 Hz, 1H), 6.25 - 6.38 (m, 2H), 4.35 - 4.46 (m, 1H), 4.06 (s, 3H), 3.13 - 3.20 (m, 2H), 2.85 - 3.06 (m, 7H), 2.80 (s, 6H), 1.28 (d, J = 6.78 Hz, 3H). MS (ESI) m/z: 630.2. [M+H]+.
4’-Methoxy-N, N-dimethyl-3’-(N-(3-((2-(3-methylbenzamido)propyl)amino)phenyl)sulfamo-yl)-[1,1’-biphenyl]-3-carboxamide (21).
Yield: 19% over four steps. 1H NMR (300 MHz, Chloroform-d) δ 8.09 (d, J = 2.45 Hz, 1H), 8.01 (s, 1H), 7.65 (dd, J = 2.45, 8.67 Hz, 1H), 7.48 - 7.59 (m, 4H), 7.41 (t, J = 7.72 Hz, 1H), 7.30 - 7.35 (m, 1H), 7.24 (s, 1H), 7.11 (s, 1H), 7.01 (d, J = 8.67 Hz, 1H), 6.90 (t, J = 8.01 Hz, 1H), 6.72 (d, J = 8.10 Hz, 1H), 6.46 (t, J = 1.98 Hz, 1H), 6.29 (dt, J = 1.79, 7.86 Hz, 2H), 4.39 - 4.47 (m, 1H), 4.02 (s, 3H), 3.00 (s, 2H), 2.80 (s, 6H), 2.32 (s, 3H), 1.25 (d, J = 6.78 Hz, 3H). MS (ESI) m/z: 601.2. [M+H]+.
tert-Butyl 4-(3-nitrophenyl)piperazine-1-carboxylate (57).
3-Nitrofluorobenzene (5.0 mmol, 35.4 mmol) and piperazine (9.16 g, 106.31 mmol) were mixed in a sealed tube and the reaction was heated at 120°C overnight. After cooling down, the volatile was evaporated under reduced pressure at 60°C. The residue was then redissolved in the mixture of THF (30 mL) and water (30 mL) followed by the addition of potassium carbonate (14.7 g, 106.2 mmol) and di-tert-butyl dicarbonate (19.3 g, 88.5 mmol). The reaction was then stirred overnight and diluted by brine (150 mL). Ethyl acetate (150 mL) was then added and the organic layer was separated and dried. The solvent was removed under reduced pressure and the residue was purified by ISCO to afford the desired product. 5.30 g as a yellow solid, yield: 49%. 1H NMR (300 MHz, Chloroform-d) δ 7.64 - 7.76 (m, 2H), 7.40 (t, J = 8.10 Hz, 1H), 7.16 - 7.24 (m, 1H), 3.54 - 3.67 (m, 2H), 3.19 - 3.30 (m, 4H), 1.49 (s, 9H). MS (ESI) m/z: 308.2. [M+H]+.
tert-Butyl 4-(3-aminophenyl)piperazine-1-carboxylate (58).
Compound 57 (5.35 g, 17.41 mmol) was dissolved in the mixture of ethanol and water (70 mL/30 mL) followed by the addition of ammonium chloride (9.31 g, 174.1 mmol) and iron powder (6.81 g, 121.8 mmol). The reaction was then heated at reflux for 3 hours. After cooling down, DCM (100 mL) was added and the mixture was filtered through celite. The organic layer was then separated and dried. The solvent was then removed under reduced pressure and the residue was purified by ISCO to afford pure desired product. 4.15 g brown oil, yield: 86%. 1H NMR (300 MHz, Chloroform-d) δ 6.98 - 7.11 (m, 1H), 6.32 - 6.39 (m, 1H), 6.20 - 6.30 (m, 2H), 3.58 - 3.85 (m, 2H), 3.49 - 3.58 (m, 4H), 3.07 - 3.14 (m, 4H), 1.47 - 1.50 (m, 9H). MS (ESI) m/z: 278.2. [M+H]+.
tert-Butyl 4-(3-((5-bromo-2-methoxyphenyl)sulfonamido)phenyl)piperazine-1-carboxylate (59).
Under the protection of nitrogen, compound 58 (3.04 g, 10.96 mmol) was dissolved in anhydrous DCM (55 mL) at 0°C, pyridine (1.06 mL, 13.15 mmol) was added followed by the addition of 2-methoxy-5-bromobenzenesulfonyl chloride (3.44 g, 12.06 mmol). The reaction was warmed up to room temperature and stirred overnight. The reaction was quenched by saturated NaHCO3 (30 mL) and DCM (100 mL) was added. The organic layer was separated and dried. The solvent was removed under reduced pressure and the residue was purified by ISCO to afford the desired product 57. 4.61 g brown solid, yield: 80%. 1H NMR (300 MHz, Chloroform-d) δ 7.94 (d, J = 2.45 Hz, 1H), 7.58 (dd, J = 2.54, 8.76 Hz, 1H), 7.26 (s, 3H), 7.07 (t, J = 8.10 Hz, 1H), 6.82 - 6.93 (m, 2H), 6.69 - 6.76 (m, 1H), 6.64 (d, J = 8.29 Hz, 1H), 6.41 (d, J = 7.91 Hz, 1H), 4.01 (s, 3H), 3.48 - 3.62 (m, 4H), 3.00 - 3.16 (m, 4H), 1.48 (s, 9H). MS (ESI) m/z: 528.2. [M+H]+.
General procedure for the synthesis of 22 and 23:
Compound 59 (1.0 eq.), boronic acid (1.2 eq.), Pd(PPh3)4 (0.1 eq.) and potassium carbonate (2.0 eq.) were placed in round-bottomed flask with an efficient condenser. The system was then flushed with nitrogen and a mixture of 1,4-dioxane/water (4/1, 0.1 M) was added. The reaction was refluxed for 2 hours. After cooling down, DCM (50 ml) was added and the organic layer was separated and dried. The solvent was then removed and the residue (compound 58) was dissolved in 4 N HCl in 1,4-dioxane (10 eq.). The reaction was stirred for 2 hours at room temperature and the solvent was then removed under reduced pressure. The residue was then dissolved in DMF (0.1 M) followed by the addition of the corresponding benzoic acid (1.1 eq.), HATU (1.2 eq) and DIPEA (1.5 eq.). The reaction was stirred overnight at room temperature and quenched by saturated NaHCO3. DCM (50 mL) was added and the organic layer was separated and dried. The solvent was removed under reduced pressure to afford the crude product that was purified by ISCO to give the desired products 22-23.
3’-(N-(3-(4-(2-(dimethylamino)benzoyl)piperazin-1-yl)phenyl)sulfamoyl)-4’-methoxy-N,N-dimethyl-[1,1’-biphenyl]-3-carboxamide (22).
Yield: 36% over three steps. 1H NMR (300 MHz, Chloroform-d) δ 8.04 (d, J = 2.07 Hz, 1H), 8.03 (d, J1 = 9.0 Hz, J2 = 177 Hz, 1H), 7.72 (d, J = 8.67 Hz, 1H), 7.52 (d, J = 12.06 Hz, 2H), 7.42 (t, J = 7.54 Hz, 1H), 7.30 - 7.37 (m, 2H), 7.22 (d, J = 7.16 Hz, 1H), 7.02 - 7.12 (m, 2H), 6.89 - 6.99 (m, 3H), 6.77 (s, 1H), 6.62 (d, J = 8.10 Hz, 1H), 6.48 (d, J = 7.54 Hz, 1H), 4.08 (s, 3H), 3.93 (br. s., 1H), 3.84 (br. s., 1H), 2.94 - 3.22 (m, 10H), 2.73 - 2.86 (m, 8H). MS (ESI) m/z: 642.2. [M+H]+.
4’-Methoxy-N,N-dimethyl-3’-(N-(3-(4-(3-methylbenzoyl)piperazin-1-yl)phenyl)sulfamoyl)-[1,1’-biphenyl]-3-carboxamide (23).
Yield: 41% over three steps. 1H NMR (300 MHz, Chloroform-d) δ 8.04 (d, J = 2.26 Hz, 1H), 7.71 (dd, J = 2.35, 8.57 Hz, 1H), 7.48 - 7.60 (m, 2H), 7.42 (t, J = 7.54 Hz, 1H), 7.31 - 7.37 (m, 1H), 7.21 - 7.30 (m, 4H), 7.17 (d, J = 7.16 Hz, 1H), 7.08 (d, J = 8.85 Hz, 2H), 6.97 - 7.04 (m, 1H), 6.78 (s, 1H), 6.62 (d, J = 8.29 Hz, 1H), 6.49 (d, J = 7.72 Hz, 1H), 4.07 (s, 3H), 3.83 (br. s., 2H), 3.54 (br. s., 2H), 2.90 - 3.25 (m, 10H), 2.38 (s, 3H). MS (ESI) m/z: 613.2. [M+H]+.
N1-(3-nitrophenyl)ethane-1, 2-diamine hydrochloride (61).
Compound 45 was dissolved in minimum of ethyl acetate and 4N HCl in dioxane (50 mL) was added. The mixture was then stirred for 2 hours until no bubbles were released. Hexane (100 mL) was added to precipitate any solid and the suspension was filtered. The solid collected was rinsed with diethyl ether and dried in vacuum overnight to give the desired product. 17 g tan solid, yield: 78%. 1H NMR (300 MHz, DMSO-d6) δ 8.12 (br. s., 4H), 7.29 - 7.50 (m, 3H), 6.91 - 7.17 (m, 1H), 3.38 (t, J = 6.40 Hz, 2H), 2.87 - 3.06 (m, 2H). MS (ESI) m/z: 182.2. [M+H]+.
N-(2-((3-aminophenyl)amino)ethyl)-3-methylbenzamide (63).
