

Graphical abstract
Binding energies and dissociation constants of sartans bound to RBD/ACE2 complex.
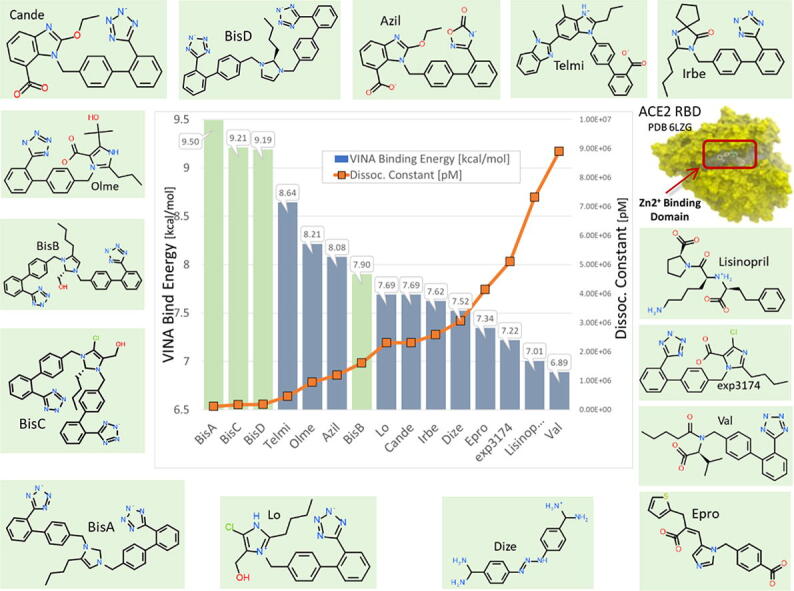
Abbreviations: Cande, Candesartan; Telmi, Telmisartan; Olme, Olmesartan; Azil, Azilsartan; Lo, Losartan; Irbe, Irbesartan; DIZE, deminazene aceturate; Epro, Eprosartan; EXP3174, losartan carboxylic acid; Val, Valsartan; BisA, 4-Butyl-N,N0-bis{[[20-(2H-tetrazol-5-yl)]biphenyl-4-yl] methyl}imidazolium bromide; BisB, 4-Butyl-2-hydroxymethyl-N,N0-bis{[20-(2H-tetrazol-5-yl)- biphenyl-4-yl]methyimidazolium bromide; BisC, 2-Butyl-4-chloro-5-hydroxymethyl-N,N0-bis{[20-(2H-tetrazol- 5-yl)biphenyl-4-yl]methyl}imidazolium bromide; BisD, 2-Butyl-N,N0-bis{[20-(2H-tetrazol-5-yl)biphenyl-4-yl]methyl} imidazolium bromide
Keywords: Angiotensin receptors, SARS-CoV-2 spike/ACE2 complex blockers, Sartans, Bisartans tetrazole, Docking RBD/ACE2 studies, Animal AT1 receptor studies, Bisartan NMR studies
Abstract
The discovery and facile synthesis of a new class of sartan-like arterial antihypertensive drugs (angiotensin receptor blockers [ARBs]), subsequently referred to as “bisartans” is reported. In vivo results and complementary molecular modelling presented in this communication indicate bisartans may be beneficial for the treatment of not only heart disease, diabetes, renal dysfunction, and related illnesses, but possibly COVID-19. Bisartans are novel bis-alkylated imidazole sartan derivatives bearing dual symmetric anionic biphenyl tetrazole moieties. In silico docking and molecular dynamics studies revealed bisartans exhibited higher binding affinities for the ACE2/spike protein complex (PDB 6LZG) compared to all other known sartans. They also underwent stable docking to the Zn2+ domain of the ACE2 catalytic site as well as the critical interfacial region between ACE2 and the SARS-CoV-2 receptor binding domain. Additionally, semi-stable docking of bisartans at the arginine-rich furin-cleavage site of the SARS-CoV-2 spike protein (residues 681–686) required for virus entry into host cells, suggest bisartans may inhibit furin action thereby retarding viral entry into host cells. Bisartan tetrazole groups surpass nitrile, the pharmacophoric “warhead” of PF-07321332, in its ability to disrupt the cysteine charge relay system of 3CLpro. However, despite the apparent targeting of multifunctional sites, bisartans do not inhibit SARS-CoV-2 infection in bioassays as effectively as PF-07321332 (Paxlovid).
1. Introduction
This study has been undertaken with the knowledge that cardiovascular disease is related to COVID-19 in terms of mechanisms that trigger the disease [1]. The storm of cytokines released in COVID-19 patients with pneumonia is related to the over-expression of toxic angiotensin II (ANG II) in the renin-angiotensin system (RAS) and clinical studies have shown that morbidity and mortality rate was lower in hypertensive patients infected by SARS-CoV-2, who are taking angiotensin-converting enzyme inhibitors and angiotensin II receptor blockers (ARBs) compared to patients not taking these drugs. ANG II, the major RAS component, and SARS-CoV-2 spike protein cleavage by furin and 3CLpro that initiate infection operate through similar charge relay system (CRS) mechanisms [2]. Tyrosinate in ANG II, serinate in furin, and cysteinate in 3CLpro are anions created through the CRS mechanism and trigger activity via their nucleophile anions. Bisartans bearing two biphenyl tetrazole moieties were found in in silico studies to be stronger inhibitors compared to current sartans [3]. Similar CRS mechanisms spark and mediate the action of ANG II [4] and the serine-like proteases furin and 3CL [2].
Two states of the angiotensin type 1 (AT1) receptor, agonist and inverse agonist states, interact, respectively, with surmountable and insurmountable ANG II non-peptide angiotensin receptor blockers (ARBs). Neutralization of tyrosinate in the ANG II analog [Sar1Ile8]ANG II (sarilesin) (by methylation) or the conversion of hydroxymethyl group to carboxylate in ARBs (e.g. losartan to EXP3174), converts inverse agonist to a surmountable antagonist in both cases, thus illustrating equivalent roles for peptide tyrosinate and ARB carboxylate at angiotensin receptors. The behavioural bias of agonists/surmountable antagonists versus [insurmountable] inverse agonists appears to depend on the nature of the bond (i.e., ion:dipole versus salt bridge) that forms with Arg167 of the receptor. These insights have provided the foundation for a new class of ARBs called bisartans with bis-alkylated imidazole bearing two tetrazoles moieties essential for binding with arginines of the Delta mutations of SARS-CoV-2. This class may be beneficial in the treatment of SARS-CoV-2 infection as it has the most suitable structural requirements between AT1 antagonists.
Biased agonism at ANG II receptors: The interaction of ANG II with its G protein-coupled AT1 receptor in isolated tissues is characterized by positive cooperativity (Hill coefficient, nH > 1 < 2), indicative of a receptor dimerization mechanism for amplifying the contractile response. Blocking of receptors by [Sar1Ile8]ANG II (sarilesin) invokes negative cooperativity (nH < 1), indicative of inverse agonism. The knowledge of the site-specific mutations (SSMs) in the AT1 receptor has demonstrated effects on receptor bioactivity. Furthermore, crystallography data investigating interactions between AT1 receptor bound to nonpeptide receptor blockers (ARB) illustrates that the inverse agonism of ARBs derives primarily from a salt bridge between ARB carboxylate and Arg167 guanidino on the receptor. This locks the receptor into an inverse agonist state mediated by an alternative second messenger, which is slow to reverse. Neutralization of tyrosinate in sarilesin (by methylation) or the oxidation of hydroxymethyl group of Losartan to carboxylate to derive EXP3174 converts inverse agonists to surmountable antagonists in both cases. These simple chemical modifications highlight equivalent roles for peptide tyrosinate and ARB carboxylate at angiotensin receptors. A CRS (composed of Tyr--His--carboxyl) in ANG II, which creates the bioactive tyrosinate anion essential for agonist [and inverse agonist] activity, may activate the receptor by CRS exchange of the Tyr [4] of ANG II with Tyr35 of the receptor. The behavioural bias of agonists and surmountable antagonists versus inverse agonists ultimately depends on the nature of the bond (i.e., ion:dipole bond versus salt bridge) that ANG II forms with R167 of the receptor. Screening of the human genome has identified a peptide (EGVYVHPV) encoded by mRNA complementary to that encoding ANG II itself, which is a naturally occurring inverse agonist [5]. Thus, opposite strands of DNA encode peptides with opposite effects of AT1 receptors.
This study describes the discovery and the in silico effects of a new generation of sartans called bisartans bearing two anionic biphenyl tetrazole moieties, which bind more strongly to the AT1 receptor compared to all eight currently available sartans. Bisartans, discovered in our laboratories, are classified as bisartans A and B where the aliphatic butyl group is located at position 4 of the imidazole ring, and bisartans C and D with the butyl group attached at position 2 as in losartan. The ability of these bisartans to interact with various strategic components of the SARS-CoV-2 viral infection process (ACE2, spike protein RBD, furin enzyme, 3CL processing enzyme) were investigated in silico, and compared to bioactivity in COVID-19 assays. Molecular docking studies (Fig. 7) reveal binding of bisartans with arginines in the SARS-Cov-2 spike protein furin cleavage site (residues 681–686), suggesting their potential as inhibitors in the treatment of COVID-19. Docking studies of bisartan A (butyl group at position 4) in the delta mutation (P681R) of spike furin cleavage site RRRAR-S (residues 681–686) resulted in a stronger binding (energy: 6,5 kcal/mol) compared to the wild type site, PRRAR-S (5,5 kcal/mol). The two negatively charged bisartan A tetrazoles make contact with positively charged residues Arg681, Arg682, Arg683, and His655. These docking studies reveal an increased number of arginines and histidines as residue contacts in the bisartan/receptor-binding domain (RBD)/angiotensin-converting enzyme-2 (ACE2) complex compared to the sartans/RBD/ACE2 complex. Coordination of ACE2 Zn2+ with ANG II and mutants involving four residues Asp (D), Tyr (Y), His (H), and the C-terminus carboxylate has been recently reported [6] in line with the coordination of spike/ACE2 Zn2+ with bisartans shown in our studies. We sought and identified new ANG II inhibitors that possess a high binding affinity to ACE2 using computational methods. Bisartans A, B, C, and D bear two acidic tetrazoles, which are isosteres of carboxylates, are able to inhibit the interaction between the spike protein of the virus and ACE2 without abolishing the ability of the enzyme to reduce ANG II levels, a beneficial factor in the RAS to maintain homeostasis and equilibrium of RAS components. Bisartans were found to be stronger binders to the RBD/ACE2 complex compared to current sartans and represent a promising new class of synthetic and cost-effective bioactive antihypertensive drugs that concomitantly act at multiple sites required for SARS-CoV-2 infection, with particular relevance for the Delta variant and furin cleavage site, both arginine-rich.
Fig. 7.