3-Methyl benzoic acid (2.73 g, 20 mmol) and 1,1′-Carbonyldiimi-dazole (3.25 g, 20 mmol) were mixed in DCM and stirred for 15 min. Compound 59 (2.62 g, 10 mmol) was then added in one portion followed by the addition of DIPEA (10.5 mL, 60 mmol). The reaction was then monitored by TLC. After completion, sat. NaHCO3 was added to quench the reaction. The organic layer was then separated and dried by anhydrous MgSO4. The solvent was removed under reduced pressure and the residue (compound 62) was then redissolved in the mixture of EtOH (80 mL) and water (30 mL) followed by the addition of ammonium chloride (10.7 g, 0.2 mol) and iron powder (7.84 g, 0.14 mol). The reaction was then brought to reflux for 2 hours. After cooling down to room temperature, the reaction mixture was filtered and the filtrate was concentrated. Ethyl acetate (200 mL) and brine (200 mL) was added to the residue and the organic layer was separated and dried. The solvent was removed under reduced pressure and the residue was subjected to ISCO to give the desired product. 3.33 g, brown oil, yield: 62%. 1H NMR (300 MHz, d6-DMSO) δ 8.56 (m, 2H), 7.54 - 8.22 (m, 3H), 7.10 - 7.50 (m, 2H), 7.02 (br. s, 2H), 3.60-3.20 (m, 4H), 2.35 (s, 3H). MS (ESI) m/z: 270.2. [M+H]+.
N-(2-((3-((5-bromo-2-methoxyphenyl)sulfonamido)phenyl)amino)ethyl)-3-methylbenzamide. (64).
Compound 63 (3.33 g, 12.4 mmol) was dissolved in DCM (100 mL) and triethylamine (3.5 mL, 24.7 mmol) was added, followed by the addition of catalytic DMAP (302 mg, 2.47 mmol). The mixture was then cooled to 0°C and 5-bromo-2-methoxy benzenesulfonyl chloride (3.86 g, 13.0 mmol) in THF (10 mL) was added slowly over a period of 20 minutes. The reaction was warmed up to room temperature and stirred overnight. The reaction was quenched by sat. NaHCO3 (50 mL) and ethyl acetate (100 mL) was added. The organic layer was separated and dried. The solvent was removed under reduced pressure and the residue was subjected to ISCO to give the desired product. 4.84 g, light yellow foam, yield: 76%. 1H NMR (300 MHz, Chloroform-d) δ 7.94 (d, J = 2.45 Hz, 1H), 7.47 - 7.60 (m, 3H), 7.29 - 7.36 (m, 2H), 6.97 (t, J = 8.01 Hz, 1H), 6.80 - 6.92 (m, 2H), 6.45 (d, J = 2.07 Hz, 2H), 6.33 - 6.40 (m, 1H), 6.29 (d, J = 9.23 Hz, 1H), 4.21 (br. s., 1H), 4.00 (s, 3H), 3.66 (q, J = 5.90 Hz, 2H), 3.32 (t, J = 5.65 Hz, 2H), 2.39 (s, 3H). MS (ESI) m/z: 519.2. [M+H]+.
General procedure for the Miyaura borylation reaction:
Aromatic bromides or iodides (1.0 equiv.) were dissolved in 1,4-dioxane (0.1 M) and Bis(pinacolato))diboron (1.5 equiv.) was added followed by PdCl2(dppf) (0.1 equiv.) and KOAc (2 equiv.). The reaction was then heated at 90°C overnight. After cooling to room temperature, the reaction mixture was filtered by Celite and the filtrate was concentrated under reduced pressure. The residue was subjected to ISCO to afford the desired product or used in the next Suzuki reaction without further purification.
General procedure for the Suzuki coupling reaction:
The boronic acid pinacol ester (1.0 equiv.), aryl halide (1.0 equiv.) and K2CO3 (2.0 equiv.) were dissolved in a mixture of 1,4-dioxane and water (v/v = 4 :1, 0.04 M). The mixture was degassed and purged with nitrogen three times. Pd(PPh3)4 (0.1 equiv.) was then added and the reaction was stirred at 90°C for 1 hour. The reaction was then cooled down and quenched with brine. Ethyl acetate was then added and the organic layer was separated and dried. The solvent was removed under reduced pressure and the residue was subjected to ISCO to give pure desired product.
N-(2-((3-((2-methoxy-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)sulfonamido)p-henyl)amino)ethyl)-3-methylbenzamide (65).
Prepared according to general procedure for the Miyaura borylation reaction using compound 64 as the starting material. yield: 87%. 1H NMR (300 MHz, Chloroform-d) δ 8.29 (d, J = 1.51 Hz, 1H), 8.15 - 8.27 (m, 1H), 7.85 - 7.95 (m, 1H), 7.45 - 7.65 (m, 2H), 7.28 - 7.34 (m, 2H), 6.89 - 7.00 (m, 3H), 6.84 (s, 1H), 6.20 - 6.53 (m, 3H), 4.10 - 4.22 (m, 1H), 4.03 (s, 3H), 3.64 (d, J = 6.03 Hz, 2H), 3.33 (br. s., 2H), 2.34 - 2.41 (m, 3H), 1.26 - 1.31 (m, 12H). MS (ESI) m/z: 566.2. [M+H]+.
Compound 24-32 and 33-42 were prepared according to general procedure for the Suzuki coupling reaction using compound 65 and the corresponding halogenated aromatic amide as starting materials:
2-(4-Methoxy-3-(N-(3-((2-(3-methylbenzamido)ethyl)amino)phenyl)sulfamoyl)phenyl)-N, N-dimethylisonicotinamide (24).
yield:78%. 1H NMR (300 MHz, Chloroform-d) δ 8.64 (d, J = 4.90 Hz, 1H), 8.43 (d, J = 2.45 Hz, 1H), 8.25 (dd, J = 2.35, 8.76 Hz, 1H), 7.67 (s, 1H), 7.55 (s, 1H), 7.47 (d, J = 6.59 Hz, 1H), 7.33-7.20 (m, 2H), 7.15 (dd, J = 1.41, 4.99 Hz, 1H), 7.08 (d, J = 8.85 Hz, 1H), 6.91 - 6.98 (m, 2H), 6.84 - 6.90 (m, 1H), 6.46 - 6.52 (m, 1H), 6.31 (d, J = 8.10 Hz, 1H), 6.23 (d, J = 9.04 Hz, 1H), 4.27 - 4.44 (m, 1H), 4.07 (s, 3H), 3.61 (d, J = 5.46 Hz, 2H), 3.25 (t, J = 5.56 Hz, 2H), 3.15 (s, 3H), 2.98 (s, 3H), 2.35 (s, 3H). MS (ESI) m/z: 588.2. [M+H]+.
5-(4-Methoxy-3-(N-(3-((2-(3-methylbenzamido)ethyl)amino)phenyl)sulfamoyl)phenyl)-N,N-dimethylnicotinamide (25).
yield: 80%. 1H NMR (300 MHz, Chloroform-d) δ 8.78 (d, J = 2.26 Hz, 1H), 8.60 (d, J = 1.88 Hz, 1H), 8.08 (d, J = 2.45 Hz, 1H), 7.89 (t, J = 2.17 Hz, 1H), 7.70 (dd, J = 2.45, 8.67 Hz, 1H), 7.57 (s, 1H), 7.50 (d, J = 6.41 Hz, 1H), 7.09 (d, J = 8.67 Hz, 1H), 6.95 - 7.03 (m, 1H), 6.86 - 6.95 (m, 1H), 6.46 - 6.51 (m, 1H), 6.34 (d, J = 8.10 Hz, 1H), 6.25 (d, J = 7.72 Hz, 1H), 4.07 (s, 3H), 3.60 - 3.70 (m, 2H), 3.26 (t, J = 5.75 Hz, 2H), 3.15 (s, 3H), 3.06 (s, 3H), 2.35 (s, 3H). MS (ESI) m/z: 588.2. [M+H]+.
4-(4-methoxy-3-(N-(3-((2-(3-methylbenzamido)ethyl)amino)phenyl)sulfamoyl)phenyl)-N,N-dimethylpicolinamide (26).
yield: 83%. 1H NMR (300 MHz, Chloroform-d) δ 8.59 (d, J = 5.27 Hz, 1H), 8.19 (d, J = 2.26 Hz, 1H), 7.77 (d, J = 2.07 Hz, 1H), 7.74 (d, J = 2.45 Hz, 1H), 7.58 (s, 1H), 7.51 (d, J = 6.41 Hz, 1H), 7.47 (dd, J = 1.88, 5.27 Hz, 1H), 7.08 (d, J = 8.67 Hz, 1H), 6.92 (t, J = 8.01 Hz, 1H), 6.86 (s, 1H), 6.48 (s, 1H), 6.32 (d, J = 8.29 Hz, 1H), 6.20 (d, J = 9.23 Hz, 1H), 4.43 - 4.59 (m, 1H), 4.07 (s, 3H), 3.61 - 3.71 (m, 2H), 3.24 (t, J = 5.56 Hz, 2H), 3.15 (d, J = 3.58 Hz, 6H), 2.34 (s, 3H). MS (ESI) m/z: 588.1. [M+H]+.