Docking of bisartans and selected common antiviral drugs to the proteo-catalytic Cys145 site of the main 3CLpro SARS-CoV-2 protease (AutoDock VINA results). BisA, B, C, and D are imidazoles containing anionic dual branching biphenyl tetrazole rings. PF-07331332 is the active antiviral component of Pfizer’s Paxlovid agent. All of the bisartan analogs (BisA-BisD) exhibited stronger binding scores compared to PF-07321332. Note that PF-07321332 remained in the non-covalently bound form for these docking exercises. Results of docking of selected antiviral ligands to the catalytic pocket of the 3CLpro main protease of SARS-CoV-2.
Furthermore, bisartans may act at multiple sites [with higher affinity than existing drugs] and have advantages over antibody treatments because they don’t discriminate significantly between variants of SARS-CoV-2. Bisartan A attacks all three of the known target proteases which SARS-CoV-2 requires for cell entry (ACE2 and furin) and viral maturation (3CLpro), and is the first potent multifunctional drug which is a serial inhibitor of multiple enzymes essential for virus activity. Although bisartan has the added [and useful] quirk of increased affinity for [the arginine-rich site on the spike protein of] the delta variant, in general drugs like bisartan, which act as proteolytic inhibitors at both the host cell and viral cell level, should not discriminate meaningfully between variants - unlike antibody treatments which are highly selective for site-specific sequences on the viral spike protein. Accordingly, drugs like bisartans may prove advantageous because they are broad-spectrum [and immediate] treatments against a continuously changing SARS-CoV-2 virus. Bisartans A, B, C, D have been found in in silico studies to bind to 3CL protease and to be stable as Pfizer’s Paxlovid combination drugs (PF-07321332 and Ritonavir) where nitrile is the warhead pharmacophoric group of antiviral PF-07321332 drug [7], [8], [9], [10]. However, despite the apparent targeting of multifunctional sites uncovered in silico, Bis D nitrile does not inhibit SARS-CoV-2 infection in bioassays, whereas Bis D tetrazole invokes 20% inhibition (compared to PF-07321332 at 95%). Bisartans bearing two pharmacophoric tetrazole groups that interact with Cys145 of the catalytic dyad, His41-Cys145, make inoperable the cysteine CRS protease mechanism of spike cleavage thus preventing infection. However, despite the apparent targeting of multifunctional sites uncovered in silico, bisartans BisA (tetrazole and nitrile) and BisD (tetrazole and nitrile) do not inhibit SARS-CoV-2 infection in bioassays as effectively as PF-07321332 (Paxlovid).
2. Results
2.1. Biased agonism at angiotensin receptors
The octapeptide ANG II (DRVYIHPF), a model for hormone-receptor interactions [4], acts on the AT1 receptor in smooth muscle and other tissues, resulting in contraction and elevated blood pressure. Lowering ANG II levels with ACE1 inhibitors or direct inhibition with ARBs provides effective treatment of hypertension. ARBs demonstrated insurmountable blocking effects at AT1 receptors, which are similar to the effects seen with certain synthetic analogs of ANG II in which the C-terminal Phe is replaced by an aliphatic amino acid, such as Ile. Initially, sarilesin was designed as a Type I desensitizing antagonist, differing from competitive surmountable Type II antagonists like [Sar1Tyr(Me)4]ANG II (sarmesin). This was due to its effects being insurmountable (i.e. lasting long after washout of tissues) and reminiscent of the desensitization/tachyphylaxis caused by supramaximal doses of the agonist. However, detailed investigation of dose–response data, transformed into Hanes-Wolff and Hill plots [11], has shown that sarilesin induces negative cooperatively (Hill coefficient nH < 1), thus, signifying negative efficacy, which is synonymous with inverse agonism (Table 1B). Peptides were classified based on their activity as follows: SUPERAGONIST (Sar-Arg-Val-Tyr-Ile-His-Pro-Phe), ANTAGONIST (SARMESIN - Sar-Arg-Val-Tyr(Me)-Ile-His-Pro-Phe - Surmountable), INVERSE AGONIST (SARILESIN - Sar-Arg-Val-Tyr-Ile-His-Pro-Ile - Insurmountable) (Table 1B). Whereas, submaximal doses of ANG II resulted in positive cooperativity (nH > 1 < 2), initiating receptor dimerization with a consequent increase in affinity for ANG II [12]. Additionally, supramaximal doses of ANG II can invoke negative cooperativity and tachyphylaxis (Fig. 1B). Hill coefficients also revealed differences in tissue responsiveness to ANG II, with receptor avidity in the order aorta > portal vein > uterus (Table 1A). The prolonged effect of sarilesin does not appear to be due to slow ligand dissociation rate from the receptor, as binding-displacement curves for 125I-ANG II and 125I-[Sar1Ile8]ANG II are superimposable. The receptor may likely be locked down as a result of slow dissociation of the second messenger, which is coupled to the “inverted” state of the receptor (Fig. 1B). It is worth noting that the receptor mechanisms shown are not exclusive to angiotensin. Receptor dimerization invoking positive and negative cooperativity/efficacy (inverse agonism) considerations apply to all G protein-coupled heptahelical transmembrane receptors, including those for nonpeptide ligands. Indeed, the threshold effects and tachyphylaxis effects, commonly observed in many pharmacological assays, are symptomatic of positive and negative cooperativity, respectively.
Table 1.
A) Significance of vasoconstriction in response to cumulative doses of ANG II between control, BV6(K+)2, BV6(Na+)2, and BV6(TFA)2 incubations obtained from Fig. 2A.
| log[ANGII], M | Control vs. BV6(K+)2 | Control vs. BV6(Na+) | Control vs. BV6(TFA) |
|---|---|---|---|
| −12.0 | No significance | No significance | No significance |
| −11.5 | No significance | No significance | No significance |
| −11.0 | No significance | * p = 0.0493 | * p = 0.0186 |
| −10.5 | *** p = 0.0002 | *** p = 0.0009 | *** p = 0.0009 |
| −10.0 | **** p < 0.0001 | **** p < 0.0001 | **** p < 0.0001 |
| −9.5 | **** p < 0.0001 | **** p < 0.0001 | **** p < 0.0001 |
| −9.0 | **** p < 0.0001 | **** p < 0.0001 | **** p < 0.0001 |
| −8.5 | **** p < 0.0001 | **** p < 0.0001 | **** p < 0.0001 |
| −8.0 | **** p < 0.0001 | **** p < 0.0001 | **** p < 0.0001 |
| −7.5 | **** p < 0.0001 | **** p < 0.0001 | **** p < 0.0001 |
| −7.0 | **** p < 0.0001 | **** p < 0.0001 | **** p < 0.0001 |
| −6.5 | No significance | *** p = 0.0003 | * p = 0.0126 |
| −6.0 | No significance | * p = 0.0135 | No significance |
| −5.5 | No significance | No significance | No significance |
| −5.0 | No significance | No significance | No significance |
|
B) Hill coefficients for angiotensin contraction of rat smooth muscle tissues in the absence and presence* of sarilesin (20nM). | ||
|---|---|---|
| Tissue | Absence of [Sar1Ile8]ANG II | Presence of [Sar1Ile8]ANG II |
| Uterus | 1.4 | 0.8* |
| Portal vein | 1.7 | 0.8* |
| Aorta | 1.9 | 0.9* |
Fig. 1.
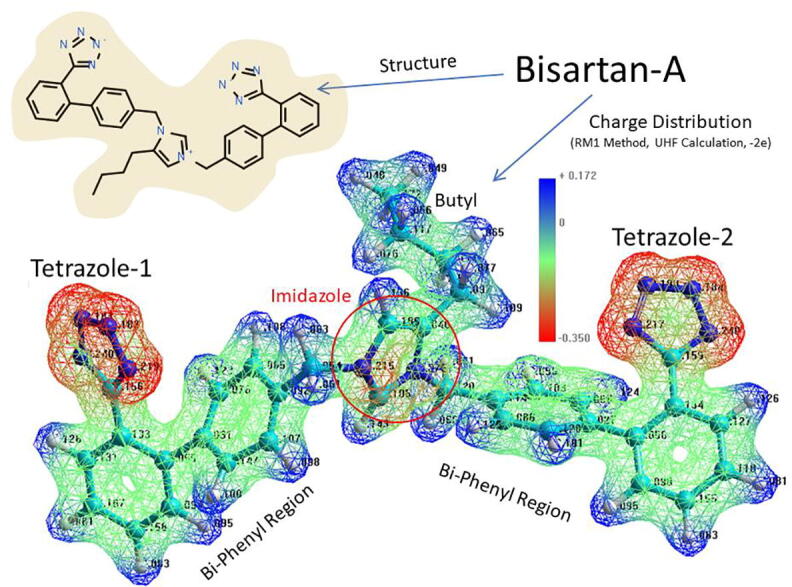
Structure (upper left) and electrostatic charge distribution (lower image) of bisartan A (BisA). Negatively charged regions = red; neutral to positive regions = green to blue. Charge distribution calculated using the RM1 semiempircal method (UHF calculation, total charge = −2.0e, geometry-optimized structure @ 0.1 kcal/mol-Å gradient).
Incredibly, the bioactivities of agonists, surmountable antagonists and insurmountable blockers (both peptides and nonpeptides), can be accounted for by an interaction with a single residue on the receptor. Thus, the quality of the bond between the ligand and the receptor Arg167 guanidino group determines the outcome, with (i) a strong salt bridge providing for insurmountable block/inverse agonism (sarilesin, or ARB with carboxylate like EXP3174); (ii) a weaker ion:dipole bond providing for surmountable antagonism (sarmesin, O-methyl-sarilesin, or ARB without carboxylate like losartan); and (iii) disrupted (exchange) bonding together with other cooperative factors (i.e. G protein binding and dimerization) leading to agonist action (ANG II). Although angiotensin may be the first peptide to have its receptor mechanism elucidated, it is not the only peptide that uses tyrosinate for signaling its receptor. A number of other “tyrosinate” hormones have been identified, including vasopressin, GnRH, and enkephalin [13]. Additionally, the presence of tyrosinate anions has been detected in oxytocin by fluorescence lifetime spectroscopy [14].
2.2. Convenient and effective synthesis of bisartans
Symmetrically bis-substituted imidazole analogs of losartan bearing at the N-1 and N-3 two biphenyl moieties ortho-substituted containing tetrazole functional groups were designed based on docking studies and synthesized by novel methods utilizing for the first time an extra hydrophobic binding cleft of AT1 receptor [3] (Fig. 1). Bisartans were synthesized by a convenient, effective, and facile method in a two-steps procedure in high yields and salt forms (K+2, N+2, TFA) and were identified by NMR methods. Two types of bisartans were synthesized and investigated, bisartans A and B with butyl group at position 4 of the imidazole ring and bisartans C and D with butyl group in position 2 as in losartan. Multiple docking studies by independent methods (including AutoDock VINA and NOVA) of the highly active bisartans clearly showed that the main core molecule containing an extra hydrophobic biphenyl binding feature, compared to prototype drug Losartan, fits to the extra hydrophobic cavity with bisartans A, C, and D, being the strongest binders (Fig. 1 and Fig. 2A). This new class of biologically active molecules synthesized through bis-alkylation of the imidazole ring by a convenient and cost-effective synthetic strategy was found highly potent in docking and animal models and constitute a new class of inhibitors that could be proven to be useful in both hypertension and COVID-19 therapeutics. In particular bisartan A, the superior spike RBD/ACE2 binder in the energy ranking of sartans, was synthesized in a high yield synthesis [3] by simultaneous bis alkylation of N-1 and N-3 imidazole nitrogens of 4(5)-butyl imidazole with alkylating reagent, 5-[4′-(Bromomethyl)-[1,1′-biphenyl]-2-yl]-2-(triphenylmethyl)-2h-tetrazole to afford trityl protected product 4-Butyl-N,N0-bis{[20-[2-(trityl)tetrazol-5-yl]biphenyl-4-yl] methyl}imidazolium bromide, which upon acidification provided the final product bisartan A (4-Butyl-N,N0-bis{[[20-(2H-tetrazol-5-yl)]biphenyl-4-yl] methyl}imidazolium bromide). Bisartan B results from hydroxymethylation of bisartan A by simple formylation at position 2 of the imidazole ring [3]. The butyl group at position 4 provides a better binding compared to counter bisartan analogs C and D with the butyl group at position 2, which is the position of the butyl group for most of the losartan-related analogs. [3] The effects of bisartans on spike RBD/ACE2 complex were related and compared with their effects on the AT1 receptor. Bisartans C, D, and B are weaker binders to RBD/ACE2 complex compared to bisartan A (Fig. 2A and Fig. 3A), since they bear at the imidazole C2 carbon substituents, hydroxyl methyl and butyl group correspondingly, which interfere between the two biphenyl tetrazole groups that bind to ACE2 or AT1 receptor, therefore, reducing their binding affinity. In a similar mode, bisartan A was found in in vitro assays (Fig. 1A) to be a stronger binder with higher affinity (−logIC50 = 9.46) and antagonist activity (pA2 = 8.45) compared to Losartan (−logIC50 = 8.25/pA2 = 8.25). Bisartan B showed lower affinity (−logIC50 = 8.37) and higher antagonist activity (pA2 = 8.58) compared to bisartan A when bound to the AT1 receptor. The lower affinity is attributed to the methylene hydroxyl group, between the two tetrazole biphenyl moieties, which increases the appropriate distance between the two tetrazoles affecting their binding affinity to the AT1 receptor. The higher antagonist activity compared to losartan is attributed to the additional negative charge provided by the hydroxylate group furthermore to the two negative charges in the two tetrazolates. Bisartan B bears three negative charges compared to ANG II, which bears two negative charges: tyrosinate and carboxylate, for approaching the AT1 receptor. Bisartans C and D were found to be very strong binders to the ARB/ACE2 complex, ranked second and third in the binding energy order after bisartan A. The two main advantages of bisartans over Sartans are the superiority in potency due to the additional tetrazole group and the facile synthesis in two steps of high yield, which render them potential low-cost drugs for treating hypertension and COVID-19.
Fig. 2.