6-(4-methoxy-3-(N-(3-((2-(3-methylbenzamido)ethyl)amino)phenyl)sulfamoyl)phenyl)-N,N-dimethylpicolinamide (27).
yield: 85%. 1H NMR (300 MHz, Chloroform-d) δ 8.58 (d, J = 2.45 Hz, 1H), 8.15 (d, J = 8.67 Hz, 1H), 7.77 (d, J = 7.72 Hz, 1H), 7.70 (s, 1H), 7.56 (s, 1H), 7.48 (d, J = 6.41 Hz, 2H), 7.06 (d, J = 8.85 Hz, 1H), 6.86 - 6.98 (m, 1H), 6.82 (s, 2H), 6.46 (s, 1H), 6.21 - 6.35 (m, 2H), 4.28 - 4.44 (m, 1H), 4.06 (s, 3H), 3.57 (d, J = 5.65 Hz, 2H), 3.22 (d, J = 5.46 Hz, 2H), 3.17 (s, 3H), 3.11 (s, 3H), 2.37 (s, 3H). MS (ESI) m/z: 588.2. [M+H]+.
4-(4-Methoxy-3-(N-(3-((2-(3-methylbenzamido)ethyl)amino)phenyl)sulfamoyl)phenyl)-N,N-dimethylthiophene-2-carboxamide (28).
Yield: 68%. 1H NMR (Chloroform-d ,300MHz): δ 8.01 - 8.04 (m, 1 H), 7.62 - 7.68 (m, 1 H), 7.55 (d, J=1.5 Hz, 2 H), 7.47 - 7.52 (m, 1 H), 7.45 (s, 1 H), 7.30 (s, 2 H), 7.00 - 7.05 (m, 1 H), 6.91 - 6.99 (m, 1 H), 6.79 - 6.83 (m, 1 H), 6.58 - 6.66 (m, 1 H), 6.46 - 6.50 (m, 1 H), 6.31 - 6.37 (m, 1 H), 6.24 - 6.30 (m, 1 H), 4.21 - 4.34 (m, 1 H), 4.05 (s, 3 H), 3.25 - 3.33 (m, 2 H), 3.10 - 3.24 (m, 2 H), 2.38 ppm (s, 3 H). MS (ESI) m/z: 593.1. [M+H]+.
5-(4-Methoxy-3-(N-(3-((2-(3-methylbenzamido)ethyl)amino)phenyl)sulfamoyl)phenyl)-N,N-dimethylthiophene-3-carboxamide (29).
Yield: 88%. 1H NMR (300 MHz, Chloroform-d) δ 8.08 (d, J = 2.26 Hz, 1H), 7.64 (dd, J1 = 2.35 Hz, J2 = 8.57 Hz, 1H), 7.56 (s, 1H), 7.46 - 7.52 (m, 1H), 7.35 (s, 1H), 7.33 - 7.34 (m, 1H), 6.99 (d, J = 8.67 Hz, 1H), 6.92 (d, J = 8.29 Hz, 1H), 6.83 - 6.88 (m, 1H), 6.81 (s, 1H), 6.48 (s, 1H), 6.30 - 6.37 (m, 1H), 6.21 - 6.27 (m, 1H), 4.34 - 4.47 (m, 1H), 4.04 (s, 3H), 3.64 (d, J = 5.84 Hz, 2H), 3.26 (s, 2H), 3.11 (br. s., 6H), 2.37 (s, 3H). MS (ESI) m/z: 593.2. [M+H]+.
4-(4-Methoxy-3-(N-(3-((2-(3-methylbenzamido)ethyl)amino)phenyl)sulfamoyl)phenyl)-N,N-dimethylthiazole-2-carboxamide (30).
Yield: 70%. 1H NMR (300 MHz, Chloroform-d) δ 8.31 (d, J = 2.26 Hz, 1H), 7.99 - 8.07 (m, 1H), 7.63 - 7.73 (m, 1H), 7.57 (s, 1H), 7.51 - 7.56 (m, 1H), 7.45 - 7.50 (m, 2H), 7.28 - 7.33 (m, 2H), 7.06 (d, J = 8.67 Hz, 1H), 6.95 (m, 1H), 6.84 (s, 1H), 6.48 (s, 1H), 6.37 - 6.43 (m, 1H), 4.08 (s, 3H), 3.57 - 3.67 (m, 5H), 3.24 - 3.33 (m, 2H), 3.17 (s, 3H), 2.38 (s, 3H). MS (ESI) m/z: 594.2. [M+H]+.
(E)-N-(2-((3-((5-(3-(dimethylamino)-3-oxoprop-1-en-1-yl)-2-methoxyphenyl)sulfonamido)-phenyl)amino)ethyl)-3-methylbenzamide (31).
Yield: 63%. 1H NMR (300 MHz, Chloroform-d) δ 8.03 (d, J = 2.26 Hz, 1H), 7.46 - 7.59 (m, 5H), 7.28 - 7.34 (m, 2H), 6.91 - 7.01 (m, 2H), 6.73 - 6.85 (m, 2H), 6.50 - 6.58 (m, 1H), 6.44 (s, 1H), 6.24 - 6.38 (m, 2H), 4.13 - 4.32 (m, 1H), 4.04 (s, 3H), 3.65 (d, J = 5.84 Hz, 2H), 3.29 (s, 2H), 3.14 (s, 3H), 3.04 (s, 3H), 2.39 (s, 3H). MS (ESI) m/z: 537.2. [M+H]+.
N-(2-((3-((5-(3-(dimethylamino)-3-oxopropyl)-2-methoxyphenyl)sulfonamido)phenyl)amin-o)ethyl)-3-methylbenzamide (32).
Yield: 67%. 1H NMR (300 MHz, Chloroform-d) δ 7.64 (d, J = 2.26 Hz, 1H), 7.58 (s, 1H), 7.52 (br. s., 1H), 7.28 - 7.36 (m, 3H), 6.95 (t, J = 8.01 Hz, 1H), 6.89 (d, J = 8.29 Hz, 1H), 6.83 (s, 1H), 6.67 - 6.75 (m, 1H), 6.43 (s, 1H), 6.35 (s, 1H), 6.28 (d, J = 7.54 Hz, 1H), 3.98 (s, 3H), 3.64 (d, J = 5.65 Hz, 2H), 3.30 (t, J = 5.75 Hz, 2H), 2.81 - 2.92 (m, 8H), 2.49 - 2.57 (m, 2H), 2.39 (s, 3H). MS (ESI) m/z: 539.2. [M+H]+.
N-(2-(dimethylamino)ethyl)-4’-methoxy-N-methyl-3’-(N-(3-((2-(3-methylbenzamido)ethyl)-amino)phenyl)sulfamoyl)-[1,1’-biphenyl]-3-carboxamide (33).
Yield: 72%. 1H NMR (300 MHz, Chloroform-d) δ 8.12 (d, J = 2.26 Hz, 1H), 7.69 (dd, J1 = 2.35 Hz, J2 = 8.57 Hz, 1H), 7.58 (br. s., 3H), 7.53 (s, 2H), 7.38 - 7.47 (m, 2H), 7.30 - 7.37 (m, 2H), 7.15 (br. s., 2H), 7.04 (d, J = 8.67 Hz, 1H), 6.93 (t, J = 8.01 Hz, 2H), 6.46 (s, 1H), 6.32 (d, J = 7.91 Hz, 1H), 6.23 (d, J = 9.23 Hz, 1H), 4.04 (s, 3H), 3.65 - 3.74 (m, 1H), 3.55 - 3.64 (m, 2H), 3.31 - 3.42 (m, 1H), 3.17 - 3.26 (m, 2H), 3.11 (br. s., 1H), 3.04 (br. s., 2H), 2.61 - 2.71 (m, 1H), 2.41 - 2.50 (m, 1H), 2.34 (s, 6H), 2.08 (br. s., 3H). MS (ESI) m/z: 644.2. [M+H]+.
N-(2-(dimethylamino)ethyl)-4’-methoxy-3’-(N-(3-((2-(3-methylbenzamido)ethyl)amino)phenyl)sulfamoyl)-[1,1’-biphenyl]-3-carboxamide (34).
Yield: 75%. 1H NMR (300 MHz, Chloroform-d) δ 8.14 (d, J = 2.45 Hz, 1H), 8.02 (s, 1H), 7.78 - 7.84 (m, 1H), 7.66 - 7.73 (m, 2H), 7.63 (s, 1H), 7.55 (br. s., 2H), 7.49 (d, J = 3.20 Hz, 2H), 7.20 (br. s., 2H), 7.04 (d, J = 8.67 Hz, 1H), 6.93 (t, J = 7.91 Hz, 2H), 6.55 (s, 1H), 6.31 (d, J = 8.10 Hz, 1H), 6.23 (d, J = 6.22 Hz, 1H), 4.03 (s, 3H), 3.65 (d, J = 5.65 Hz, 2H), 3.47 - 3.58 (m, 2H), 3.26 (br. s., 2H), 2.57 (t, J = 5.75 Hz, 1H), 2.52 (d, J = 6.03 Hz, 1H), 2.31 (s, 6H), 2.25 (s, 3H). MS (ESI) m/z: 630.2. [M+H]+; LCMS.
4’-Methoxy-N-methyl-3’-(N-(3-((2-(3-methylbenzamido)ethyl)amino)phenyl)sulfamoyl)-N-(pyridin-2-yl)-[1,1’-biphenyl]-3-carboxamide (35). Yield:
78%. 1H NMR (300 MHz, Chloroform-d) δ 8.36 - 8.40 (m, 1H), 8.24 - 8.33 (m, 2H), 8.18 - 8.22 (m, 1H), 7.70 - 7.76 (m, 1H), 7.59 - 7.62 (m, 1H), 7.56 - 7.59 (m, 1H), 7.53 - 7.56 (m, 1H), 7.50 - 7.52 (m, 1H), 7.47 - 7.50 (m, 1H), 7.44 - 7.47 (m, 1H), 7.30 - 7.39 (m, 2H), 7.21 (s, 1H), 7.18 (s, 1H), 7.02 - 7.07 (m, 1H), 6.89 - 6.96 (m, 1H), 6.79 - 6.83 (m, 1H), 6.49 - 6.58 (m, 2H), 6.27 - 6.33 (m, 1H), 6.14 - 6.22 (m, 1H), 4.04 (s, 3H), 3.90 (s, 3H), 3.63 - 3.73 (m, 2H), 3.23 - 3.35 (m, 2H), 2.28 (s, 3H). MS (ESI) m/z: 650.2. [M+H]+.