(A) Inhibitory effect of the novel bisartan B salts BV6(K+)2, BV6(Na+)2, and BV6(TFA)2 on ANG II-induced vasoconstriction responses in rabbit iliac arteries. To determine if bisartans could mimic the effect of ARBs, rabbit iliac artery rings were incubated with BV6(K+)2, BV6(Na+)2, or BV6(TFA)2 and constricted using an ANG II dose–response. Incubation with Bisartans resulted in potent inhibition of vasoconstriction at: BV6(K+)2 doses [10−10.5 M to 10−7.0 M]; BV6(Na+)2 doses [10−11.0 M to 10−6.0 M] and BV6(TFA)2 doses [10−11.0 M to 10−6.5 M]. Data represented as meanSEM, significance represented at Table 1A. (B) Receptor switching from an agonist-induced state to a desensitized inverse agonist state. The binding of submaximal doses of ANG II to its receptor induces G protein binding and dimerization of the receptor, a concerted mechanism of positive cooperativity (Hill coefficient nH > 1), resulting in an increase in agonist affinity and consequent amplification of the contractile response. Different analogs may invoke different levels of cooperativity (increasing the affinity) when compared to other analogs (e.g. weak or partial agonists). The maximum response may be limited by the available supply of G protein, without which the mode of receptor binding of ANG II at supramaximal doses changes to inverted state and induces negative cooperativity (nH < 1) synonymous with inverse agonism (tachyphylaxis). Thus, at high doses, ANG II becomes an inverse agonist, as do other partial agonists [5]. Inverse agonists, such as ARBs, sarilesin, and angiotensin “antipeptides”, are unable to activate the receptor, and bind to inverted state forming a salt bridge with Arg167 (shown as - -X). The resulting insurmountable “inverted” state of the angiotensin receptor engenders smooth muscle relaxation (vasodilation, via an alternative second messenger) for prolonged periods. This receptor lockdown effect may be due to a slow rate of dissociation of the second messenger.
Fig. 3.

Left Panel: Results of global docking of 15 ARBs (including the 4 bisartans) to the ACE2-SARS-CoV-2/RBD complex (PDB 6LZG). The docking volume comprised a non-periodic (walled) cuboid cell with boundaries 8 Å distant from any target atom. Docking was carried out using AutoDock VINA (YAMBER3 force field; https://www.Yasara.org) with 100 trials per ARB. Best poses for the docked ARBs are superimposed in this image to show preferential binding regions (domains). Five binding domains (BDs) were observed (I-V), with most ligands occupying BD #IV. Bisartans BisB and BisC docked (though comparatively weakly) into the region (BD #III) corresponding to the interface between the ACE2 receptor and the SARS-CoV-2 RBD, suggesting these two bisartans might serve as potential antagonists of virus adsorption to host tissues expressing the ACE2 receptor. Right Panel: Rotation of the ACE2-RBD complex 90O.
2.3. Molecular docking calculations of ARBs targeting the RBD-ACE2 complex
Global docking of 15 ARBs (including the four experimental bisartans A–D) to the complex between SARS-CoV-2 spike protein RBD and ACE2 (PDB 6LZG) was performed. The molecular simulations suggested that there are five main binding domains (Fig. 3): binding domains I (located on the SARS-CoV-2 RBD) and V (ACE2) exhibited the least occupancy, while domain III (ACE2, located near to the zinc cofactor) exhibited the greatest occupancy. The dissociation constant for all ARBs bound to ACE2 revealed that the interactions between bisartan A, bisartan C, and bisartan D with ACE2 (bound to spike RBD) are stronger compared to all the other ARBs (Fig. 4). Based on the VINA docking metric the order of binding was: BisA > BisC > BisD > Telmi > Olme > Azil > BisB > Lo > Cande > Irbe > Dize > Epro > Exp3174 > Lisinopril > Val. The best-docked poses of the bisartans A and B are stabilized inside the binding cavity (domain III) of ACE2 through H-bonds, salt bridges, and π stacking contacts (Fig. 5). ACE2 residue contacts for each of the 15 ARBs and binding mechanisms of the BisA, B, C, and D ligands docked in the Zn2+ pocket of the ACE2 receptor are illustrated in Fig. 4A and B and Fig. 5. A 40-ns MD simulation of the bisartan A, B, C and D/RBD complex revealed that all bisartans remained stable inside the zinc pocket of ACE2, although Bis B, C, and D exhibited greater motion compared to BisA (Fig. 6). BisB had the largest mean RMSD value (3.2 Å), which corresponded to its lower binding strength (7.9–8.25 kcal/mol) in docking studies. It is noteworthy that bisartan BisC is also docked in the interfacial region between the ACE2 receptor and the SARS-CoV-2/RBD (Fig. 6). Interestingly, docking studies revealed a weak interaction (approximately 5.5 kcal/mol) between bisartan A and the furin cleavage site (PRRARS) of the spike protein RBD. This interaction has lower binding energy than that of bound BisA in domain III of ACE2 (10.78 kcal/mol). Despite its relatively weak interaction energy, bisartan A remained stably bound at the furin site for the initial 30 ns of a 120 ns MD simulation (data not shown). Bisartan A makes contact with arginines (R681, R682 and R683) of the cleavage motif (especially with R682 and R683 closest to the tetrazole), while the Arg (R685) is oriented in the opposite direction (Fig. 9). The mechanism of AT1-Arg167 receptor interaction with ARB tetrazole and carboxylate and ANG II is depicted in Fig. 9. The facile high yield synthesis of bisartans A, B, C, and D is depicted in Fig. 10.
Fig. 4.

Docking of 15 ARBs (including the 4 bisartans) to the zinc pocket of the SARS-CoV-2 RBD-ACE2 complex (PDB 6LZG). AutoDock VINA binding energies are illustrated in the central plot with columns. Green columns = the 4 Bisartans; blue columns = other ARBs. Dissociation constants are represented by the orange line with square markers. Based on the VINA docking metric the order of binding was: BisA > BisC > BisD > Telmi > Olme > Azil > BisB > Lo > Cande > Irbe > Dize > Epro > Exp3174 > Lisinopril > Val.
Fig. 5.

(A) Cumulative stacked line graphic depicting the relative frequency of ACE2 residue contacts for each of the 15 ARBs docked into the Zn2+ pocket of the ACE2 receptor. Dominant amino acid contacts with ARBs included Asp, Glu, His, Leu, Phe, Ser, and Trp. Residues with the least contacts included Cys, Gln, Ile, Lys, Pro, Val. Bisartans A, B, and D were the only ARBs contacting the Zn2+ cofactor. (B) 2D interaction ligand-receptor diagrams of BisA, B, C, and D in the Zn2+ pocket of the ACE2 receptor (6LZG). Colour key for chemical interactions: green shading = hydrophobic regions; Blue shading = hydrogen bond acceptor; White dashed arrows = hydrogen bonds; Grey parabolas = accessible surface for large areas; Grey residues = “generic” van der Waals contact (non-hydrophobic, non-H-bond); Broken thick line around ligand = accessible surface; Size of residue ellipse = strength of the contact; 2D distance between residue label and ligand = proximity.
Fig. 6.

(A) Docked chlorinated bisartan BisC to the interfacial region between the ACE2 receptor (Van der Waals surface; yellow) and the SARS-CoV-2/RBD (PDB 6LZG). This pose resulted from the global docking of BisC to PDB entry 6LZG using AutoDock VINA. The docking domain comprised cuboidal cells with non-periodic (wall) boundaries 8 Å from any target atom. (B) The BisC binding motif primarily involved pi/pi (red lines), pi/cation (blue lines), and hydrogen bonding (thick dashed lines) interactions with the SARS-CoV-2 RBD residues Tyr505, Arg403, Phe456, and Tyr421 (yellow spheres). The binding of BisC to the ACE2 interfacial region was mainly dominated by hydrophobic interactions (green lines) to Pro389, Leu29, Val93, Lys26, and Asp30; and secondarily by pi/cation interactions (blue lines) to Arg393. (C) The bound BisC molecule was moderately stable and remained bound inside the interfacial zone (ave. RMSD = 2.46 Å) in MD simulations run out to at least 41 ns (NPT ensemble, 0.9% saline, 311°K). The green shading indicates the time period over which the RMSD values were calculated. (D) Upper panel: Frame captures from MD simulations of bisartan-A, B, C, D/ACE2 complexes. Bound bisartans are indicated by red spheres. MD simulations were run with periodic boundaries for approximately 40 ns at 311oK in physiological saline (water and NaCl ions are hidden for clarity). All bisartans remained stable inside the zinc pocket of ACE2, although Bis B, C, and D exhibited greater motion (RMSD[ave] = 3.2 Å, 2.1 Å and 2.6 Å respectively) compared to BisA (RMSD[ave] = 1.22 Å). ACE2 molecular surfaces are shown. Lower panel: RMSD values as a function of MD simulation time. The stability of the complexes is retained for at least 40 ns.
Fig. 9.

Comparison the docking behaviour of the hypothetical compound BisNitrile against bisartans A-D, Ombitavir and PF-07321332. Docking was carried out using AutoDock VINA (500 runs per ligand) targeting the 3CLpro catalytic pocket of SARS-CoV-2 (PDB 6Y2F).
Fig. 10.

(A) Binding of bisartan B to ACE2 open channel near to zinc-binding motif. The highlighted interactions are: i) π stacking between Tyr202 and an aromatic group of bisartan, ii) H-bond between OH group of Tyr202 and N atom of bisartan and iii) salt bridges between the second tetrazolate with Arg514. The distance between the second tetrazole and Lys 562 is greater than 5.5 A. It is difficult to support any contact between them. (B) Binding of bisartan A by two Arginines in wild type furin cleavage site (PRRARS) of the spike protein receptor-binding domain. One tetrazole interacts with two Arg residues (R682 and R683 which is closest to the tetrazole) located in the cleavage site cavity. The third Arg (R685) is oriented in the opposite direction. (C) Binding of bisartan A by three arginines in P681R mutated furin cleavage site (RRRARS) of the spike protein-binding domain. One tetrazole interacts with the three arginine residues R681, R682, and R683, and the second tetrazole with His655. (D) Structures of ARBs Losartan (surmountable) and EXP3174 (insurmountable). One-dimensional graphs and (E) CRS in ANG II and (F) angiotensin receptor blocker binding to the receptor. (G) Interactions between receptor and Olmesartan were determined from the crystal structure. The critical interactions of Olmesartan are with Arg167, Trp84, Tyr35, and Lys199 residues of AT1 receptor. (H) Amino acids at positions 452 (Leu) and 478 (Thr) in spike RBD were replaced by arginines in the delta variant located at the nearest negatively charged residues (Glu22, Asp38, and Glu35) in ACE2. They also depict the bound bisartan (best bitetrazole binder A) molecule and the zinc atom.
2.4. Molecular binding calculations of BisA, B, C and D targeting the 3CL catalytic site of SARS-CoV-2 protease
Docking of BisA, B, C, and D to the 3CL SARS-CoV-2 main protease was performed. Their binding sites and binding energies were compared with those of known antiviral drugs and PF-07331332 Pfizer antiviral drug. The results showed that all bisartan analogs (BisA-BisD) showed stronger binding compared to PF-07321332 (Fig. 7), and BisA and C showed stronger binding compared to all antiviral drugs except Ombitasvir. In the case of BisA binding, a tetrazole interacts strongly with the His163 ring via pi-pi resonance (Fig. 8). The second tetrazole did not show strong interactions, however, the diphenyl groups showed significant hydrophobic interactions with fairly close residues, particularly Pro168 (Fig. 8). Although BisA did not appear to interact directly with Cys145, however, the distance between a tetrazole of BisA and the Cys145 residue is less than 4 A, suggesting a weak interaction.
Fig. 8.

Comparison of docking motifs of BisA and Pfizer’s PF-07321332 antiviral drug with the Cys145 catalytic site of the main 3CLpro protease of SARS-CoV-2. (A) Details of BisA-3CLpro residue interactions. Ligand Atom Colors: C = cyan; N = blue; O = red. Tetrazole#2 of BisA interacted strongly with the His163 ring via pi-pi resonance (red lines). Tetrazole#1 exhibited no strong interactions, however, the biphenyl groups showed significant hydrophobic interactions (green lines) with several nearby residues, particularly Pro168. BisA did not appear to undergo direct interactions with Cys145, although BisA tetrazole#2 was proximal (3.74 Å) to the sulfur atom (large yellow sphere) and therefore probably interacted electrostatically. (B) As was the case for BisA binding, no direct interactions were observed between PF-07321332 and Cys145. However, it is noted that the –CN nitrile group was located proximal to the Cys145 sulfur, which is a potential site of covalent interaction with the ligand. Ligand binding was also marked by a strong hydrogen bond between the nitrile N and the Ser144 –OH. (C) Superimposed view of bound BisA and PF-07321332. (D) Frame captures at 0, 26, 40 and 68 ns from independent MD simulations of the BisA-3CLpro complex and the PF-07321332-3CLpro complex (drugs bound in the Cys145 catalytic pocket). MD conditions for both runs were: 311°K, NVT ensemble, 0.9 wt% sodium chloride (physiological saline), AMBER14 force field parameters. Bound PF-07321332 was unstable and exited the catalytic pocket beginning at about 20 ns, with complete extraction by about 25 ns. In contrast, BisA remained stably bound in the pocket for the duration of the 70-ns MD run. (E) RMSD values for: (1) BisA-3CLpro (blue line) and (2) PF-07321332-3CLpro complexes as a function of MD simulation time out to approximately 70 ns.
2.5. Bisartan tetrazoles versus bisartan nitrile and PF-07321332
Bisartans tetrazole and PF-0732133 nitrile groups are the pharmacophoric warheads of the two inhibitors to disrupt the Cys CRS catalytic center of 3CLpro preventing spike protein cleavage and infection. Bisartans have been found in in silico studies to be binders to protease 3CLpro catalytic center and stable in MD simulations (Fig. 9).
Therefore to further test the functioning roles and compare tetrazole and nitrile the two principal structural factors in the two drugs that interact with Cys145 of the catalytic dyad, Cys145 - His41, we compared bisartan A tetrazole and bisartan A nitrile, where tetrazole was replaced by nitrile, by additional docking (Fig. 9). The docking results showed that bisartans tetrazole have higher binding energy compared to bisartan nitrile and bisartan nitrile has higher binding energy compared to PF-0732133 (Fig. 2C), which indicate an additional contributing role for the biphenyl group of bisartans in the inhibition furthermore to the tetrazole moiety.
2.6. The novel bisartans potently inhibit angiotensin II-mediated vasoconstriction in rabbit iliac arteries
To determine if the newly formulated bisartans mimic the effect of ARBs, iliac artery rings collected from rabbits were incubated with bisartan A (K+)2, bisartan A (Na+)2, and bisartan A (TFA)2. To evaluate the ability of these drugs to inhibit vascular smooth muscle cell constriction an ANG II dose–response assay was performed, and results were compared to untreated control rings. Vasoconstriction responses to cumulative doses of ANG II in iliac rings were potently inhibited by all bisartan A salts investigated (Table 1A). Vasoconstriction was potently inhibited by: bisartan A (K+)2 at doses [10−10.5 M] (bisartan A (K+)2: 0.850.49% vs. control: 12.79 1.88%, p = 0.0002) to [10−7.0 M] (bisartan A (K+)2: 4.522.49% vs. control: 18.181.63%, p < 0.0001); BV6(Na+)2 at doses [10−11.0 M] (bisartan A (Na+)2: 0.910.16% vs. control: 7.830.82%, p = 0.0493) to [10−6.0 M] (bisartan A (Na+)2: 0.140.41% vs. control: 8.440.66%, p = 0.0135); and bisartan A (TFA)2 at doses [10−11.0 M] (bisartan A (TFA)2: −0.150.32% vs. control, p = 0.0186) and [10−6.5 M] (BV6(Na+)2: 3.280.95% vs. control: 11.651.32%, p = 0.0126). Our results demonstrate that the novel bisartans may act as ARBs by exerting their effects through association and inhibition of AT1 receptor function with low drug concentrations. Fig. 2A depicts the inhibitory effect of the novel bisartan A salts, bisartan A (K+)2, bisartan A (Na+)2, and bisartan A (TFA)2 on ANG II-induced vasoconstriction responses in rabbit iliac arteries.
2.7. Angiotensin antipeptides – regulators of the renin-angiotensin system
The replacement of the C-terminal Phe residue of ANG II with an aliphatic amino acid (Ile, Leu, Val, Ala) converted the molecule to an analog with [insurmountable] inverse agonist properties. It was of interest to investigate if such a molecule could predicate a naturally occurring product encoded by the human genome, designed to counteract ANG II. The sequence of human mRNA, which is complementary to that for ANG II [5], was found to encode the ANG II analog EGVTVHPV, and likewise the ANG III analog GVTVHPV. The C-terminal Val residue conferred insurmountable antagonist properties to these “antipeptides”, whereas the absence of the Arg side chain at position 2 has only modest effects on activity. Accordingly, this angiotensin antipeptide was produced as a natural countermeasure to ANG II, by binding to the inverse agonist site (Fig. 1B). Interestingly, RNA sequences encoded peptides with opposite actions. ANG II has a fundamental role in the regulation of cardiovascular function and its actions are controlled at multiple levels, including gene expression and enzymatic biosynthesis of components of the RAS. This gives rise to ANG II metabolism by ACE2, resulting in the vasodilator peptide angiotensin 1–7, which associates with AT2 receptors, as well as signaling effects at AT1 receptors. The present findings add yet another potential level of control for physiological regulation of the RAS via the production of endogenous antipeptides acting directly as inverse agonists at AT1 receptors.
2.8. SARS-CoV-2 and angiotensin receptor blockers versus angiotensin-converting enzyme inhibitors
COVID-19 patients often present with compromised lung function due to severe pulmonary edema [15]. We have noted when conducting rat pressor assays that bolus injections of EC50 doses of ANG II, carried out at 15-minute intervals, resulted in severe pulmonary edema within hours, as evidenced by the appearance of fluid within the tracheal tube. This effect may be reversed by ARBs, but not by ACE1 inhibitors. Pulmonary edema seen in SARS-CoV-2 infection may respond to treatment with ARBs counteracting the toxic effects of ANG II. It is to be expected that molecules, which resemble angiotensin and/or contain certain structural features of angiotensin, should bind to both ANG receptors and ACE as both contain active sites, which evolved to accommodate the angiotensin molecule. In the case of ACE1, which is a zinc protease (dipeptidyl carboxypeptidase) responsible for converting inactive ANG I (DRVYHPF-XX) to the bioactive octapeptide ANG II, the overlap in binding activity may be less for ACE2, which is a zinc carboxypeptidase, that recognizes ANG II and converts it to angiotensin 1–7. Therefore, it seems likely that ARBs may be superior to ACE1 inhibitors at blocking ACE2. Thus, ARBs should bind to ACE2 and effectively inhibit binding of the SARS-CoV-2 spike protein, preventing cell entry of the virus and, consequently, viral infection. This effect has been demonstrated in recent clinical trials involving the ARB telmisartan [16], [17]. Other extensive studies have also shown the clinical effects and the therapeutic potential of RAS inhibitors and in particular of ARBs in hypertensive patients with COVID-19 [18], [19], [20], [21], [22], [23], [24], [25], [26], [27], [28].
2.9. Bisartans complex with AT1 receptor and ACE2
We have developed a new generation of ARBs, called bisartans [29], the structures of which include two tetrazole groups. As previously discussed, tyrosinate in sarilesin and carboxylate in EXP3174 were both effective at creating salt bridges with R167. We wanted to determine if tetrazolate, which is a functional mimetic of carboxylate, might also suffice. By using imidazole as the template for mounting two biphenyl tetrazole groups, we conjectured that the resulting positive charge on the imidazole may be the correct distance from the two tetrazoles to subsume the space normally occupied by the Arg2 guanidino group of ANG II when the peptide is bound to the receptor (Arg2 is predicted to interact with Asp263 and Asp281 of the receptor) [30]. Results determined that bisartans are potent insurmountable antagonists of AT1 receptors [31], and are the first ARBs discovered that do not use a carboxylate for binding to Arg167. Instead, bisartans use a second tetrazolate to form the critical salt bridge with Arg167, which appears to provide for insurmountable activity (Fig. 1B). This study complements results from previously published literature [31] by demonstrating the ability of bisartans to act as ARBs through interactions with the AT1 receptor by inhibiting constriction in response to ANG II. An ANG II dose–response was performed on iliac artery rings collected from male New Zealand White rabbits subsequent to incubation with the following bisartan A salts: BV6(K+)2, BV6(Na+)2, and BV6(TFA)2. Results from this study determined that bisartan A salts were able to potently abolish ANG II-induced vasoconstriction (Table 1A and Fig. 2A). Bisartans likely exhibit significant bioactivity at AT1 receptors because of tight binding to Arg167 due to the particular geometry of the double tetrazole unit surrounding the Arg167 guanidino group. Essentially, bisartan can enfold the Arg167 guanidino group in the tight embrace of two tetrazole moieties (Fig. 10). In addition to their properties as potent ARBs, bisartans are expected to be inhibitors of zinc proteases, such as ACE2. Zinc proteases are inhibited by chelating agents, which bind to the zinc atom at the active site of this class of enzymes. For example, ACE2 can be completely inhibited by the chelating agent 1,10-phenanthroline [31], which due to the juxtaposition of its two nitrogens, can have a bivalent interaction with zinc. Likewise, bisartans contain two powerful chelators in the form of anionic tetrazole groups, which can effectively embrace the zinc atom at the active site of zinc proteases like ACE2 in a quadrivalent interaction, as was observed in docking models for BisD in particular (see Fig. 5, Fig. 10 and Fig. 11). Inhibition of ACE2 enzyme activity would be expected to result in interference with the binding of SARS-cov2 spike protein to ACE2, and viral entry into cells. The two tetrazoles in bisartans are in the form of potassium salts, as also the tetrazole of commercial Losartan, after treatment with KBr. The two tetrazole protons should be taken by bromide to create the salt. Then tetrazole is negatively charged and traps positively charged groups as guanidino group of arginine or imidazole of histidine. The positive charge of imidazole appears since the second nitrogen lone pair of electrons is given to make a bond with the methylene biphenyl tetrazole, after bis alkylation with an alkylating reagent which is bromine methylene biphenyl tetrazole. The positive charge of imidazole is neutralized with the negative charge of bromine anion and can then imidazole complex with positive atoms as Zn2+.
Fig. 11.