N-Benzyl-4’-methoxy-N-methyl-3’-(N-(3-((2-(3-methylbenzamido)ethyl)amino)phenyl)sulfa-moyl)-[1,1’-biphenyl]-3-carboxamide (36).
Yield: 81%. 1H NMR (300 MHz, Chloroform-d) δ 8.13 (br. s., 1H), 7.48 - 7.75 (m, 5H), 7.30 - 7.46 (m, 6H), 6.99 - 7.21 (m, 3H), 6.93 (t, J = 7.91 Hz, 1H), 6.85 (s, 1H), 6.45 (s, 1H), 6.32 (d, J = 8.10 Hz, 1H), 6.23 (d, J = 7.72 Hz, 1H), 4.49 - 4.82 (m, 2H), 4.04 (s, 3H), 3.58 (br. s., 2H), 3.21 (br. s., 2H), 2.76 - 3.11 (m, 3H), 2.33 (br. s., 3H). MS (ESI) m/z: 663.2. [M+H]+.
4’-Methoxy-N-methyl-3’-(N-(3-((2-(3-methylbenzamido)ethyl)amino)phenyl)sulfamoyl)-N-(-pyridin-2-ylmethyl)-[1,1’-biphenyl]-3-carboxamide (37).
Yield: 84%. 1H NMR (300 MHz, Chloroform-d) δ 8.56 - 8.69 (m, 1H), 8.00 - 8.17 (m, 1H), 7.67 - 7.77 (m, 2H), 7.47 - 7.66 (m, 5H), 7.42 (br. s., 3H), 7.01 - 7.13 (m, 2H), 6.93 (t, J = 8.01 Hz, 1H), 6.84 (br. s., 1H), 6.46 (s, 1H), 6.32 (d, J = 8.10 Hz, 1H), 6.17 - 6.27 (m, 1H), 4.88 (s, 1H), 4.62 (s, 1H), 4.04 (br. s., 3H), 3.59 (br. s., 2H), 3.21 (br. s., 2H), 3.09 (d, J = 15.82 Hz, 3H), 2.34 (br. s., 3H). MS (ESI) m/z: 664.2. [M+H]+.
4’-Methoxy-3’-(N-(3-((2-(3-methylbenzamido)ethyl)amino)phenyl)sulfamoyl)-N-(pyridin-2-ylmethyl)-[1,1’-biphenyl]-3-carboxamide (38).
Yield: 62%. 1H NMR (300 MHz, Chloroform-d) δ 8.52 - 8.60 (m, 1H), 8.14 (d, J = 2.45 Hz, 1H), 8.04 (s, 1H), 7.78 (d, J = 7.72 Hz, 2H), 7.61 - 7.72 (m, 3H), 7.44 - 7.56 (m, 3H), 7.06 - 7.14 (m, 1H), 6.98 - 7.05 (m, 2H), 6.88 - 6.98 (m, 1H), 6.55 (s, 1H), 6.27 - 6.36 (m, 1H), 6.19 - 6.26 (m, 1H), 4.72 (d, J = 4.90 Hz, 2H), 4.25 - 4.48 (m, 1H), 4.00 (s, 3H), 3.65 (d, J = 5.46 Hz, 2H), 3.29 (br. s., 2H), 2.30 (s, 3H). MS (ESI) m/z: 650.2. [M+H]+.
4’-Methoxy-N-methyl-3’-(N-(3-((2-(3-methylbenzamido)ethyl)amino)phenyl)sulfamoyl)-N-(pyridin-3-ylmethyl)-[1,1’-biphenyl]-3-carboxamide (39).
Yield: 68%. 1H NMR (300 MHz, Methanol-d4) δ 8.40 - 8.62 (m, 1H), 7.86 - 8.07 (m, 1H), 7.31 (s, 12H), 7.10 - 7.23 (m, 1H), 6.84 - 6.92 (m, 1H), 6.45 - 6.51 (m, 1H), 6.35 - 6.42 (m, 1H), 6.24 - 6.35 (m, 1H), 3.99 (s, 3H), 3.40 - 3.48 (m, 2H), 3.21 (m, 2H), 2.94 - 3.08 (m, 3H), 2.36 (s, 3H). MS (ESI) m/z: 664.1. [M+H]+.
4’-Methoxy-N-methyl-3’-(N-(3-((2-(3-methylbenzamido)ethyl)amino)phenyl)sulfamoyl)-N-(pyridin-4-ylmethyl)-[1,1’-biphenyl]-3-carboxamide (40).
Yield: 80%. 1H NMR (300 MHz, Methanol-d4) δ 8.49 (br. s., 2H), 7.91 - 8.09 (m, 1H), 7.02 - 7.78 (m, 12H), 6.86 (t, J = 8.10 Hz, 1H), 6.50 (br. s., 1H), 6.40 (d, J = 6.59 Hz, 1H), 6.23 - 6.33 (m, 1H), 4.49 - 4.83 (m, 2H), 3.95 (br. s., 3H), 3.36 - 3.49 (m, 2H), 3.18 (t, J = 6.03 Hz, 2H), 2.89 - 3.10 (m, 3H), 2.33 (s, 3H). 13C NMR (75 MHz, Chloroform-d) δ 174.1 (1C, J = 45 Hz), 170.7, 157.9, 150.7, 150.5, 148.9, 141.0, 139.8, 139.5, 137.6, 135.6, 134.3, 133.3, 130.6, 130.4, 130.2, 129.5, 129.3, 128.8, 128.5, 126.9, 126.4, 126.2, 125.8, 125.4, 124.3, 123.4, 114.2, 110.5, 110.2, 105.6, 56.9, 53.3 (1 C, J = 307.5 Hz), 44.1, 40.4, 36.4 (1C, J = 307.5 Hz), 21.4. MS (ESI) m/z: 664.2. [M+H]+.
4’-Methoxy-N-methyl-3’-(N-(3-((2-(3-methylbenzamido)ethyl)amino)phenyl)sulfamoyl)-N-(2-(pyridin-2-yl)ethyl)-[1,1’-biphenyl]-3-carboxamide (41).
Yield: 85 %. 1H NMR (300 MHz, Methanol-d4) δ 8.25 (dd, J1 = 159.0 Hz, J2 = 3.0 Hz, 1H), 7.95 (d, J = 33.0 Hz, 1H), 7.73 - 7.84 (m, 1H), 7.58 (dd, J1 = 63.0 Hz, J2 = 9.0 Hz, 1H), 7.18 - 7.62 (m, 9H), 7.03 (t, J = 6.0 Hz, 1H), 6.91 (t, J = 6.0 Hz, 1H), 6.48-6.83 (m, 2H), 6.42 (t, J = 9.0 Hz, 1H), 6.26-6.36 (m, 1H), 4.03 (d, J = 12.0 Hz, 3H), 3.91 (t, J = 6.0 Hz, 1H), 3.71 (t, J = 6.0 Hz, 1H), 3.35-3.58 (m, 2H), 3.08-3025 (m, 7H), 2.37 (s, 3H). MS (ESI) m/z: 678.2. [M+H]+.
4’-Methoxy-N-methyl-3’-(N-(3-((2-(3-methylbenzamido)ethyl)amino)phenyl)sulfamoyl)-N-(2-(pyridin-4-yl)ethyl)-[1,1’-biphenyl]-3-carboxamide (42).
Yield: 70%. 1H NMR (300 MHz, Chloroform-d) δ 8.55 (br. s., 1H), 8.25 - 8.36 (m, 1H), 7.95 - 8.11 (m, 1H), 7.30 - 7.75 (m, 6H), 6.68 - 7.22 (m, 8H), 6.49 (br. s., 1H), 6.31 (br. s., 2H), 4.04 (s, 3H), 3.83 (br. s., 1H), 3.59 (br. s., 4H), 3.12 - 3.29 (m, 3H), 3.03 (br. s., 1H), 2.90 (br. s., 3H), 2.34 (s, 3H). MS (ESI) m/z: 678.1. [M+H]+.
General procedure for the synthesis of compounds 43 and 44:
Intermediate 63 (539 mg, 2.0 mmol) was dissolved in DCM (20 mL) and triethylamine (0.56 mL, 4.0 mmol) was added. Then catalytic DMAP (25 mg, 0.4 mmol) was introduced and the reaction mixture was cooled down to 0°C. At this temperature, 5-bromo-2-methoxybenzenecarboxylic chloride (499 mg, 2.0 mmol) in THF (10 mL) was added slowly. The reaction was then stirred at this temperature for 1 hour and allowed to warm up to room temperature overnight. Saturated sodium bicarbonate (50 mL) was added to quench the reaction and the reaction was extracted with ethyl acetate (50 mL). The organic layer was separated and dried. The solvent was removed under reduced pressure and the residue was subjected to ISCO to give desired product 66.
5-Bromo-2-methoxy-N-[3-({2-[(3-methylphenyl)formamido]ethyl}amino)phenyl]benzamide (66).
579 mg, yield: 60%. 1H NMR (300 MHz, Chloroform-d) δ 9.61 (s, 1H), 8.35 (d, J = 2.64 Hz, 1H), 7.47 - 7.62 (m, 3H), 7.21 - 7.35 (m, 4H), 7.12 (t, J = 8.01 Hz, 1H), 6.90 (d, J = 8.85 Hz, 1H), 6.75 (d, J = 7.91 Hz, 1H), 6.65 (br. s., 1H), 6.43 (dd, J = 1.70, 8.10 Hz, 1H), 4.15 - 4.27 (m, 1H), 4.02 (s, 3H), 3.69 (q, J = 5.97 Hz, 2H), 3.32 - 3.50 (m, 2H), 2.36 (s, 3H). MS (ESI) m/z: 484.2. [M+H]+.