(A) Binding of ARB to angiotensin receptor. The binding of ARB results in a strong salt bridge (hatched bond) with Arg167 (and Lys199), preventing binding of the receptor to the G protein (which in turn prevents receptor dimerization), and thereby locking the receptor in the inverted state (Fig. 1B). An alternative second messenger (possibly B-arrestin or a different G protein) binds to the intracellular domain of the receptor leading to inverse agonism (relaxation). Binding and activation of ANG II receptor. (B) The conformation of ANG II determined by various spectroscopic techniques is characterized by the presence of a CRS and clustering of the three aromatic rings, including a Phe:His ring:ring interaction. The CRS relays the negative charge at the C-terminus of ANG II to the Tyr hydroxylate, distributing the charge across the CRS, and steering the ANG II molecule to align with two corresponding positive charges (Arg167 and Lys199) on the receptor. (C) Salt bridge formation (hatched bond) with Arg167 and Lys199 enables an induced fit of key peptide and receptor-based groups, allowing Tyr35 to H-bond with the imidazole N of His6 of ANG II. This releases the salt bridge between Tyrosinate4 of ANG II and Arg167. Agonist activity may derive from the exchange of the hydroxylate of Tyr4 of ANG II with that of Tyr35 of the receptor, releasing the intramolecular charge relay interactions in ANG II and replacing it with an intermolecular interaction with the receptor. These dynamic interactions are transduced by a cooperative mechanism involving G protein binding and receptor dimerization leading to amplification (nH > 1, Table 1B) of the contractile response. Simultaneous interaction of the aromatic ring of Tyr35 with the rings of Phe8 and His6 of ANG II may be a critical factor in this receptor triggering process, perhaps by aligning Tyr35 for bonding to the imidazole N of ANG II [NOTE: an aromatic ring has a quadrupole moment which allows it to form a slipped parallel plate or perpendicular plate electrostatic interaction with another ring]. Accordingly, sarilesin, which lacks the necessary aromatic ring for electrostatic interaction with the Tyr35 ring, is unable to exchange its tyrosinate with that of Tyr35 and elicit the response. Instead, it maintains a strong salt bridge anchor with Arg167 rendering sarilesin insurmountable. In contrast, the nonpeptide losartan and the peptide analog sarmesin are surmountable antagonists because they cannot form this salt bridge and form a weaker ion:dipole bond with Arg167, as does the ANG II TyrOH when released by Tyr35. (D) Binding of bisartan to the active site zinc atom of ACE2 (E) and Arg167 of the AT1 receptor.
2.10. Severe acute respiratory syndrome coronavirus 2019 cleavage site 681–686 (PRRARS) and the role of arginines in the cleavage by furin
Inhibition of ACE2 enzyme activity may be due to allosteric interference with the binding of SARS-CoV-2 spike protein to ACE2, even though the proteolytic site is distant from the spike RBD, and therefore may only partially inhibit entry of the virus into the cell. There is also the possibility that bisartans could attach to the furin cleavage site PRRARS (residues 681–686 of spike protein), due to the preponderance of Arg residues (as per Fig. 10B), which gives rise to subunits S1 and S2. These subunits assist in the viral cell entry process, offering an additional potential mechanism for inhibiting the virus [32], [33]. A mutation at P681R in the cleavage site 681–686 (PRRARS) has been observed in the new fatally spreading delta variant globally, increasing the number of arginines (RRRARS) and further facilitating cleavage. Bisartans, through their negative tetrazolate groups, have an additional receptor-binding target, forming salt bridges and blocking cleavage and consequently infection. This interaction occurs between the two tetrazoles of bisartan A with the guanidino groups of Arginines P681R, R682, and R683 in the furin basic cleavage site (PRRARS) of the spike protein RBD (Fig. 10C). An increase of arginines in the furin cleavage site, mutation P681R, increases the basicity of the cleavage site which facilitates cleavage thus infectivity. Moreover, in silico global docking studies have shown that ARBs bind to the spike protein-ACE2 complex (Fig. 11A), and bisartans are an effective ARB in this regard (Fig. 2). Bisartans are anticipated to be effective inhibitors of viral infection as these novel drugs have multiple actions at the viral spike protein-ACE2 complex and may be able to treat the detrimental pulmonary edema effects of ANG II, resulting from SARS-CoV-2 infection. Bisartans have shown greater potency when compared to the ARB Losartan (−logIC50 = 8.25, pA2 = 8.25) in in vitro studies (i.e. rat uterus and human embryonic kidney cells), and may be an ideal potential therapy due to their low cost and efficient syntheses [3]. In particular, bisartan A displayed higher antagonistic activities (pA2 values) and binding affinities (elogIC50 values) in comparison to the ANG II AT1 receptor blocker losartan. The potassium (−logIC50 = 9.04), the sodium (−logIC50 = 8.54) salts of 4-butyl-N,N0-bis{[20-(2H-tetrazol-5-yl)biphenyl-4-yl]methyl} imidazolium bromide (bisartan A) as well as its free acid (−logIC50 = 9.46) demonstrated higher binding affinity for the AT1 receptor and higher antagonistic activity (potency) (Fig. 2). Bisartans contain most of the pharmacological segments of sartans and an additional biphenyl tetrazole moiety where tetrazole is superior to carboxylates of sartans [34].
The significant role of the bisartan two tetrazoles and ANG II receptor blockers containing tetrazole and a hydroxylate/carboxylate group is well depicted when comparing the binding affinities of bisartan A (ΔG = −16.21 kcal/mol) and Losartan (ΔG = −12.30 kcal/mol) [3]. Bisartans have an additional tetrazole which is superior to hydroxyl or carboxyl group in sartans resulting in higher affinity [3].
COVID-19 bioassay: The ability of bisartans to prevent SARS-CoV-2 infection in host cells was investigated. Bisartan D nitrile did not inhibit infection whereas bisartan D tetrazole demonstrated 20% inhibition compared to 95% inhibition by PF-07321332 (Fig. 15). Overall there are two approaches to inhibit the affinity of spike protein with ACE2 and subsequently to attenuate infection; one is by blocking the affinity of spike RBD with ACE2 and the second to prevent cleavage of spike protein by the furin enzyme. In both cases, arginine is the key amino acid that drives infection and therefore the arginine blockers as bisartans constitute a new class of potential drugs for the treatment of COVID-19. Bisartans through the tetrazolate were found to be the strongest binders with ACE2-RBD complex where an arginine mutant, either in RBD or cleavage cavity site, appears to be the critical amino acid for the infectivity of SARS-CoV-2. Dominating mutations in Alpha, Beta, Gamma, Delta variants have been identified with arginine mutant to be predominant [35], [36], [37], [38], [39].
Fig. 15.

COVID-19 assay comparing NTV (PF07321332), 2-BDN (BisD nitrile), 2-BV6 (BisD tetrazole). Vero cells infected with SARS-Cov-2 were treated with the different compounds. The cytopathic effect (CPE) was measured under an inverted microscope and expressed as a percentage to the CPE observed in untreated cells. The 2-BDN and 2-BV6 bisartans bear the butyl group at position 2 of the imidazole ring as in losartan. Details of the study are seen in the supplementary material.
3. Discussion
Burgeoning evidence from chemical reactivity, structure–activity, NMR, and fluorescence lifetime studies [13], [29], [40], [41] suggest that activation of AT1 receptors may be driven by the formation of a CRS in ANG II, encompassing TyrOH -- His -- carboxylate (analogous to serine proteases) [2], which generates tyrosinate anion species for activating the receptor (Fig. 10F). Alternatively, the C-terminal carboxylate could interact directly with the TyrOH of ANG II to create a tyrosinate anion. Notably, methylation of the Tyr hydroxyl group of the superagonist [Sar1]ANG II or of the inverse agonist sarilesin, results in reversible surmountable antagonists. Thus, implying that the Tyr hydroxyl has a critical role in the agonist and the inverse agonist states of the receptor. It is the amino acid side chain at position 8 that ultimately determines which receptor state is in play, with the Phe ring being essential for agonist activity. The N-terminal portion of ANG II is thought to have a largely supportive role in maintaining the CRS [41]. Accordingly, the role of Arg2 in ANG II is to help stabilize the CRS and mainly act as a chaperone of the C-terminal of the molecule [40], [41]. Its role is essentially replaced or supplanted by Arg167 of the receptor when ANG II binds to its receptor. The role of Asp at position 1 of ANG II is considered non-obligatory and possibly detrimental, as its replacement in [Sar1]ANG II results in a superagonist [29].
The first nonpeptide ARB to be discovered was the surmountable antagonist Losartan, which is metabolized to the [insurmountable] inverse agonist EXP3174 in the bloodstream (Fig. 10D and E). This transformation involves the conversion of an imidazole-based neutral hydroxymethyl group of Losartan to the carboxylate anion found in losartan carboxylate (Fig. 10F). Similar considerations apply to the carboxamide and carboxylate versions of Olmesartan, which are likewise a surmountable antagonist and an [insurmountable] inverse agonist respectively [42]. As outlined above, the peptide equivalents are the TyrOMe and TyrO− species in surmountable sarmesin and insurmountable sarilesin, respectively. Overlay of peptide and ARB structures clearly demonstrates the equivalence of tyrosinate and carboxylate functions, respectively [13].
SSM of Y35A of the AT1 receptor results in an inability to bind ANG II or ARBs, exposing a critical role for the phenolic side chain of Tyr35. Crystallography studies [42], [43] have elaborated aspects of the binding of the ARB inverse agonist Olmesartan to AT1 receptors, wherein an intermolecular CRS-like structure is formed with Tyr35 of the receptor (Fig. 10G). Although CRS formation in ANG II involves an intramolecular interaction, the parallel structural features of ARB and ANG II are unmistakable (Fig. 10E and F). Details of the crystal structure of ARB bound to the AT1 receptor demonstrate that ARB binding involves salt bridges between the carboxylate and tetrazole groups of ARBs and the Arg167 guanidino group [and Lys199] of the receptor (Fig. 9A) [42], [43], [44]. The salt bridge between Arg167 and carboxylate is solely responsible for the insurmountable effects of ARBs, as evident by surmountable losartan. Similarly, the peptide antagonist sarmesin cannot form this salt bridge but can form a weaker ion: dipole bond, same as losartan, and is therefore a surmountable antagonist. In contrast, the peptide sarilesin is able to form the necessary salt bridge between its tyrosinate anion and Arg167 and is therefore an insurmountable inverse agonist (Table 1B). Methylation of the TyrOH of sarilesin converts this peptide from insurmountable to surmountable antagonist, as expected.
The main role of the CRS in ANG II is the creation of two partial negative charges, TyrO− and CO2−, the spacing of which is reproduced by the two acid groups in insurmountable ARBs (Fig. 11B). This is the only way the superagonist [Sar1]ANG II, which contains a single naked anion, can achieve a two-point attachment to the receptor. These partial anion pairs in ANG II can form salt bridges with Arg167 and Lys199 of the receptor in a similar manner to ARBs (Fig. 10G and Fig. 11B and C). It follows that if Tyr35 is close enough to form a bond with the imidazole N of ARB (Fig. 8B and C), then it must also be in the right place to potentially interact with the imidazole N of His6 in ANG II (Fig. 11A, B, and C). Therefore, receptor activation may be driven by a charge relay exchange mechanism between peptide and receptor (Fig. 8A, B and C), wherein the Tyr4 of ANG II and the Tyr35 of the receptor can alternate within the CRS. Thus, the incursion of Tyr35 effectively neutralizes Tyr4 of ANG II by converting O− to OH. Thus, it can no longer form a salt bridge with Arg167 on the receptor, and can only form an ion:dipole bond. The receptor dynamic may be characterized by competition between the two tyrosines of receptor and ANG II. For agonist activity, the G protein-linked receptor dimerization process, which results in amplification of the contractile response, requires the presence of the Phe8 ring of ANG II, which is notably absent in ARBs. Accordingly, the triggering process may also require interaction of the Phe8 ring with the ring of Tyr35 of the receptor (Fig. 10G and Fig. 11B and C), an interaction that is not feasible for insurmountable Sarilesin. Clustering of the aromatic rings of ANG II has been observed by NMR [40], [41], which may be reproduced at the receptor, but with Tyr35 substituting for Tyr4 of ANG II (see Fig. 7 E and F for details). This interaction, which is based on the quadrupole moments of aromatic rings, may be reinforced by the preexisting Phe:His ring interaction in ANG II (see Fig. 8 B and C for details), and part of a concerted mechanism involving G protein binding/receptor dimerization [29] for activating AT1 receptor. In contrast, selectivity for AT2 receptor GPCR invokes [Y6]ANG II which is a negative regulator of AT1 receptor signaling [45].
Also, Trp84 of the receptor interacts with the imidazole of ARBs [42], [43] and by inference may bond with the His6 ring of ANG II, suggesting that there is ring stacking of Tyr35. In stark contrast to ANG II, the binding of ARB to the receptor is static (Fig. 11B and C) involving a strong salt bridge with Arg167, rendering it unable to stimulate G protein binding and receptor dimerization required for the contractile response. The same considerations apply to sarilesin because the electrostatic properties of aromatic rings prevent interaction of the hydrophobic Ile8 side chain of sarilesin with either His4 of ANG II or Tyr35 of the receptor, deeming these interactions necessary for agonist activity (Fig. 11A, B and C). Sarilesin and ARBs (e.g. EXP3174) form a stable salt bridge with Arg167 and act as insurmountable ligands/inverse agonists. Moreover, the inverted receptor state binds a second messenger, which invokes a relaxation response. The exact identity of this messenger is not known; however, possible candidates include B-arrestin, bradykinin receptor or a different G protein [42], [43].
Knowledge from previous studies have allowed the construction of this model for angiotensin action (Fig. 11B and C), which is entirely consistent with known facts, namely SSMs effects on activity and the crystal structure of AT1 bound to ARB [42], [43] show that (i) Tyr35 is essential for binding of ARBs and ANG II; (ii) Arg167 salt bridge anchor is essential for the insurmountable effects of ARBs; (iii) Lys199 strengthens binding of ARBs; (iv) Tyr35 phenolic group bonds to the imidazole N of ARBs; (v) Trp84 forms a ring:ring interaction with the imidazole of ARBs; and (vi) Trp253 bonds to the biphenyl group of ARBs.
The receptor triggering event may involve a cascade or ripple effect through the jellyfish-like tentacles of the 7 transmembrane helices of the receptor and may involve the salt bridging network Asn46-Asp74-Asn 295 located in the base of the binding domain [43]. Unraveling of the helix may be due to disaggregation of salt bridges, which maintain the helical structure, as has been seen by NMR for the conversion of helical peptide TFA salts to their acetate salts [40], [41]. This would occur simultaneously with G protein binding and receptor dimerization. Recent studies of sarilesin binding suggest that peptides, being longer molecules than ARBs, penetrate deeper into the membrane receptor binding pocket [30]. Although, the present findings show that these critical interactions providing for bioactivity must remain the same for peptides and ARBs.
The role of arginines in the basic furin cleavage site 681–686 (RRRARS) catalysing the Arg685-Ser686 cleavage which triggers infection is intriguing. It is very likely that arginines in the spike cleavage 681–686 site act as enzyme mimics, through their guanidinium ion, and catalyse the cleavage by furin at site 685–686 to S1 and S2 subunits triggering SARS-CoV-2 infection. This may occur through a mechanism similar to that proposed by List and MacMillan in their independent pioneer research where they discovered that proline or proline/ imidazole based small molecules catalyse through iminium- enamine anion intermediates the asymmetric carbon–carbon aldol condensation suggesting also proline simple aminoacid organocatalysis furthermore to known metal and enzyme catalysis [46], [47], [48]. Furthermore, the arginines in the basic cleavage site 681–686 of the spike are thought to have a supportive role in maintaining the CRS of furin, a serine protease, which cleaves the Arg685-Ser686 bond. However bisartans are less potent than PF-07321332 in COVID-19 bioassays (Fig. 15). Bisartans block the furin catalysis of spike protein which occurs through a CRS mechanism involving the triad Asp-His-Ser. Therefore, we thought to test if bisartans block as well 3CL protease which cleaves spike protein in a similar CRS mechanism through dyad His41-Cys145 which is the catalytic center of the protease. We compared our bisartans with Pfizer’s new oral drug PF-07321332 which blocks the spike cleavage through the nitrile group, the warhead of the drug which disrupts the cleavage mechanism. Bisartan A was found to bind to 3CLpro stronger than PF-07321332 and to be more stable (Fig. 7 and Fig. 8). Overall, bisartans block the spike cleavages through the three cell entries (ACE2, furin, 3CLpro) rendering them promising drugs for clinical trials.
General conclusions which can be drawn from MD and the docking studies are 1) that bisartans bind strongly to 3-CL protease active site, 2) bisartans have higher affinities than ligands containing nitrile, namely PF07321332 and bisartan- nitrile, 3) the tetrazole and nitrile “warheads” of these ligands appear to be close enough to Cys145 thiol to form a (transient) covalent, and 4) bisartans have the added desirable property of interfering with the ACE2/SPD interaction in addition to 3-CL protease. It is also possible that bisartans, due to their zwitterionic nature, do not readily permeate membranes, and demonstrate reduced COVID-19 bioassay inhibition due to poor cell permeation/access.
4. Conclusion
In SARS-CoV-2 infection, ARBs may prevent ANG II-induced pulmonary edema and may inhibit cell entry of the virus by binding to ACE2. Based on our modelling of receptor mechanisms of biased agonism, we have designed and synthesized a new generation of ARBs called bisartans bearing two tetrazoles. These novel drugs result in inhibition of both the AT1 receptor and ACE2 enzyme through association with their respective active sites, guanidino group and zinc atom, between two symmetrically arranged tetrazole groups. Furthermore, bisartans also bind directly to multiple loci on the spike protein of SARS-CoV-2, invoking additional potential mechanisms for viral inhibition. This study reveals a common mechanism for the binding of sartans with AT1 receptor and the spike/ACE2 complex in agreement with other studies reporting the ACE2 Zn2+ coordination with ANG II and mutants and in particular with aspartic acid, tyrosine, histidine, and C-terminal carboxylate of the peptide [6]. However in COVID-19 bioassays (Fig. 15), bisartan (Bis D) was not as effective as PF-07321332 (Paxlovid), and the utility of ARB bisartans in COVID-19 therapy may derive more from blockade of ANG II, inhibiting pulmonary edema and the release of inflammatory cytokines mediated by AT1 receptors on regulatory T cells, rather than from the viral infection process itself. Arginine seems to be the critical amino acid in the Delta variant which enhances affinity with ACE2, due to the strong positive charge shared by the three nitrogen atoms of the guanidino group [49], suggesting focusing on arginine blockers in the treatment of COVID-19 like bisartans. Furthermore, bisartans bearing two pharmacophoric tetrazole groups can block both furin cleavage at position P685-S686 of the basic cleavage site 681–686 (RRRARS) and hydrolysis of spike protein at glutamine positions by 3CL protease which triggers infection. In the second case one tetrazole of bisartan interacts with Cys145 of the catalytic dyad, His41-Cys145 of 3CL protease and disrupts the cysteine CRS mechanism of spike cleavage thus preventing infection. Docking studies show that tetrazole of bisartans appears to be superior to nitrile, the pharmacophoric warhead group of PF-07321332 (Pfizer’s antiviral drug), in its ability to disorder and put off the Cysteine CRS of 3CL protease, rendering bisartans as a novel new class of multifunctional antiviral compounds for treating COVID-19. Bisartans in silico studies can block the cleavage of spike protein through the three cell entries (ACE2, furin, 3CLpro) and are promising antiviral drugs for further investigation.
5. Methods
5.1. Synthesis of tetrazole bisartans A, B, C, and D
The synthesis of bisartan A was a two-step procedure: a) bis-alkylation of 4(5) butyl imidazole using alkylating reagent 4′-(bromomethyl)-[1,1′-biphenyl]-2-(trityl)tetrazol, b) removal of trityl chloride group with novel methods described earlier [3]. Bisartan B was synthesized by formylation of bisartan A at position 2 using 37% formalin and diisopropylethylamine in DMF at 80 °C for 1 h. In bisartans A and B, the butyl group is at position 4 which is favourable for better binding of this lipophilic group with the hydrophobic cleft site of the AT1 receptor. Bisartan BisC (bis alkylated Losartan), designed and included in the in silico study for docking comparion, is under preparation using methods previously described [3]. Its synthesis will be published elsewhere. Bisartan D was synthesized as BisA with bis alkylation of 2(5) butyl imidazole [3] (Fig. 12).
Fig. 13.