Compound 67 was synthesized according to general procedure for Miyaura borylation reaction using 66 as starting material. yield: >99%.
2-Methoxy-N-[3-({2-[(3-methylphenyl)formamido]ethyl}amino)phenyl]-5-(tetramethyl-1,3-,2-dioxaborolan-2-yl)benzamide (67).
Yield: > 99%. 1H NMR (300 MHz, Chloroform-d) δ 9.57 (s, 1H), 8.70 (d, J = 1.51 Hz, 1H), 7.91 (dd, J = 1.60, 8.19 Hz, 1H), 7.51 - 7.66 (m, 2H), 7.42 (s, 1H), 7.22 - 7.35 (m, 3H), 7.12 (t, J = 8.01 Hz, 1H), 7.01 (d, J = 8.29 Hz, 1H), 6.73 (d, J = 7.91 Hz, 1H), 6.63 (br. s., 1H), 6.44 (d, J = 7.91 Hz, 1H), 4.05 (s, 3H), 3.72 (q, J = 5.65 Hz, 2H), 3.37 - 3.52 (m, 2H), 2.37 (s, 3H), 1.33 (s, 12H). MS (ESI) m/z: 530.2. [M+H]+.
Compounds 43 and 44 were synthesized according to general procedure for Suzuki coupling reaction using compound 67 as starting material:
4-Methoxy-N3’,N3’-dimethyl-N3-(3-((2-(3-methylbenzamido)ethyl)amino)phenyl)-[1,1’-biph-enyl]-3,3’-dicarboxamide (43).
Yield: 78%. 1H NMR (300 MHz, Chloroform-d) δ 9.77 (s, 1H), 8.53 (d, J = 2.26 Hz, 1H), 7.73 (dd, J = 2.45, 8.48 Hz, 1H), 7.66 (s, 2H), 7.58 (s, 1H), 7.54 (br. s., 1H), 7.47 (t, J = 7.91 Hz, 1H), 7.34 - 7.42 (m, 3H), 7.29 (d, J = 4.71 Hz, 2H), 7.09 - 7.21 (m, 3H), 6.79 (d, J = 8.48 Hz, 1H), 6.46 (d, J = 8.10 Hz, 2H), 4.13 - 4.22 (m, 1H), 4.10 (s, 3H), 3.68 - 3.78 (m, 2H), 3.47 (t, J = 5.75 Hz, 3H), 2.93 - 3.19 (m, 7H), 2.38 (s, 3H). MS (ESI) m/z: 551.2. [M+H.
4-Methoxy-N3’-methyl-N3-(3-((2-(3-methylbenzamido)ethyl)amino)phenyl)-N3’-(pyridin-4-ylmethyl)-[1,1’-biphenyl]-3,3’-dicarboxamide (44).
Yield: 72%. 1H NMR (300 MHz, Chloroform-d) δ 9.75 (br. s., 1H), 8.62 (d, J = 6.03 Hz, 2H), 8.40 - 8.57 (m, 1H), 7.69 (d, J = 7.72 Hz, 3H), 7.36 - 7.60 (m, 5H), 7.28 - 7.34 (m, 3H), 7.06 - 7.22 (m, 3H), 6.72 - 6.83 (m, 1H), 6.46 (d, J = 7.91 Hz, 1H), 4.78 (br. s., 2H), 4.10 (s, 3H), 3.74 (q, J = 5.84 Hz, 2H), 3.41 - 3.53 (m, 2H), 2.90 - 3.16 (m, 3H), 2.37 (s, 3H). MS (ESI) m/z: 628.2. [M+H]+.
OX1R and OX2R calcium mobilization assays.
Activity of the target compounds at the human OX1 and OX2 receptors was determined utilizing CHO RD-HGA16 cells (Molecular Devices) engineered to stably express either the human OX1 or the human OX2 receptor. Cells were maintained in Ham’s F12 supplemented with 10% fetal bovine serum, 100 units of penicillin and streptomycin, and 100 μg/mL normocin™. To conduct the assay, cells were plated at 25,000 cells/well into 96-well black wall/clear bottom assay plates and incubated overnight at 37°C, 5% CO2. The next day prior to the assay, Calcium 5 dye (Molecular Devices) was reconstituted according to the manufacturer’s instructions. The reconstituted dye was diluted 1:40 in pre-warmed (37°C) assay buffer (1X HBSS, 20 mM HEPES, 2.5 mM probenecid, pH 7.4 at 37°C). Growth medium was removed and the cells were gently washed with 100 μL of pre-warmed (37°C) assay buffer. The cells were incubated for 45 minutes at 37°C, 5% CO2 in 200 μL of the diluted Calcium 5 dye. Serial dilutions of test compounds and the agonist control orexin A were prepared at 10x the desired final concentration in 0.25% BSA/1% DMSO/assay buffer, aliquoted into 96-well polypropylene plates, and warmed to 37°C for 15 min. After the dye-loading incubation period, cells were pretreated with 25 µL of 9% DMSO/assay buffer and incubated for 15 min at 37°C. After the pre-treatment incubation period, the plate was read with a FlexStation II (Molecular Devices). Calcium-mediated changes in fluorescence were monitored every 1.52 seconds over a 60 second time period, with the FlexStation II adding 25 μL of the test compound/control serial dilutions at the 19 second time point (excitation at 485 nm, detection at 525 nm). Peak kinetic reduction (SoftMax, Molecular Devices) relative fluorescent units (RFU) were plotted against the log of compound concentration and nonlinear regression analysis was used to generate EC50 values (GraphPad Prism, GraphPad Software, Inc., San Diego, CA).
Computational Methods:
Preparation of initial full-length OX2R and OX1R models.
Initial full-length models of human OX2R were prepared based on the reported coordinates for 5 (PDBID: 7L1V) and orexin-B bound OX2R (PDBID: 7L1U) cryo-EM structures,50 with restoration of missing residues in loops and reversal of truncation of ICL3 and the N- and C-termini. In addition, the miniGsqi/Gβ1γ2 segments that were absent in the 5 and orexin-B bound cryogenic structural solutions were completed using MODELLER.62 Thus, the initial conformations of the ICL3/N- and C-termini of the OX2R were generated self consistently with the initial full-length G-protein. To accomplish these steps, a sequence alignment between the sequence present in the cryogenic construct and the human OX2R sequence was performed, employing CLUSTALX75 and a GONNET 250 scoring matrix and a GAP penalty of 10. This sequence and the cryogenic coordinates of OX2R were input to MODELLER and the best zDOPE scored models collected to select the top scored model for input into SCWRL,76 where new (non-clashing) rotameric states of non-conserved residues were generated for the newly modeled residues. We then employed the Epik function embodied in the Schrodinger Small Molecule Suite to make self-consistent protonation assignments along with Schrodinger protein preparation steps. The model was then energy minimized for 2500 conjugate gradients (PBCG) steps following implementing a disulfide constraint between cysteine127 and cysteine210. The completed model then underwent Nudged Elastic Band pulls of the N-/C-termini to generate compact initial coordinates (primarily to move away from fully extended initial N-/C- terminal conformations) and the full model immersed in an initial 200 × 200 × 200 Å3 1,2-Dioleoyl-sn-glycero-3-phosphocholine (DOPC) lipid box with 50 Å layer of water/KCl above and below the lipid bilayer. The system was then parameterized using the AMBER LIPD1466 and the AMBER1877 ffSB14 forcefields including TIP3P model parameters. The initial simulation membrane system/GPCR complex was obtained by immersing the GPCR complex (excluding the G-protein portion for this study) in the membrane/water/ion system and deleting all lipids/water/ions within 3.5 angstroms of any GPCR-complex atom. The system was then energy minimized using 8000 steps of AMBER sander conjugate gradient steps, heated with all atoms constrained for 5 ns and then equilibrated unconstrained for ca. 30 ns. In the case of 40, we continued with production molecular dynamics out to 180 ns using AMBER PMEMD (Particle Mesh Ewald Molecular Dynamics) for the purpose of examining time dependent interactions between the Schrodinger induced fit poses and residues in the orthosteric binding site.
Docking, induced fit and molecular dynamics studies.
The initial full length models following equilibration and energy minimization were employed in GLIDE SP/XP and induced fit studies. Our docking protocol was to collect 120 post docking energy-minimized poses which were then distilled to the top 5 poses. We found simple GLIDE SP/XP docking with rigid receptor was often inadequate to get good self-docking (i.e. docking the native ligand from the crystal or cryo-EM structures) with GPCRs such as OX1R and OX2R, which is often improved with introduction of receptor flexibility. To his end, we employed MODELLER annealing of initial cryogenic structural solution coordinates and verified adequate docking and RMSD’s using GLIDE SP, which has a more exhaustive configuration sampling than GLIDE-XP which uses a more conservative anchor and grow (CONFGEN78) sampling approach. A simple GLIDE-SP docking of 5 in the OX2R gave a root mean square root (RMSD) of 2.5 Å for the best Emodel scored pose, demonstrating structural similarity to the cryo-EM structures and adequate pose reproduction. In all remaining cases we employed induced fit and/or MD-equilibration starting with initial GLIDE-XP poses. The top Emodel docked pose was then used as the seed structure for Schrodinger-Prime induced-fit modeling with flexibility of 5-7 Å around any atom in the ligand. We used the 7 Å extended region, in particular, to include Arg and Lys residues at the top of the OX2R and OX1R full-length models and explicitly included R3397.28 (OX2R) and R3337.29 (OX1R) in order to enhance sampling of variable conformations including those residues in GLIDE-XP redocking into the Prime variable backbone/sidechain conformation sampled OX2R and OX1R configurations in an induced fit procedure.79 Top scoring induced fit poses were then analyzed for salient interactions and these employed in following MD studies. All ligand models for use in Schrodinger GLIDE SP/XP docking were prepared with the LIGPREP module.