Interaction of bisartan negative tetrazoles with positive guanidino group of AT1 receptor (Arg167). 4-butyl imidazole bisartan (A) and 2-butyl imidazole bisartan, dialkylated bisartan (B) interacting with Arg167 of AT1 receptor.
Fig. 14.

CRS Protease mechanisms. SARS-CoV-2 spike protein can be cleaved by furin at positions 685–686 and 3CLpro at glutamine positions through CRS mechanisms. The catalytic center of furin is the triad Asp – His – Ser and for 3CL protease is the dyad Cys145 – His41. [James J. Neitzel, Enzyme Catalysis: The Serine Proteases, Nature Education 3(9):21 (2010)].
Fig. 12.

Bisartans are in the form of tetrazole potassium salts. (A) Two-step facile synthesis of bisartan A (B) Facile two-step high yield synthesis of bisartan A, B, and D. Bisartan BisC (bis alkylated Losartan), designed and included in the in silico study for docking comparison, is under preparation.
5.2. Synthesis of nitrile bisartans – general procedure for bi-alkylation
A solution of 4(5)-butylimidazole (0.8 mmol, 0.1 g) and alkylating reagent 4′-(bromomethyl)-[1,1′-biphenyl]-2-carbonitrile (1.69 mmol, 0,46 g) in 20 mL of acetonitrile was refluxed for 8 h and the reaction was monitored by RP-HPLC. After completion, acetonitrile was removed under reduced pressure and the oily residue was dissolved in 50 mL of dichloromethane. The organic solution was extracted with 20 mL of 1 M NaOH aqueous solution and, after separation, the organic layer was dried over anhydrous Na2SO4, filtered and dichloromethane was evaporated in vacuo. In the oily residue, diethylether was added (50 mL) and a pale yellow solid was precipitated. The solid was afforded after filtration, in high yield (75%) and purity (95%).
5.3. In silico evaluation studies
5.3.1. Docking calculations
Eight ARBs were built in 3D coordinates and their best most stable (lowest energy) conformation was detected by geometrical optimization of its structure in the gas phase, as implemented in the Spartan '14 Molecular Modeling program suite (Spartan '08 v.1.1.4, Wavefunction Inc). The structure of the molecule was initially optimized (via energy minimization) by conformational search using the Monte Carlo method with the MMFF94 17 molecular mechanics model, included in the Spartan '14 program suite. Geometry optimization (leading to the most stable conformer with the lowest energy) was accomplished via quantum-chemical calculations by utilizing the ab initio Hartree-Fock method with a 6- 31G* basis set. All ARBs were globally docked to the SAR-CoV-2-ACE2 complex (PDB 6LZG) using the open-source program AutoDock VINA [50], [51], using default parameters with point charges initially assigned according to the AMBER03 force field [52], and then damped to mimic the less polar Gasteiger charges used to optimize the AutoDock scoring function. Docking was performed using non-periodic (walled) boundaries that confined ligands to an approximately 8 to 10-Å buffer zone surrounding the receptor. The setup was implemented using the YASARA molecular modeling program [53]. The best hits and ligand conformational poses, expressed as kcal/mol free energy of binding, resulting from a minimum of 200 and a maximum of 500 runs were reported. UCSF Chimera and/or Yasara were used to visualize the molecules and the results of the docking and to construct the molecular models [54].
5.3.2. Molecular dynamics
MD simulations were run with YASARA [55]. The setup included an optimization of the hydrogen bonding network [56] to increase the solute stability, and a pKa prediction to fine-tune the protonation states of protein residues at the chosen pH of 7.4 [57]. NaCl ions were added with a physiological concentration of 0.9%, with an excess of either Na or Cl to neutralize the cell. After steepest descent and simulated annealing minimizations to remove clashes, the simulation was run for up to 120 ns using the AMBER14 force field [58] for the solute, GAFF2 [59], and AM1BCC [60] for ligands and TIP3P for water. The cutoff was 8 Å for Van der Waals forces (the default used by AMBER [61], no cutoff was applied to electrostatic forces (using the Mesh Ewald algorithm [62]). The equations of motion were integrated with multiple timesteps of 2.5 fs for bonded intermolecular interactions and 5.0 fs for non-bonded interactions at a temperature of 311°K and a pressure of 1 atm (NPT ensemble) using algorithms described in detail previously [63], [64].
5.3.3. SWISS-MODEL repository
Full-sequence experimental SARS-CoV-2 spike protein assembly structures are currently only available based on electron microscopy (EM) analysis. One drawback of such structures is that a number of residue segments are not well resolved or missing in the electron density maps. Among the omitted residue sequences for such EM structures is the 14-residue section (approximately) bracketed by residues 675 and 687. For this reason, EM-based structures could not be directly used for mutational screening and analysis in this region. This was especially problematic for the P681H variant. To resolve this issue we obtained from the SWISS-MODEL Repository a high-quality homo-trimeric homology model (SM#05; sequence similarity = 0.62) of the SARS-CoV-2 spike protein assembly built from an EM structure (PDB entry 6XR8) resolved at 2.90 Å. The “global model quality estimation” (GMQE) for this model was 0.7. The GMQE is expressed as a number between 0 and 1, reflecting the expected accuracy of a model built with that alignment and template.
5.3.4. Virtual ligand screening (docking) methods
Two independent methods were used to perform virtual ligand screening (VLS) targeting the receptor-binding domain of the ACE2 receptor (PDB 6LZG). These methods included (1) Internal Coordinate Mechanics (ICM) algorithms and associated software (Molsoft, LLC;) described by Abagyan et al. (1994; see http://www.molsoft.com/docking.html); and (2) AutoDock VINA [19] as implemented in the Yasara software suite [55].
ICM Docking: In the ICM approach five interaction potentials describe the receptor pocket subspace, including (i) van der Waals potential for a hydrogen atom probe; (ii) van der Waals potential for a heavy-atom probe (i.e., a generic carbon atom with 1.7 Å radius); (iii) an optimized electrostatic term; (iv) hydrophobic terms; and (v) a loan-pair-based potential accounting for directional preferences in hydrogen bonding. The energy terms are based on the ECEPP/3 force field [65], [66], an all-atom vacuum force field with appended terms for solvation free energy and entropic contributions. Conformational sampling is based on the biased probability Monte Carlo procedure [67] which randomly selects a conformation in the ICM grid space and then makes a step to a new random position independent of the previous one but according to a predefined continuous probability distribution. The ICM method invokes global optimization of a flexible ligand in the receptor field and combines large-scale random moves with conjugate-gradient local minimizations. Although Molsoft offers three independent algorithms for scoring ligand binding, the “mfscore” method (statistical potential of mean force; [68]) was used in the current study since results of this scoring method best correlated with: (i) docking scores generated using AutoDock VINA (see below) and (ii) RMSD and binding energy results from MD simulations of drug-receptor complexes.
VINA Docking: Docking of ligands to the ACE2 receptor was also performed using AutoDock VINA [51] with default parameters. Point charges and dihedral barriers were initially assigned according to the YAMBER14 force field [57]; however, YAMBER point charges were damped to mimic less polar Gasteiger charges used to optimize the AutoDock scoring function. Docking was performed using non-periodic (walled) boundaries that effectively confined ligands to an approximately 17 × 17 × 32 Å cuboid volume encompassing the Zn2+ domain (pocket) in the main channel of the ACE2 receptor molecule (PDB 6LZG). The setup was implemented using the YASARA molecular modeling program [53]. The best hits and ligand conformational poses, expressed as kcal/mol free energy of binding, resulting from a minimum of 100 runs per ligand were reported.
5.4. Pharmacological evaluation
5.4.1. Cell culture
Bisartans and sartans were evaluated in human embryonic kidney (HEK 293) cells which were grown in DMEM/F12 (1:1) containing 3.15 g/L glucose and 10% bovine calf serum at 37 °C and 5% CO2 (21). They were also evaluated in iliac arteries as described below.
5.4.2. In vitro rabbit blood vessel functional study
5.4.2.1. Animal model
Male New Zealand White rabbits (n = 4) at 7 weeks of age were purchased from Flinders City University (SA, AUS). Upon arrival, animals were housed individually at the Victoria University Werribee Campus Animal Facilities and allowed a 7-day acclimatization period. Food and water were supplied ad libitum, and animals were kept on a 12-hour day/night cycle and maintained at a constant temperature of 21 °C and relative humidity between 40 and 70%. Rabbits were kept until 16 weeks of age. All experimental procedures were approved by the Victoria University Animal Ethics Committee (VUAEC#17/013) and were conducted in accordance with the National Health and Medical Research Council ‘Australia Code of Practice for the Care and Use of Animals for Scientific Purposes’ (8th edition, 2013).
5.4.2.2. Sedation and anesthesia protocol
Animals were first sedated using a subcutaneous injection (0.25 mg/kg) of medetomidine at the scruff of the neck and then anaesthetized using isoflurane (4%). An incision was made at the lower abdomen and the subcutaneous tissues and muscles were dissected to expose the inferior vena cava. The inferior vena cava was perforated and exsanguination was allowed for 3 min or until loss of colour and dilation of the pupils was observed. A T-tube connected to a syringe was introduced distal to the aortic arch to allow for the flushing of the aorta and iliac arteries with cold oxygenated Krebs-Henseleit solution (Krebs). Both iliac arteries were retrieved from each animal and were cleaned of connective tissue and fat under a light microscope. Rings were dissected into 2–3 mm rings in preparation for isometric tension analysis.
5.4.2.3. Isometric tension analysis and bisartan incubations
We show they undergo stable in vitro binding to (and inactivation of) the AT1 receptor. The antihypertensive activity of bisartans was demonstrated using an in vitro rabbit iliac arterial model in which potent dose-responsive (10−6 to 10−11 M) inhibition of vasoconstriction was observed following exposure to Na+ or K+ salts of selected bisartans. Iliac artery rings were immediately and sequentially placed into adjacent organ baths (Zultek Engineering, VIC, AUS) filled with 5 mL of Krebs solution. To replicate a physiologically relevant environment, baths were maintained at 37 °C and continuously bubbled with 95% carbogen. After a 15-minute acclimatization period, rings were mounted between two metal organ hooks attached to force displacement transducers, stretched to 0.5 g, and equilibrated for 15 min. Rings were re-stretched, refreshed, and equilibrated for a further 15-minutes before drug incubations. Iliac artery rings were left to rest for 10 min (control; n = 4) or incubated with bisartan A (K+)2 (BV6(K+)2; n = 3), bisartan A (Na+) (BV6 Na+); n = 3) or bisartan A (TFA) (BV6(TFA); n = 3) [10−15.0 M] for 10 min. An ANG II (Cat#51480, Mimitopes, VIC, AUS) dose–response [10−12.0 M – 10−5.0 M] was performed to determine the ability of the novel bisartans to inhibit ANG II-mediated vasoconstriction. Following the completion of the ANG II dose–response, rings were washed, allowed to return to baseline tension, and were then constricted with KPSS [125 mM] to determine standard vasoconstriction abilities.
5.5. COVID-19 assay
COVID-19 assay for BisD (tetrazole), BisD (nitrile), and NTV (PF07321332) were carried out by methods previously described [69], [70]. Briefly, the cell culture assay was done as follows. The virus stock was prepared by infecting fully confluent Vero E6 cells in DMEM, 10% fetal bovine serum, with antibiotics, at 37 °C, 5% CO2. Infections were carried out in 96‐well plates, using SARS‐CoV‐2 (m.o.i. of 0.1) on Vero E6 cells. Cells were treated with different compounds, in a volume of 15 μl, per 150 μl of medium, for 48 h. Cell morphology was observed with phase contrast, in an inverted microscope, to record CPE.
5.6. Statistical analysis
GraphPad Prism (version 9) was utilized for statistical analysis of isometric tension data. The significant p-value was set at p < 0.05, and a two-way ANOVA followed by Sidak’s multiple comparisons post hoc test was performed to determine significance. All data are represented as meanSEM.
6. In Brief
This study describes the discovery and the multiple benefits as revealed by docking studies of a new class of sartans, “bisartans”, bearing symmetric anionic tetrazoles that bind more strongly to the SARS-CoV-2 RBD/ACE2 complex compared to other common sartans. Bisartans, in a similar mode as in the interaction of ARBs with AT1 receptor (Arg167), block the critical amino acid arginine in the furin cleavage site 681–686, which catalyzes the cleavage of the spike protein and triggers COVID-19. Bisartans also exhibit stronger binding to the 3CLpro main protease of SARS-CoV-2 and are stable in molecular dynamics simulations. Since bisartans act at three targets essential for viral infection and replication (i.e., ACE2, furin, 3CLpro) they are promising chemistries for clinical trials.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
Acknowledgments
G.J.M. would like to thank PepMetics for its support. J.M.M. would like to thank the General Secretariat for Research and Technology, the Patras Science Park, the Region of Western Greece (Research and Technology), and the pharmaceutical companies Eli Lilly Greece and Uni-Pharma for supporting his research in multiple sclerosis, hypertension, and COVID-19. A.Z., V.A., and L.K.G. would like to thank the Institute for Health and Sport, Victoria University, Melbourne, Australia for their support. V.A. and H.R. were supported by a Planetary Health Grant PH098 from Victoria University. V.A. would like to thank the Greek Orthodox Archdiocese of Australia Funds, The Thelma and Paul Constantinou Foundation, The Pappas Family, and the VU Vaccine appeal Funds, whose generous philanthropic support made possible the research of this paper. L.K.G. was supported by a Victoria University postgraduate scholarship. V.G.G. received support from the National Public Investment Program of the Ministry of Development and Investment/General Secretariat for Research and Technology, in the framework of the Flagship Initiative to address SARS-CoV-2 (2020ΣΕ01300001); the Welfare Foundation for Social & Cultural Sciences (KIKPE), Athens, Greece; H. Pappas donation; grants no. 775 (Hippo) and 3782 (PACOREL) from the Hellenic Foundation for Research and Innovation (HFRI); and NKUA-SARG grant 70/3/8916. I.K and V.G.G. are thankful to Bodosakis Foundation for funding the work in Biosafety level 3 facility. TM an IL were supported by Special Account for Research Grants (SARG), National Kapodistrian University of Athens (NKUA).
We thank Mr. Nikolaos Maniotis for typing and editing this manuscript.
Author contributions
Conceptualization and coordination, G.J.M. and J.M.M.; molecular dynamic calculations, H.R., T.M., I.L., and C.C.; experimental work and methodology, I.K., V.G.G., K.K., L.K.G., A.Z., and V.A.; writing—original draft preparation, G.J.M., J.M.M.; writing—review and editing, A.Z., V.A., ST, V.G.G. and J.M.M. All authors have read and agreed to the published version of the manuscript.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.csbj.2022.04.010.
Contributor Information
Harry Ridgway, Email: ridgway@vtc.net.
Graham J. Moore, Email: mooregj@shaw.ca.
Thomas Mavromoustakos, Email: tmavrom@chem.uoa.gr.
Konstantinos Kelaidonis, Email: k.kelaidonis@gmail.com.
Christos T. Chasapis, Email: cchasapis@upatras.gr.
Laura Kate Gadanec, Email: laura.gadanec@live.vu.edu.au.
Anthony Zulli, Email: anthony.zulli@vu.edu.au.
Vasso Apostolopoulos, Email: vasso.apostolopoulos@vu.edu.au.
Russell Petty, Email: r.petty@dundee.ac.uk.
Ioannis Karakasiliotis, Email: ioakarak@med.duth.gr.
Vassilis G. Gorgoulis, Email: vgorg@med.uoa.gr.
John M. Matsoukas, Email: imats@upatras.gr.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- 1.Guo J., Huang Z., Lin L., Lv J. Coronavirus disease 2019 (COVID-19) and cardiovascular disease: A viewpoint on the potential influence of angiotensin-converting enzyme inhibitors/angiotensin receptor blockers on onset and severity of severe acute respiratory syndrome coronavirus 2 infection. J Am Heart Assoc. 2020;9 doi: 10.1161/JAHA.120.016219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Blow D.M., Birktoft J.J., Hartley B.S. Role of a buried acid group in the mechanism of action of chymotrypsin. Nature. 1969;221:337–340. doi: 10.1038/221337a0. [DOI] [PubMed] [Google Scholar]
- 3.Agelis G., et al. Rational design, efficient syntheses and biological evaluation of N, N ′-symmetrically bis-substituted butylimidazole analogs as a new class of potent Angiotensin II receptor blockers. Eur J Med Chem. 2013;62:352–370. doi: 10.1016/j.ejmech.2012.12.044. [DOI] [PubMed] [Google Scholar]
- 4.Moore G.J., Matsoukas J.M. Angiotensin as a model for hormone – receptor interactions. Biosci Rep. 1985;5:407–416. doi: 10.1007/BF01116558. [DOI] [PubMed] [Google Scholar]
- 5.Moore G.J., Ganter R.C., Franklin K.J. Angiotensin ‘antipeptides’: (−)messenger RNA complementary to human angiotensin II (+)messenger RNA encodes an angiotensin receptor antagonist. Biochem Biophys Res Commun. 1989;160:1387–1391. doi: 10.1016/s0006-291x(89)80157-3. [DOI] [PubMed] [Google Scholar]
- 6.Fatouros PR, Roy U, Sur S. Modeling substrate coordination to Zn-bound angiotensin converting enzyme 2. bioRxiv, preprint (2021).
- 7.Berteotti A., et al. Predicting the reactivity of nitrile-carrying compounds with cysteine: A combined computational and experimental study. ACS Med Chem Lett. 2014;5:501–505. doi: 10.1021/ml400489b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ahmad B., Batool M., Ain Q.u., Kim M.S., Choi S. Exploring the binding mechanism of PF-07321332 SARS-CoV-2 protease inhibitor through molecular dynamics and binding free energy simulations. Int J Mol Sci. 2021;22:9124. doi: 10.3390/ijms22179124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ferreira J.C., Fadl S., Villanueva A.J., Rabeh W.M. Catalytic dyad residues His41 and Cys145 Impact the catalytic activity and overall conformational fold of the main SARS-CoV-2 protease 3-chymotrypsin-like protease. Front Chem. 2021;9 doi: 10.3389/fchem.2021.692168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Owen D.R., et al. An oral SARS-CoV-2 M pro inhibitor clinical candidate for the treatment of COVID-19. Science. 2021 doi: 10.1126/science.abl4784. [DOI] [PubMed] [Google Scholar]
- 11.Moore G.J. Methods for analyzing and interpreting cooperativity in dose-response curves—II. Partial agonists acting on muscarinic receptors in smooth muscle. Gen Pharmacol Vasc Syst. 1989;20:199–203. doi: 10.1016/0306-3623(89)90015-3. [DOI] [PubMed] [Google Scholar]
- 12.Scanlon M.N., Koziarz P., Moore G.J. The relationship between homotropic and heterotropic cooperativity for angiotensin receptors in smooth muscle. General Pharmacology: The Vascular System. 1990;21:59–65. doi: 10.1016/0306-3623(90)90596-e. [DOI] [PubMed] [Google Scholar]
- 13.Moore G.J., Smitht J.R., Baylis B.W., Matsoukas J.M. Design and pharmacology of peptide mimetics. Proc West Pharmacol Soc. 1995;33:91–141. doi: 10.1016/s1054-3589(08)60667-5. [DOI] [PubMed] [Google Scholar]
- 14.Turner R.J., Matsoukas J.M., Moore G.J. Tyrosinate fluorescence lifetimes for oxytocin and vasopressin in receptor-simulating environments: Relationship to biological activity and 1H-NMR data. Biochem Biophys Res Commun. 1990;171:996–1001. doi: 10.1016/0006-291x(90)90782-i. [DOI] [PubMed] [Google Scholar]
- 15.Cui X., et al. Pulmonary edema in COVID-19 patients: mechanisms and treatment potential. Front Pharmacol. 2021;12 doi: 10.3389/fphar.2021.664349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Duarte M., et al. Telmisartan for treatment of Covid-19 patients: An open multicenter randomized clinical trial. EClinicalMedicine. 2021;37 doi: 10.1016/j.eclinm.2021.100962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kakuta H., Sudoh K., Sasamata M., Yamagishi S. Telmisartan has the strongest binding affinity to angiotensin II type 1 receptor: comparison with other angiotensin II type 1 receptor blockers. Int J Clin Pharmacol Res. 2005;25:41–46. [PubMed] [Google Scholar]
- 18.Abassi Z.A., Skorecki K., Heyman S.N., Kinaneh S., Armaly Z. Covid-19 infection and mortality: a physiologist’s perspective enlightening clinical features and plausible interventional strategies. Am J Physiol-Lung Cell Mol Physiol. 2020;318:L1020–L1022. doi: 10.1152/ajplung.00097.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Abassi Z.A., Skorecki K., Heyman S.N., Kinaneh S., Armaly Z. Reply to Letter to the Editor: “Don’t judge too RAShly: the multifaceted role of the renin-angiotensin system and its therapeutic potential in COVID-19”. Am J Physiol-Lung Cell Mol Physiol. 2020;318:L1029–L1030. doi: 10.1152/ajplung.00133.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dambha-Miller H., et al. Currently prescribed drugs in the UK that could upregulate or downregulate ACE2 in COVID-19 disease: a systematic review. BMJ Open. 2020;10 doi: 10.1136/bmjopen-2020-040644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rico-Mesa J.S., White A., Anderson A.S. Outcomes in patients with COVID-19 infection taking ACEI/ARB. Curr Cardiol Reports. 2020;22 doi: 10.1007/s11886-020-01291-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shete A. Urgent need for evaluating agonists of angiotensin-(1–7)/Mas receptor axis for treating patients with COVID-19. Int J Infect Dis. 2020;96:348–351. doi: 10.1016/j.ijid.2020.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Meng J., et al. Renin-angiotensin system inhibitors improve the clinical outcomes of COVID-19 patients with hypertension. Emerg Microbes Infect. 2020;9:757–760. doi: 10.1080/22221751.2020.1746200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sriram K., Loomba R., Insel P.A. Targeting the renin−angiotensin signaling pathway in COVID-19: Unanswered questions, opportunities, and challenges. Proc Natl Acad Sci. 2020;117:29274–29282. doi: 10.1073/pnas.2009875117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Warner Fiona J., Rajapaksha H., Shackel N., Herath C.B. ACE2: from protection of liver disease to propagation of COVID-19. Clin Sci. 2020;134:3137–3158. doi: 10.1042/CS20201268. [DOI] [PubMed] [Google Scholar]
- 26.Ni W., et al. Role of angiotensin-converting enzyme 2 (ACE2) in COVID-19. Crit Care. 2020;24 doi: 10.1186/s13054-020-03120-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang P., et al. Association of inpatient use of angiotensin-converting enzyme inhibitors and angiotensin II receptor blockers with mortality among patients with hypertension hospitalized with COVID-19. Circ Res. 2020;126:1671–1681. doi: 10.1161/CIRCRESAHA.120.317134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vaduganathan M., Vardeny O., Michel T., McMurray J.J.V., Pfeffer M.A., Solomon S.D. Renin–angiotensin–aldosterone system inhibitors in patients with covid-19. N Engl J Med. 2020;382:1653–1659. doi: 10.1056/NEJMsr2005760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Moore G.J., et al. Receptor interactions of angiotensin II and angiotensin receptor blockers—relevance to COVID-19. Biomolecules. 2021;11:979. doi: 10.3390/biom11070979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wingler L.M., McMahon C., Staus D.P., Lefkowitz R.J., Kruse A.C. Distinctive activation mechanism for angiotensin receptor revealed by a synthetic nanobody. Cell. 2019;176:479–490.e412. doi: 10.1016/j.cell.2018.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Koziarz P., Moore G.J. Inhibition of enzymatic degradation of angiotensin II in membrane binding assays: utility of 1,10-phenanthroline. Biochem Cell Biol. 1990;68:218–220. doi: 10.1139/o90-028. [DOI] [PubMed] [Google Scholar]
- 32.Lan J., et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature. 2020;581:215–220. doi: 10.1038/s41586-020-2180-5. [DOI] [PubMed] [Google Scholar]
- 33.Bhattarai A., Pawnikar S., Miao Y. Mechanism of ligand recognition by human ACE2 receptor. J Phys Chem Lett. 2021;12:4814–4822. doi: 10.1021/acs.jpclett.1c01064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Agelis G., et al. Interactions of the potent synthetic AT1 antagonist analog BV6 with membrane bilayers and mesoporous silicate matrices. Biochim Biophys Acta (BBA) - Biomembr. 2013;1828:1846–1855. doi: 10.1016/j.bbamem.2013.03.009. [DOI] [PubMed] [Google Scholar]
- 35.Cherian S., et al. SARS-CoV-2 spike mutations, L452R, T478K, E484Q and P681R, in the second wave of COVID-19 in Maharashtra, India. Microorganisms. 2021;9:1542. doi: 10.3390/microorganisms9071542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu Y, et al. Delta spike P681R mutation enhances SARS-CoV-2 fitness over Alpha variant. bioRxiv, preprint (2021). [DOI] [PMC free article] [PubMed]
- 37.McCallum M., et al. SARS-CoV-2 immune evasion by the B.1.427/B.1.429 variant of concern. Science. 2021;373:648–654. doi: 10.1126/science.abi7994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lemmin T., Kalbermatter D., Harder D., Plattet P., Fotiadis D. Structures and dynamics of the novel S1/S2 protease cleavage site loop of the SARS-CoV-2 spike glycoprotein. J Struct Biol: X. 2020;4 doi: 10.1016/j.yjsbx.2020.100038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Peacock T.P., et al. The furin cleavage site in the SARS-CoV-2 spike protein is required for transmission in ferrets. Nat Microbiol. 2021;6:899–909. doi: 10.1038/s41564-021-00908-w. [DOI] [PubMed] [Google Scholar]
- 40.Matsoukas J.M., Bigam G., Zhou N., Moore G.J.I. 1H-NMR studies of [Sar1]angiotensin II conformation by nuclear Overhauser effect spectroscopy in the rotating frame (ROESY): Clustering of the aromatic rings in dimethylsulfoxide. Peptides. 1990;11:359–366. doi: 10.1016/0196-9781(90)90093-k. [DOI] [PubMed] [Google Scholar]
- 41.Matsoukas J.M., et al. Role of the NH2-terminal domain of angiotensin II (ANG II) and [Sar1]angiotensin II on conformation and activity. NMR evidence for aromatic ring clustering and peptide backbone folding compared with [des-1,2,3]angiotensin II. J Biol Chem. 1994;269:5303–5312. [PubMed] [Google Scholar]
- 42.Zhang H., et al. Structural basis for ligand recognition and functional selectivity at angiotensin receptor. J Biol Chem. 2015;290:29127–29139. doi: 10.1074/jbc.M115.689000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Takezako T., Unal H., Karnik S.S., Node K. Current topics in angiotensin II type 1 receptor research: Focus on inverse agonism, receptor dimerization and biased agonism. Pharmacol Res. 2017;123:40–50. doi: 10.1016/j.phrs.2017.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang H., et al. Structure of the angiotensin receptor revealed by serial femtosecond crystallography. Cell. 2015;161:833–844. doi: 10.1016/j.cell.2015.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Magnani F., et al. Electronic sculpting of ligand-GPCR subtype selectivity: the case of angiotensin II. ACS Chem Biol. 2014;9:1420–1425. doi: 10.1021/cb500063y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ahrendt K.A., Borths C.J., MacMillan D.W.C. New strategies for organic catalysis: the first highly enantioselective organocatalytic Diels−Alder reaction. J Am Chem Soc. 2000;122:4243–4244. [Google Scholar]
- 47.List B., Lerner R.A., Barbas C.F. Proline-catalyzed direct asymmetric aldol reactions. J Am Chem Soc. 2000;122:2395–2396. [Google Scholar]
- 48.List B., Pojarliev P., Biller W.T., Martin H.J. The proline-catalyzed direct asymmetric three-component Mannich reaction: scope, optimization, and application to the highly enantioselective synthesis of 1,2-amino alcohols. J Am Chem Soc. 2002;124:827–833. doi: 10.1021/ja0174231. [DOI] [PubMed] [Google Scholar]
- 49.Courvoisier E., Williams P.A., Lim G.K., Hughes C.E., Harris K.D.M. The crystal structure of l-arginine. Chem Commun. 2012;48:2761. doi: 10.1039/c2cc17203h. [DOI] [PubMed] [Google Scholar]
- 50.Pettersen E.F., et al. UCSF Chimera?A visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 51.Trott O., Olson A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2009 doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Duan Y., et al. A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J Comput Chem. 2003;24:1999–2012. doi: 10.1002/jcc.10349. [DOI] [PubMed] [Google Scholar]
- 53.Krieger E., Koraimann G., Vriend G. Increasing the precision of comparative models with YASARA NOVA-a self-parameterizing force field. Proteins Struct Funct Bioinf. 2002;47:393–402. doi: 10.1002/prot.10104. [DOI] [PubMed] [Google Scholar]
- 54.Bertoni M., Kiefer F., Biasini M., Bordoli L., Schwede T. Modeling protein quaternary structure of homo- and hetero-oligomers beyond binary interactions by homology. Sci Rep. 2017;7 doi: 10.1038/s41598-017-09654-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Krieger E., Vriend G. YASARA View—molecular graphics for all devices—from smartphones to workstations. Bioinformatics. 2014;30:2981–2982. doi: 10.1093/bioinformatics/btu426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Krieger E, Dunbrack RL, Hooft RWW, Krieger B. Assignment of protonation states in proteins and ligands: combining pKa prediction with hydrogen bonding network optimization. 819, 405-421 (2012). [DOI] [PubMed]
- 57.Krieger E., Nielsen J.E., Spronk C.A.E.M., Vriend G. Fast empirical pKa prediction by Ewald summation. J Mol Graph Model. 2006;25:481–486. doi: 10.1016/j.jmgm.2006.02.009. [DOI] [PubMed] [Google Scholar]
- 58.Maier J.A., Martinez C., Kasavajhala K., Wickstrom L., Hauser K.E., Simmerling C. ff14SB: improving the accuracy of protein side chain and backbone parameters from ff99SB. J Chem Theory Comput. 2015;11:3696–3713. doi: 10.1021/acs.jctc.5b00255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang J., Wolf R.M., Caldwell J.W., Kollman P.A., Case D.A. Development and testing of a general amber force field. J Comput Chem. 2004;25:1157–1174. doi: 10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]
- 60.Jakalian A., Jack D.B., Bayly C.I. Fast, efficient generation of high-quality atomic charges. AM1-BCC model: II. Parameterization and validation. J Comput Chem. 2002;23:1623–1641. doi: 10.1002/jcc.10128. [DOI] [PubMed] [Google Scholar]
- 61.Hornak V., Abel R., Okur A., Strockbine B., Roitberg A., Simmerling C. Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins Struct Funct Bioinf. 2006;65:712–725. doi: 10.1002/prot.21123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Essmann U, Perera L, Berkowitz ML, Darden T, Lee H, Pedersen LG. A smooth particle mesh Ewald method. J Chem Phys 1995; 103:8577-8593.
- 63.Krieger E., Vriend G. New ways to boost molecular dynamics simulations. J Comput Chem. 2015;36:996–1007. doi: 10.1002/jcc.23899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wang Y., Liu M., Gao J. Enhanced receptor binding of SARS-CoV-2 through networks of hydrogen-bonding and hydrophobic interactions. Proc Natl Acad Sci. 2020;117:13967–13974. doi: 10.1073/pnas.2008209117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Arnautova Y.A., Jagielska A., Scheraga H.A. A new force field (ECEPP-05) for peptides, proteins, and organic molecules. J Phys Chem B. 2006;110:5025–5044. doi: 10.1021/jp054994x. [DOI] [PubMed] [Google Scholar]
- 66.Arnautova Y.A., Vorobjev Y.N., Vila J.A., Scheraga H.A. Identifying native-like protein structures with scoring functions based on all-atom ECEPP force fields, implicit solvent models and structure relaxation. Proteins Struct Funct Bioinf. 2009;77:38–51. doi: 10.1002/prot.22414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Abagyan R., Totrov M. Biased probability Monte Carlo conformational searches and electrostatic calculations for peptides and proteins. J Mol Biol. 1994;235:983–1002. doi: 10.1006/jmbi.1994.1052. [DOI] [PubMed] [Google Scholar]
- 68.Muegge I. PMF scoring revisited. J Med Chem. 2005;49:5895–5902. doi: 10.1021/jm050038s. [DOI] [PubMed] [Google Scholar]
- 69.Evangelou K., et al. Pulmonary infection by SARS-CoV-2 induces senescence accompanied by an inflammatory phenotype in severe COVID-19: possible implications for viral mutagenesis. Eur Respir J. 2022:2102951. doi: 10.1183/13993003.02951-2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Panagiotopoulos A., et al. p-cymene impairs SARS-CoV-2 and Influenza A (H1N1) viral replication: In silico predicted interaction with SARS-CoV-2 nucleocapsid protein and H1N1 nucleoprotein. Pharmacol Res Perspect. 2021;9 doi: 10.1002/prp2.798. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.