Sleep studies:
Animals:
Twelve-month-old male mice (bred at Minneapolis VA Health Care System, n = 6) were housed individually in solid bottom cages with corncob bedding and a chewing substrate (Nylabone, natural flavor, BioServ, Frenchtown, NJ). Throughout the study, a 12-h light/12-h dark cycle (lights on at 06:00) in a temperature-controlled environment (21–22°C) was used for experiments. Rodent chow (Harlan Teklad 8604) and water were allowed ad libitum. Studies were approved by the Institutional Animal Care and Use Committee at the Minneapolis VA Health Care System.
Surgery:
Mice were anesthetized with a ketamine/xylazine mixture (15 mg/kg; 1.5 mg/kg), and implanted with a radiotelemetric transmitter and EEG/EMG electrodes to record vigilance states (F20-EET, Data Sciences International [DSI], St. Paul, MN). Stereotaxic coordinates for the EE electrodes were as follows: −0.1 mm anterior, 2.0 mm lateral to bregma, and inserted to predrilled holes to touch the dura. The EMG leads were secured in the nuchal musculature. Animals were allowed to recover from surgery for at least 7–10 days before experimental trials began.
Injections:
Mice were injected with either 40 (RTOXA-43, 40 mg/kg i.p. in 0.3ml saline) or vehicle (saline, n=5) in a counter-balanced design. Doses were chosen based on results from sleep studies on YNT-185.46 Mice were randomly assigned to a treatment group, each animal received each treatment once, and all treatments were represented on each day. Injections were performed between 10:00 and 10:30 (zeitgeber time 4–4.5) with ≥ 48 h between treatments. Continuous EEG/EMG recordings were obtained for 24h post-injection.
EEG/EMG Recording and Behavioral State Determination:
To allow freely moving polysomnogram recordings, a receiver (PhysioTel RPC-1, DSI, St. Paul, MN) was placed beneath the testing cage to detect EEG/EMG signals from the implanted transmitter. Briefly, signals were digitized by a Data Exchange Matrix connected to a PC with Dataquest A.R.T 4.1 software (DSI, St. Paul, MN). Electroencephalogram signals (0.3–30.0 Hz bandpass) and EMG signals (1.0–100.0 Hz bandpass) were stored on a computer, visualized with Neuroscore software (version 2.0.1, DSI, St. Paul, MN), and sleep and wakefulness states were scored manually in accordance with previously described methods. Briefly, consecutive 10-s epochs of EEG and EMG signals were classified into one of the following 3 behavioral states: NREM sleep, REM sleep, or wakefulness, and percent time spent in each state was then calculated from the scored data. The following dependent variables were quantified for each recording session: (a) percent time spent in wakefulness, NREM sleep, REM sleep; (b) total number of episodes for each vigilance state; (c) mean duration of episodes for each vigilance state; and (d) total number of state transitions.
Supplementary Material
Scheme 5.
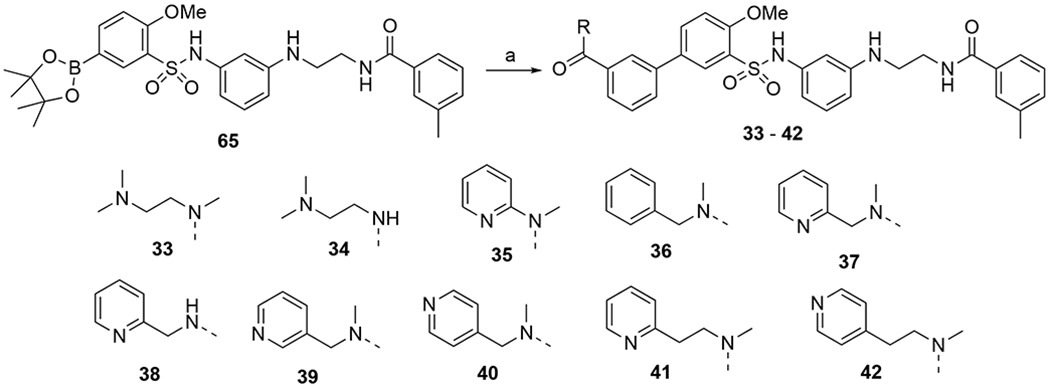
Synthesis of compounds 33-42:
Funding Sources
This work was supported by National Institute on Drug Abuse, National Institutes of Health, U.S. (Grants DA040693 to Y.Z), and the US Department of Veteran Affairs (grants 1IO1BX003004-01A2 and 1IO1BX003687-01A1 to C.M.K).
ABBREVIATIONS
- 1,2-DCE
1,2-dichloroethane
- DCM
dichloromethane
- DME
1,2-dimethoxyethane
- DOPC
1,2-Dioleoyl-sn-glycero-3-phosphocholine
- EC50
Half maximal effective concentration
- FLIPR
fluorometric imaging plate reader
- GPCR
G protein-coupled receptor
- HPLC
high performance liquid chromatography
- KCl
potassium chloride
- MD
molecular dynamics
- MS
mass spectrometry
- NMR
nuclear magnetic resonance
- OX1R
orexin-1 receptor
- OX2R
orexin-2 receptor
- RMSD
root mean square deviation
- SAR
structure–activity relationship
- TLC
thin-layer chromatography
Footnotes
The authors declare no competing financial interest.
Supporting Information
HPLC analysis results of target compounds and summary table of molecular formula strings with biological data. The Supporting Information is available free of charge on the ACS Publications website.
REFERENCES
- 1.de Lecea L; Kilduff TS; Peyron C; Gao X; Foye PE; Danielson PE; Fukuhara C; Battenberg ELF; Gautvik VT; Bartlett FS, 2nd; Frankel WN; van den Pol AN; Bloom FE; Gautvik KM; Sutcliffe JG, The hypocretins: Hypothalamus-specific peptides with neuroexcitatory activity. Proc Natl Acad Sci U S A 1998, 95 (1), 322–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sakurai T; Amemiya A; Ishii M; Matsuzaki I; Chemelli RM; Tanaka H; Williams SC; Richardson JA; Kozlowski GP; Wilson S; Arch JR; Buckingham RE; Haynes AC; Carr SA; Annan RS; McNulty DE; Liu WS; Terrett JA; Elshourbagy NA; Bergsma DJ; Yanagisawa M, Orexins and orexin receptors: A family of hypothalamic neuropeptides and g protein-coupled receptors that regulate feeding behavior. Cell 1998, 92 (4), 573–585. [DOI] [PubMed] [Google Scholar]
- 3.Peyron C; Tighe DK; van den Pol AN; de Lecea L; Heller HC; Sutcliffe JG; Kilduff TS, Neurons containing hypocretin (orexin) project to multiple neuronal systems. J Neurosci 1998, 18 (23), 9996–10015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Date Y; Ueta Y; Yamashita H; Yamaguchi H; Matsukura S; Kangawa K; Sakurai T; Yanagisawa M; Nakazato M, Orexins, orexigenic hypothalamic peptides, interact with autonomic, neuroendocrine and neuroregulatory systems. Proc Natl Acad Sci U S A 1999, 96 (2), 748–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nambu T; Sakurai T; Mizukami K; Hosoya Y; Yanagisawa M; Goto K, Distribution of orexin neurons in the adult rat brain. Brain Res 1999, 827 (1–2), 243–260. [DOI] [PubMed] [Google Scholar]
- 6.Sutcliffe JG; de Lecea L, The hypocretins: Setting the arousal threshold. Nat Rev Neurosci 2002, 3 (5), 339–349. [DOI] [PubMed] [Google Scholar]
- 7.Sakurai T, Roles of orexin/hypocretin in regulation of sleep/wakefulness and energy homeostasis. Sleep Med Rev 2005, 9 (4), 231–241. [DOI] [PubMed] [Google Scholar]
- 8.de Lecea L; Sutcliffe JG, The hypocretins and sleep. FEBS J 2005, 272 (22), 5675–5688. [DOI] [PubMed] [Google Scholar]
- 9.Kilduff TS; Peyron C, The hypocretin/orexin ligand-receptor system: Implications for sleep and sleep disorders. Trends Neurosci 2000, 23 (8), 359–365. [DOI] [PubMed] [Google Scholar]
- 10.Ohno K; Sakurai T, Orexin neuronal circuitry: Role in the regulation of sleep and wakefulness. Front Neuroendocrinol 2008, 29 (1), 70–87. [DOI] [PubMed] [Google Scholar]
- 11.Adamantidis AR; Zhang F; Aravanis AM; Deisseroth K; de Lecea L, Neural substrates of awakening probed with optogenetic control of hypocretin neurons. Nature 2007, 450 (7168), 420–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.de Lecea L, Optogenetic control of hypocretin (orexin) neurons and arousal circuits. Curr Top Behav Neurosci 2015, 25, 367–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Furlong TM; Vianna DM; Liu L; Carrive P, Hypocretin/orexin contributes to the expression of some but not all forms of stress and arousal. Eur J Neurosci 2009, 30 (8), 1603–1614. [DOI] [PubMed] [Google Scholar]
- 14.Johnson PL; Truitt W; Fitz SD; Minick PE; Dietrich A; Sanghani S; Traskman-Bendz L; Goddard AW; Brundin L; Shekhar A, A key role for orexin in panic anxiety. Nat Med 2010, 16 (1), 111–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Johnson PL; Federici LM; Fitz SD; Renger JJ; Shireman B; Winrow CJ; Bonaventure P; Shekhar A, Orexin 1 and 2 receptor involvement in co2 -induced panic-associated behavior and autonomic responses. Depress Anxiety 2015, 32 (9), 671–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bonaventure P; Yun S; Johnson PL; Shekhar A; Fitz SD; Shireman BT; Lebold TP; Nepomuceno D; Lord B; Wennerholm M; Shelton J; Carruthers N; Lovenberg T; Dugovic C, A selective orexin-1 receptor antagonist attenuates stress-induced hyperarousal without hypnotic effects. J Pharmacol Exp Ther 2015, 352 (3), 590–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kessler BA; Stanley EM; Frederick-Duus D; Fadel J, Age-related loss of orexin/hypocretin neurons. Neuroscience 2011, 178, 82–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nixon JP; Mavanji V; Butterick TA; Billington CJ; Kotz CM; Teske JA, Sleep disorders, obesity, and aging: The role of orexin. Ageing Res Rev 2015, 20, 63–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang L; Zou B; Xiong X; Pascual C; Xie J; Malik A; Xie J; Sakurai T; Xie XS, Hypocretin/orexin neurons contribute to hippocampus-dependent social memory and synaptic plasticity in mice. J Neurosci 2013, 33 (12), 5275–5284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mavanji V; Butterick TA; Duffy CM; Nixon JP; Billington CJ; Kotz CM, Orexin/hypocretin treatment restores hippocampal-dependent memory in orexin-deficient mice. Neurobiol Learn Mem 2017, 146, 21–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mahoney CE; Cogswell A; Koralnik IJ; Scammell TE, The neurobiological basis of narcolepsy. Nat Rev Neurosci 2019, 20 (2), 83–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nishino S; Ripley B; Overeem S; Lammers GJ; Mignot E, Hypocretin (orexin) deficiency in human narcolepsy. Lancet 2000, 355 (9197), 39–40. [DOI] [PubMed] [Google Scholar]
- 23.Peyron C; Faraco J; Rogers W; Ripley B; Overeem S; Charnay Y; Nevsimalova S; Aldrich M; Reynolds D; Albin R; Li R; Hungs M; Pedrazzoli M; Padigaru M; Kucherlapati M; Fan J; Maki R; Lammers GJ; Bouras C; Kucherlapati R; Nishino S; Mignot E, A mutation in a case of early onset narcolepsy and a generalized absence of hypocretin peptides in human narcoleptic brains. Nature Medicine 2000, 6 (9), 991–997. [DOI] [PubMed] [Google Scholar]
- 24.Thannickal TC; Moore RY; Nienhuis R; Ramanathan L; Gulyani S; Aldrich M; Cornford M; Siegel JM, Reduced number of hypocretin neurons in human narcolepsy. Neuron 2000, 27 (3), 469–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mavanji V; Perez-Leighton CE; Kotz CM; Billington CJ; Parthasarathy S; Sinton CM; Teske JA, Promotion of wakefulness and energy expenditure by orexin-a in the ventrolateral preoptic area. Sleep 2015, 38 (9), 1361–1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tsujino N; Sakurai T, Role of orexin in modulating arousal, feeding, and motivation. Front Behav Neurosci 2013, 7, 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fadel JR; Jolivalt CG; Reagan LP, Food for thought: The role of appetitive peptides in age-related cognitive decline. Ageing Res Rev 2013, 12 (3), 764–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Davies J; Chen J; Pink R; Carter D; Saunders N; Sotiriadis G; Bai B; Pan Y; Howlett D; Payne A; Randeva H; Karteris E, Orexin receptors exert a neuroprotective effect in alzheimer’s disease (ad) via heterodimerization with gpr103. Sci Rep 2015, 5, 12584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fronczek R; van Geest S; Frolich M; Overeem S; Roelandse FW; Lammers GJ; Swaab DF, Hypocretin (orexin) loss in alzheimer’s disease. Neurobiol Aging 2012, 33 (8), 1642–1650. [DOI] [PubMed] [Google Scholar]
- 30.Fronczek R; Overeem S; Lee SY; Hegeman IM; van Pelt J; van Duinen SG; Lammers GJ; Swaab DF, Hypocretin (orexin) loss in parkinson’s disease. Brain 2007, 130 (Pt 6), 1577–1585. [DOI] [PubMed] [Google Scholar]
- 31.Thannickal TC; Lai YY; Siegel JM, Hypocretin (orexin) cell loss in parkinson’s disease. Brain 2007, 130 (Pt 6), 1586–1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Thannickal TC; Lai YY; Siegel JM, Hypocretin (orexin) and melanin concentrating hormone loss and the symptoms of parkinson’s disease. Brain 2008, 131 (Pt 1), e87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chow M; Cao M, The hypocretin/orexin system in sleep disorders: Preclinical insights and clinical progress. Nat Sci Sleep 2016, 8, 81–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mieda M; Sakurai T, Orexin (hypocretin) receptor agonists and antagonists for treatment of sleep disorders. Rationale for development and current status. CNS Drugs 2013, 27 (2), 83–90. [DOI] [PubMed] [Google Scholar]
- 35.Lebold TP; Bonaventure P; Shireman BT, Selective orexin receptor antagonists. Bioorganic and Medicinal Chemistry Letters 2013, 23 (17), 4761–4769. [DOI] [PubMed] [Google Scholar]
- 36.Perrey DA; Zhang Y, Therapeutics development for addiction: Orexin-1 receptor antagonists. Brain Res 2020, 1731, 145922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Andrews SP; Aves SJ; Christopher JA; Nonoo R, Orexin receptor antagonists: Historical perspectives and future opportunities. Curr Top Med Chem 2016, 16 (29), 3438–3469. [DOI] [PubMed] [Google Scholar]
- 38.Boss C; Roch C, Orexin research: Patent news from 2016. Expert Opin Ther Pat 2017, 27 (10), 1123–1133. [DOI] [PubMed] [Google Scholar]
- 39.Roecker AJ; Cox CD; Coleman PJ, Orexin receptor antagonists: New therapeutic agents for the treatment of insomnia. J Med Chem 2016, 59 (2), 504–530. [DOI] [PubMed] [Google Scholar]
- 40.Boutrel B; Kenny PJ; Specio SE; Martin-Fardon R; Markou A; Koob GF; de Lecea L, Role for hypocretin in mediating stress-induced reinstatement of cocaine-seeking behavior. Proc Natl Acad Sci U S A 2005, 102 (52), 19168–19173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.James MH; Mahler SV; Moorman DE; Aston-Jones G, A decade of orexin/hypocretin and addiction: Where are we now? Curr Top Behav Neurosci 2017, 33, 247–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bonaventure P; Dugovic C; Shireman B; Preville C; Yun S; Lord B; Nepomuceno D; Wennerholm M; Lovenberg T; Carruthers N; Fitz SD; Shekhar A; Johnson PL, Evaluation of jnj-54717793 a novel brain penetrant selective orexin 1 receptor antagonist in two rat models of panic attack provocation. Front Pharmacol 2017, 8, 357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yangisawa M Small-molecule agonists for type-2 orexin receptor. Us 2010/0150840 a1. 2010. [Google Scholar]
- 44.Turku A; Rinne MK; Boije Af Gennas G; Xhaard H; Lindholm D; Kukkonen JP, Orexin receptor agonist yan 7874 is a weak agonist of orexin/hypocretin receptors and shows orexin receptor-independent cytotoxicity. PLoS One 2017, 12 (6), e0178526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nagahara T; Saitoh T; Kutsumura N; Irukayama-Tomobe Y; Ogawa Y; Kuroda D; Gouda H; Kumagai H; Fujii H; Yanagisawa M; Nagase H, Design and synthesis of non-peptide, selective orexin receptor 2 agonists. J Med Chem 2015, 58 (20), 7931–7937. [DOI] [PubMed] [Google Scholar]
- 46.Irukayama-Tomobe Y; Ogawa Y; Tominaga H; Ishikawa Y; Hosokawa N; Ambai S; Kawabe Y; Uchida S; Nakajima R; Saitoh T; Kanda T; Vogt K; Sakurai T; Nagase H; Yanagisawa M, Nonpeptide orexin type-2 receptor agonist ameliorates narcolepsy-cataplexy symptoms in mouse models. Proc Natl Acad Sci U S A 2017, 114 (22), 5731–5736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Toyama S; Shimoyama N; Tagaito Y; Nagase H; Saitoh T; Yanagisawa M; Shimoyama M, Nonpeptide orexin-2 receptor agonist attenuates morphine-induced sedative effects in rats. Anesthesiology 2018, 128 (5), 992–1003. [DOI] [PubMed] [Google Scholar]
- 48.Yukitake H; Fujimoto T; Ishikawa T; Suzuki A; Shimizu Y; Rikimaru K; Ito M; Suzuki M; Kimura H, Tak-925, an orexin 2 receptor-selective agonist, shows robust wake-promoting effects in mice. Pharmacol Biochem Behav 2019, 187, 172794. [DOI] [PubMed] [Google Scholar]
- 49.Bogen SL; Clausen DJ; Guiadeen DG; rudd MT; Yang D 5-alkyl pyrrolidine orexin receptor agonists. US 2020/0255403A1, 2020. [Google Scholar]
- 50.Hong C; Byrne NJ; Zamlynny B; Tummala S; Xiao L; Shipman JM; Partridge AT; Minnick C; Breslin MJ; Rudd MT; Stachel SJ; Rada VL; Kern JC; Armacost KA; Hollingsworth SA; O’Brien JA; Hall DL; McDonald TP; Strickland C; Brooun A; Soisson SM; Hollenstein K, Structures of active-state orexin receptor 2 rationalize peptide and small-molecule agonist recognition and receptor activation. Nat Commun 2021, 12 (1), 815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Marcus JN; Aschkenasi CJ; Lee CE; Chemelli RM; Saper CB; Yanagisawa M; Elmquist JK, Differential expression of orexin receptors 1 and 2 in the rat brain. J Comp Neurol 2001, 435 (1), 6–25. [DOI] [PubMed] [Google Scholar]
- 52.Lu J; Bjorkum AA; Xu M; Gaus SE; Shiromani PJ; Saper CB, Selective activation of the extended ventrolateral preoptic nucleus during rapid eye movement sleep. J Neurosci 2002, 22 (11), 4568–4576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nixon JP; Mavanji V; Butterick TA; Billington CJ; Kotz CM; Teske JA, Sleep disorders, obesity, and aging: The role of orexin. Ageing research reviews 2015, 20, 63–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mieda M; Hasegawa E; Kisanuki YY; Sinton CM; Yanagisawa M; Sakurai T, Differential roles of orexin receptor-1 and −2 in the regulation of non-rem and rem sleep. J Neurosci 2011, 31 (17), 6518–6526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Willie JT; Chemelli RM; Sinton CM; Tokita S; Williams SC; Kisanuki YY; Marcus JN; Lee C; Elmquist JK; Kohlmeier KA; Leonard CS; Richardson JA; Hammer RE; Yanagisawa M, Distinct narcolepsy syndromes in orexin receptor-2 and orexin null mice: Molecular genetic dissection of non-rem and rem sleep regulatory processes. Neuron 2003, 38 (5), 715–730. [DOI] [PubMed] [Google Scholar]
- 56.Chemelli RM; Willie JT; Sinton CM; Elmquist JK; Scammell T; Lee C; Richardson JA; Williams SC; Xiong Y; Kisanuki Y; Fitch TE; Nakazato M; Hammer RE; Saper CB; Yanagisawa M, Narcolepsy in orexin knockout mice: Molecular genetics of sleep regulation. Cell 1999, 98 (4), 437–451. [DOI] [PubMed] [Google Scholar]
- 57.Willie JT; Chemelli RM; Sinton CM; Yanagisawa M, To eat or to sleep? Orexin in the regulation of feeding and wakefulness. Annu Rev Neurosci 2001, 24, 429–458. [DOI] [PubMed] [Google Scholar]
- 58.Gotter AL; Forman MS; Harrell CM; Stevens J; Svetnik V; Yee KL; Li X; Roecker AJ; Fox SV; Tannenbaum PL; Garson SL; Lepeleire ID; Calder N; Rosen L; Struyk A; Coleman PJ; Herring WJ; Renger JJ; Winrow CJ, Orexin 2 receptor antagonism is sufficient to promote nrem and rem sleep from mouse to man. Sci Rep 2016, 6, 27147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Winrow CJ; Gotter AL; Cox CD; Doran SM; Tannenbaum PL; Breslin MJ; Garson SL; Fox SV; Harrell CM; Stevens J; Reiss DR; Cui D; Coleman PJ; Renger JJ, Promotion of sleep by suvorexant-a novel dual orexin receptor antagonist. J Neurogenet 2011, 25 (1–2), 52–61. [DOI] [PubMed] [Google Scholar]
- 60.Morairty SR; Revel FG; Malherbe P; Moreau JL; Valladao D; Wettstein JG; Kilduff TS; Borroni E, Dual hypocretin receptor antagonism is more effective for sleep promotion than antagonism of either receptor alone. PLoS One 2012, 7 (7), e39131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.German NA; Decker AM; Gilmour BP; Thomas BF; Zhang Y, Truncated orexin peptides: Structure-activity relationship studies. ACS Med Chem Lett 2013, 4 (12), 1224–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Webb B; Sali A, Protein structure modeling with modeller. Methods Mol Biol 2017, 1654, 39–54. [DOI] [PubMed] [Google Scholar]
- 63.Kolossvary I; Keseru G, Hessian-free low-mode conformational search for large-scale protein loop optimization: Application to c-jun n-terminal kinase jnk3. J. Comput. Chem. 2001, 22 (1), 21–30. [Google Scholar]
- 64.Case DA; Ben-Shalom IY; Brozell SR; Cerutti DS; T.E. Cheatham I; Cruzeiro VWD; Darden TA; Duke RE; Ghoreishi D; Gilson MK; Goetz HGW; Greene D; Harris R; Homeyer N; Izadi S; Kovalenko A; Kurtzman T; Lee TS; LeGrand S; Li P; Lin C; Liu J; Luchko T; Luo R; Mermelstein DJ; Merz KM; Miao Y; Monard f.; Nguyen C; Nguyen H; Omelyan I; Onufriev A; Pan F; Qi R; Roe DR; Roitberg A; Sagui C; Schott-Verdugo S; Shen J; Simmerling CL; Smith J; Salomon-Ferrer R; Swails J; Walker RC; Wang J; Wei H; Wolf RM; Wu X; Xiao L; York DM; Kollman PA Amber 2018, 2018. [Google Scholar]
- 65.Case DA, Cerutti RMB,DS, Cheatham TE III, Darden TA, Duke RE, Giese TJ, Gohlke H, Goetz AW, Homeyer N, Izadi S, Janowski P, Kaus J, Kovalenko A, Lee TS, LeGrand S, Li P, Lin C, Luchko T, Luo R Madej B, Mermelstein D, Merz KM, Monard G, Nguyen H, Nguyen HT, Omelyan I, Onufriev A, Roe DR, Roitberg A, Sagui C, Simmerling CL, Botello-Smith WM, Swails J, Walker RC, Wang J, Wolf RM, Wu X, Xiao Land Kollman PA (2016), AMBER 2016, University of California, San Francisco. Amber 2016, UCSF, San Francisco, 2016. [Google Scholar]
- 66.Dickson CJ; Madej BD; Skjevik AA; Betz RM; Teigen K; Gould IR; Walker RC, Lipid14: The amber lipid force field. J Chem Theory Comput 2014, 10 (2), 865–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Amato GS; Manke A; Harris DL; Wiethe RW; Vasukuttan V; Snyder RW; Lefever TW; Cortes R; Zhang Y; Wang S; Runyon SP; Maitra R, Blocking alcoholic steatosis in mice with a peripherally restricted purine antagonist of the type 1 cannabinoid receptor. J Med Chem 2018, 61 (10), 4370–4385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Friesner RA; Murphy RB; Repasky MP; Frye LL; Greenwood JR; Halgren TA; Sanschagrin PC; Mainz DT, Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J Med Chem 2006, 49 (21), 6177–6196. [DOI] [PubMed] [Google Scholar]
- 69.Sherman W; Day T; Jacobson MP; Friesner RA; Farid R, Novel procedure for modeling ligand/receptor induced fit effects. J Med Chem 2006, 49 (2), 534–553. [DOI] [PubMed] [Google Scholar]
- 70.Yin J; Mobarec JC; Kolb P; Rosenbaum DM, Crystal structure of the human ox2 orexin receptor bound to the insomnia drug suvorexant. Nature 2015, 519 (7542), 247–250. [DOI] [PubMed] [Google Scholar]
- 71.Suno R; Kimura KT; Nakane T; Yamashita K; Wang J; Fujiwara T; Yamanaka Y; Im D; Horita S; Tsujimoto H; Tawaramoto MS; Hirokawa T; Nango E; Tono K; Kameshima T; Hatsui T; Joti Y; Yabashi M; Shimamoto K; Yamamoto M; Rosenbaum DM; Iwata S; Shimamura T; Kobayashi T, Crystal structures of human orexin 2 receptor bound to the subtype-selective antagonist empa. Structure 2018, 26 (1), 7–19 e15. [DOI] [PubMed] [Google Scholar]
- 72.Sakurai T, The neural circuit of orexin (hypocretin): Maintaining sleep and wakefulness. Nat Rev Neurosci 2007, 8 (3), 171–181. [DOI] [PubMed] [Google Scholar]
- 73.Takenoshita S; Sakai N; Chiba Y; Matsumura M; Yamaguchi M; Nishino S, An overview of hypocretin based therapy in narcolepsy. Expert Opin Investig Drugs 2018, 27 (4), 389–406. [DOI] [PubMed] [Google Scholar]
- 74.Seigneur E; de Lecea L, Hypocretin (orexin) replacement therapies. Medicine in Drug Discovery In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sievers F; Higgins DG, Clustal omega for making accurate alignments of many protein sequences. Protein Sci 2018, 27 (1), 135–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wang Q; Canutescu AA; Dunbrack RL Jr., Scwrl and molide: Computer programs for side-chain conformation prediction and homology modeling. Nat Protoc 2008, 3 (12), 1832–1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lee TS; Cerutti DS; Mermelstein D; Lin C; LeGrand S; Giese TJ; Roitberg A; Case DA; Walker RC; York DM, Gpu-accelerated molecular dynamics and free energy methods in amber18: Performance enhancements and new features. J Chem Inf Model 2018, 58 (10), 2043–2050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Watts KS; Dalal P; Murphy RB; Sherman W; Friesner RA; Shelley JC, Confgen: A conformational search method for efficient generation of bioactive conformers. J Chem Inf Model 2010, 50 (4), 534–546. [DOI] [PubMed] [Google Scholar]
- 79.Miller EB; Murphy RB; Sindhikara D; Borrelli KW; Grisewood MJ; Ranalli F; Dixon SL; Jerome S; Boyles NA; Day T; Ghanakota P; Mondal S; Rafi SB; Troast DM; Abel R; Friesner RA, Reliable and accurate solution to the induced fit docking problem for protein-ligand binding. J Chem Theory Comput 2021, 17 (4), 2630–2639. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.